

Abstract
Objectives
In patients with severe combined immunodeficiency (SCID), the immune system often fails to eradicate maternal cells that enter the foetus via the placenta, resulting in transplacental maternal engraftment (TME) syndrome. However, the clinical significance of TME has not been comprehensively elucidated.
Methods
Here, we describe a patient with SCID with a novel frameshift IL2RG mutation associated with maternal engrafted CD8+ T cells that had been expanded by viral infection. To evaluate the origin of the expanded T cells, we HLA‐typed the myeloid and T cells of the patient and analysed the immunological characteristics of the expanded CD8+ T cells using T‐cell receptor (TCR) repertoire and flow cytometry analysis.
Results
In our patient, the maternal engrafted CD8+ T cells expanded and exerted in vitro antiviral function against human cytomegalovirus (CMV) infection before and after haematopoietic cell transplantation (HCT). After haploidentical HCT from the maternal donor, maternal engrafted CMV‐specific CD8+ T cells were maintained, successfully proliferated and activated against CMV. We found no evidence of acute graft‐versus‐host disease or infectious complications other than recurrent episodes of CMV viraemia, which were well controlled by ganciclovir and, possibly by, the maternal engrafted CMV‐specific CD8+ T cells.
Conclusion
Our findings elucidate a possible functional role of TME in controlling CMV infection in patient with SCID and suggest an optimal strategy for donor selection in patients with SCID with TME.
Keywords: severe combined immunodeficiency, transplacental maternal engraftment, CD8+ T cells, cytomegalovirus infection, haploidentical haematopoietic stem cell transplantation
We describe a patient with severe combined immunodeficiency (SCID) with a novel frameshift IL2RG mutation associated with maternal engrafted CD8+ T cells that expanded and exerted antiviral function against human cytomegalovirus (CMV) infection.

Objectives
Severe combined immunodeficiency (SCID) is a group of heterogeneous genetic diseases characterised by fatal immune deficiency. 1 The most common cause of SCID in humans is variants of the interleukin‐2 receptor γ (IL2RG) gene. 2 , 3 , 4 , 5 Although some variations exist depending on the degree of function of the variant alleles, SCID often causes death during early infancy because of life‐threatening infections.
During pregnancy, maternal cells may transfer to the foetus through the placenta, resulting in microchimerism. 6 In a healthy foetus, the immune system eliminates these engrafted cells, but the immune system in SCID may lack the functional capacity to eliminate engrafted cells, resulting in transplacental maternal engraftment (TME). 7 In patients with SCID, TME may result in graft‐versus‐host disease (GVHD) even before haematopoietic stem cell transplantation (HCT). 8 However, the clinical significance of TME in SCID remains poorly understood.
Currently, the standard treatment for SCID is allogeneic HCT in early life. 9 , 10 As an alternative to fully human leukocyte antigen (HLA)‐matched donors, parental haploidentical donors are usually a more readily available source of stem cells. However, deciding which parental donor is better and the suitable degree of conditioning for HCT are unclear for patients with SCID. 11 Furthermore, because of HCT‐related morbidity and mortality, accurate diagnosis of SCID is necessary before HCT is performed. Here, we describe a patient with SCID with a novel IL2RG mutation who had TME, specifically engraftment of human cytomegalovirus (CMV)‐specific maternal CD8+ T cells. The engrafted CD8+ T cells responded to CMV infection in the patient with SCID by robustly expanding and exerting antiviral function before and after maternal HCT.
