

Abstract
In this article, we broadly review the application of cfDNA analysis to the diagnosis and management of lymphoma. We introduce the advantages of cfDNA measurement over conventional tissue biopsy and describe how cfDNA may be utilized for both genotyping and detection of minimal residual disease. First, we discuss genotyping, beginning with differences in identifying mutations from the blood plasma vs. from circulating cells. We review the technical distinctions between PCR- and NGS-based assays and describe two important applications of NGS-based cfDNA tests, namely the identification of resistance mutations and classification of disease subtype. We discuss difficulties in genotyping diseases with low burden of tumor cells and the application of cfDNA assays in these contexts. Second, we describe the utility of ctDNA measurement in assessing MRD. We cover recent advances in the assessment of pre-treatment disease burden as a prognostic biomarker, detection of molecular response to therapy, and early detection of relapsing disease. Third, we explore select emerging areas of research in ctDNA technologies that show promise in boosting the performance of existing ctDNA-based assays. These include cell-free DNA fragment structure analysis or ‘fragmentomics’, epigenetic modifications, and novel circulating analytes such as tumor-educated platelets and extracellular vesicular DNA. We also discuss alternative analytes to blood plasma for tumor detection, such as urine, saliva, and stool. Finally, we present a case that highlights potential applications of ctDNA approaches to the management of patients with lymphoma, while also defining important prerequisite advances before this can be fully realized. We close with a look to the future of cfDNA applications, outlining one potential timeline and path forward towards routine clinical application.
Keywords: (6), Precision medicine, Cell-free DNA, Lymphoma, Liquid biopsy, Minimal Residual Disease, Circulating tumor DNA
Introduction:
‘Precision Medicine’ requires detailed knowledge of the molecular profile of a patient’s lymphoma. Currently, this information is typically obtained from a tissue biopsy; however, invasive biopsies have significant limitations. Tissue biopsies carry procedural risks and cannot account for spatial inter- and intra-tumor heterogeneity (1, 2) due to sampling from only one location in a single tumor lesion. ‘Liquid biopsies’ – an emerging class of methods to detect and characterize tumors through a blood draw – can potentially improve on these limitations. Analysis of circulating tumor DNA, or ctDNA, is the leading liquid biopsy approach in lymphomas, allowing for assessment of tumor-derived DNA through analysis of cell-free DNA (cfDNA) from a typical blood plasma collection. Liquid biopsy via ctDNA can capture the average of a patient’s tumor mass at any location, including its clonal architecture. Furthermore, given procedural risks and small residual lymphoma masses after therapy, re-biopsy can be challenging. Hence, a reliance on tissue biopsy often fails to reflect temporal heterogeneity and the emergence of treatment-resistant clones (3). Considering this, there is a strong case for liquid biopsies as a tool to characterize lymphoma on a molecular basis (i.e., genotype) before and during treatment.
The identification of specific mutations and molecular subtypes is a key feature of a liquid biopsy; however, it is only one aspect of ctDNA analysis. The ability of ctDNA to quantitatively assess disease burden has significant potential clinical utility, not only at the time of diagnosis, but also during and after therapy. Indeed, detection of minimal residual disease (MRD) from peripheral blood has become a powerful tool in many hematologic malignancies, including chronic myelogenous leukemia (CML) and chronic lymphocytic leukemia (CLL) (4, 5). As most lymphomas do not present with circulating lymphoma cells, few patients would harbor circulating tumor cells (CTC) in high enough levels for MRD detection from peripheral blood. This makes disease quantification from plasma-derived cfDNA attractive in many lymphoma subtypes, with significant clinical data already published in diffuse large B-cell lymphoma (DLBCL), classical Hodgkin lymphoma (cHL), mantle cell lymphoma (MCL), and peripheral T-cell lymphoma (PTCL) (6–14).
Here, we will assess the current state-of-the-art methods for both disease characterization and quantification from ctDNA-based liquid biopsies. We will explore emerging methodologies aiming to extend the utility of liquid biopsies in diverse malignancies including lymphomas. Finally, we present a real-world clinical case highlighting how ctDNA analysis can move forward and impact patient care in the clinic today and in the near future.
1. Lymphoma Genotyping
There are two ways in which genetic information from lymphoma tissue can be detected in the blood: analysis of CTCs or circulating cfDNA. The analysis of CTCs is only possible in some lymphomas where circulating tumor load exists, such as MCL, follicular lymphoma (FL), marginal zone lymphoma, small lymphocytic lymphoma, and a subset of Burkitt lymphoma. In contrast, DLBCL (15) and cHL do not typically harbor CTCs. However, cfDNA regularly contains low levels of detectable lymphoma-derived ctDNA (16). Therefore, ctDNA-based liquid biopsy has emerged as a platform to genotype diverse lymphomas.
Two main types of assays exist to genotype ctDNA: polymerase chain reaction (PCR)-based assays, often using droplet digital PCR (ddPCR), and next-generation sequencing (NGS)-based assays. Although (dd)PCR-based assays are cheap, relatively easy to perform, and have a short turnaround time, they can only genotype recurrent, truncal mutation hotspots. These assays quickly lose their advantages if multiple, non-recurrent loci of interest exist. In contrast, NGS-based assays can target hundreds of genes representing a target region also known as a selector. This selector is designed based on prior knowledge of a specific lymphoma’s common genetic aberrations, enabling NGS-based assays to identify most somatic genetic changes that define a lymphoma including single nucleotide variants (SNV), insertions/deletions, and structural variants (9, 10, 17–19). Despite the often high amount of total plasma cfDNA in patients with lymphoma (16), the proportion of tumor-derived ctDNA can be low. This highlights the importance of high-sensitivity assays with suppression of technical (e.g., PCR-induced) background errors (20) and simultaneous sequencing of germline DNA. These methods enable differentiation of true, tumor-derived changes from constitutional variants or clonal hematopoiesis of indeterminate potential (CHIP) (21, 22).
To judge the quality of genotyping from cfDNA, mutations identified by liquid biopsy and tissue genotyping have been compared. Several studies have estimated the concordance of tissue and liquid biopsy-based genotyping in lymphoma to be greater than 80%. (10, 23, 24). Interestingly, studies usually identify variants from liquid biopsies that were not identified in tumor tissue. At least one study performed in-depth validation of variants identified in liquid biopsies from HL using nested PCR amplification and Sanger sequencing, thereby validating all variants (25). This highlights the ability of liquid biopsy-based assays to identify sub-clones that might be missed in a single-site tumor biopsy.
An example for potential clinical use of liquid biopsy-based genotyping in DLBCL is the recent finding by Wilson et al. that co-occurring mutations in MYD88 and CD79B can predict response to Ibrutinib (26). Considering the overall low response rates in DLBCL to Ibrutinib (26), such a predictive molecular profile is relevant for patient and therapy selection. Identifying such targets using liquid biopsies will not only be useful for selecting treatment options, but also for the identification of resistance mutations by repetitive genotyping. A small series of FL patients has already shown that this is feasible, where Scherer et al. (2) identified emergent resistance mutations to Ibrutinib noninvasively from cfDNA.
Genotyping lymphomas using liquid biopsies can also classify disease to individualize treatment. In DLBCL, this includes classification of the activated B-cell-like (ABC) and germinal center B-cell-like (GCB) cell-of-origin (COO) groups, originally developed based on gene expression profiling (GEP) (27). Other tissue-based methods of COO classification have been developed, utilizing immunohistochemistry (Hans algorithm) (28) or gene-expression profiling of only a few targets with high discriminative power (29). COO classification based on the identification of mutations using liquid biopsies has recently been tested in one study with an 80% concordance rate with the Hans algorithm (9). More recently, two novel classifications based on the presence or absence of mutations and structural variations have been proposed by Schmitz et al. (30) and Chapuy et al. (31). While the clinical relevance of these classifications is not yet established, it is encouraging that at least one study was able to categorize DLBCL into the classification proposed by Schmitz et al. (30) using only liquid biopsy-derived information with high concordance (32).
Genotyping Lymphoma with low tumor cell burden
Hodgkin Lymphoma
Genotyping of HL using tissue-based methods is difficult due to the low tumor cell content – often only 1% – of the lymphoma tissue. Previous studies have used laser microdissection (33) and flow sorting (34) of the malignant Hodgkin-Reed-Sternberg (HRS) cells to molecularly profile HL, but these technically challenging methods have resulted in less than 100 HL genomes being profiled to date. Recently, two studies developed targeted NGS assays to genotype HL from liquid biopsies (24, 25). Both studies demonstrated the feasibility of liquid biopsy genotyping in HL and identified similar recurrently mutated genes, such as STAT6, GNA13, ITPKB, SOCS1 and TNFAIP3 (24, 25).
