

Abstract
Background:
Sickle cell disease (SCD) is characterized by chronic hemolytic anemia, vaso-occlusive crises, chronic inflammation, and activation of coagulation. The clinical complications such as painful crisis, stroke, pulmonary hypertension, nephropathy and venous thromboembolism lead to cumulative organ damage and premature death. High molecular weight kininogen (HK) is a central cofactor for the kallikrein-kinin and intrinsic coagulation pathways, which contributes to both coagulation and inflammation.
Objective:
We hypothesize that HK contributes to the hypercoagulable and pro-inflammatory state that causes end-organ damage and early mortality in sickle mice.
Methods:
We evaluated the role of HK in the Townes mouse model of SCD.
Results/Conclusions:
We found elevated plasma levels of cleaved HK in sickle patients compared to healthy controls, suggesting ongoing HK activation in SCD. We used bone marrow transplantation to generate wild type and sickle cell mice on a HK-deficient background. We found that short-term HK deficiency attenuated thrombin generation and inflammation in sickle mice at steady state, which was independent of bradykinin signaling. Moreover, long-term HK deficiency attenuates kidney injury, reduces chronic inflammation, and ultimately improves survival of sickle mice.
Keywords: anemia, sickle cell, blood coagulation, inflammation, kidney disease, kininogen, high molecular weight
1 |. INTRODUCTION
Recent advances in preventive care, such as hydroxyurea and prophylactic antibiotics, have reduced the mortality of sickle cell disease (SCD) patients in developed countries. Yet, the continual hemolytic anemia and recurrent vaso-occlusive crises result in chronic systemic inflammation and coagulopathy. Markers of coagulation activation correlate with painful crises, acute chest syndrome, stroke, venous thromboembolism, pulmonary hypertension, left ventricular diastolic heart disease, and sickle nephropathy.1,2 These complications result in end-organ failure that results in increased morbidity and mortality in SCD. We and others have observed tissue factor (TF)-dependent activation of coagulation in sickle cell patients and mouse models,3–7 and reported that this pathway contributes to inflammation and end-organ damage in sickle mice.3,8–13
High molecular weight kininogen (HK) circulates in complex with factor XI (FXI) or prekallikrein (PK) and serves as a non-enzymatic cofactor for activation of both proteins by factor XIIa (FXIIa). HK is a 120-kD α-globulin comprised of six domains. Domains 1 to 3 are known to inhibit atrial natriuretic factor (ANF), calpain, and papain, respectively. Domain 4 contains the bradykinin (BK) peptide. Domain 5 binds to cells via interactions with CD11b/CD18 (Mac-1),14 urokinase plasminogen activator receptor (uPAR), cytokeratin 1, globular C1q receptor (gC1qR), and heparin sulfate,15–18 and domain 6 binds FXI and kallikrein.19 Kallikrein-mediated cleavage of HK results in the generation of cleaved HK (cHK) and the release of BK, a nine amino acid peptide. BK is a potent inflammatory mediator and vasodilator that exerts its biological effects through one of two G-protein coupled receptors, BK receptor 1 and 2 (B1R and B2R). B1R is inducible upon tissue injury and its activation promotes inflammation via activation of NF-κB.20 B2R is constitutively expressed and is responsible for smooth muscle relaxation and vasodilation.20 Fragments of cHK are connected via a disulfide bond between D1 and D6 of HK.21 cHK binds Mac-1 on monocytes via D5, resulting in cytokine release that induces TF expression and activity.22
High molecular weight kininogen deficient mice have a prolonged activated partial thromboplastin time (aPTT) and time to thrombosis in the Rose Bengal carotid artery thrombosis model.23,24 In the model of ischemic stroke, HK deficiency reduced the brain infarct size, intracerebral thrombosis, inflammation, and blood-brain barrier disruption, and significantly prolonged survival after stroke.25 Compared to other components of the contact pathway, HK deficiency offers more robust protection from cerebral ischemia, probably due to the central role of HK in multiple pathways including inflammation, thrombosis, and vascular permeability.26,27
Decreased levels of FXII, PK, and HK zymogens have been reported in the plasma of sickle cell patients compared to healthy controls, indicating an increased consumption of these factors due to activation of the contact pathway.28 Because HK plays a central role for both the intrinsic coagulation and contact pathways, we hypothesized that HK activation may contribute to the hypercoagulable state, chronic inflammation, and organ damage in SCD.
2 |. METHODS
2.1 |. Patient samples
The study was approved by the University of North Carolina’s Institutional Review Board for human subjects. We recruited 54 African American outpatients with SCD and 23 healthy African American controls. Inclusion criteria were that patients were in steady state (at least 1 month from last pain crisis); had not received blood products within 3 months; and were not on oral contraceptive, anticoagulant, or antiplatelet therapy. Blood samples were obtained by clean venipuncture using a 21G butterfly needle and the first 3 mL were discarded. Platelet-poor plasma was prepared from blood drawn into 3.2% sodium citrate (9:1), and frozen at −80°C until analysis.
2.2 |. Detection of cleaved HK in patient samples
To detect changes in HK and cHK in human plasma using a sandwich enzyme-linked immunosorbent assay (ELISA), we used a capture antibody able to detect both HK and cHK (3E8), and two detection antibodies specific for intact HK (2B7) or cHK (4B12) as previously described.29 Antibodies were biotinylated using EZ-Link® Sulfo-NHS-LC-Biotin (Thermo Scientific) according to the manufacturer’s instructions. Nunc Maxisorp plates were coated with capture antibody 3E8 (100 ng/well) in sodium bicarbonate buffer (eBiosciences) overnight at 4°C. Plasma was thawed at 37°C for 8 minutes prior to analysis of cHK and intact HK (iHK); the samples had not previously undergone any freeze-thaw cycles. Plasma samples were diluted at 1:500 and 1:1000 using Hank’s balanced salt solution (Gibco) with 1% bovine serum albumin (BSA) prior to loading, and incubated for 60 minutes at room temperature (RT). After washing, biotinylated 2B7 or 4B12 were added to replicate wells to detect iHK or cHK, respectively, and incubated for 60 minutes at RT. After washing, horseradish peroxidase-conjugated streptavidin (R&D Systems) was added to all wells according to manufacturer specifications. After a final wash, color was developed by adding 3,3′,5,5′-tetramethylbenzidine (TMB) substrate for 30 minutes at RT, and the reaction was stopped with 0.2 mol/L sulfuric acid. Results were measured by absorbance at 450 nm on a plate reader (Biotek Synergy H1).
2.3 |. Mouse models
The Townes knock-in model of SCD (hα/hα, hγ/ hγ, hβA/hβS) was used for these studies.30,31 Mice were bred at the University of North Carolina by intercrossing HbAS parents to generate either HbSS sickle mice (SS) or HbAA wild type (AA) controls that exclusively express the human globins α, γ, and either βS (HbSS) or βA (HbAA). Hemoglobin electrophoresis was used to phenotype AA and SS mice (Helena Laboratories). HK−/− mice were generated as previously described on a C57 BL/6J background.23 To create chimeras, bone marrow cells were isolated from the femurs and tibias of 2-month-old AA and SS mice. Approximately 5 × 106 donor cells were transplanted via retro-orbital injection into lethally irradiated 2-month-old HK−/− or C57 BL/6J control recipients. In a separate study, bone marrow was transplanted into mice deficient in both B1R and B2R (Bdkrb1−/−/Bdkrb2−/−, referred to as B1RB2R-null) and their littermate controls (B1RB2R-wt).32 Donor chimerism for human HbAA or HbSS was confirmed by hemoglobin electrophoresis. In the short-term study, mice were aged for 4 months after bone marrow transplantation (BMT), and in the long-term study mice were aged for 8 months after BMT. All studies were in accordance with the University of North Carolina Animal Care and Use Committee.
At the endpoints of the studies, mice were anesthetized with isoflurane and whole blood was collected from the inferior vena cava (IVC) into syringes containing 3.8% sodium citrate (final blood to citrate ratio was 9:1). Platelet-poor plasma was collected by centrifugation (4000 × g, 15 minutes), aliquoted, and stored at −80°C until use. Organs were collected, weighed, and fixed for immunohistochemistry.
