

Abstract
The poor metabolic stability of the HIV-1 CA inhibitor PF-74 is a major concern in its development towards clinical use. To improve on the metabolic stability, we employed a novel multistep computationally driven workflow, which facilitated the rapid design of improved PF-74 analogs in an efficient manner. Using this workflow, we designed three compounds that interact specifically with the CA interprotomer pocket, inhibit HIV-1 infection, and demonstrated enantiomeric preference. Moreover, using this workflow, we were able to increase the metabolic stability 204-fold in comparison to PF-74 in only three analog steps. These results demonstrate our ability to rapidly design CA compounds using a novel computational workflow that has improved metabolic stability over the parental compound. This workflow can be further applied to the redesign of PF-74 and other promising inhibitors with a stability shortfall.
Keywords: Bioisosteres, HIV-1 capsid, antiviral, surface plasmon resonance, computer-aided drug design, metabolic stability
Graphical Abstract

INTRODUCTION
With the emergence of combination antiretroviral therapy (cART), the mortality rate associated with human immunodeficiency virus type-1 (HIV-1) infection has decreased. However, due to the emergence of drug-resistant strains and toxicities associated with current therapies, the demand remains for new inhibitors of HIV-1 replication. The capsid (CA) protein a broadly conserved protein that is essential for HIV-1 replication, playing roles in both the early and late stages of the viral life cycle, and has emerged as a promising new therapeutic target.1–4 During the maturation process, the CA protomers rearrange to form a conical core surrounding the viral contents that is composed of approximately 250 CA hexamers and 12 CA pentamers.5
Within the structure of assembled CA multimers exists a conserved interprotomer pocket that has been shown to interact with host-cell proteins such as cleavage and polyadenylation specificity factor-6 (CPSF6) and nucleoporin 153 (NUP153).6–9 Disrupting these interactions via mutagenesis can interfere with HIV-1 replication, indicating that this interprotomer pocket is a prime target for the design of inhibitors. In agreement with this postulation, this interprotomer pocket has been identified as the mature binding site of the CA-targeted small molecule inhibitor PF-3450074 (PF-74).6, 7 However, despite its attractive properties and mechanism of action, we were the first group to demonstrate that PF-74 is extremely metabolically labile, which limits its clinical utility.10 Since this demonstration, a number of other groups have taken it upon themselves to improve this deficit in metabolic stability of PF-74, often employing large analoging strategies and testing selected compounds for increases in metabolic stability.11–18 Unfortunately, some of the most promising compounds derived from PF-74 have complicated chemical syntheses and would be difficult to scale up for clinical production.17, 18 As such, it would be advantageous if metabolic stability could be integrated into a design workflow for new synthetically tractable compounds, rather than a consequence of brute force analoging.
Therefore, in this brief article, we describe the use of a multistep computational workflow that can be used during the design of compounds to rapidly make analogs of a parental compound with improved metabolic stability. Using this design workflow, we created three enantioselective inhibitors (CX15, CX16, and CX17) that specifically bound to the CA hexamer and inhibited HIV-1 infection. Upon metabolic stability analysis, we demonstrated that the senantiomeric form of one of the compounds, in agreement with computational prediction, had a 204-fold increase in half-life (t1/2) in comparison to PF-74. This finding, along with another study employing this approach for metabolic stability improvement,19 now solidifies the general utility of including steps to address metabolic stability during the computational design process.
RESULTS AND DISCUSSION
Multistep computational design of CX15, CX16, and CX17.
Driven by the deficits in the current HIV-1 CA inhibitor class, our group has previously targeted the interprotomer pocket of the assembled hexameric HIV-1 CA to find novel inhibitors of HIV-1 replication. We have used three different approaches to do this – (1) high content pharmacophore-based screening,20 (2) chemistry-driven analoging around the PF-74 scaffold,13–16 and (3) computationally guided bioisosteric replacement.10 These efforts have provided compounds that target the hexameric configuration of the HIV-1 CA but lacked either the requisite potency and/or metabolic stability to be viable leads.
The incredible metabolic lability of PF-74 is a significant issue with this inhibitor.10, 12, 14 However, we have recently described a novel computational workflow that allowed us to identify a unique HIV-1 entry inhibitor scaffold that has improved metabolic stability.19 Therefore, we sought to see whether this workflow (Figure 1) could be applied to the rapid improvement of an HIV-1 CA inhibitor-based upon PF-74. Briefly, we first performed bioisosteric replacement experiments on the bioactive conformation of PF-74 (PDB ID 4XFZ) using Spark (Spark version 10.5.6, Cresset®, Litlington, Cambridgeshire, UK; http://www.cresset-group.com/spark/).21–23 The results of the bioisosteric replacement experiments were then imported into SeeSAR 10.0 (BioSolveIT Gmbh, Germany) and assessed for predicted affinity using the HYdrogen bond and DEhydration (HYDE) energy scoring function.24 Compounds with high predicted affinity were then imported into StarDrop 6.6 (Optibrium Ltd., UK) and assessed for absorption, distribution, metabolism, and excretion/pharmacokinetics (ADME/PK) properties, such as logS and logP, using the oral non-central nervous system (oral non-CNS) drug scoring profile.25 We also used StarDrop to assess for metabolic vulnerability to the major cytochrome P450 (CYP) enzymes using the P450 module (Optibrium Ltd., UK).26 This analysis predicted the major CYP isoform most likely to metabolize these compounds, and PF-74, was CYP isoform 3A4. One caveat with the metabolic liability prediction is that it is based upon the assumption that all compounds interact with the CYP isoform with the same affinity. Therefore, to further refine the metabolic prediction analysis, the compounds were then docked and assessed for their binding affinities to CYP isoform 3A4 (PDB ID 4D78).27 Finally, compounds with better predicted drug-like metrics and inferred improved metabolic stability were then redocked into the HIV-1 CA hexamer using AutoDock 4.0,28 as implemented in DockingServer (www.dockingserver.com)29 and were determined to have very similar binding poses as PF-74, indicating the robust choices of bioisosteric replacements (Figure 2 B and C). As a quality control, PF-74 was docked to the HIV-1 CA protein in the interprotomer pocket to ensure the derived prediction matched the crystal structure and to provide evidence that we could have confidence in our docking procedure (Figure 2C). From the final list of compounds that successfully passed through the above workflow, a compound in which a 1H-indazol-7-amine group replaced the methylindole headgroup of PF-74 (designated CX15), and a compound in which a 1H-indazol group replaced the benzene tail group of PF-74 (designated CX16) were chosen. Interestingly, a chimeric compound containing both of these replacements (designated CX17) was predicted to retain the potency of PF-74 but with much improved metabolic stability. All three compounds were chosen for synthesis as racemates before the assessment of enantioselectivity (Figure 2A). Figure 3 highlights the improvement of the drug-like properties of our compounds as compared to PF-74, as assessed in StarDrop (Optibrium Ltd., UK). Within this analysis, CX15 and CX16 showed a substantial improvement in the oral non-CNS scoring function as compared to PF-74, whereas CX17 scored similarly with the drug-like function but displayed increased predicted aqueous solubility (logS).
Figure 1. Computational workflow.

Workflow chart highlighting the steps used in the computational design process of compounds.
Figure 2. Docking of racemic CX15, CX16, and CX17.

(A) Compounds chosen for analysis. Regions that were modified on PF-74 during the bioisosteric replacement are highlighted in pink (CX15), yellow (CX16), and green (CX17). The chemical structures were drawn with ChemAxon software (Budapest, Hungary). (B) Crystal structure of HIV-1 CA with compounds, PF-74 in blue (PDB ID 4XFZ) and docked CX15 in pink, CX16 in yellow, CX17 in green in the interprotomer pocket (highlighted by black box) of a CA hexamer (for simplicity, only an extracted dimer is shown). (C) Magnification of each docked compound, CX15 in pink, CX16 in yellow, CX17 in green, and PF-74 in grey in the predicted binding site aligned with the PF-74-bound crystal structure (blue).
Figure 3. Computational analysis of drug-like properties of CX15, CX16, and CX17.

Oral Non-CNS scoring profile of CX15, CX16, CX17, and PF-74 calculated with StarDrop (Optibrium Ltd., UK).
Affinity and potency determination of racemic CX15, CX16, and CX17.
After synthesis and prior to antiviral testing, we sought to establish that the new compounds retained the target specificity of the parental PF-74 compound to the CA hexamer. We chose to demonstrate this via surface plasmon resonance (SPR). Figure 4 shows the representative sensorgrams for each compound interacting with the disulfide stabilized HIV-1NL4-3 capsid hexamer,20 and Table 1 shows the kinetic parameters obtained after analysis. SPR analysis showed that each of the three novel compounds interacts with the immobilized disulfide stabilized CA hexamer robustly and with a broadly similar kinetic signature as PF-74 (Figure 4). All four compounds have a very rapid on-rate, but with slightly different off-rates, with CX16 having the slowest off-rate (Table 1). These different off-rates leads to the different affinities observed for each compound (Table 1). Despite their different off-rates, all compounds had affinities in the nM range within 5-fold of PF-74, with CX16 showing a marginally improved KD over PF-74 (43.9nM and 95.4nM, respectively). The SPR analysis results confirm that the newly designed compounds maintain the target specificity of the parental compound.
Figure 4. Representative sensorgrams depicting the interaction of compounds with the CA hexamer.

SPR sensorgrams showing (A) PF-74 (5μM starting concentration with 1:4 serial dilutions), (B) CX15 (5μM starting concentration with 1:4 serial dilutions), (C) CX16 (5μM starting concentration with 1:4 serial dilutions), (D) CX17 (2μM starting concentration with 1:4 serial dilutions) interacting with immobilized disulfide-stabilized NL4-3 CA hexamer. Colored lines represent actual data collected from the dilution series, whereas black lines signify the fits to a 1:1 binding model. Interaction parameters derived from 5 sets of data are given in Table 1.
Table 1.
Kinetics and affinity of racemic compounds for disulfide stabilized HIVNL4-3 CA hexamer.
| Compound | ka (M−1s−1) | kd (s−1) | KD (nM) |
|---|---|---|---|
| PF-74 | 3.07 ± 0.103 × 105 | 2.93 ± 0.182 × 10−2 | 95.4 |
| CX15 | 5.62 ± 0.299 × 105 | 5.62 ± 0.229 × 10−2 | 189 |
| CX16 | 2.75 ± 0.100 × 105 | 1.21 ± 0.037 × 10−2 | 43.9 |
| CX17 | 2.20 ± 0.083 × 105 | 1.06 ± 0 × 10−1 | 494 |
KD values were derived using the average ka and kd values.
After confirmation that each of the new compounds retains their target specificity, we sought to determine their antiviral activities. Using a single-round infectious HIV-1 pseudoviral assay,20 the potencies of each compound for inhibition of the virus were tested in the early stages of the HIV-1 lifecycle. This analysis revealed that two of the compounds inhibited at concentrations in agreement with both their predicted and experimentally determined affinities. However, CX17 had an IC50 approximately 5-fold higher than its predicted and experimentally determined affinity, perhaps hinting towards an enantiomer bias within the racemic mixture (Table 2). Simultaneous analysis of the toxicity of each compound half-maximal cytotoxic concentration values (CC50) all higher that the parental PF-74 compound. Taken together, this demonstrates that our design strategy results in compounds that retain key properties of the parental compound, namely target specificity and antiviral action but reduced toxicity.
Table 2:
Potency and toxicity values of racemic PF-74, CX15, CX16, and CX17 against HIV-1 pseudoviruses.
| Compound | IC50 (nM) | CC50 (μM) | Therapeutic Index (CC50/IC50) |
|---|---|---|---|
| PF-74 | 55.5 ± 9.5 | 36.8 ± 16.4 | 664 |
| CX15 | 97.2 ± 12.2 | 83.5 ± 19.5 | 859 |
| CX16 | 38.3 ± 4.96 | 76.9 ± 9.5 | 2008 |
| CX17 | 2420.0 ± 305.0 | 415.0 ± 207.0 | 171 |
Binding site mapping.
As we have demonstrated antiviral activity and target specificity to the CA hexamer, we next wanted to verify that the new compounds shared the same interprotomer binding site as PF-74 and to validate our docking hypotheses. With no complex structure in hand at this time, binding site verification and validation of contributing residues crucial for binding provides essential information for further optimization for our HIV-1 CA inhibitor class. To achieve this, we chose to perform mutagenesis of residues within the predicted binding pocket, guided by our docking models. When analyzing the 2D ligand-protein interaction network (Figure S1) from our docking models, all compounds are predicted to make slightly different contacts. Therefore, we chose to prioritize the mutagenesis of residues that contribute the most to the overall predicted binding energies for each compound (Figure 5 and Table S1). Specifically, we chose residues N53, L56, N57, M66, L69, K70, and I73, as the side chains of these residues have the potential to form hydrogen bonds, cation-π, or hydrophobic interactions with our compounds (Figure S1). We chose to alter the selected residues to alanine as substitution with alanine eliminates side-chain interactions but with minimal effects upon main-chain conformation, hence preserving overall protein structure.
Figure 5. Percent maximal response of compounds PF-74, CX15, CX16, CX17 in the CA Hexamer interprotomer pockets of the wild-typed as compared to mutants.

Compounds were tested at a single concentration of 5μM to ensure saturation of each surface. Responses were normalized to percent theoretical maximal binding response to allow for cross-comparison. The red dashed line represents the average of the wild-type (wt) binding responses. Error bars represent the standard deviation of at least three replicates.
The recombinant HIV-1 CA proteins with introduced alanine mutations at positions 53, 56, 57, 66, 69, 70, and 73 were overproduced and purified as previously described.10, 20, 30, 31 However, the hexameric CA fractions were subsequently isolated using size-exclusion chromatography before SPR analysis.
If our docking models are correct, we expect that performing alanine mutagenesis would allow us to identify residues involved in the compound-protein interaction. As expected, we consistently saw a decrease in the percent binding upon mutation of our chosen residues to varying degrees. However, with mutants N57A, M66A, and K70A, we observed the most significant reduction in percent binding across all of the compounds as compared to the wild-type (wt) CA (Figure 5).
To understand the reasons for the pronounced effect of the N57A, M66A, and K70A CA mutants on the interactions with the compounds, we completed a computational assessment. The electrostatic complementarity (EC) of a ligand-receptor complex can provide important insights into the nature and strength of the complex.32 Since electrostatic interactions are key contributors to the free energy of biological binding processes (enthalpic component ΔG), we chose to look at the EC-map of each compound in the context of our models of the wt and mutant CA proteins. Analysis of the EC of these compounds within the binding sites from our docked models indicated that these mutants increased the electrostatic clash (or decreased the EC) for each of the compounds (Figure 6) as compared to the wt HIV-1 CA protein. This finding likely contributes to the reduction in binding observed with our compounds for these mutant CA proteins.
Figure 6: Electrostatic complementarity (EC) analysis of PF-74, CX15, CX16, and CX17 within wild-type (wt) and HIV-1 capsid (NL4-3) mutants.

