

Abstract
Background.
HIV treatment of neonates requires identifying appropriate antiretroviral dosing regimens. Our aims were to characterize raltegravir elimination kinetics in low birth weight (LBW) neonates after maternal dosing and to develop a pharmacokinetic model to predict raltegravir plasma concentrations for term and preterm neonates.
Methods.
Mothers living with HIV who received raltegravir during pregnancy and their LBW neonates participated in IMPAACT P1097 study. Up to six serial plasma samples were collected from each infant over the first two postnatal weeks to characterize raltegravir elimination. Safety laboratory evaluations were obtained and infants were monitored for 6 weeks for signs of raltegravir toxicity. An integrated maternal-neonatal pharmacokinetic model was developed to predict neonatal raltegravir plasma concentrations.
Results.
Sixteen mothers and their 18 LBW neonates were enrolled. Median (range) raltegravir elimination half-life was 24.4 (10.1–83) hours (N=17 neonates). No adverse events related to raltegravir in utero exposure were observed. Pharmacokinetic modeling revealed that raltegravir clearance in full term LBW neonates was well-described by allometric scaling but clearance in preterm LBW neonates was better described using slower clearance maturation kinetics. Simulations suggest receipt of the current dosing recommendations in a 34-week gestation neonate would result in plasma concentrations up to 2.5-fold higher than those observed in full-term LBW infants.
Conclusions.
Modeling suggests that prematurity reduces raltegravir clearance and a modified raltegravir dosing regimen will be necessary to avoid elevated plasma raltegravir concentrations.
Keywords: raltegravir, pharmacokinetics, human immunodeficiency virus, low birth weight, preterm neonate
SUMMARY:
Postnatal raltegravir clearance was slower in preterm neonates than full term neonates. A pharmacokinetic model incorporating slower clearance in preterm neonates demonstrates that a reduction in raltegravir dosing is required in preterm neonates.
INTRODUCTION
Limited data exist to provide dosing recommendations for combination antiretroviral regimens to prevent or treat HIV infection in neonates. The Department of Health and Human Services Perinatal Guidelines recommend the administration of a three-drug antiretroviral regimen for presumptive treatment of newborns at highest risk of HIV acquisition and for treatment of neonates with documented HIV infection.[1] Only 6 antiretrovirals, zidovudine, lamivudine, emtricitabine, stavudine, nevirapine and raltegravir, are approved by the Food and Drug Administration (FDA) for use in full term neonates aged less than 14 days. Zidovudine is the only antiretroviral with robust pharmacokinetic and safety data in preterm infants with gestational age (GA) of less than 37 weeks [2].
Pregnant women living with HIV have an increased risk of delivering a low birth weight (LBW) newborn (birth weight less than 2500 g). The overall frequency of LBW worldwide is 14.6%, ranging from 7.0% in North America, Europe, Australia, and New Zealand to 14.0% in Sub-Saharan Africa and 26.4% in Southern Asia.[3] Rates of LBW are increased in infants born to mothers living with HIV, with rates reported as high as 41% in South Africa and 34% in India.[4-6]. Drug disposition may differ significantly in LBW infants due to their impaired in utero growth and/or prematurity.
Raltegravir is a potent antiretroviral agent and in 2017 became the first HIV integrase strand transfer inhibitor (INSTI) approved for use in full term infants. Raltegravir blocks the establishment of post-integration HIV latency and could play an important role in both prophylaxis and treatment of neonates. The raltegravir oral granule formulation is scalable and can provide accurate small doses suitable for use in premature infants. However, the pharmacokinetics and safety of raltegravir have not been studied in LBW or premature neonates [Gestational Age (GA) < 37 weeks].
Raltegravir is primarily metabolized by uridine diphosphate glucuronosyltransferase 1A1 (UGT1A1), the activity of which is well known to be low at birth and to increase nearly 100-fold during the first 14 weeks of life.[7] Consistent with this increase, raltegravir clearance has been shown to increase dramatically over the first weeks of life. An eight-fold daily dosing increase from 1.5 mg/kg once daily during the first week of life; to 3 mg/kg twice daily weeks during weeks 2, 3, and 4; and then to 6 mg/kg twice daily at 4 weeks of life is required to maintain therapeutic yet safe raltegravir plasma concentrations in full term infants.[8]
Avoiding excessive raltegravir plasma concentrations in neonates is important because raltegravir and unconjugated bilirubin compete for binding to albumin. When raltegravir plasma concentrations are extremely high, raltegravir can displace unconjugated bilirubin from albumin [9]. This free, unconjugated bilirubin can cross the blood-brain barrier, leading to bilirubin-induced neurologic damage.[10, 11] Preterm infants have reduced bilirubin metabolism from decreased UGT1A1 activity and a more permeable blood brain barrier, placing them at higher risk for bilirubin toxicity than full term infants.[12] Raltegravir metabolism is also decreased if UGT1A1 activity is low and high plasma raltegravir plasma exposures may further increase the risk for bilirubin toxicity in preterm infants by displacing unconjugated bilirubin from albumin.