Primary Central Nervous System Lymphoma and Vitreoretinal Lymphoma
Genotyping of PCNSL using tissue biopsies has the disadvantage of requiring an often challenging stereotactic biopsy. In addition to blood, cerebrospinal fluid (CSF) and vitreous fluid are compartments that are easier to access than brain parenchyma and might be enriched for ctDNA. In vitreoretinal lymphoma (VRL), direct biopsy of the vitreous fluid typically allows for cytology and flow cytometry that aid in the diagnosis. However, molecular analysis of vitreous fluid could also detect cfDNA that gives diagnostic information by confirming a disease-specific genotype. PCNSL and VRL are defined by the pathognomonic, highly recurrent MYD88 L265P mutation occurring in up to 85% of patients (35, 36). As this mutation is exceedingly rare in non-lymphoma central nervous system cancers, it can be used to differentiate lymphoma from non-lymphoma lesions. Through targeted sequencing or (dd)PCR assays of plasma, CSF, or vitreous humor cfDNA, multiple studies have identified this and other mutations previously described in tissue biopsies from PCNSL or VRL (37–40). However, the cohort sizes in existing studies are still limited, and assays are not yet sufficiently standardized (Fig. 1) to judge the value of liquid biopsy-based genotyping in PCNSL and VRL.
Figure 1. Circulating Tumor DNA Applications in Lymphoma.
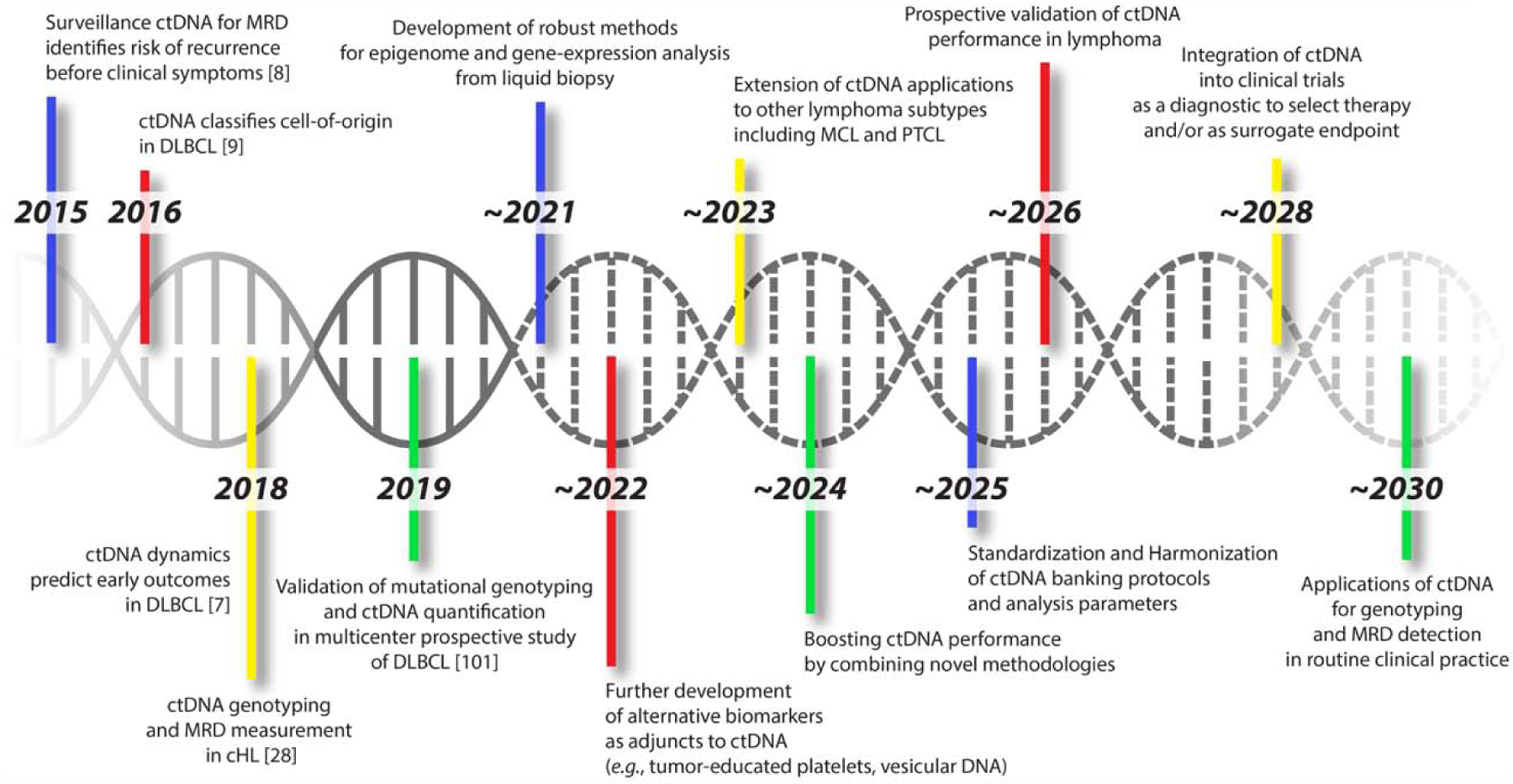
This figure shows a timeline of select past, present, and expected key milestones in liquid biopsies as applied to lymphoma. We show seminal studies marking currently achieved milestones, as well as attempt to identify one potential roadmap to the routine application of ctDNA in clinical practice.
2. Disease burden and minimal residual disease monitoring
2.1. Assessment of pre-treatment disease burden as a prognostic biomarker
Pre-treatment burden of disease is an established risk-factor in lymphomas, where larger volume disease portends worse outcomes. Multiple prior risk factors are related to tumor burden, including the International Prognostic Index (IPI) (41) and more directly, total metabolic tumor volume (TMTV) from PET/CT scans (42). In addition to mutational genotyping and molecular characterization, the ability to quantitatively measure disease burden is a key advantage of ctDNA-based liquid biopsies. While bulk cfDNA – which contains DNA derived from both tumor cells and healthy cells – has been proposed as a biomarker in DLBCL (43), its detection is not sufficiently specific to detect MRD and low burden disease, as all patients have some amount of cfDNA in their blood plasma. Thus, quantitation of tumor-specific ctDNA using sequencing or PCR-based approaches outlined above has become the mainstay of disease quantification from liquid biopsies. Accordingly, pre-treatment levels of ctDNA in DLBCL measured by Cancer Personalized Profiling by Deep Sequencing (CAPP-Seq) are significantly correlated with TMTV and IPI (7). Similar correlations between ctDNA and TMTV also exist with alternative amplicon-based approaches (44). Importantly, quantitative ctDNA levels are also predictive of outcomes including event-free survival (EFS) (7). Pre-treatment ctDNA levels have clinical utility in other lymphomas, including correlations with TMTV in classical Hodgkin lymphoma (14) and the ability to help differentiate between lymphoma subtypes, such as indolent and transformed follicular lymphoma (9, 45).
2.2. Detection of molecular response to therapy
In addition to pretreatment levels, quantitative assessment of therapeutic response from ctDNA can be highly prognostic in diverse lymphoma subtypes. The concept of detecting a target threshold or drop in MRD levels by a given landmark – a so-called ‘molecular response’ – was first described in CML (46). Accordingly, multiple studies have sought to establish a ‘molecular response’ in ctDNA-based MRD to predict outcomes in lymphoma. Similar to pre-treatment ctDNA levels, most of the data supporting this approach come from DLBCL.
An initial approach in DLBCL utilized ctDNA detection by immunoglobulin high throughput sequencing (IgHTS) in 126 patients with untreated DLBCL receiving dose-adjusted EPOCH +/− Rituximab therapy (8). Here, detection of ctDNA prior to cycle 3 of therapy identified a group with inferior time-to-progression. Similarly, in a study of over 200 subjects, Kurtz et al. demonstrated superior EFS in patients who have at least a 2-log reduction in ctDNA after one cycle of therapy compared to pre-treatment levels. (7). Interestingly, this 2-log reduction in ctDNA after 1 cycle of therapy was prognostic in both front-line and salvage therapeutic regimens. This landmark reduction in ctDNA was therefore termed an Early Molecular Response (EMR). Similarly, a 2.5-log reduction in ctDNA after 2 cycles of therapy was also found to be highly prognostic and was termed a Major Molecular Response (MMR). Notably, both EMR and MMR had higher prognostic value than pre-treatment ctDNA levels. Furthermore, molecular response by ctDNA remained prognostic for both EFS and overall survival (OS) even when controlling for other relevant factors in multivariate analysis, including the IPI, cell-of-origin, and interim PET/CT scans. Given the prognostic utility of interim PET/CT scans in DLBCL, albeit with suboptimal utility to select therapy for patients (47), the independence of ctDNA molecular response suggests significant value as a prognostic biomarker alone and potentially, in combination with these scans (48).
