

Abstract
Post-traumatic headache (PTH) following TBI is a common and often persisting pain disability. PTH is often associated with a multimodal central pain sensitization on the skin surface described as allodynia. However, the particular neurobiology underlying cTBI-induced pain disorders are not known. These studies were performed to assess trigeminal sensory sensitization and to determine if sensitization measured behaviorally correlated with detectable changes in portions of the trigeminal sensory system (TSS), particularly trigeminal nucleus, thalamus, and sensory cortex. Thermal stimulation is particularly well suited to evaluate sensitization and was used in these studies. Recent advances in the use of reward/conflict paradigms permit use of operant measures of behavior, versus reflex-driven response behaviors, for thermal sensitization studies. Thus, to quantitate facial thermal sensitization (allodynia) in the setting of acute TBI, the current study utilized an operant orofacial pain reward/conflict testing paradigm to assess facial thermal sensitivity in uninjured control animals compared with those two weeks after cTBI in a rodent model. Significant reductions in facial contact/lick behaviors were observed in the TBI animals using either cool or warm challenge temperatures compared with behaviors in the normal animals. These facial thermal sensitizations correlated with detectable changes in multiple levels of the TSS. The immunohistochemical (IHC) studies revealed significant alterations in the expression of the serotonin (5-HT), neurokinin 1 receptor (NK1R), norepinephrine (NE), and gamma-aminobutyric acid (GABA) in the caudal trigeminal nucleus, thalamic VPL/VPM nucleus, and sensory cortex of the orofacial pain pathways. There was a strong correlation between increased expression of certain IHC markers and increased behavioral markers for facial sensitization. The authors conclude that TBI-induced changes observed in the TSS are consistent with the expression of generalized facial allodynia following cTBI. To our knowledge, this is the first report of orofacial sensitization correlated with changes in selected neuromodulators/neurotransmitters in the TSS following experimental mild TBI.
Keywords: Closed head TBI, Facial allodynia, Trigeminal sensitization, Operant conditioning
1. Introduction
Each year more than 1.3 million individuals in the US experience TBI and are treated and released from hospital emergency departments (Faul et al., 2010). TBI can initiate a wide array of cognitive, motor, and sensory disabilities (Alwis et al., 2012; Cook and Hawley, 2014). Headache is common among mild/moderate traumatic brain injury (TBI) patients (Nampiaparampil, 2008; Theeler et al., 2013). The form of this PTH is predominantly migraine and tension type headache (Baandrup and Jensen, 2005; Haas, 1996; Lucas et al., 2012). Migraine patients and patients with PTH often develop increased sensitization in the affected region called allodynia (Burstein et al., 2010) in which a non- or mildly noxious stimulus is perceived as painful (MacGregor et al., 2013; Nampiaparampil, 2008). However, the particular neurobiology of the TSS underlying cTBI-induced acute and progressing pain disorders is not known.
Reports from clinical studies of a variety of orofacial pain disorders indicate that these disabilities are often characterized by enhanced multimodal sensitivities and may reflect changes at multiple levels of the TSS (Burstein et al., 2010; Guven et al., 2013). It is known that cTBI produces diffuse axonal injury (DAI) in the brain and brainstem (Bose et al., 2013) and these injuries initiate acute and progressing disabilities (Smith et al., 2013). The TBI related facial pain and headache could potentially result from DAI and microvascular injuries that involve primary, secondary, and tertiary portions of the TSS (Haggman-Henrikson et al., 2011; Sterling et al., 2003), although specific alterations induced by TBI are not known.