2.4 |. Hematologic assessment
Complete blood count was determined in whole blood by Hemavet HV950 analyzer (Drew Scientific) or an Element HT5 veterinary complete blood count analyzer (Heska).
2.5 |. Plasma and urine analysis
Platelet-poor plasma was used for analysis of thrombin-antithrombin (TAT) complexes (Siemens Diagnostica), interleukin-6 (IL-6; R&D Systems), and soluble vascular cell adhesion molecular (sVCAM-1; R&D Systems). Urine was collected 240 days after BMT for 24 hours in metabolic cages (Hatteras Instruments). Urine analysis was performed as previously described.33
2.6 |. Echocardiography analysis
Echocardiography was performed 245 days after BMT using a Vevo 2100 ultrasonographic system (VisualSonics). The left ventricle (LV) function was measured by M-mode echocardiography on the long-axis parasternal plane at the mid-ventricular level. LV volume (LV Vol), LV internal diameter (LVID), and interventricular septum thickness (IVS) were measured at diastole and systole, and the ejection fraction (EF%) was calculated from the measured dimensions.
2.7 |. Histopathologic analysis
Livers and kidneys were fixed in 10% formalin and embedded in paraffin. Kidneys were sectioned (5 micron), stained with Masson’s Trichrome, periodic acid Schiff-hematoxylin, and Prussian blue using standard protocols and analyzed by a blinded pathologist (MK) for glomerulosclerosis, mesangial expansion, glomerular hypertrophy, interstitial fibrosis, iron accumulation, and brush border loss as previously described.33 For immunohistochemistry assay, kidney sections were stained for Wilms’ tumor 1 (WT-1) with anti-Wilms’ tumor protein antibody (CAN-R9 [IHC]-56–2; ab89901, 1:600, Abcam) and macrophages infiltration (F4/80) with anti-F4/80 antibody (MCA497GA, 1;200; BioRad) in 2.5% goat serum (Vector Laboratories). Cortical renal sections were blindly quantified (MK) using 10 microscopic fields under 200× magnification using QuPath v0.2.0 and ImageJ version 1.52 software. Livers were sectioned (5 micron), stained with hematoxylin and eosin, and scored by two blinded pathologists (ES and MH; Appendix S1 in supporting information).
2.8 |. Statistics
Clinical sample data are presented as a scatter plot of median ± standard deviation (SD). Linear regression analysis was used to determine the relationship between cHK/iHK levels and markers of anemia in sickle patients. Mouse data are presented as mean ± standard error of the mean (SEM). Significance was assessed by two-way analysis of variance (ANOVA) with Tukey post-test for parametric data, and by the Mann-Whitney test for non-parametric data. Difference in survival was assessed by the Mantel-Cox and Gehan-Breslow-Wilcoxon tests. We considered P < .05 as statistically significant.
3 |. RESULTS
3.1 |. Cleavage of kininogen is increased in sickle cell patients
Participant demographic and laboratory values indicate that African American controls and SCD patients had similar age and gender distribution (Table 1). Sickle cell patients had significantly lower hematocrit and hemoglobin levels than controls, as well as increased platelet counts and white blood cell counts (Table 1). Previous studies reported a decrease in levels of zymogens FXII, PK, and HK in sickle patients;28,34 however, this is not a direct measure of accelerated HK turnover. Therefore, we determined levels of cHK in the plasma samples of sickle patients and healthy controls by ELISA as previously described.29 We found that the levels of cHK were significantly increased in sickle cell patient samples (Figure 1A), whereas levels of total intact HK (iHK) were not different between the two groups (Figure 1B). Moreover, the ratio of cHK/iHK was also elevated in SCD patient plasma (Figure 1C). Linear regression analysis between cHK/iHK with hematocrit did not reveal a relationship between these parameters (Table 1). We also compared the mean cHK/iHK ratios of sickle patients with and without a history of venous thromboembolism (VTE), and again, no difference between these two groups was apparent (data not shown). A table listing the cHK/iHK level for each patient, along with hydroxyurea treatment status and common organ involvement is included in Table S1 in supporting information.
TABLE 1.
Demographic and descriptive data of patient samples
| Parameter | HbAA (n = 23) | HbSS (n = 54) |
|---|---|---|
| Age: Median (range) | 28 (19–54) | 33.3 (19.1–59.4) |
| Gender | 16 female, 7 male | 28 female, 26 male |
| Hematocrit (%) | 39 (34–45) | 27.5 (19.5–34.4)**** |
| Hgb (g/dL) | 13 (10.8–14.8) | 9.4 (6.5–11.5)**** |
| MCV | 89 (80.8–97) | 98.5 (69–122)* |
| Platelet (109/µL) | 253 (167–352) | 329 (191–695)** |
| White blood cells (103/µL) | 5.4 (3.2–8.5) | 8.75 (7.1–11.9)**** |
| HCT: cHK/HK linear regression | N/A | Slope: −0.027, R2: .034, P = .22 |
Abbreviations: cHK, cleaved HK; HCT, hematocrit; Hgb, hemoglobin; HK, high molecular weight kininogen; MCV, mean corpuscular volume.
P < .05.
P < .01.
P < .0001 versus control samples via unpaired Student’s t-test.
FIGURE 1.
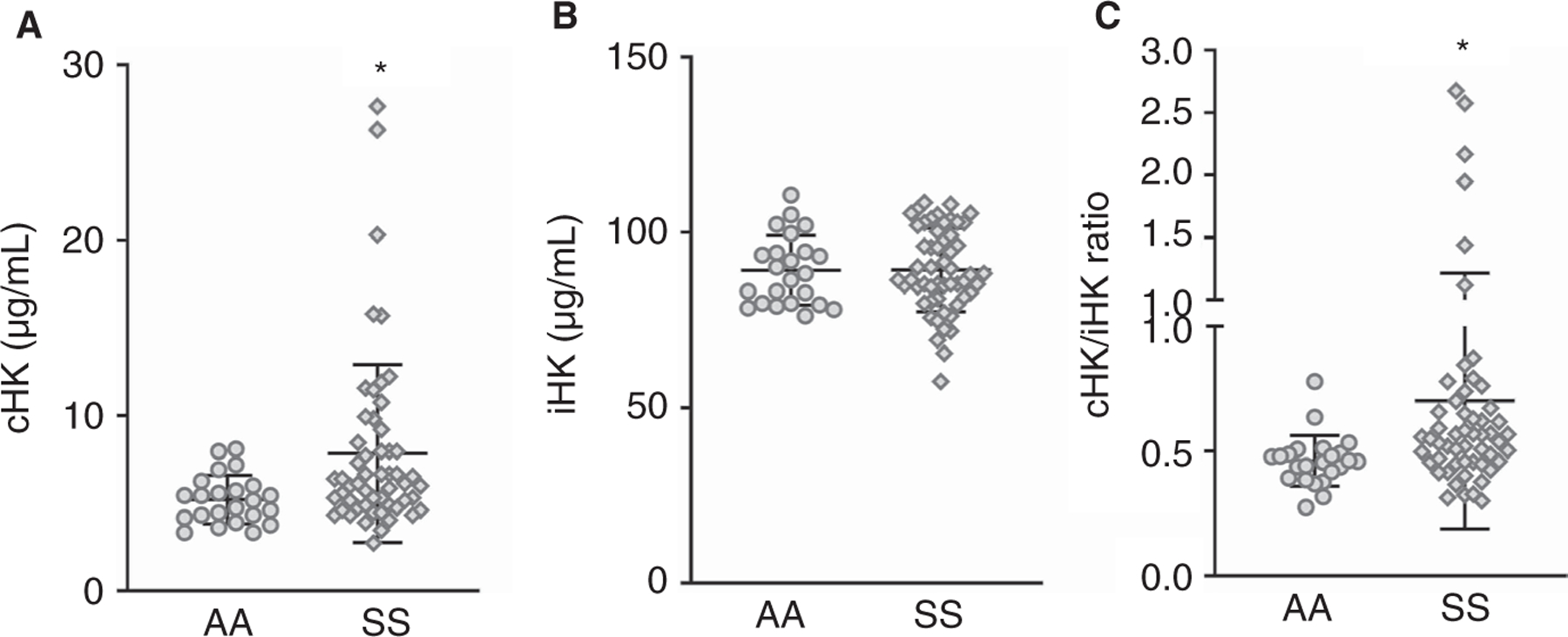
Levels of cleaved high molecular weight kininogen (HK) are higher in sickle patients than in healthy controls. Plasma from HbAA (n = 23) and HbSS (n = 54) patients was analyzed for (A) cleaved (cHK) and (B) intact HK (iHK). C, The ratio of cHK/iHK was also calculated. Data are presented as mean ± standard deviation. *P < .05 versus HbAA by Mann-Whitney test
3.2 |. HK deficiency attenuates coagulation and inflammation in sickle cell mice
To investigate the role of HK in sickle pathology, bone marrow from Townes AA and SS mice was transplanted into lethally irradiated WT (C57 BL/6J) and HK−/− mice. Four months after transplantation, plasma samples were evaluated for biomarkers of thrombin generation (TAT complexes), inflammation (Il-6), and endothelial cell activation (sVCAM). The elevated levels of both TAT and IL-6 in SS/WT mice were significantly lower in HK−/− sickle chimeras (SS/HK−/−; Figure 2A and B). In contrast, HK deficiency did not reduce the increased plasma levels of sVCAM in sickle mice (Figure 2C). Similarly, HK deficiency had no effect on anemia or leukocytosis observed in SS mice (Table 2). To ensure that hematopoietic cells do not produce HK in HK−/− chimeras, we performed western blot for HK in the plasma of all groups of mice. HK was not detected in the plasma of AA/HK−/− and SS/HK−/− mice (Figure S1 in supporting information).
