

Abstract

The metal-free reduction of a range of phosphine(V) oxides employing oxalyl chloride as an activating agent and hexachlorodisilane as reducing reagent has been achieved under mild reaction conditions. The method was successfully applied to the reduction of industrial waste byproduct triphenylphosphine(V) oxide, closing the phosphorus cycle to cleanly regenerate triphenylphosphine(III). Mechanistic studies and quantum chemical calculations support the attack of the dissociated chloride anion of intermediated phosphonium salt at the silicon of the disilane as the rate-limiting step for deprotection. The exquisite purity of the resultant phosphine(III) ligands after the simple removal of volatiles under reduced pressure circumvents laborious purification prior to metalation and has permitted the facile formation of important transition metal catalysts.
Introduction
Applications of Phosphine(III) Ligands and Synthesis
Phosphines and their derivatives are of significant importance to both academic and industrial chemistry. In particular, within organic chemistry phosphine(III) compounds have a distinguished history, mediating classical transformations such as the Appel,1 Mitsunobu,2 and Wittig3,4 reactions. Additionally, the ready modulation of electronic and steric properties of phosphine(III) has made them excellent ligands for the formation of well-defined transition metal complexes,5 although recalcitrant phosphine(V) oxides arise, when phosphine(III) compounds are employed as labile ligands6 or the metal complexes are simply decomposed, in the presence of a suitable oxidant.7 Arguably, the stoichiometric formation of phosphine(V) oxide waste from the above-named organic reactions presents an even greater issue, especially on the industrial scale,3,4 as the conversion of P(V)=O to the P(III) oxidation state is nontrivial (vide infra).
Direct Reduction of Phosphine(V) Oxide
Given the significance of phosphine(III) compounds, a variety of anaerobic syntheses have been reported.8,9 However, the sensitivity of phosphine(III) to oxidation (requiring only minutes to hours) has led to the widespread use of “protected” phosphines,10 such as phosphine–borane adducts11,12 and phosphine(V) sulfides13,14 but predominantly phosphine(V) oxides.15−17 These precursors tolerate the reaction conditions necessary to construct more complex architectures18 although the protection must be removed in the penultimate12,19 or final20,21 step of the ligand synthesis. Thus, much attention has been focused on the conversion of P(V)=O to P(III)15,16 (Scheme 1a), including the use of silanes and siloxanes such as HSiCl3,22−25 HSiCl3/Ph3P,26 Si2Cl6,24,27 Si2Me6 with CsF/TBAF,28 HSi(OEt)3/Ti(O-i-Pr)4,29 PhSiH3,30−32 1,1,3,3-tetramethyldisiloxane (TMDS) with CuX2,33 polymethylhydrosiloxane (PMHS),34,35 1,3-diphenyldisiloxane (DPDS),36 and (EtO)2MeSiH/(RO)2P(O)OH;37 aluminum hydrides such as LiAlH4,38,39 LiAlH4/CeCl3,40 AlH3,41 and HAl(i-Bu)2;42 low-valent metals such as SmI2/HMPA (hexamethylphosphoramide)43 or Cp2TiCl2/Mg;44 hydrocarbon/activated carbon;45 and electrochemical reduction.46−48 A mild iodine-catalyzed reduction of phosphine(V) oxides employing a sacrificial electron-rich phosphine was developed by Laven and Kullberg,49 while Li et al.50 employed less expensive phosphite, although in both cases P(V)=O-containing contaminants must be removed from the final products. Thus, disadvantages of these procedures include harsh reaction conditions, toxic and/or highly reactive, potentially explosive reducing agents, narrow scope or undesirable side reactions, e.g., C–P,51,52 C–O,52 or P–N53−56 bond cleavage, and laborious column chromatography to purify the desired phosphine(III).
Scheme 1. Phosphine Synthesis: Background and This Work.

Left: (a) direct reduction of P(V)=O or P(V)=S affording P(III); (b) conversion of P(V)=O or P(V)=S to activated phosphonium salt; (c) reduction of activated phosphonium salt to P(III); (d) conversion of activated phosphonium salt to phosphine–borane; (e) deprotection of phosphine–borane affording P(III). Right-top: (f) BASFs conversion of Ph3PO to Ph3P using phosgene and silicon. Right-bottom: (g) Paradies et al. recent conversion of Ph3PO to Ph3P using oxalyl chloride and pressurized hydrogen. Center-bottom: (h) this work.
Reduction of Activated Chlorophosphonium Salts
The inherent stability of the P(V)=O has compelled others to explore sequential activation reduction methods, i.e., the conversion of the phosphine(V) oxide to more reactive chlorophosphonium salts (CPS) and subsequent reduction (Scheme 1b,c). Horner, Hoffmann, and Beck first published the reduction of chlorotriphenylphosphonium chloride (Ph3PCl2) in 1958,57 with both LiAlH4 and sodium. The following year a sequential activation and deprotection was published, converting triphenylphosphine(V) oxide (Ph3PO) first to activated CPS, Ph3PCl2, before it was reduced to triphenylphosphine (Ph3P) with sodium metal.58 Being readily afforded via inexpensive chlorinating reagents,59 CPSs have also been reduced with aluminum/metal salts,60 alkali metals,57,58 LiAlH4,57,61,62 thiols/Et3N,63 activated carbon,45 Hantzsch ester/Et3N,64 electrochemically,46−48,65,66 elemental aluminum67,68 or silicon,69 and hydrogenolysis,70 which may be catalyzed by frustrated Lewis pairs (FLPs).71,72 Harsh metal bases and Grignard reagents have even been used to deprotect certain CPSs.73 Alternatively, CPS can be converted to phosphine–boranes by either NaBH474,75 or LiBH4,76−79 although ultimately the borane “protecting group” itself requires removal (Scheme 1b,d,e).
Motivation to Develop a New Facile Reduction of Phosphine(V) Oxides
Our interest in phosphine(V) oxides reduction originates from our desire to explore bulky N-phosphinomethyl-functionalized N-heterocyclic carbene ligands (NHCPs)80,81 as potential ligands for new olefin metathesis catalyst (Scheme 2).19 Progress has been severely hampered due to difficulties accessing azolium salt 5, with the problematic reduction of 4 being achievable only with a large excess of trichlorosilane (27.0 equiv) in anhydrous degassed chlorobenzene at elevated temperature over 2 days.19 As well as the lengthy reaction time, we experienced some reproducibility issues, with the unsuccessful reduction being accompanied by the decomposition of the precious azolinium 4, previously obtained via a multistep synthesis.19 In light of this, a simple procedure for the conversion of 4 to 5 would be a great advantage. Such a process might also permit access to other challenging phosphine(III) and metal catalysts as well as permitting the recovery of the valuable phosphine(III) ligands: “closing the phosphorus cycle” is of increasing importance due to environmental and availability concerns.82−84 Herein, we report a new activation/deprotection of phosphine(V) oxides without the use of harsh reaction conditions, metals, or sacrificial phosphanes. Intermediate CPSs are directly converted to desired phosphines by reaction with hexachlorodisilane. Mechanistic details have been elucidated by experimentation and supported by computation. The “one-pot” procedure affords excellent yields of pure phosphine(III) ligands that can be telescoped into formation of transition metal catalysts without the prior need for silica gel chromatography.
Scheme 2. Problematic Reduction of NHCP Precursor.

Synthesis of NHCP 6via the challenging reduction of phosphine(V) oxide in azolium salt 4 to phosphine(III) 5.
