

Abstract
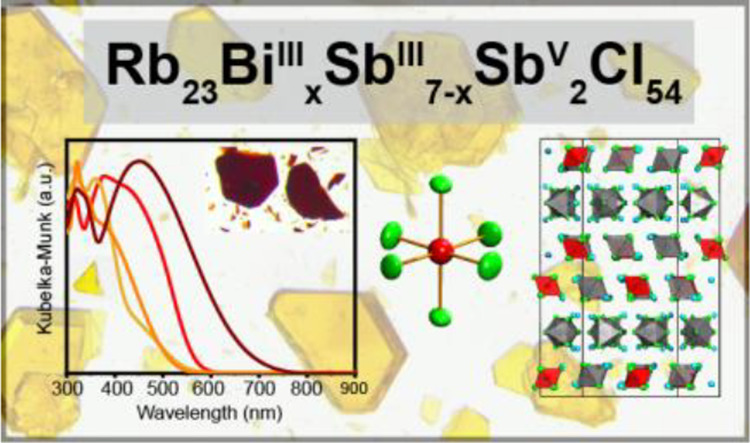
Mixed-valent metal-halides containing ns2 lone pairs may exhibit intense visible absorption, while zero-dimensional (0D) ns2-based metal-chlorides are generally colorless but have demonstrated promising optoelectronic properties suitable for thermometry and radiation detection. Here, we report solvothermally synthesized mixed-valent 0D metal-halides Rb23BiIIIxSbIII7–xSbV2Cl54 (0 ≤ x ≤ 7). Rb23SbIII7SbV2Cl54 crystallizes in an orthorhombic space group (Cmcm) with a unique, layered 0D structure driven by the arrangement of the 5s2 lone pairs of the SbIIICl6 octahedra. This red material is likely the true structure of a previously reported monoclinic “Rb2.67SbCl6” phase, the structure of which was not determined. Partially or fully substituting SbIII with isoelectronic BiIII yields the series Rb23BiIIIxSbIII7–xSbV2Cl54 (0 < x ≤ 7), which exhibits a similar layered 0D structure but with additional disorder that yields a trigonal crystal system with an enantiomorphic space group (R32). Second harmonic generation of 532 nm light from a 1064 nm laser using Rb23BiIII7SbV2Cl54 powder confirms the noncentrosymmetry of this space group. As with the prototypical mixed-valent pnictogen halides, the visible absorption bands of the Rb23BiIIIxSbIII7–xSbV2Cl54 family are the result of intervalent SbIII–SbV and mixed-valent BiIII–SbV charge transfer bands (CTB), with a blueshift of the absorption edge as BiIII substitution increases. No PL is observed from this family of semiconductors, but a crystal of Rb23BiIII7SbV2Cl54 exhibits a high resistivity of 1.0 × 1010 Ω·cm and X-ray photoconductivity with a promising μτ product of 8.0 × 10–5 cm2 s–1 V–1. The unique 0D layered structures of the Rb23BiIIIxSbIII7–xSbV2Cl54 family highlight the versatility of the ns2 lone pair in semiconducting metal-halides, pointing the way toward new functional 0D metal-halide compounds.
Introduction
The vibrant colors of mixed-valent solid-state compounds have captivated chemists since the discovery of the famous Fe(II,III) pigment, Prussian blue, in the early 18th century. Subsequent studies have developed a rich chemistry of compounds with transition or post-transition metals in two distinct oxidation states,1 which exhibit unusual behavior including superconductivity,2,3 magnetic phase transitions,4,5 and an intervalence charge transfer band (CTB) in absorption.6−8
While such mixed-valent materials come in a variety of structures and compositions, mixed-valent pnictogen halides exclusively exhibit cubic or tetragonal unit cells with the general formula, A4MIIISbVX12 (A = K, Rb, Cs; MIII = Sb, Bi, In, Tl; X = Cl, Br; Figure 1). These can be considered as transmutations of the vacancy-ordered double perovskites, A2MIVX6 (e.g., MIV = Sn,9,10 Te,11 Pb12)—a well-known 0D system in which MIVX6 octahedra are separated by regular vacancies and arranged with cations, such as Cs or Rb, into an antifluorite structure.13,14 In the mixed-valent pnictogen halides, the transmutation of the 4+ charge is achieved by doubling the cell and splitting half of the filled sites into a 3+ and the other half into a 5+, i.e., doubling the formula from A2MIVX6 to A4MIIIMVX12 (often written A2MIII0.5MV0.5X6 to maintain the connection to the parent structure type). The first reported mixed-valent main group metal-halide of this type was the intensely indigo-colored Cs2SbCl6, first reported in 1882.15
Figure 1.
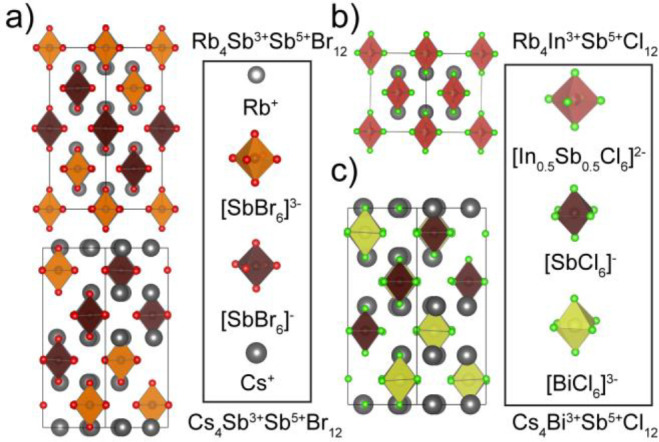
Structures of mixed-valent metal halides at RT: (a) (Rb/Cs)4Sb3+Sb5+Br12; (b) Rb4InSbCl12; and (c) Cs4BiSbCl12.
The strong color of these compounds was unexpected because undoped, low-dimensional ternary chlorides containing either trivalent or pentavalent pnictogen metal-centers tend to be colorless both in solution and in the solid state.
The earliest reports on such phases mistakenly described the indigo-black Cs2SbCl6 and, later, the black Cs2SbBr6, as containing a tetravalent Sb cation;16,17 however, this oxidation state is unknown for Sb and the mixed-valent hypothesis began to gain traction. Magnetic measurements later demonstrated these materials to be diamagnetic, thereby confirming the mixed-valent composition Cs4SbIIISbVX12,18 and the optical features of this mixed valency were first described in 1963 by Peter Day.7
These materials were then intensely investigated in the 20th century as a prototypical example of charge transfer (CT) in the solid state, due in part to the accessibility of their optical properties which lie in the visible spectrum.19,20 At this point, a growing library of A4MIIIMVX12 mixed-valent materials began to develop with various combinations of SbV and MIII (MIII = Sb, Bi, In, Tl, Fe, and Rh) as well as bromides and chlorides,1,7,21−24 allowing for comparative studies of the optical and electrical properties as well as structures. On the basis of these observations and other works, Robin and Day published a seminal paper that put forth the Robin-Day classification of mixed-valence compounds, in which class I compounds have distinct sites with very stable, trapped valencies that lack mixed-valent transitions in the visible range, class II mixed-valent compounds offer distinguishable valences that possess sufficient orbital overlap to induce a visible mixed-valent absorption transition, and class III compounds have shared valencies on indistinguishable sites with maximal orbital delocalization.1
It was demonstrated that for the intensely colored A4MIIIMVX12 compositions containing BiIII and SbIII, the CT occurs from the ns2 orbital of the MIII cation to the 5s0 orbital of the SbV cation, thereby explaining the shift in the CTB into the visible range.1,7,19 In contrast, InIII and TlIII lack the lone pair and thus the nd10 to ns0 transition occurs at much higher energy, yielding a pale yellow color and a corresponding change from a Robin-Day class II mixed-valent system for A4MIIIMVX12 compounds containing SbIII and BiIII to a class I system in those with InIII and TlIII.1,7,19 The crucial nature of the lone pair was further demonstrated in a series of experiments in Cs4SbIIIxInIII1–xSbVCl12 showing that the intensity of the CTB is proportional to the product of [SbIII] and [MV] concentrations, and hence was at its maximum intensity when the ratio [SbIII]:[MV] = 1 in both the solid state as well as in solution.1
The through-space or outer-sphere nature of the energy transfer was supported by both electrical conductivity and resistivity measurements as well as structural analysis. These materials form disconnected structures with nearly cubic symmetry, and the phases all tend to adopt either cubic structures, in which MIII and SbV sites are identical, or tetragonal structures with unique sites for MIII and SbV (Figure 1; Table 1).24 Importantly, there are no extended paths or chains through these 0D structures for CT; additionally, there are no aggregates such as dimers or trimers that could provide inner-sphere CT mechanism.1,28 This outer-sphere mechanism was further supported with the observation of high resistivities (107–1015 Ω·cm) and negligible photoconductivity (Table S1 of the Supporting Information, SI).8,20,22 Electrical measurements within the series Cs4SbIIIxInIII1–xSbVCl12 again showcased the importance of the ns2 lone pair in this outer-sphere conduction pathway, as compositions with x > ∼0.1 demonstrated electronic conductivity proportional to the product of [SbIII] and [SbV] concentrations, while the pure InIII composition, and those lacking sufficient SbIII to generate an outer-sphere charge pathway, exhibited high resistance and ionic conductivity.20
Table 1. Summary of A4MIIISbVX12 Phases.