FIGURE 2.
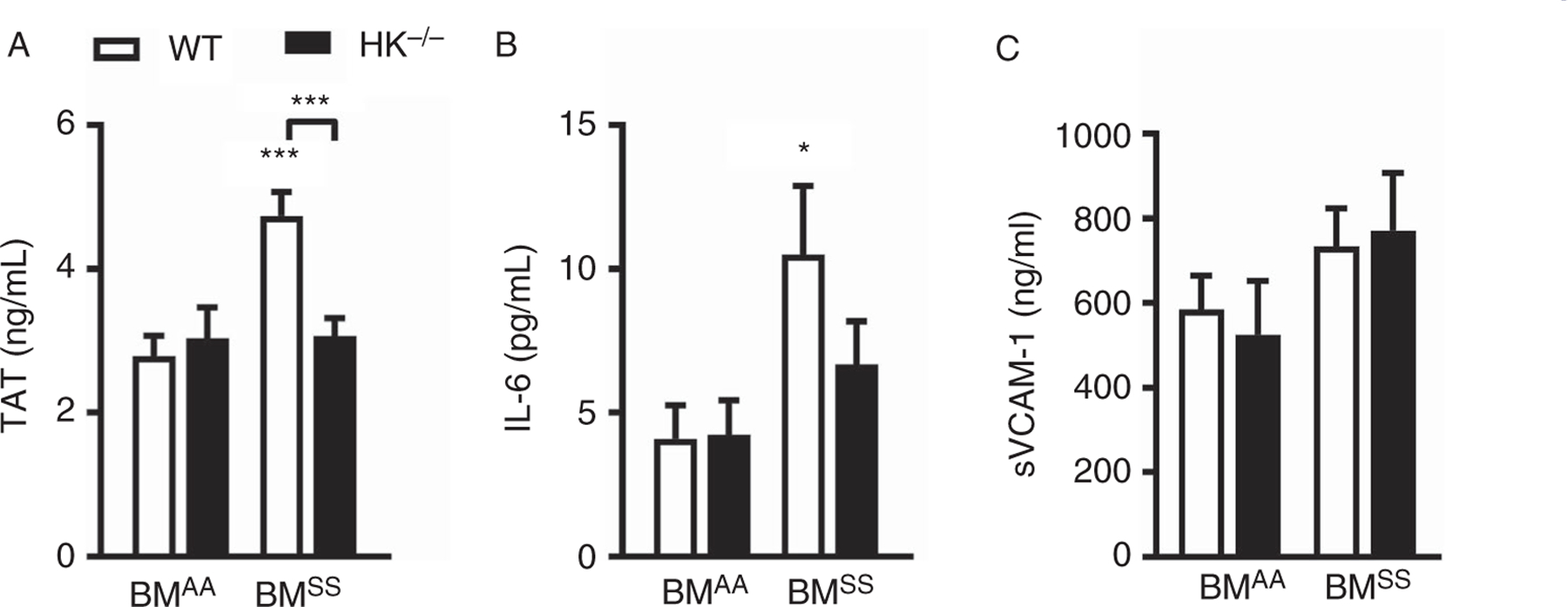
High molecular weight kininogen (HK) contributes to thrombin generation and inflammation in sickle cell disease. Wild type (WT) and HK−/− mice were lethally irradiated and transplanted with bone marrow from AA (n = 15–22) and SS mice (n = 25–28). Four months later plasma was collected for analysis of (A) thrombin-antithrombin (TAT), (B) interleukin 6 (IL-6), and (C) soluble vascular cell adhesion molecular (sVCAM). Data are presented as mean ± standard error of the mean and were analyzed by two-way analysis of variance and Tukey’s multiple comparison post-hoc analysis. *P < .05 and ***P < .001 versus AA/WT. Asterisks above lines indicate significance between SS/HK−/− and SS/WT
TABLE 2.
CBC analysis in WT and HK−/− sickle mice
| Parameter | AA/WT | AA/HK−/− | SS/WT | SS/HK−/− |
|---|---|---|---|---|
| RBC × 106/µL | 9.31 ± 0.19 | 9.73 ± 0.13 | 5.79 ± 0.11*** | 5.89 ± 0.17*** |
| MCV | 36.56 ± 0.99 | 38.17 ± 1.27 | 50.88 ± 0.34*** | 51.28 ± 0.59*** |
| Hemoglobin (g/dL) | 9.73 ± 0.26 | 9.73 ± 0.13 | 7.11 ± 0.18*** | 7.28 ± 0.24*** |
| Hematocrit (%) | 35.27 ± 0.68 | 37.07 ± 1.07 | 29.46 ± 0.56*** | 30.02 ± 0.94*** |
| WBC × 103/µL | 15.54 ± 2.21 | 11.36 ± 0.86 | 33.22 ± 1.67**** | 31.09 ± 2.72**** |
| Neutrophil × 103/µL | 2.82 ± 0.59 | 1.27 ± 0.14* | 4.96 ± 0.46* | 4.13 ± 0.59** |
| Lymphocyte × 103/µL | 9.16 ± 0.50 | 8.59 ± 0.73 | 24.67 ± 1.25**** | 26.06 ± 2.06**** |
| Monocyte × 103/µL | 1.03 ± 0.18 | 0.53 ± 0.04 | 1.99 ± 0.18* | 1.94 ± 0.20* |
Abbreviations: CBC, complete blood count; Hgb, hemoglobin; HK, high molecular weight kininogen; MCV, mean corpuscular volume; RBC, red blood cell; WBC, white blood cell; WT, wild type.
P < .05.
P < .01.
P < .001.
P < .0001 vs AA/WT or AA/HK−/− by two-way analysis of variance.
3.3 |. The protective effect of HK deficiency is not mediated by BK signaling
We hypothesized that the reduction in thrombin generation and inflammation in sickle mice deficient in HK was due to absence of BK-mediated activation of the B1R and B2R receptors. To test this hypothesis, we investigated the effect of deficiency of both BK receptors on thrombin generation and inflammation in sickle mice. Interestingly, plasma levels of TAT and IL-6 were not different between SS/B1RB2R-WT and SS/B1RB2R-null mice (Figure S2 in supporting information), indicating that HK-dependent thrombin generation and inflammation are not mediated by BK signaling in sickle cell mice. Expression of BK receptors also did not affect anemia or leukocytosis in SS mice (Table S2 in supporting information).