Results and Discussion
Reduction of Activated CPSs with Disilane
In 1996, BASF reported the generation of tetrachlorosilane (SiCl4) when the CPS, Ph3PCl2 (2), was heated with elemental silicon at 185 °C.69 Not wanting to expose our ligand precursor to such harsh reaction conditions, we hypothesized that hexachlorodisilane might serve as a suitable surrogate for elemental silicon and similarly generate 2 equiv of SiCl4 on reactions with a CPS. The abundant industrial byproduct Ph3PO (1) appeared to be the ideal test substrate,3,4 and was easily converted to activated Ph3PCl2 (2) with inexpensive oxalyl chloride.59 Gratifyingly on reaction with 1.1 equiv of hexachlorodisilane (Si2Cl6) at room temperature, both 1H NMR and 31P NMR indicated the immediate, clean, and complete formation of Ph3P (3), with 29Si NMR showing only the formation of tetrachlorosilane, SiCl4 (δ = −18.8 ppm). Motivated by the ability of Si2Cl6 to reduce 2, we chose to explore other disilanes (Table 1, entries 2–10): 1,1,2,2-tetrachloro-1,2-dimethyldisilane (Si2Me2Cl4), hexamethyldisilane (Si2Me6), and hexaphenyldisilane (Si2Ph6), which might generate the corresponding attractive byproducts trichloromethylsilane (MeSiCl3), trimethylsilyl chloride (Me3SiCl), or triphenylsilyl chloride (Ph3SiCl). However, the more electron-rich and sterically hindered disilanes generated the desired phosphines in either lower yield, over extended reaction times or not at all. For instance, the addition of a single electron-donating methyl group to each of the silicon atoms in Si2Me2Cl4 drastically decreased the rate of reaction, with only a 28% conversion to 3 after 24 h, eventually reaching completion after 144 h. In contrast, the reaction with Si2Cl6 was complete in under 5 min.a No reaction was observed for even more electron-rich and sterically shielded Si2Ph6 or Si2Me6.
Table 1. Reaction of Phosphonium Salts with Disilanes.

| entry | CPS 2a–c, X = | disilane | equiv | time | conv to 3 [%]a |
|---|---|---|---|---|---|
| 1 | Cl | Si2Cl6 | 1.1 | 5 min | 100 |
| 2 | Cl | Si2Me2Cl4 | 1.1 | 5 min | 0 |
| 3 | Cl | Si2Me2Cl4 | 1.1 | 1 day | 28 |
| 4 | Cl | Si2Me2Cl4 | 1.1 | 2 days | 55 |
| 5 | Cl | Si2Me2Cl4 | 1.1 | 3 days | 72 |
| 6 | Cl | Si2Me2Cl4 | 1.1 | 4 days | 78 |
| 7 | Cl | Si2Me2Cl4 | 1.1 | 5 days | 83 |
| 8 | Cl | Si2Me2Cl4 | 1.1 | 6 days | 100 |
| 9 | Cl | Si2Me6 | 1.0 | 1 day | 0 |
| 10 | Cl | Si2Ph6 | 1.0 | 1 day | 0 |
| 11 | OTf | Si2Cl6 | 1.1 | 10 min | 7 |
| 12 | OTf | Si2Cl6 | 1.1 | 1 day | 80 |
| 13 | OTf | Si2Cl6 | 1.1 | 2 days | 100 |
| 14 | BArCl | Si2Cl6 | 4 | 2 days | 0 |
Conversion judged by 31P NMR of 2a–c relative to 3.
Scope of the New Procedure
With Si2Cl6 proving to be the reductant of choice, we expanded the application of the procedure to other phosphine(III) compounds.b Aliphatic tricyclohexylphosphine (7) was afforded in 97% yield, in contrast to the recently reported hydrogenation at 130 °C, which notably afforded none of the desired phosphine(III) complexes.71 Cyclic alkene 2-phospholene oxide was also converted to P(III) 2-phospholene (8)77 (98%) without the reduction or isomerization of the C=C bond. Reduction of phosphinamides without the P–N bond scission is particularly challenging;53−56 while Gilheany et al. synthesized “protected” aminophosphine–borane adducts from CPSs in excellent yields,75 we were able to furnish the free aminophosphine 9 directly (89%). The dimethylamino group in DavePhos 11 (93%) was also tolerated well, with fellow Buchwald ligand CyJohnPhos 10 being cleanly afforded in 95% yield. Chiral phosphines8 are still of great significance, and we chose to explore binaphthyl systems as the CPSs of P-chirogenic phosphines are known to racemize.85 The oxides of chiral phosphepines permit structure elaboration,86 and our new method rapidly afforded (S)-Ph-BINEPINE (12)87 (96% yield). (R)-MeO-MOP (13)88 was also readily synthesized (99%). It is of note that the direct reaction of MeO–MOP oxide with Si2Cl6 in acetonitrile led exclusively to scission of the C–O bond without reduction of P(V)=O,52 highlighting the divergence in the reactivity of the activated P(V)Cl2 compared to recalcitrant P(V)=O. Moreover, we observed no racemization in the case of either 12 or 13.
Having established the optimal conditions for the generation of a range of phosphine(III) compounds, we turned our attention back to azolinium 5. The reaction of 4 with excess oxalyl chloride yielded a new chlorophosphonium bearing azolinium salt 15 (after removal of 4-toluenesulfonyl chloride produced by chlorination of the 4-toluenesulfonate; see the Supporting Information) which was readily transformed to the desired azolium 5 with hexachlorodisilane (1.5 equiv). The identity of both salts 5 and 15 was established by single-crystal X-ray diffraction analysis. Crystals suitable for this purpose were obtained by layering methylene chloride with hexane and storing at −30 °C. The salts crystallize in the monoclinic P21/c (CPS 15) and P21/n (azolium 5) space group, respectively. Graphical representation of molecular structure of both compounds is shown in Figure 1. The tetravalent phosphorus atom effectively means each molecule of CPS 15 has two dissociated chloride counteranions: one for each of the cationic phosphonium and the azolinium constituent parts. Interestingly, the asymmetric unit of the crystal lattice of 15 also contained a molecule of hydrochloride (Figure S2).c The additional chloride counterion has important implications for the deprotection of 15, which thus requires 1.5 equiv of hexachlorodisilane to fully convert the CPS to P(III)5: presumably, the extra Cl– counterion of the imidazolium moiety also reacts with Si2Cl6 (vide infra). Finally, mesityl-substituted 5 could be facilely synthesized in an excellent 94% yield, without implementing harsh reaction conditions. In addition, we further demonstrated the usefulness of the new procedure at generating phosphine-bearing azolium salts with the synthesis of the 2,6-diisopropylphenyl analogue 14, in a comparable 92% yield. More details concerning the crystal structure of CPS 15 and azolium 5 can be found in the Supporting Information (Figures S58–S66).
Figure 1.
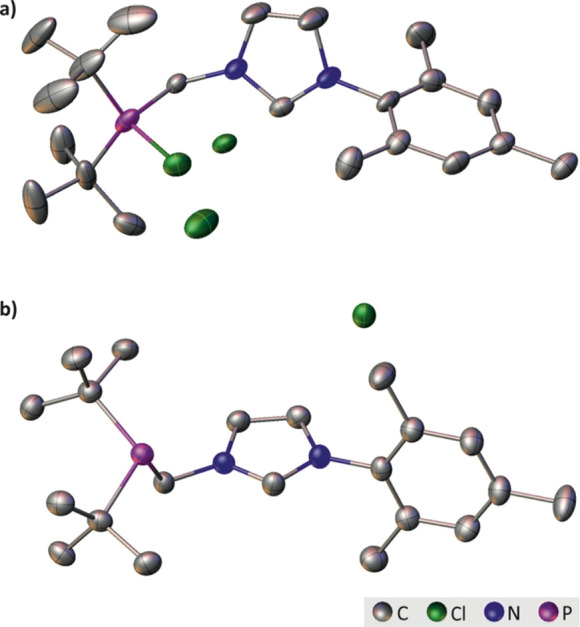
Graphical representation of molecular structure, where (a) CPS 15 and (B) azolium 5. Displacement ellipsoids are drawn at the 50% probability level. The H atoms, the HCl molecule (CPS 15), and the ionic pair “B” (azolium 5) were omitted for clarity.