The International Maternal Pediatric Adolescent AIDS Clinical Trials (IMPAACT) P1097 study examined washout elimination of raltegravir in LBW neonates exposed to raltegravir after maternal dosing during pregnancy. A modified integrated maternal-neonatal pharmacokinetic model was developed to characterize raltegravir clearance pharmacokinetics in preterm and full term neonates and to predict raltegravir plasma concentrations after postnatal dosing.
METHODS
Patients and sample collection
IMPAACT P1097 study was designed to describe the washout pharmacokinetics and safety of in utero/intrapartum exposure to raltegravir in neonates born to pregnant women living with HIV who received raltegravir-based antiretroviral therapy during pregnancy. Results from an initial cohort of full term neonates have been published previously; this report presents the data from a second cohort of LBW neonates.[13] Infants with a birth weight of < 2500 grams born to a mother who had received at least one dose of raltegravir within 2 to 24 hours prior to delivery were eligible for enrollment in version 2 of the protocol. Weight at the time of delivery was used as the enrollment criterion rather than gestational age (GA) because of the difficulty in accurate determination of gestational age in settings where routine ultrasounds are not available and registration for prenatal care is generally late. It was anticipated that some full term infants might be enrolled who were LBW ( < 2500 grams). The study was conducted at IMPAACT Network sites in Brazil, South Africa, USA, Tanzania and Thailand. Local institutional review boards approved the protocol at all participating sites, and signed informed consent was obtained from the mothers of all study subjects before participation. All procedures were performed in accordance with the ethical principles outlined in the 1964 Helsinki Declaration and its later amendments.
Pregnant women living with HIV who had received at least one dose of raltegravir 400 mg within 2 to 24 hours prior to delivery and their neonates were enrolled prior to delivery or up to 48 hours after birth. Maternal medical histories were abstracted from the medical record, and maternal plasma and cord blood for raltegravir assay were obtained at the time of delivery. Neonatal physical exams were performed shortly after birth and neonatal medical histories were obtained. Gestational age (GA) at birth was determined by obstetrical history and neonatal exam. Neonatal blood sampling for raltegravir assay was performed if birth weight was < 2500 g, no medications that might induce UGT1A1 activity were received, and no severe congenital anomaly or life-threatening medical condition was present. Serial neonatal plasma samples were collected at 1–6 hours, 12–24 hours, 36–48 hours, 72–84 hours, 108–132 hours and 7-14 days after birth. Infants could enroll up to 48 hours after birth and the collection window for any samples scheduled for collection prior to the time of enrollment was extended by 12 hours before the sample was skipped. Study neonates had blood drawn for complete blood counts and total and direct bilirubin at 36–48 hours after birth, liver transaminases, creatinine and bilirubin at 72-84 hours after birth, and complete blood counts, liver transaminases, creatinine and bilirubin at 1-2 weeks after birth. Dried blood spots were obtained from newborns for determination of UGT1A1 genetic polymorphisms. Infants were monitored until 6 weeks after birth for signs of raltegravir toxicity. The target sample size was 15 infants with collection of 3 postnatal pharmacokinetic samples for raltegravir assay.
Pharmacokinetic analysis
Raltegravir plasma concentrations were measured using a modified version of a previously published method.[14] A simple protein precipitation method using acetonitrile-containing raltegravir internal standard was employed to extract raltegravir from human plasma. The method was fully validated and used isocratic, reverse-phase high-performance liquid chromatography-tandem mass spectrometry on an AB SCIEX 5500. The linear calibration range was 10-10,000 ng/mL from a 10 μL plasma sample.
Descriptive statistics were determined for plasma raltegravir concentrations from maternal delivery, cord blood, and neonatal samples. Regression analysis was used to determine the neonatal raltegravir terminal elimination half-life (t1/2) with calculations performed in Excel. Following natural log transformation, the last 2 to 3 measured concentration-time points were used to determine the slope of the elimination curve to estimate the elimination rate constant. The t1/2 was calculated as ln2 divided by the elimination rate constant.