The left panel shows the docking poses of CX15, CX16, CX17 (colors scheme as in figure 1), and the co-crystal structure of HIV-1 NL4-3 capsid bound to PF-74 (PDB ID 4XFZ). Green labeled residues are contributing hydrogen bonds (dashed green lines), and purple labeled residues contribute to cation-pi interactions to stabilize the complex (both if double-colored). The letter F next to the residue highlights that the adjacent HIV-1 capsid protomer contributes residues for complex stabilization. The letter O next to the residue highlights that a main chain carbonyl is involved in hydrogen bonding. The right panel shows the EC-surface map of each compound in the context of wt or indicated in silico generated and energy minimized mutant. Each compound is shown from the front view (left) and in a 180° view (right) for wt and mutant. Regions of the ligand surface with perfect electrostatic complementarity with the protein are colored green, while the areas with perfect electrostatic clash are colored red. Neutral EC areas are colored white.
As we have affinity, potency, and kinetic information for the interaction of these compounds with wt and mutant CA proteins, we sought to see any correlations we could observe between these parameters and EC. All compounds displayed similar on-rate kinetics. However, the affinity (KD) and the off-rates differed substantially between all four compounds (Table 1), with CX16 displaying the slowest and CX17 the fastest off-rate. In the context of the wt HIV-1 CA, we could observe a correlation trend between the EC-scores and the affinities as well as the off-rates, showing higher EC-scores for compounds with lower KD and slower off-rates and lower EC-scores for compounds with higher KD or faster off-rates (Figure 7A and B with CX16, PF-74, CX15, and CX17, in this order). Interestingly, this EC/KD and EC/off-rate correlation was disrupted by introducing the N57A and K70A mutations (data not shown), showing the importance of these residues in contributing electrostatic contacts for compound interaction. We have previously noted such correlations with our novel HIV-1 entry inhibitors.33 The correlations between the EC, affinity (KD) and off-rate of our compounds (including PF-74) to its target site can be utilized as powerful tools in the design and screening processes to help further our optimization efforts to improve upon the CX compounds.
Figure 7. Affinity and off-rates correlate with electrostatic complementarity.

(A) The dissociation constant (KD) of CX16, PF-74, CX15, and CX17 (in this order) correlates with the EC. (B) Off-rates of CX16, PF-74, CX15, and CX17 (in this order) correlates with the EC in the wt HIV-1 CA context. SCI: Normalized surface integral of the complementarity score, Pearson R: Pearson correlation coefficient, Spearman rho rank: Spearman rank correlation coefficient.
Conservation analysis of the CA interprotomer pocket.
Since we observed that we could introduce mutations within the interprotomer pocket that reduce the binding and presumably the potency of the compounds, we next sought to gauge the likelihood of these mutations arising as compound resistance changes. First, we performed a conservation analysis of 5,774 CA sequences from the Los Alamos Database (www.hiv.lanl.gov) to look at the probability of these residues to be naturally mutated. This analysis showed that the residues within the interprotomer pocket are highly conserved (Figure 8, mutated residues highlighted in blue). This extreme conservation indicates that these residues have critical structural and/or functional roles within the virus and that these contact points are not likely to mutate in isolation due to selective pressure during drug treatment. It is known that CA is a highly conserved and genetically fragile protein with about 70% of random point mutations throughout the protein causing replication-defective virus.3 Specifically, previous extensive mutagenesis analysis of residues N57, M66, L69, and I73 revealed that mutation of these residues produced non-viable viruses.3 Despite the genetic fragility of CA, a PF-74 resistant virus named 5Mut, which contains 5 mutations in the NTD (both in and outside of the PF-74 binding site), has been isolated. This mutant was obtained by serial passaging the virus in the presence of increasing concentrations of a closely related analog of PF-74.34 Notably, 5Mut contains a mutation at K70, which individually has been observed to decrease viral fitness; however, this fitness is restored upon the presence of the other compensatory mutations.35 This indicates that multiple mutations are required to both confer PF-74 resistance and maintain viral fitness; therefore, we did not incorporate our single mutations into the viral backbone for infectivity assays as the resultant virus would likely be attenuated. However, this combined information again highlights the attractive nature of this interprotomer pocket for therapeutic targeting.
Figure 8. Conservation of residues within the CA interprotomer binding pocket.