| composition | space group | a | c | Abs max (nm) | ref. |
|---|---|---|---|---|---|
| Cs4SbIIISbVCl12 | I41/amd | 10.4650 (2) | 21.0095 (7) | 540.5 | (19,24,25) |
| Cs4BiIIISbVCl12 | I41/amd | 10.492 (2) | 21.1017 (6) | 417 | (24,25) |
| Cs4InIIISbVCl12 | Fm-3m | 9.9093 (1)a | 590 | (19,24) | |
| Rb4SbIIISbVBr12 | I41/amd | 10.706 (3)a | 21.695 (22) | “black” | (21,23) |
| Rb4InIIISbVCl12 | Fm-3m | 10.0613 (1)a | (24) | ||
| Cs4SbIIISbVBr12 | I41/amd | 10.842 (3) | 21.91 (15) | “black” | (18,24,26) |
| (NH4)4SbIIISbVBr12 | I41/amd | 10.66 | 21.52 | 1053 | (21,25,27) |
| “Rb4SbIIISbVCl12” | 10.14 (2) | 510.2 | (19,21,25) |
Value at 4.7 K.
Although there is a large body of work related to the structures and properties of this family of materials, the structure and properties of the Rb–SbIII–BiIII–SbV–Cl phases remain underexplored. While the tetragonal Rb4SbIIISbVBr12 is known (Table 1),22,23 the corresponding chloride has been reported to be either cubic,21 cubic yet slightly disordered,18 or to decompose into a reportedly monoclinic Rb2.67SbCl6 phase for which the structure is unknown.28,29 Furthermore, no reports exist on Rb4BiIIISbVCl12 or any related phase.
In this work, we report the mixed-valent pnictogen halide series Rb23BiIIIxSbIII7–xSbV2Cl54 (0 ≤ x ≤ 7) and describe the structural and optoelectronic properties of these materials. The red Rb23SbIII7SbV2Cl54 crystallizes in a unique, layered 0D structure driven by the arrangement of the 5s2 lone pairs of SbIII. Substitution with Bi3+ generates a solid solution of disordered variants of this structure, the Rb23BiIIIxSbIII7–xSbV2Cl54 (0 < x ≤ 7) family, which exhibit large unit cells (V ≈ 16 000 Å3) that are demonstrated to be noncentrosymmetric through second harmonic generation (SHG) measurements. The visible absorption of these materials is driven by CTBs of the MIIIns2 orbitals to SbV 5s0 orbitals although electrical measurements show Rb23BiIII7SbV2Cl54 to be a resistive semiconductor (1.0 × 1010 Ω·cm) with a weak X-ray photoresponse (μτ product of 8.0 × 10–5 cm2V–1).
Experimental Section
Materials
Rubidium chloride (RbCl, 99+%) and hydrogen peroxide (H2O2, > 30%) were purchased from ACROS. Antimony(III) oxide (Sb2O3, 99%), bismuth(III) oxide (Bi2O3, 99.9%), and nitric acid (HNO3, ≥ 65%) were purchased from Sigma-Aldrich. Hydrochloric acid (HCl, 37%) was purchased from VWR. All solids and acids were stored and handled under ambient conditions, while H2O2 was kept refrigerated. Inductively coupled plasma mass spectrometry (ICP–MS) reference standards of Rb, In, Sb, Ho, Ir, and Bi were purchased from Inorganic Ventures or Merck and were used to prepare tuning, calibration, and sample solutions. Concentrated nitric acid (>65 wt %; purified by double sub-boiling distillation), ultrapure Normatom hydrochloric acid (32–35 wt % purchased from VWR chemicals) and purified water (≥18.2 MΩ·cm, Millipore, Billerica, U.S.A.) were used for the sample preparation. All chemicals were used as received without additional purification. Stainless steel autoclaves from Parr instruments and Amar Equipment were utilized with PTFE containers for all solvothermal syntheses.
Synthesis of Rb23Sb9Cl54
In a typical synthesis of Rb23Sb9Cl54, three solutions are first prepared. The first solution, containing Sb(V), is prepared by dissolving the appropriate amount of Sb2O3 in HCl (typically 1 mL, 0.6 M in Sb3+). Any visibly insoluble material was separated via filtration before proceeding. Once a clear, colorless solution had been obtained, the Sb(III) was oxidized to Sb(V) by the addition of a large molar excess of H2O2. As H2O2 was added, the solution changed color to yellow, indicating the coexistence of Sb(III) and Sb(V). To ensure complete oxidation, the solution was heated to 80–100 °C. Once the solution had become completely colorless (after 20–30 min), this solution was left to cool to RT. At this time two additional solutions were prepared, one containing Sb2O3 dissolved in HCl (2 mL, 1.04 M in Sb3+) and another containing RbCl dissolved in HCl (4 mL, 1.71 M). The solution containing Sb(III) was added to the Sb(V) solution to again produce a yellow solution; this additional step allows for more precise control of the Sb(III):Sb(V) ratio in the reaction.
This solution, containing Sb(III) and Sb(V), was then slowly pipetted into the RbCl/HCl solution resulting in the formation of a colored precipitate. To obtain single crystals, the precipitate was not separated from the mother liquor. Instead, the entire mixture was added to a Teflon lined autoclave and heated to 160 °C at 50 °C h–1. This temperature was held for 48 h before cooling back to RT at 2–5 °C h–1. The resulting crystals could be separated from the mother liquor by vacuum filtration. To ensure that they were dry enough for storage, the crystals were kept under dynamic vacuum for a minimum of 30 min or until they were visibly dry and free-flowing.
Synthesis of Rb23BiIIIxSbIII7–xSbV2 Cl54:
The procedure to obtain Bi3+ substituted materials, is the same as above except that the Sb(III) solution (unoxidized) is replaced with a Sb(III)/Bi(III) solution prepared with the desired Sb(III):Bi(III) ratio. If a white precipitate forms immediately upon the addition of the Sb(III)/Bi(III)/Sb(V) solutions to the RbCl/HCl solution, then the reaction should be stirred until a color change has been observed.
Single Crystal X-ray Diffraction
Single-crystal X-ray diffraction (XRD) measurements were conducted at room temperature on a Bruker Smart, Bruker Smart Apex 2, and Oxford Xcalibur S. Each diffractometer is equipped with a molybdenum sealed-tube X-ray source (Mo Kα; λ = 0.71073 Å) and graphite monochromators. Crystals were tip-mounted with Lithelen or paraffin oil. Multiscan CrysAlis PRO 1.171.39.31d (Rigaku Oxford Diffraction, 2017) was used for the absorption correction. Empirical absorption corrections were performed using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm. Data was processed and integrated with CrysAlis PRO 1.171.39.31d (Rigaku OD, 2017), solved using SHELXT,30 and refined using SHELXL30 through the Olex231 interface. Crystal structure images were generated using Vesta32 and Diamond33 software packages.
Powder X-ray Diffraction (PXRD) and Temperature-Dependent PXRD
A STADI P diffractometer (STOE & Cie GmbH, Darmstadt, Germany) in transmission mode (Debye–Scherrer geometry) was used to collect powder diffraction patterns. This diffractometer is equipped with a silicon strip MYTHEN 1K detector (Fa. DECTRIS), a curved Ge (111)-monochromator, and a Cu X-ray source (Cu Kα1, λ = 1.54056 Å). For temperature-dependent PXRD measurements, a STADI P diffractometer (STOE & Cie GmbH, Darmstadt, Germany) in transmission mode (Debye–Scherrer geometry) was used to collect powder diffraction patterns. This diffractometer is equipped with a silicon strip MYTHEN 1K detector (Fa. DECTRIS), a curved Ge (111)-monochromator, and a Molybdenum X-ray source (λ = 0.70930 Å). The sample was sealed in a glass capillary, and diffraction patterns were measured at −173, −73, 25, and 200 °C.
Inductively Coupled Plasma Mass Spectrometry (ICPMS)
The analysis was carried out using a sector-field inductively coupled plasma mass spectrometer (Element XR, ThermoFisher, Bremen, Germany). The solutions were introduced using a micro concentric nebulizer (200 μL min–1, borosilicate glass, glass expansion) combined with a cyclonic spray chamber (borosilicate glass, glass expansion), quartz injector, torch with guard electrode, sampler, and skimmer made of nickel. Further instrumental parameters were adjusted as follows: a nebulizer gas flow of 1.06 L min–1, a Plasma gas flow of 16 L min–1, an auxiliary gas flow of 1 L min–1, and a power of 1350 W.