While most of the evidence for ctDNA-based molecular response assessment in lymphomas comes from DLBCL, similar approaches have been examined in other lymphomas. Indeed, a similar 2-log threshold for decrease in ctDNA was found to be prognostic in 24 cHL patients receiving ABVD therapy (49). Both interim and end-of-therapy ctDNA detection by IgHTS were found to be prognostic for outcomes in MCL patients undergoing first-line therapy (11).
2.3. Early detection of relapse
In addition to monitoring treatment response, ctDNA has utility for early detection of relapsing disease. While many patients with aggressive lymphomas are treated with curative intent, a significant fraction will experience relapse. In some centers, surveillance strategies with radiographic imaging (i.e., CT or PET/CT scans) are used; however, their utility is controversial (50) due to suboptimal specificity (47, 51) and radiation exposure. In contrast, ctDNA-based methods have high specificity due to their reliance on tumor-specific DNA sequences (6, 8, 9), making them attractive candidates for disease surveillance.
In two independent proof-of-concept studies, monitoring DLBCL patients in remission with IgHTS from ctDNA demonstrated a ~3–3.5 month ‘lead-time’ for recurrent disease over radiographic imaging (6, 8). Improvements in this lead-time could potentially be achieved with more sensitive methodologies (Fig. 1). Indeed, by following multiple somatic mutations per patient, Scherer et. al were able to detect relapse of DLBCL an average of ~6 months prior to clinical relapse in a cohort of 11 patients (9). Beyond DLBCL, studies have demonstrated earlier detection of relapsing disease from ctDNA in diverse lymphoma types, including in MCL by using IgHTS (13) and T-cell receptor sequencing in peripheral T-cell lymphomas (12).
3. Novel Methods in Cell-Free DNA Applications
Over the past decade, studies of ctDNA have consistently shown impressive capabilities for non-invasive cancer detection and monitoring of MRD. Many of the early issues with ctDNA analysis stemmed from both biological factors such as low recovery of cfDNA molecules in some samples, and non-biological factors such as technical errors that accumulate throughout the processes of library preparation, hybridization, and sequencing (20). Techniques to maximize depth of sequencing and integrate error-suppression algorithms using molecular biology and in silico designs have resulted in substantial strides forward for ctDNA analysis (20). Extensive reviews have already been conducted to document the limits to which ctDNA analysis has been pushed, both biologically and bioinformatically, without help from any adjunct analytes (52–55). Furthermore, significant limitations – including sensitivity limited by biological background, difficult application to solid tumors, and dependency on total number of mutations — still present a major obstacle, and translation of the liquid biopsy into the clinic has remained elusive in many cases. A variety of novel techniques and methodologies are under active development to address these limitations by focusing on additional aspects of cfDNA biology to improve performance of ctDNA detection, beyond ctDNA quantitation and mutational analysis (Fig. 1). Unlike prior approaches discussed above, this work focuses on leveraging additional analytes to boost ctDNA performance beyond what is currently possible. We here review four nascent realms of research in non-invasive cancer detection: (1) cfDNA fragment structure, (2) epigenetic modifications, (3) the circulating microenvironment, and (4) alternative anatomic compartments for non-invasive detection.
3.1. Cell-free DNA fragment structure
Correlation of cfDNA fragment size with cancer activity has been an active area of interest in recent years. This field of ‘fragmentomics’ focuses on utilizing patterns in healthy cell-free DNA to help identify non-conforming molecules as potentially tumor-derived, without knowledge of any potential mutations within those molecules.
Fragment length itself has been extensively studied. Observations that cfDNA typically shows peaks at multiples of ~166 base pairs in length have been used as evidence that apoptosis serves as a major mechanism for cfDNA release (56, 57); however, exact mechanisms remain unclear, and other mechanisms of DNA release (e.g., necrosis or cellular stress) are possible. A key finding in cfDNA fragmentomics is that the difference between tumor-derived and healthy-derived cfDNA fragments is not limited to alterations in sequence, but can also entail fragment length. One study demonstrated that tumor-derived cfDNA molecules are on average 3–6 base pairs shorter than those of healthy origin, and also generally exhibit greater variability in their lengths (58, 59). This gave rise to the idea that describing molecules by size could improve the detection of DNA molecules of interest, as has been shown in fetal cfDNA studies (60, 61). A genome-wide study of fragmentation features showed that size-selecting for fragments 90–150 base pairs in length significantly improved sensitivity of detecting copy-number alterations using ctDNA (57).
The size distribution of fragmented molecules has also been used to make inferences about nucleosome positioning, relating to gene expression and cell-of-origin. In principle, cfDNA should be present in abundance around nucleosomes, where DNA is wrapped around histones and protected from endogenous nucleases; thus, low-depth regions should indicate ‘open’ or transcriptionally-active DNA, and therefore correspond to areas of increased gene expression. Analysis of these ‘nucleosome-occupancy maps’ has been shown to improve detection when used to augment canonical ctDNA variant calling and enable inference of transcription-factor binding activity (62, 63). Additionally, these in vivo nucleosome footprints have been used to describe cell-of-origin, potentially enabling non-invasive diagnosis of cancer type by hinting at the interactions between DNA and intracellular proteins (62). Indeed, in one initial study, this nucleosome footprinting was used to determine the cell-of-origin in DLBCL subjects from cfDNA (64). It has also been speculated that fragment end patterns could differ between disease states (65).
3.2. Epigenetic modifications
Rising interest in the relationship between cancer cells and their microenvironments has contributed to a new focus on epigenetic alterations seen in cfDNA. The theory behind this concept is that cancer cells should leave behind a visible trace of their growth in the form of epigenetic alterations to their genomes. This is a process that occurs naturally over time in healthy cells, but proceeds with distinct aberrations in cancer cells. Importantly, studying epigenetic shifts does not rely on the recovery of mutational information in cfDNA molecules. Thematically similar to the idea of fragmentomics, ‘epigenetic sequencing’ involves leveraging the entire pool of cfDNA rather than limiting analysis to somatically-mutated ctDNA alone (66). Multiple liquid biopsy studies have been performed utilizing DNA methylation markers in various cancer types (67–72). In the context of lymphoma, abnormal methylation patterns detected in cfDNA have been shown to be an independent poor prognostic factor for 5-year overall survival in DLBCL (73, 74).
DNA methylation is an important mechanism of modulating gene expression in both normal and cancer cells (75), adding ‘punctuation’ to the genetic code. Addition of methyl groups to transcription start site 5’-Cytosine-phosphate-Guanine-3’ segments of DNA, also known as ‘CpG islands’, allows for regulation of expression of the downstream gene. This occurs in distinct patterns that can be used to identify specific cell types and lineages (76); moreover, the methylation events of interest occur at known, stereotyped positions, making them relatively easy to target for sequencing (77). The utility of cfDNA-derived methylation analysis has been demonstrated in cancer as well as in other diseases such as multiple sclerosis and myocardial infarction, that do not always feature reliable genetic mutations to track by ‘conventional’ ctDNA analysis methods (76, 78). Furthermore, tissue-specific DNA methylation patterns have been shown to enable accurate determination of cell-of-origin from cfDNA and non-invasive cancer classification (79–81). Multiple probabilistic models have been developed for cancer detection from cfDNA methylation analysis (82, 83). 5-hydroxymethylcytosine is also being actively investigated as a more specific marker of methylation in cfDNA, having been employed to accurately predict cancer type and stage in one study (84). DNA methylation analysis has thus been pursued as both an adjunct and a competitor to ctDNA in the early detection, characterization, and monitoring of various cancer types.
A key biological limitation of this approach is that in principle, healthy cells should also participate in the epigenetic modification of their environment and thus contribute their own unique epigenetic signal (66, 76). Analogous to the concept of filtering out germline single-nucleotide polymorphisms or clonal hematopoiesis variants from mutational analyses, these normal epigenetic changes need to be precisely distinguished from the effects of cancer cells. Combining epigenetic analysis of the entire cfDNA pool with mutational analysis of circulating tumor DNA molecules is a potentially promising avenue of study (Fig. 1).