Results
Initial clinical course and diagnosis of SCID with a IL2RG frameshift mutation
A 3‐month‐old Korean male infant was admitted to the hospital with fever (Figure 1a). He was born at full term from non‐consanguineous parents (Figure 1b), weighing 3250 g, with no dysmorphic features. No family history of early childhood death or recurrent infection was noted. Immunisations were performed as scheduled, including subcutaneous Bacillus Calmette–Guérin (BCG) and oral rotavirus vaccines. Upon physical examination, the infant’s general appearance was normal and his vital signs were stable except for the fever (≥ 39°C). We found no lymphadenopathy, rash or hepatosplenomegaly. Laboratory tests revealed leukopenia (white blood cell count 2170 µL−1) and lymphopenia (absolute lymphocyte count [ALC] 940 µL−1), and thymic shadow was absent on a chest radiograph. Despite empiric antimicrobial treatment, the patient’s fever persisted for more than a week. Further laboratory tests revealed persistent lymphopenia and elevated alanine aminotransferase (ALT, 346 IU L−1), lactate dehydrogenase (LDH, 2156 IU L−1), ferritin (> 16 500 ng mL−1) levels, hypertriglyceridaemia (279 mg dL−1), elevated soluble CD25 (5790 U mL−1) and low NK cell activity. Considering the possibility of haemophagocytic lymphohistiocytosis (HLH), a bone marrow biopsy was performed, revealing haemophagocytosis. However, some clinical and laboratory features, such as a normal C‐reactive protein level and no clinical symptoms other than fever, were not compatible with the diagnosis of HLH. Real‐time PCR was performed for Epstein–Barr virus (EBV) and CMV to evaluate fever with unknown origin, and early postnatal CMV infection was diagnosed (Figure 1a, Table 1). We decided to treat the CMV infection first. After starting intravenous ganciclovir, the fever gradually disappeared and the CMV levels decreased rapidly. Lymphocyte subset analysis upon initiation of the CMV treatment revealed low T‐cell (22 µL−1) and NK cell (31 µL−1) counts; however, B‐cell counts (557 µL−1) were relatively maintained in the normal range. Thus, we performed T‐cell receptor excision circle (TREC) screening and genetic testing for primary immunodeficiency. The levels of TRECs were extremely low, and targeted gene sequencing revealed a novel variant of IL2RG (c.681del, p.Phe227LeufsTer46) that resulted in a premature termination codon in the extracellular domain of interleukin‐2 receptor γ (Figure 1c, d, Supplementary table 1).
Figure 1.
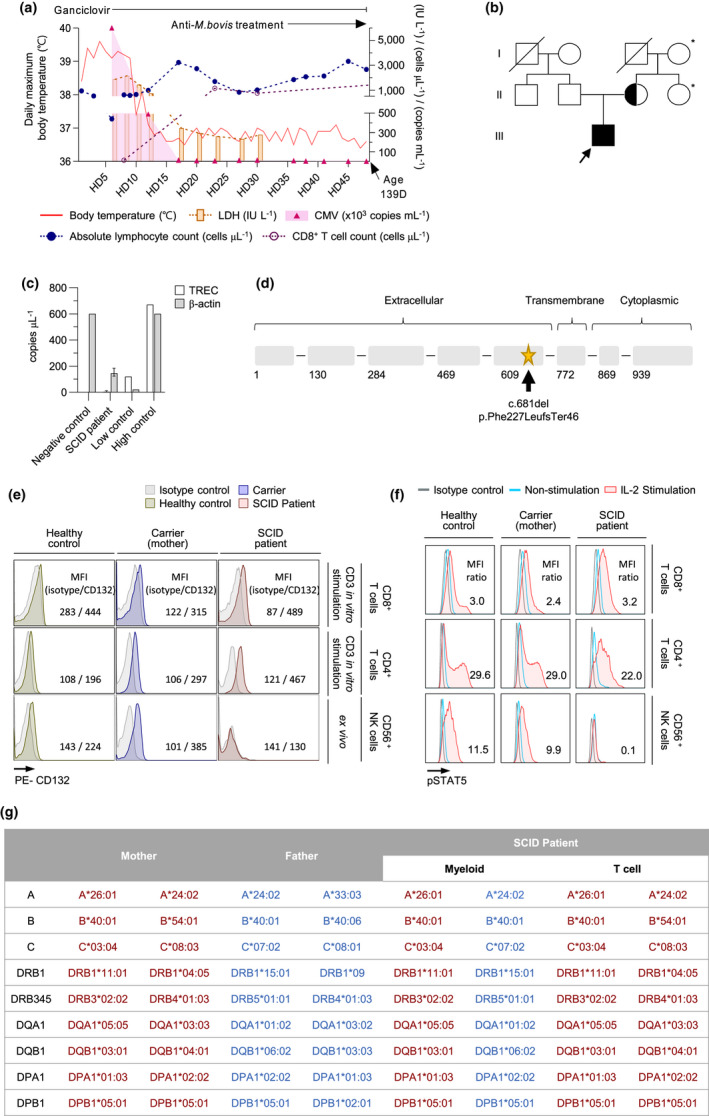