The isotopes 85Rb, 115In, 121Sb, 165Ho, and 209Bi were measured in the low resolution (m/Δm = 300) mode using the e-scan mode. Six runs and six passes were performed yielding a total measurement time of approximately 2 min per sample.
An external calibration was carried out. The limits of detection (LODs) for Rb, In, Sb, and Bi were determined to be 19.5 ng L–1, 2.4 ng L–1, 12.7 ng L–1 and 0.7 ng L–1, respectively. To validate the method and exclude any matrix effects, some samples were spiked with Rb, In, Sb, and Bi ICP reference standards. For additional details, see SI Note S1.
UV–vis Absorption Measurements
A Jasco V670 spectrophotometer equipped with an integrating sphere (ILN-725) was used to record diffuse reflectance spectra of microcrystalline powders. The absorption spectra were then calculated using a Kubelka–Munk (KM) transformation.
Second Harmonic Generation (SHG)
Samples for SHG measurements were ground in a mortar and pestle and pressed between quartz slides, which were held together at the edges by a UV-curable resin. A Nd:YAG laser (Duetto from Time-bandwidth) with a wavelength of 1064 nm (10 ps pulses) was focused to a spot size of 50–100 μm on the sample. The focused laser was then moved along the powdered sample until a bright SHG active spot could be located. Second harmonic light was then detected with a fiber-coupled CCD spectrometer (CCS200 from Thorlabs).
X-ray Photoconductivity and I–V Measurements
A Keithley 236 source measurement unit was used to record I–V curves, crystals were contacted using Ag paint. For the characterization with X-rays, the Keithley 237 source measurement unit was used to apply a bias voltage while X-rays were generated using a Mini–X Amptek X-ray tube with an accelerating voltage of 50 kV and an X-ray tube current of 50 μA.
Results and Discussion
Red, hexagonal plates of Rb23SbIII7SbV2Cl54 were grown solvothermally from an HCl solution (Figure 2a). The crystals tend to grow together and stack, resulting in varying color intensities. Previously, two phases, the cubic Rb4SbIIISbVCl12 and the monoclinic Rb2.67SbCl6 (equivalent to Rb24Sb9Cl54 or Rb24SbIII7.5SbV1.5Cl54, not far off from the true stoichiometry), were reported to form in the Rb–SbIII/V–Cl compositional space. While the former was suggested to be cubic, it was also reported to decompose spontaneously at room-temperature to the monoclinic phase, which was observed to grow as hexagonal red platelets. At first glance, this agrees with the appearance of our crystals except that they do not exhibit birefringence, which would suggest either the presence of twins or a higher symmetry axis such as that found in a nearly cubic system.
Figure 2.

Structural characterization of Rb23SbIII7SbV2Cl54. (a) Photo of the crystalline material obtained from a typical solvothermal synthesis; scale bar is 1 mm in length. (b) The unit cell viewed along (9b) the (210) plane and (c) the b-axis. (d) Comparison of [SbCl6]3– and [SbCl6]− octahedra, as emphasized in (b), with purple bonds showing the lengthened bonds (greater than 2.75 Å) along the direction of SbIII lone pair expression. Note that Sb1 is rotated to highlight the distorted disphenoidal geometry of the 4 short bonds (yellow). (e) Arrangement of SbCl6 octahedra as viewed along the b-axis, with long SbIII–Cl bonds shown in purple and Rb atoms removed for clarity. [SbCl6]3– octahedra (gray), [SbCl6]− octahedra (red), and Rb+ cations (light blue); all thermal ellipsoids in this work are drawn at 35% probability.
Surprisingly, single crystal XRD at room temperature reveals that Rb23SbIII7SbV2Cl54 adopts an orthorhombic structure with the Cmcm space group (a = 22.3435(7) Å, b = 12.9057(4) Å, c = 37.0625(13) Å; Figure 2; Tables S2–S5). This structure, while also consisting of zero-dimensional MX6 octahedra separated by Rb+, significantly deviates from those observed for other MIII-SbV systems, in which the structures adopt a cubic or tetragonal unit cell with all octahedra effectively aligned along one of the unit cell axes (Figure 1). In contrast, the octahedra in Rb23SbV7SbIII2Cl54 arrange into layers with distinct orientations: these layers stack along c in a repeating ABC-C′B′A′ sequence, where C′B′A′ are related to ABC by a glide plane (Figure 2b). In this stacking, the SbIIICl6 and SbVCl6 octahedra in slab I and slab III are aligned with their faces along the a-b plane while the octahedra in slab II (comprised entirely of SbIIICl6) are rotated so that they lie on their edges, and slabs III′, II′, and I′ are related by symmetry to slabs I, II, III (best viewed along the (210) plane, Figure 2b). Slabs I and III are also differentiated from slab II by their contents: each contains one SbVCl6 octahedra for every three SbIIICl6 octahedra (aligned along the b-axis) while Slab II has none, yielding the composition Rb23SbIII7SbV2Cl54 (Figure 2c). This composition was further supported by ICPMS measurements (Table S6).
There are four antimony sites in this structure with three distinct geometries and sizes: sites Sb1 and Sb2 comprise slab II and exhibit similar disphenoidal distortions due to the stereoactive 5s2 lone pair of SbIII, while Sb3 and Sb4 make up slabs I and III. Sb3 exhibits a trigonal lone pair distortion and Sb4 (the SbV site) possesses a nearly perfect octahedron (Figure 2d). The average Sb–Cl bond lengths of Sb1 (2.660 Å), Sb2 (2.644 Å), and Sb3 (2.647 Å) are consistent with those of other SbIIICl6 octahedra in the literature (e.g., 2.637 Å in Rb7Sb3Cl1634), while that of Sb4 is significantly smaller at 2.345 Å, supporting the assignment of SbV on this site (Table S7). Within slabs I and III the [SbVCl6]− octahedra of Sb4 are surrounded by six [SbCl6]3– octahedra, with an average Sb···Sb distance of 7.50 Å and no Rb cations directly between them (Figure S1). The SbIII octahedra in Rb23SbIII7SbV2Cl54 are slightly more distorted than those in Rb7Sb3Cl16, suggesting greater lone-pair stereoactivity.34
The origin of the layered Rb23SbIII7SbV2Cl54 structure is the confluence of the lone-pair repulsion between the SbIII ions and the templating effect of the smaller Rb+ cation. The SbIII sites exhibit longer bonds in the direction of the active 5s2 lone-pair,35 shown in purple to highlight the direction of the lone-pair expression, and the repulsion of these lone-pairs leads to the arrangement shown in Figure 2e. The lone-pair expression of the SbIII octahedra in slabs I and III are aligned against each other pointing toward slab 2, pushing the SbV octahedra slightly in the other direction.
This two-layer ordering precludes the addition of a third layer with trigonal SbIII slabs, as the resulting distortion would express lone pairs toward one or the other layers and cause repulsion. Thus, slab II consists of disphenoidally distorted SbIII octahedra (an unusual coordination for SbIII, usually only observed as part of an edge-shared Sb2X10 dimer, e.g., Rb7Sb3X1636) with octahedra oriented differently than the other two slabs, these effects express the lone pairs in-plane so as to avoid repulsion with those of the other two slabs, thus driving the overall 3-fold ordering of this complex structure.
To further understand the origin of this new structure type, which deviates significantly from those of the A4MIIIMVX12 phases, we examined the coordination environments of Rb in this structure, as the A-site cations are considered the structure-directing atoms in related systems such as the halide perovskites.37 By assigning each unique Rb-site a different color according to its coordination number (Figure S2), the structure-directing effect of the Rb polyhedra becomes evident (Figure S3a). Given the much stronger bonding between Sb–Cl as compared to Rb–Cl, some Rb atoms are forced to sacrifice their coordination number to satisfy the overall stoichiometry, resulting in coordination numbers ranging from 6 to 12 (Table S8). The 6-fold coordinated Rb are squeezed into a trigonal prism around which the surrounding layer is ordered (Figure S3b,c). Although Rb is known to adopt 6-fold octahedral coordination in simple Rb-halide binary compounds, 6-fold coordination is not commonly observed in ternary metal halides. Furthermore, trigonal prismatic 6-fold coordination is even rarer. One such example is Rb4CdBr6 (ICSD 39621), which exhibits pseudotrigonal prismatic 6-fold coordination as a result of a 5° twist. We consider this unusual Rb-halide geometry to most likely occur in Rb23SbIII7SbV2Cl54 as a result of lone-pair expression in Sb3+ octahedra. Note that the lone pairs of the three neighboring SbIIICl6 octahedra all point toward this special site (Figure S3b,c), while the bond valence sums of these disphenoidal Sb sites are less than 3 (Table S7). This indicates that the excess charge of the expressed lone pair is partially shared by this seemingly under-coordinated Rb site, permitting the unusually low Rb coordinations that serve to template this offset slab II. This 6-fold coordinate Rb, and the gradient in Rb-coordination that results, generates the overall ordering and corrugation in the unit cell.