Genetic analysis
To assess genetic polymorphisms that might be associated with elevated bilirubin and raltegravir concentrations, genetic variants of UGT1A1 (rs5839491), the enzyme primarily responsible for bilirubin and raltegravir metabolism were evaluated.[15-17] Genotyping for UGT1A1 polymorphisms was performed on dried blood spots on filter paper that could be obtained with any blood sample if maternal consent was provided. Polymorphisms in UGT1A1 were determined by real-time PCR on DNA extracted from dried blood spots using QIAamp DNA Mini Kit (Qiagen, Valencia, CA). The median raltegravir clearance (CL/F) for the wild type [(TA)6/(TA)6 genotype] versus mutation [heterozygous (TA)6/(TA)7 and (TA)5/(TA)7 or homozygous (TA)5/(TA)5 and (TA)7/(TA)7 genotypes] UGT1A1 groups was analyzed using the Wilcoxon Rank Sum test as previously described.[13]
Safety evaluation
Infant safety data, including adverse birth outcomes, signs and symptoms, diagnoses and laboratory test results from evaluations specified in the protocol and additional evaluations done as part of the infant’s clinical care, were evaluated. In addition, the number and proportion of infants who received any therapy to treat elevated bilirubin were determined.
Pharmacokinetic modeling
An integrated maternal-neonatal population pharmacokinetic model was developed, as previously described [18]. For purposes of modeling, full term was defined as GA > 38 weeks; transitional GA > 35 weeks to < 38 weeks; preterm GA ≤ 35 weeks. To account for potential differences between full term and preterm neonates, a covariate term (COVCLτ) was added to the clearance maturation function (CL[t]) based on the GA:
Preterm (GA ≤ 35 weeks): COVCLτ = θ
Transitional (35 weeks < GA ≤ 38 weeks):
Full term (GA > 38 weeks): COVCLτ = 1
where CLmax is the maximum increase in apparent clearance from birth, CLτ is the first-order rate constant for the age-related changes in apparent clearance, and t is the age of the neonate in years. Age in years was used to facilitate combining data from this study with those from studies in older infants.
Key assumptions of the modified model are the following: (1) raltegravir pharmacokinetics for preterm infants can be described by solely reducing the clearance maturation rate. The allometric scaling parameters are assumed to be sufficient for adjusting the volumes of distribution and clearances to describe the pharmacokinetics of both preterm and full term LBW neonates. (2) The gastrointestinal tract for preterm infants follows similar development as full term infants. (3) The fetal and neonatal physiological development of preterm infants is not affected by in utero raltegravir exposure.
Simulation studies were carried out in R using the mgrsolve package (version 0.8.12) [19]. Growth of preterm neonates was based on FENTON growth curves (boys) [20] (https://www.ucalgary.ca/fenton/) and approximated by:
where WTbirth is the body weight at birth in kg and Age the age in years.
For the simulation study comparing the predicted plasma raltegravir concentrations for preterm and full term infants, the following FDA-approved dosing regimen for full term infants was used: 1.5 mg/kg once daily during the first week after birth; 3 mg/kg twice daily during weeks 2, 3 and 4; and 6 mg/kg twice daily after week 4. The body weight increase in full term infants was based on an empirically derived function [18]:
where WTbirth = 2.94 kg, WTmax = 9.0 kg, α = 1.07 year−1 and Age is the age in years. The preterm neonate was characterized to have a GA of 34 weeks with a weight at birth of 2.2 kg, as derived from the FENTON growth curve. Simulations were also preformed to predict pharmacokinetic profiles in two published case studies in which therapeutic drug monitoring was used to guide dosing of raltegravir in preterm infants.[21, 22]
SAS version 9.4 (SAS Institute, Cary, NC) was used for data set construction. Nonlinear Mixed Effects Modelling (ICON Inc., USA) was used for the pharmacokinetic modeling and simulations. R version 3.5.0 (R Foundation for Statistical Computing, Vienna, Austria) was used for post-processing and to generate graphs.
To determine goodness-of-fit, the model estimates were compared to the plasma raltegravir concentration data for the neonates for whom the precise maternal dosing information was available. Six plasma concentration measurements for LBW neonates were below the assay lower limit of quantification (LLOQ) and were imputed to 0.5*LLOQ = 11.25 nM.
RESULTS
Sixteen mothers living with HIV and their 18 infants with birth weight < 2500 grams (including 2 sets of twins) were enrolled in IMPAACT P1097 between January 2015 and March 2018. All mother-infant pairs enrolled after delivery. Infants were enrolled in Brazil (8 infants), South Africa (4 infants), USA (4 infants), Tanzania (1 infant) and Thailand (1 infant). There were 12 female and 6 male infants. For PK evaluable infants, the median (range) duration of maternal raltegravir treatment prior to delivery was 11.9 (0.4–309.0) weeks and the median (range) time between the last raltegravir dose and delivery was 6.5 (0.3–13.5) hours. Baseline characteristics of PK evaluable infants include median (range) birth weight of 2010 (1700–2485) grams and the median (range) gestational age at birth was 35.7 (32.7–40.0) weeks. (Table 1) All 18 neonates were included in the safety analysis.