The probability of conservation of residues from 5,774 sequences within the Los Alamos Database is shown for the residues within the interprotomer pocket of (A) Protomer F and (B) Protomer G. Residue numbers correspond to their placement within Hxbc2. Blue letters indicate residues that were mutated during the binding site mapping. The size of the letter indicates the probability of conservation. Generated using the AnalyzeAlign tool on the Los Alamos Database (www.hiv.lanl.gov).
Synthesis of enantiomerically pure compounds.
Initially, each of these compounds were synthesized as racemic mixtures to ensure that the computational design workflow used led to the generation of active compounds that bound to the interprotomer pocket of the CA hexamer. When looking at the potencies derived for each compound, especially CX17, in comparison to their affinities, we noticed that the IC50 values seemed to be slightly higher than expected. This, of course, could be due to several reasons; however, in the case of compounds with chirality, such a discrepancy may be attributed to enantioselectivity.36 When a chiral compound is biologically active as a racemate, one enantiomer can be more active than the other, with the more active enantiomer being called the eutomer and the less active or inactive compound being called the distomer.36, 37 Enantioselectivity is an important but often underappreciated factor in the drug discovery process. Therefore, this led us to establish if our CX compounds (and PF-74) displayed enantioselectivity and if one enantiomeric form of our compounds was more active than the other. Therefore, we synthesized the enantiomerically pure forms of each compound and confirmed their structures by NMR and purity by LC-MS (Figure S2-S12).
Potency and affinity determination of enantiomerically pure compounds against HIV-1 pseudoviruses.
After synthesizing and isolating the pure enantiomers, we wanted to test their activities. We again began by testing their binding via SPR to the disulfide stabilized CA hexamer to determine the affinity and specificity of each compound. To our surprise, there were little to no binding interactions observed with each of the R-enantiomers (R-PF-74, R-CX15, R-CX16, and R-CX17), while each of the S-enantiomers (S-PF-74, S-CX15, S-CX16, and S-CX17) showed robust binding with similar binding profiles as the racemic compounds (Figure 9). Moreover, by isolating the S-enantiomers, we saw an improvement in the affinity for each compound in comparison to the racemic mixtures (Table 3). Again, showing that we can improve upon the affinity of the parental PF-74 as our highest affinity compound S-CX16 showed over a 2-fold improvement in affinity over S-PF-74.
Figure 9: Sensorgrams depicting the interaction of enantiomerically pure compounds with CA hexamers.
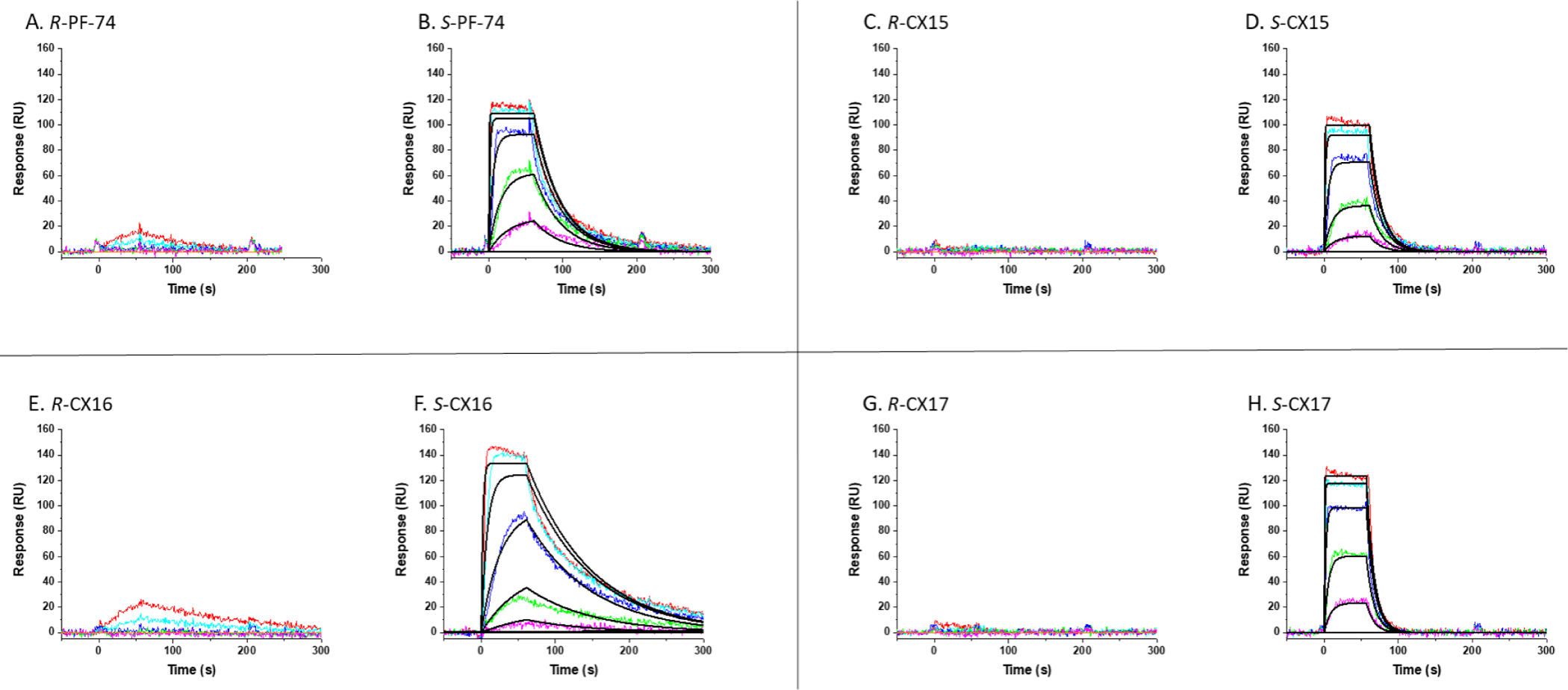
SPR sensorgrams showing (A) R-PF-74 (5μM starting concentration with 1:4 serial dilutions), (B) S-PF-74 (5μM starting concentration with 1:4 serial dilutions), (C) R-CX15 (5μM starting concentration with 1:4 serial dilutions), (D) S-CX15 (5μM starting concentration with 1:4 serial dilutions), (E) R-CX16 (5μM starting concentration with 1:4 serial dilutions), (F) S-CX16 (5μM starting concentration with 1:4 serial dilutions), (G) R-CX17 (2μM starting concentration with 1:4 serial dilutions), (H) S-CX17 (2μM starting concentration with 1:4 serial dilutions) interacting with immobilized disulfide-stabilized NL4-3 CA hexamer. Colored lines represent actual data collected from the dilution series, whereas black lines signify the fits to a 1:1 binding model. Interaction parameters derived from 5 sets of data are given in Table 3.
Table 3:
Kinetics and affinity of enantiomerically pure compounds for disulfide stabilized HIVNL4-3 CA hexamer.
| Compound | ka (M−1s−1) | kd (s−1) | KD (nM) |
|---|---|---|---|
| S-PF-74 | 4.75 ± 0.169 × 105 | 2.92 ± 0.111 × 10−2 | 61.5 |
| S-CX15 | 4.76 ± 0.216 × 105 | 6.60 ± 0.328 × 10−2 | 139 |
| S-CX16 | 4.37 ± 0.158 × 105 | 1.09 ± 0.043 × 10−2 | 25 |
| S-CX17 | 2.86 ± 0.041 × 105 | 9.66 ± 0.145 × 10−2 | 337 |
KD values were derived using the average ka and kd values.
Using the single-round infectious HIV-1 pseudotyped virus, we determined the potency for each of the enantiomerically pure compounds in the early stages of the HIV-1 lifecycle. It was clear that each compound had an active form and an inactive one since the R-enantiomers, which showed no binding to the CA hexamer via SPR, also had no activity in the early-stage antiviral assay (Table 4). To our delight, we also observed an improvement in the IC50 values obtained from the active enantiomers in comparison to the racemic compounds, with S-CX16 showing improved potency in relation to the active S-PF-74 (20.6nM vs. 80nM, respectively). This data shows that there is an enantiomeric preference for our compounds, with the S-enantiomers being the active eutomer and the R-enantiomers being the inactive distomer. By isolating the active eutomer of each compound, we were also able to improve upon the affinity and potency in relation to the starting racemic mixtures and in relation to the parental compound, PF-74. These values are also more in line with the predicted affinities from the DockingServer.28, 29 This resolves the inconsistencies observed between the IC50 values and affinities of the racemic compounds, indicates that the docked models utilized the active enantiomer, and confirms the enantioselective properties seen for these compounds. Moreover, although slightly diminished, the antiviral potency of S-CX17 was changed on only of the order of 3.5-fold, demonstrating that this computational workflow can be used to maintain the potency of the parental compound largely.
Table 4:
Potency and toxicity of enantiomerically pure compounds against HIV-1 pseudoviruses.
| Compound | IC50 (nM) | CC50 (μM) | Therapeutic Index (CC50/IC50) |
|---|---|---|---|
| R-PF-74 | NA | NA | NA |
| S-PF-74 | 80.0 ± 5.7 | 66.7 ± 6.8 | 834 |
| R-CX15 | NA | NA | NA |
| S-CX15 | 62.1 ± 8.2 | 101.0 ± 30.0 | 1626 |
| R-CX16 | NA | NA | NA |
| S-CX16 | 20.6 ± 1.8 | 46.9 ± 12.8 | 2281 |
| R-CX17 | NA | NA | NA |
| S-CX17 | 279.0 ± 52.6 | 102.0 ± 33.5 | 365 |
NA = not active.
Metabolic stability of enantiomerically pure CX compounds versus PF-74.
Since a large pitfall of PF-74 is its metabolic lability, one of our main goals while designing these compounds was to improve upon the metabolic half-life.6 After the characterization of the enantiomerically pure compounds, we sought to test their metabolic stability. Utilizing a human liver microsome assay, we determined the half-life of each compound.10, 19 As we had predicted from the computational workflow, each of our novel compounds had improved metabolic stability over the active S-PF-74, with S-CX17 showing the most dramatic improvement with a 204-fold increase in its half-life (Table 5 and Table S1). Overall this result shows that considering the metabolic vulnerability to the major CYP enzymes within the computational workflow can be a useful tool for multiple compound classes to consider the metabolic stability of newly designed compounds and improve upon this parameter rapidly in fewer analog steps.
Table 5:
Metabolic stability of active enantiomerically pure compounds derived from a human liver microsome assay.
| Compound | T1/2 (min) | Fold Increase (over S-PF-74) |
|---|---|---|
| S-PF-74 | 0.5 | 1.0 |
| S-CX15 | 15.4 | 30.8 |
| S-CX16 | 1.0 | 2.0 |
| S-CX17 | 102.0 | 204 |
CONCLUSIONS
PF-74 is a promising small molecule for biochemical studies that inhibits the proper function of the CA protein; however, PF-74 has limitations, including its poor metabolic stability. Using our novel computational workflow, we were able to optimize this class of CA-targeting compounds and improve the potency 4-fold and the metabolic stability 204-fold in few analog steps and simple substitutions. Furthermore, we found new correlations between the EC, off-rate, and potency of these compounds. Implementing these correlations into our computational workflow will allow for further optimization of this class of compounds to bring them towards the realm of clinical utility. Moreover, implementing this workflow will also enable us to improve upon compounds from other inhibitor classes and compounds for different targets. In summary, this study shows that we can rapidly improve upon the affinity, potency, and metabolic stability of PF-74 by designing synthetically tractable compounds without performing extensive analoging.
EXPERIMENTAL
Synthesis of Compounds
The purity of all compounds is ≥95% as determined by LC-MS (Figure S2-S12).
1. Synthesis of CX15
1.1. Preparation of tert-butyl(1-(methyl(phenyl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate (intermediate 1)
At 0 °C, to a solution of (tert-butoxycarbonyl)phenylalanine (1.0 g, 3.8 mmol) in DMF(10 mL) was added N-methylaniline (444 mg, 4.2 mmol), HATU (1.7 g, 4.5 mmol) and DIPEA (974.0 mg, 7.5 mmol). After the reaction was stirred at RT for 1 h, the mixture was diluted with ethyl acetate (30 mL), and then washed with brine (10 mL × 3). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (PE/EA = 3:1, v/v) to give tert-butyl (1-(methyl(phenyl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate as yellow oil (1.3 g, Y = 97.0%). LC-MS (ESI): m/z (M+H)+ 355.19.
1.2. Preparation of 2-amino-N-methyl-N,3-diphenylpropanamide hydrochloride (intermediate 2)
A mixture of tert-butyl (1-(methyl(phenyl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate (1.3 g, 3.7 mmol) in HCl/dioxane (4M, 15 mL) was stirred at RT for 1 h. Then the reaction solvents were removed under vacuum to give crude 2-amino-N-methyl-N,3-diphenylpropan-amide hydrochloride as yellow oil (1.1 g), which was used in the next step without purification. LC-MS (ESI): m/z (M+1)+ = 255.08.
1.3. Preparation of N,N-dimethyl-1-(7-nitro-1H-indol-3-yl)methanamine (intermediate 3)
A mixture of N,N,N’,N’-Tetramethyldiaminomethane (1.6 g, 15.6 mol) dissolved in acetic acid (30 mL) was added dropwise to a solution of 7-nitro-1H-indole (2.3 g, 14.2 mol) in acetic acid (30 mL) over 60 min. After the reaction mixture was stirred at RT for 3.5 h, the reaction mixture was cooled to 0 °C, and adjusted to pH = 11 with 20 % aqueous sodium hydroxide. Then the reaction mixture was extracted with CHCl3 (3 × 300 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure to give crude N,N-dimethyl-1-(7-nitro-1H-indol-3-yl)methanamine as yellow solid (3.1 g, Y = 99.7%), which was used in the next step without purification. LC-MS (ESI): m/z (M+1)+ = 220.08; 1H NMR (400 MHz, CDCl3) δ 9.83 (br. s,1H), 8.16 (d, J = 8.0 Hz, 1H), 8.10 (d, J = 8.0 Hz, 1H), 7.30 (d, J = 2.0 Hz, 1H), 7.21 (t, J = 8.0 Hz, 1H), 3.64 (s, 2H), 2.28 (s, 6H).
1.4. Preparation of 2-(7-nitro-1H-indol-3-yl)acetonitrile (intermediate 4)
A flask was charged with N,N-dimethyl-1-(7-nitro-1H-indol-3-yl)methanamine (100.0 mg, 0.46 mmol) and iodomethane (162.0 mg, 1.14 mmol) in DMF/H2O/THF (1: 1: 50, V/V; 5 mL). The reaction mixture was refluxed for 15 min until a white precipitate was formed and then potassium cyanide (149.0 mg, 2.28 mmol) was added to the reaction mixture. The resulted mixture was refluxed for another 2 h. After cooled to RT, the resulted reaction mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was triturated with MeOH to give 2-(7-nitro-1H-indol-3-yl)acetonitrile as yellow solid (50.0 mg, Y = 54.5%). LC-MS (ESI): m/z (M-1)− = 199.93; 1H NMR (400 MHz, DMSO-d6) δ 11.93 (br. s,1H), 8.13–8.18 (m, 2H), 7.55 (d, J = 2.0 Hz, 1H), 7.32 (t, J = 8.0 Hz, 1H), 4.17 (s, 2H).
1.5. Preparation of 2-(7-nitro-1H-indol-3-yl)acetic acid (intermediate 5)
A mixture of 2-(7-nitro-1H-indol-3-yl)acetonitrile (50.0 mg, 0.25 mmol) in 3 N aqueous HCI (5 mL) was stirred at 95°C for 72 h. After cooled to RT, the resulted mixture was adjusted to pH = 11 with 10 N aqueous NaOH solution and washed with EtOAc (10 mL). The aqueous layer was adjusted to pH = 2 with 1 N aqueous HCI solution and extracted with EtOAc (10 mL × 3). The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo to give crude 2-(7-nitro-1H-indol-3-yl)acetic acid as yellow solid (55.0 mg), which was used in the next step without purification. LC-MS (ESI): m/z (M-1)− =218.89; 1H NMR (400 MHz, DMSO-d6) δ 12.27 (br. s,1H), 11.81 (s, 1H), 8.17 (d, J = 8.0 Hz, 1H), 8.10 (d, J = 8.0 Hz, 1H), 7.50 (d, J = 2.0 Hz, 1H), 7.30 (t, J = 8.0 Hz, 1H), 3.82 (s, 2H).
1.6. Preparation of N-methyl-2-(2-(7-nitro-1H-indol-3-yl)acetamido)-N,3-diphenylpropanamide (intermediate 6)
At 0 °C, to a mixture of 2-(7-nitro-1H-indol-3-yl)acetic acid (55.0 mg, 0.25 mmol) in DMF(5 mL) was added 2-amino-N-methyl-N,3-diphenylpropanamide (70.0 mg, 0.28 mmol), HATU (114.0 mg, 0.3 mmol) and DIPEA (65.0 mg, 0.5 mmol), then the reaction mixture was stirred at RT for 1 h. Then reaction mixture was diluted with ethyl acetate (10 mL), and washed with brine (5 mL × 3). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (PE/EA = 3:1, v/v) to give N-methyl-2-(2-(7-nitro-1H-indol-3-yl)-acetamido)-N,3-diphenylpropanamide as yellow solid (45.0 mg, Y = 39.5%). LC-MS (ESI): m/z (M+1)+ 457.15; 1H NMR (400 MHz, DMSO-d6) δ 11.67 (s, 1H), 8.51 (d, J = 8.0 Hz, 1H), 8.06 (d, J = 8.0 Hz, 1H), 7.82 (d, J = 8.0 Hz, 1H), 7.43–7.37 (m, 3H), 7.31 (d, J = 2.0 Hz, 1H), 7.24–7.22 (m, 2H), 7.15–7.11 (m, 4H), 6.78–6.77 (m, 2H), 4.47–4.40 (m, 1H), 3.59–3.51 (m, 2H), 3.15 (s, 3H), 2.87–2.83 (m, 1H), 2.69–2.63 (m, 1H).
1.7. Preparation of 2-(2-(7-amino-1H-indol-3-yl)acetamido)-N-methyl-N,3-diphenylpropanamide (CX15)
To a mixture of N-methyl-2-(2-(7-nitro-1H-indol-3-yl)acetamido)-N,3-diphenylpropan-amide (45.0 mg, 0.1 mmol) in EtOH / H2O (5 mL, 4:1, V/V) was added Fe powder (28.0 mg, 0.5 mmol), NH4CI (27.0 mg, 0.5 mmol), then the reaction mixture was stirred at 80 °C for 1 h. After cooled down to RT, the reaction mixture was filtered by Celite. The filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (C18, 40–100% MeCN in H2O with 0.1% formic acid) to give 2-(2-(7-amino-1H-indol-3-yl)acetamido)-N-methyl-N,3-diphenylpropanamide as white solid (11.0 mg, Y = 26.2%). 1H NMR (400 MHz, CD3OD) δ 7.52 (d, J = 8.0 Hz, 1H), 7.25–7.26 (m, 3H), 6.97–7.08 (m, 3H), 6.93–6.88 (m, 2H), 6.79–6.71 (m, 2H), 6.65–6.63 (m, 2H) 6.45–6.43 (m, 1H), 4.52–4.57 (m, 1H), 3.47 (d, J = 5.2 Hz, 2H), 3.06 (s, 3H), 2.72–2.79 (m, 1H), 2.50–2.56 (m, 1H); LC-MS (ESI): m/z (M+1)+ 427.18 (Figure S2).
2. Synthesis of CX16
2.1. Preparation of tert-butyl (1-((1H-indazol-5-yl)amino)- 1-oxo-3-phenylpropan -2-yl) carbamate