In our initial attempts to synthesize single crystals of Rb23SbIII7SbV2Cl54, it also became evident that the choice of oxidant during the synthesis plays a crucial role in obtaining a phase-pure product. Nitric acid, for example, is known to be a powerful oxidant. When used for the synthesis of Rb23SbIII7SbV2Cl54, large, colorless rods are formed as a side product. These were found to be the RbSbCl6 phase (Figure S4; Tables S9–S12). To avoid this excess oxidation, the oxidant was substituted for H2O2, which could be more readily decomposed.
PXRD demonstrates that the measured crystals of Rb23SbIII7SbV2Cl54 were representative of the entire reaction outcome (Figure S5). Additionally, temperature dependent PXRD measured from −150 to 150 °C only demonstrated lattice contraction and expansion without any evidence for a phase change; we were unable to obtain the reported metastable cubic Rb4SbIIISbVCl12 phase (Figure S6).
The deeply colored appearance of these crystals arises from CT that is expected to occur from the 5s2 state of the SbIII to the 5s0 of the SbV.1 While clearly separated, this distance is reasonable for outer sphere electron transfer38 as distances of up to 20 Å can be surmounted in biological systems through this process.39 However, the rates of electron transfer typically have an inverse exponential dependence on the distance.40 For this reason, the shortest donor–acceptor distance, or in this case SbIII···SbV distance, is likely to be the most important. While the SbIII···SbV distance within each slab (I or III) is approximately 7.50 Å, the interslab (i.e., between I and I′ or III and III′) SbIII···SbV distance can be as low as 7.085(3) Å (Figure 2e). Additionally, the octahedra between these two layers are aligned with respect to each other, whereas they have different orientations when compared with the SbIII-only layer.
Noting the absence of any reports for a Rb2BiIIISbVCl12 phase, we sought to substitute Sb3+ with Bi3+ and investigate how the structure and CTB would be altered by the expected change in unit cell size, octahedral separation, and electronic configuration. Our initial attempts were centered on obtaining Sb3+ rich phases (approximately 2:1 SbIII:BiIII) and occasionally resulted in crystals that appeared brown. Upon closer examination, it became evident that in these cases two phases were present and that colorless Rb7Bi3–3xSb3xCl16 crystals had nucleated and served as a substrate for the intergrowth of smaller, red crystals (Figure 3a).34
Figure 3.

Structural and optical properties of the Rb23BixSb7−xSbV2Cl54 phase. (a) Optical images of the Rb23BixSb7−xSbV2Cl54 series with the color changing from red to yellow as x increases, becoming more Bi-rich. (b) The unit cell of Rb23Bi6.62Sb0.38SbV2Cl54 displayed along the (210) plane. (c) Metal-halide octahedral sites of the ordered and disordered layers, as emphasized in (b), with purple bonds showing the lengthened bonds along the direction of BiIII(SbIII) lone pair expression. Note that the Cl atoms of site Bi6 are 1/3 occupied, reflecting the 3-fold rotational disorder of the octahedra. d) Structure field diagram of mixed-valent metal-halide phases and Rb23BixSb7−xSbV2Cl54 phases. (e) KM spectrum for Rb23BiIII7SbV2Cl54.
The structure of this red material was determined by single crystal X-ray diffraction on a crystal similar to that shown in Figure 3a. This material was found to crystallize in a trigonal crystal system with the R32 space group with a unit cell that is unexpectedly large for a fully inorganic material (a = b = 12.95670(10) Å, c = 111.7130(14) Å, V = 16241.4(3) Å3, Tables S13–S15). We note that an inversion twin law was used to solve this enantiomorphic structure, as this compound crystallizes in a racemic mixture. Refining mixed occupancies on each MIII site in the structure (Table S16), we determined the composition of this red phase to be Rb23Bi2.50Sb6.50Cl54, with the Bi:Sb ratio corroborated by ICPMS analysis (Table S6).
Further adjusting the Sb3+ and Bi3+ precursor ratios, we were able to synthesize a solid solution that exists among all the M3+ octahedra of this structure to obtain the series Rb23BixSb7–xSbV2Cl54 (0 < x ≤ 7), which were all found to crystallize in the same trigonal space group as the mixed-metal samples. More specifically, the fully Bi3+ substituted endmember, Rb23BiIII7SbV2Cl54, crystallizes into pale orange hexagonal plates with the R32 space group with a = b = 12.9752(6) Å, c = 112.438(8) Å, V = 16393.5(18) Å3 (Figure 3a,b; Figure S7; Tables S17–S20). As before, an inversion twin law was used to refine this structure as a racemic mixture of two enantiomorphs. Note that while the single crystal refinement yields a stoichiometry of Rb23Bi6.62Sb2.38Cl54, the ICPMS results (Table S6) yield a Bi:Sb ratio of 3.5(1), corresponding to the Bi-saturated composition Rb23BiIII7SbV2Cl54.
This 0D structure contains metal-halide octahedra and adopts layers that stack in 3-slab units akin to those of Rb23SbIII7SbV2Cl54, but this structure exhibits disorder in every other 3-slab unit (Figure 3b). These slabs alternate in the rhombohedral symmetry in the sequence O–D–O′–D–O″–D to yield the tripled unit cell, where O denotes an ordered 3-slab layer and D denotes a disordered layer. This disorder is characterized by a 3-fold rotationally disordered site with 1/3 occupancy of the Cl atoms in three disphenoidally distorted Bi3+ octahedra (Bi6), with the neighboring site (Bi4/Sb4) mixed such that the location of the Sb5+ octahedra in these slabs cannot be identified except by the significantly shorter average bonds of the site (Figure 3c, Table S5). The smallest of the octahedra in the ordered layers are again those containing only Sb5+, which have an average bond length of 2.322 Å, and the remaining two sites exhibit trigonal and disphenoidal distortions due to the 5s2 lone pair (Figure 3c, Table S20).
Given that the previously mentioned and published mixed-valent structures all exhibited either cubic or tetragonal unit cells at room temperature, we created a structure field diagram to observe how these new structures could be related to the other known compositions. This was prepared by comparing the A/X and B/X size ratios for different symmetries and compositions (Figure 3d).41 On the basis of this diagram it is clear that the nonexistent “Rb4SbIIISbVCl12” and the newly found Rb23BiIIIxSbIII7–xSbV2Cl54 series occupy an otherwise empty region distinct from the other cubic and tetragonal phases. Furthermore, these crystals, like the red Rb23Sb9Cl54, exhibit a strong absorption band in the visible range, unlike either pure Bi3+, Sb3+, or pure Sb5+ materials (Figure 3e).
To understand the structural similarities and demonstrate a one-to-one mapping of the 0D units between these two structures, we first identified the composition of each plane of atoms that constitute the Rb23Sb9Cl54 structure based on a convention introduced by Mattfeld and Meyer and previously utilized by Ruck et al. (Figure 4a).42 Each set of numbers directly references the relative amount of A, M, or X ions in the plane. For example, the first and third layers of octahedra (slabs I and III) in Rb23Sb9Cl54 have planes with compositions 103 and 010, which indicates that (i) the Rb cations are in the same plane as the halides and (ii) that these layers are effectively equivalent to the (111) index for cubic structures or the (112) index for tetragonal structures (Table 1). This additionally emphasizes the relation of the Rb23Sb9Cl54 structure to the other known mixed-valent compositions.
Figure 4.

Structural relationship between orthorhombic Rb23Sb9Cl54 and trigonal Rb23Bi7Sb2Cl54. (a) Layer-by-layer breakdown showing one-to-one mapping of MCl6 octahedra in each layer, with Mattfeld notation denoting the occupancy of AMX composition in each octahedral environment. (b) Unit cell of each structure viewed along the b-axis with Rb atoms removed for clarity, highlighting the offset stacking of SbV octahedra in Rb23Bi7Sb2Cl54 that yields a tripled lattice constant c for the rhombohedral structure relative to orthorhombic Rb23Sb9Cl54. (c) Unit cells of each structure’s disphenoidal layer (II) viewed along the c-axis, showing the mapping of the a and b axes of both unit cells.
The layer between I and III is given the designation “slab II” as it has differently oriented octahedra. The atom counting of this layer is slightly more complex than the previous case; however, the same counting rules apply; i.e., only atoms within the unit cell are counted and atoms on faces, edges, or corners are counted accordingly while also taking occupancy into account. Within layer II, planes are composed of 5/302 and 1/312 atoms and, incidentally, this layer contains the undercoordinated Rb that permits the formation of the superstructure. The following layers are identified as I′, II′, and III′ and they are related to their counterparts by a glide plane.
The overall structure of Rb23Bi7Sb2Cl54 has several repeating layers, one three-layer block resembling that of Rb23SbIII7SbV2Cl54 (“O”) and a second three-layer block with disordered octahedra sandwiched by octahedra with large thermal parameters (“D”), these units arrange similarly to Rb23SbIII7SbV2Cl54 (Figure 4a). From unwrapped images taken along the b-axis, it becomes clear that the disorder occurring in every other slab (marked as D) is intrinsic based on the diffuse scattering in the h0l and h1l images (Figure S8).