3.3. The circulating microenvironment
In addition to cfDNA, an abundance of other types of tumor-derived cells and molecules circulate in the bloodstream, most obviously CTCs. These and other circulating analytes could theoretically be leveraged to improve liquid biopsy performance. An immunoglobulin high-throughput sequencing approach previously showed that cfDNA more accurately represented global tumor burden compared to bulk CTCs (6), pointing to the robustness of the cell-free compartment as a biomarker, but single-cell approaches are currently being explored that may improve the utility of CTC sequencing. In particular, reliable tumor-specific signals have been detected through single-cell whole-genome and whole-exome approaches, notably for metastatic tumors; however, challenges remain concerning heterogeneity, technical error, and cost prohibitive of routine clinical usage (85–88). Aside from cfDNA and CTCs, this ‘circulating microenvironment’ is a relatively unexplored compartment that could contribute valuable information to cancer diagnosis and monitoring. Of this microenvironment, tumor-educated platelets and extracellular vesicular DNA have been most robustly investigated.
Platelets circulating in the tumor microenvironment are known to promote growth, enhance metastasis, and induce phenotypic changes in cancer cells (89). Conversely, platelets have been shown to bear identifiable marks of encounters with tumor cells, mostly relating to RNA splice events and ingestion of local tumor-derived mRNA (90). These tumor-educated platelets have been used for cancer detection and monitoring throughout therapy, much like ctDNA. Multiple studies of RNA sequencing of these platelet contents have been performed and show promising results for detection and classification (90–92); one study discriminated metastatic cancer cases from healthy controls with 96% accuracy and localized primary tumors with 71% accuracy across 6 tumor types (90). Because these marked platelets appear to be created by a mechanism distinct from that which generates ctDNA, it is logical to suppose that they could provide information independent from ctDNA and thus serve as a useful adjunct for non-invasive cancer studies.
Tumor-derived extracellular vesicular nucleic acids have been isolated in recent years, nuancing our understanding of the mechanism of release of cfDNA from apoptotic cells; however, further research into this compartment is needed. In particular, it is currently unclear to what degree exosomes vs. other extracellular vesicles, including apoptotic bodies, contain healthy vs. tumor-derived DNA, and in what proportions (66, 93). In one recent analysis in lung cancer, the addition of exosomal tumor-derived RNA sequencing to ctDNA analysis resulted in mutant EGFR detection of 98%, superior to 84% by ctDNA sequencing agnostic to exosomes (94). Another study explored the ability of HL-specific exosomal microRNAs (miRNAs) to inform treatment response in HL. The authors found that specificity and sensitivity of exosomal miRNAs were superior to protein-bound miRNAs with regard to HL detection (95). Interactions between cfDNA circulating in the blood and other vesicular cfDNA molecules have yet to be elucidated and are an exciting focus of future work (Fig. 1).
3.4. Other anatomical compartments
Thus far, we have discussed approaches to liquid biopsies that all fundamentally rely on detection from the blood plasma. While these approaches are now proven in multiple studies, there are limitations to blood-based liquid biopsies as well, including limitations on blood volume collection and the need for phlebotomy. Additional anatomical compartments for non-invasive biopsies and detection of tumor DNA have therefore been explored in diverse malignancies, including stool (96), urine (97–100), and saliva (101). Perhaps the furthest developed of these analytes is stool, where analysis of stool DNA has improved on detection rates for colon cancer using fecal immunochemical testing alone (96). Indeed, this test has now been commercialized as the Cologuard™ assay for colon cancer screening. DNA from urine is an attractive analyte for genitourinary malignancies, with high sensitivity for bladder cancer detection in multiple reports (97, 100) and emerging data in renal cell carcinoma (98, 99). A similar anatomically associated analyte-malignancy pair is saliva and head and neck cancer, where both somatic mutations and HPV viral DNA have been detected in the saliva (101). These anatomic compartments for non-invasive biopsies have not been widely explored in lymphoid malignancies, but provide a potential tool to expand on more traditional blood-based ctDNA detection.
These novel areas of investigation in non-invasive cancer detection provide new insights into the circulating compartment that potentially augment the quantitative and mutational information that somatic ctDNA analysis already provides. An effective combination of any of these modalities with somatic mutational ctDNA analysis has promise to revolutionize liquid biopsies in the future.
4. Clinical Applications and Future Directions
Liquid biopsy in the clinic: an illustrative case
A 78 year old female was diagnosed with grade 1–2 follicular NHL (FL) from a core biopsy of an inguinal node biopsy after presentation with widespread lymphadenopathy. Due to symptoms of obstructive jaundice with compressive porta-hepatis lymphadenopathy, (Figure 2) she was transferred to our specialist unit for further diagnostic work-up. However, the patient subsequently suffered collapse and hematemesis. An endoscopy and biopsy of gastric ulceration confirmed germinal center DLBCL with BCL-2/MYC double-expression (Figure 2). FISH for MYC and BCL2 could not be performed on the sample. Staging investigations included PET/CT which showed Stage IV disease (Figure 2). Her CNS-IPI at diagnosis was high (score 5, >10% 2-year risk of CNS progression) (102). She proceeded with 6 cycles R-mini CHOP treatment as she was unfit for intensive therapy based on her ECOG 2 and age. Interim CT re-staging demonstrated a very good partial response (VGPR) and end-of-therapy PET confirmed near complete remission. Consolidative radiotherapy to abdominal bulk was planned, however shortly after chemotherapy completion, the patient re-presented with recurrent falls. A CT-head and MRI-brain confirmed two periventricular lesions suspicious for lymphoma relapse (Figure 2). An LP could not be performed due to significant vasogenic edema and risk of uncinate herniation. After urgent steroids, the patient went on to have a stealth-guided brain biopsy. The biopsy was subsequently found to be non-diagnostic. The patient had made initial clinical and radiological improvement after short-term steroid exposure and was able to recover enough functional independence to return home. This created a difficult clinical situation, and after the patient was counselled, a decision was made for close observation.
Figure 2: Tumor heterogeneity and response to treatment.
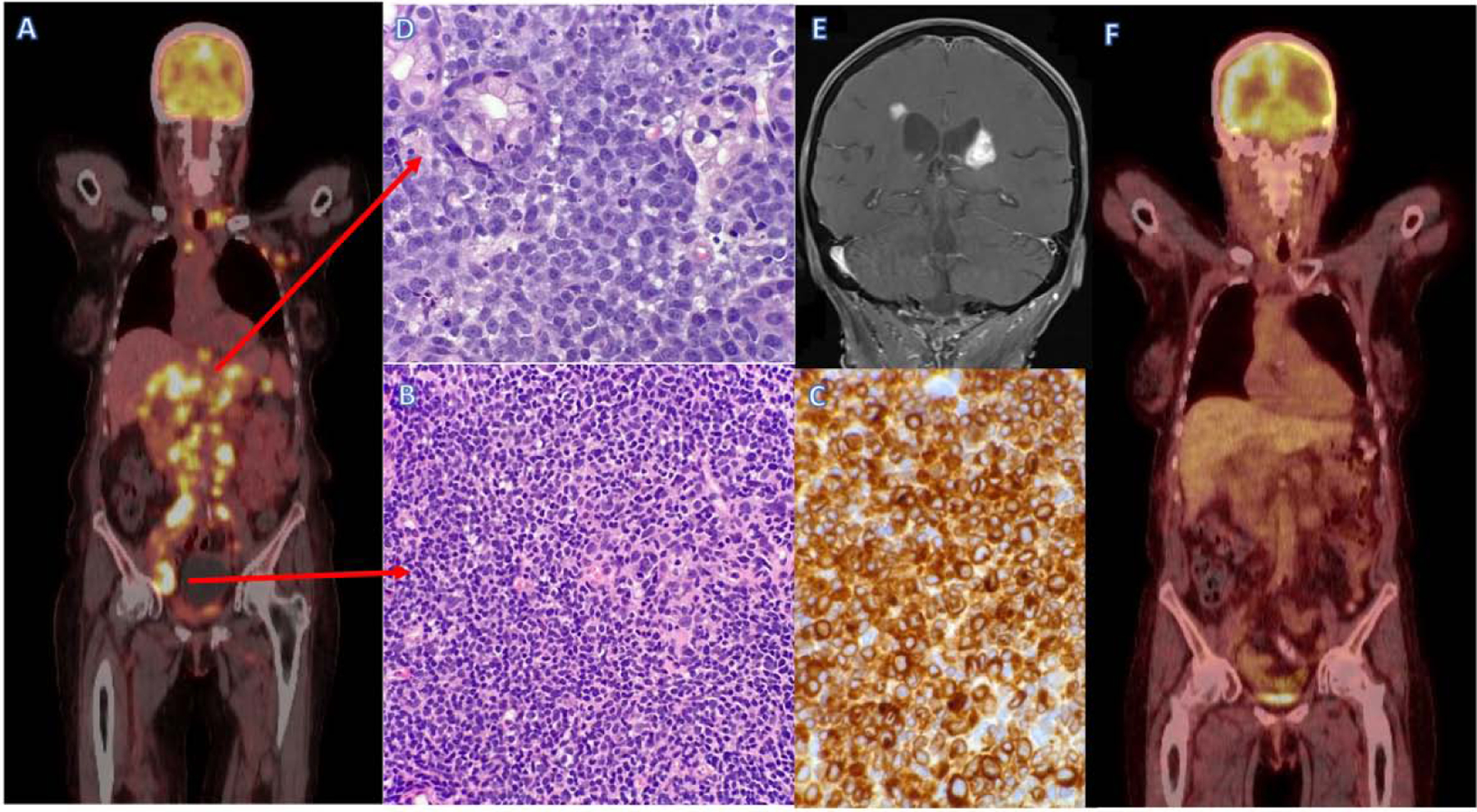
A; Pre-treatment PET/CT scan in newly diagnosed lymphoma demonstrates advanced stage disease.