At room temperature, to a mixture of 1H-indazol-5-amine (1.5 g, 11.26 mmol) in DMF(20 mL) was added (tert-butoxycarbonyl)phenylalanine (3.29 g, 12.4 mmol), HATU (5.14 g, 13.5 mmol) and DIPEA (4.36 g, 33.75 mmol), then the reaction mixture was stirred at RT for 3 h. Then the reaction mixture was diluted with ethyl acetate and washed with brine. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (PE/EA = 3:1, v/v) to give tert-butyl (1-((1H-indazol-5-yl)amino)-1-oxo-3- phenylpropan-2-yl)carbamate as yellow solid (3.2 g, 75 % yield). LC-MS (ESI): m/z (M+1)+ = 381.25.
2.2. Preparation of tert-butyl 5-(2-((tert-butoxycarbonyl)amino) -3-phenylpropanamido)-1H-indazole-1-carboxylate
At room temperature, to a mixture of tert-butyl (1-((1H-indazol-5-yl)amino)-1-oxo- 3-phenylpropan-2-yl)carbamate (1.7 g, 4.47 mmol) in THF(20 mL) was added Boc2O (2 g, 8.94 mmol) and DMAP (20 mg), then the reaction mixture was stirred at RT for 2 h. Then the reaction mixture was diluted with ethyl acetate and washed with brine. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (PE/EA = 3:1, v/v) to give tert-butyl 5-(2-((tert-butoxycarbonyl)amino) -3-phenylpropanamido)-1H-indazole-1-carboxylate as yellow solid (1.8 g, 84 % yield). LC-MS (ESI): m/z (M+1)+ = 479.38.
2.3. Preparation tert-butyl 5-(2-((tert-butoxycarbonyl)amino) -N-methyl- 3-phenylpropanamido)-1H-indazole-1-carboxylate
At room temperature, to a mixture of tert-butyl 5-(2-((tert-butoxycarbonyl)amino)- 3-phenylpropanamido)-1H-indazole-1-carboxylate (1.4 g, 2.92 mmol) in MeCN (20 mL) was added Cs2CO3 (2.84 g, 8.76 mmol) and MeI (0.62 g, 5.84 mmol), then the reaction mixture was stirred at RT for 4 h. Then the reaction mixture was diluted with ethyl acetate and washed with brine. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (PE/EA = 3:1, v/v) to give tert-butyl 5-(2-((tert-butoxycarbonyl)amino)-N-methyl-3-phenylpropanamido)-1H-indazole-1-carboxylate as yellow solid (1.15 g, 80 % yield). LC-MS (ESI): m/z (M+1)+= 495.28.
2.4. Preparation of 2-amino-N-(1H-indazol-5-yl)-N-methyl-3 -phenylpropanamide
To a mixture of tert-butyl 5-(2-((tert-butoxycarbonyl)amino)-N-methyl- 3-phenylpropanamido)-1H-indazole-1-carboxylate (300 mg, 0.61 mmol) in DCM was added TFA then the reaction mixture was stirred at rt for 1 h. The reaction mixture was concentrated to be used in next step without purification. LC-MS (ESI): m/z (M+1)+ = 295.19.
2.5. Preparation of N-(1H-indazol-5-yl)-N-methyl-2-(2-(2-methyl-1H-indol-3-yl) acetamido)-3-phenylpropanamide (CX16)
At room temperature, to a mixture of 2-amino-N-(1H-indazol-5-yl)-N-methyl-3-phenylpropanamide (83 mg, 0.28 mmol) in DMF(5 mL) was added 2-(2-methyl-1H-indol-3-yl)acetic acid (59 mg, 0.31 mmol), HATU (129.0 mg, 0.34 mmol) and DIPEA (110.0 mg, 0.85 mmol), then the reaction mixture was stirred at RT for 2 h. Then reaction mixture was filtered by Celite. The filtrate was concentrated under reduced pressure. The residue was purified by prep-HPLC (C18, 40–100% MeCN in H2O with 0.1% formic acid) to give N-(1H-indazol-5-yl)-N-methyl-2-(2-(2-methyl-1H-indol-3-yl)acetamido)-3-phenylpropanamide as white solid (20 mg, 15 % yield). 1H NMR (400 MHz, DMSO) δ 13.21 (s, 1H), 10.69 (s, 1H), 8.23 (d, J = 7.9 Hz, 1H), 8.01 (s, 1H), 7.55 – 7.39 (m, 2H), 7.28 (d, J = 7.8 Hz, 1H), 7.20 – 7.03 (m, 5H), 6.96 – 6.90 (m, 1H), 6.86 – 6.80 (m, 1H), 6.76 (d, J = 7.0 Hz, 2H), 4.40 – 4.30 (m, 1H), 3.45 – 3.38 (m, 2H), 3.16 (s, 3H), 2.88 (dd, J = 13.3, 4.7 Hz, 1H), 2.66 (dd, J = 13.3, 9.2 Hz, 1H), 2.22 (s, 3H); LC-MS (ESI): m/z (M+1)+ 466.29 (Figure S3).
3. Synthesis of CX17
3.1. Preparation of tert-butyl (1-((1H-indazol-5-yl)amino)- 1-oxo-3-phenylpropan -2-yl) carbamate
At room temperature, a solution of 7-nitro-1H-indazole (3.0 g, 18.4 mmol), iodine (7 g, 27.6 mmol) and potassium hydroxide (2.0 g, 36.8 mmol) in DMF (30 mL) was stirred overnight. After quenched with 6 mol/L hydrochloric acid (7 mL), the mixture was diluted with ethyl acetate (30 mL) and then washed with brine (90 mL × 3). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to give 3-iodo-7-nitro-1H-indazole as yellow solid (5 g, yield: 94%) which was used in next step without purification. LC-MS (ESI): m/z (M+H)+ 289.86.
3.2. Preparation of 3-iodo-1-(4-methoxybenzyl)-7-nitro-1H-indazole
At room temperature, a solution of 3-iodo-7-nitro-1H-indazole (1.77 g, 6.12 mmol), potassium hydroxide (687 mg, 12.24 mmol) and PMBCl (1.44 g, 9.18 mmol) in acetone (25 mL) and water (5 mL) was stirred overnight under N2. After quenched with 6 mol/L hydrochloric acid to pH=6~7, the mixture was diluted with ethyl acetate (30 mL), and then washed with brine (30 mL × 3). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (0~20% ethyl acetate in petroleum ether) to give 3-iodo-1-(4-methoxybenzyl)-7-nitro-1H-indazole as yellow solid (980 mg, yield: 39%). LC-MS (ESI): m/z (M+1)+410.23.
3.3. Preparation of dimethyl 2-(1-(4-methoxybenzyl)-7-nitro-1H-indazol-3-yl)malonate
At room temperature, a suspension of 3-iodo-1-(4-methoxybenzyl)-7-nitro-1H-indazole (930 mg, 2.27 mmol), dimethyl malonate (450 mg, 3.41mmol), 2-picolinic acid (56 mg, 0.45 mmol), copper(I) iodide (86 mg, 0.45 mmol) and cesium carbonate (1.48 g, 4.5 mmol) in dioxane (10 mL) was heated up to 100°C and stirred for 5 hours under N2. The suspension was concentrated under reduced pressure to give the crude product which was purified by silica gel column chromatography (0~30% ethyl acetate in petroleum ether) to give 2-(1-(4-methoxybenzyl)-7-nitro-1H-indazol-3-yl)malonate as yellow solid (688 mg, yield: 73%). LC-MS (ESI): m/z (M+1)+414.21.
3.4. Preparation of 2-(1-(4-methoxybenzyl)-7-nitro-1H-indazol-3-yl)acetic acid
At room temperature, a suspension of 2-(1-(4-methoxybenzyl) -7-nitro-1H-indazol-3-yl)malonate (688 mg, 1.66 mmol) and potassium hydroxide (140 mg, 2.49 mmol) in ethanol (10 mL) was heated up to 80°C and stirred overnight under N2. After cooling down to room temperature and acidified by 1 mol/L hydrochloric acid to pH=3~4, the solution was diluted with ethyl acetate (30 mL), and then washed with brine (30 mL × 3). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to give the product 2-(1-(4-methoxybenzyl)-7-nitro-1H-indazol-3-yl)acetic acid as brown solid (530 mg, yield:93%) which was used in next step without purification. LC-MS (ESI): m/z (M+1)+ = 342.15.
3.5. Preparation of 2-(7-nitro-1H-indazol-3-yl)acetic acid
At room temperature, a suspension of 2-(1-(4-methoxybenzyl)-7-nitro -1H-indazol-3-yl) acetic acid (500 mg, 1.46 mmol), trifluoroacetic acid (0.5 mL) and Trifluoromethanesulfonic acid (0.5 mL) in 1,2-dichloroethane (5 mL) was heated up to reflux and stirred for 1 hour under N2. After cooling down to room temperature, the solution was concentrated under reduced pressure to give the product 2-(7-nitro-1H-indazol-3-yl)acetic acid as black solid (400 mg) which was used in next step without purification. LC-MS (ESI): m/z (M+1)+ = 222.09.
3.6. Preparation of 2-(7-amino-1H-indazol-3-yl)acetic acid
At room temperature, a suspension of 2-(7-nitro-1H-indazol-3-yl)acetic acid (400 mg, 2.27 mmol) and Pd(OH)2/C (100 mg) in methanol (5 mL) was stirred for 2 hours under H2.After the reaction mixture filtered by Celite, the filtrate was concentrated under reduced pressure to give the crude product which was purified by prep-HPLC (C18, 40–100% acetonitrile in water with 0.1% formic acid) to afford 2-(7-amino-1H-indazol-3-yl)acetic acid as white solid (97.0 mg, two steps yield: 35%). LC-MS (ESI): m/z (M+1)+ = 192.13. 1H NMR (400 MHz, DMSO) δ 12.36 (s, 1H), 6.87 (d, J = 7.6 Hz, 1H), 6.80 (t, J = 7.6 Hz, 1H), 6.45 (d, J = 6.8 Hz, 1H), 5.32 (s, 2H), 3.80 (s, 2H).
3.7. Preparation of 2-(2-(7-amino-1H-indazol-3-yl)acetamido)-N-(1H-indazol-5-yl)-N-methyl-3-phenylpropanamide (CX17)
At room temperature, a suspension of 2-(7-amino-1H-indazol-3-yl)acetic acid (50 mg, 0.26 mmol), 2-amino-N-(1H-indazol-5-yl)-N-methyl-3-phenylpropanamide, HATU, (198.8 mg, 0.52 mmol) and DIPEA (101 mg, 0.78 mmol) in DMF (5 mL) was stirred for 2 hours. Then the reaction mixture was diluted with ethyl acetate(20 mL) and washed with brine (10 ml × 3). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (0~30% ethyl acetate in petroleum ether) to give the crude product which was purified by prep-HPLC (C18, 10~100% acetonitrile in water with 0.1% formic acid) to afford After the reaction mixture filtered by Celite, the filtrate was concentrated under reduced pressure to give the crude product which was purified by prep-HPLC (C18, 40–100% acetonitrile in water with 0.1% formic acid) to afford 2-(2-(7-amino-1H-indazol-3-yl)acetamido)-N-(1H-indazol-5-yl)-N-methyl-3-phenylpropanamide as a white solid (18 mg, yield: 15%). 1H NMR (400 MHz, DMSO) δ 13.19 (s, 1H), 12.27 (s, 1H), 8.49 (d, J = 7.9 Hz, 1H), 8.00 (s, 1H), 7.50 (d, J = 8.7 Hz, 1H), 7.41 – 7.27 (m, 1H), 7.19 – 7.03 (m, 4H), 6.80 (d, J = 6.8 Hz, 2H), 6.72 – 6.64 (m, 2H), 6.40 (dd, J = 5.9, 1.9 Hz, 1H), 5.21 (s, 2H), 4.44 – 4.36 (m, 1H), 3.66 (s, 2H), 3.16 (s, 3H), 2.92 (dd, 1H), 2.68 (dd, J = 13.3, 8.7 Hz, 1H); LC-MS (ESI): m/z (M+1)+ = 468.66 (Figure S4).
4. Synthesis of S-PF-74
4.1. Preparation of (S)-Tert-butyl 1-(methyl(phenyl)amino)-1-oxo-3-phenylpropan-2-ylcarbamate (AP-5-5)
To a stirred solution of (S)-2-(tert-butoxycarbonylamino)-3-phenylpropanoic acid (1.0 g; 3.77 mmol) and N-methylaniline (0.40 g; 3.77 mmol) in 20 mL of dry CH2Cl2 at 0 °C were added diisopropyl ethyl amine (1.21 g; 9.42 mmol) and T3P 50% solution in CH2Cl2 by weight (3.12 g; 9.43 mmol) dropwise simultaneously. The reaction mixture was allowed to stir for 30 min at 0 °C. Completion of the reaction was confirmed by LC–MS. The reaction mixture was quenched with cold water and the product was extracted with CH2Cl2 and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the crude product was purified by flash column chromatography to afford title compound (1.13 g; 3.12 mmol, 85%). The product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, CDCl3) δ 7.44–7.28 (m, 3H), 7.24–7.15 (m, 3H), 6.98–6.78 (m, 4H), 5.18 (d, J = 7.6 Hz, 1H), 4.53 (d, J = 7.1 Hz, 1H), 3.21 (s, 3H), 2.87 (dd, J = 13.1, 7.4 Hz, 1H), 2.68 (dd, J = 12.6, 6.7 Hz, 1H), 1.38 (s, 9H). Mass m/z: calcd for C21H26N2NaO3+ [M + Na]+, 377.2; found, 377.2.
4.2. Preparation of (S)-2-Amino-N-methyl-N,3-diphenylpropanamide (AP-5-9)
To a stirred solution of (S)-tert-butyl 1-(methyl(phenyl)amino)-1-oxo-3-phenylpropan-2-ylcarbamate (1.0 g; 2.82 mmol) in 15 mL CH2Cl2 at 0 °C was added 5 mL of TFA. The reaction mixture was then warmed to room temperature and stirred for 2 h. Completion of the reaction was confirmed by LC– MS. Volatiles were evaporated under reduced pressure to yield the crude product which was co-distilled with 20 mL of toluene to afford title compound as trifluoracetic acid salt (1.02 g; 2.77 mmol, 98%) as a viscous liquid. The product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, CDCl3) δ 7.52 – 7.35 (m, 3H), 7.32 – 7.21 (m, 2H), 7.21 – 7.11 (m, 1H), 6.99 (dd, J = 46.4, 19.9 Hz, 2H), 6.86 (dd, J = 7.5, 1.7 Hz, 2H), 4.24 (t, J = 7.2 Hz, 1H), 3.24 (s, 2H), 3.04 (dd, J = 13.9, 6.3 Hz, 1H), 2.95 – 2.77 (m, 1H).
1H NMR (400 MHz, CDCl3) δ 7.45 – 7.36 (m, 3H), 7.31 – 7.21 (m, 3H), 7.20 – 7.13 (m, 1H), 6.97 (s, 2H), 6.86 (dd, J = 7.5, 1.7 Hz, 2), 4.24 (t, J = 7.2 Hz, 3H), 3.24 (s, 8H), 3.04 (dd, J = 13.9, 6.3 Hz, 3H), 2.97 – 2.82 (m, 3H). Mass m/z: calcd for C16H18N2NaO+ [M + Na]+, 277.1; found, 277.1.
4.3. Preparation of (S)-N-methyl-2-(2-(2-methyl-1H-indol-3-yl)acetamido)-N,3-diphenylpropanamide (S-PF-74)
To a stirred solution of 2-(2-methyl-1H-indol-3-yl)acetic acid (63 mg; 0.33 mmol) and (S)-2-amino-N-methyl-N,3-diphenylpropanamide 2,2,2-trifluoroacetate (136 mg; 0.37 mmol) in 6 mL of dry CH2Cl2 at 0 °C were added diisopropyl ethyl amine (108 mg; 0.83 mmol) and T3P 50% solution in CH2Cl2 by weight (275 mg; 0.43 mmol) dropwise simultaneously. The reaction mixture was allowed to stir for 1 h at 0 °C. Completion of the reaction was confirmed by LC–MS. The reaction mixture was quenched with cold water and the product was extracted with CH2Cl2 and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the crude product was purified by flash column chromatography to afford title compound (113 mg; 0.26 mmol, 80%). The product was confirmed by 1H NMR and MS (Figure S5).
1H NMR (400 MHz, CDCl3) δ 8.00 (s, 1H), 7.41 – 7.31 (m, 4H), 7.29 (d, J = 8.0 Hz, 1H), 7.18 – 7.11 (m, 2H), 7.11 – 7.03 (m, 3H), 6.93 (d, J = 5.5 Hz, 2H), 6.66 (d, J = 7.1 Hz, 2H), 6.18 (d, J = 8.1 Hz, 1H), 4.78 (dd, J = 15.2, 7.1 Hz, 1H), 3.65 – 3.51 (m, 2H), 3.16 (s, 3H), 2.72 (dd, J = 13.3, 6.9 Hz, 1H), 2.52 (dd, J = 13.3, 7.0 Hz, 1H), 2.31 (s, 3H). Mass m/z: calcd for C27H27N3NaO2+ [M + Na]+, 448.2; found, 448.2.
5. Synthesis of R-PF-74
5.1. Preparation of (R)-Tert-butyl 1-(methyl(phenyl)amino)-1-oxo-3-phenylpropan-2-ylcarbamate (AP-5-4)
To a stirred solution of (R)-2-(tert-butoxycarbonylamino)-3-phenylpropanoic acid (1.0 g; 3.77 mmol) and N-methylaniline (0.4 g; 3.77 mmol) in 20 mL of dry CH2Cl2 at 0 °C were added diisopropyl ethyl amine (1.21 g; 9.42 mmol) and T3P 50% solution in CH2Cl2 by weight (3.1 g; 4.9 mmol) dropwise simultaneously. The reaction mixture was allowed to stir for 30 min at 0 °C. Completion of the reaction was confirmed by LC–MS. The reaction mixture was quenched with cold water and the product was extracted with CH2Cl2 and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the crude product was purified by flash column chromatography to afford title compound (1.20 g; 3.39 mmol, 90%). The product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, CDCl3) δ 7.44–7.27 (m, 3H), 7.24–7.15 (m, 3H), 6.97–6.76 (m, 4H), 5.18 (d, J = 7.4 Hz, 1H), 4.53 (d, J = 6.9 Hz, 1H), 3.21 (s, 3H), 2.87 (dd, J = 13.1, 7.3 Hz, 1H), 2.68 (dd, J = 12.5, 6.6 Hz, 1H), 1.37 (s, 9H). Mass m/z: calcd for C21H26N2NaO3+ [M + Na]+, 377.2; found, 377.2.
5.2. Preparation of (R)-2-amino-N-methyl-N,3-diphenylpropanamide (AP-5-8)
To a stirred solution of (R)-tert-butyl 1-(methyl(phenyl)amino)-1-oxo-3-phenylpropan-2-ylcarbamate (1.1 g; 2.82 mmol) in 15 mL CH2Cl2 at 0 °C was added 5 mL of TFA. The reaction mixture was then warmed to room temperature and stirred for 2 h. Completion of the reaction was confirmed by LC–MS. Volatiles were evaporated under reduced pressure to yield the crude product which was co-distilled with 20 mL of toluene to afford title compound as trifluoracetic acid salt (1.1 g; 2.80 mmol, 97%) as a viscous liquid. The product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, CDCl3) δ 7.95 (s, 2H), 7.44 – 7.35 (m, 3H), 7.29 – 7.22 (m, 3H), 7.19 – 7.13 (m, 1H), 6.98 (d, J = 12.4 Hz, 1H), 6.86 (dd, J = 7.6, 1.7 Hz, 2H), 4.25 (t, J = 7.2 Hz, 1H), 3.23 (s, 3H), 3.04 (dd, J = 13.9, 6.4 Hz, 1H), 2.98 – 2.84 (m, 1H). Mass m/z: calcd for C16H18N2NaO+ [M + Na]+, 277.1; found, 277.1.
5.3. Preparation of (R)-N-methyl-2-(2-(2-methyl-1H-indol-3-yl)acetamido)-N,3-diphenylpropanamide (R-PF-74)