Table 1.
Pharmacokinetic analysis data set
| PN | Population | nID | nObs | MinWeek | MaxWeek | Males | Females | MinBW | MaxBW | MinGA | MaxGA |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1097 | Neonates (LBW) | 17 | 84 | 0 | 1.9 | 6 | 11 | 1.7 | 2.4 | 32.7 | 40.0 |
| 1097 | Mothers (of LBW) | 16 | 6 | 0 | 17 | ||||||
| 1097 | Neonates | 19 | 75 | 0.01 | 0.2 | 14 | 5 | 2.2 | 4.1 | 37.1 | 40.0 |
| 1097 | Mothers | 19 | 19 | 0 | 19 | ||||||
| 1110 | Neonates | 50 | 522 | 0.03 | 6 | 27 | 23 | 2.1 | 4.2 | 37 | 41.3 |
| 1110 | Mothers | 14 | 0 | 0 | 14 | ||||||
| 1066 | Infants | 24 | 244 | 5.4 | 125 | 15 | 9 | NA | NA | NA | NA |
PN, study ID number; nID, number of subjects; nObs, number of pharmacokinetic samples; minWeek, first observation (weeks of age); maxWeek, last observation (weeks of age); BW, birth weight (kg); GA, gestational age (weeks), NA, data not available
Raltegravir washout pharmacokinetics could be determined for 17 neonates; one infant had an insufficient number of samples with a quantifiable raltegravir concentration to allow determination of washout elimination half-life. Raltegravir plasma concentrations (ng/mL) versus time from birth are shown in Figure 1: 1A mean (SE); 1B individual results for all 17 neonates. Included in Table 1S are the raltegravir concentrations obtained during the sampling windows: 1–6 hours (N = 5), 12–24 hours (N = 11), 36–48 hours (N = 16), 72–84 hours (N = 16), 108–132 hours (N = 16), and 7-14 days (N = 16). The median (range) terminal elimination t1/2 for raltegravir in these infants was 24.4 (10.1–83) hours. UGT1A1 genotyping was available for 16 infants, of whom 7 were wild type and 9 had genotypes associated with hyperbilirubinemia. No significant differences were seen when raltegravir concentrations or elimination t1/2 in the infants with the wild type genotype were compared to the infants with other genotypes. The highest grade of adverse events observed was Grade 3 in 6 infants and Grade 4 in 3 infants. No adverse events were considered related to raltegravir exposure. Serum total bilirubin levels were highest at 72–84 hours after birth with a median (range) of 10.7 (6.5–12.6) mg/dL. Four infants received phototherapy for treatment of elevated bilirubin levels per site standard of care.
Figure 1. Raltegravir elimination kinetics.
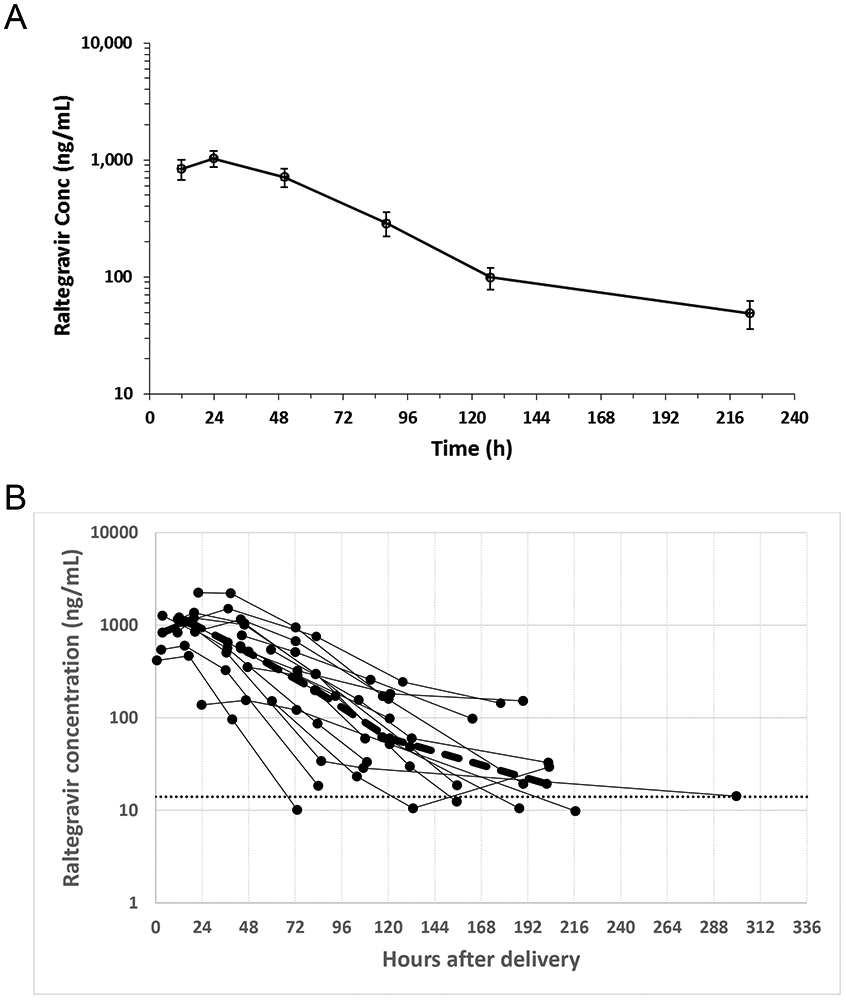
(A) Mean (SE) raltegravir plasma concentration (ng/mL) versus time from birth. The sampling time intervals are 1–6 hours (N = 5), 12–24 hours (N = 11), 36–48 hours (N = 16), 72–84 hours (N = 16), 108–132 hours (N = 16), and 7-14 days (N = 16). (B) raltegravir plasma concentration over time for individual low birth weight (LBW) neonates.
Population Pharmacokinetics Modeling and Simulations