Diagnosis of SCID with a novel IL2RG mutation and engrafted transplacental maternal T cells. (a) First episode of CMV infection in this patient. He was initially admitted to the hospital with a fever accompanied by leukopenia. (b) Pedigree showing affected individuals harbouring a novel IL2RG mutation. Solid symbols indicate affected persons who were hemizygous for the mutant allele; half‐solid symbols indicate heterozygous individuals; void symbols indicate unaffected persons; circles represent female family members; and squares represent male family members. *Genetic screening was not performed. (c) TREC results for SCID screening. < 40 copies μL−1 was defined as negative. 23 (d) Location of the IL2RG mutation in our patient. The patient had a novel hemizygous frameshift mutation altering the extracellular domain (arrow) of the interleukin‐2 receptor γ protein. (e) Flow cytometry analysis of CD132 expression by in vitro anti‐CD3/CD‐28‐stimulated CD8+/CD4+ T cells and ex vivo CD56+ NK cells from a healthy donor, heterozygous carrier (patient’s mother) and the patient. The mean fluorescence intensities (MFIs) of isotype and CD132 are shown. (f) STAT5 phosphorylation after IL‐2 stimulation (50 ng mL−1). The ratio of the MFI of phospho‐STAT5 (pSTAT5) after IL‐2 stimulation to isotype is shown. (g) HLA typing of myeloid cells and T cells from the patient and both parents.
Table 1.
Laboratory findings during first cytomegalovirus infection
| Age | 92D | 97 ~ 99D | 108D | 114D | 121 ~ 122D | 132D | 139D | |
|---|---|---|---|---|---|---|---|---|
| Laboratory | HD Normal | HD 1 | HD 6 ~ 8 | HD 17 | HD 23 | HD 30 ~ 31 | HD 41 | HD 48 |
| WBC | 6000–14 000, µL−1 | 2170 | 1680 | 5360 | 2940 | 2540 | 3250 | 3810 |
| ALC | 1500–3000, µL−1 | 940 | 620 | 3230 | 1700 | 1010 | 2130 | 2670 |
| Hb | 10.5–14.0, g dL−1 | 11.6 | 8.8 | 9.9 | 9.9 | 10.4 | 9.0 | 8.7 |
| PLT | 150–400, ×103 µL−1 | 178 | 187 | 327 | 314 | 286 | 296 | 300 |
| CRP | 0–8, mg dL−1 | 0.4 | ‐ | < 0.3 | < 0.3 | < 0.3 | < 0.3 | < 0.3 |
| TB | 0.2–0.8, mg dL−1 | 0.4 | 0.4 | 0.3 | 0.3 | 0.2 | 0.2 | < 0.15 |
| AST | 13–34, IU L−1 | 141 | 892 | 76 | 62 | 95 | 249 | 127 |
| ALT | 5–46, IU L−1 | 192 | 346 | 85 | 60 | 96 | 155 | 122 |
| LDH | 119–247, IU L−1 | ‐ | 2156 | 345 | 259 | 276 | ‐ | ‐ |
| Ferritin | 22–322, ng mL−1 | ‐ | > 16 500 | 1063.6 | 466.2 | 348 | ‐ | ‐ |
| CD3+ | 2284–4776, µL−1 | ‐ | 22 | ‐ | 1183 | ‐ | ‐ | ‐ |
| CD4+ | 1523–3472, µL−1 | ‐ | 9 | ‐ | 19 | 14 | ‐ | ‐ |
| CD8+ | 524–1583, µL−1 | ‐ | 13 | ‐ | 1150 | 744 | ‐ | ‐ |
| CD19+ | 776–2238, µL−1 | ‐ | 557 | ‐ | 502 | 246 | ‐ | ‐ |
| CD56+ | 230–801, µL−1 | ‐ | 31 | ‐ | 7 | 4 | ‐ | ‐ |
| IgG | 176–601, mg dL−1 | ‐ | ‐ | ‐ | ‐ | ‐ | < 18 | ‐ |
| IgA | 4.4–84, mg dL−1 | ‐ | ‐ | ‐ | ‐ | ‐ | < 5 | ‐ |
| IgM | 17–105, mg dL−1 | ‐ | ‐ | ‐ | ‐ | ‐ | < 5 | ‐ |
| sCD25 | 158–623, U mL−1 | ‐ | 5790 | ‐ | ‐ | ‐ | ‐ | ‐ |
| CMV | < 500, copies mL−1 | 60 255 000 | 18 450 | 4880 | 11 900 | < 500 | < 500 | |
WBC, white blood cell; ALC, absolute lymphocyte count; Hb, haemoglobin; PLT, platelet count; CRP, C‐reactive protein; TB, total bilirubin; AST, aspartate aminotransferase; ALT, alanine aminotransferase; LDH, lactate dehydrogenase; Ig, immunoglobulin; sCD25, soluble interleukin‐2 receptor; CMV, cytomegalovirus; D, day; HD, hospitalisation day.