To a stirred solution of 2-(2-methyl-1H-indol-3-yl)acetic acid (100 mg; 0.53 mmol) and (R)-2-amino-N-methyl-N,3-diphenylpropanamide 2,2,2-trifluoroacetate (214 mg; 0.58 mmol) in 6 mL of dry CH2Cl2 at 0 °C were added diisopropyl ethyl amine (171 mg; 1.32 mmol) and T3P 50% solution in CH2Cl2 by weight (438 mg; 0.69 mmol) dropwise simultaneously. The reaction mixture was allowed to stir for 30 min at 0 °C. Completion of the reaction was confirmed by LC–MS. The reaction mixture was quenched with cold water and the product was extracted with CH2Cl2 and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the crude product was purified by flash column chromatography to afford title compound (184 mg; 0.43 mmol, 82%). The product was confirmed by 1H NMR and MS (Figure S6).
1H NMR (400 MHz, CDCl3) δ 8.00 (s, 1H), 7.41 – 7.31 (m, 4H), 7.29 (d, J = 8.0 Hz, 1H), 7.18 – 7.11 (m, 2H), 7.11 – 7.03 (m, 3H), 6.93 (d, J = 5.7 Hz, 2H), 6.66 (d, J = 7.1 Hz, 2H), 6.18 (d, J = 8.3 Hz, 1H), 4.78 (dd, J = 15.2, 7.1 Hz, 1H), 3.67 – 3.46 (m, 2H), 3.16 (s, 3H), 2.72 (dd, J = 13.3, 6.9 Hz, 1H), 2.52 (dd, J = 13.3, 7.0 Hz, 1H), 2.31 (s, 3H). Mass m/z: calcd for C27H27N3NaO2+ [M + Na]+, 448.2; found, 448.2.
6. Synthesis of S-CX15
6.1. Preparation of (S)-N-Methyl-2-(2-(7-nitro-1H-indol-3-yl)acetamido)-N,3-diphenylpropanamide (AP-5-47)
To a stirred solution of 2-(7-nitro-1H-indol-3-yl)acetic acid (100 mg; 0.5 mmol) and (S)-2-amino-N-methyl-N,3-diphenylpropanamide 2,2,2-trifluoroacetate (184 mg; 0.55 mmol) in 20 mL of dry CH2Cl2 at 0 °C were added diisopropyl ethyl amine (193 mg; 1.5 mmol) and T3P 50 % solution in CH2Cl2 by weight (413 mg; 0.65 mmol) dropwise simultaneously. The reaction mixture was allowed to stir for 30 min at 0 °C. Completion of the reaction was confirmed by LC–MS. The reaction mixture was quenched with cold water and the product was extracted with CH2Cl2 and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the crude product was purified by flash column chromatography to afford 9 (171 mg; 0.37 mmol, 75 %). The product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, CDCl3) δ 9.85 (s, 1H), 8.17 (d, J = 8.0 Hz, 1H), 7.78 (d, J = 7.8 Hz, 1H), 7.42 – 7.31 (m, 3H), 7.21 – 7.12 (m, 2H), 7.08 (t, J = 7.2 Hz, 2H), 6.96 (t, J = 11.2 Hz, 2H), 6.72 (d, J = 7.0 Hz, 2H), 6.20 (d, J = 8.1 Hz, 1H), 4.83 (dd, J = 15.2, 7.3 Hz, 1H), 3.74 – 3.59 (m, 2H), 3.20 (s, 3H), 2.80 (dd, J = 13.5, 6.8 Hz, 1H), 2.80 (dd, J = 13.5, 6.8 Hz, 1H), 2.59 (dd, J = 13.5, 7.5 Hz, 1H). Mass m/z: calcd for C26H25N4O4+ [M + H]+, 457.2; found, 457.3.
6.2. Preparation of (S)-2-(2-(7-Amino-1H-indol-3-yl)acetamido)-N-methyl-N,3-diphenylpropanamide (S-CX15)
To as stirred solution of (S)-N-methyl-2-(2-(7-nitro-1H-indol-3-yl)acetamido)-N,3-diphenylpropanamide (90 mg, 0.2 mmol) in EtOH (15 mL) at room temperature Pd/C (10%, 30 mg) was added and stirred under Hydrogen atmosphere at room temperature for 16 h. Completion of the reaction confirmed LC-MS, the reaction was filtered over Celite, and the filter bed was washed with EtOH (20 mL). The combined organic portions were evaporated crude product was purified by flash column chromatography to afford title compound (63 mg, 0.15 mmol, 75%). The product was confirmed by 1H NMR and MS (Figure S7).
1H NMR (400 MHz, DMSO) δ 10.35 (s, 1H), 8.19 (d, J = 7.7 Hz, 1H), 7.53 – 7.30 (m, 3H), 7.26 – 7.03 (m, 5H), 6.96 (s, 1H), 6.79 (d, J = 3.6 Hz, 2H), 6.68 – 6.54 (m, 2H), 6.38 – 6.06 (m, 1H), 4.93 (s, 2H), 4.44 (d, J = 4.4 Hz, 1H), 3.51 – 3.34 (m, 2H), 3.13 (s, 3H), 2.81 (dd, J = 13.3, 4.4 Hz, 1H), 2.71 – 2.55 (m, 1H). Mass m/z: calcd for C26H27N4O2+ [M + H]+, 427.2; found, 427.3.
7. Synthesis of R-CX15
7.1. Preparation of (R)-N-Methyl-2-(2-(7-nitro-1H-indol-3-yl)acetamido)-N,3-diphenylpropanamide (AP-5-46)
To a stirred solution of 2-(7-nitro-1H-indol-3-yl)acetic acid (100 mg; 0.45 mmol) and (R)-2-amino-N-methyl-N,3-diphenylpropanamide 2,2,2-trifluoroacetate (184 mg; 0.5 mmol) in 20 mL of dry CH2Cl2 at 0 °C were added diisopropyl ethyl amine (175 mg; 1.36 mmol) and T3P 50 % solution in CH2Cl2 by weight (376 mg; 1.36 mmol) dropwise simultaneously. The reaction mixture was allowed to stir for 1h at 0 °C. Completion of the reaction was confirmed by LC–MS. The reaction mixture was quenched with cold water and the product was extracted with CH2Cl2 and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the crude product was purified by flash column chromatography to afford 9 (144 mg; 0.32 mmol, 70%). The product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, CDCl3) δ 9.85 (s, 1H), 8.17 (d, J = 8.0 Hz, 1H), 7.78 (d, J = 7.8 Hz, 1H), 7.43 – 7.28 (m, 3H), 7.21 – 7.12 (m, 2H), 7.08 (t, J = 7.2 Hz, 2H), 6.96 (t, J = 11.2 Hz, 2H), 6.72 (d, J = 7.0 Hz, 2H), 6.21 (d, J = 8.1 Hz, 1H), 4.83 (dd, J = 15.2, 7.3 Hz, 1H), 3.76 – 3.59 (m, 2H), 3.20 (s, 3H), 2.80 (dd, J = 13.4, 6.7 Hz, 1H), 2.59 (dd, J = 13.5, 7.4 Hz, 1H). Mass m/z: calcd for C26H25N4O4+ [M + H]+, 457.2; found, 457.3.
7.2. Preparation of (R)-2-(2-(7-Amino-1H-indol-3-yl)acetamido)-N-methyl-N,3-diphenylpropanamide (R-CX15)
To as stirred solution of (R)-N-methyl-2-(2-(7-nitro-1H-indol-3-yl)acetamido)-N,3-diphenylpropanamide (90 mg, 0.2 mmol) in EtOH (15 mL) at room temperature Pd/C (10%, 30 mg) was added and stirred under Hydrogen atmosphere at room temperature for 16 h. Completion of the reaction confirmed LC-MS, the reaction was filtered over Celite, and the filter bed was washed with EtOH (20 mL). The combined organic portions were evaporated crude product was purified by flash column chromatography to afford title compound (60 mg, 0.14 mmol, 70%). The product was confirmed by 1H NMR and MS (Figure S8).
1H NMR (400 MHz, DMSO) δ 10.35 (s, 1H), 8.19 (d, J = 7.8 Hz, 1H), 7.52 – 7.26 (m, 3H), 7.17 (dd, J = 26.0, 5.1 Hz, 5H), 6.97 (s, 1H), 6.79 (d, J = 3.6 Hz, 2H), 6.65 (dd, J = 7.6, 5.4 Hz, 2H), 6.28 (dd, J = 8.2, 4.4 Hz, 1H), 4.92 (s, 2H), 4.55 – 4.31 (m, 1H), 3.49 – 3.34 (m, 2H), 3.13 (s, 3H), 2.81 (dd, J = 13.3, 4.5 Hz, 1H), 2.64 (dd, J = 13.3, 9.5 Hz, 1H). Mass m/z: calcd for C26H27N4O2+ [M + H]+, 427.3; found, 427.3.
8. Synthesis of S-CX16
8.1. Preparation of N-methyl-1H-indazol-5-amine (AP-5-69)
To a stirred solution of tert-butyl 5-(2,2,2-trifluoro-N-methylacetamido)-1H-indazole-1-carboxylate (prepared as reported procedure JMC, 2016, 59, 3793–3807) (5.84 g; 17.01 mmol) in methanol (90 mL), and water (45 mL) at room temperature, potassium carbonate (11.75 g; 85.05 mmol) was added. The reaction was heated to 85 °C and stirred overnight with a condenser. Cooled down reaction and neutralized with 1 N HCl. Extracted the aqueous layer with ethyl acetate (2×100 mL), washed with aq NaCl (2×100 mL), dried over Na2SO4, and concentrated. The crude product was purified by flash column chromatography to afford title compound a white solid (1.5 g; 10.2 mmol, 60%). The product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, DMSO) δ 12.63 (s, 1H), 7.74 (d, J = 15.2 Hz, 1H), 7.28 (t, J = 9.3 Hz, 1H), 6.82 – 6.74 (m, 1H), 6.63 – 6.50 (m, 1H), 5.38 (t, J = 9.8 Hz, 1H), 2.68 (d, J = 4.7 Hz, 3H). Mass m/z: calcd for C8H10N3+ [M + H]+, 147.0; found, 147.0.
8.2. Preparation of (S)-Tert-butyl 1-((1H-indazol-5-yl)(methyl)amino)-1-oxo-3-phenylpropan-2-ylcarbamate (AP-5-93)
To a stirred solution of N-methyl-1H-indazol-5-amine (600 mg; 4.08 mmol) and (S)-2-(tert-butoxycarbonylamino)-3-phenylpropanoic acid (1.08 g; 4.08 mmol) in 15 mL of dry CH2Cl2 at 0 °C were added diisopropyl ethyl amine (1.58 g; 12.24 mmol) and T3P 50% solution in CH2Cl2 by weight (3.11 g; 4.89 mmol) dropwise simultaneously. The reaction mixture was allowed to stir for 1 h min at 0 °C. Completion of the reaction was confirmed by LC–MS. The reaction mixture was quenched with cold water and the product was extracted with CH2Cl2 and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the crude product was purified by flash column chromatography to afford title compound (1.05 g; 2.65 mmol, 65%). The product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, CDCl3) δ 10.41 (s, 1H), 8.13 – 7.73 (m, 1H), 7.51 – 7.33 (m, 1H), 7.31 – 7.09 (m, 5H), 7.01 – 6.71 (m, 3H), 5.15 (d, J = 57.7 Hz, 1H), 4.45 (d, J = 7.3 Hz, 1H), 3.22 (m, 3H), 2.91 (dt, J = 17.3, 9.4 Hz, 1H), 2.74 (dd, J = 12.7, 5.9 Hz, 1H), 1.39 (s, 9H). Mass m/z: calcd for C22H27N4O3+ [M + H]+, 395.2; found, 395.3.
8.3. Preparation of (S)-2-Amino-N-(1H-indazol-5-yl)-N-methyl-3-phenylpropanamide (AP-5-99)
To a stirred solution of (S)-tert-butyl 1-((1H-indazol-5-yl)(methyl)amino)-1-oxo-3-phenylpropan-2-ylcarbamate (1.04 g; 2.64 mmol) in 16 mL CH2Cl2 at 0 °C was added 4 mL of TFA. The reaction mixture was then warmed to room temperature and stirred for 2 h. Completion of the reaction was confirmed by LC–MS. Volatiles were evaporated under reduced pressure to yield the crude product which was co-distilled with 20 mL of toluene to afford title compound as TFA salt (1.07 mg; 2.64 mmol, 100%) as a viscous liquid. The product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, DMSO) δ 13.31 (s, 1H), 8.30 (s, 2H), 8.17 – 7.93 (m, 1H), 7.70 – 7.45 (m, 1H), 7.47 – 7.05 (m, 4H), 6.94 – 6.74 (m, 2H), 3.18 (m, 3H), 3.06 – 2.91 (m, 1H), 2.88 – 2.73 (m, 1H). Mass m/z: calcd for C17H19N4O+ [M + H]+, 295.2; found, 295.1.
8.4. Preparation of (S)-N-(1H-Indazol-5-yl)-N-methyl-2-(2-(2-methyl-1H-indol-3-yl)acetamido)-3-phenylpropanamide (S-CX16)
To a stirred solution of 2-(2-methyl-1H-indol-3-yl)acetic acid (70 mg; 0.37 mmol) and (S)-2-amino-N-(1H-indazol-5-yl)-N-methyl-3-phenylpropanamide 2,2,2-trifluoroacetate (166 mg; 0.41 mmol) in 7 mL of dry CH2Cl2 at 0 °C were added diisopropyl ethyl amine (143 mg; 1.11 mmol) and T3P 50% solution in CH2Cl2 by weight (282 mg; 0.44 mmol) dropwise simultaneously. The reaction mixture was allowed to stir for 30 min at 0 °C. Completion of the reaction was confirmed by LC–MS. The reaction mixture was quenched with cold water and the product was extracted with CH2Cl2 and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the crude product was purified reverse phase HPLC to afford title compound (69 mg; 0.15 mmol, 40%). The product was confirmed by 1H NMR and MS (Figure S9).
1H NMR (400 MHz, CDCl3) δ 10.87 (s, 1H), 8.33 – 8.09 (m, 1H), 8.05 – 7.76 (m, 1H), 7.43 – 7.30 (m, 1H), 7.31 – 7.23 (m, 3H), 7.20 (dd, J = 9.2, 5.6 Hz, 1H), 7.16 – 7.04 (m, 4H), 6.73 (t, J = 11.0 Hz, 2H), 6.51 – 6.18 (m, 1H), 4.78 (dt, J = 14.9, 7.4 Hz, 1H), 3.70 – 3.48 (m, 2H), 3.20 (s, 3H), 2.80 (dt, J = 23.6, 11.8 Hz, 1H), 2.62 (dd, J = 13.1, 6.5 Hz, 1H), 2.37 – 2.15 (m, 3H). Mass m/z: calcd for C28H28N5O2+ [M + H]+, 466.2; found, 466.3.
9. Synthesis of R-CX16
9.1. Preparation of (R)-Tert-butyl 1-((1H-indazol-5-yl)(methyl)amino)-1-oxo-3-phenylpropan-2-ylcarbamate (AP-5-72)
To a stirred solution of (R)-2-(tert-butoxycarbonylamino)-3-phenylpropanoic acid (1.2 g; 4.52 mmol) and N-methyl-1H-indazol-5-amine (665 mg; 4.52 mmol) in 20 mL of dry CH2Cl2 at 0 °C were added diisopropyl ethyl amine (1.46 g; 11.3 mmol) and T3P 50% solution in CH2Cl2 by weight (3.74 g; 5.87 mmol) dropwise simultaneously. The reaction mixture was allowed to stir for 30 min at 0 °C. Completion of the reaction was confirmed by LC–MS. The reaction mixture was quenched with cold water and the product was extracted with CH2Cl2 and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the crude product was purified by flash column chromatography to afford title compound (0.74 g; 1.89 mmol, 45%). The product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, CDCl3) δ 10.59 (s, 1H), 8.04 – 7.87 (m, 1H), 7.43 – 7.29 (m, 2H), 7.29 – 7.16 (m, 4H), 6.94 (d, J = 6.6 Hz, 2H), 6.87 – 6.78 (m, 1H), 5.30 (t, J = 4.3 Hz, 1H), 4.47 (d, J = 7.1 Hz, 1H), 4.17 – 4.04 (m, 1H), 3.23 (s, 3H), 2.95 (dd, J = 12.9, 8.4 Hz, 1H), 2.89 (s, 1H), 2.76 (dd, J = 12.8, 6.0 Hz, 1H), 1.42 (d, J = 23.6 Hz, 8H). Mass m/z: calcd for C22H26N4NaO3+ [M + Na]+, 417.2; found, 417.3.
9.2. Preparation of (R)-2-Amino-N-(1H-indazol-5-yl)-N-methyl-3-phenylpropanamide (AP-5-73)
To a stirred solution of (R)-tert-butyl 1-((1H-indazol-5-yl)(methyl)amino)-1-oxo-3-phenylpropan-2-ylcarbamate (720 mg; 1.82 mmol) in 16 mL CH2Cl2 at 0 °C was added 4 mL of TFA. The reaction mixture was then warmed to room temperature and stirred for 2 h. Completion of the reaction was confirmed by LC–MS. Volatiles were evaporated under reduced pressure to yield the crude product which was co-distilled with 20 mL of toluene to afford title compound as TFA salt (745 mg; 1.79 mmol, 98%) as a viscous liquid. The product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, MeOD) δ 8.18 – 7.79 (m, 1H), 7.74 – 7.08 (m, 5H), 6.90 (d, J = 7.2 Hz, 2H), 3.92 (s, 1H), 3.27 (s, 3H), 3.16 – 3.00 (m, 1H), 2.84 (dd, J = 13.2, 6.4 Hz, 1H). Mass m/z: calcd for C17H18N4NaO+ [M + Na]+, 317.1; found, 317.1.
9.3. Preparation of (R)-N-(1H-Indazol-5-yl)-N-methyl-2-(2-(2-methyl-1H-indol-3-yl)acetamido)-3-phenylpropanamide (R-CX16)
To a stirred solution of 2-(2-methyl-1H-indol-3-yl)acetic acid (70 mg; 0.37 mmol) and (R)-2-amino-N-(1H-indazol-5-yl)-N-methyl-3-phenylpropanamide 2,2,2-trifluoroacetate (151 mg; 0.37 mmol) in 7 mL of dry CH2Cl2 at 0 °C were added diisopropyl ethyl amine (143 mg; 1.11 mmol) and T3P 50% solution in CH2Cl2 by weight (282 mg; 0.44 mmol) dropwise simultaneously. The reaction mixture was allowed to stir for 30 min at 0 °C. Completion of the reaction was confirmed by LC–MS. The reaction mixture was quenched with cold water and the product was extracted with CH2Cl2 and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the crude product was purified reverse phase HPLC to afford title compound (74 mg; 0.16 mmol, 43%). The product was confirmed by 1H NMR and MS (Figure S10).
1H NMR (400 MHz, CDCl3) δ 10.86 (s, 1H), 8.16 (s, 1H), 7.95 (s, 1H), 7.37 (t, J = 9.2 Hz, 1H), 7.31 – 7.23 (m, 3H), 7.20 (t, J = 7.4 Hz, 3H), 7.16 – 7.05 (m, 3H), 6.75 (d, J = 7.2 Hz, 2H), 6.36 (d, J = 8.4 Hz, 2H), 4.77 (dd, J = 14.8, 7.9 Hz, 2H), 3.74 – 3.52 (m, 2H), 3.20 (s, 3H), 2.81 (dd, J = 13.1, 7.9 Hz, 1H), 2.62 (dd, J = 13.1, 6.5 Hz, 1H), 2.29 (s, 3H). Mass m/z: calcd for C28H28N5O2+ [M + H]+, 466.2; found, 466.3.
10. Synthesis of S-CX17
10.1. Preparation of (S)-N-(1H-indazol-5-yl)-N-methyl-2-(2-(7-nitro-1H-indazol-3-yl)acetamido)-3-phenylpropanamide (102)
To a stirred solution of 2-(7-nitro-1H-indazol-3-yl)acetic acid (100 mg; 0.45 mmol) and (S)-2-amino-N-(1H-indazol-5-yl)-N-methyl-3-phenylpropanamide 2,2,2-trifluoroacetate (203 mg; 0.49 mmol) in 6 mL of dry CH2Cl2 at 0 °C were added diisopropyl ethyl amine (175 mg; 1.36 mmol) and T3P 50% solution in CH2Cl2 by weight (374 mg; 0.58 mmol) dropwise simultaneously. The reaction mixture was allowed to stir for 30 min at 0 °C. Completion of the reaction was confirmed by LC–MS. The reaction mixture was quenched with cold water and the product was extracted with CH2Cl2 and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the crude product was purified by flash column chromatography to afford title compound (115 mg; 0.23 mmol, 51%). The product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, DMSO) δ 13.63 (s, 1H), 13.14 (d, J = 35.9 Hz, 1H), 8.72 (d, J = 7.9 Hz, 1H), 8.30 (dd, J = 7.8, 0.7 Hz, 1H), 8.03 (s, 1H), 7.97 (d, J = 7.7 Hz, 1H), 7.50 (t, J = 12.2 Hz, 1H), 7.28 – 7.18 (m, 1H), 7.18 – 7.01 (m, 4H), 6.87 – 6.66 (m, 2H), 4.53 – 4.34 (m, 1H), 3.85 (s, 2H), 3.23 – 3.11 (m, 3H), 3.02 – 2.85 (m, 1H), 2.70 (dd, J = 13.4, 9.2 Hz, 1H). Mass m/z: calcd for C26H24N7O4+ [M + H]+, 498.2; found, 498.2.