3.4 |. Kininogen contributes to early mortality of sickle mice
Because HK deficiency attenuated plasma TAT and IL-6 levels in a short-term experiment, we hypothesized that long-term attenuation of coagulation and inflammation might reduce organ dysfunction and early mortality, as previously shown.8,10 To test this hypothesis, we generated SS/WT and SS/HK−/− chimeras and tracked survival for 8 months. SS/HK−/− showed significantly improved survival compared with SS/WT mice (Figure 3A). Long-term HK deficiency had no effect on anemia or leukocytosis in sickle mice (Table S3 in supporting information). We collected urine, blood, and organs from the mice that survived 8 months (8 of 27 SS/WT and 17 of 21 SS/HK−/−) to evaluate markers of inflammation and organ function. SS/WT mice had an increase in the neutrophil-lymphocyte ratio (Figure 3B), which was attenuated in SS/HK−/− chimeras, indicating that HK contributes to chronic inflammation. Despite a modest reduction in liver hypertrophy, no significant differences were observed in this organ between SS/WT and SS/HK−/− mice with respect to the severity of liver injury, inflammatory infiltrates, or sinusoidal congestion (Figure S3 in supporting information).
FIGURE 3.
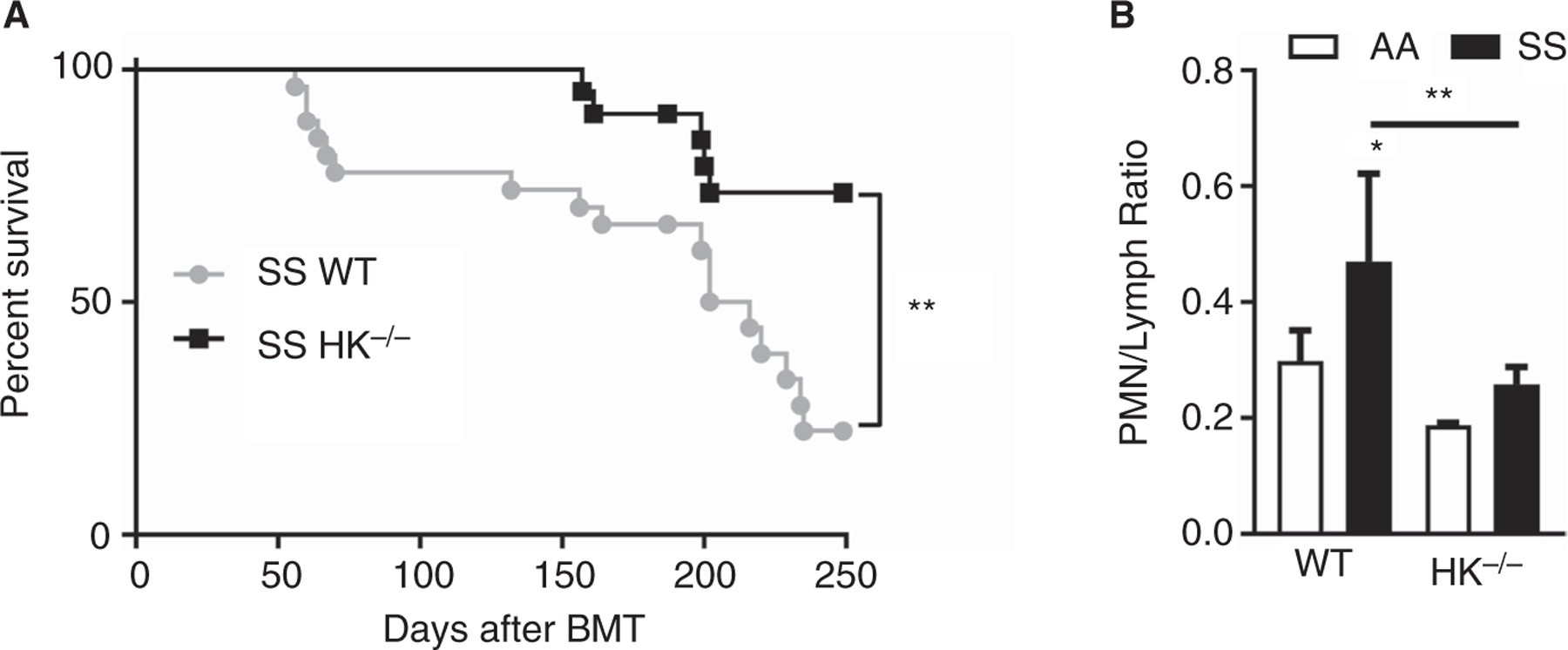
Long-term high molecular weight kininogen (HK) deficiency prolongs survival and reduces inflammation in sickle mice. Two-month-old wild type (WT) and HK−/− mice were lethally irradiated and transplanted with bone marrow from AA and SS mice. A, Kaplan-Meier survival curve of SS/WT (gray, n = 27 at day 0) and SS/HK−/− (blue, n = 21 at day 0) mice after bone marrow transplantation, and analyzed by Mantel-Cox test, with a significant difference in survival of **P < .01. After 8 months, blood and was collected for analysis of (B) neutrophil-lymphocyte ratio. *P < .05 and **P < .01 versus AA/WT; asterisks above lines indicate difference from SS/WT
Echocardiography revealed that LV mass was increased in both SS/WT and SS/HK−/− mice (Figure 4A), although there was no change in ejection fraction (Figure 4B). Moreover, LV volume was significantly increased in both SS/WT and SS/HK−/− mice at both diastole and systole (Figure 4C and D). The LV internal diameter was increased at diastole (Figure 4E) but not systole (Figure 4F) in both SS/ WT and SS/HK−/− mice. However, the interventricular septum thickness was significantly increased in SS/WT but not in SS/HK−/− mice compared to relevant AA controls (Figure 4G and H). Overall, these data indicate that HK deficiency had no effect on pathologic heart remodeling observed in sickle mice.
Figure 4:
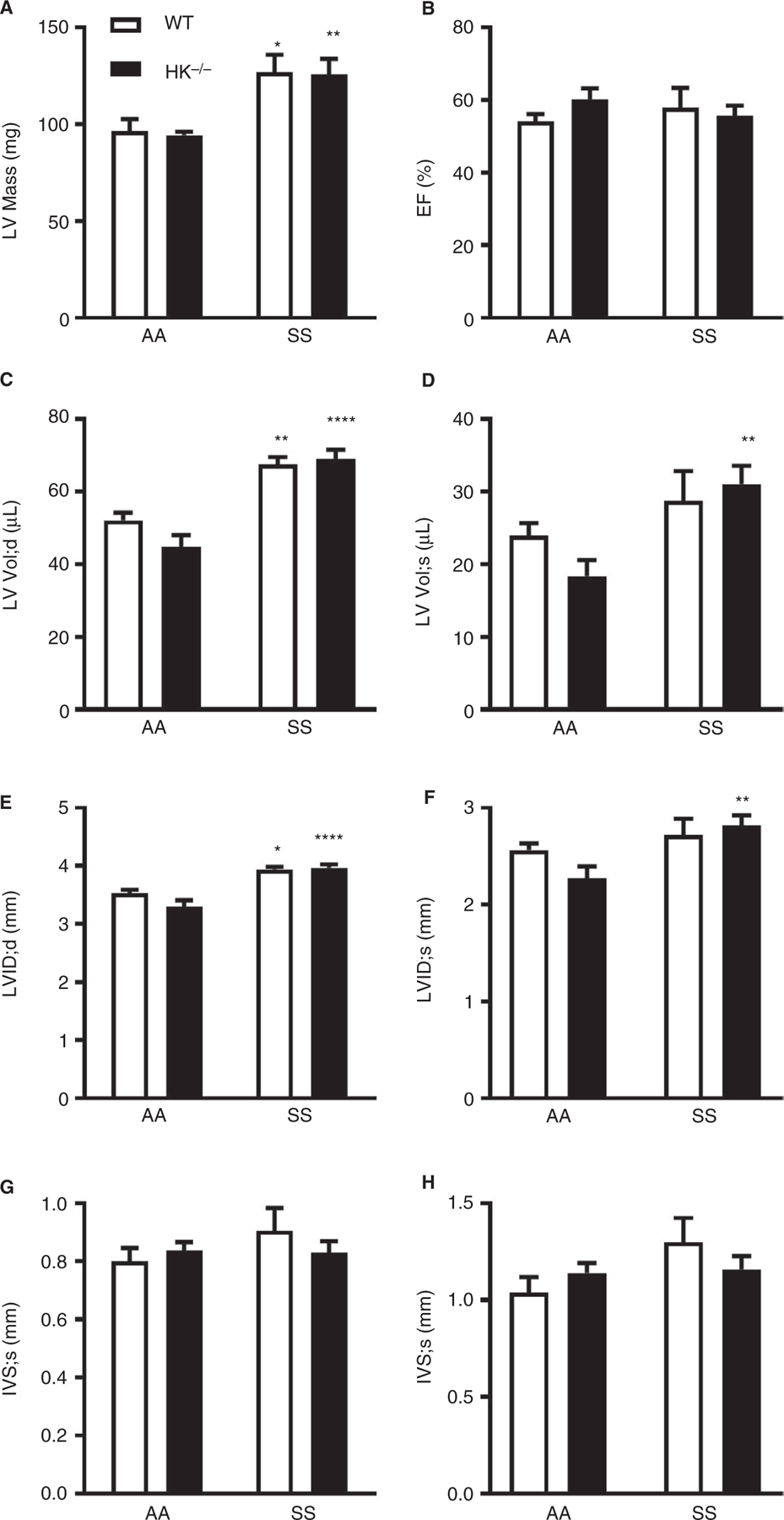
Sickle mice develop mild cardiovascular dysfunction. Echocardiography of the LV was performed to evaluate (A) LV Mass, (B) ejection fraction, LV volume at (C) diastole and (D) systole, LV internal diameter at (E) diastole and (F) systole, and IVS distance at (G) diastole and (H) systole. Data are presented as mean ± SEM and analyzed by Two-way ANOVA with post-hoc analysis by Tukey’s multiple comparison test. *p<0.05, **p<0.01, and ***p<0.001 versus AA/WT or AA/HK−/−.