During the course of treatment with intravenous ganciclovir, elevated liver enzymes reappeared after the initial normalisation. Moreover, the BCG injection site presented mild suppuration, and splenomegaly with mesenteric lymphadenopathy was observed on abdomen ultrasonography. Treatment with anti‐tubercular drugs (isoniazid, rifampin and ethambutol) was initiated with the impression of BCGitis, and the serum liver enzymes were normalised.
Engraftment of maternal CD8+ T cells in the patient with SCID
Intriguingly, the patient’s ALC and CD3+ T‐cell counts, particularly CD8+ T‐cell counts, rapidly normalised after hospitalisation day (HD) 8. On HD 23, the CD3+ and CD8+ T‐cell counts were 1183 µL−1 and 1150 µL−1, respectively (Figure 1a, Table 1). Next, we examined common γ‐chain (γC, CD132) expression and IL‐2‐induced STAT5 phosphorylation. T cells obtained from the patient on HD 43 expressed γC, similar to those from a healthy control and the mother (carrier; Figure 1e). IL‐2‐induced phospho‐STAT5 was detected not only in T cells from the healthy control and the carrier, but also in the patient (Figure 1f). In contrast, γC and IL‐2‐induced phospho‐STAT5 were not detected in NK cells from the patient. We hypothesised that the expanded T cells originated from the mother and transferred to the patient via the TME. HLA typing of the patient and his parents showed that T cells from the patient had identical haplotypes as his mother, whereas myeloid cells had one maternal haplotype and one paternal haplotype, confirming that the expanded CD8+ T cells were engrafted from the mother (Figure 1g).
Immunological characteristics of maternal engrafted CD8+ T cells
We examined CD8+ T‐cell cytokine production following stimulation with various pathogenic antigens. CD8+ T cells obtained from the carrier produced IFN‐γ and/or TNF‐α in response to CMV pp65, CMV IE1, EBV BARF1 and purified protein derivative (PPD), but did not respond to influenza A virus (IAV; H1N1) MP1 or respiratory syncytial virus (RSV) MSG. Interestingly, CD8+ T cells obtained from the patient on HD 43 produced IFN‐γ and/or TNF‐α in response to only CMV pp65 and CMV IE1, indicating that CMV‐specific maternal CD8+ T cells expanded in the patient in vivo during CMV infection (Figure 2a). T‐cell receptor (TCR) repertoire analysis revealed that the top five clonotypes represented ~ 45% of the total CD8+ T cells obtained from the patient on HD 43, whereas the top five clonotypes contributed to < 5% of the total CD8+ T cells obtained from the healthy control and the carrier, indicating vigorous expansion of oligoclones in the patient (Figure 2b). Vigorous expansion of oligoclones was also observed in the γδ T cells, although the proportion of γδ T cells in the patient decreased relative to that in the healthy control and the carrier (Supplementary figure 1a–c). These data indicate the oligoclonal expansion of CMV‐specific maternal CD8+ T cells in the patient. These findings were supported by immunophenotyping of naïve/memory cells. In the carrier, the percentage of effector/effector memory cells and CD57+CD28− replicative senescent cells were 37.5% and 9.0%, respectively, among the total CD8+ T cells. However, these values increased up to 97.8% and 66.7%, respectively, of the patient’s total CD8+ T cells on HD 43 (Figure 2c).