We created an integrated maternal-neonatal model as described, incorporating the PK data from IMPAACT studies P1097 (full term and LBW neonates and mothers), P1110 (full term neonates and mothers), and P1066 (infants > 4 weeks of age and < 2 years of age) as shown in Table 1.[18, 23, 24] The analysis data set included 86 neonates, 24 infants, and 50 mothers, with a total of 681 pharmacokinetic samples for the neonates, 244 for the infants, and 25 for the mothers. Parameter estimates for the integrated maternal-neonatal pharmacokinetic model for raltegravir clearance are shown in Table 2S.The birth weights ranged from 1.7 to 4.2 kg and the GA ranged from 32.7 to 41.3 weeks. Using allometric scaling for body weight, we determined that the model accurately predicted raltegravir clearance for full term neonates that were LBW, but poorly predicted the clearance for preterm infants (Figure S1). Adding a covariate term to account for delayed clearance maturation in preterm infants greatly improved the overall fit. (Table 2S)
Figure 2 shows all LBW neonate comparisons of the model predictions to the observed raltegravir concentrations.
Figure 2. Comparison of the modified maternal-neonatal model results to empirical raltegravir clearance data from LBW neonates.

Shown are the raltegravir plasma concentrations for individual LBW neonates (blue symbols) superimposed on the integrated maternal-neonatal model predictions for the mother (green lines) and full term neonates (blue lines). GA, gestational age (weeks); BW, birth weight (kg). Preterm: ≤ 35 weeks; transitional preterm: 35 weeks < gestational age ≤ 38 weeks, full term: > 38 weeks.
We next performed simulations using the modified integrated maternal-neonatal model to examine the relationship between the model predictions and the observed plasma raltegravir concentrations in two published case reports describing RAL plasma concentrations after raltegravir dosing of preterm LBW infants using real-time raltegravir concentration assay results to guide timing of dosing. Figure 3A shows the raltegravir plasma concentrations in a neonate (GA: 34 weeks; birth weight: 1.91 kg) who received two oral doses (1.57 mg/kg) of raltegravir [22]. The timing of the second dose was based on the half-life calculated from plasma samples collected at 48 and 72 hours after the first dose. Figure 3B shows the raltegravir plasma concentrations in a second neonate (GA: 25 weeks; birth weight 0.800 kg) receiving recurrent dosing with raltegravir and whose dosing intervals in the first weeks of life were adjusted based on serial raltegravir concentration sampling [21]. For both cases, we also present the results of a simulation using the modified integrated maternal-neonatal model to predict the plasma raltegravir concentrations based on the published dosing procedures. The agreement between predicted and observed plasma raltegravir concentrations is high across the entire observation period for both cases. These results support the assertion that the modified integrated maternal-neonatal model can be extrapolated to predict the plasma raltegravir concentrations in response to postnatal oral dosing for preterm neonates up to 4-5 weeks of life.