3.5 |. Kininogen deficiency significantly attenuates sickle nephropathy
To determine if HK contributes to glomerulopathy in sickle mice, we evaluated albuminuria, a marker of glomerular injury, as well as renal histology. Compared to AA/WT controls, SS/WT mice did not exhibit kidney hypertrophy (Figure 5A), but had albuminuria (Figure 5B) that was prevented by HK deficiency. Histological analysis of glomeruli confirmed significant glomerular hypertrophy (Figure 5C), glomerulosclerosis (Figure 5D and E), mesangial expansion (Figure 5F and G), and loss of podocyte density (Figure 5H and I) in SS/WT mice compared to AA/WT controls. Preserved podocyte density (Figure 5H and I) and markedly reduced glomerular sclerosis (Figure 5D and E) was observed in SS/HK−/− mice.
FIGURE 5.
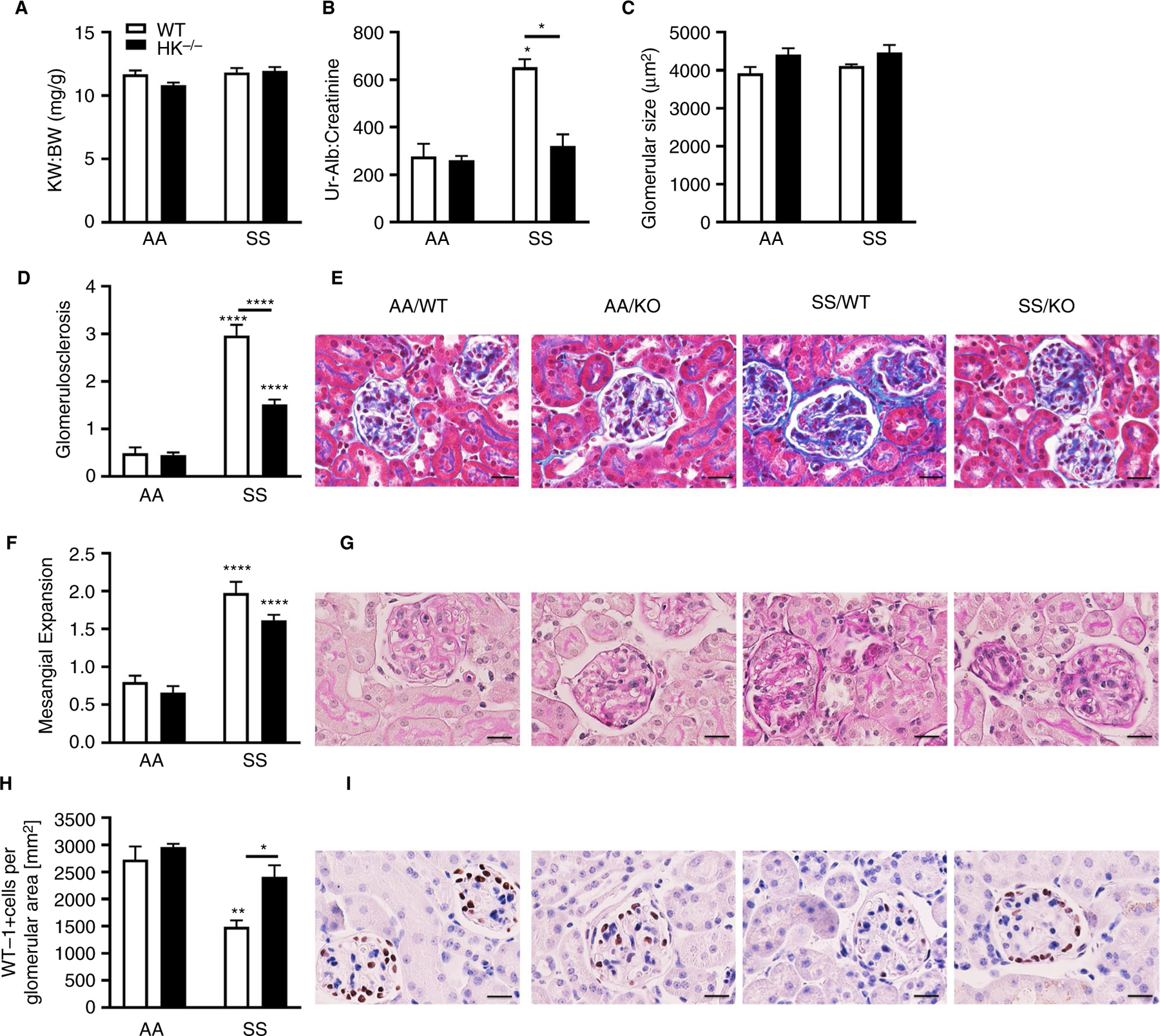
Long-term high molecular weight kininogen (HK) deficiency protects sickle mice from glomerular injury. Kidneys were collected and weighed, and (A) the ratio of kidney to body weight was calculated. Urine was collected for 24 hours and analyzed for (B) albumin normalized to creatinine. Data are presented as mean ± standard error of the mean (SEM) and analyzed by two-way analysis of variance and Tukey’s multiple comparisons post hoc analysis. Formalin-fixed and paraffin-embedded kidneys were analyzed for (C) glomerular size, presented as mean area of glomeruli (µm2) ± SEM. D, Quantification of glomerulosclerosis represented as sclerosis index score and (E) representative Masson trichrome-stained sections of glomeruli from all four groups of mice. Original magnification ×120; scale bar = 50 µm. F, Quantification of mesangial expansion score and (G) representative periodic acid Schiff-hematoxylin-stained sections of kidneys from all four groups of mice. Original magnification ×120; scale bar = 50 µm. H, Quantification of WT-1+-stained glomerular sections, represented as number of WT-1+-stained cells per glomerular area and (I) representative WT-1 staining from all four groups of mice. Original magnification ×120; scale bars = 50 µm. All data are presented as mean ± SEM. Data were analyzed by two-way analysis of variance; *P < .05, **P < .01, ****P < .0001 versus AA/WT. Asterisks above lines indicate difference from SS/WT
We also evaluated whether HK contributes to tubular dysfunction and injury. Indeed, SS/WT mice had hyposthenuria (Figure 6A) and elevated urinary levels of kidney injury marker-1 (KIM-1) compared to AA/WT mice (Figure 6B). Immunohistochemistry revealed increased interstitial fibrosis (Figure 6C and D), infiltration of F4/80 positive cells (Figure 6E and F), decreased brush border thickness (Figure 6G and H), and extensive tubular iron accumulation (Figure 6I and J). Urinary (Figure 6A and B) and structural markers of tubular dysfunction (Figure 6G) were within normal limits in SS/HK−/− mice. Moreover, renal fibrosis (Figure 6D), inflammation (Figure 6E), and iron deposition (Figure 6I) were significantly reduced in HK-deficient SS mice.