Figure 2.
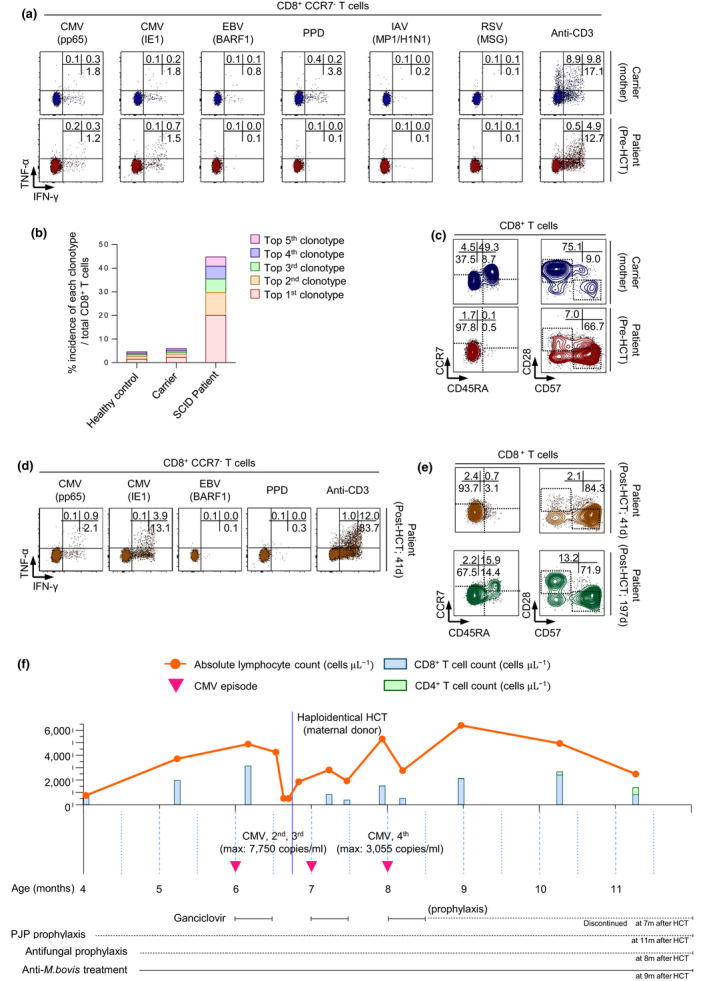
Maternal engrafted CD8+ T cells exert antiviral functions against CMV before and after HCT in the patient with SCID. (a) Maternal engrafted CD8+ T cells secreted TNF‐α and/or IFN‐γ in response to CMV antigen (pp65, IE1), but not to other antigens. (b) TCR repertoire analysis of CD8+ T cells from a healthy donor, mother (carrier) and the patient showed vigorous expansion of the oligoclonotypes of engrafted CD8+ T cells from the patient. (c) Immunophenotyping demonstrating an increased frequency of effector/effector memory (CD45RA−CCR7−) and replicative senescent (CD28−CD57+) CD8+ T cells from the patient. (d) Engrafted CD8+ T cells secreted higher levels of TNF‐α and/or IFN‐γ in response to CMV antigen after HCT than before HCT, and (e) exhibited a terminally differentiated immunophenotype, including effector/effector memory and replicative senescent populations. (f) Schematic of the time course of disease and treatments. The patient underwent haploidentical allogeneic HCT at the age of 6 months. Following HCT, there was no evidence of acute GVHD or disseminated infections, even with persistent CMV viraemia.
Course before and after HCT
The patient underwent haploidentical allogeneic HCT at 6 months of age. In the absence of a fully matched donor, the maternal donor was selected, and the patient was pretreated only with antithymocyte globulin. Six weeks after HCT, we examined cytokine production and immunophenotypes of the patient’s CD8+ T cells. CD8+ T cells continued to produce IFN‐γ and/or TNF‐α in response to CMV pp65 and CMV IE1, but not to other antigens. In particular, the cytokine response to CMV IE1 increased vigorously 41 days after HCT (Figure 2d, Supplementary figure 2a). The percentage of effector/effector memory cells among total CD8+ T cells was maintained at a high level (93.7%), and the percentage of CD57+CD28− replicative senescent cells increased to 84.3% 41 days after HCT (Figure 2e, upper). However, the percentage of effector/effector memory cells among CD8+ T cells was only 67.5% 197 days after HCT, and the percentage of naïve cells increased to 15.9%. Furthermore, the percentage of CD57+ CD28− replicative senescent cells decreased to 71.9% (Figure 2e, lower). The total CD4+ T‐cell count (585 µL−1 104 days after HCT) and percentage of naïve cells among CD4+ T cells (51.9% 197 days after HCT) also increased long‐term after HCT (Supplementary figure 2b, Supplementary table 2). Collectively, these findings indicate successful haematopoietic stem cell engraftment, resulting in the reconstitution of both CD4+ and CD8+ T cells.