10.2. Preparation of (S)-2-(2-(7-amino-1H-indazol-3-yl)acetamido)-N-(1H-indazol-5-yl)-N-methyl-3-phenylpropanamide (S-CX17)
To as stirred solution of (S)-N-(1H-indazol-5-yl)-N-methyl-2-(2-(7-nitro-1H-indazol-3-yl)acetamido)-3-phenylpropanamide (110 mg, 0.22 mmol) in EtOH (10 mL) at room temperature Pd/C (10%, 30 mg) was added and stirred under Hydrogen atmosphere at room temperature for 16 h. Completion of the reaction confirmed LC-MS, the reaction was filtered over Celite, and the filter bed was washed with EtOH (20 mL). The combined organic portions were evaporated crude product was purified by flash column chromatography to afford title compound (80 mg, 0.17 mmol, 77.6 %). The product was confirmed by 1H NMR and MS (Figure S11).
1H NMR (400 MHz, DMSO) δ 13.14 (d, J = 36.9 Hz, 1H), 12.24 (s, 1H), 8.48 (d, J = 7.9 Hz, 1H), 8.03 (d, J = 24.3 Hz, 1H), 7.47 (dd, J = 21.3, 11.4 Hz, 1H), 7.25 – 6.96 (m, 4H), 6.78 (t, J = 13.5 Hz, 2H), 6.75 – 6.61 (m, 2H), 6.40 (dd, J = 5.8, 2.2 Hz, 1H), 5.20 (s, 2H), 4.51 – 4.32 (m, 1H), 3.66 (s, 2H), 3.15 (d, J = 5.2 Hz, 3H), 2.92 (dd, J = 13.4, 5.4 Hz, 1H), 2.78 – 2.62 (m, 1H). Mass m/z: calcd for C26H26N7O2+ [M + H]+, 468.2; found, 468.2.
11. Synthesis of R-CX17
11.1. Preparation of (R)-N-(1H-indazol-5-yl)-N-methyl-2-(2-(7-nitro-1H-indazol-3-yl)acetamido)-3-phenylpropanamide (AP-5-104)
To a stirred solution of 2-(7-nitro-1H-indazol-3-yl)acetic acid (150 mg; 0.68 mmol) and (R)-2-amino-N-(1H-indazol-5-yl)-N-methyl-3-phenylpropanamide 2,2,2-trifluoroacetate (305 mg; 0.75 mmol) in 7 mL of dry CH2Cl2 at 0 °C were added diisopropyl ethyl amine (263 mg; 2.04 mmol) and T3P 50% solution in CH2Cl2 by weight (562 mg; 0.88 mmol) dropwise simultaneously. The reaction mixture was allowed to stir for 30 min at 0 °C. Completion of the reaction was confirmed by LC–MS. The reaction mixture was quenched with cold water and the product was extracted with CH2Cl2 and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the crude product was purified by flash column chromatography to afford title compound (237 mg; 0.47 mmol, 70%). The product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, DMSO) δ 13.63 (s, 1H), 13.19 (s, 1H), 8.72 (d, J = 8.0 Hz, 1H), 8.30 (d, J = 7.2 Hz, 1H), 8.03 (s, 1H), 7.97 (d, J = 7.5 Hz, 1H), 7.59 – 7.39 (m, 2H), 7.23 (t, J = 7.9 Hz, 1H), 7.19 – 7.04 (m, 3H), 6.80 (d, J = 6.6 Hz, 2H), 4.43 (d, J = 5.1 Hz, 1H), 3.84 (d, J = 12.0 Hz, 2H), 3.21 – 3.10 (m, 3H), 2.95 (dd, J = 13.3, 5.1 Hz, 1H), 2.70 (dd, J = 13.5, 9.3 Hz, 1H). Mass m/z: calcd for C26H24N7O4+ [M + H]+, 498.2; found, 498.3.
11.2. Preparation of (R)-2-(2-(7-amino-1H-indazol-3-yl)acetamido)-N-(1H-indazol-5-yl)-N-methyl-3-phenylpropanamide (R-CX17)
To as stirred solution of (R)-N-(1H-indazol-5-yl)-N-methyl-2-(2-(7-nitro-1H-indazol-3-yl)acetamido)-3-phenylpropanamide (160 mg, 0.32 mmol) in EtOH (10 mL) at room temperature Pd/C (10%, 40 mg) was added and stirred under Hydrogen atmosphere at room temperature for 16 h. Completion of the reaction confirmed LC-MS, the reaction was filtered over Celite, and the filter bed was washed with EtOH (20 mL). The combined organic portions were evaporated crude product was purified by flash column chromatography to afford title compound (100 mg, 0.21 mmol, 66.7 %). The product was confirmed by 1H NMR and MS (Figure S12).
1H NMR (400 MHz, DMSO) δ 13.18 (s, 1H), 12.24 (s, 1H), 8.48 (d, J = 7.9 Hz, 1H), 8.00 (s, 1H), 7.50 (d, J = 8.7 Hz, 1H), 7.35 (d, J = 39.0 Hz, 1H), 7.22 – 6.99 (m, 4H), 6.80 (d, J = 6.7 Hz, 2H), 6.74 – 6.63 (m, 2H), 6.46 – 6.36 (m, 1H), 5.20 (s, 2H), 4.49 – 4.32 (m, 1H), 3.66 (s, 2H), 3.22 – 3.09 (m, 4H), 2.92 (dd, J = 13.3, 5.4 Hz, 1H), 2.69 (dd, J = 13.3, 8.8 Hz, 1H). Mass m/z: calcd for C26H26N7O2+ [M + H]+, 468.3; found, 468.2.
Metabolic stability evaluation of PF-74 in human liver microsomes.
Metabolic stability analysis was performed in human liver microsomes as outlined in Lu et al.38 and was performed by HD Biosciences (Shanghai, China).
Cells
Human embryonic kidney 293T (a gift from Dr. Irwin Chaiken, Drexel University, Philadelphia, PA) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM), 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin and 2mM l-glutamine. Human astroglioma U87 cells stably expressing CD4/CCR5 or CD4/CXCR4 (obtained from Prof. Hongkui Deng, Peking University, and Prof. Dan Littman, New York University, USA, through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH)39, 40 were cultured in DMEM supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin and 2mM l-glutamine, 300μg/ml G418 (Thermo Scientific, Waltham, MA) and 1μg/ml Puromycin (Thermo Scientific). Cells were incubated continuously, unless otherwise stated, at 37°C in a humidified 5% CO2 / 95% air environment.
Proteins
IgG b12 anti HIV-1 gp120; was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: Anti-HIV-1 gp120 Monoclonal (IgG1 b12) from Dr. Dennis Burton and Carlos Barbas); p24 was produced in-house as previously described.41 Briefly, a vector containing C-terminally His-tagged HIV-1NL4-3CA (a gift from Dr. Eric Barklis, Oregon Health and Science University, Portland, OR) was transformed into BL21-Codon Plus (DE3)-RIL Competent Cells (Agilent Technologies, Wilmington, DE) and grown up in autoinduction ZYP-5052 medium overnight with shaking (225 rpm) at 30°C42. Bacterial cultures were spun down at 7000 rpm, and the supernatant was discarded. Cell pellets were resuspended in PBS and lysed via sonication. The resultant supernatant was clarified and immediately applied to a Talon cobalt resin affinity column (Clonetech Laboratories, Mountain View, CA). Protein was eluted using 1X PBS with 250mM imidazole. Purified CA-H6 monomers were dialyzed overnight into 20mM Tris-HCl pH 8.0 at 4°C, concentrated to 120μM, flash-frozen in liquid nitrogen, aliquoted and stored at −80°C. The CA hexamer was generated by introducing mutations at the following sites: A14C, E45C, W184A, and M185A through site-directed mutagenesis (Stratagene). The CA hexamer construct was expressed and purified following the same protocol as described above. After purification, the CA-H6 hexamers were dialyzed into 200mM ßME followed by sequential dialyzes to slowly remove the ßME to allow for hexamer assembly.
Production of mutant CA hexamers
A PCR was performed following Platinum SuperFi DNA Polymerase protocol (Invitrogen) to introduce site-directed mutations. Primers for site-directed mutagenesis used can be found in Table S3. After PCR amplification, the samples were treated with 1 μl/μg of DpnI for an hour and a half to ensure digestion. The digested amplicon and digested plasmid DNA were transformed, and mutations were confirmed via sanger sequencing through Genewiz. Protein expression and purification were performed as described above. Briefly, mutant CA was transformed into Bl21 DE3 (RIL) (Agilent Technologies) and expressed in ZYP5052 overnight at 30°C with shaking at 250rpm. The cells were then sonicated and the clarified lysate was purified via Cobalt-affinity chromatography. After the sequential dialysis, the assembled CA hexamers underwent size exclusion chromatography using a Superdex, 200 Increase 10/300 GL column to isolate pure hexamers.
Production of pseudotyped viruses
Single-round infectious envelope-pseudotyped luciferase-reporter viruses were produced by dual transfection of two vectors (3μg of vector 1 and 4μg of vector 2) in 6-well plated 293T cells (1×106 cells/well).40 Vector 1 is an envelope-deficient HIV-1 pNL4-3-Luc+R-E plasmid that carries the luciferase-reporter gene.43 Vector 2 is a plasmid expressing the HIV-1BG505 gp160 Env.44 Transfections of these vectors were carried out via calcium phosphate (ProFection Mammalian Transfection System, Promega, Madison, WI) for 5 h. Following the 5 h transfection incubation, DNA-containing medium was removed, cells were washed with DMEM and replenished with fresh culture media. Supernatants containing pseudovirus were collected 72 h post-transfection, clarified, filtered, aliquoted and stored at −80°C.
ELISA-based quantification of p24 content
ELISA plate was coated with 50ng of mouse anti-p24 (Abcam, ab9071) overnight at 4°C, blocked with 3% BSA for 2 h at room temperature and washed with 0.5% Tween in PBS. Pseudoviral stocks were lysed with 0.1% Triton X-100 (Sigma-Aldrich, St. Louis, MO) at 37°C for 1 h and added to the plate overnight at 4°C. Simultaneously, p24 protein (produced, purified, and validated as described above) was added for the generation of a standard curve. Following the overnight incubation, the plate was washed with PBST and 1:5000 dilution of rabbit anti-p24 (Abcam, ab63913) was added for 2 h at room temperature. After washing the unbound rabbit anti-p24 off the plate with PBST, goat, anti-rabbit-HRP at a 1:5000 dilution was added for 1 h at room temperature. The plate was then extensively washed with PBST. Subsequently, a solution of 0.4mg/ml o-phenylenediamine in a phosphate-citrate buffer with sodium perborate (Sigma-Aldrich) was added and incubated in the dark for 30 min. Optical densities were then obtained at 450 nm in a Multiskan™ GO Microplate Spectrophotometer (Thermo Scientific).
Single-round infection assay
The details of the single-round HIV-1 infection assay have been published previously.43, 45, 46 Briefly, U87.CD4.CCR5/CXCR4 (1.2 × 104 cells/well) target cells were seeded in 96-well luminometer-compatible tissue culture plates (Greiner Bio-one). After 24 hours compound, DMSO (vehicle control for compounds, Sigma) were mixed with pseudotyped viruses (normalized to p24 content) and the mixture was added to the target cells and incubated for 48 hours at 37°C. Following this, the media was removed from each well, and the cells were lysed by the addition of 50 μl/well of luciferase lysis buffer (Promega) and one freeze-thaw cycle. A GloMax 96 microplate luminometer (Promega) was used to measure the luciferase activity of each well after the addition of 50 μl/well of luciferase assay substrate (Promega).
Cellular toxicity
The viability of the U87.CD4.CCR5/CXCR4 cells was determined by the Cell Counting Kit-8 cell proliferation and cytotoxicity assay (Dojindo Molecular Technologies, Rockville, MD) per the manufacturer’s instructions. U87.CD4.CCR5/CXCR4 cells were seeded at a density of 1.2 × 104 cells/well in 96 well tissue culture plates (Olympus plastics) 24 hours before treatment. Cells were treated with compound (1mM-0.3μM) or DMSO (Sigma) for 48 hours at 37°C. Subsequently, 10μl of the CCK-8 solution was added to each well and allowed to incubate at 37°C for 4 h before absorbance at 450nm was measured in a Multiskan™ GO Microplate Spectrophotometer (Thermo Scientific). Untreated cells were used as a medium control, and 0.1% SDS treated cells served as a positive control. The 50% cytotoxicity (CC50) value was defined as the concentration of the compounds that reduced the absorbance of treated cells by 50% as compared with control cells.
SPR direct interaction analysis
All binding assays were performed on a ProteOn XPR36 SPR Protein Interaction Array System (Bio-Rad Laboratories, Hercules, CA). The instrument temperature was set at 25 °C for all kinetic analyses. ProteOn GLH sensor chips were preconditioned with two short pulses each (10 s) of 50mM NaOH, 100mM HCl, and 0.5% sodium do-decyl sulfide. Then the system was equilibrated with PBS-T buffer (20mM sodium phosphate, 150mM NaCl, and 0.005% polysorbate 20, pH 7.4). The surface of a GLH sensor chip was activated with a 1:100 dilution of a 1:1 mixture of 1-ethyl-3-(3- dimethylaminopropyl) carbodiimide hydrochloride (0.2 M) and sulfo-N-hydroxysuccinimide (0.05 M). Immediately after chip activation, the HIV-1 NL4-3 capsid protein constructs, purified as described above, were prepared at a concentration of 100 μg/mL in 10mM sodium acetate, pH 5.0 and injected across ligand flow channels for 5 min at a flow rate of 30 ul/min. Then, after unreacted protein had been washed out, excess active ester groups on the sensor surface were capped by a 5 min injection of 1M ethanolamine HCl (pH 8.0) at a flow rate of 5 μl/min. A reference surface was similarly created by immobilizing a non-specific protein (IgG b12 anti-HIV-1 gp120; was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: Anti-HIV-1 gp120 Monoclonal (IgG1 b12) from Dr. Dennis Burton and Carlos Barbas) and was used as a background to correct nonspecific binding.To prepare a compound for direct binding analysis, compound stock solutions, along with 100% DMSO, and totaling 30 μl was made to a final volume of 1 mL by addition of sample preparation buffer (PBS, pH 7.4). Preparation of analyte in this manner ensured that the concentration of DMSO was matched with that of running buffer with 3% DMSO. Serial dilutions were then prepared in the running buffer (PBS, 3% DMSO, 0.005% polysorbate 20, pH 7.4) and injected at a flow rate of 100 μl/min, for a 1 min association phase, followed by up to a 5 min dissociation phase using the “one-shot kinetics” capability of the Proteon instrument. Data were analyzed using the ProteOn Manager Software version 3.0 (Bio-Rad). The responses from the reference flow cell were subtracted to account for the nonspecific binding and injection artifacts. Experimental data were fitted to a simple 1:1 binding model (where applied). The average kinetic parameters (association [ka] and dissociation [kd] rates) generated from 3–6 data sets were used to define the equilibrium dissociation constant (KD).
Binding Site Analysis via SPR was conducted similarly as in Kortagere et al31. Wild-type and mutant HIV-1 CA hexamer proteins were attached to the surface of a GLH sensor chip by standard amine chemistry as described above. Compounds were injected over these surfaces at a concentration of 50 μM at a flow rate of 100 μL/min, for a 1 min association phase, and the response at equilibrium recorded. For comparison and to take into account minor differences in the ligand density of the mutant surfaces, responses were normalized to the theoretical maximum response (Rmax) for a given surface, assuming a 1:1 interaction. The responses from at least 3 replicate injections over each CA surface were analyzed.
Molecular docking and electrostatic complementarity (EC) analysis
Electrostatic complementarity analysis was achieved using Flare 2.0.1 (Cresset®, Litlington, Cambridgeshire, UK, http://www.cresset-group.com/forge/) for wt and in silico mutated HIV-1 NL4-3 Capsid protein.
Supplementary Material
Scheme 1.