The relationship between “O” slabs in Rb23BiIII7SbV2Cl54 and the Rb23Sb9Cl54 structure are
clear as these slabs are identical to one another. By describing the
compositions of the constituent planes in the “D” slabs
with the same Mattfeld notation, it also becomes apparent that the
subunits of each slab are equivalent (Figure 4a). Slabs IV and VI of the D-trilayer reduce
to the same 103 and 010 compositions as I′ and III′
in Rb23Sb9Cl54; furthermore, slab
V has a composition of 506 and 136 (when considering occupancy), which
reduces to the same 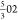 and
and 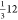 atoms as slab II′
in Rb23Sb9Cl54 (Figure 4a). The relations between each
crystallographic site
in these two structures is described in the SI Note S2, Tables S7, S16, and S20.
atoms as slab II′
in Rb23Sb9Cl54 (Figure 4a). The relations between each
crystallographic site
in these two structures is described in the SI Note S2, Tables S7, S16, and S20.
The tripling of the c-axis of the R32 structure relative to that of Rb23Sb9Cl54 is a result of the different stacking of the SbV and SbIII octahedra along the a-axis, with the orthorhombic structure deriving from aligned SbV octahedra while those of Rb23Bi7Sb2Cl54 shift by one octahedral unit (1/3 of the structure), such that it takes three repetitions to achieve an equivalent layer (Figure 4b). Finally, the relations of the a and b axes can be observed by examining the mapping of the ordered slab II with each unit cell (Figure 4c). Clearly, the disorder present within the structure forces the selection of an alternate unit cell and changes the symmetry, but it does not affect the underlying structural building blocks or the overall composition. This may be because both BiIII and SbIII possess ns2 lone pairs, thus substitution alters the size of individual MIIICl6 octahedra while the ordering of the MIIICl6 and SbVCl6 octahedra is maintained.
Given the unusually large unit cells of the structures in the Rb23BixSbIII7–xSb2Cl54 series and the enantiomorphic nature of this space group, we measured SHG to check whether we had correctly ascribed the R32 space group to Rb23Bi7SbV2Cl54. Briefly, R32 is one of the space groups that allows for the occurrence of piezoelectricity. Effects such as piezoelectricity and SHG are related to the symmetry of the structure rather than the chemical composition, and hence serve as a good evidence for noncentrosymmetry in a structure. To examine this property, we exposed a polycrystalline powder of Rb23BiIII7SbV2Cl54 to a pulsed 1064 nm laser (10 ps pulse duration).
A powder is well suited as it contains all possible crystal orientations thus there will be some grains where crystal axes orient toward the incident beam according to a phase-matched condition, required for the SHG process. While scanning the focused laser beam (50–100 μm spot size) across the sample, some areas would produce visible green spots. Spectrally, this SHG emission occurs as a sharp line located exactly at 532 nm (Figure 5a). Furthermore, the intensity of this peak is expected to have a quadratic power dependence. When log(intensity) was plotted against power, a line with a slope of 2 was found, further confirming the lack of centrosymmetry in Rb23Bi7Sb2Cl54 and supporting the assignment of the R32 space group (Figure 5b).43,44
Figure 5.
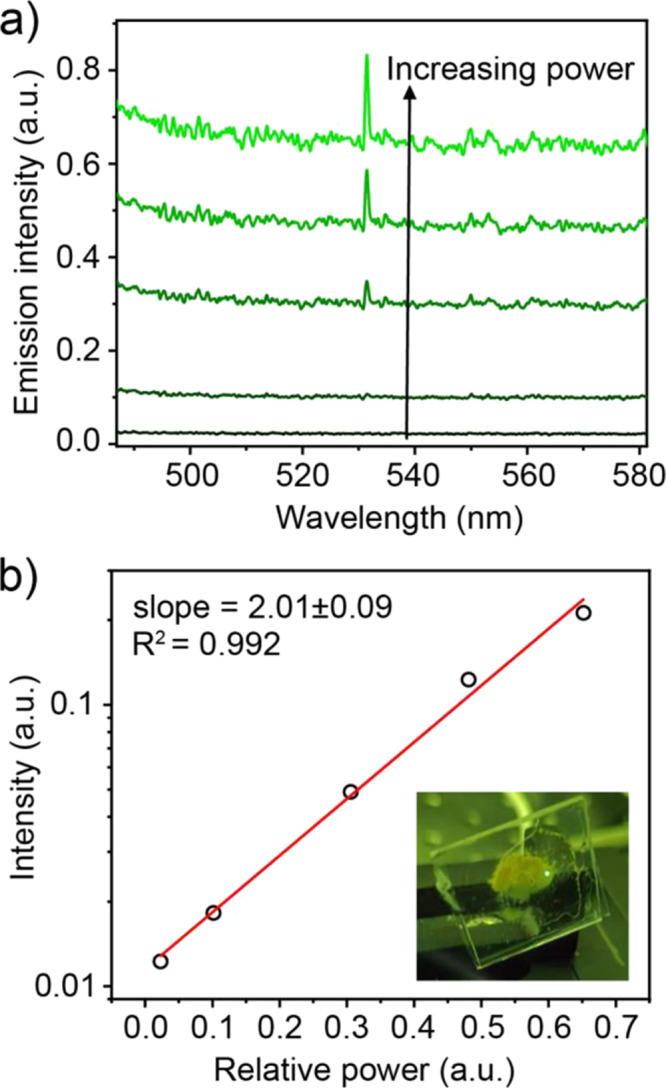
Second harmonic generation in Rb23Bi7Sb2Cl54. (a) Spectral view of power dependence. (b) Linear fit of log(SHG intensity) vs power.
Structurally, very little changes between the Bi3+ only, Bi3+ rich, and Bi3+ poor samples, as observed by the similarities of the crystallographic sites in each structure (Tables S7, S16, and S20). The Sb5+ octahedra remain in the same position and the overall superstructure corrugation is still observable. This solid solution that exists within Rb23BixSbIII7–xSb2Cl54 is demonstrated by the linear dependence of the c-lattice parameter on the ratio of BiIII:SbIII (Figure 6a). While the changes to composition within the Rb23BixSbIII7–xSb2Cl54 series do not strongly affect the structure, they do affect the energy of the CTB that gives these phases their intense colors (Figure 6b). For example, Rb23Bi7Sb2Cl54 is a light-yellow crystal (inset image in Figure 6a); this can also be observed from the KM spectra measured for a series of compositions in the Rb23BixSbIII7–xSb2Cl54 series (Figure 6b). From these, it can be concluded that Bi3+–Sb5+ and SbIII–SbV CT interactions occur at visibly different energies, with the SbIII–SbV interaction at lower energy. However, they appear to be too close in energy and too broad to deconvolute the spectra and meaningfully describe the contributions of each transition to the overall spectral shape.
Figure 6.
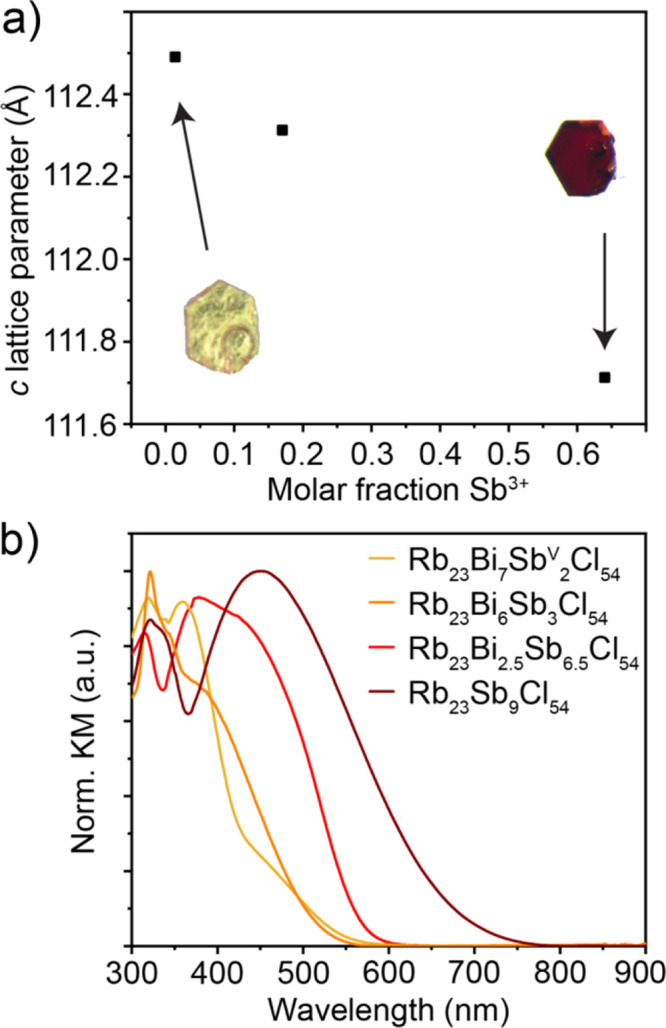
Tunability in the Rb23BixSbIII7–xSb2Cl54 system. (a) c-lattice parameter vs molar fraction of Sb3+. (b) CTB tunability.
Having confirmed the structures of these materials and having observed CTB tunability, we turned our attention toward potential applications for these materials. Considering the existence of the visible CTB, we further examined if any of these materials exhibited photoluminescence (PL). While no reports have indicated that MIII/SbV mixed-valent materials exhibit any PL, previous studies on Wolfram’s red salt (WRS; [Pt(C2H5NH2)4][Pt(C2H5NH2)4Cl2]Cl4·4H2O), and N,N-dimethylaminobenzylidene-1,3-indandione (DMABI) have found evidence of emission from localized self-trapped CT excitons (STCTEs). In the case of DMABI, this was observed around 2.5 eV at RT; whereas, WRS exhibited an emission peak around 1.15 eV at 4.2 K.45,46 In both cases, the emission was reported to be very weak (<0.1% quantum yield for DMABI).47 Furthermore, recent reports of efficient luminescence from 5s2 lone pairs35 encouraged the investigation of potential emission in the Rb23BixSbIII7–xSb2Cl54 family. Rb23BixSbIII7–xSb2Cl54 samples with various compositions were cooled to 12 K and excited in the UV as well as in the CTB region to search for visible PL (450–850 nm). No PL was observed in this range, regardless of excitation (ranging from 250 to 600 nm), at any temperature down to 12 K. Therefore, we also examined the possibility of IR emission at 77 K. Samples were cooled in a cryostat and illuminated at specific wavelengths from 250 to 600 nm with a Xe lamp equipped with a monochromator. An IR detector (In1–xGaxAs) was utilized with several long pass filters to exclude scattered light from the lamp. No emission was observed from any Rb23BixSbIII7–xSb2Cl54 samples. Given the lack of visible emission in these systems, we also investigated the dilution of Sb3+ centers with both In3+ and Bi3+ by targeting the Rb23BixInySb7–x–ySbV2Cl54 system. One reaction incidentally yielded a new oxychloride phase, Rb2SbCl5O (Figure S9; Tables S22–S25). Aside from this, pure phases seldom formed, and no PL was observed down to 77 K in all cases.
We then investigated the electrical properties of these mixed-valent semiconductors. Previously, Day et al. had characterized the electrical properties of several A4MIIIMVX12 phases and investigated photoconductivity.20
They found that all investigated phases were highly resistive with conductivity that increased with temperature, identifying these materials as wide-gap semiconductors (Table S1). The character of the conductivity shifts from electronic in the A4SbIIISbVCl12 compounds, where it is driven by an outer-sphere charge transfer process from SbVCl6 to SbIVCl6 followed by SbIVCl6 excited state migration among SbIIICl6 octahedra, to ionic in the case of Cs4InIIISbVCl12, which lacks the active lone pair and thus has significantly higher resistivity.20 Also, no significant photoconductivity or changes to conductivity were observed under O2 or H2.
I–V curves were only measured for Rb23BiIII7SbV2Cl54 as single crystals of suitable size could not be grown for other compositions. This material has a resistivity of 1.0 × 1010 Ω·cm, 1–4 orders of magnitude lower than the A4MIIIMVX12 compounds (Table S1), indicating that the greater percentage of ns2-containing MIII in the Rb23BixSbIII7–xSb2Cl54 structure promotes the formation and migration of charge-transfer electronic bands (Figure S10a). Instead, the resistivity of Rb23BiIII7SbV2Cl54 is on par with values measured for other low-dimensional ns2-based pnictogen-halides such as Rb7Sb3Cl16 or the A3M2X9 series.34,48,49 Following these measurements, visible light photoconductivity was also examined but, similarly to previously reported mixed-valent compounds, no significant response could be detected.
We additionally considered the utility of these phases in X-ray detection. In several recent reports, highly resistive metal-halides such as the 0D Cs3Bi2I9 or the 2D (NH4)3Bi2I9 have been presented as possible alpha-particle and X-ray detector materials (ρ = 2.79 × 1010 Ω·cm for Cs3Bi2I9).50−52 The benefit of using low- and zero-dimensional materials of this type is that they will have very low noise under bias due to the intrinsically high resistivity.34,48 Exposing the Bi-rich single crystal Rb23BiIII7SbV2Cl54 (with a thickness of 0.36 mm) to X-rays from a Ag X-ray source operating at 50 kV resulted in a measurable photoresponse that could be fit with the single-carrier Hecht model (Figure S10b). Doing so yields a mobility-lifetime (μτ) product of 8.0 × 10–5 cm2 V–1. The quality of the fit to the single-carrier Hecht model indicates that one carrier type is dominant in this material, similar to the case of the leading semiconductor γ radiation detector Cd1–xZnxTe (CZT), which exhibits a significant difference in the charge transport of electrons and holes.53 Note that we cannot distinguish whether our μτ value corresponds to the charge transport properties of holes or electrons on the basis of this measurement alone due to the high penetration of X-rays that generate electron–hole pairs throughout the detector thickness. Surprisingly, while this unoptimized μτ product does not match that of CZT at 4 × 10–3 cm2 V–1,53 it is quite competitive with other candidate radiation detector materials such as PbI2,53 Hg3Se2I2,54 and BiI355 which have each demonstrated spectroscopic gamma-ray response despite low μτ products on the order of 10–5 cm2 s–1 V–1. Compared with leading 0D materials, the μτ product of Rb23BiIII7SbV2Cl54 is only one order lower than the best value reported for Cs3Bi2I9 (7.97 × 10–4 cm2 V–1).52 The sensitivity and detection limit for Rb23BiIII7SbV2Cl54 at 1.1 V μm–1 were determined to be 32.2 μC Gy1–air cm–2 and 25.3 μGyair s–1, respectively. For comparison, a-Se, which is utilized commercially for X-ray detection, has a sensitivity and detection limit of 20 μC Gy1–air cm–2 and 5.5 μGyair s–1, respectively at 10 V μm–1; CZT exhibits a sensitivity and detection limit of 318 μC Gy1–air cm–2 and 50 μGyair s–1, respectively, at 0.1–1 V μm–1.56 Although this performance is not state-of-the-art, it is the first-of-its-kind for mixed-valent semiconductors and may offer a new route toward discovering X-ray semiconductor detector materials. Further performance improvements are expected from optimization of the size and quality of Rb23BixSbIII7–xSb2Cl54 single crystals, as well as from improvements to device architecture, and future work toward this end is underway.
Conclusions
After over a century of research on the A4MIIIMVX12 family of mixed-valent alkali pnictogen halides, new phases can still be discovered within this compositional space. We demonstrated that the combination of Rb, SbIII, SbV, and Cl results in the orthorhombic Rb23SbIII7SbV2Cl54, a 0D mixed-valent compound with a unique layered structure type with octahedral ordering defined by the orientation of the stereoactive 5s2 lone pairs. Substitution of SbIII by BiIII generates a family of Rb23BixSbIII7–xSb2Cl54 (0 < x ≤ 7) materials that crystallize in the noncentrosymmetric R32 space group. This noncentrosymmetry was confirmed by the observation of SHG with a 1064 nm laser. Furthermore, the charge-transfer absorption band in the visible range was tunable from dark red to pale yellow through M(III) substitution. Finally, Rb23BiIII7SbV2Cl54 single crystals were demonstrated to be high resistivity semiconductors that unexpectedly exhibited X-ray photoresponse with a μτ product of 8.0 × 10–5 cm2 V–1 despite no efforts to optimize the crystal quality or device structure.
These compounds present a wholly new structure type with prominent lone-pair expression coexisting with mixed-valency, highlighting the importance of the sizes of Rb+ and ns2-based MCl6 octahedra that together induces the formation of this structure over the high-symmetry octahedral environments of the mixed-valent pnictogen halides, A4MIIIMVX12. The novel composition and ordering in the Rb23BixSbIII7–xSb2Cl54 family leads to different orbital overlap relative to their high-symmetry contemporaries, as evidenced by the disparate colors of red Rb23SbIII7SbV2Cl54 and indigo Cs4SbIIISbVCl12, indicating nontrivial differences in the outer-sphere charge transfer processes of these new compounds. The X-ray detection exhibited by unoptimized Rb23BiIII7SbV2Cl54 also indicates that mixed-valent materials may have potential in high-energy radiation detection applications. Beyond simply adding a new entry to the growing library of 0D metal halides, the complexity of this structure offers ripe opportunities for tuning and improving these materials; namely, the ability to tune both lone-pair expression as well as the charge transfer absorption band characteristic of mixed-valent materials within the same structure.
Acknowledgments
This work was financially supported by ETH Zurich through the ETH+ Project SynMatLab and by the European Union through Horizon 2020 research and innovation programme [ERC Consolidator Grant, agreement No. (819740), project SCALE-HALO].
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.chemmater.0c04491.