Following HCT, we found no evidence of acute GVHD or other complications. CMV viraemia was detected three times immediately before and after HCT. However, the CMV titres (maximal titre, 3055–7750 copies mL−1) were much lower than those at the time of the initial infection (> 5 × 106 copies mL−1) and were successfully reduced by ganciclovir treatment without complications. Anti‐mycobacterial treatment for BCGitis was discontinued 9 months after HCT (Figure 2f, Supplementary table 3).
These data indicate that engrafted CMV‐specific maternal CD8+ T cells harbouring antiviral activity further proliferated and differentiated during the course of HCT in the patient. The patient is currently undergoing outpatient follow‐up 13 months after HCT and is in good condition without antimicrobial treatment or prophylaxis.
Discussion
Here, we describe a patient with SCID with a novel frameshift IL2RG mutation associated with maternal engraftment of CD8+ T cells. TME expansion in response to specific antigens, including EBV, has been reported, but little is known about the clinical significance and immunological characteristics of TME. 12 , 13 , 14 CMV infection in patients with SCID during early infancy can be a life‐threatening complication. 15 There have been many trials on controlling CMV infection using donor‐derived CMV‐specific CD8+ T cells, especially in recipients post‐HCT. 16 , 17 , 18 In our patient, naturally engrafted CD8+ T cells expanded and exerted presumed in vivo antiviral function against CMV infection prior to HCT. These findings indicate that engrafted CD8+ T cells expanded in response to cognate antigens and potentially engaged in a protective role against CMV infection. Further studies, such as cytotoxic assays against virus‐infected cells, are required to investigate the antiviral function of TME CD8+ T cells against various viral infections.
In our patient, haploidentical HCT from the maternal donor was performed. During the early post‐transplant course of HCT, CMV‐specific maternal engrafted CD8+ T cells successfully maintained their antiviral effector functions, indicating that they may contribute to the control of CMV infection in the patient.
In summary, this case reinforces the importance of TREC screening and early diagnosis for SCID. 2 In addition, it provides knowledge about the possible roles of maternal engrafted T cells in patients with SCID for improved management and would be informative for HCT in patients with SCID.
Methods
This study was reviewed and approved by the Institutional Review Board of Severance Hospital (Seoul, Republic of Korea; 4‐2020‐0220). Informed consent was obtained from the patient’s parents.
T‐cell receptor excision circles (TRECs)
Blood collection was performed on a filter paper (903 ProteinSaver Snap‐Apart Card; GE Healthcare, Westborough, MA, USA) using the heel‐stick technique. A single punch was taken from a dried blood spot using a 1.5‐mm puncher (Perkin Elmer, Waltham, MA, USA). The assay was performed according to the instructions provided in the EnLite Neonatal TREC Kit (Perkin Elmer).