B; Inguinal lymph node biopsy demonstrating low-grade follicular NHL, H&E stain (x40 magnification).
C; Inguinal lymph node biopsy demonstrating low-grade follicular NHL, BCL-2 IHC stain (x60 magnification).
D; Gastric ulcer biopsy demonstrating high grade B-cell lymphoma DLBCL, H&E stain (x60 magnification).
E; End of treatment PET/CT scan demonstrates near complete remission.
F; MRI scan after NHL treatment demonstrates new peri-ventricular white matter lesions suggestive of NHL relapse.
Approximately 1 month later, the patient was readmitted with increasing confusion and a repeat CT identified a new left-sided brain lesion. The patient chose empirical lymphoma treatment (declining repeat biopsy) and received one cycle of IV Methotrexate without clinical or radiological improvement. The patient was then treated with palliative Rituximab and Temozolomide, but subsequently died after 2 cycles due to progressive disease.
Practical applications of liquid biopsy
This case highlights several potential future applications of ctDNA techniques. In the diagnostic evaluation of this case, the patient was found on a single lymph node core biopsy to have low-grade FL. However, a subsequent gastric biopsy revealed a focus of high-grade disease transformation. Ideally a lymph node excision is performed as part of the diagnostic work-up in all cases, but due to clinical deterioration of this patient a repeat (endoscopic) biopsy had already been performed. We can only speculate on the additional information that liquid biopsy might have given in this particular case, where the mutational profile of the involved sites was likely to be heterogenous. It is clear from early work in FL that pretreatment ctDNA is likely to be detectable and correlates well with disease outcome and TMTV (103). In DLBCL, we now know that the majority of patients will have mutations detectable from peripheral blood cfDNA, with detection rates dependent on the applied molecular technique (104). Hence for those patients with localized transformation of FL, ctDNA might be extremely valuable at the time of diagnosis and for MRD response assessment.
In this case, there was a very short time between the end of therapy and development of progressive disease. Hence ctDNA monitoring might have been a useful adjunct to interim and end-of-treatment PET/CT in this scenario. As described above, Kurtz et al. defined a 2-log reduction in MRD after 1 cycle of therapy as an excellent predictor to response, known as EMR (18). However, future prospective studies are needed to determine if early response assessment by ctDNA (such as after 1–2 cycles of therapy) is useful as a risk-adaptive strategy towards treatment. In DLBCL, risk-adapted therapy based on interim PET/CT assessment is not routine, although local practices may differ (105). There appears to be a trend towards risk-adapted therapy in other lymphomas such as HL (106–108), and clinicians are increasingly looking for methods to personalize cancer medicine, optimize treatment outcomes and avoid unnecessary toxicity.
At the time of presumed CNS relapse, this patient underwent an invasive (and potentially hazardous) procedure that ultimately failed to yield a diagnosis. If ctDNA assessment was readily available at clinical relapse, a peripheral blood sample (or potentially CSF sample) could have been compared to a prior tissue sample and perhaps avoided the need for such a procedure. In CNS lymphoma, it is likely that cfDNA collected from CSF samples are likely to have higher mutational load than blood, although current data is limited (39). Recurrent cfDNA mutations associated with both PCNSL and secondary CNS lymphoma have been reported from patient CSF samples (40, 109). Further prospective studies could examine the correlation between cfDNA in peripheral blood and CSF (at diagnosis and/or end of treatment) and in the case of secondary CNS relapse for selected DLBCL patients. Incorporating cfDNA in combination with standard CSF cytology and flow cytometry techniques could potentially improve diagnostic accuracy and reduce the need for invasive biopsy in many patients.
While the concept is promising, there is currently a lack of data to define the positive and negative predictive value of cfDNA from CSF samples in lymphoma. There is also an absence of defined test specifications which would be applicable to a clinical assay, such as minimum sample volume. Thus, it would be useful to study the feasibility of CSF and plasma ctDNA detection and monitoring in patients with PCNSL, VRL, and DLBCL with moderate to high CNS-IPI risk in larger prospective studies in the future.
Lastly, this case highlights a sub-population of patients with FL who present with localized DLBCL transformation. There is scant information as to the molecular signature of this type of patient, and it is especially uncommon to have sites of low-grade (grade 1–2) FL concurrent with DLBCL. Here, the use of liquid biopsy opens avenues for new disease characterization at diagnosis and at relapse (Fig. 1). Indeed, early data suggests that identification of transformation of follicular lymphoma to DLBCL from ctDNA analysis is possible (9, 23).
Conclusion:
In the era of precision medicine, patient-centered therapy is an elusive goal, and liquid biopsy will likely be an important tool in our armamentarium. We have here reviewed the molecular methodologies and clinical applications of non-invasive tumor detection and monitoring in lymphoma. Our ability to detect vanishingly small quantities of ctDNA and accurately classify mutations is improving through advances in our understanding of ctDNA biology, bioinformatics and NGS. Synthesis of improved ctDNA analysis frameworks with novel methodologies, aided by intelligent machine-learning algorithms, has the potential to dramatically improve the test characteristics of liquid biopsy-based assays. With these rapid strides forward, we expect to see prospective evaluation of the role of ctDNA in lymphoma genotyping and response monitoring as well as integration into clinical trials in the very near future (Fig. 1).
Highlights:
Liquid Biopsy:
Can detect small amounts of circulating tumor DNA from a single blood test
Emerges as a viable method of tumor genotyping in most lymphoma subtypes
Has the potential to predict relapse before clinically detectable tumor growth
Is evolving towards practical clinical use with new technologies and strategies
Acknowledgments:
The authors wish to thank Dr Agatino Calogero, Dr Stephanie Clugston and particularly Dr Michael Armstrong for their contribution to the clinical case presented here.
Funding:
S.B.: Supported by Else Kröner-Fresenius-Stiftung, Grant No. 2016-Kolleg-19, Deutsche Forschungsgemeinschaft (DFG), Grant No. EN 179/13-1 and Frauke Weiskam + Christel Ruranski-Stiftung, Grant No. T 0136 – 33.661.
A.F.M.C.: supported by a Physician-Scientist Career Development Award from the American Society of Hematology, Washington, D.C., USA.