FIGURE 6.
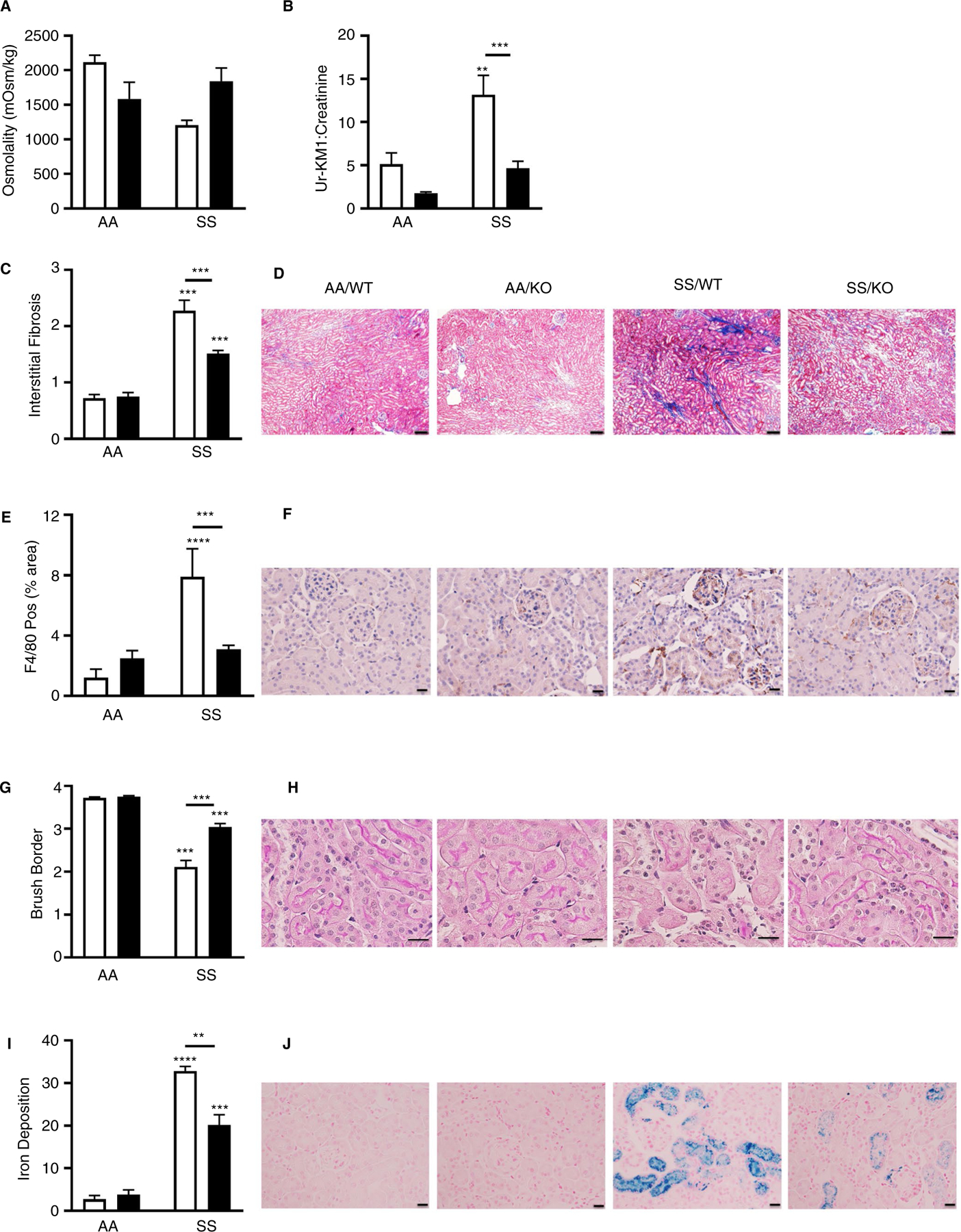
Long-term high molecular weight kininogen (HK) deficiency attenuates tubular injury in sickle mice. Urine was collected for 24 hours from AA/WT, AA/KO, SS/WT, and SS/KO mice 240 days after bone marrow transplant and analyzed for (A) osmolality and (B) kidney injury marker-1 (KIM-1) levels normalized to creatinine. Data are presented as mean ± standard error of the mean (SEM). C, Quantification of interstitial fibrosis index score and (D) representative images of Picro Sirius-red stained sections. Original magnification ×20; scale bar = 100 µm. E, Quantification of F4/80+-stained area and (F) representative images of F4/80+-stained glomerular sections. Original magnification ×40; scale bar = 50 µm. G, Quantification of brush border loss index score and (H) representative images from hematoxylin and eosin-stained sections. Original magnification ×120; scale bar = 50 µm. (I) Quantification of Prussian blue staining representing iron deposition and (J) representative images of Prussian blue stained sections. Original magnification ×40; scale bar = 50 µm. Data are presented as mean ± SEM, and analyzed by two-way analysis of variance. *P < .05, **P < .01, ***P < .001 and ****P < .0001 versus AA/WT. Asterisks above lines indicate difference from SS/WT
4 |. DISCUSSION
High molecular weight kininogen is a non-enzymatic cofactor that promotes FXI and PK activation, which are the key components of the intrinsic coagulation and contact pathways, respectively. Although others have demonstrated reduced plasma levels of FXII, PK, and HK zymogens in sickle cell patients,28,34 it is possible that the lower levels of zymogens are a result of decreased production due to the pathological state and not due to accelerated consumption. To the best of our knowledge, this is the first study to demonstrate increased levels of cleaved HK in sickle patients at steady state. The levels of cHK/iHK were not different between patients with a history of VTE and those without. We also did not observe a consistent end organ involvement in the patients with the highest cHK/iHK levels. Currently, there are no validated and widely accepted scoring systems that define disease severity in SCD.35 This may be because of the myriad complications of the disease, and possibly because clinical subphenotypes may depend on heterogeneous mechanisms of disease as proposed for example by Gladwin and Vichinsky.36 Given this limitation, clinical studies that use quantifiable biomarkers (a) to define mechanisms of a specific complication of SCD, and/or (b) to predict a specific complication of SCD continue to be important. Because of the challenging complexity of SCD, our approach has been to combine human and mouse studies where possible, because each has its own strengths and limitations. In the current study, for example, the findings from the mouse experiments predict that HK plays an important role in renal pathophysiology, but in order to show this might be clinically relevant in humans, we begin by demonstrating that HK turnover is indeed accelerated. However, we acknowledge that given the limited size of the patient cohort and the disease heterogeneity, the data can at best be viewed as hypothesis-generating, and that further larger scale prospective studies will be needed before cHK or another similar analyte in the contact pathway can be recommended as a mechanistic or prognostic biomarker.
We also demonstrate that the chronic activation of coagulation observed in sickle mice is markedly reduced by HK deficiency, which may be explained, in part, by the absence of cofactor functions of HK and subsequent reduction of thrombin generation. Consistent with this hypothesis, FXIIa-mediated activation of FXI and PK is markedly reduced in the absence of HK,37 and HK−/− mice have blunted, pro-thrombotic responses.23,25 However, HK cleavage could also contribute to the hypercoagulable state observed in SCD via two other mechanisms: BK-mediated increases in vascular permeability or increased TF expression on leukocytes induced by cHK. Increased vascular permeability has been reported in mouse models of sickle and wild type mice challenged with free heme,38–40 which may result in the exposure of perivascular TF. We have previously demonstrated that perivascular TF significantly contributes to thrombin generation in lipopolysaccharide (LPS)- and heme-challenged mice.38,41 However, we found that neither TAT nor IL-6 were reduced in sickle mice deficient in both BK receptors, indicating that BK-dependent vascular permeability is not involved in thrombin generation and inflammation in sickle mice. Another possibility is increased TF expression on leukocytes. Indeed, the increased exposure of domain 5 following HK cleavage leads to increased binding to Mac-1 on leukocytes.26 Binding of cHK to Mac-1 on monocytes has been shown to increase the expression of TF and the procoagulant activity of these cells.22 Previous data from our laboratory indicates that the heme-mediated increase in monocyte TF expression contributes to thrombin generation in sickle mice.3,8,38 We hypothesized that cHK binding to monocyte Mac-1 could enhance pathologic TF expression mediated by heme. Indeed, in a pilot experiment we observed that inhibition of Mac-1 with M1/70 antibody for 1 week reduced thrombin generation in sickle mice (data not shown).