Next‐generation sequencing (NGS) panel
The xGen Inherited Diseases Panel (Integrated DNA Technologies, Coralville, IA, USA) comprising 4503 genes, including genes associated with various syndromic and non‐syndromic immunodeficiencies, was used. Genomic DNA was extracted using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). Approximately 500 ng of genomic DNA was used, and the library was sequenced on a NextSeq 550 system (Illumina, San Diego, CA, USA) with 2 × 151 bp reads. Reads were aligned to human genomic reference sequences (GRCh37) using the Burrows–Wheeler alignment (BWA) tool (0.7.12). 19 HaplotypeCaller in the Genome Analysis Toolkit (GATK) package (3.8‐0) was used for variant calling, and ANNOVAR and VEP (87) software were used for annotation. 20 , 21 , 22
Surface antibody staining for flow cytometry
Cryopreserved peripheral blood mononuclear cells (PBMCs) were thawed and stained with fluorochrome‐conjugated antibodies against surface markers for 10 min at room temperature. CD132 expression was evaluated after 6 h of anti‐CD3/CD28 stimulation (100 ng mL−1 and 1 μg mL−1, respectively). Dead cells were excluded using LIVE/DEAD red/aqua fluorescent reactive dye (Invitrogen, Waltham, MA, USA). Multicolour flow cytometry was performed using an LSR II instrument (BD Biosciences, Franklin Lakes, NJ, USA), and the data were analysed using FlowJo software (BD Biosciences; Supplementary table 4).
Cell sorting
T cells and myeloid cells were isolated from PBMCs for TCR repertoire analysis and HLA typing of each subpopulation. After staining with fluorochrome‐conjugated antibodies, T cells and myeloid cells were isolated using a FACSAria II instrument (BD Biosciences).
TCR repertoire analysis
T‐cell receptor repertoire diversity was analysed using the LymphoTrack Dx TRG Assay Panel (Invivoscribe, Inc., San Diego, CA, USA) and LymphoTrack Dx TRB Assay Panel (Invivoscribe) on the NextSeq 550 system (Illumina).
HLA typing
HLA typing was performed using the AllType NGS Kit (One Lambda, Inc., Los Angeles, CA, USA) on the NextSeq 550 system (Illumina). TypeStream Visual NGS Analysis Software (One Lambda) was used for data analysis.
Antigen stimulation and intracellular cytokine staining
Thawed PBMCs (approximately 1 × 106 cells) were stimulated with soluble anti‐CD3 (100 ng mL−1; Miltenyi Biotec) and overlapping peptides (EBV; BARF1, IFNA; H1N1, RSV; MSG, and CMV; pp65, IE1 4 μg mL−1 and JPT; PPD 10 μg mL−1; Rockville, USA). After 13 h of stimulation, brefeldin A (BD Biosciences) and monensin (BD Biosciences) were added and culturing was continued for 11 h. The cultured cells were then washed and stained with fluorochrome‐conjugated antibodies against surface markers. After permeabilisation, intracellular cytokines were stained and evaluated using flow cytometry.
Methanol fixation for analysis of phosphorylated STAT5
Peripheral blood mononuclear cells (approximately 1 × 106 cells) were stained with antibodies against surface markers. After washing, cells were treated with IL‐2 (50 ng mL−1) or PBS for 10 min at 37°C, followed by incubation with IC fixation buffer (Invitrogen) for 10 min at room temperature and fixation with 100% cold methanol for 40 min at 4°C. Cells were then stained with anti‐human phosphor‐STAT5 antibodies for 1 h at room temperature, followed by flow cytometry.
Author contribution
June‐Young Koh: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Visualization; Writing‐original draft; Writing‐review & editing. Sang‐Bo Lee: Data curation; Formal analysis. Borahm Kim: Data curation; Methodology. Younhee Park: Methodology. Jong Rak Choi: Methodology. Sohee Son: Methodology. Yae‐Jean Kim: Conceptualization; Data curation. Seung Min Hahn: Methodology; Resources. Jong Gyun Ahn: Methodology; Resources. Ji‐Man Kang: Conceptualization; Data curation; Funding acquisition; Investigation; Methodology; Project administration; Supervision; Validation; Visualization; Writing‐original draft; Writing‐review & editing. Eui‐Cheol Shin: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Supervision; Validation; Visualization; Writing‐original draft; Writing‐review & editing.
Conflict of interest
The authors have no conflicting financial interests.
Supporting information
Acknowledgments
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education [Grant Number: 2019032869].
Contributor Information
Ji‐Man Kang, Email: umi87c@yuhs.ac.
Eui‐Cheol Shin, Email: ecshin@kaist.ac.kr.