D.M.K: Supported by the Damon Runyon Cancer Research Foundation (PST#09-16) and the National Cancer Institute (1-K08-CA241076-01).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Araf S, Wang J, Korfi K, Pangault C, Kotsiou E, Rio-Machin A, et al. Genomic profiling reveals spatial intra-tumor heterogeneity in follicular lymphoma. Leukemia. 2018;32(5):1261–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scherer F, Kurtz DM, Newman AM, Craig A, Stehr H, Zhou L, et al. Noninvasive Detection of Ibrutinib Resistance in Non-Hodgkin Lymphoma Using Cell-Free DNA. Blood. 2016;128(22):1752–. [Google Scholar]
- 3.Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. The New England journal of medicine. 2012;366(10):883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kovacs G, Robrecht S, Fink AM, Bahlo J, Cramer P, von Tresckow J, et al. Minimal Residual Disease Assessment Improves Prediction of Outcome in Patients With Chronic Lymphocytic Leukemia (CLL) Who Achieve Partial Response: Comprehensive Analysis of Two Phase III Studies of the German CLL Study Group. J Clin Oncol 2016;34(31):3758–65. [DOI] [PubMed] [Google Scholar]
- 5.Kwok M, Rawstron AC, Varghese A, Evans PA, O’Connor SJ, Doughty C, et al. Minimal residual disease is an independent predictor for 10-year survival in CLL. Blood. 2016;128(24):2770–3. [DOI] [PubMed] [Google Scholar]
- 6.Kurtz DM, Green MR, Bratman SV, Scherer F, Liu CL, Kunder CA, et al. Noninvasive monitoring of diffuse large B-cell lymphoma by immunoglobulin high-throughput sequencing. Blood. 2015;125(24):3679–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kurtz DM, Scherer F, Jin MC, Soo J, Craig AFM, Esfahani MS, et al. Circulating Tumor DNA Measurements As Early Outcome Predictors in Diffuse Large B-Cell Lymphoma. J Clin Oncol 2018:JCO2018785246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roschewski M, Dunleavy K, Pittaluga S, Moorhead M, Pepin F, Kong K, et al. Circulating tumour DNA and CT monitoring in patients with untreated diffuse large B-cell lymphoma: a correlative biomarker study. The Lancet Oncology. 2015;16(5):541–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scherer F, Kurtz DM, Newman AM, Stehr H, Craig AF, Esfahani MS, et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Science translational medicine. 2016;8(364):364ra155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rossi D, Diop F, Spaccarotella E, Monti S, Zanni M, Rasi S, et al. Diffuse large B-cell lymphoma genotyping on the liquid biopsy. Blood. 2017;129(14):1947–57. [DOI] [PubMed] [Google Scholar]
- 11.Lakhotia R, Melani C, Pittaluga S, Dunleavy K, Saba NS, Lucas AN, et al. Circulating Tumor DNA Dynamics during Therapy Predict Outcomes in Mantle Cell Lymphoma. Blood. 2018;132:147–. [Google Scholar]
- 12.Melani C, Pittaluga S, Yee L, Lucas A, Shovlin M, Jacob A, et al. Next-Generation Sequencing Based Monitoring of Circulating-Tumor DNA in Untreated Peripheral T-Cell Lymphoma. Blood. 2017;130(Suppl 1):2728–.28935695 [Google Scholar]
- 13.Roschewski MJ, Melani CJ, Pittaluga S, Dunleavy K, Saba NS, Grant C, et al. Circulating tumor DNA to predict timing of relapse in mantle cell lymphoma. Journal of Clinical Oncology. 2018;36(15_suppl):7576–. [Google Scholar]
- 14.Camus V, Viennot M, Viailly P, Bohers E, Bessi L, Marcq B, et al. Targeted genotyping of circulating tumor DNA for classical Hodgkin lymphoma monitoring: a prospective study. Hematological Oncology. 2019;37:187–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muringampurath-John D, Jaye DL, Flowers CR, Saxe D, Chen Z, Lechowicz MJ, et al. Characteristics and outcomes of diffuse large B-cell lymphoma presenting in leukaemic phase. British journal of haematology. 2012;158(5):608–14. [DOI] [PubMed] [Google Scholar]
- 16.Hohaus S, Giachelia M, Massini G, Mansueto G, Vannata B, Bozzoli V, et al. Cell-free circulating DNA in Hodgkin’s and non-Hodgkin’s lymphomas. Annals of oncology : official journal of the European Society for Medical Oncology. 2009;20(8):1408–13. [DOI] [PubMed] [Google Scholar]
- 17.Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nature medicine. 2014;20(5):548–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurtz DM, Scherer F, Jin MC, Soo J, Craig AFM, Esfahani MS, et al. Circulating Tumor DNA Measurements As Early Outcome Predictors in Diffuse Large B-Cell Lymphoma. J Clin Oncol 2018;36(28):2845–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rossille D, Gressier M, Damotte D, Maucort-Boulch D, Pangault C, Semana G, et al. High level of soluble programmed cell death ligand 1 in blood impacts overall survival in aggressive diffuse large B-Cell lymphoma: results from a French multicenter clinical trial. Leukemia. 2014;28(12):2367–75. [DOI] [PubMed] [Google Scholar]
- 20.Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nature biotechnology. 2016;34(5):547–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nature medicine. 2014;20(12):1472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Razavi P, Li BT, Brown DN, Jung B, Hubbell E, Shen R, et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nature medicine. 2019;25(12):1928–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scherer F, Kurtz DM, Newman AM, Stehr H, Liu CL, Zhou L, et al. Noninvasive Genotyping and Assessment of Treatment Response in Diffuse Large B Cell Lymphoma. Blood. 2015;126(23):114–. [Google Scholar]
- 24.Spina V, Bruscaggin A, Cuccaro A, Martini M, Di Trani M, Forestieri G, et al. Circulating tumor DNA reveals genetics, clonal evolution and residual disease in classical Hodgkin lymphoma. Blood. 2018. [DOI] [PubMed] [Google Scholar]
- 25.Desch AK, Hartung K, Botzen A, Brobeil A, Rummel M, Kurch L, et al. Genotyping circulating tumor DNA of pediatric Hodgkin lymphoma. Leukemia. 2020;34(1):151–66. [DOI] [PubMed] [Google Scholar]
- 26.Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nature medicine. 2015;21(8):922–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503–11. [DOI] [PubMed] [Google Scholar]
- 28.Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004;103(1):275–82. [DOI] [PubMed] [Google Scholar]
- 29.Scott DW, Wright GW, Williams PM, Lih CJ, Walsh W, Jaffe ES, et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood. 2014;123(8):1214–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. The New England journal of medicine. 2018;378(15):1396–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nature medicine. 2018;24(5):679–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Esfahani MS, Alig S, Kurtz DM, Soo J, Jin MC, Macaulay C, et al. Towards Non-Invasive Classification of DLBCL Genetic Subtypes By Ctdna Profiling. Blood. 2019;134(Supplement_1):551–. [Google Scholar]
- 33.Tiacci E, Ladewig E, Schiavoni G, Penson A, Fortini E, Pettirossi V, et al. Pervasive mutations of JAK-STAT pathway genes in classical Hodgkin lymphoma. Blood. 2018;131(22):2454–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reichel J, Chadburn A, Rubinstein PG, Giulino-Roth L, Tam W, Liu Y, et al. Flow sorting and exome sequencing reveal the oncogenome of primary Hodgkin and Reed-Sternberg cells. Blood. 2015;125(7):1061–72. [DOI] [PubMed] [Google Scholar]
- 35.Braggio E, Van Wier S, Ojha J, McPhail E, Asmann YW, Egan J, et al. Genome-Wide Analysis Uncovers Novel Recurrent Alterations in Primary Central Nervous System Lymphomas. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(17):3986–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vater I, Montesinos-Rongen M, Schlesner M, Haake A, Purschke F, Sprute R, et al. The mutational pattern of primary lymphoma of the central nervous system determined by whole-exome sequencing. Leukemia. 2015;29(3):677–85. [DOI] [PubMed] [Google Scholar]
- 37.Hiemcke-Jiwa LS, Leguit RJ, Snijders TJ, Bromberg JEC, Nierkens S, Jiwa NM, et al. MYD88 p.(L265P) detection on cell-free DNA in liquid biopsies of patients with primary central nervous system lymphoma. British journal of haematology. 2019;185(5):974–7. [DOI] [PubMed] [Google Scholar]