Crystallographic Information File for Rb23SbIII7SbV2Cl54 at 293 K (CIF)
Crystallographic Information File for Rb23BiIII2.50SbIII3.50SbV2Cl54 at 293 K (CIF)
Crystallographic Information File for Rb23BiIII6.62SbIII0.38SbV2Cl54 at 293 K (CIF)
Crystallographic Information File for RbSbVCl6 at 293 K (CIF)
Crystallographic Information File for Rb2SbVCl5O at 293 K (CIF)
Additional crystal structure figures, coordination environment tables, temperature-dependent PXRD plot, and crystallographic refinement tables (PDF)
Author Contributions
§ These authors contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Robin M. B.; Day P., Mixed Valence Chemistry-A Survey and Classification. In Advances in Inorganic Chemistry and Radiochemistry; Emeléus H. J., Sharpe A. G., Eds.; Academic Press: New York, 1968; Vol. 10, pp 247–422. [Google Scholar]
- Cava R. J.; Batlogg B.; Krajewski J. J.; Farrow R.; Rupp L. W.; White A. E.; Short K.; Peck W. F.; Kometani T. Superconductivity near 30 K without copper: the Ba0.6K0.4BiO3 perovskite. Nature 1988, 332 (6167), 814–816. 10.1038/332814a0. [DOI] [Google Scholar]
- Schoop L. M.; Müchler L.; Felser C.; Cava R. J. Lone Pair Effect, Structural Distortions, and Potential for Superconductivity in Tl Perovskites. Inorg. Chem. 2013, 52 (9), 5479–5483. 10.1021/ic400381g. [DOI] [PubMed] [Google Scholar]
- Coey J. M. D.; Viret M.; von Molnár S. Mixed-valence manganites. Adv. Phys. 1999, 48 (2), 167–293. 10.1080/000187399243455. [DOI] [Google Scholar]
- Stitzer K. E.; Smith M. D.; Gemmill W. R.; zur Loye H.-C. Novel Mixed-Valent (V/VI) Triple Perovskite Ruthenates: Observation of a Complex Low-Temperature Structural and Magnetic Transition. J. Am. Chem. Soc. 2002, 124 (46), 13877–13885. 10.1021/ja0271781. [DOI] [PubMed] [Google Scholar]
- Allen G. C.; Hush N. S., Intervalence-Transfer Absorption. Part 1. Qualitative Evidence for Intervalence-Transfer Absorption in Inorganic Systems in Solution and in the Solid State. In Prog. Inorg. Chem.; John Wiley & Sons, Inc.: 2007; pp 357–389. [Google Scholar]
- Day P. Spectra and Constitution of Antimony(III) Antimony(V) Hexahalide Salts and Related Compounds. Inorg. Chem. 1963, 2 (3), 452–456. 10.1021/ic50007a006. [DOI] [Google Scholar]
- Day P.; Diggle P. J.; Griffiths G. A. Charge-transfer Spectra of Post-transition-metal Halide Complexes. J. Chem. Soc., Dalton Trans. 1974, 13, 1446–1452. 10.1039/dt9740001446. [DOI] [Google Scholar]
- Lee B.; Stoumpos C. C.; Zhou N.; Hao F.; Malliakas C.; Yeh C. Y.; Marks T. J.; Kanatzidis M. G.; Chang R. P. Air-stable molecular semiconducting iodosalts for solar cell applications: Cs2SnI6 as a hole conductor. J. Am. Chem. Soc. 2014, 136 (43), 15379–85. 10.1021/ja508464w. [DOI] [PubMed] [Google Scholar]
- Kaltzoglou A.; Antoniadou M.; Kontos A. G.; Stoumpos C. C.; Perganti D.; Siranidi E.; Raptis V.; Trohidou K.; Psycharis V.; Kanatzidis M. G.; Falaras P. Optical-Vibrational Properties of the Cs2SnX6 (X = Cl, Br, I) Defect Perovskites and Hole-Transport Efficiency in Dye-Sensitized Solar Cells. J. Phys. Chem. C 2016, 120 (22), 11777–11785. 10.1021/acs.jpcc.6b02175. [DOI] [Google Scholar]
- Peresh E. Y.; Sidei V. I.; Zubaka O. V.; Stercho I. P. K2(Rb2,Cs2,Tl2)TeBr6(I6) and Rb3(Cs3)Sb2(Bi2)Br9(I9) perovskite compounds. Inorg. Mater. 2011, 47 (2), 208–212. 10.1134/S0020168511010109. [DOI] [Google Scholar]
- Engel G. Die Kristallstrukturen einiger Hexachlorokomplexsalze. Z. Kristallogr. Cryst. Mater. 1935, 90 (1–6), 341–373. 10.1524/zkri.1935.90.1.341. [DOI] [Google Scholar]
- Cai Y.; Xie W.; Ding H.; Chen Y.; Thirumal K.; Wong L. H.; Mathews N.; Mhaisalkar S. G.; Sherburne M.; Asta M. Computational Study of Halide Perovskite-Derived A2BX6 Inorganic Compounds: Chemical Trends in Electronic Structure and Structural Stability. Chem. Mater. 2017, 29 (18), 7740–7749. 10.1021/acs.chemmater.7b02013. [DOI] [Google Scholar]
- Maughan A. E.; Ganose A. M.; Scanlon D. O.; Neilson J. R. Perspectives and Design Principles of Vacancy-Ordered Double Perovskite Halide Semiconductors. Chem. Mater. 2019, 31 (4), 1184–1195. 10.1021/acs.chemmater.8b05036. [DOI] [Google Scholar]
- Setterberg C. om några Csesium- och Rubidium-föreningar. Oefversigt K. Vetensk, Akad. Forhandl. 1882, 39 (6), 23–29. [Google Scholar]
- Wells H. L.; Metzger F. J. On a Salt of Quadrivalent Antimony. Am. Chem. J. 1901, 26, 268–271. [Google Scholar]
- Weinland R. F.; Schmid H. Ueber Halogendoppelsalze des vierwerthigen Antimons. Ber. Dtsch. Chem. Ges. 1905, 38 (1), 1080–1087. 10.1002/cber.190503801196. [DOI] [Google Scholar]
- Tovborg Jensen A.; Rasmussen S. E. A Note on the Alleged Tetravalency of Antimony. Acta Chem. Scand. 1955, 9, 7. [Google Scholar]
- Atkinson L.; Day P. Charge transfer in mixed-valence solids. Part IV. Electronic spectra of hexachloroantimonates(III,V). J. Chem. Soc. A 1969, 0, 2423–2431. 10.1039/j19690002423. [DOI] [Google Scholar]
- Atkinson L.; Day P. Charge transfer in mixed-valence solids. Part V. Semiconductivity of hexachloroantimonates(III,V). J. Chem. Soc. A 1969, 0, 2432–2436. 10.1039/j19690002432. [DOI] [Google Scholar]
- Jensen K. A. Die Kristallstruktur der Verbindungen (NH4)2SbBr6, Rb2SbBr6 und Rb2SbCI6. Z. Anorg. Allg. Chem. 1937, 232, 193–201. 10.1002/zaac.19372320210. [DOI] [Google Scholar]
- Hackert M. L.; Lawton S. L.; Jacobson R. A. Properties of lntervalence Antimony Bromides. Proc. Iowa Acad. Sci. 1968, 75 (1), 97–108. [Google Scholar]
- Hubbard C. R.; Jacobson R. A. The Crystal Structure of Rb4SbIIISbVBr12. Proc. Iowa Acad. Sci. 1968, 75 (1), 85–96. [Google Scholar]
- Prassides K.; Day P.; Cheetham A. K. Crystal structures of mixed-valency and mixed-metal salts A2MIII0.5SbV0.5X6 (A = Rb, Cs; M = Sb, Bi, In, Tl, Fe, Rh; X = Cl, Br). A powder neutron diffraction study. Inorg. Chem. 1985, 24 (4), 545–552. 10.1021/ic00198a023. [DOI] [Google Scholar]
- Clark R. J. H.; Trumble W. R. Resonance Raman spectra of some mixed-valence halogeno-compounds of antimony and lead. J. Chem. Soc., Dalton Trans. 1976, 12, 1145–1149. 10.1039/dt9760001145. [DOI] [Google Scholar]
- Combs V. E.; Oswald I. W. H.; Neilson J. R. Hydrothermal Crystal Growth of Mixed Valence Cs2SbBr6. Cryst. Growth Des. 2019, 19 (7), 4090–4094. 10.1021/acs.cgd.9b00481. [DOI] [Google Scholar]
- Lawton S. L.; Jacobson R. A. The Crystal Structure of Ammonium Hexabromoantimonate, (NH4)4SbIIISbVBr12. Inorg. Chem. 1966, 5 (5), 743–749. 10.1021/ic50039a011. [DOI] [Google Scholar]
- Prassides K.; Day P.; Cheetham A. K. Anion ordering in mixed valence dicesium hexachloroantimonate (Cs2SbCl6) and related salts. J. Am. Chem. Soc. 1983, 105 (10), 3366–3368. 10.1021/ja00348a085. [DOI] [Google Scholar]