Preparation of tert-butyl(1-(methyl(phenyl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate (intermediate 1)
Scheme 2.

Preparation of 2-amino-N-methyl-N,3-diphenylpropanamide hydrochloride (intermediate 2)
Scheme 3.

Preparation of N,N-dimethyl-1-(7-nitro-1H-indol-3-yl)methanamine (intermediate 3)
Scheme 4.
Preparation of 2-(7-nitro-1H-indol-3-yl)acetonitrile (intermediate 4)
Scheme 5.

Preparation of 2-(7-nitro-1H-indol-3-yl)acetic acid (intermediate 5)
Scheme 6.

Preparation of N-methyl-2-(2-(7-nitro-1H-indol-3-yl)acetamido)-N,3-diphenylpropanamide (intermediate 6)
Scheme 7.

Preparation of 2-(2-(7-amino-1H-indol-3-yl)acetamido)-N-methyl-N,3-diphenylpropanamide (CX15)
Scheme 8.

Preparation of tert-butyl (1-((1H-indazol-5-yl)amino)- 1-oxo-3-phenylpropan -2-yl) carbamate
Scheme 9.

Preparation of tert-butyl 5-(2-((tert-butoxycarbonyl)amino) -3-phenylpropanamido)-1H-indazole-1-carboxylate
Scheme 10.

Preparation tert-butyl 5-(2-((tert-butoxycarbonyl)amino) -N-methyl- 3-phenylpropanamido)-1H-indazole-1-carboxylate
Scheme 11.

Preparation of 2-amino-N-(1H-indazol-5-yl)-N-methyl-3 -phenylpropanamide
Scheme 12.

Preparation of N-(1H-indazol-5-yl)-N-methyl-2-(2-(2-methyl-1H-indol-3-yl) acetamido)-3-phenylpropanamide (CX16)
Scheme 13.

Preparation of tert-butyl (1-((1H-indazol-5-yl)amino)- 1-oxo-3-phenylpropan -2-yl) carbamate
Scheme 14.

Preparation of 3-iodo-1-(4-methoxybenzyl)-7-nitro-1H-indazole
Scheme 15.

Preparation of dimethyl 2-(1-(4-methoxybenzyl)-7-nitro-1H-indazol-3-yl)malonate
Scheme 16.

Preparation of 2-(1-(4-methoxybenzyl)-7-nitro-1H-indazol-3-yl)acetic acid
Scheme 17.

Preparation of 2-(7-nitro-1H-indazol-3-yl)acetic acid
Scheme 18.

Preparation of 2-(7-amino-1H-indazol-3-yl)acetic acid
Scheme 19.

Preparation of 2-(2-(7-amino-1H-indazol-3-yl)acetamido)-N-(1H-indazol-5-yl)-N-methyl-3-phenylpropanamide (CX17)
Scheme 20.

Preparation of (S)-Tert-butyl 1-(methyl(phenyl)amino)-1-oxo-3-phenylpropan-2-ylcarbamate (AP-5-5)

Scheme 21. Preparation of (S)-2-Amino-N-methyl-N,3-diphenylpropanamide (AP-5-9)
Scheme 22.

Preparation of (S)-N-methyl-2-(2-(2-methyl-1H-indol-3-yl)acetamido)-N,3-diphenylpropanamide (S-PF-74)
Scheme 23.

Preparation of (R)-Tert-butyl 1-(methyl(phenyl)amino)-1-oxo-3-phenylpropan-2-ylcarbamate (AP-5-4)
Scheme 24.

Preparation of (R)-2-amino-N-methyl-N,3-diphenylpropanamide (AP-5-8)
Scheme 25.

Preparation of (R)-N-methyl-2-(2-(2-methyl-1H-indol-3-yl)acetamido)-N,3-diphenylpropanamide (R-PF-74)
Scheme 26.

Preparation of (S)-N-Methyl-2-(2-(7-nitro-1H-indol-3-yl)acetamido)-N,3-diphenylpropanamide (AP-5-47)
Scheme 27.
Preparation of (S)-2-(2-(7-Amino-1H-indol-3-yl)acetamido)-N-methyl-N,3-diphenylpropanamide (S-CX15)
Scheme 28.

Preparation of (R)-N-Methyl-2-(2-(7-nitro-1H-indol-3-yl)acetamido)-N,3-diphenylpropanamide (AP-5-46)
Scheme 29.

Preparation of (R)-2-(2-(7-Amino-1H-indol-3-yl)acetamido)-N-methyl-N,3-diphenylpropanamide (R-CX15)
Scheme 30.

Preparation of N-methyl-1H-indazol-5-amine (AP-5-69)
Scheme 31.

Preparation of (S)-Tert-butyl 1-((1H-indazol-5-yl)(methyl)amino)-1-oxo-3-phenylpropan-2-ylcarbamate (AP-5-93)
Scheme 32.

Preparation of (S)-2-Amino-N-(1H-indazol-5-yl)-N-methyl-3-phenylpropanamide (AP-5-99)
Scheme 33.

Preparation of (S)-N-(1H-Indazol-5-yl)-N-methyl-2-(2-(2-methyl-1H-indol-3-yl)acetamido)-3-phenylpropanamide (S-CX16)
Scheme 34.

Preparation of (R)-Tert-butyl 1-((1H-indazol-5-yl)(methyl)amino)-1-oxo-3-phenylpropan-2-ylcarbamate (AP-5-72)
Scheme 35.

Preparation of (R)-2-Amino-N-(1H-indazol-5-yl)-N-methyl-3-phenylpropanamide (AP-5-73)
Scheme 36.

Preparation of (R)-N-(1H-Indazol-5-yl)-N-methyl-2-(2-(2-methyl-1H-indol-3-yl)acetamido)-3-phenylpropanamide (R-CX16)
Scheme 37.

Preparation of (S)-N-(1H-indazol-5-yl)-N-methyl-2-(2-(7-nitro-1H-indazol-3-yl)acetamido)-3-phenylpropanamide (102)
Scheme 38.

Preparation of (S)-2-(2-(7-amino-1H-indazol-3-yl)acetamido)-N-(1H-indazol-5-yl)-N-methyl-3-phenylpropanamide (S-CX17)
Scheme 39.

Preparation of (R)-N-(1H-indazol-5-yl)-N-methyl-2-(2-(7-nitro-1H-indazol-3-yl)acetamido)-3-phenylpropanamide (AP-5-104)
Scheme 40.