- 38.Hiemcke-Jiwa LS, Ten Dam-van Loon NH, Leguit RJ, Nierkens S, Ossewaarde-van Norel J, de Boer JH, et al. Potential Diagnosis of Vitreoretinal Lymphoma by Detection of MYD88 Mutation in Aqueous Humor With Ultrasensitive Droplet Digital Polymerase Chain Reaction. JAMA ophthalmology. 2018;136(10):1098–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hickmann A-K, Frick M, Hadaschik D, Battke F, Bittl M, Ganslandt O, et al. Molecular tumor analysis and liquid biopsy: a feasibility investigation analyzing circulating tumor DNA in patients with central nervous system lymphomas. BMC Cancer. 2019;19(1):192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zorofchian S, Lu G, Zhu JJ, Duose DY, Windham J, Esquenazi Y, et al. Detection of the MYD88 p.L265P Mutation in the CSF of a Patient With Secondary Central Nervous System Lymphoma. Frontiers in oncology. 2018;8:382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.International Non-Hodgkin’s Lymphoma Prognostic Factors P. A predictive model for aggressive non-Hodgkin’s lymphoma. The New England journal of medicine. 1993;329(14):987–94. [DOI] [PubMed] [Google Scholar]
- 42.Sasanelli M, Meignan M, Haioun C, Berriolo-Riedinger A, Casasnovas RO, Biggi A, et al. Pretherapy metabolic tumour volume is an independent predictor of outcome in patients with diffuse large B-cell lymphoma. Eur J Nucl Med Mol Imaging. 2014;41(11):2017–22. [DOI] [PubMed] [Google Scholar]
- 43.Andrade-Campos MM, Salar A, Sanchez-Gonzalez B, Fernández-Rodríguez C, Gimeno E, Ruiz L, et al. Assessment of Cell-Free DNA (cfDNA) in 221 Patients with Lymphoproliferative Malignancies at Diagnosis and during Follow-up. Blood. 2019;134(Supplement_1):492–.31371395 [Google Scholar]
- 44.Bohers E, Viailly PJ, Becker S, Marchand V, Ruminy P, Maingonnat C, et al. Non-invasive monitoring of diffuse large B-cell lymphoma by cell-free DNA high-throughput targeted sequencing: analysis of a prospective cohort. Blood Cancer J 2018;8(8):74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schroers-Martin JG, Kurtz DM, Soo J, Jin M, Scherer F, Craig A, et al. Determinants of Circulating Tumor DNA Levels across Lymphoma Histologic Subtypes. Blood. 2017;130(Supplement 1):4018–. [Google Scholar]
- 46.Hughes TP, Kaeda J, Branford S, Rudzki Z, Hochhaus A, Hensley ML, et al. Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. The New England journal of medicine. 2003;349(15):1423–32. [DOI] [PubMed] [Google Scholar]
- 47.Moskowitz CH. Interim PET-CT in the management of diffuse large B-cell lymphoma. Hematology Am Soc Hematol Educ Program. 2012;2012(1):397–401. [DOI] [PubMed] [Google Scholar]
- 48.Kurtz DM, Esfahani MS, Scherer F, Soo J, Jin MC, Liu CL, et al. Dynamic Risk Profiling Using Serial Tumor Biomarkers for Personalized Outcome Prediction. Cell. 2019;178(3):699–713 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spina V, Bruscaggin A, Cuccaro A, Martini M, Di Trani M, Forestieri G, et al. Circulating tumor DNA reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood. 2018;131(22):2413–25. [DOI] [PubMed] [Google Scholar]
- 50.Thompson CA, Ghesquieres H, Maurer MJ, Cerhan JR, Biron P, Ansell SM, et al. Utility of routine post-therapy surveillance imaging in diffuse large B-cell lymphoma. J Clin Oncol 2014;32(31):3506–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moskowitz CH, Schoder H, Teruya-Feldstein J, Sima C, Iasonos A, Portlock CS, et al. Risk-adapted dose-dense immunochemotherapy determined by interim FDG-PET in Advanced-stage diffuse large B-Cell lymphoma. J Clin Oncol 2010;28(11):1896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Condoluci A, Rossi D. The future of cell-free DNA testing to guide therapeutic decisions in B-cell lymphomas. Curr Opin Hematol 2019;26(4):281–7. [DOI] [PubMed] [Google Scholar]
- 53.Crombie J, Armand P. The Emerging Role of Liquid Biopsies in Lymphoproliferative Disorders. Curr Hematol Malig Rep 2019;14(1):11–21. [DOI] [PubMed] [Google Scholar]
- 54.Melani C, Wilson WH, Roschewski M. Liquid biopsy in non-Hodgkin’s lymphoma. Hematol Oncol 2019;37 Suppl 1:70–4. [DOI] [PubMed] [Google Scholar]
- 55.Sriram D, Lakhotia R, Fenske TS. Measurement of circulating tumor DNA to guide management of patients with lymphoma. Clin Adv Hematol Oncol 2019;17(9):509–17. [PubMed] [Google Scholar]
- 56.Bronkhorst AJ, Wentzel JF, Aucamp J, van Dyk E, du Plessis L, Pretorius PJ. Characterization of the cell-free DNA released by cultured cancer cells. Biochimica et biophysica acta. 2016;1863(1):157–65. [DOI] [PubMed] [Google Scholar]
- 57.Mouliere F, Chandrananda D, Piskorz AM, Moore EK, Morris J, Ahlborn LB, et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Science translational medicine. 2018;10(466). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cristiano S, Leal A, Phallen J, Fiksel J, Adleff V, Bruhm DC, et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature. 2019;570(7761):385–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mouliere F, Robert B, Arnau Peyrotte E, Del Rio M, Ychou M, Molina F, et al. High fragmentation characterizes tumour-derived circulating DNA. PloS one. 2011;6(9):e23418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu SC, Chan KC, Zheng YW, Jiang P, Liao GJ, Sun H, et al. Size-based molecular diagnostics using plasma DNA for noninvasive prenatal testing. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(23):8583–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chan KCA, Zhang J, Hui ABY, Wong N, Lau TK, Leung TN, et al. Size distributions of maternal and fetal DNA in maternal plasma. Clin Chem 2004;50(1):88–92. [DOI] [PubMed] [Google Scholar]
- 62.Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell. 2016;164(1–2):57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ulz P, Perakis S, Zhou Q, Moser T, Belic J, Lazzeri I, et al. Inference of transcription factor binding from cell-free DNA enables tumor subtype prediction and early detection. Nature communications. 2019;10(1):4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mehrmohamadi M, Esfahani MS, Soo J, Scherer F, Schroers-Martin JG, Chen B, et al. Distinct Chromatin Accessibility Profiles of Lymphoma Subtypes Revealed By Targeted Cell Free DNA Profiling. Blood. 2018;132(Supplement 1):672–. [Google Scholar]
- 65.Chandrananda D, Thorne NP, Bahlo M. High-resolution characterization of sequence signatures due to non-random cleavage of cell-free DNA. BMC medical genomics. 2015;8:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van der Pol Y, Mouliere F. Toward the Early Detection of Cancer by Decoding the Epigenetic and Environmental Fingerprints of Cell-Free DNA. Cancer cell. 2019;36(4):350–68. [DOI] [PubMed] [Google Scholar]
- 67.Melson J, Li Y, Cassinotti E, Melnikov A, Boni L, Ai J, et al. Commonality and differences of methylation signatures in the plasma of patients with pancreatic cancer and colorectal cancer. International journal of cancer. 2014;134(11):2656–62. [DOI] [PubMed] [Google Scholar]
- 68.Fackler MJ, Lopez Bujanda Z, Umbricht C, Teo WW, Cho S, Zhang Z, et al. Novel methylated biomarkers and a robust assay to detect circulating tumor DNA in metastatic breast cancer. Cancer research. 2014;74(8):2160–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wen L, Li J, Guo H, Liu X, Zheng S, Zhang D, et al. Genome-scale detection of hypermethylated CpG islands in circulating cell-free DNA of hepatocellular carcinoma patients. Cell research. 2015;25(11):1250–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Herbst A, Vdovin N, Gacesa S, Ofner A, Philipp A, Nagel D, et al. Methylated free-circulating HPP1 DNA is an early response marker in patients with metastatic colorectal cancer. International journal of cancer. 2017;140(9):2134–44. [DOI] [PubMed] [Google Scholar]
- 71.Widschwendter M, Evans I, Jones A, Ghazali S, Reisel D, Ryan A, et al. Methylation patterns in serum DNA for early identification of disseminated breast cancer. Genome medicine. 2017;9(1):115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gordevicius J, Krisciunas A, Groot DE, Yip SM, Susic M, Kwan A, et al. Cell-Free DNA Modification Dynamics in Abiraterone Acetate-Treated Prostate Cancer Patients. Clinical cancer research : an official journal of the American Association for Cancer Research. 2018;24(14):3317–24. [DOI] [PubMed] [Google Scholar]
- 73.Wedge E, Hansen JW, Garde C, Asmar F, Tholstrup D, Kristensen SS, et al. Global hypomethylation is an independent prognostic factor in diffuse large B cell lymphoma. American journal of hematology. 2017;92(7):689–94. [DOI] [PubMed] [Google Scholar]