In our long-term study, HK deficiency had no effect on cardiac hypertrophy and LV dysfunction as measured by echocardiography. However, the degree of cardiac dysfunction in our aged SS/WT mice was not as severe as in other reports.42 This could potentially be explained by the less severe phenotype displayed by sickle chimeras generated by bone marrow transplantation compared to genetic sickle mice. Another reason is that cardiovascular function was only measured in the 22% of SS/WT mice that survived the 240-day aging period. It is reasonable to postulate that the mice that died earlier succumbed to cardiovascular dysfunction, including pulmonary hypertension and lung edema, whereas survivors had a less severe phenotype.
It has been reported that long-term hypoprothrombinemia in sickle mice prevented cardiovascular dysfunction,10 which we did not observe in the sickle mice lacking HK. It is important to point out that the kallikrein-kinin system has been demonstrated to play a protective role in various models of cardiovascular dysfunction. cHK, and in particular the D5 domain, has been shown to inhibit neointima formation after injury to the femoral artery, due to its anti-adhesive properties.43 BK administration reduced infarct size in an isolated perfused mouse heart model of ischemia-reperfusion.44 Moreover, mice deficient in B2R developed LV enlargement, fibrosis, and cardiovascular dysfunction by 180 days of age.45 This protection can be attributed to the reduction in blood pressure, cardiac infarction, and cardiac remodeling modulated by BK-dependent B2R signaling.46 Indeed, the kinin system plays an important role in blood pressure regulation; however, we did not evaluate blood pressure in our mice, and this is a limitation of this study. It is possible that the lack of HK signaling in SS/HK−/− mice can minimize the potential cardiovascular benefit of the reduction in coagulation and inflammation.
In our long-term study, all surviving sickle mice presented with nephropathy, and deficiency of HK significantly attenuated all urinary markers of kidney injury. The renal pathology of SCD is one of the most serious organ-specific complications, and affects approximately two thirds of sickle patients, with nearly one fifth of patients dying from renal disease.33 Sickle nephropathy is characterized by structural and functional pathologies, including damage to and loss of the glomerular filtration barrier, tubulointerstitial injury, dysfunctional urine concentrating ability, and defective distal nephron function.47 The kidney pathology of SCD has been linked to oxidative stress,48 inflammatory signaling through the receptor for advanced glycation end products (RAGE)49 and TLR4,50 endothelin signaling,33 and thrombin.10 We observed that SS/HK−/− mice exhibited less inflammatory cell infiltration, reduced iron accumulation, and interstitial fibrosis, as well as preserved brush border thickness and number of podocytes. The protection mediated by HK deficiency on these structural changes to the glomerulus and tubules resulted in preserved kidney function in the SS/KO mice. It is reasonable to conclude that the reduction in renal pathology is secondary to reduced hyperactivation of chronic coagulation and inflammation. This could also be attributed to an improvement in endothelial function and blood flow, or other yet unknown, HK-dependent mechanisms.
In our study, HK deficiency attenuates thrombin generation, which ultimately leads to less fibrinogen cleavage and deposition of fibrin. A recent report demonstrated that SS mice that express Fibγ390−396A, a mutant form of fibrinogen that cannot engage Mac-1 on leukocytes, had significantly reduced circulating levels of inflammatory cytokines IL-6 and TNFα.51 Interestingly, and consistent with the data presented herein, the SS/Fibγ390−396A mice had preserved renal structure and function, in the absence of protection from cardiopulmonary and hepatic injury.
In summary, our study demonstrates that HK is a central mediator of thrombin generation and inflammation in SCD. HK deficiency attenuated sickle nephropathy and ultimately improved survival of sickle mice. Together, these data suggest that therapeutic targeting of HK might be beneficial for sickle cell patients.
Supplementary Material
Essentials.
Hypercoagulability and inflammation driven by extrinsic coagulation contribute to end-organ damage and early mortality in SCD.
The role of the intrinsic and contact activation pathways in the pathology of sickle cell disease was investigated.
An increased cleavage of high molecular weight kininogen (HK) is observed in plasma of SCD patients compared to healthy controls.
HK deficiency in a mouse model of SCD attenuates thrombin generation, inflammation, kidney injury, and early mortality.
ACKNOWLEDGMENTS
The UNC Chapel Hill Animal Metabolism Phenotyping Core provided metabolic cages for these studies (NIH Grant DK056350).
Funding information
RP and EMS are supported by R01HL142604. EMS was supported by 16POST30230002. SS was supported by R01NS102721, the Alzheimer’s Association, and the Cure Alzheimer’s Fund. MWH, PE, and NSK are supported by R01HL146226. DMP was supported by U01HL117684. MK and PRD are supported by K99HL144817. PE was supported by T32 HL007149.
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicts of interest to disclose.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
REFERENCES
- 1.Ataga KI, Brittain JE, Desai P, et al. Association of coagulation activation with clinical complications in sickle cell disease. PLoS One 2012;7(1):e29786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ataga KI. Hypercoagulability and thrombotic complications in hemolytic anemias. Haematologica 2009;94(11):1481–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chantrathammachart P, Mackman N, Sparkenbaugh E, et al. Tissue factor promotes activation of coagulation and inflammation in a mouse model of sickle cell disease. Blood 2012;120(3):636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Setty BN, Betal SG, Zhang J, Stuart MJ. Heme induces endothelial tissue factor expression: potential role in hemostatic activation in patients with hemolytic anemia. J Thromb Haemost 2008;6(12):2202–2209. [DOI] [PubMed] [Google Scholar]
- 5.Ataga KI, Key NS. Hypercoagulability in sickle cell disease: new approaches to an old problem. Hematology Am Soc Hematol Educ Program 2007;2007(1):91–96. [DOI] [PubMed] [Google Scholar]
- 6.Solovey A, Kollander R, Shet A, et al. Endothelial cell expression of tissue factor in sickle mice is augmented by hypoxia/reoxygenation and inhibited by lovastatin. Blood 2004;104(3):840–846. [DOI] [PubMed] [Google Scholar]
- 7.Shet AS, Aras O, Gupta K, et al. Sickle blood contains tissue factor-positive microparticles derived from endothelial cells and monocytes. Blood 2003;102(7):2678–2683. [DOI] [PubMed] [Google Scholar]
- 8.Sparkenbaugh EM, Chantrathammachart P, Chandarajoti K, Mackman N, Key NS, Pawlinski R. Thrombin-independent contribution of tissue factor to inflammation and cardiac hypertrophy in a mouse model of sickle cell disease. Blood 2016;127(10):1371–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sparkenbaugh EM, Chantrathammachart P, Mickelson J, et al. Differential contribution of FXa and thrombin to vascular inflammation in a mouse model of sickle cell disease. Blood 2014;123(11):1747–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arumugam PI, Mullins ES, Shanmukhappa SK, et al. Genetic diminution of circulating prothrombin ameliorates multiorgan pathologies in sickle cell disease mice. Blood 2015;126(15):1844–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gavins FN, Russell J, Senchenkova EL, et al. Mechanisms of enhanced thrombus formation in cerebral microvessels of mice expressing hemoglobin-S. Blood 2011;117(15):4125–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whelihan MF, Lim MY, Mooberry MJ, et al. Thrombin generation and cell-dependent hypercoagulability in sickle cell disease. J Thromb Haemost 2016;14(10):1941–1952. [DOI] [PubMed] [Google Scholar]
- 13.Hillery C, Foster TD, Holzhauer SL, et al. Tissue factor deficiency decreases sickle cell-induced vascular stasis in a hematopoietic stem cell transplant model of murine sickle cell disease. Blood 2004;104:236. [Google Scholar]
- 14.Sheng N, Fairbanks MB, Heinrikson RL, et al. Cleaved high molecular weight kininogen binds directly to the integrin CD11b/CD18 (Mac-1) and blocks adhesion to fibrinogen and ICAM-1. Blood 2000;95(12):3788–3795. [PubMed] [Google Scholar]
- 15.Renne T, Dedio J, David G, Müller-Esterl W. High molecular weight kininogen utilizes heparan sulfate proteoglycans for accumulation on endothelial cells. J Biol Chem 2000;275(43):33688–33696. [DOI] [PubMed] [Google Scholar]
- 16.Mahdi F, Shariat-Madar Z, Kuo A, Carinato M, Cines DB, Schmaier AH. Mapping the interaction between high molecular mass kininogen and the urokinase plasminogen activator receptor. J Biol Chem 2004;279(16):16621–16628. [DOI] [PubMed] [Google Scholar]
- 17.Joseph K, Shibayama Y, Nakazawa Y, Peerschke EI, Ghebrehiwet B, Kaplan AP. Interaction of factor XII and high molecular weight kininogen with cytokeratin 1 and gC1qR of vascular endothelial cells and with aggregated Abeta protein of Alzheimer’s disease. Immunopharmacology 1999;43(2–3):203–210. [DOI] [PubMed] [Google Scholar]
- 18.Joseph K, Ghebrehiwet B, Kaplan AP. Cytokeratin 1 and gC1qR mediate high molecular weight kininogen binding to endothelial cells. Clin Immunol 1999;92(3):246–255. [DOI] [PubMed] [Google Scholar]
- 19.Colman RW, Schmaier AH. Contact system: a vascular biology modulator with anticoagulant, profibrinolytic, antiadhesive, and proinflammatory attributes. Blood 1997;90(10):3819–3843. [PubMed] [Google Scholar]
- 20.Renne T. The procoagulant and proinflammatory plasma contact system. Semin Immunopathol 2012;34(1):31–41. [DOI] [PubMed] [Google Scholar]
- 21.Guo YL, Colman RW. Two faces of high-molecular-weight kininogen (HK) in angiogenesis: bradykinin turns it on and cleaved HK (HKa) turns it off. J Thromb Haemost 2005;3(4):670–676. [DOI] [PubMed] [Google Scholar]
- 22.Khan MM, Liu Y, Khan ME, Gilman ML, Khan ST, Bromberg M, Colman RW. Upregulation of tissue factor in monocytes by cleaved high molecular weight kininogen is dependent on TNF-alpha and IL-1beta. Am J Physiol Heart Circ Physiol 2010;298(2):H652–H658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merkulov S, Zhang WM, Komar AA, et al. Deletion of murine kininogen gene 1 (mKng1) causes loss of plasma kininogen and delays thrombosis. Blood 2008;111(3):1274–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kokoye Y, Ivanov I, Cheng Q, et al. A comparison of the effects of factor XII deficiency and prekallikrein deficiency on thrombus formation. Thromb Res 2016;140:118–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langhauser F, Göb E, Kraft P, et al. Kininogen deficiency protects from ischemic neurodegeneration in mice by reducing thrombosis, blood-brain barrier damage, and inflammation. Blood 2012;120(19):4082–4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmaier AH. The contact activation and kallikrein/kinin systems: pathophysiologic and physiologic activities. J Thromb Haemost 2016;14(1):28–39. [DOI] [PubMed] [Google Scholar]
- 27.Schmaier AH, McCrae KR. The plasma kallikrein-kinin system: its evolution from contact activation. J Thromb Haemost 2007;5(12):2323–2329. [DOI] [PubMed] [Google Scholar]
- 28.Gordon EM, Klein BL, Berman BW, Strandjord SE, Simon JE, Coccia PF. Reduction of contact factors in sickle cell disease. J Pediatr 1985;106(3):427–430. [DOI] [PubMed] [Google Scholar]
- 29.Yamamoto-Imoto H, Zamolodchikov D, Chen ZL, et al. A novel detection method of cleaved plasma high-molecular-weight kininogen reveals its correlation with Alzheimer’s pathology and cognitive impairment. Alzheimers Dement 2018;10:480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu LC, Sun CW, Ryan TM, Pawlik KM, Ren J, Townes TM. Correction of sickle cell disease by homologous recombination in embryonic stem cells. Blood 2006;108(4):1183–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ryan TM, Ciavatta DJ, Townes TM. Knockout-transgenic mouse model of sickle cell disease. Science 1997;278(5339):873–876. [DOI] [PubMed] [Google Scholar]
- 32.Kakoki M, Sullivan KA, Backus C, et al. Lack of both bradykinin B1 and B2 receptors enhances nephropathy, neuropathy, and bone mineral loss in Akita diabetic mice. Proc Natl Acad Sci USA 2010;107(22):10190–10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kasztan M, Fox BM, Speed JS, et al. Long-term endothelin-a receptor antagonism provides robust renal protection in humanized sickle cell disease mice. J Am Soc Nephrol 2017;28(8):2443–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verma PS, Adams RG, Miller RL. Reduced plasma kininogen concentration during sickle cell crisis. Res Commun Chem Pathol Pharmacol 1983;41(2):313–322. [PubMed] [Google Scholar]
- 35.Quinn CT. Minireview: clinical severity in sickle cell disease: the challenges of definition and prognostication. Exp Biol Med 2016;241(7):679–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. N Engl J Med 2008;359(21):2254–2265. [DOI] [PubMed] [Google Scholar]
- 37.De Maat S, Hofman ZLM, Maas C. Hereditary angioedema: the plasma contact system out of control. J Thromb Haemost 2018;16(9):1674–1685. [DOI] [PubMed] [Google Scholar]
- 38.Sparkenbaugh EM, Chantrathammachart P, Wang S, et al. Excess of heme induces tissue factor-dependent activation of coagulation in mice. Haematologica 2015;100(3):308–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vinchi F, Gastaldi S, Silengo L, Altruda F, Tolosano E. Hemopexin prevents endothelial damage and liver congestion in a mouse model of heme overload. Am J Pathol 2008;173(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghosh S, Tan F, Ofori-Acquah SF. Spatiotemporal dysfunction of the vascular permeability barrier in transgenic mice with sickle cell disease. Anemia 2012;2012:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pawlinski R, Mackman N. Cellular sources of tissue factor in endotoxemia and sepsis. Thromb Res 2010;125(Suppl 1):S70–S73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bakeer N, James J, Roy S, et al. Sickle cell anemia mice develop a unique cardiomyopathy with restrictive physiology. Proc Natl Acad Sci USA 2016;113(35):E5182–E5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Daniel JM, Reich F, Dutzmann J, et al. Cleaved high-molecular-weight kininogen inhibits neointima formation following vascular injury. Thromb Haemost 2015;114(3):603–613. [DOI] [PubMed] [Google Scholar]
- 44.Bell RM, Yellon DM. Bradykinin limits infarction when administered as an adjunct to reperfusion in mouse heart: the role of PI3K, Akt and eNOS. J Mol Cell Cardiol 2003;35(2):185–193. [DOI] [PubMed] [Google Scholar]
- 45.Emanueli C, Maestri R, Corradi D, et al. Dilated and failing cardiomyopathy in bradykinin B(2) receptor knockout mice. Circulation 1999;100(23):2359–2365. [DOI] [PubMed] [Google Scholar]
- 46.Chao J, Chao L. Kallikrein-kinin in stroke, cardiovascular and renal disease. Exp Physiol 2005;90(3):291–298. [DOI] [PubMed] [Google Scholar]
- 47.Ataga KI, Orringer EP. Renal abnormalities in sickle cell disease. Am J Hematol 2000;63(4):205–211. [DOI] [PubMed] [Google Scholar]
- 48.Zahr RS, Chappa P, Yin H, Brown LA, Ataga KI, Archer DR. Renal protection by atorvastatin in a murine model of sickle cell nephropathy. Br J Haematol 2018;181(1):111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Charrin E, Faes C, Sotiaux A, et al. Receptor for advanced glycation end products antagonism blunts kidney damage in transgenic townes sickle mice. Front Physiol 2019;10:880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nath KA, Belcher JD, Nath MC, et al. Role of TLR4 signaling in the nephrotoxicity of heme and heme proteins. Am J Physiol Renal Physiol 2018;314(5):F906–F914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nasimuzzaman M, Arumugam PI, Mullins ES, et al. Elimination of the fibrinogen integrin alphaMbeta2-binding motif improves renal pathology in mice with sickle cell anemia. Blood Adv 2019;3(9):1519–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.