- Prassides K.; Day P. Analysis of the intervalence absorption band edge in the mixed-valency SbIII,V salt Rb2.67SbCl6. J. Chem. Soc., Faraday Trans. 2 1985, 81 (8), 1259–1268. 10.1039/f29858101259. [DOI] [Google Scholar]
- Sheldrick G. M. A short history of SHELX. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64 (1), 112–122. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42 (2), 339–341. 10.1107/S0021889808042726. [DOI] [Google Scholar]
- Momma K.; Izumi F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44 (6), 1272–1276. 10.1107/S0021889811038970. [DOI] [Google Scholar]
- Putz H.; Brandenburg K. DIAMOND-Crystal and molecular structure visualization. Crystal Impact-GbR. Kreuzherrenstr 2006, 102, 53227. [Google Scholar]
- Benin B. M.; McCall K. M.; Wörle M.; Morad V.; Aebli M.; Yakunin S.; Shynkarenko Y.; Kovalenko M. V. The Rb7Bi3–3xSb3xCl16 family: A Fully Inorganic Solid Solution with Room-Temperature Luminescent Members. Angew. Chem., Int. Ed. 2020, 59 (34), 14490–14497. 10.1002/anie.202003822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall K. M.; Morad V.; Benin B. M.; Kovalenko M. V. Efficient Lone-Pair-Driven Luminescence: Structure–Property Relationships in Emissive 5s2 Metal Halides.. ACS Mater. Lett. 2020, 2 (9), 1218–1232. 10.1021/acsmaterialslett.0c00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall K. M.; Benin B. M.; Wörle M.; Vonderach T.; Günther D.; Kovalenko M. V. Expanding the 0D Rb7M3X16 (M = Sb, Bi; X = Br, I) Family: Dual-Band Luminescence in Rb7Sb3Br16. Helv. Chim. Acta 2020, 104 (1), e2000206. 10.1002/hlca.202000206. [DOI] [Google Scholar]
- Mao L.; Stoumpos C. C.; Kanatzidis M. G. Two-Dimensional Hybrid Halide Perovskites: Principles and Promises. J. Am. Chem. Soc. 2019, 141 (3), 1171–1190. 10.1021/jacs.8b10851. [DOI] [PubMed] [Google Scholar]
- Launay J.-P. Long-distance intervalence electron transfer. Chem. Soc. Rev. 2001, 30 (6), 386–397. 10.1039/b101377g. [DOI] [Google Scholar]
- Gray H. B.; Winkler J. R. Electron Flow through Proteins. Chem. Phys. Lett. 2009, 483 (1–3), 1–9. 10.1016/j.cplett.2009.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Launay J.-P. Mixed-Valent Compounds and their Properties - Recent Developments. Eur. J. Inorg. Chem. 2020, 2020 (4), 329–341. 10.1002/ejic.201901180. [DOI] [Google Scholar]
- Beck H. P.; Milius W. Study on A4BX6 compounds. I. Structure Refinement of Ternary Cd Halides A4CdX6 (A = NH4, K, Rb, In, Tl; X = Cl, I). Z. Z. Anorg. Allg. Chem. 1986, 539 (8), 7–17. 10.1002/zaac.19865390802. [DOI] [Google Scholar]
- Chang J.-H.; Doert T.; Ruck M. The crystal structures of α-Rb7Sb3Br16, α- and β-Tl7Bi3Br16 and their relationship to close packings of spheres. Z. Kristallogr. - Cryst. Mater. 2020, 235 (8–9), 255–261. 10.1515/zkri-2020-0013. [DOI] [Google Scholar]
- Stoumpos C. C.; Frazer L.; Clark D. J.; Kim Y. S.; Rhim S. H.; Freeman A. J.; Ketterson J. B.; Jang J. I.; Kanatzidis M. G. Hybrid germanium iodide perovskite semiconductors: active lone pairs, structural distortions, direct and indirect energy gaps, and strong nonlinear optical properties. J. Am. Chem. Soc. 2015, 137 (21), 6804–19. 10.1021/jacs.5b01025. [DOI] [PubMed] [Google Scholar]
- Li Y.; Rao Y.; Mak K. F.; You Y.; Wang S.; Dean C. R.; Heinz T. F. Probing symmetry properties of few-layer MoS2 and h-BN by optical second-harmonic generation. Nano Lett. 2013, 13 (7), 3329–33. 10.1021/nl401561r. [DOI] [PubMed] [Google Scholar]
- Tanino H.; Kobayashi K. Relaxation of Electron-Phonon system in optically excited Quasi-1-D Mixed Valence Crystal Wolffram’s Red Salt. J. Phys. Soc. Jpn. 1983, 52 (4), 1446–1456. 10.1143/JPSJ.52.1446. [DOI] [Google Scholar]
- Jursenas S.; Gruodis A.; Kodis G.; Chachisvilis M.; Gulbinas V.; Silinsh E. A.; Valkunas L. Free and Self-Trapped Charge-Transfer Excitons in Crystals of Dipolar Molecules of N,N-Dimethylaminobenzylidene 1,3-Indandione. J. Phys. Chem. B 1998, 102, 1086–1094. 10.1021/jp971991e. [DOI] [Google Scholar]
- Jursenas S.; Gulbinas V.; Gruodis A.; Kodis G.; Kovalevskij V.; Valkunas L. Spectroscopy of self-trapped charge-transfer excitons in polar Đlms and crystals of N,N-dimethylaminobenzylidene 1,3-indandione (DMABI). Phys. Chem. Chem. Phys. 1999, 1, 1715–1718. 10.1039/a808611g. [DOI] [Google Scholar]
- McCall K. M.; Stoumpos C. C.; Kostina S. S.; Kanatzidis M. G.; Wessels B. W. Strong Electron–Phonon Coupling and Self-Trapped Excitons in the Defect Halide Perovskites A3M2I9 (A = Cs, Rb; M = Bi, Sb). Chem. Mater. 2017, 29 (9), 4129–4145. 10.1021/acs.chemmater.7b01184. [DOI] [Google Scholar]
- Timmermans C. W. M.; Cholakh S. O.; van der Woude R. L.; Blasse G. Van der Woude, R. L.; Blasse, G., Some Optical and Electrical Measurements on Cs3Bi2Br9 single crystals. Phys. Status Solidi B 1983, 115 (1), 267–271. 10.1002/pssb.2221150130. [DOI] [Google Scholar]
- McCall K. M.; Liu Z.; Trimarchi G.; Stoumpos C. C.; Lin W.; He Y.; Hadar I.; Kanatzidis M. G.; Wessels B. W. α-Particle Detection and Charge Transport Characteristics in the A3M2I9 Defect Perovskites (A = Cs, Rb; M = Bi, Sb). ACS Photonics 2018, 5 (9), 3748–3762. 10.1021/acsphotonics.8b00813. [DOI] [Google Scholar]
- Zhuang R.; Wang X.; Ma W.; Wu Y.; Chen X.; Tang L.; Zhu H.; Liu J.; Wu L.; Zhou W.; Liu X.; Yang Y. Highly sensitive X-ray detector made of layered perovskite-like (NH4)3Bi2I9 single crystal with anisotropic response. Nat. Photonics 2019, 13 (9), 602–608. 10.1038/s41566-019-0466-7. [DOI] [Google Scholar]
- Zhang Y.; Liu Y.; Xu Z.; Ye H.; Yang Z.; You J.; Liu M.; He Y.; Kanatzidis M. G.; Liu S. F. Nucleation-controlled growth of superior lead-free perovskite Cs3Bi2I9 single-crystals for high-performance X-ray detection. Nat. Commun. 2020, 11 (1), 2304. 10.1038/s41467-020-16034-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens A.; Peacock A. Compound semiconductor radiation detectors. Nucl. Instrum. Methods Phys. Res., Sect. A 2004, 531 (1–2), 18–37. 10.1016/j.nima.2004.05.071. [DOI] [Google Scholar]
- He Y.; Kontsevoi O. Y.; Stoumpos C. C.; Trimarchi G.; Islam S. M.; Liu Z.; Kostina S. S.; Das S.; Wessels B. W.; Kanatzidis M. G.; Kim J.-I.; Lin W. Defect anti-perovskite compounds Hg3Q2I2 (Q = S, Se and Te) for Room Temperature Hard Radiation Detection. J. Am. Chem. Soc. 2017, 139 (23), 7939–7951. 10.1021/jacs.7b03174. [DOI] [PubMed] [Google Scholar]
- Saito T.; Iwasaki T.; Kurosawa S.; Yoshikawa A.; Den T. BiI3 single crystal for room-temperature gamma ray detectors. Nucl. Instrum. Methods Phys. Res., Sect. A 2016, 806, 395–400. 10.1016/j.nima.2015.10.036. [DOI] [Google Scholar]
- Wei H.; Huang J. Halide lead perovskites for ionizing radiation detection. Nat. Commun. 2019, 10 (1), 1066. 10.1038/s41467-019-08981-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.