- 74.Kristensen LS, Hansen JW, Kristensen SS, Tholstrup D, Harslof LB, Pedersen OB, et al. Aberrant methylation of cell-free circulating DNA in plasma predicts poor outcome in diffuse large B cell lymphoma. Clinical epigenetics. 2016;8(1):95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Estreller M Epigenetic Changes in Cancer: The study of how covalent marks on DNA and histones are involved in the origin and spread of cancer cells is also leading to new therapeutic strategies. F1000 Biol Reports. 2011. [Google Scholar]
- 76.Zemmour H, Planer D, Magenheim J, Moss J, Neiman D, Gilon D, et al. Non-invasive detection of human cardiomyocyte death using methylation patterns of circulating DNA. Nature communications. 2018;9(1):1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hesson A, editor. Clinical Epigenetics. Singapore: Springer Nature Singapore Ltd.; 2019. [Google Scholar]
- 78.Lehmann-Werman R, Neiman D, Zemmour H, Moss J, Magenheim J, Vaknin-Dembinsky A, et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(13):E1826–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sun K, Jiang P, Chan KC, Wong J, Cheng YK, Liang RH, et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(40):E5503–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moss J, Magenheim J, Neiman D, Zemmour H, Loyfer N, Korach A, et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nature communications. 2018;9(1):5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chan KC, Jiang P, Chan CW, Sun K, Wong J, Hui EP, et al. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(47):18761–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li W, Li Q, Kang S, Same M, Zhou Y, Sun C, et al. CancerDetector: ultrasensitive and non-invasive cancer detection at the resolution of individual reads using cell-free DNA methylation sequencing data. Nucleic acids research. 2018;46(15):e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kang S, Li Q, Chen Q, Zhou Y, Park S, Lee G, et al. CancerLocator: non-invasive cancer diagnosis and tissue-of-origin prediction using methylation profiles of cell-free DNA. Genome biology. 2017;18(1):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Song CX, Yin S, Ma L, Wheeler A, Chen Y, Zhang Y, et al. 5-Hydroxymethylcytosine signatures in cell-free DNA provide information about tumor types and stages. Cell research. 2017;27(10):1231–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ni X, Zhuo M, Su Z, Duan J, Gao Y, Wang Z, et al. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(52):21083–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lohr JG, Adalsteinsson VA, Cibulskis K, Choudhury AD, Rosenberg M, Cruz-Gordillo P, et al. Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nature biotechnology. 2014;32(5):479–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Navin NE. Cancer genomics: one cell at a time. Genome biology. 2014;15(8):452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gawad C, Koh W, Quake SR. Single-cell genome sequencing: current state of the science. Nature reviews Genetics. 2016;17(3):175–88. [DOI] [PubMed] [Google Scholar]
- 89.Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer cell. 2011;20(5):576–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Best MG, Sol N, Kooi I, Tannous J, Westerman BA, Rustenburg F, et al. RNA-Seq of Tumor-Educated Platelets Enables Blood-Based Pan-Cancer, Multiclass, and Molecular Pathway Cancer Diagnostics. Cancer cell. 2015;28(5):666–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Calverley DC, Phang TL, Choudhury QG, Gao B, Oton AB, Weyant MJ, et al. Significant downregulation of platelet gene expression in metastatic lung cancer. Clinical and translational science. 2010;3(5):227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Best MG, Sol N, In 't Veld S, Vancura A, Muller M, Niemeijer AN, et al. Swarm Intelligence-Enhanced Detection of Non-Small-Cell Lung Cancer Using Tumor-Educated Platelets. Cancer cell. 2017;32(2):238–52.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gahan PB, Stroun M. The virtosome-a novel cytosolic informative entity and intercellular messenger. Cell biochemistry and function. 2010;28(7):529–38. [DOI] [PubMed] [Google Scholar]
- 94.Krug AK, Enderle D, Karlovich C, Priewasser T, Bentink S, Spiel A, et al. Improved EGFR mutation detection using combined exosomal RNA and circulating tumor DNA in NSCLC patient plasma. Annals of oncology : official journal of the European Society for Medical Oncology. 2018;29(10):2143. [DOI] [PubMed] [Google Scholar]
- 95.van Eijndhoven MA, Zijlstra JM, Groenewegen NJ, Drees EE, van Niele S, Baglio SR, et al. Plasma vesicle miRNAs for therapy response monitoring in Hodgkin lymphoma patients. JCI insight. 2016;1(19):e89631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Imperiale TF, Ransohoff DF, Itzkowitz SH, Levin TR, Lavin P, Lidgard GP, et al. Multitarget stool DNA testing for colorectal-cancer screening. The New England journal of medicine. 2014;370(14):1287–97. [DOI] [PubMed] [Google Scholar]
- 97.Dudley JC, Schroers-Martin J, Lazzareschi DV, Shi WY, Chen SB, Esfahani MS, et al. Detection and Surveillance of Bladder Cancer Using Urine Tumor DNA. Cancer Discov 2019;9(4):500–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nuzzo PV, Berchuck JE, Korthauer K, Spisak S, Nassar AH, Abou Alaiwi S, et al. Detection of renal cell carcinoma using plasma and urine cell-free DNA methylomes. Nature medicine. 2020;26(7):1041–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Smith CG, Moser T, Mouliere F, Field-Rayner J, Eldridge M, Riediger AL, et al. Comprehensive characterization of cell-free tumor DNA in plasma and urine of patients with renal tumors. Genome medicine. 2020;12(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Springer SU, Chen CH, Rodriguez Pena MDC, Li L, Douville C, Wang Y, et al. Noninvasive detection of urothelial cancer through the analysis of driver gene mutations and aneuploidy. Elife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang Y, Springer S, Mulvey CL, Silliman N, Schaefer J, Sausen M, et al. Detection of somatic mutations and HPV in the saliva and plasma of patients with head and neck squamous cell carcinomas. Science translational medicine. 2015;7(293):293ra104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schmitz N, Zeynalova S, Nickelsen M, Kansara R, Villa D, Sehn LH, et al. CNS International Prognostic Index: A Risk Model for CNS Relapse in Patients With Diffuse Large B-Cell Lymphoma Treated With R-CHOP. J Clin Oncol 2016;34(26):3150–6. [DOI] [PubMed] [Google Scholar]
- 103.Delfau-Larue MH, van der Gucht A, Dupuis J, Jais JP, Nel I, Beldi-Ferchiou A, et al. Total metabolic tumor volume, circulating tumor cells, cell-free DNA: distinct prognostic value in follicular lymphoma. Blood advances. 2018;2(7):807–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Arzuaga-Mendez J, Prieto-Fernandez E, Lopez-Lopez E, Martin-Guerrero I, Garcia-Ruiz JC, Garcia-Orad A. Cell-free DNA as a biomarker in diffuse large B-cell lymphoma: A systematic review. Critical reviews in oncology/hematology. 2019;139:7–15. [DOI] [PubMed] [Google Scholar]
- 105.Moskowitz CH, Schöder H, Teruya-Feldstein J, Sima C, Iasonos A, Portlock CS, et al. Risk-adapted dose-dense immunochemotherapy determined by interim FDG-PET in Advanced-stage diffuse large B-Cell lymphoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28(11):1896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Borchmann P, Goergen H, Kobe C, Lohri A, Greil R, Eichenauer DA, et al. PET-guided treatment in patients with advanced-stage Hodgkin’s lymphoma (HD18): final results of an open-label, international, randomised phase 3 trial by the German Hodgkin Study Group. Lancet (London, England). 2018;390(10114):2790–802. [DOI] [PubMed] [Google Scholar]
- 107.Radford J, Illidge T, Counsell N, Hancock B, Pettengell R, Johnson P, et al. Results of a Trial of PET-Directed Therapy for Early-Stage Hodgkin’s Lymphoma. New England Journal of Medicine. 2015;372(17):1598–607. [DOI] [PubMed] [Google Scholar]
- 108.Johnson P, Federico M, Kirkwood A, Fosså A, Berkahn L, Carella A, et al. Adapted Treatment Guided by Interim PET-CT Scan in Advanced Hodgkin’s Lymphoma. New England Journal of Medicine. 2016;374(25):2419–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hiemcke-Jiwa LS, Leguit RJ, Snijders TJ, Jiwa NM, Kuiper JJW, de Weger RA, et al. Molecular analysis in liquid biopsies for diagnostics of primary central nervous system lymphoma: Review of literature and future opportunities. Critical reviews in oncology/hematology. 2018;127:56–65. [DOI] [PubMed] [Google Scholar]