References
- 1. Shearer WT, Dunn E, Notarangelo LD et al. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: the Primary Immune Deficiency Treatment Consortium experience. J Allergy Clin Immunol 2014; 133: 1092–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kwan A, Abraham RS, Currier R et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA 2014; 312: 729–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by gamma(c) family cytokines. Nat Rev Immunol 2009; 9: 480–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cirillo E, Cancrini C, Azzari C et al. Clinical, immunological, and molecular features of typical and atypical severe combined immunodeficiency: report of the italian primary immunodeficiency network. Front Immunol 2019; 10: 1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fischer A, Notarangelo LD, Neven B, Cavazzana M, Puck JM. Severe combined immunodeficiencies and related disorders. Nat Rev Dis Primers 2015; 1: 15061. [DOI] [PubMed] [Google Scholar]
- 6. Kinder JM, Stelzer IA, Arck PC, Way SS. Immunological implications of pregnancy‐induced microchimerism. Nat Rev Immunol 2017; 17: 483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Muller SM, Ege M, Pottharst A, Schulz AS, Schwarz K, Friedrich W. Transplacentally acquired maternal T lymphocytes in severe combined immunodeficiency: a study of 121 patients. Blood 2001; 98: 1847–1851. [DOI] [PubMed] [Google Scholar]
- 8. Denianke KS, Frieden IJ, Cowan MJ, Williams ML, McCalmont TH. Cutaneous manifestations of maternal engraftment in patients with severe combined immunodeficiency: a clinicopathologic study. Bone Marrow Transplant 2001; 28: 227–233. [DOI] [PubMed] [Google Scholar]
- 9. Gatti RA, Meuwissen HJ, Allen HD, Hong R, Good RA. Immunological reconstitution of sex‐linked lymphopenic immunological deficiency. Lancet 1968; 2: 1366–1369. [DOI] [PubMed] [Google Scholar]
- 10. Dorsey MJ, Dvorak CC, Cowan MJ, Puck JM. Treatment of infants identified as having severe combined immunodeficiency by means of newborn screening. J Allergy Clin Immunol 2017; 139: 733–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wahlstrom J, Patel K, Eckhert E et al. Transplacental maternal engraftment and posttransplantation graft‐versus‐host disease in children with severe combined immunodeficiency. J Allergy Clin Immunol 2017; 139: 628–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thompson LF, O'Connor RD, Bastian JF. Phenotype and function of engrafted maternal T cells in patients with severe combined immunodeficiency. J Immunol 1984; 133: 2513–2517. [PubMed] [Google Scholar]
- 13. Meyer‐Olson D, Shoukry NH, Brady KW et al. Limited T cell receptor diversity of HCV‐specific T cell responses is associated with CTL escape. J Exp Med 2004; 200: 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Touzot F, Dal‐Cortivo L, Verkarre V et al. Massive expansion of maternal T cells in response to EBV infection in a patient with SCID‐Xl. Blood 2012; 120: 1957–1959. [DOI] [PubMed] [Google Scholar]
- 15. Pai SY, Logan BR, Griffith LM et al. Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N Engl J Med 2014; 371: 434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Blyth E, Clancy L, Simms R et al. Donor‐derived CMV‐specific T cells reduce the requirement for CMV‐directed pharmacotherapy after allogeneic stem cell transplantation. Blood 2013; 121: 3745–3758. [DOI] [PubMed] [Google Scholar]
- 17. Neuenhahn M, Albrecht J, Odendahl M et al. Transfer of minimally manipulated CMV‐specific T cells from stem cell or third‐party donors to treat CMV infection after allo‐HSCT. Leukemia 2017; 31: 2161–2171. [DOI] [PubMed] [Google Scholar]
- 18. Tzannou I, Papadopoulou A, Naik S et al. Off‐the‐Shelf virus‐specific T cells to treat BK virus, human herpesvirus 6, cytomegalovirus, Epstein‐Barr virus, and adenovirus infections after allogeneic hematopoietic stem‐cell transplantation. J Clin Oncol 2017; 35: 3547–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li H. Toward better understanding of artifacts in variant calling from high‐coverage samples. Bioinformatics 2014; 30: 2843–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cibulskis K, Lawrence MS, Carter SL et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013; 31: 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res 2010; 38: e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 2010; 26: 2069–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Blom M, Pico‐Knijnenburg I, Sijne‐van Veen M et al. An evaluation of the TREC assay with regard to the integration of SCID screening into the Dutch newborn screening program. Clin Immunol 2017; 180: 106–110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials