

Abstract
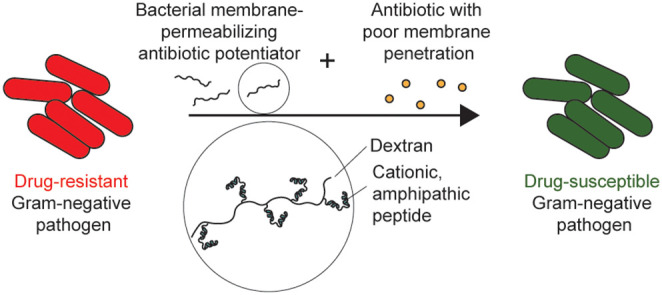
The drug-impermeable bacterial membrane in Gram-negative pathogens limits antibiotic access to intracellular drug targets. To expand our rapidly waning antibiotic arsenal, one approach is to improve the intracellular delivery of drugs with historically poor accumulation in Gram-negative bacteria. To do so, we engineered macromolecular potentiators to permeabilize the Gram-negative membrane to facilitate drug influx. Potentiators, known as WD40, were synthesized by grafting multiple copies of a cationic α-helical antimicrobial peptide, WLBU2, onto a dextran polymer scaffold. WD40 enabled drug uptake in the model pathogen P. aeruginosa, a capability that was not observed with unmodified WLBU2 peptide. WD40 was able to reduce minimum inhibitory concentrations of a drug panel by up to 3 orders of magnitude. Hydrophobic and highly three-dimensional antibiotics exhibited the greatest potentiation. Antibiotic activity was potentiated in several clinical strains and resulted in sensitization of drug-resistant strains to rifampin, a drug not previously used for Gram-negative infections.
Keywords: Gram-negative bacteria, drug resistance, antibiotic potentiator, antibiotic adjuvant, antimicrobial peptides, multivalent
Antibiotic drug resistance is a serious threat to global public health. Among the biggest threats are the ESKAPE pathogens (i.e., Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinobacter baumanii, Pseudomonas aeruginosa, and the Enterobacter species), which are bacteria with a rapidly growing frequency of drug resistance and the most common causes of hospital-acquired infections.1 Of the six pathogens, four are Gram-negative bacteria. Gram-negative bacteria are innately resistant to many antibiotics due to their highly drug-impermeable cell wall, which consists of two membranes, an outer membrane and inner membrane, sandwiching a periplasmic space. The outer membrane, in particular, contributes to poor drug penetration due to its lipopolysaccharide (LPS)-dense outer leaflet. Tight packing of the saturated lipid chains in adjacent LPS molecules and the polyanionic surface charge conferred by the LPS oligosaccharides hinder the passive diffusion of small molecules.2 Thus, Gram-negative pathogens are particularly difficult to kill. Antimicrobial leads found to engage robustly with intracellular targets in high-throughput screens have generally exhibited poor membrane penetration.3 Thus, many potential therapeutics are lost in the drug development pipeline due to the membrane barrier. Given the scarcity of new drug classes and the waning efficacy of existing drugs, new strategies must be established to address this clinical gap.
One such strategy is to repurpose drugs that have historically only been used to treat Gram-positive infections. The limited activity of these drugs in Gram-negative pathogens is largely due to the membrane barrier. Therefore, by permeabilizing the Gram-negative membrane, these drugs can enter the periplasm or cytoplasm to engage with their drug targets. This can be achieved by coadministration of antibiotics with an antibiotic adjuvant that selectively disrupts the Gram-negative bacterial membrane to prevent off-target damage and toxicity to mammalian cells (Figure 1). However, there are currently no FDA-approved antibiotic adjuvants with this capability. Chelating agents (e.g., EDTA) can permeabilize the outer membrane by sequestering the divalent cations that electrostatically cross-link LPS (i.e., Ca2+, Mg2+).4 However, these agents do not act selectively on bacterial membranes. Cationic antimicrobial peptides (AMPs) destabilize the outer membrane by binding anionic oligosaccharides and by subsequently displacing divalent cations or by forming pores.5 However, AMPs also have poor therapeutic indices due to their off-target toxicity at bacteria-killing concentrations.6 In the pharmaceutical pipeline, Spero Therapeutics has developed SPR741, a systemically administered outer membrane-disrupting peptide agent derived from polymyxin B, which has gone through Phase 1 clinical trials for safety.7−9 By removing the lipid tail and reducing the cationic charge in polymyxin B, they were able to mitigate off-target toxicity at membrane-disrupting concentrations.
Figure 1.
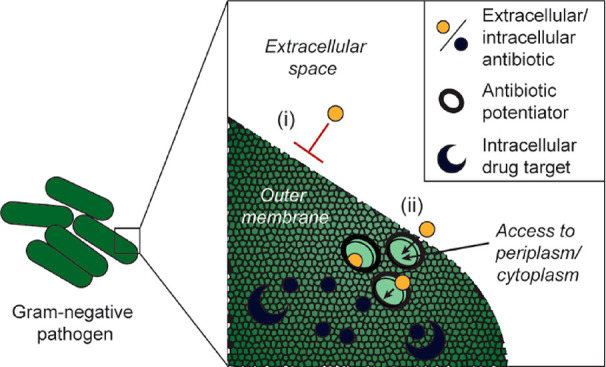
Schematic of antibiotic potentiator approach. (i) The Gram-negative bacterial membrane is a barrier for drug entry. (ii) Antibiotic potentiators that destabilize the bacterial membrane allow small molecule drug entry into the periplasm or cytoplasm, where the drug can bind its intracellular target.
In this work, we explore an alternative approach using multivalent AMP display on polymeric scaffolds to develop membrane-disrupting antibiotic potentiators. Multivalency is a strategy that has been previously used to increase AMP activity at lower concentrations.10,11 Multivalent AMPs can be more potent than their monovalent form due to (1) simultaneous engagement of multiple binding sites, (2) enhanced oligomerization for pore-forming AMPs, (3) greater charge density to maintain activity at physiological salt concentrations, and (4) improved stability against protease degradation.10,11 We are particularly interested in antibiotic potentiators for respiratory infections because multidrug-resistant Gram-negative pathogens are prevalent causes in ventilator-associated pneumonia (VAP).12 Here we developed a multivalent antibiotic potentiator that reduces the minimum inhibitory concentration (MIC) for a diverse range of antibiotics in P. aeruginosa, a common VAP pathogen. We further show the clinical utility of the potentiator by sensitizing drug-resistant clinical strains to rifampin, a drug not previously used for Gram-negative infections.
Prior to our appreciation for the practicality and modularity of potentiator agent-antibiotic coformulations, we initially sought to design peptide-drug conjugates for improved drug delivery into Gram-negative bacteria. Cell-penetrating peptides (CPPs) have played a large role in delivery systems in mammalian cells.13 Therefore, we conjectured that peptides with selectivity for the bacterial cell membrane over mammalian cell membrane could serve a similar purpose for delivery of a drug payload into the bacterial periplasm or cytoplasm. Peptide-drug conjugates were initially investigated for their ability to traverse the Gram-negative membrane, and our findings led us to explore codelivery of antibiotics with multivalent antibiotic potentiators. In this initial work, a linezolid variant (LZDvar) was used as a model drug with limited intracellular accumulation due to the outer membrane barrier and efflux pumps.14,15 Linezolid is a bacteriostatic antibiotic in the oxazolidinone class, which inhibits bacterial protein synthesis by binding the 50S subunit of the prokaryotic ribosome. In our assay, LZDvar was conjugated to one of nine different peptides reported to interact with bacterial membranes and/or to possess antimicrobial activity (Figure 2A). Each conjugate was then evaluated for its ability to inhibit bacterial growth in vitro relative to the drug or peptide alone to determine the effect of the peptide on drug trafficking into the bacteria (Figure 2B). LZDvar is a piperazine variant of linezolid and was used to synthesize conjugates because the nitrogen substitution of the morpholine oxygen in the 4′-position of the C-ring enables alkylation with an azide linker as previously reported16 (Figure S1). Alteration of this position with different functional groups is known to be well-tolerated without significant loss of activity.16,17 Following alkylation with an azide linker containing a 6-carbon chain, the completed azido-functionalized linezolid variant (azido-LZDvar) was conjugated via a dibenzylcyclooctyne (DBCO)-maleimide linker to the peptides (Figure 2A). The peptide panel includes several cationic amphipathic peptides with α-helical secondary structure that disrupt or cross the bacterial membrane through a range of mechanisms (e.g., transient toroidal pore formation, membrane micellization, and transporter-dependent translocation) (Table S1).5 To assess the effect of the peptide on antibiotic trafficking into the cell, the model Gram-negative pathogen, Pseudomonas aeruginosa (strain PA14), was incubated with peptide-LZDvar conjugates and peptides alone in microdilution assays to determine the concentrations at which 90% of bacterial growth is inhibited (MIC90). The ratio of MIC90 values for the peptide alone over the conjugate, MIC90Peptide/MIC90Conjugate, was used as a functional readout for antibiotic cell entry (Figure 2C). Azido-LZDvar does not inhibit growth at the tested concentrations due to the membrane barrier (Figure 2D). Therefore, if a peptide helps the conjugated LZDvar traffic across the bacterial membrane, the MIC90 of the conjugate is expected to be lower than that of the peptide alone (i.e., MIC90 ratio >1) because of the combined activity of the peptide and the now-active drug. Of the nine conjugates evaluated, the conjugate containing WLBU2 peptide had the greatest MIC90 ratio, >4-fold difference between MIC90Peptide and MIC90Conjugate (Figure 2C). WLBU2 is a 24-residue peptide derived from lentiviral lytic peptide 1 (LLP1), a peptide with broad spectrum activity and a sequence corresponding to the C-terminal region of the HIV-1 gp41 envelope protein.18 To increase the membrane affinity of LLP1, arginine and tryptophan substitutions were used on the cationic and hydrophobic faces, respectively, to form WLBU2, an idealized amphipathic, helical peptide.19 While WLBU2 increased cell entry of the conjugated LZDvar, comparison of the MIC curves for the conjugate versus the physical mixture of WLBU2 and azido-LZDvar indicates that it does so only when linezolid is covalently bound (Figure 2D). This suggests that the peptide is not permeabilizing the outer membrane for drug entry, but rather the entire conjugate is trafficking across the intact membrane. Using super-resolution microscopy to visualize GFP-expressing PA14 fixed after a 10 min incubation with rhodamine-labeled WLBU2, we observed that WLBU2 does, in fact, enter the bacterial cytoplasm with the fluorophore cargo in addition to rapidly localizing to the bacterial membrane (Figure 2E). No intracellular signal was observed with a rhodamine-only control. Taken together, these results indicate that WLBU2 has a high affinity for the Gram-negative membrane, although its interaction with the membrane does not disrupt the barrier function sufficiently to increase azido-LZDvar uptake at sub-MIC concentrations. Antibiotic conjugation to WLBU2 can therefore be used to increase drug uptake. However, we realized this strategy was impractical given that each antibiotic of interest would require modification with a reactive handle while working within the constraints of the drug-specific structure–activity relationship (SAR). In contrast, a more modular strategy would involve coadministration of a bacterial membrane-disruptive antibiotic adjuvant with a poorly penetrating antibiotic. When we tested mixtures of peptides and linezolid, no MIC changes were observed for the tested concentration range (Table S2). We hypothesized this was due to limited potency of peptides in monovalent form. Therefore, we sought to investigate if we could leverage its membrane affinity and modify WLBU2 into a membrane-disruptive multivalent form that could promote antibiotic influx into Pseudomonas aeruginosa.
Figure 2.

Identification of antibiotic-potentiating peptide WLBU2 in functional assays. (A) Structure of the membrane-impermeable model antibiotic (azido-functionalized linezolid variant, or azido-LZDvar) and peptide-LZDvar conjugates used to identify peptides that can facilitate transmembrane antibiotic transport. Conjugates were synthesized by attaching azido-LZDvar to one of nine candidate peptides via copper-free click chemistry with a DCBO-maleimide linker. Percent yields of peptide-LZDvar conjugates were calculated by dividing actual yield by theoretical yield. (B) Schematic of microdilution assays in 96-well plate format used to assess bacterial growth inhibition with peptide-LZDvar conjugates versus peptides alone. MIC90 is the peptide or conjugate concentration inhibiting 90% bacterial growth. When MIC90Conjugate < MIC90Peptide (as shown with the blue peptide), this indicates improved antibiotic trafficking into the cytoplasm. Alternatively, when MIC90Conjugate = MIC90Peptide (as shown with the red peptide), this indicates no improvement in antibiotic drug trafficking into the cytoplasm. (C) The ratio, MIC90Peptide/MIC90Conjugate, was used to rank peptides from most to least effective for intracellular drug delivery. Three technical replicates were averaged to determine MIC90Peptide and MIC90Conjugate for each candidate peptide. > indicates that the MIC90 ratio is greater than the indicated value because MIC90Peptide was greater than the maximum tested concentration. The MIC90 ratio for TAT was not determined (n.d.) because both MIC90Peptide and MIC90Conjugate were greater than the maximum tested concentration. (D) Bacterial growth inhibition curves for azido-LZDvar alone, WLBU2 peptide alone, a physical mixture of WLBU2 peptide and azido-LZDvar, and the WLBU2-LZDvar conjugate (mean ± s.d., n = 3). (E) Fluorescent images collected using a super-resolution microscope showing localization of rhodamine-labeled WLBU2 peptide in the membrane (yellow arrow) and cytoplasm (white arrow) of GFP-expressing P. aeruginosa. Images are representative of observations from two independent imaging experiments. All experiments were completed using P. aeruginosa (strain PA14).
Kumagai et al. previously showed that WLBU2 increases stiffness and chain order of bacterial membrane mimics at low concentrations and softens and increases the disorder of the lipids at high concentrations.20 Differences in local peptide concentration at the bacterial membrane are thus thought to lead to juxtapositioning of membrane domains with different stiffness and order, leading to leakage at the domain boundaries. Using a multivalent form of WLBU2, we hypothesized that this membrane disruption would be more exaggerated given the greater differential in local peptide concentrations. To test this hypothesis, multivalent WLBU2 constructs were synthesized by grafting WLBU2 onto 10 kDa and 40 kDa linear dextrans, which we will refer to as Dextran 10 and Dextran 40, respectively. We chose hydrophilic dextran to offset the hydrophobicity of WLBU2 peptide and to investigate the effect of scaffold size on membrane disruption. To introduce amine groups for peptide conjugation, dextrans were first oxidized using sodium periodate to convert hydroxyl groups into reactive aldehydes and subsequently reacted with sodium cyanoborohydride and ethylenediamine for reductive amination (Figure 3A). Reaction conditions were optimized, and overnight reaction of 25:1 and 50:1 molar ratio of sodium periodate to Dextran 10 and Dextran 40, respectively, followed by reductive amination produced an average of ∼22 and ∼27 amine groups per dextran (Table S3). 5, 10, and 20 copies of peptide were reacted per dextran size to produce a total of six multivalent potentiator candidates (referred to as WD10 and WD40 potentiator candidates for WLBU2-conjugated Dextran 10 and Dextran 40, respectively). 280 nm absorbance showed that WD10 potentiator candidates contained an average of 7, 13, or 22 peptides per Dextran 10 and the WD40 potentiator candidates contained an average of 5, 8, or 14 peptides per Dextran 40 (Figure 3B, Figure S2). As expected, multivalent display increased peptide potency. While the MIC90 for WLBU2 peptide was greater than 40–80 μM (Figure 2D, Figure 3B), MIC90 values for the potentiator candidates were as low as 5 μM by peptide concentration (Figure 3B). Regardless of peptide valency, WD40 MICs were reduced 2-fold when combined in a physical mixture with free linezolid in microdilution assays with PA14, indicating linezolid uptake (Figure 3B). This was also true with trimethoprim, another antibiotic with poor membrane penetration that targets the bacterial folate pathway.21 In contrast, WD10 MICs were not consistently reduced when combined with free antibiotics, suggesting weaker membrane disruption. From the potentiator panel, we subsequently focused on WD10 with 7 peptides and WD40 with 5 peptides (i.e., the candidates with the fewest peptides) because higher-valency potentiator candidates precipitated during microdilution assays. In imaging experiments to assess membrane permeabilization, PA14 was incubated with a mixture of potentiator candidates and propidium iodide (PI), a small molecule dye that fluoresces upon intercalation with DNA. PI uptake was observed in bacteria treated with WD40 but not WD10 (Figure 3C). Therefore, we moved forward with WD40 for all subsequent experiments. TEM imaging of PA14 after 5 min exposure to WD40 confirmed membrane disruption, which was more severe than that caused by free peptide at equal concentration (Figure 3D).
Figure 3.

Potentiator candidate WD40 selectively disrupts the bacterial membrane to allow small molecule influx into the cytoplasm. (A) Synthetic scheme for potentiator candidates—WD10 and WD40. (B) Table showing MIC90 values for WD10 and WD40 containing different number of WLBU2 peptides (n = 3). MIC90 values were determined in a microdilution assay with P. aeruginosa (strain PA14) in which different dilutions of potentiator candidates were combined with a fixed drug concentration—5 μM free linezolid (LZD) or 5 μM trimethoprim (TMP). (C) Fluorescent images acquired using a super-resolution microscope to visualize PI influx in PA14 with WD10 or WD40 treatment. Images are representative of observations from two independent imaging experiments. (D) Transmission electron microscopy (TEM) images of the PA14 untreated control and PA14 after a 5 min incubation with Am-Dextran 40 (1 μM), free WLBU2 peptide (5 μM), or WD40 (5 μM). Images show disruption of the bacterial membrane by free peptide and WD40. (E) Comparison of the concentration-dependent hemolytic activity of WD40 versus free WLBU2 peptide (n = 3). No significant difference was observed between the two treatments. Repeated measures ANOVA was used to determine the p-value. (F) Comparison of the concentration-dependent cytotoxicity of WD40 versus free WLBU2 peptide in HEK293T cells (n = 3). WD40 is significantly less toxic than free WLBU2 peptide. Repeated measured ANOVA was used to determine p-value. Lethal concentration for 50% cells (LC50) was determined using nonlinear regression to fit a dose–response curve.
Selectivity of antibiotic potentiators for bacterial membranes over mammalian membranes is necessary to prevent off-target toxicity. Concentration-dependent WD40 toxicity in mammalian cells was measured via a hemolysis assay in which lysis of red blood cells (RBCs) was quantified after 1 h incubation with WD40 or free WLBU2 peptide. WD40 exhibited comparable hemolytic activity as free peptide (Figure 3E, p = 0.2616). WD40 cytotoxicity in HEK293T cells, an immortalized human embryonic kidney cell line, was also assessed using an MTS assay. WD40 exhibited significantly less toxicity to cells at the same concentration as the free peptide after 24 h incubation (Figure 3F, p = 0.0041). The ratio of LC50/MIC90 can be used as a selectivity index (SI),22 where LC50 is the concentration lethal to 50% of mammalian cells in the MTS assay (Figure 3F). A higher SI is reflective of a better therapeutic index. For the free peptide, SI < 0.2. For WD40, SI = 12.0. Therefore, WD40 has ∼60-fold greater bacterial selectivity than the WLBU2 peptide. Taken together, these results indicate that WD40 can potentially disrupt bacterial membranes to increase small molecule uptake while leaving host cell membranes intact.
Several classes of narrow spectrum antibiotics exist that only have activity in Gram-positive bacteria. Our goal was to create a potentiator for coadministration with these drugs to create broad spectrum activity. To determine which drugs would benefit most from this approach, we completed standard checkerboard assays in which WD40 was combined with 1 of 32 different antibiotics to determine if combinations were synergistic, additive, or antagonistic for bacterial growth inhibition (drug class and physicochemical properties shown in Table S4). We chose this panel to represent a variety of antibiotic classes with different drug targets. Furthermore, we included drugs historically known to accumulate poorly in Gram-negative pathogens (i.e., vancomycin, novobiocin, mupirocin, fusidic acid, rifampin, clindamycin)23 as well as drugs with high accumulation (i.e., those from antipseudomonal drug classes such as the aminoglycosides, fluoroquinolones, and select beta-lactams). Using the checkerboard assay results, the fractional inhibitory concentration index (FICI) was derived for each pairing, where FICI ≤ 0.5 indicates synergism, 0.5 < FICI ≤ 4.0 indicates an additive effect, and FICI > 4.0 indicates antagonism. WD40 demonstrated additive effects with 9 of the 32 antibiotics and synergistic effects with 23 of the 32 antibiotics (Figure S3). Representative FIC curves are shown for additive and synergistic combinations in Figure 4.
Figure 4.

WD40 has additive and synergistic activity with antibiotics in P. aeruginosa (strain PA14). Checkerboard assays were used to determine the fractional inhibitory concentrations (FICs) of the tested potentiator-antibiotic pairs. FIC is the MIC of the compound in combination divided by the MIC of the compound alone. Fractional inhibitory concentration indices (FICIs) are calculated by summing FICWD40 and FICAntibiotic to determine if the pairing is synergistic (FICI ≤ 0.5, blue area in graph), additive (0.5 < FICI ≤ 4.0, gray area in graph), or antagonistic (FICI > 4.0, area not shown in graph). (A, B) Representative graphs of additive potentiator-drug relationships. (C) Representative graph of a synergistic potentiator-drug relationship. Results were reproduced in two independent experiments. For each experiment, n = 1 per concentration combination.
In checkerboard assays, when antibiotics were combined with sub-MIC levels of WD40 (5 μM equivalent WLBU2 concentration), MIC fold change for the antibiotic panel ranged from no change to up to 4096-fold change (Figure 5A). Using these values as measures of potentiation, we observed that activities of low-accumulation drugs are the most improved with WD40, whereas high-accumulation drugs are the least improved. The slimmer margin of improvement for the latter group makes sense given that they already possess potent activity in Gram-negative bacteria. Using this data set, we sought to identify the drug physicochemical properties that are most predictive of potentiation with WD40. In a prior survey of over 180 diverse compounds, Richter et al. discovered that molecules with sterically unencumbered, ionizable nitrogens (generally primary amines), low three-dimensionality (globularity ≤ 0.25), and high rigidity (≤5 rotatable bonds) have the greatest intracellular accumulation in E. coli due to transport through porin channels.23,24 Prior to this work, molecular weight and hydrophobicity (reflected by ClogP, the calculated octanol:water distribution coefficient) were canonically viewed to be the most important factors for drug accumulation in Gram-negative pathogens.25 Here, we assessed the correlation between potentiation and molecular weight, ClogP, three-dimensionality (using globularity and plane-of-best-fit or PBF), and the number of rotatable bonds (Figure 5B–F). We found that ClogP was the strongest predictor of potentiation (Figure 5C, r = 0.676, p < 0.0001). Globularity and PBF are two different metrics used to describe a molecule’s three-dimensionality. While globularity did not strongly correlate with potentiation (Figure 5D, r = 0.266, p = 0.1559), PBF did (Figure 5E, r = 0.480, p = 0.0073). Molecular weight showed weak but insignificant correlation with potentiation (Figure 5B, r = 0.314, p = 0.0906), and the number of rotating bonds (i.e., flexibility) showed no significant correlation with potentiation (Figure 5F, r = 0.285, p = 0.1276). These analyses demonstrate that hydrophobic and highly three-dimensional antibiotics are best potentiated by WD40 in the PA14 strain and is potentially useful for predicting best potentiator-drug pairings.
Figure 5.

WD40 improves the activity of hydrophobic and nonplanar antibiotics in P. aeruginosa (strain PA14). (A) Bar graph showing MIC fold changes for antibiotics in combination with WD40. The names of drugs are colored blue for those with reported antipseudomonal activity. To identify which physicochemical properties are best predictors for potentiation, dot plots were generated of MIC fold change versus (B) molecular weight, (C) ClogP, (D) globularity, (E) plane-of-best-fit (PBF), and (F) number of rotating bonds. (B–F) Drugs with reported antipseudomonal activity are indicated by blue dots. Only small molecule antibiotics were included in correlation analysis. Therefore, colistin and vancomycin are excluded from the shown graphs. Pearson’s correlation coefficients (r) and p-values were calculated using linear regression analysis. The results were reproduced in two independent experiments. For each experiment, n = 1 per antibiotic.
For clinical applicability, we sought to determine if antibiotic potentiation was also observed in P. aeruginosa clinical isolates, including strains with drug resistance against standard antipseudomonal drugs (Figure 6A). In this assessment, we focused on fusidic acid, clindamycin, and rifampin, the three drugs with the greatest potentiation by WD40 in prior studies. Interestingly, the MIC of WD40 itself was consistently 2-fold higher in these isolates (20 μM) compared to that in PA14. When combined with WD40 at sub-MIC concentrations (10 μM), drug MICs were generally reduced by 64–512-fold (Figure 6B–D), which is lower than the 1024–2048-fold change observed in PA14 using 5 μM WD40. These observations suggest that clinical isolates have a more robust membrane barrier compared to PA14. Despite the synergistic activity of WD40 with the three drugs in nearly all clinical isolates (Figure S4), MICs for fusidic acid and clindamycin did not meet their clinical breakpoints, the MIC threshold below which a strain is defined to be drug-susceptible (Figure 6B,C). WD40 did, however, reduce rifampin MICs to below its clinical breakpoint in all the tested isolates (Figure 6D). This is particularly interesting as BWH006 represents a multi-drug-resistant strain that would be difficult to treat in a clinical setting. However, using WD40, we can sensitize it to rifampin and provide a new potential therapeutic solution.
Figure 6.

WD40 potentiates antibiotics in P. aeruginosa clinical isolates. (A) Clinical isolates with varying susceptibilities to antipseudomonal drugs. “S” indicates susceptible (MIC < clinical breakpoint) and “R” indicates resistant (MIC > clinical breakpoint). (B–D) MIC95 values for fusidic acid, clindamycin, and rifampin in clinical isolates in the absence or presence of 10 μM WD40. Clinical breakpoints for Gram-positive S. aureus, S. pneumoniae, and Streptococcus spp. (source: EUCAST) are indicated in graphs by dotted lines. < symbol indicates MIC95 values lower than the lowest value on the y-axis. The results were reproduced in two independent experiments. For each experiment, n = 1 per experimental condition.
While clinical isolates were already more sensitive to rifampin compared to fusidic acid and clindamycin (MICIsolates,Rifampin = 8–16 μg/mL, MICIsolates,Fusidic acid = 512–1024 μg/mL, MICIsolates,Clindamycin = 1024–2048 μg/mL), our results suggest that initial sensitivity alone does not dictate whether the clinical breakpoint is met when the drug is combined with the potentiator. The strain PA14 is much less sensitive to rifampin than the clinical isolates (MICPA14,Rifampin = 512 μg/mL), yet WD40 was still able to reduce the MIC to the clinical breakpoint. Furthermore, fusidic acid and clindamycin MICs were comparable in clinical isolates and PA14 (Table S4), yet clinical breakpoints were met for both drugs in PA14 and not in the clinical isolates (Figure 6B,C). Taken together, these results demonstrate more consistent potentiation of rifampin in different P. aeruginosa strains compared to fusidic acid and clindamycin. This could potentially be attributed to differences in drug mechanism (rifampin is an RNA polymerase inhibitor while fusidic acid and clindamycin inhibit protein synthesis via binding of the elongation factor G and 50s ribosomal subunit, respectively). Synergistic combinations of rifampin and membrane-disruptive agents (colistin, oligo-acyl-lysyls, SPR741) have been previously reported in several scientific reports for drug-resistant Gram-negative pathogens.26−28 With the addition of our independent findings, there is now even greater support for pairing rifampin with a membrane-destabilizing antibiotic adjuvant.
The Gram-negative bacterial membrane is a delivery barrier for several classes of antibiotics. In this work, we demonstrated that a monovalent peptide with affinity for the Gram-negative membrane and exhibiting no membrane-disruptive activity can be modified into a membrane-disruptive agent via multivalent display on a polymer scaffold. At sub-MIC concentrations, the resulting construct, WD40, potentiated the antimicrobial activity of previously ineffective antibiotics by enabling drug uptake into the bacterial cell. Membrane permeabilization is likely a result of higher, more localized concentrations of WLBU2 at the membrane surface, which can lead to juxtapositioning of membrane domains with different stiffness and order.20 Through an in vitro screen, we found that highly hydrophobic and three-dimensional antibiotics are best potentiated by WD40. Rifampin is an example of a drug fitting these criteria for which we were able to reduce MICs below the clinical breakpoint in drug-resistant clinical isolates. In addition to potentiating established drugs such as rifampin, we can potentially rescue the activity of drug candidates that have fallen out of the drug development pipeline due to poor membrane penetration.
In summary, we have demonstrated the feasibility of using engineered macromolecular constructs to overcome drug resistance, which can provide more favorable pharmacokinetics compared to small molecules and single peptide agents. Antibiotic potentiation was demonstrated in vitro using standard broth dilution assays. While these methods are well-accepted, additional in vitro investigation in simulated lung fluid may be helpful in light of the possible instability of maleimide–thiol conjugates caused by in vivo thiol exchange reactions.29 Furthermore, while selectivity was studied in the context host versus pathogen, additional investigation is needed to determine selectivity for Gram-negative pathogens over Gram-negative commensals that are found in healthy human airways. Finally, in vivo assessment of therapeutic efficacy of potentiator-drug pairings will be completed in future work.
Methods
Peptide Synthesis
The peptide panel used for identification of Gram-negative membrane-penetrating peptides (Table S1) was synthesized with C-terminal cysteines for linezolid conjugation. Peptides were synthesized to ≥80% purity by the Koch Institute Swanson Biotechnology Center. For subsequent studies, WLBU2 peptide was synthesized to ≥90% purity by CPC Scientific.
Synthesis of Peptide-Linezolid Conjugate Panel
Linezolid was modified with a C6 linker terminating in an azido reactive group for covalent conjugation to the peptide panel via copper-free click chemistry. Linezolid conjugation was completed by first reacting dibenzylcyclooctyne-maleimide linker (Sigma-Aldrich, Cat. No. 760668) with peptide at a 4:1 molar ratio in PBS pH 7.4 for 2 h. Unreacted linker was removed using desalting spin columns with 1000 MWCO (G Biosciences SpinOUT GT-600). Azido-functionalized linezolid was then reacted with the DBCO-modified peptide at a 3:1 molar ratio in PBS pH 7.4 overnight. The product was HPLC purified and confirmed using MALDI-MS.
Microdilution Assays to Determine Minimum Inhibitory Concentrations (MICs) for Single Agents
Samples (peptides, peptide-linezolid conjugates, peptide-linezolid mixtures) were diluted from stock solutions into Mueller–Hinton broth (MHB) for a maximum working concentration of up to 160 μM peptide. Serial 1:1 dilutions were completed in MHB to prepare 7 different concentrations plus one blank MHB control. Each sample was plated in triplicate into a 96 well-plate in 50-μL volumes. 50 μL of a bacterial suspension (1 × 106 cfu/mL P. aeruginosa (strain PA14) in MHB) prepared fresh from a secondary culture was then added to each well. Plates were incubated in a humidified chamber at 37 °C overnight for 16 h and absorbance at 600 nm was read the following morning using a plate reader. PA14 was provided by the laboratory of Deborah Hung.
Visualization of Fluorophore-Labeled WLBU2 in P. aeruginosa (Strain PA14)
For microscopic visualization of WLBU2 in peptide-treated bacteria, WLBU2 with a C-terminal cysteine was labeled with Rodamine Red C2 maleimide (ThermoFisher Scientific, Cat. No. R6029). 1 × 108 cfu PA14 was incubated with 22.5 μM labeled WLBU2 or labeled Slides were imaged on the DeltaVision-OMX super-resolution microscope.
Synthesis of WLBU2 Potentiator Candidates
Dextran μ and Dextran 40 (Sigma-Aldrich Cat. No. 9260) were first functionalized with amine groups for peptide conjugation through a series of reactions and purification steps. To produce 20–30 amine groups per dextran, the dextrans were first oxidized to produce reactive aldehyde groups. 0.5 g Dextran 10 was dissolved in 12.5 mL 100 mM sodium periodate in DI water, and 0.5 g Dextran 40 was dissolved in 18.75 mL 100 mM sodium periodate in DI water. After reacting in the dark overnight under stirring conditions, the reactions were transferred to Spectra/Por dialysis tubing with 500–1000 Da MWCO (for Dextran 10) and 10 kDa MWCO (for Dextran 40) to remove the sodium periodate. After dialyzing against DI water for 24h with 6 changes of DI water, the oxidized dextrans were lyophilized. For amine functionalization, the oxidized dextrans were each dissolved in 12.5 mL 3 M ethylenediamine in 0.1 M sodium phosphate buffer, pH 7.4, 150 mM NaCl, and 2.5 mL 1 M sodium cyanoborohydride (prepared fresh) was added under stirring conditions. After reacting overnight, the amine-functionalized dextrans (Am-Dextran 10 and Am-Dextran 40) were isolated from the other reagents by running the reaction solutions twice each through PD-10 size exclusion columns (GE Healthcare). For more exhaustive purification, the column-filtered products were dialyzed against DI water for 24 h with 6 changes of DI water in dialysis tubing with the same corresponding MWCO as before. Following dialysis, the final products were lyophilized. Amine quantification was completed using a TNBS assay. WLBU2 peptides with C-terminal cysteines were conjugated to amine-functionalized dextrans in a one-pot reaction with SIA linkers (ThermoFisher Scientific, Cat. No. 22349). The conjugation reaction was completed at a 1:n:n molar ratio of amine groups to SIA linkers to WLBU2 peptide in pH 8.2 borate buffer, where n = 5, 10, or 20. After overnight reaction, unreacted linkers and peptide were removed using dialysis. To prevent precipitation of reactants/products, the reaction solution was dialyzed against pH 7.4 PBS the first day and against DI water the second and third day. The finished products were lyophilized for long-term storage. For visualization in bacteria, fluorescent W2 potentiator candidates were synthesized by first fluorophore-labeling Am-Dextrans followed by peptide conjugation as describe above. Briefly, Am-Dextran 10 and Am-Dextran 40 were reacted with AlexaFluor 488 NHS ester (ThermoFisher Scientific, Cat. No. A20000) in pH 7.4 PBS at a 3:1 molar ratio of fluorophore to dextran overnight and subsequently dialyzed and lyophilized.
Visualization of Propidium Uptake in P. aeruginosa (Strain PA14)
Standard secondary culture preparation as described above was used to grow bacteria to log phase with OD = 0.5–0.7. Bacteria were pelleted by centrifugation (2000 rcf for 5 min) and resuspended in PBS. 1 × 108 cfu PA14 were aliquoted into Eppendorf tubes, spun down to remove the PBS, and resuspended in a solution containing 500 nM PI (ThermoFisher Scientific, Cat. No. P3566) and W2-Dextran 10/W2-Dextran 40 (10 μM by peptide concentration). After 15 min incubation time, bacteria were pelleted and washed twice with PBS. Bacteria were then fixed in 4% chilled paraformaldehyde for 10 min, pelleted, and resuspended in 30 μL PBS. The sample was then mounted on a glass slide by placing one drop of bacterial suspension on top of a single drop of Fluoromount-G (SouthernBiotech, Cat. No. 0100-01) and coverslipping. Slides were imaged using a DeltaVision-OMX super-resolution microscope.
TEM Imaging
A secondary culture of P. aeruginosa (strain PA14) was grown to OD600 −0.5. Bacteria was washed twice with PBS and 1 × 106 cfu was aliquoted and pelleted in 1.5 cc Eppendorf tubes. Bacteria was then resuspended in 100 μL PBS (control) or a solution of Am-Dextran 40 (1 μM), free WLBU2 peptide (5 μM), or WD40 (5 μM). After 5 min incubation, bacteria were pelleted by centrifugation (2000 rcf for 5 min) and supernatant was removed. The bacteria pellet was then fixed in glutaraldehyde and subsequently dehydrated with acetone. After embedding in epoxy resin, ultrathin sections were placed on Formvar-coated grids and stained with 3% uranyl acetate. Sections were imaged using the FEI Technai Spirit transmission electron microscope.
Hemolysis Assay
All animal studies were approved by the Massachusetts Institute of Technology’s Committee on Animal Care and were completed in accordance with the National Institutes on Health Guide for the Care and Use of Laboratory Animals. 3–4 mL total blood was collected from two 8-week old female CD-1 mice (Charles River) via cardiac puncture using a 22G needle and 5 cm3 syringe containing an anticoagulant solution (1 cm3 50 mM EDTA). To prepare a red blood cell (RBC) suspension for the hemolysis assay, the collected blood was processed through a series of wash steps and then diluted. The blood was first centrifuged in a 15 mL Falcon tube at 1500 rpm for 5 min to pellet the RBCs. After centrifugation, lines were drawn on the tube to mark the level of the RBCs and the plasma. The plasma was aspirated and discarded in bleach. The RBCs were then washed 2 times by first gentle resuspension in a volume of 150 mM NaCl solution equal to that of the discarded plasma followed by centrifugation at 1500 rpm for 5 min and aspiration of the NaCl supernatant. The RBCs were then washed once with PBS pH 7.4 and then gently resuspended in PBS pH 7.4 to replace the plasma volume. Finally, the RBC suspension was diluted 1:50 in PBS pH 7.4. For the hemolysis assay, 20 μL 1% TritonX-100 (positive control for hemolysis), PBS (negative control for hemolysis), samples diluted in PBS (WD40 and free WLBU2 peptide at concentrations equivalent to 6.25, 12.5, 25, 50, 100, 200, and 400 μM peptide) were plated in triplicate in a conical-bottom 96-well plate. 180 μL of the diluted RBC suspension was then added to each well. After 1 h incubation at 37 °C, the plate was centrifuged at 1500 rpm for 5 min, and 100 μL supernatant from each well was transferred to a flat-bottom 96-well plate for absorbance measurements at 541 nm wavelength using a plate reader. Percent hemolysis was calculated using the following equation: % Hemolysis = (A541,Sample – A541,Buffer)/(A541,Triton – A541,Buffer) × 100.
MTS Assay for Mammalian Cell Cytotoxicity
10 000 HEK293T cells in 80 μL complete media (DMEM with 10% FBS and 1% penicillin/streptomycin) were plated per well in a 96-well plate. After 24 h incubation to allow cells to adhere, 20 μL 1% TritonX-100 (positive control for cell death), complete media (negative control for cell death), and samples diluted in complete media (WD40 and free WLBU2 peptide at concentrations equivalent to 3.125, 6.25, 12.5, 25, 50, 100, 200 μM peptide) were added to the wells in triplicate. Wells containing only complete media with no cells were included for background measurements. After 24 h incubation, 20 μL MTS reagent (Promega Aqueous One Proliferation Assay Kit) was added to each well. After 1 h incubation, absorbance at 490 nm wavelength was measured using a plate reader. Percent cell viability was calculated using the following equation: % Viability = (A490,Sample – A490,Background)/(A490,Media – A490,Background) × 100.
Checkerboard Assays to Assess Antimicrobial Activity of Potentiator-Antibiotic Pairs in P. aeruginosa (Strain PA14 and Clinical Isolates)
Checkerboard assays were completed in 96 well-plates with increasing potentiator concentration going up the plate (up to 20 μM by peptide concentration) and increasing drug concentration from the left to right side of the plate. Drug solutions were prepared in MHB with less than 1% DMSO and were plated in the right-most column of the plate for 1:1 serial dilution using a multichannel pipet (25 μL per well). Potentiator solutions were prepared by diluting a volume of the stock solution in MHB to 80 μM and lower concentrations were prepared by serial 1:1 dilution in Eppendorf tubes. Twenty-five μL of the potentiator solution was added per well. After plating drugs and potentiators, 50 μL of a bacterial suspension (1 × 106 cfu/mL) prepared fresh from a secondary culture was added to each well. Plates were incubated in a humidified chamber at 37 °C overnight for 16 h and absorbance at 600 nm was read the following morning using a plate reader. FIC values were calculated using the formula: FICA = MICA,combination/MICA,alone, where A is either the drug or potentiator. The FICI is the sum of FIC values for the drug and potentiator. Clinical isolates (BWH006, BWH012, BWH013, BWH021, and BWH027) were provided by the laboratory of Deborah Hung.
Analysis of Potentiation and Drug Physicochemical Properties
For each antibiotic in the drug panel, values for physicochemical properties were collected from various online databases and plotted against log2(MIC fold change) in GraphPad Prism 8.1.0 to determine the Pearson’s correlation coefficients (r) and p-values via linear regression analysis. ClogP values predicted by ChemAxon were collected from the DrugBank database. Globularity, PBF, and number of rotatable bonds was collected from Entryway, an online tool for predicting drug accumulation in Gram-negative bacteria based on the publication by Richter et al.23 (www.entry-way.org). Globularity values from Entryway are determined using an open-source method as opposed to using molecular operating environment (MOE) software. Entryway defines rotatable bonds as nonterminal single bonds between heavy atoms not in a ring and does not count N–C bonds in amides as rotatable. PBF is the average distance to the plane of best fit and is calculated by Entryway using a custom Python program. This custom Python program uses single value decomposition to determine the plane of best fit defined by a set of coordinate points for the heavy atoms in the molecule.
Statistical Analysis
For microdilution assays, single compounds are tested in triplicate for each concentration, and mode MIC values are reported. Checkerboard assays are more time-intensive and require substantial amounts of potentiator. Therefore, each potentiator-antibiotic pairing is tested with an n = 1 per experiment, and two independent experiments are completed to ensure reproducibility. Repeated measures ANOVA in GraphPad Prism 8.1.0 was used to calculate p-values for comparison of WD40 versus free peptide curves for hemolysis and cytotoxicity. Using the same software, LC50 was determined using nonlinear regression to fit a dose–response curve to data from the MTS assay. As described in the prior section, to determine the magnitude of association between log2(MIC fold change) and various physicochemical properties of antibiotics, Pearson’s correlation coefficient (r) and p-values were calculated using linear regression analysis in GraphPad Prism 8.1.0.
Acknowledgments
We thank H. Fleming (MIT) for critical editing of the manuscript. We thank the Koch Institute’s Robert A. Swanson (1969) Biotechnology Center for technical support, specifically the Biopolymers & Proteomics Core Facility and the Microscopy Core Facility. Special thanks to A. Austin, A. Leshinsky, H. Amoroso, and R. Cook for peptide synthesis, HPLC purification, and mass spectrometry services, and E. Vasile for assistance with super-resolution microscopy. We also thank N. Watson at the Whitehead Institute W. M. Keck Microscopy Facility for her TEM imaging services. This study was supported in part by a Broad next 10 gift from the Broad Institute and by grant R01 AI132413 from the National Institute of Allergy and Infectious Diseases. L.W.C. acknowledges support from the National Institutes of Health Pathway to Independence Award (K99 EB028311). S.N.B. is a Howard Hughes Institute Investigator.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.0c00805.
Candidate peptides for peptide-antibiotic conjugates (Table S1), MIC values for peptides alone or in a physical mixture with linezolid (Table S2), Test reaction conditions for amine-functionalization of 0.5-g dextran quantities (Table S3), Class, clinical breakpoints, and physicochemical properties of drugs in the tested antibiotic panel (Table S4), Synthetic scheme of azido-LZDvar (Figure S1), Quantification of conjugated WLBU2 peptides in potentiator candidates (Figure S2), FIC graphs for antibiotics in combination with WD40 in PA14 (Figure S3), FIC graphs for fusidic acid, clindamycin, and rifampin in combination with WD40 in clinical isolates (Figure S4) (PDF)
The authors declare the following competing financial interest(s): S.N.B., L.W.C., and E.J.K. are listed as inventors on patent applications related to the content of this work. S.N.B. holds equity in Glympse Bio and Impilo Therapeutics; is a director at Vertex; consults for Cristal, Maverick and Moderna; and receives sponsored research funding from Johnson and Johnson.
Supplementary Material
References
- Mulani M. S.; Kamble E. E.; Kumkar S. N.; Tawre M. S.; Pardesi K. R. (2019) Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 10, 1–24. 10.3389/fmicb.2019.00539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcour A. H. (2009) Outer Membrane Permeability and Antibiotic Resistance. Biochim. Biophys. Acta, Proteins Proteomics 1794, 808–816. 10.1016/j.bbapap.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis K. (2013) Platforms for Antibiotic Discovery. Nat. Rev. Drug Discovery 12, 371–387. 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]
- Vaara M. (1992) Covalent Structure of Two Novel Neutrophile Leucocyte-Derived Proteins of Porcine and Human Origin. Microbiol. Rev. 56, 535–547. [DOI] [PubMed] [Google Scholar]
- Wimley W. C. (2010) Describing the Mechanism of Antimicrobial Peptide Action with the Interfacial Activity Model. ACS Chem. Biol. 5, 905–917. 10.1021/cb1001558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. H.; Lu T. K. (2020) Development and Challenges of Antimicrobial Peptides for Therapeutic Applications. Antibiotics 9, 1–20. 10.3390/antibiotics9010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaara M.; Siikanen O.; Apajalahti J.; Fox J.; Frimodt-Møller N.; He H.; Poudyal A.; Li J.; Nation R. L.; Vaara T. (2010) A Novel Polymyxin Derivative That Lacks the Fatty Acid Tail and Carries Only Three Positive Charges Has Strong Synergism with Agents Excluded by the Intact Outer Membrane. Antimicrob. Agents Chemother. 54, 3341–3346. 10.1128/AAC.01439-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg P. B.; Lister T.; Walpole S.; Keutzer T.; Utley L.; Tomayko J.; Kopp E.; Farinola N.; Coleman S. (2019) Safety, Tolerability, Pharmacokinetics, and Drug Interaction Potential of SPR741, an Intravenous Potentiator, after Single and Multiple Ascending Doses and When Combined with β-Lactam Antibiotics in Healthy Subjects. Antimicrob. Agents Chemother. 63, 1–12. 10.1128/AAC.00892-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French S.; Farha M.; Ellis M. J.; Sameer Z.; Côté J. P.; Cotroneo N.; Lister T.; Rubio A.; Brown E. D. (2020) Potentiation of Antibiotics against Gram-Negative Bacteria by Polymyxin B Analogue SPR741 from Unique Perturbation of the Outer Membrane. ACS Infect. Dis. 6, 1405–1412. 10.1021/acsinfecdis.9b00159. [DOI] [PubMed] [Google Scholar]
- Liu S. P.; Zhou L.; Lakshminarayanan R.; Beuerman R. W. (2010) Multivalent Antimicrobial Peptides as Therapeutics: Design Principles and Structural Diversities. Int. J. Pept. Res. Ther. 16, 199–213. 10.1007/s10989-010-9230-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnusch C. J.; Branderhorst H.; Kruijff B.; De Liskamp R. M. J.; Breukink E.; Pieters R. J. (2007) Enhanced Membrane Pore Formation by Multimeric/Oligomeric Antimicrobial Peptides. Biochemistry 46, 13437–13442. 10.1021/bi7015553. [DOI] [PubMed] [Google Scholar]
- Erfani Y.; Rasti A.; Janani L. (2016) Prevalence of Gram-Negative Bacteria in Ventilator-Associated Pneumonia in Neonatal Intensive Care Units: A Systematic Review and Meta-Analysis Protocol. BMJ. Open 6, 1–5. 10.1136/bmjopen-2016-012298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder E. L.; Dowdy S. F. (2004) Cell Penetrating Peptides in Drug Delivery. Pharm. Res. 21, 389–392. 10.1023/B:PHAM.0000019289.61978.f5. [DOI] [PubMed] [Google Scholar]
- Livermore D. M. (2003) Linezolid in Vitro: Mechanism and Antibacterial Spectrum. J. Antimicrob. Chemother. 51, 9–16. 10.1093/jac/dkg249. [DOI] [PubMed] [Google Scholar]
- Schumacher A.; Trittler R.; Bohnert J. A.; Kuemmerer K.; Pages J.-M.; Kern W. V. (2007) Intracellular Accumulation of Linezolid in Escherichia Coli, Citrobacter Freundii and Enterobacter Aerogenes: Role of Enhanced Efflux Pump Activity and Inactivation. J. Antimicrob. Chemother. 59, 1261–1264. 10.1093/jac/dkl380. [DOI] [PubMed] [Google Scholar]
- Phetsang W.; Blaskovich M. A. T.; Butler M. S.; Huang J. X.; Zuegg J.; Mamidyala S. K.; Ramu S.; Kavanagh A. M.; Cooper M. A. (2014) Bioorganic & Medicinal Chemistry An Azido-Oxazolidinone Antibiotic for Live Bacterial Cell Imaging and Generation of Antibiotic Variants. Bioorg. Med. Chem. 22, 4490–4498. 10.1016/j.bmc.2014.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalska K.; Karpiuk I.; Król M.; Tyski S. (2013) Bioorganic & Medicinal Chemistry Recent Development of Potent Analogues of Oxazolidinone Antibacterial Agents. Bioorg. Med. Chem. 21, 577–591. 10.1016/j.bmc.2012.11.036. [DOI] [PubMed] [Google Scholar]
- Tencza S. B.; Douglass J. P.; Creighton D. J.; Montelaro R. C.; Mietzner T. A. (1997) Novel Antimicrobial Peptides Derived from Human Immunodeficiency Virus Type 1 and Other Lentivirus Transmembrane Proteins. Antimicrob. Agents Chemother. 41, 2394–2398. 10.1128/AAC.41.11.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deslouches B.; Phadke S. M.; Lazarevic V.; Cascio M.; Islam K.; Montelaro R. C.; Mietzner T. A. (2005) De Novo Generation of Cationic Antimicrobial Peptides: Influence of Length and Tryptophan Substitution on Antimicrobial Activity. Antimicrob. Agents Chemother. 49, 316–322. 10.1128/AAC.49.1.316-322.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A.; Dupuy F. G.; Arsov Z.; Elhady Y.; Moody D.; Ernst R. K.; Deslouches B.; Montelaro R. C.; Di Y. P.; Tristam-Nagle S. (2019) Elastic Behavior of Model Membranes with Antimicrobial Peptides Depends on Lipid Specificity and D-Enantiomers. Soft Matter 15, 1860–1868. 10.1039/C8SM02180E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner R. G.; Goeth H. (1984) Trimethoprim, Failure to Penetrate into Pseudomonas Aeruginosa Cells. FEMS Microbiol. Lett. 23, 201–204. 10.1111/j.1574-6968.1984.tb01063.x. [DOI] [Google Scholar]
- Lee M.-Y.; Park S.-C.; Jung M.; Shin M.-K.; Kang H.-L.; Baik S.-C.; Cheong G.-W.; Jang M.-K.; Lee W.-K. (2018) Cell-Selectivity of Tryptophan and Tyrosine in Amphiphilic a -Helical Antimicrobial Peptides against Drug-Resistant Bacteria. Biochem. Biophys. Res. Commun. 505, 478–484. 10.1016/j.bbrc.2018.09.095. [DOI] [PubMed] [Google Scholar]
- Richter M. F.; Drown B. S.; Riley A. P.; Garcia A.; Shirai T.; Svec R. L.; Hergenrother P. J. (2017) Predictive Compound Accumulation Rules Yield a Broad-Spectrum Antibiotic. Nature 545, 299–304. 10.1038/nature22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M. F.; Hergenrother P. J. (2019) The Challenge of Converting Gram-Positive-Only Compounds into Broad-Spectrum Antibiotics. Ann. N. Y. Acad. Sci. 1435, 18–38. 10.1111/nyas.13598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea R.; Moser H. E. (2008) Physicochemical Properties of Antibacterial Compounds: Implications for Drug Discovery. J. Med. Chem. 51, 2871–2878. 10.1021/jm700967e. [DOI] [PubMed] [Google Scholar]
- Lee H. J.; Bergen P. J.; Bulitta J. B.; Tsuji B.; Forrest A.; Nation R. L.; Li J. (2013) Synergistic Activity of Colistin and Rifampin Combination against Multidrug-Resistant Acinetobacter Baumannii in an In Vitro Pharmacokinetic/Pharmacodynamic Model. Antimicrob. Agents Chemother. 57, 3738–3745. 10.1128/AAC.00703-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jammal J.; Zaknoon F.; Kaneti G.; Goldberg K.; Mor A. (2015) Sensitization of Gram-Negative Bacteria to Rifampin and OAK Combinations. Sci. Rep. 5, 9216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurawski D. V.; Reinhart A. A.; Alamneh Y. A.; Pucci M. J.; Si Y.; Abu-Taleb R.; Shearer J. P.; Demons S. T.; Tyner S. D.; Lister T. (2017) SPR741, an Antibiotic Adjuvant, Potentiates the In Vitro and In Vivo Activity of Rifampin against Clinically Relevant Extensively Drug-Resistant Acinetobacter Baumannii. Antimicrob. Agents Chemother. 61, 1–6. 10.1128/AAC.01239-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine S. D.; Reid R.; Robinson L.; Ashley G. W.; Santi D. V. (2015) Long-Term Stabilization of Maleimide-Thiol Conjugates. Bioconjugate Chem. 26, 145–152. 10.1021/bc5005262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.