Table 2. Conversion of Phosphine(V) Oxides to Phosphine(III) Ligands via CPS Intermediates.


The azolium salts were reacted with 5.0 equiv (COCl)2. The resultant CPS was separated from TsCl and then reacted with 1.5–1.6 equiv of Si2Cl6.
Experimental and Computational Mechanism Studies
CPSs in methylene chloride form a cationic phosphonium with a noncoordinated anionic chloride counteranion,89−93 while it has been demonstrated that Cl– (e.g., from ammonium salts) leads to scission of the Si–Si bond in Si2Cl6 (Scheme 3).94−99 This lead us to surmise that the reaction is initiated by the attack of chloride anion at silicon of Si2Cl6 generating an equivalent tetrachlorosilane (SiCl4) and a reactive transient trichlorosilanide anion [:SiCl3]− which then abstracts the remaining phosphorus bound chloride from intermediated 17 to generate the second and final equivalent of SiCl4.
Scheme 3. Reaction Mechanism of Si2Cl6 with Dissociated Chloride Anions.

Known formation of anion [:SiCl3]– from Si2Cl6.
Stepwise reaction mechanism (bottom left).
Concerted mechanism (bottom right).
To explore this mechanistic proposal, chlorotriphenylphosphonium triflate (Ph3PClOTf) 2b was synthesized.100 The triflate anion is a superb nucleofuge, being a far more stable leaving group than chloride;101 therefore, the dissociated triflate ion (TfO–) of 2b would be expected to react much slower with hexachlorodisilane than Cl– of 2a. Indeed, after reaction for 10 min, 31P NMR indicated 5b had generated only 7% Ph3P 6, progressing to 80% and 100% after 24 and 48 h, respectively (Table 1, entries 11–13; Figure 2), significantly slower than the dichloride analogue 2a which appears to react instantly. As with 2a, 29Si NMR analysis of the reaction mixture of monotriflate 2b with Si2Cl6 showed the generated of SiCl4 (singlet at δ = −18.8 ppm) but in addition a singlet at δ = −38.2 ppm. 13C NMR spectra showed a quartet at δ = 118 ppm (J = 320 Hz) and 19F NMR a singlet at δ = −75.6 ppm; these signals are tentatively attributed to trichlorosilyl triflate, SiCl3OTf (see the Supporting Information). Finally, CPS 2c bearing the non-nucleophilic tetrakis(3,5-dichlorophenyl)borate anion, [BArCl]−, was mixed with Si2Cl6 in methylene chloride. As anticipated, no triphenylphosphine 3 was formed, even with an excess of Si2Cl6, demonstrating that the reaction is initiated by the attack of a dissociated anion at silicon.
Figure 2.
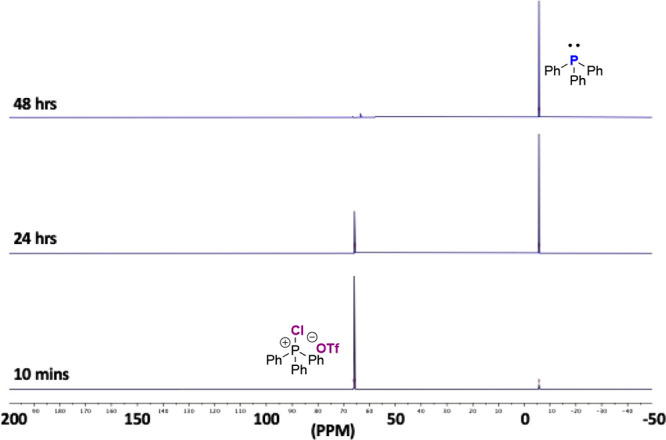
31P NMR (CD2Cl2) of Ph3PClOTf (2b) + Si2Cl6 → PPh3 (3). Reaction times = 10 min (bottom), 24 h (center), and 48 h (top).
To gain further insight, quantum-chemical calculations employing the TURBOMOLE program were performed to study the thermodynamics and kinetics of the reaction. By use of the harmonic oscillator and rigid rotator approximation with a reference pressure of 1 bar, Gibbs free energies are given at the PBE0-D3/def2-TZVPP//PBE-D3/dhf-SV(P) level of theory.102−109 Our calculations show that the disproportionation of CPS into free phosphine with liberation of chlorine is uphill in free energy by 94 kJ/mol; similarly, formation of (unstabilized):SiCl2 by disproportionation of Si2Cl6 is also expected to be very unfavorable, ΔG = 107 kJ/mol. However, the formation of the free phosphine with Si2Cl6 releasing two SiCl4 molecules is thermodynamically favorable, ΔG = −246 kJ/mol (Scheme 3b,c).
Table 3. Conversion of Phosphine(V) Oxides to Their Corresponding Phosphine(III) Ligands and Metal Complexes.


Activated with 3.0 equiv of (COCl)2, deprotected with 2.1 equiv of Si2Cl6.
Activated with 6.0 equiv of (COCl)2, deprotected with 4.1 equiv of Si2Cl6.
A Telescoped Synthesis of Metal Complexes from Their Corresponding Phosphine(V) Oxides
With the new method of generating phosphine(III) ligands with high yield and purity in hand, we attempted to telescope110 the procedure for the synthesis of organometallic catalysts. As such, after deprotection and removal of SiCl4 by evaporation, “intermediate” phosphine(III) compounds were filtered through Celite and then reacted with a suitable metal precursor to yield a selection of prominent phosphine-bearing catalysts. The resultant monodentate triphenylphosphine, tricyclohexylphosphine, and CyJohnPhos were reacted with the dichloro(p-cymene)ruthenium(II) dimer, Umicore M31, and (η3-allyl)palladium(II) dichloride to afford the versatile dichloro(p-cymene)(triphenylphosphine)ruthenium(II) catalyst, 18,111 olefin metathesis catalyst Umicore M2 (Grubbs catalyst M202), 19,112 and the palladium Buchwald complex, CyJohnPhos(η3-allyl)PdCl, 20,113 respectively, in excellent yields (91–98%). Moreover, the oxides of multidentate ligands where similarly reduced and successfully metalated, thus affording bidentate nickel 21(114) and tetradentate palladium complexes 22(115) in good yields of 83% and 86%, respectively.
Conclusions
We have developed a simple mild one-pot activation/deprotection procedure in which phosphine(V) oxides are converted to their corresponding phosphine(III) ligands cleanly and efficiently at ambient temperature without the use of metals or the need for silica gel chromatography. The reduction of activated CPS 2 was investigated with a range of disilanes, and Si2Cl6 was demonstrated to be the best reductant. A reaction mechanism for the transformation has been elucidated through experimentation and supported by computation calculations, with the reduction being initiated by attack of the CPS’s dissociated chloride anion at the silicon of hexachlorodisilane. The new method was successfully applied to a range of aryl and alkyl phosphines, including state-of-the-art ligands, and found to be compatible with alkene, ether, and amine function groups. Challenging phosphine-bearing azolium salts were readily furnished. Furthermore, the high purity of resultant phosphine(III) compounds allowed the procedure to be telescoped for the formation of some prominent transition metal catalysts. We believe this research will facilitate the synthesis of both known and novel new phosphine(III) ligands as well as their corresponding complexes, while the catalytic use, reuse, or recycling of valuable phosphine(III)-based reagents is of importance for sustainability and is likely to be of only greater significance as increased demands or restrictions are placed upon finite phosphorus resources.82−84
Acknowledgments
P.I.J. is grateful to the POLONEZ project financed from the National Science Centre, Poland, on the basis of decision DEC-2016/21/P/ST5/04016. This project received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 665778. The study was performed at the Biological and Chemical Research Centre, University of Warsaw, established within the project cofinanced by the European Union from the European Regional Development Fund under the Operational Programme Innovative Economy, 2007–2013. The X-ray diffraction data collection was collected at the Core Facility for crystallographic and biophysical research to support the development of medicinal products established within the TEAM-TECH Core Facility programme of the Foundation for Polish Science cofinanced by the European Union under the European Regional Development Fund. In addition, P.I.J. is grateful to Sebastian Planer, Michał Da̧browski, and Volodymyr Semeniuchenko for assistance with data acquisition and Marcin Kałek for donation of (S)-Ph-BINEPINE. P.N.P. acknowledges support by the state of Baden-Württemberg through bwHPC (bwUnicluster and JUSTUS, RV bw17D01) and support from the Helmholtz Association is also gratefully acknowledged. P.I.J. and P.H. worked at CaRLa (Catalysis Research Laboratory) of Heidelberg University, being cofinanced by the University of Heidelberg, the State of Baden-Wuerttemberg, and BASF SE.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.organomet.0c00788.