Figure 3. Validation of the pharmacokinetic model with empirical data from case studies.

The predicted raltegravir concentrations from the pharmacokinetic model (blue lines) are shown in response to the dosing regimen described by (A) Kreutzwiser et al.[22] or (B) Trahan et al.[21] Case A: GA= 34 weeks; BW= 1.91 kg. Case B: GA= 25 weeks; BW= 0.800 kg. Red symbols indicate empirical data.
Figure 4 shows the results of a simulation using the modified integrated maternal-neonatal model to predict the expected plasma raltegravir concentrations in a 34 week gestation infant receiving the current dose regimen approved for full term neonates.[25] The predicted plasma concentrations for preterm infants exceed those predicted for full term infants by up to 2.5-fold.
Figure 4. Simulation of the response to neonatal raltegravir dosing.

The neonatal raltegravir dosing regimen approved for full term neonates (week 1: 1.5 mg/kg QD; weeks 2-4: 3 mg/kg BID; weeks 5-6: 6 mg/kg BID) was used in a simulation to predict the plasma raltegravir concentrations. Blue = full term; red = preterm.
DISCUSSION
Raltegravir elimination in preterm and LBW neonates is slow, with a median washout t1/2 of 24.4 hours. Washout elimination could be predicted in full term LBW neonates with allometric scaling of body weight. Accurate prediction of raltegravir elimination in preterm neonates required the addition of a covariate that reduced clearance associated with lower gestational age, which allowed the model to more accurately predict terminal raltegravir clearance in preterm neonates after maternal dosing prior to delivery. This modified integrated maternal-neonatal model accurately described the data from two case studies reporting postnatal raltegravir concentrations in the first month after birth in preterm infants exposed to raltegravir in utero. Furthermore, a simulation using the modified integrated maternal-neonatal model predicted that use of the full term dosing regimen in 34 week gestation infants would result in accumulation of raltegravir plasma concentrations to elevated and potentially dangerous levels. These differences in raltegravir clearance associated with prematurity are consistent with the described reduction in UGT1A1 activity in preterm infants compared to full term infants.[7]
Overall, raltegravir elimination kinetics in full term but LBW neonates exposed to raltegravir in utero are consistent with the results of a prior study in full term neonates with birth weight above 2000 grams.[13] As in that study, plasma raltegravir concentrations increased over the first 12-24 hours after delivery for about half of the neonates and then began to decline. This increase after removal from the source of additional exogenous raltegravir could be caused by redistribution of raltegravir among body compartments, as suggested by our mother-term infant population pharmacokinetic model [17]. Deconjugation of raltegravir glucuronide in the newborn gut and reabsorption of the resulting unconjugated raltegravir, known as enterohepatic recirculation and well described for bilirubin in newborns, may also contribute to the increases in raltegravir concentration observed after birth.[13, 18, 26].
High variability in raltegravir elimination kinetics has been observed in full term neonates, with a subset of neonates having very slow elimination.[13] A similar phenomenon was observed in this study, and complete elimination of raltegravir was not achieved for all neonates during the study period of at least 1 week. The mechanisms underlying incomplete elimination are not clear but could reflect low neonatal UGT1A1 activity combined with redistribution of in utero acquired drug and enterohepatic recirculation. The current study also found that the UGT1A1 genotype did not predict raltegravir clearance. Genetic variations are associated with UGT1A1 gene expression and bilirubin concentrations later in life.[27] Although our analysis may have been underpowered due to the small sample size, it also may be these genetic variations do not impact neonatal raltegravir clearance in premature neonates because of the low level of UGT1A1 enzyme activity in this population.
The slower clearance of raltegravir in preterm LBW neonates compared to full term neonates is similar to that reported for zidovudine.[2] Zidovudine is metabolized by another uridine diphosphate glucuronyl transferase isoenzyme, UGT2B7. In term infants, zidovudine clearance is reported to double during the first few days of life and quadruple over the first month of life, parallel to the increase in UGT1A1 activity and bilirubin metabolism.[7, 28] However, in premature infants, zidovudine clearance is lower at birth and matures at a slower rate than in term infants.[28] At week 5 after birth, the maturation of UGT2B7 in preterm infants is estimated to be about 50% of that observed in full term infants. Our current analysis indicates that the relative maturation of UGT1A1 metabolism of raltegravir in preterm infants is even slower and reaches only about 30% of the maturation observed for full term infants at week 5 after birth.