Preparation of (R)-2-(2-(7-amino-1H-indazol-3-yl)acetamido)-N-(1H-indazol-5-yl)-N-methyl-3-phenylpropanamide (R-CX17)
ACKNOWLEDGMENT
This work was supported by NIH/NIAID grant AI118415 (Cocklin, PI) and NIH/NIAID grant R01AI150491 (Cocklin, PI, Salvino, Co-I). M.E.M. is also partially supported by NIH training grant T32-MH079785. A.D. is partially funded by NS089435 (Cocklin, Co-I [Nonnemacher, PI]). We’d like to thank Cresset (UK; http://www.cresset-group.com/) for access to their software via an academic license.
ABBREVIATIONS
- HIV-1
human immunodeficiency virus type-1
- CA
Capsid
- cART
combination antiretroviral therapy
- CPSF6
cleavage and polyadenylation specificity factor-6
- NUP153
nucleoporin 153
- PF-3450074
PF-74
- t1/2
half-life
- HYDE
Hydrogen bond and dehydration
- ADME/PK
absorption, distribution, metabolism and excretion/pharmacokinetics
- oral non-CNS
oral non-central nervous system
- CYP
cytochrome P450, SPR, Surface Plasmon Resonance
- IC50
half-maximal inhibitory concentration
- CC50
half-maximal cytotoxic concentration
- wt
wild-type
- EC
electrostatic complementarity
- NMR
nuclear magnetic resonance
Footnotes
Supporting Information
Supplemental figures including the 2D interaction schemes and the LC-MS and 1H NMR spectra of each synthesized compound as well as tables summarizing the predicted interaction energy for binding site residues, metabolic stability parameters, and primers used for mutagenesis can be found in the Supporting Information.
The molecular formula strings, the potency and toxicity values, and the kinetic and affinity data are available.
The PDBs of the docking models for each compound are available.
This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Campbell EM; Hope TJ, HIV-1 capsid: the multifaceted key player in HIV-1 infection. Nat Rev Microbiol 2015, 13 (8), 471–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sundquist WI; Krausslich HG, HIV-1 assembly, budding, and maturation. Cold Spring Harb Perspect Med 2012, 2 (7), a006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rihn SJ; Wilson SJ; Loman NJ; Alim M; Bakker SE; Bhella D; Gifford RJ; Rixon FJ; Bieniasz PD, Extreme genetic fragility of the HIV-1 capsid. PLoS Pathog 2013, 9 (6), e1003461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rossi E; Meuser ME; Cunanan CJ; Cocklin S, Structure, function, and interactions of the HIV-1 capsid protein. 2021, 11 (2), 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li S; Hill CP; Sundquist WI; Finch JT, Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature 2000, 407 (6802), 409–13. [DOI] [PubMed] [Google Scholar]
- 6.Bhattacharya A; Alam SL; Fricke T; Zadrozny K; Sedzicki J; Taylor AB; Demeler B; Pornillos O; Ganser-Pornillos BK; Diaz-Griffero F; Ivanov DN; Yeager M, Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc Natl Acad Sci U S A 2014, 111 (52), 18625–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Price AJ; Jacques DA; McEwan WA; Fletcher AJ; Essig S; Chin JW; Halambage UD; Aiken C; James LC, Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog 2014, 10 (10), e1004459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Price AJ; Fletcher AJ; Schaller T; Elliott T; Lee K; KewalRamani VN; Chin JW; Towers GJ; James LC, CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS Pathog 2012, 8 (8), e1002896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matreyek KA; Yucel SS; Li X; Engelman A, Nucleoporin NUP153 phenylalanine-glycine motifs engage a common binding pocket within the HIV-1 capsid protein to mediate lentiviral infectivity. PLoS Pathog 2013, 9 (10), e1003693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu JP; Francis AC; Meuser ME; Mankowski M; Ptak RG; Rashad AA; Melikyan GB; Cocklin S, Exploring Modifications of an HIV-1 Capsid Inhibitor: Design, Synthesis, and Mechanism of Action. J Drug Des Res 2018, 5 (2). [PMC free article] [PubMed] [Google Scholar]
- 11.Wang L; Casey MC; Vernekar SKV; Do HT; Sahani RL; Kirby KA; Du H; Hachiya A; Zhang H; Tedbury PR; Xie J; Sarafianos SG; Wang Z, Chemical profiling of HIV-1 capsid-targeting antiviral PF74. Eur J Med Chem 2020, 200, 112427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vernekar SKV; Sahani RL; Casey MC; Kankanala J; Wang L; Kirby KA; Du H; Zhang H; Tedbury PR; Xie J; Sarafianos SG; Wang Z, Toward Structurally Novel and Metabolically Stable HIV-1 Capsid-Targeting Small Molecules. Viruses 2020, 12 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun L; Huang T; Dick A; Meuser ME; Zalloum WA; Chen CH; Ding X; Gao P; Cocklin S; Lee KH; Zhan P; Liu X, Design, synthesis and structure-activity relationships of 4-phenyl-1H-1,2,3-triazole phenylalanine derivatives as novel HIV-1 capsid inhibitors with promising antiviral activities. Eur J Med Chem 2020, 190, 112085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun L; Dick A; Meuser ME; Huang T; Zalloum WA; Chen CH; Cherukupalli S; Xu S; Ding X; Gao P; Kang D; De Clercq E; Pannecouque C; Cocklin S; Lee KH; Liu X; Zhan P, Design, Synthesis, and Mechanism Study of Benzenesulfonamide-Containing Phenylalanine Derivatives as Novel HIV-1 Capsid Inhibitors with Improved Antiviral Activities. J Med Chem 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang X; Wu G; Zalloum WA; Meuser ME; Dick A; Sun L; Chen CH; Kang D; Jing L; Jia R; Cocklin S; Lee KH; Liu X; Zhan P, Discovery of novel 1,4-disubstituted 1,2,3-triazole phenylalanine derivatives as HIV-1 capsid inhibitors. RSC Adv 2019, 9 (50), 28961–28986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu G; Zalloum WA; Meuser ME; Jing L; Kang D; Chen CH; Tian Y; Zhang F; Cocklin S; Lee KH; Liu X; Zhan P, Discovery of phenylalanine derivatives as potent HIV-1 capsid inhibitors from click chemistry-based compound library. Eur J Med Chem 2018, 158, 478–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Link JO; Rhee MS; Tse WC; Zheng J; Somoza JR; Rowe W; Begley R; Chiu A; Mulato A; Hansen D; Singer E; Tsai LK; Bam RA; Chou CH; Canales E; Brizgys G; Zhang JR; Li J; Graupe M; Morganelli P; Liu Q; Wu Q; Halcomb RL; Saito RD; Schroeder SD; Lazerwith SE; Bondy S; Jin D; Hung M; Novikov N; Liu X; Villaseñor G; Cannizzaro CE; Hu EY; Anderson RL; Appleby TC; Lu B; Mwangi J; Liclican A; Niedziela-Majka A; Papalia GA; Wong MH; Leavitt SA; Xu Y; Koditek D; Stepan GJ; Yu H; Pagratis N; Clancy S; Ahmadyar S; Cai TZ; Sellers S; Wolckenhauer SA; Ling J; Callebaut C; Margot N; Ram RR; Liu YP; Hyland R; Sinclair GI; Ruane PJ; Crofoot GE; McDonald CK; Brainard DM; Lad L; Swaminathan S; Sundquist WI; Sakowicz R; Chester AE; Lee WE; Daar ES; Yant SR; Cihlar T, Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh K; Gallazzi F; Hill KJ; Burke DH; Lange MJ; Quinn TP; Neogi U; Sonnerborg A, GS-CA Compounds: First-In-Class HIV-1 Capsid Inhibitors Covering Multiple Grounds. Front Microbiol 2019, 10, 1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karadsheh R; Meuser ME; Cocklin S, Composition and Orientation of the Core Region of Novel HIV-1 Entry Inhibitors Influences Metabolic Stability. Molecules 2020, 25 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu JP; Branson JD; Lawrence R; Cocklin S, Identification of a small molecule HIV-1 inhibitor that targets the capsid hexamer. Bioorg Med Chem Lett 2016, 26 (3), 824–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheeseright T; Mackey M; Rose S; Vinter A, Molecular field extrema as descriptors of biological activity: definition and validation. J Chem Inf Model 2006, 46 (2), 665–76. [DOI] [PubMed] [Google Scholar]
- 22.Cheeseright TJ; Mackey MD; Scoffin RA, High content pharmacophores from molecular fields: a biologically relevant method for comparing and understanding ligands. Curr Comput Aided Drug Des 2011, 7 (3), 190–205. [DOI] [PubMed] [Google Scholar]
- 23.Gres AT; Kirby KA; KewalRamani VN; Tanner JJ; Pornillos O; Sarafianos SG, STRUCTURAL VIROLOGY. X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability. Science 2015, 349 (6243), 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reulecke I; Lange G; Albrecht J; Klein R; Rarey M, Towards an integrated description of hydrogen bonding and dehydration: decreasing false positives in virtual screening with the HYDE scoring function. ChemMedChem 2008, 3 (6), 885–97. [DOI] [PubMed] [Google Scholar]
- 25.Tuyishime M; Danish M; Princiotto A; Mankowski MK; Lawrence R; Lombart HG; Esikov K; Berniac J; Liang K; Ji J; Ptak RG; Madani N; Cocklin S, Discovery and optimization of novel small-molecule HIV-1 entry inhibitors using field-based virtual screening and bioisosteric replacement. Bioorg Med Chem Lett 2014, 24 (23), 5439–5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hunt PA; Segall MD; Tyzack JD, WhichP450: a multi-class categorical model to predict the major metabolising CYP450 isoform for a compound. J Comput Aided Mol Des 2018, 32 (4), 537–546. [DOI] [PubMed] [Google Scholar]
- 27.Kaur P; Chamberlin AR; Poulos TL; Sevrioukova IF, Structure-Based Inhibitor Design for Evaluation of a CYP3A4 Pharmacophore Model. J Med Chem 2016, 59 (9), 4210–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huey R; Morris GM; Olson AJ; Goodsell DS, A semiempirical free energy force field with charge-based desolvation. J Comput Chem 2007, 28 (6), 1145–52. [DOI] [PubMed] [Google Scholar]
- 29.Bikadi Z; Hazai E, Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J Cheminform 2009, 1, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pornillos O; Ganser-Pornillos BK; Banumathi S; Hua Y; Yeager M, Disulfide bond stabilization of the hexameric capsomer of human immunodeficiency virus. J Mol Biol 2010, 401 (5), 985–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kortagere S; Xu JP; Mankowski MK; Ptak RG; Cocklin S, Structure-activity relationships of a novel capsid targeted inhibitor of HIV-1 replication. J Chem Inf Model 2014, 54 (11), 3080–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bauer MR; Mackey MD, Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein–Ligand Complexes. Journal of Medicinal Chemistry 2019, 62 (6), 3036–3050. [DOI] [PubMed] [Google Scholar]
- 33.Meuser ME; Murphy MB; Rashad AA; Cocklin S, Kinetic Characterization of Novel HIV-1 Entry Inhibitors: Discovery of a Relationship between Off-Rate and Potency. Molecules 2018, 23 (8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blair WS; Pickford C; Irving SL; Brown DG; Anderson M; Bazin R; Cao J; Ciaramella G; Isaacson J; Jackson L; Hunt R; Kjerrstrom A; Nieman JA; Patick AK; Perros M; Scott AD; Whitby K; Wu H; Butler SL, HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog 2010, 6 (12), e1001220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi J; Zhou J; Halambage UD; Shah VB; Burse MJ; Wu H; Blair WS; Butler SL; Aiken C, Compensatory substitutions in the HIV-1 capsid reduce the fitness cost associated with resistance to a capsid-targeting small-molecule inhibitor. J Virol 2015, 89 (1), 208–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen LA; He H; Pham-Huy C, Chiral drugs: an overview. Int J Biomed Sci 2006, 2 (2), 85–100. [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang JK; Shen M; Thomas CJ; Boxer MB, Chiral kinase inhibitors. Curr Top Med Chem 2011, 11 (7), 800–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu C; Li P; Gallegos R; Uttamsingh V; Xia CQ; Miwa GT; Balani SK; Gan LS, Comparison of intrinsic clearance in liver microsomes and hepatocytes from rats and humans: evaluation of free fraction and uptake in hepatocytes. Drug Metab Dispos 2006, 34 (9), 1600–1605. [DOI] [PubMed] [Google Scholar]
- 39.Bjorndal A; Deng H; Jansson M; Fiore JR; Colognesi C; Karlsson A; Albert J; Scarlatti G; Littman DR; Fenyo EM, Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J Virol 1997, 71 (10), 7478–7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zentner I; Sierra LJ; Fraser AK; Maciunas L; Mankowski MK; Vinnik A; Fedichev P; Ptak RG; Martin-Garcia J; Cocklin S, Identification of a small-molecule inhibitor of HIV-1 assembly that targets the phosphatidylinositol (4,5)-bisphosphate binding site of the HIV-1 matrix protein. ChemMedChem 2013, 8 (3), 426–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kortagere S; Madani N; Mankowski MK; Schon A; Zentner I; Swaminathan G; Princiotto A; Anthony K; Oza A; Sierra LJ; Passic SR; Wang X; Jones DM; Stavale E; Krebs FC; Martin-Garcia J; Freire E; Ptak RG; Sodroski J; Cocklin S; Smith AB 3rd, Inhibiting early-stage events in HIV-1 replication by small-molecule targeting of the HIV-1 capsid. J Virol 2012, 86 (16), 8472–8481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Studier FW, Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 2005, 41 (1), 207–234. [DOI] [PubMed] [Google Scholar]
- 43.Connor RI; Chen BK; Choe S; Landau NR, Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 1995, 206 (2), 935–944. [DOI] [PubMed] [Google Scholar]
- 44.Hoffenberg S; Powell R; Carpov A; Wagner D; Wilson A; Kosakovsky Pond S; Lindsay R; Arendt H; DeStefano J; Phogat S; Poignard P; Fling SP; Simek M; LaBranche C; Montefiori D; Wrin T; Phung P; Burton D; Koff W; King CR; Parks CL; Caulfield MJ, Identification of an HIV-1 clade A envelope that exhibits broad antigenicity and neutralization sensitivity and elicits antibodies targeting three distinct epitopes. Journal of Virology 2013, 87 (10), 5372–5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cocklin S; Gopi H; Querido B; Nimmagadda M; Kuriakose S; Cicala C; Ajith S; Baxter S; Arthos J; Martin-Garcia J; Chaiken IM, Broad-spectrum anti-human immunodeficiency virus (HIV) potential of a peptide HIV type 1 entry inhibitor. J Virol 2007, 81 (7), 3645–3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He J; Choe S; Walker R; Di Marzio P; Morgan DO; Landau NR, Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J Virol 1995, 69 (11), 6705–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.