Experimental procedures and characterization data (PDF)
Accession Codes
CCDC 2023530–2023531 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge viawww.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare the following competing financial interest(s): A patent on this research has been applied for. The Polish patent application number is P.426256.
This paper was published ASAP on March 5, 2021, with a typographical error in the title of the paper. The corrected version was reposted on March 9, 2021.
Footnotes
We believe the deprotection occurs immediately, although 5 min had elapsed between addition of Si2Cl6 and acquisition of NMR data.
In the case of Ph3P=O 1, we were able perform a one-pot process without removal of excess oxalyl chloride in vacou: as little as 1.01 equiv of oxalyl chloride was reacted with 1 in dry degassed methylene chloride before 1.04 equiv of Si2Cl6 was added to the intermediate CPS 2, thus completely converting 1 to Ph3P 3in situ. However, for expediency we decided use 1.5 equiv of oxalyl chloride and then strip the excess chlorinating reagent and solvent in vacuo before the CPS salt was once again dissolved in dry methylene chloride and deprotected with 1.04–1.10 equiv of Si2Cl6 (see the Supporting Information for details). It should be noted that residual oxalyl chloride appears to react vigorously with hexachlorodisilane leading to discoloration of phosphine(III) and even undesired byproducts.
The molecule of HCl is likely to arise from oxalyl chloride.
Supplementary Material
References
- Wang Z.Appel Reaction. In Comprehensive Organic Name Reactions and Reagents; American Cancer Society: 2010; pp 95–99. [Google Scholar]
- Fletcher S. The Mitsunobu Reaction in the 21 St Century. Org. Chem. Front. 2015, 2 (6), 739–752. 10.1039/C5QO00016E. [DOI] [Google Scholar]
- Pommer H. The Wittig Reaction in Industrial Practice. Angew. Chem., Int. Ed. Engl. 1977, 16 (7), 423–429. 10.1002/anie.197704233. [DOI] [Google Scholar]
- Eggersdorfer M.; Laudert D.; Létinois U.; McClymont T.; Medlock J.; Netscher T.; Bonrath W. One Hundred Years of Vitamins—A Success Story of the Natural Sciences. Angew. Chem., Int. Ed. 2012, 51 (52), 12960–12990. 10.1002/anie.201205886. [DOI] [PubMed] [Google Scholar]
- Crabtree R. H.The Organometallic Chemistry of the Transition Metals, 6th ed.; Wiley: Hoboken, NJ, 2014. [Google Scholar]
- Ton S. J.; Fogg D. E. The Impact of Oxygen on Leading and Emerging Ru-Carbene Catalysts for Olefin Metathesis: An Unanticipated Correlation Between Robustness and Metathesis Activity. ACS Catal. 2019, 9 (12), 11329–11334. 10.1021/acscatal.9b03285. [DOI] [Google Scholar]
- Borguet Y.; Sauvage X.; Zaragoza G.; Demonceau A.; Delaude L. Synthesis and Catalytic Evaluation in Olefin Metathesis of a Second-Generation Homobimetallic Ruthenium–Arene Complex Bearing a Vinylidene Ligand. Organometallics 2011, 30 (10), 2730–2738. 10.1021/om2001074. [DOI] [Google Scholar]
- Fernández-Pérez H.; Etayo P.; Panossian A.; Vidal-Ferran A. Phosphine–Phosphinite and Phosphine–Phosphite Ligands: Preparation and Applications in Asymmetric Catalysis. Chem. Rev. 2011, 111 (3), 2119–2176. 10.1021/cr100244e. [DOI] [PubMed] [Google Scholar]
- Musina E. I.; Balueva A. S.; Karasik A. A. Phosphines: Preparation, Reactivity and Applications. Organophosphorus Chemistry 2019, 48, 1–63. 10.1039/9781788016988-00001. [DOI] [Google Scholar]
- Pietrusiewicz K. M.; Zablocka M. Preparation of Scalemic P-Chiral Phosphines and Their Derivatives. Chem. Rev. 1994, 94 (5), 1375–1411. 10.1021/cr00029a009. [DOI] [Google Scholar]
- Imamoto T.; Kusumoto T.; Suzuki N.; Sato K. Phosphine Oxides and Lithium Aluminum Hydride-Sodium Borohydride-Cerium(III) Chloride: Synthesis and Reactions of Phosphine-Boranes. J. Am. Chem. Soc. 1985, 107 (18), 5301–5303. 10.1021/ja00304a061. [DOI] [Google Scholar]
- Brill M.; Kühnel E.; Scriban C.; Rominger F.; Hofmann P. A Short and Modular Synthesis of Bulky and Electron-Rich N -Phosphinomethyl-Functionalised N-Heterocyclic Carbene Complexes. Dalton Trans. 2013, 42 (36), 12861–12864. 10.1039/c3dt51777b. [DOI] [PubMed] [Google Scholar]
- Maier L. Organische Phosphorverbindungen XVI. Reduktion von Phosphinsulfiden Zu Den Entsprechenden Dreiwertigen Phosphorverbindungen. Helv. Chim. Acta 1964, 47 (8), 2137–2140. 10.1002/hlca.19640470806. [DOI] [Google Scholar]
- Omelanczuk J.; Mikolajczyk M. Optically Active Trivalent Phosphorus Compounds. 2. Reactivity of Alkylthio- and Alkylselenophosphonium Salts. The First Stereospecific Synthesis of a Chiral Phosphinite. J. Am. Chem. Soc. 1979, 101 (24), 7292–7295. 10.1021/ja00518a026. [DOI] [Google Scholar]
- Hérault D.; Hanh Nguyen D.; Nuel D.; Buono G. Reduction of Secondary and Tertiary Phosphine Oxides to Phosphines. Chem. Soc. Rev. 2015, 44 (8), 2508–2528. 10.1039/C4CS00311J. [DOI] [PubMed] [Google Scholar]
- Kovacs T.; Keglevich G. The Reduction of Tertiary Phosphine Oxides by Silanes. Curr. Org. Chem. 2017, 21 (7), 569–585. 10.2174/1385272821666161108121532. [DOI] [Google Scholar]
- Chrzanowski J.; Krasowska D.; Drabowicz J. Synthesis of Optically Active Tertiary Phosphine Oxides: A Historical Overview and the Latest Advances. Heteroat. Chem. 2018, 29 (5–6), e21476. 10.1002/hc.21476. [DOI] [Google Scholar]
- Corey E. J.; Chen Z.; Tanoury G. J. A New and Highly Enantioselective Synthetic Route to P-Chiral Phosphines and Diphosphines. J. Am. Chem. Soc. 1993, 115 (23), 11000–11001. 10.1021/ja00076a072. [DOI] [Google Scholar]
- Salem H.; Schmitt M.; Herrlich (née Blumbach) U.; Kühnel E.; Brill M.; Nägele P.; Bogado A. L.; Rominger F.; Hofmann P. Bulky N-Phosphinomethyl-Functionalized N-Heterocyclic Carbene Chelate Ligands: Synthesis, Molecular Geometry, Electronic Structure, and Their Ruthenium Alkylidene Complexes. Organometallics 2013, 32 (1), 29–46. 10.1021/om300487r. [DOI] [Google Scholar]
- Muci A. R.; Campos K. R.; Evans D. A. Enantioselective Deprotonation as a Vehicle for the Asymmetric Synthesis of C2-Symmetric P-Chiral Diphosphines. J. Am. Chem. Soc. 1995, 117 (35), 9075–9076. 10.1021/ja00140a028. [DOI] [Google Scholar]
- Qiu L.; Kwong F. Y.; Wu J.; Lam W. H.; Chan S.; Yu W.-Y.; Li Y.-M.; Guo R.; Zhou Z.; Chan A. S. C. A New Class of Versatile Chiral-Bridged Atropisomeric Diphosphine Ligands: Remarkably Efficient Ligand Syntheses and Their Applications in Highly Enantioselective Hydrogenation Reactions. J. Am. Chem. Soc. 2006, 128 (17), 5955–5965. 10.1021/ja0602694. [DOI] [PubMed] [Google Scholar]
- Fritzsche H.; Hasserodt U.; Korte F. Reduktion organischer Verbindungen des fünfwertigen Phosphors zu Phosphinen, II. Reduktion tertiärer Phosphinoxyde zu tertiären Phosphinen mit Trichlorsilan. Chem. Ber. 1965, 98 (1), 171–174. 10.1002/cber.19650980122. [DOI] [Google Scholar]
- Horner L.; Balzer W. D. Phosphororganische verbindungen IXL zum sterischen verlauf der desoxygenierung von tertiären phosphinoxyden zu tertiären phosphinen mit trichlorsilan. Tetrahedron Lett. 1965, 6 (17), 1157–1162. 10.1016/S0040-4039(01)83990-3. [DOI] [Google Scholar]
- Naumann K.; Zon G.; Mislow K. Use of Hexachlorodisilane as a Reducing Agent. Stereospecific Deoxygenation of Acyclic Phosphine Oxides. J. Am. Chem. Soc. 1969, 91 (25), 7012–7023. 10.1021/ja01053a021. [DOI] [Google Scholar]
- Krenske E. H. Theoretical Investigation of the Mechanisms and Stereoselectivities of Reductions of Acyclic Phosphine Oxides and Sulfides by Chlorosilanes. J. Org. Chem. 2012, 77 (8), 3969–3977. 10.1021/jo300346g. [DOI] [PubMed] [Google Scholar]
- Wu H.-C.; Yu J.-Q.; Spencer J. B. Stereospecific Deoxygenation of Phosphine Oxides with Retention of Configuration Using Triphenylphosphine or Triethyl Phosphite as an Oxygen Acceptor. Org. Lett. 2004, 6 (25), 4675–4678. 10.1021/ol048227c. [DOI] [PubMed] [Google Scholar]
- Krenske E. H. Reductions of Phosphine Oxides and Sulfides by Perchlorosilanes: Evidence for the Involvement of Donor-Stabilized Dichlorosilylene | The Journal of Organic Chemistry. J. Org. Chem. 2012, 77 (1), 1–4. 10.1021/jo202370x. [DOI] [PubMed] [Google Scholar]
- Gevorgyan A.; Mkrtchyan S.; Grigoryan T.; Iaroshenko V. O. Disilanes as Oxygen Scavengers and Surrogates of Hydrosilanes Suitable for Selective Reduction of Nitroarenes, Phosphine Oxides and Other Valuable Substrates. Org. Chem. Front. 2017, 4 (12), 2437–2444. 10.1039/C7QO00566K. [DOI] [Google Scholar]
- Coumbe T.; Lawrence N. J.; Muhammad F. Titanium (IV) Catalysis in the Reduction of Phosphine Oxides. Tetrahedron Lett. 1994, 35 (4), 625–628. 10.1016/S0040-4039(00)75855-2. [DOI] [Google Scholar]
- Marsi K. L. Stereochemistry of Some Reactions of Phospholane Derivatives. J. Am. Chem. Soc. 1969, 91 (17), 4724–4729. 10.1021/ja01045a025. [DOI] [Google Scholar]
- Marsi K. L. Phenylsilane Reduction of Phosphine Oxides with Complete Stereospecificity. J. Org. Chem. 1974, 39 (2), 265–267. 10.1021/jo00916a041. [DOI] [Google Scholar]
- Schirmer M.-L.; Jopp S.; Holz J.; Spannenberg A.; Werner T. Organocatalyzed Reduction of Tertiary Phosphine Oxides. Adv. Synth. Catal. 2016, 358 (1), 26–29. 10.1002/adsc.201500762. [DOI] [Google Scholar]
- Li Y.; Das S.; Zhou S.; Junge K.; Beller M. General and Selective Copper-Catalyzed Reduction of Tertiary and Secondary Phosphine Oxides: Convenient Synthesis of Phosphines. J. Am. Chem. Soc. 2012, 134 (23), 9727–9732. 10.1021/ja301764m. [DOI] [PubMed] [Google Scholar]
- Fritzsche H.; Hasserodt U.; Korte F. Reduktion organischer Verbindungen des fünfwertigen Phosphors zu Phosphinen, I. Reduktion tertiärer Phosphinoxyde zu tertiären Phosphinen mit Silanen. Chem. Ber. 1964, 97 (7), 1988–1993. 10.1002/cber.19640970729. [DOI] [Google Scholar]
- Nicolas E.; Guerriero A.; Lyaskovskyy V.; Peruzzini M.; Lammertsma K.; Gonsalvi L.; Slootweg J. C. Metal-Free Reduction of Phosphine Oxides Using Polymethylhydrosiloxane. Inorganics 2016, 4 (4), 34. 10.3390/inorganics4040034. [DOI] [Google Scholar]
- Buonomo J. A.; Eiden C. G.; Aldrich C. C. Chemoselective Reductions of Phosphine Oxides by 1,3-Diphenyl-Disiloxane. Chem. - Eur. J. 2017, 23 (58), 14434–14438. 10.1002/chem.201703875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Lu L.-Q.; Das S.; Pisiewicz S.; Junge K.; Beller M. Highly Chemoselective Metal-Free Reduction of Phosphine Oxides to Phosphines. J. Am. Chem. Soc. 2012, 134 (44), 18325–18329. 10.1021/ja3069165. [DOI] [PubMed] [Google Scholar]
- Hein F.; Issleib K.; Rabold H. Über die Reduktion von tertiären Phosphinoxyden bzw. -sulfiden mit Lithium- bzw. Calciumalanat zu den entsprechenden Phosphinen. Z. Anorg. Allg. Chem. 1956, 287 (4–6), 208–213. 10.1002/zaac.19562870406. [DOI] [Google Scholar]
- Henson P. D.; Naumann K.; Mislow K. Stereomutation of Phosphine Oxides by Lithium Aluminum Hydride. J. Am. Chem. Soc. 1969, 91 (20), 5645–5646. 10.1021/ja01048a041. [DOI] [Google Scholar]
- Imamoto T.; Takeyama T.; Kusumoto T. Facile Reduction of Organic Halides and Phosphine Oxides with LiAlH4–CeCl3. Chem. Lett. 1985, 14 (10), 1491–1492. 10.1246/cl.1985.1491. [DOI] [Google Scholar]
- Griffin S.; Heath L.; Wyatt P. Alane — A Novel Way to Reduce Phosphine Oxides. Tetrahedron Lett. 1998, 39 (24), 4405–4406. 10.1016/S0040-4039(98)00748-5. [DOI] [Google Scholar]
- Busacca C. A.; Raju R.; Grinberg N.; Haddad N.; James-Jones P.; Lee H.; Lorenz J. C.; Saha A.; Senanayake C. H. Reduction of Tertiary Phosphine Oxides with DIBAL-H. J. Org. Chem. 2008, 73 (4), 1524–1531. 10.1021/jo7024064. [DOI] [PubMed] [Google Scholar]
- Handa Y.; Inanaga J.; Yamaguchi M. Rapid and Mild Deoxygenation of Organoheteroatom Oxides with the Efficient Electron Transfer System SmI 2 – Tetrahydrofuran–Hexamethylphosphoric Triamide. J. Chem. Soc., Chem. Commun. 1989, 0 (5), 298–299. 10.1039/C39890000298. [DOI] [Google Scholar]
- Mathey F.; Maillet R. Reduction Des Oxydes de Phosphines Par Le Systeme Cp2TiCl2-Mg. Tetrahedron Lett. 1980, 21 (26), 2525–2526. 10.1016/0040-4039(80)80117-1. [DOI] [Google Scholar]
- Dockner T. Reduction and Hydrogenation with the System Hydrocarbon/Carbon [New Synthetic Methods (70)]. Angew. Chem., Int. Ed. Engl. 1988, 27 (5), 679–682. 10.1002/anie.198806791. [DOI] [Google Scholar]
- Elias J. S.; Costentin C.; Nocera D. G. Direct Electrochemical P(V) to P(III) Reduction of Phosphine Oxide Facilitated by Triaryl Borates. J. Am. Chem. Soc. 2018, 140 (42), 13711–13718. 10.1021/jacs.8b07149. [DOI] [PubMed] [Google Scholar]
- Chakraborty B.; Menezes P. W.; Driess M. Beyond CO2 Reduction: Vistas on Electrochemical Reduction of Heavy Non-Metal Oxides with Very Strong E—O Bonds (E = Si, P, S). J. Am. Chem. Soc. 2020, 142 (35), 14772–14788. 10.1021/jacs.0c05862. [DOI] [PubMed] [Google Scholar]
- Manabe S.; Wong C. M.; Sevov C. S. Direct and Scalable Electroreduction of Triphenylphosphine Oxide to Triphenylphosphine. J. Am. Chem. Soc. 2020, 142 (6), 3024–3031. 10.1021/jacs.9b12112. [DOI] [PubMed] [Google Scholar]
- Laven G.; Kullberg M.. A Process for the Reduction of a Tertiary Phosphine Oxide to the Corresponding Tertiary Phosphine in the Presence of a Catalyst and Use of a Tertiary Phosphine for Reducing a Tertiary Phosphine Oxide in the Presence of a Catalyst. WO2011123037 (A1), October 6, 2011.
- Li P.; Wischert R.; Métivier P. Mild Reduction of Phosphine Oxides with Phosphites To Access Phosphines. Angew. Chem., Int. Ed. 2017, 56 (50), 15989–15992. 10.1002/anie.201709519. [DOI] [PubMed] [Google Scholar]
- Quin L. D.; Rao N. S. 1-Phenyl-Cis-3a,7a-Dihydrophosphindole and Its Properties. J. Org. Chem. 1983, 48 (21), 3754–3759. 10.1021/jo00169a029. [DOI] [Google Scholar]
- Higham L. J.; Clarke E. F.; Müller-Bunz H.; Gilheany D. G. P-Chirogenic Phosphines. MOP/DiPAMP Hybrids, Their Oxide Crystal Structures, Reduction Studies and Alternative Syntheses. J. Organomet. Chem. 2005, 690 (1), 211–219. 10.1016/j.jorganchem.2004.09.015. [DOI] [Google Scholar]
- Henson P. D.; Ockrymiek S. B.; Markham R. E. Reductive Cleavage of Phosphinanilides with Lithium Aluminum Hydride. J. Org. Chem. 1974, 39 (15), 2296–2298. 10.1021/jo00929a039. [DOI] [Google Scholar]
- Quin L. D.; Keglevich G. Stereochemistry of the Reaction of Oxygen Nucleophiles with a Phosphinous Chloride in the 7-Phosphanorbornene Series. J. Chem. Soc., Perkin Trans. 2 1986, 2 (7), 1029–1034. 10.1039/p29860001029. [DOI] [Google Scholar]
- Szewczyk J.; Quin L. D. Stereochemical Studies with Aminophosphines and Related Compounds Having the 7-Phosphanorbornene Structure. J. Org. Chem. 1987, 52 (7), 1190–1196. 10.1021/jo00383a003. [DOI] [Google Scholar]
- Kuroboshi M.; Kita T.; Aono A.; Katagiri T.; Kikuchi S.; Yamane S.; Kawakubo H.; Tanaka H. Reduction of Phosphine Oxides to the Corresponding Phosphine Derivatives in Mg/Me3SiCl/DMI System. Tetrahedron Lett. 2015, 56 (7), 918–920. 10.1016/j.tetlet.2014.12.132. [DOI] [Google Scholar]
- Horner L.; Hoffmann H.; Beck P. Phosphororganische Verbindungen, XVI. Wege zur Darstellung primärer, sekundärer und tertiärer Phosphine. Chem. Ber. 1958, 91 (8), 1583–1588. 10.1002/cber.19580910803. [DOI] [Google Scholar]
- Horner L.; Beck P.; Hoffmann H. Phosphororganische Verbindungen, XIX. Reduktion von Phosphorverbindungen mit Alkalimetallen. Chem. Ber. 1959, 92 (9), 2088–2094. 10.1002/cber.19590920920. [DOI] [Google Scholar]
- Rajendran K. V.; Kennedy L.; O’Connor C. T.; Bergin E.; Gilheany D. G. Systematic Survey of Positive Chlorine Sources in the Asymmetric Appel Reaction: Oxalyl Chloride as a New Phosphine Activator. Tetrahedron Lett. 2013, 54 (51), 7009–7012. 10.1016/j.tetlet.2013.10.044. [DOI] [Google Scholar]
- Yano T.; Hoshino M.; Kuroboshi M.; Tanaka H. A Practical One-Pot Transformation of Triphenylphosphine Oxide to Triphenylphosphine by Reduction of in Situ Generated Triphenylphosphine Dichloride. Synlett 2010, 2010 (5), 801–803. 10.1055/s-0029-1219194. [DOI] [Google Scholar]
- Byrne P. A.; Rajendran K. V.; Muldoon J.; Gilheany D. G. A Convenient and Mild Chromatography-Free Method for the Purification of the Products of Wittig and Appel Reactions. Org. Biomol. Chem. 2012, 10 (17), 3531–3537. 10.1039/c2ob07074j. [DOI] [PubMed] [Google Scholar]
- Carr D. J.; Kudavalli J. S.; Dunne K. S.; Müller-Bunz H.; Gilheany D. G. Synthesis of 2,3-Dihydro-1-Phenylbenzo[b]Phosphole (1-Phenylphosphindane) and Its Use as a Mechanistic Test in the Asymmetric Appel Reaction: Decisive Evidence against Involvement of Pseudorotation in the Stereoselecting Step. J. Org. Chem. 2013, 78 (20), 10500–10505. 10.1021/jo401318g. [DOI] [PubMed] [Google Scholar]
- Masaki M.; Fukui K. Reaction of Tertiary Phosphine Dichlorides with Thiols in the Presence of Triethylamine. a Convenient Method for the Reduction of Phosphine Oxides to Phosphines. Chem. Lett. 1977, 6 (2), 151–152. 10.1246/cl.1977.151. [DOI] [Google Scholar]
- Zhang T.-X.; Zhang W.-X.; Luo M.-M. Metal-Free Reduction of Tertiary Phosphine Oxides with Hantzsch Ester. Chin. Chem. Lett. 2014, 25 (1), 176–178. 10.1016/j.cclet.2013.09.007. [DOI] [Google Scholar]
- Yano T.; Kuroboshi M.; Tanaka H. Electroreduction of Triphenylphosphine Dichloride and the Efficient One-Pot Reductive Conversion of Phosphine Oxide to Triphenylphosphine. Tetrahedron Lett. 2010, 51 (4), 698–701. 10.1016/j.tetlet.2009.11.115. [DOI] [Google Scholar]
- Kuroboshi M.; Yano T.; Kamenoue S.; Kawakubo H.; Tanaka H. Electroreduction of Tetra-Coordinate Phosphonium Derivatives; One-Pot Transformation of Triphenylphosphine Oxide into Triphenylphosphine. Tetrahedron 2011, 67 (32), 5825–5831. 10.1016/j.tet.2011.05.044. [DOI] [Google Scholar]
- Bassler P.; Hammes P.; Hermeling D.; Hugo R.; Lechtken P.; Siegel H.. Preparation of Triphenylphosphine. US5527966A, June 18, 1996.
- Hugo R.; Bassler P. D.; Hammes P. D.; Hermeling D. D.; Lechtken P. D.; Siegel H. D.. Process for the Preparation of Triphenyl Phosphine. EP0638580B1, December 30, 1998.
- Wettling T. D.Process for the Preparation of Tertiary Phosphines. EP0548682B1, November 6, 1996.
- Masaki M.; Kakeya N. Hydrogenolysis of Trisubstituted Dichlorophosphoranes—A New Method for Deoxygenation of Oxophosphoranes. Angew. Chem., Int. Ed. Engl. 1977, 16 (8), 552–553. 10.1002/anie.197705522. [DOI] [Google Scholar]
- Stepen A. J.; Bursch M.; Grimme S.; Stephan D. W.; Paradies J. Electrophilic Phosphonium Cation-Mediated Phosphane Oxide Reduction Using Oxalyl Chloride and Hydrogen. Angew. Chem., Int. Ed. 2018, 57 (46), 15253–15256. 10.1002/anie.201809275. [DOI] [PubMed] [Google Scholar]
- Zhu H.; Qu Z.-W.; Grimme S. Reduction of Phosphine Oxide by Using Chlorination Reagents and Dihydrogen: DFT Mechanistic Insights. Chem. - Eur. J. 2019, 25 (18), 4670–4672. 10.1002/chem.201900379. [DOI] [PubMed] [Google Scholar]
- Vetter A. C.; Nikitin K.; Gilheany D. G. Long Sought Synthesis of Quaternary Phosphonium Salts from Phosphine Oxides: Inverse Reactivity Approach. Chem. Commun. 2018, 54 (46), 5843–5846. 10.1039/C8CC02173B. [DOI] [PubMed] [Google Scholar]
- Rajendran K. V.; Gilheany D. G. Simple Unprecedented Conversion of Phosphine Oxides and Sulfides to Phosphine Boranes Using Sodium Borohydride. Chem. Commun. 2012, 48 (6), 817–819. 10.1039/C1CC14856G. [DOI] [PubMed] [Google Scholar]
- Kenny N. P.; Rajendran K. V.; Jennings E. V.; Gilheany D. G. Cleavage of P=O in the Presence of P–N: Aminophosphine Oxide Reduction with In Situ Boronation of the PIII Product. Chem. - Eur. J. 2013, 19 (42), 14210–14214. 10.1002/chem.201302907. [DOI] [PubMed] [Google Scholar]
- Al Sulaimi S. S.; Rajendran K. V.; Gilheany D. G. Lithium Borohydride for Achiral and Stereospecific Reductive Boronation at Phosphorus: Lack of Electronic Effects on Stereoselective Formation of Alkoxyphosphonium Salts. Eur. J. Org. Chem. 2015, 2015 (27), 5959–5965. 10.1002/ejoc.201500521. [DOI] [Google Scholar]
- Herbay R.; Bagi P.; Fogassy E.; Keglevich G. Preparation of P-Heterocyclic Phosphine Boranes and Optically Active Phosphine Oxides via Phosphonium Salts. Phosphorus, Sulfur Silicon Relat. Elem. 2016, 191 (11–12), 1656–1657. 10.1080/10426507.2016.1225060. [DOI] [Google Scholar]
- Herbay R.; Bagi P.; Mucsi Z.; Mátravölgyi B.; Drahos L.; Fogassy E.; Keglevich G. A Novel Preparation of Chlorophospholenium Chlorides and Their Application in the Synthesis of Phospholene Boranes. Tetrahedron Lett. 2017, 58 (5), 458–461. 10.1016/j.tetlet.2016.12.059. [DOI] [Google Scholar]
- Bagi P.; Herbay R.; Ábrányi-Balogh P.; Mátravölgyi B.; Fogassy E.; Keglevich G. Dynamic Kinetic Resolution of 1-Substituted-3-Methyl-3-Phospholene Oxides via the Formation of Diastereomeric Alkoxyphospholenium Salts. Tetrahedron 2018, 74 (40), 5850–5857. 10.1016/j.tet.2018.07.058. [DOI] [Google Scholar]
- Weskamp T.; Böhm V. P. W.; Herrmann W. A. Combining N-Heterocyclic Carbenes and Phosphines: Improved Palladium(II) Catalysts for Aryl Coupling Reactions. J. Organomet. Chem. 1999, 585 (2), 348–352. 10.1016/S0022-328X(99)00237-5. [DOI] [Google Scholar]
- Hofmann P.; Brill M.. NHCP Ligands for Catalysis. In Molecular Catalysts; John Wiley & Sons, Ltd.: 2014; pp 207–234. [Google Scholar]
- Withers P. J. A.; Elser J. J.; Hilton J.; Ohtake H.; Schipper W. J.; van Dijk K. C. Greening the Global Phosphorus Cycle: How Green Chemistry Can Help Achieve Planetary P Sustainability. Green Chem. 2015, 17 (4), 2087–2099. 10.1039/C4GC02445A. [DOI] [Google Scholar]
- Withers P. J. A. Closing the Phosphorus Cycle. Nat. Sustain. 2019, 2 (11), 1001–1002. 10.1038/s41893-019-0428-6. [DOI] [Google Scholar]
- Keijer T.; Bakker V.; Slootweg J. C. Circular Chemistry to Enable a Circular Economy. Nat. Chem. 2019, 11 (3), 190–195. 10.1038/s41557-019-0226-9. [DOI] [PubMed] [Google Scholar]
- Nikitin K.; Jennings E. V.; Al Sulaimi S.; Ortin Y.; Gilheany D. G. Dynamic Cross-Exchange in Halophosphonium Species: Direct Observation of Stereochemical Inversion in the Course of an SN2 Process. Angew. Chem., Int. Ed. 2018, 57 (6), 1480–1484. 10.1002/anie.201708649. [DOI] [PubMed] [Google Scholar]
- Enthaler S.; Erre G.; Junge K.; Michalik D.; Spannenberg A.; Marras F.; Gladiali S.; Beller M. Enantioselective Rhodium-Catalyzed Hydrogenation of Enol Carbamates in the Presence of Monodentate Phosphines. Tetrahedron: Asymmetry 2007, 18 (11), 1288–1298. 10.1016/j.tetasy.2007.06.001. [DOI] [Google Scholar]
- Kalek M.; Fu G. C. Phosphine-Catalyzed Doubly Stereoconvergent γ-Additions of Racemic Heterocycles to Racemic Allenoates: The Catalytic Enantioselective Synthesis of Protected α,α-Disubstituted α-Amino Acid Derivatives. J. Am. Chem. Soc. 2015, 137 (29), 9438–9442. 10.1021/jacs.5b05528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Q.; Zeng H.; Yang Z. New Route for Synthesis of MeO-MOP. Synth. Commun. 2011, 41 (23), 3556–3560. 10.1080/00397911.2010.519095. [DOI] [Google Scholar]
- Al-Juboori M. A. H. A.; Gates P. N.; Muir A. S. Ionic–Molecular Isomerism in Chlorophenylphosphoranes Ph n PCl 5 – n (1 ⩽ n ⩽ 3). J. Chem. Soc., Chem. Commun. 1991, 0 (18), 1270–1271. 10.1039/C39910001270. [DOI] [Google Scholar]
- Godfrey S. M.; McAuliffe C. A.; Sheffield J. M. Structural Dependence of the Reagent Ph3PCl2 on the Nature of the Solvent, Both in the Solid State and in Solution; X-Ray Crystal Structure of Trigonal Bipyramidal Ph3PCl2, the First Structurally Characterised Five-Coordinate R3PCl2 Compound. Chem. Commun. 1998, 8, 921–922. 10.1039/a800820e. [DOI] [Google Scholar]