The main strength of this study is that it provides pharmacokinetic data on an understudied population, LBW full term and premature neonates, allowing us to build a pharmacokinetic model to predict raltegravir plasma concentrations. The multicenter study included neonates from five countries, minimizing bias in sample acquisition. The small sample size limits the generalizability of our findings. Mother-infant pairs could enroll up to 48 hours after birth so maternal plasma concentrations and cord blood samples at the time of delivery are not available for all study participants. Raltegravir was not delivered directly to the neonates in this study, limiting the ability to predict neonatal absorption of raltegravir. Despite these limitations, the model effectively predicted the plasma raltegravir concentrations observed in two case studies describing multiple weeks of oral raltegravir dosing in premature infants. While the pharmacokinetic modeling supports the assertion that differences in clearance maturation underlie the differences in raltegravir plasma concentrations between preterm and full term infants, we cannot rule out the possibility that other mechanisms may influence raltegravir clearance, including the presence of comorbidities in either the mother or the infant. Finally, our simulation predicts that administration of the raltegravir dosing regimen approved for use in full term neonates to 34-week gestation neonates could lead to accumulation of elevated and potentially toxic raltegravir plasma concentrations.
The results of our modelling and simulation suggest that it will be unsafe to dose raltegravir in preterm infants by simply scaling the full term infant neonatal raltegravir dose for their smaller body weights. Safe and effective use of raltegravir in preterm neonates requires a dosing regimen that incorporates an understanding of the impact of physiologic differences associated with prematurity on pharmacology to avoid exposure of the neonate to elevated and potentially toxic raltegravir plasma concentrations. A pharmacokinetic and safety study in preterm neonates must be conducted before raltegravir can safely be used in this population. Use of population pharmacokinetic modeling and simulations would enhance the safety and efficiency of this study.
Supplementary Material
ACKNOWLEDGEMENTS
Nancy Linford, PhD, (Synchrogenix, a Certara company) provided content development assistance.
FUNDING
Overall support for the International Maternal Pediatric Adolescent AIDS Clinical Trials Network (IMPAACT) was provided by the National Institute of Allergy and Infectious Diseases (NIAID) with co-funding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and the National Institute of Mental Health (NIMH), all components of the National Institutes of Health (NIH), under Award Numbers UM1AI068632 (IMPAACT LOC), UM1AI068616 (IMPAACT SDMC) and UM1AI106716 (IMPAACT LC), and by NICHD contract number HHSN275201800001I. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
REFERENCES
- 1.Panel on Treatment of HIV-Infected Pregnant Women and Prevention of Perinatal Transmission. Recommendations for Use of Antiretroviral Drugs in Pregnant HIV-1-Infected Women for Maternal Health and Interventions to Reduce Perinatal HIV Transmission in the United States. Available at http://aidsinfo.nih.gov/contentfiles/lvguidelines/PerinatalGL.pdf. Accessed February 9, 2020.
- 2.Capparelli EV, Mirochnick M, Dankner WM, et al. Pharmacokinetics and tolerance of zidovudine in preterm infants. J Pediatr 2003; 142(1): 47–52. [DOI] [PubMed] [Google Scholar]
- 3.Blencowe H, Krasevec J, de Onis M, et al. National, regional, and worldwide estimates of low birthweight in 2015, with trends from 2000: a systematic analysis. Lancet Glob Health 2019; 7(7): e849–e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao PL, Zhou YB, Chen Y, et al. Association between maternal HIV infection and low birth weight and prematurity: a meta-analysis of cohort studies. BMC Pregnancy Childbirth 2015; 15: 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gibango N, Mda S, Ntuli T. Factors associated with delivering premature and/or low birth weight infants among pregnant HIV-positive women on antiretroviral treatment at Dr George Mukhari Hospital, South Africa. Southern African Journal of Infectious Diseases 2018; 33(2): 42–5. [Google Scholar]
- 6.Darak S, Darak T, Kulkarni S, et al. Effect of highly active antiretroviral treatment (HAART) during pregnancy on pregnancy outcomes: experiences from a PMTCT program in western India. AIDS patient care and STDs 2013; 27(3): 163–70. [DOI] [PubMed] [Google Scholar]