- Godfrey S. M.; Hinchliffe A.; Mkadmh A. Ab Initio Studies on the Reagent Ph3PCl2. J. Mol. Struct.: THEOCHEM 2005, 719 (1), 85–88. 10.1016/j.theochem.2005.01.027. [DOI] [Google Scholar]
- Nikitin K.; Müller-Bunz H.; Gilheany D. Direct Evidence of a Multicentre Halogen Bond: Unexpected Contraction of the P–XXX–P Fragment in Triphenylphosphine Dihalides. Chem. Commun. 2013, 49 (14), 1434–1436. 10.1039/c2cc38363b. [DOI] [PubMed] [Google Scholar]
- Vetter A. C.; Nikitin K.; Gilheany D. G. Exploring an Umpolung Strategy for Quaternization of Phosphorus. Phosphorus, Sulfur Silicon Relat. Elem. 2019, 194 (4–6), 339–342. 10.1080/10426507.2018.1541242. [DOI] [Google Scholar]
- Wilkins C. J. 682. The Reactions of Hexachlorodisilane with Ammonium Halides and Trimethylamine Hydrohalides. J. Chem. Soc. 1953, 3409–3412. 10.1039/jr9530003409. [DOI] [Google Scholar]
- Cooper G. D.; Gilbert A. R. Cleavage and Disproportionation of Polychlorodisilanes, Trichloromethylchlorosilanes and Hexachlorodisiloxane by Amines and Ammonium Salts. J. Am. Chem. Soc. 1960, 82 (19), 5042–5044. 10.1021/ja01504a008. [DOI] [Google Scholar]
- Tillmann J.; Meyer L.; Schweizer J. I.; Bolte M.; Lerner H.-W.; Wagner M.; Holthausen M. C. Chloride-Induced Aufbau of Perchlorinated Cyclohexasilanes from Si2Cl6: A Mechanistic Scenario. Chem. - Eur. J. 2014, 20 (30), 9234–9239. 10.1002/chem.201402655. [DOI] [PubMed] [Google Scholar]
- Teichmann J.; Bursch M.; Köstler B.; Bolte M.; Lerner H.-W.; Grimme S.; Wagner M. Trapping Experiments on a Trichlorosilanide Anion: A Key Intermediate of Halogenosilane Chemistry. Inorg. Chem. 2017, 56 (15), 8683–8688. 10.1021/acs.inorgchem.7b00216. [DOI] [PubMed] [Google Scholar]
- Georg I.; Teichmann J.; Bursch M.; Tillmann J.; Endeward B.; Bolte M.; Lerner H.-W.; Grimme S.; Wagner M. Exhaustively Trichlorosilylated C1 and C2 Building Blocks: Beyond the Müller–Rochow Direct Process. J. Am. Chem. Soc. 2018, 140 (30), 9696–9708. 10.1021/jacs.8b05950. [DOI] [PubMed] [Google Scholar]
- Teichmann J.; Wagner M. Silicon Chemistry in Zero to Three Dimensions: From Dichlorosilylene to Silafullerane. Chem. Commun. 2018, 54 (12), 1397–1412. 10.1039/C7CC09063C. [DOI] [PubMed] [Google Scholar]
- Hwang S. J.; Powers D. C.; Maher A. G.; Nocera D. G. Tandem Redox Mediator/Ni(II) Trihalide Complex Photocycle for Hydrogen Evolution from HCl. Chem. Sci. 2015, 6 (2), 917–922. 10.1039/C4SC02357A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhakal B.; Bohé L.; Crich D. Trifluoromethanesulfonate Anion as Nucleophile in Organic Chemistry. J. Org. Chem. 2017, 82 (18), 9263–9269. 10.1021/acs.joc.7b01850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer A.; Horn H.; Ahlrichs R. Fully Optimized Contracted Gaussian Basis Sets for Atoms Li to Kr. J. Chem. Phys. 1992, 97 (4), 2571–2577. 10.1063/1.463096. [DOI] [Google Scholar]
- Schäfer A.; Huber C.; Ahlrichs R. Fully Optimized Contracted Gaussian Basis Sets of Triple Zeta Valence Quality for Atoms Li to Kr. J. Chem. Phys. 1994, 100 (8), 5829–5835. 10.1063/1.467146. [DOI] [Google Scholar]
- Eichkorn K.; Treutler O.; Öhm H.; Häser M.; Ahlrichs R. Auxiliary Basis Sets to Approximate Coulomb Potentials. Chem. Phys. Lett. 1995, 240 (4), 283–290. 10.1016/0009-2614(95)00621-A. [DOI] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77 (18), 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Häser M.; Patzelt H.; Ahlrichs R. RI-MP2: Optimized Auxiliary Basis Sets and Demonstration of Efficiency. Chem. Phys. Lett. 1998, 294 (1), 143–152. 10.1016/S0009-2614(98)00862-8. [DOI] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7 (18), 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Weigend F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8 (9), 1057–1065. 10.1039/b515623h. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132 (15), 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Hayashi Y. Pot Economy and One-Pot Synthesis. Chem. Sci. 2016, 7 (2), 866–880. 10.1039/C5SC02913A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fürstner A.; Liebl M.; Lehmann C. W.; Picquet M.; Kunz R.; Bruneau C.; Touchard D.; Dixneuf P. H. Cationic Ruthenium Allenylidene Complexes as Catalysts for Ring Closing Olefin Metathesis. Chem. - Eur. J. 2000, 6 (10), 1847–1857. 10.1002/(SICI)1521-3765(20000515)6:10<1847::AID-CHEM1847>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Boeda F.; Clavier H.; Nolan P. S. Ruthenium – Indenylidene Complexes: Powerful Tools for Metathesis Transformations. Chem. Commun. 2008, 0 (24), 2726–2740. 10.1039/b718287b. [DOI] [PubMed] [Google Scholar]
- Kisanga P.; Widenhoefer R. A. Development, Synthetic Scope, and Mechanistic Studies of the Palladium-Catalyzed Cycloisomerization of Functionalized 1,6-Dienes in the Presence of Silane. J. Am. Chem. Soc. 2000, 122 (41), 10017–10026. 10.1021/ja001730+. [DOI] [Google Scholar]
- Jarrett P. S.; Sadler P. J. Nickel(II) Bis(Phosphine) Complexes. Inorg. Chem. 1991, 30 (9), 2098–2104. 10.1021/ic00009a028. [DOI] [Google Scholar]
- Aizawa S.; Iida T.; Funahashi S. Mechanistic Studies on Halo-Ligand Substitution of Five-Coordinate Trigonal-Bipyramidal Palladium(II) Complexes of Tris(2-(Diphenylphosphino)Ethyl)Phosphine with Trimethyl Phosphite in Chloroform at Various Temperatures and Pressures. Inorg. Chem. 1996, 35 (18), 5163–5167. 10.1021/ic9601596. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.