- 7.Kawade N, Onishi S. The prenatal and postnatal development of UDP-glucuronyltransferase activity towards bilirubin and the effect of premature birth on this activity in the human liver. Biochem J 1981; 196(1): 257–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clarke DF, Mirochnick M, Acosta EP, et al. Use of Modeling and Simulations to Determine Raltegravir Dosing in Neonates: A Model for Safely and Efficiently Determining Appropriate Neonatal Dosing Regimens: IMPAACT P1110. Journal of Acquired Immune Deficiency Syndromes 2019; 82(4): 392–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clarke DF, Wong RJ, Wenning L, Stevenson DK, Mirochnick M. Raltegravir in vitro effect on bilirubin binding. Pediatr Infect Dis J 2013; 32(9): 978–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahlfors CE. Unbound bilirubin associated with kernicterus: a historical approach. J Pediatr 2000; 137(4): 540–4. [DOI] [PubMed] [Google Scholar]
- 11.Bhutani VK, Johnson-Hamerman L. The clinical syndrome of bilirubin-induced neurologic dysfunction. Semin Fetal Neonatal Med 2015; 20(1): 6–13. [DOI] [PubMed] [Google Scholar]
- 12.Bhutani VK, Wong RJ, Stevenson DK. Hyperbilirubinemia in Preterm Neonates. Clin Perinatol 2016; 43(2): 215–32. [DOI] [PubMed] [Google Scholar]
- 13.Clarke DF, Acosta EP, Rizk ML, et al. Raltegravir pharmacokinetics in neonates following maternal dosing. Journal of Acquired Immune Deficiency Syndromes 2014; 67(3): 310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Long MC, Bennetto-Hood C, Acosta EP. A sensitive HPLC-MS-MS method for the determination of raltegravir in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci 2008; 867(2): 165–71. [DOI] [PubMed] [Google Scholar]
- 15.Yang H, Wang Q, Zheng L, et al. Multiple Genetic Modifiers of Bilirubin Metabolism Involvement in Significant Neonatal Hyperbilirubinemia in Patients of Chinese Descent. PLoS One 2015; 10(7): e0132034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alencastro de Azevedo L, Reverbel da Silveira T, Carvalho CG, Martins de Castro S, Giugliani R, Matte U. UGT1A1, SLCO1B1, and SLCO1B3 polymorphisms vs. neonatal hyperbilirubinemia: is there an association? Pediatr Res 2012; 72(2): 169–73. [DOI] [PubMed] [Google Scholar]
- 17.Yueh MF, Chen S, Nguyen N, Tukey RH. Developmental, Genetic, Dietary, and Xenobiotic Influences on Neonatal Hyperbilirubinemia. Mol Pharmacol 2017; 91(5): 545–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lommerse J, Clarke D, Kerbusch T, et al. Maternal-Neonatal Raltegravir Population Pharmacokinetics Modeling: Implications for Initial Neonatal Dosing. CPT Pharmacometrics Syst Pharmacol 2019; 8(9): 643–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing, 2017. [Google Scholar]
- 20.Fenton TR, Kim JH. A systematic review and meta-analysis to revise the Fenton growth chart for preterm infants. BMC Pediatr 2013; 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trahan MJ, Lamarre V, Metras ME, Kakkar F. Use of Triple Combination Antiretroviral Therapy With Raltegravir as Empiric HIV Therapy in the High-risk HIV-exposed Newborn. Pediatr Infect Dis J 2019; 38(4): 410–2. [DOI] [PubMed] [Google Scholar]
- 22.Kreutzwiser D, Sheehan N, Dayneka N, et al. Therapeutic drug monitoring guided raltegravir dosing for prevention of vertical transmission in a premature neonate born to a woman living with perinatally acquired HIV. Antiviral Therapy 2017; 22(6): 545–9. [DOI] [PubMed] [Google Scholar]
- 23.Nachman S, Alvero C, Acosta EP, et al. Pharmacokinetics and 48-Week Safety and Efficacy of Raltegravir for Oral Suspension in Human Immunodeficiency Virus Type-1-Infected Children 4 Weeks to 2 Years of Age. Journal of the Pediatric Infectious Diseases Society 2015; 4(4): e76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clarke DF, Acosta EP, Cababasay M, et al. Raltegravir (RAL) in neonates: Dosing, pharmacokinetics (PK), and safety in HIV-1 exposed neonates at risk of infection (IMPAACT P1110). Journal of Acquired Immune Deficiency Syndromes 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Isentress package insert. Merck Sharp & Dohme Corp. Whitehouse Station, NJ 08889, USA. January 2019. [Google Scholar]
- 26.Dennery PA, Seidman DS, Stevenson DK. Neonatal hyperbilirubinemia. The New England journal of medicine 2001; 344(8): 581–90. [DOI] [PubMed] [Google Scholar]
- 27.Miners JO, McKinnon RA, Mackenzie PI. Genetic polymorphisms of UDP-glucuronosyltransferases and their functional significance. Toxicology 2002; 181-182: 453–6. [DOI] [PubMed] [Google Scholar]
- 28.Mirochnick M, Capparelli E, Connor J. Pharmacokinetics of zidovudine in infants: a population analysis across studies. Clin Pharmacol Ther 1999; 66(1): 16–24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.