A study using a controlled cortical impact model of TBI in mice reported that focal injury, detected at the level of the sensory/motor cortex, induced an acute onset of periorbital allodynia (Elliott et al., 2012). This allodynia was associated with increased expression of central neuropeptides substance P (SP) and calcitonin gene related peptide (CGRP) in the caudal trigeminal nucleus (Elliott et al., 2012). Accordingly, it would be important to evaluate such changes in a model of closed head TBI-induced sensitization. In addition, it would be important to evaluate the expression of other key neurotransmitters and neuromodulators known to be associated with pain transmission to identify potential mechanistic bases for TBI-induced facial allodynia. In particular, an important candidate would be altered expression of serotonin, which was reported to cause hyperalgesia in rats by activating 5-HT1A receptor in a model of temporal mandibular joint (TMJ) pain and inflammation (Taiwo and Levine, 1992). Another key candidate is the SP high affinity receptor neurokinin-1 (NK1R). In a neuropathic and inflammatory orofacial pain model, injection of SP produced heat hyperalgesia by binding with NK1 R (Teodoro et al., 2013). In post-synaptic dorsal horn neurons this receptor has been shown to regulate heat hyperalgesia by enhancing transient vallinoid receptors 1 (TRPV1) (Zhang et al., 2007). In a model of TMJ inflammation model, suppression of NK1 receptor attenuated central sensitization (Takeda et al., 2012). In addition to these excitatory agents, it would be important to evaluate factors associated with inhibitory regulation of pain transmission such as norepinephrine (NE) and GABA which function as major inhibitory neurotransmitters. Perhaps, a more detailed study of the distribution of these molecules within the multiple levels of the trigeminal pathways could provide insight into the neurobiology of TBI-induced sensitization and frequently associated pain disabilities such as headache pain and facial allodynia.
An animal model of cTBI provided an opportunity for direct inquiry into TBI-induced alterations associated with a facial sensitization. Accordingly, the purpose of this study was to quantitate changes in operant avoidance in response to thermal stimulation of the orofacial region using mildly noxious challenge temperatures as a measure of trigeminal sensitization. To our knowledge, this is the first report of comprehensive evidence in a cTBI rodent model for acute orofacial hypersensitivity correlated with changes in selected neuromodulators/neurotransmitters in primary, secondary, and tertiary levels of the TSS.
2. Materials and methods
2.1. Animal subjects
Fourteen Sprague Dawley specific pathogen free (SPF) female rats (12 weeks old, weighing 240–270 g at the start of this study; Charles River laboratory, USA) were used in this study. All procedures were performed in accordance with the U.S Government Principle for the Utilization and Care of Vertebrate Animals, specifically following the National Institutes of Health “Guide for the Care and Use of Laboratory Animals”. Experimental protocols were approved by the Institutional Animal Care & Use Committee (IACUC) at the North Florida/South Georgia Veterans Health System, and the University of Florida.
2.2. Surgical approach for mild closed head TBI
This procedure used a standardized weight-drop method to produce controlled levels of cTBI as described previously (Foda and Marmarou, 1994; Marmarou et al., 1994) and in our previous publication (Bose et al., 2013). This method reproduced many of the injury patterns observed in humans after a noninvasive TBI (Morales et al., 2005; Xiong et al., 2009). Briefly, the animal’s body temperature was monitored and maintained at 37–39 °C using a sanitized, temperature-controlled blanket. The animal and the surrounding surgery field were covered with a sterile drape. cTBI procedures were conducted under aseptic surgical techniques. The dorsal surface of the head was shaved and the incision site was infiltrated with 0.25% bupivacaine (3 mg/kg). The surgical area was then cleaned with repeating cycles of betadine scrub (7.5%) followed by a 70% alcohol wipe and a final application of betadine solution (5.0%). A skin incision was made along the dorsal midline of the cranium. Fascia and soft tissue were retracted to expose the two landmarks, the lambda and bregma, on the cranium. A stainless disk (10 mm diameter, 2 mm height) was attached to the exposed skull with dental acrylic. The animal was then placed on a foam pad, and the cortical impact trauma device was maneuvered over the animal and aligned with the stainless disk. A 450-g impactor, with a 6-mm-diameter tip, was dropped through a thermoplastic (Delrin) tube from a 1.25-m height. The impactor struck the stainless disk affixed to the cranium of the animal to produce an impact-acceleration injuries that are uniformly reported as a mild level of closed head traumatic brain injury (cTBI) (Foda and Marmarou, 1994; Marmarou et al., 1994). The stainless disk served to diffuse the force of the impactor, thus decreasing the probability of a skull fracture. Further, the elastic properties of the foam pad, on which the head was resting, produced a controlled acceleration/rotation during the closed head diffuse brain trauma injury (Bose et al., 2013; Marmarou et al., 1994). After injury, the stainless disk was removed from the cranium and the skin was closed with sterile sutures.
2.3. Thermal nociception assay
A reward-conflict operant testing paradigm was used to test for orofacial allodynia as we described previously (Anderson et al., 2013; Mustafa et al., 2013; Neubert et al., 2005). In this paradigm, the rat may choose to experience a nociceptive thermal stimulation in order to obtain a sweetened milk reward or choose not to experience nociception by abstaining from the reward. Rats were trained on the Orofacial Pain Assessment Device (OPAD, Stoelting, Co., Wood Dale, IL) to press their faces into peltier devices whose temperature can be heated or cooled to aversive temperatures. To access to a reward bottle filled with diluted sweetened condensed milk (1:2, milk:water), the animals needed to press their faces into the heated or cooled peltier device. Food-fasted rats (15 ± 1 h) were trained for this task at a neutral adaptive temperature (32 °C) as described in thermal testing procedure in TBI patients (Ofek and Defrin, 2007). An aversive temperature was selected, either 42 °C (hot) or 22 °C (cold), based on several previously published studies (Anderson et al., 2013; Murphy et al., 2014; Mustafa et al., 2013). Next, baseline measurements were obtained for two temperatures, normal and hot/cold, within a single session. This was accomplished by ramping the temperature up from normal to aversive, and then aversive to normal over a course of 7 min as illustrated in Fig. 1A and B. After baseline data were collected, rats underwent the TBI surgical procedure and allowed to recover from initial trauma for two weeks. To rule out decreased performance due to post-injury memory dysfunction, rats were allowed to do the same licking task at neutral temperature (32 °C) for two sessions before running the aversive temperature test. Among the several outcomes of the test, we considered the ratio of total number of active reward taking event (licking) against the total number of active contact event at selected aversive temperature to score their facial allodynia, as described earlier with video reference (Anderson et al., 2013).
Fig. 1.
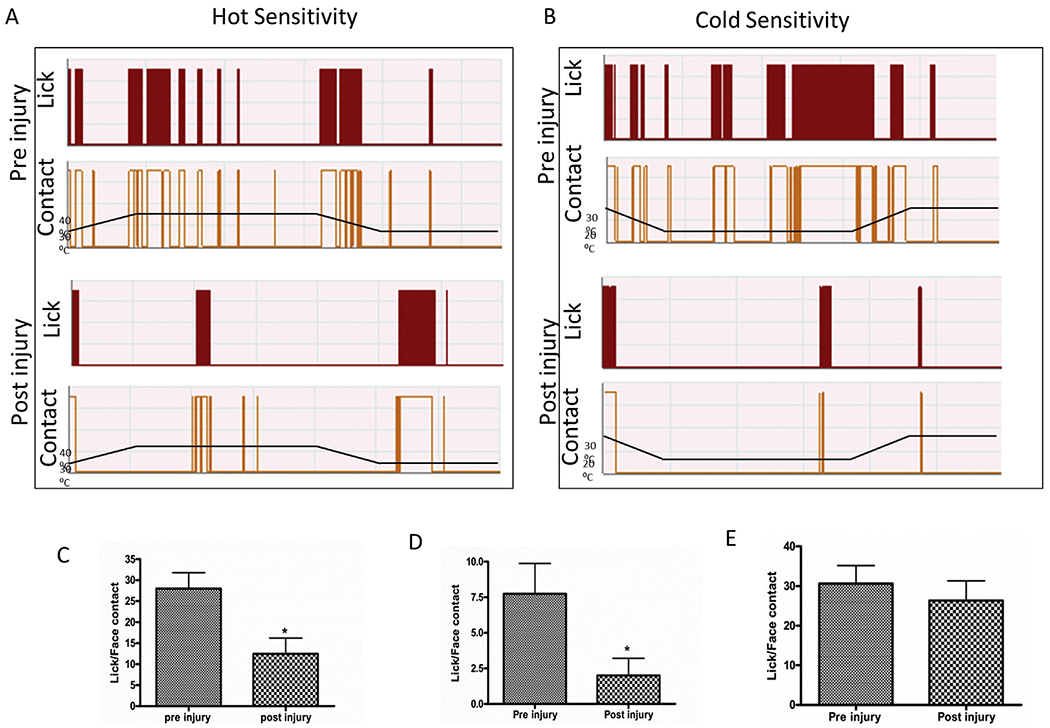
Graphical representation of the reward conflict behavior in OPAD system. Rats (n = 8) were trained in OPAD system in both temperature with a ramp up and ramp down protocol as described in methods for a baseline hot sensitivity (A) and cold sensitivity (B). Lick and face contact ratio indicating the sensitivity in hot at 42 °C (C), cold 22 °C (D) noxious temperatures and 37 °C (E) neutral temperature and in pre- and post-TBI rat. “*” indicates P < 0.05 after 2 weeks of TBI in a paired t-test.
2.4. Immunohistochemistry (IHC) and histology
Following completion of behavioral testing, animals (cTBI, n = 8; age- and sex-matched normal control, n = 6) were deeply anesthetized with ketamine and sacrificed by transcardiac perfusion with phosphate-buffered saline (PBS; 0.01 M, pH 7.4), followed by 4% paraformaldehyde (PFA) in phosphate buffer (0.1 M, pH 7.4, 4 °C) for IHC and histology as reported recently (Bose et al., 2013; Hou et al., 2014). Briefly, brain were dissected and postfixed with 4% PFA for 24–48 h. The whole brain was placed into 30% sucrose for 3 days and then cut serially into 40-μm sections rostrocaudally by Cryostat (Leica CM 1850, Leica Biosystems, USA) approximately from Bregma (B:) −1.80 to B: −3.12 for primary somatosensory area of upper lip (S1ULP) and secondary somatosensory cortex (S2); B: −2.52 to B: −3.36 for VPL and VPM; B: −14.28 to B: −15.48 for spinal trigeminal nucleus, caudal (SP5C) according to Paxinos and Watson rat brain Atlas (Sixth edition). SP5C was used in IHC studies due to the fact that the Aδ and C trigeminal primary afferents which carry pain and thermal sensations from all the three divisions of trigeminal nerve (Opthalmic (V1), maxillary (V2), and mandibuller (V3)) terminate to the SP5C (Lazarov, 2012). A total of four sets of serial sections were made for four antigens (the 67-kDa isoform of glutamate decarboxylase [GAD67], serotonin [5-HT], dopamine beta hydroxylase (DβH) for norepinephrine, and Neurokinin 1 receptor [NK1R]). One additional set of sections used for conventional histology. Each set contained equally spaced sections. For IHC, floating sections were washed three times with PBS (pH 7.4) and blocked in 5% goat serum in PBS for 1 h at room temperature. Sections were then incubated with primary antibodies (rabbit anti-5-HT, 1:8000 [Immunostar, Hudson, WI]; rabbit anti-NK1R, 1:1000 [Millipore]; mouse anti-DβH 1:6000 [Millipore]; mouse anti-GAD67, 1:4000 [Millipore]) overnight at 4 °C in 1% goat serum in PBS. Sections were then washed three times in PBS to remove unbound primary antibodies. Sections were incubated with secondary antibodies (Alexa Fluor 594 goat anti-rabbit immunoglobulin G [IgG], 1:2000 for 5-HT and NK1R; Alexa Fluor 594 goat anti-mouse IgG, 1:2000 for GAD67) for 2 h at room temperature. In control experiments, the primary antibody was either preabsorbed with an excess amount of the immunogenic peptide (1 μm/ml) or replaced by pre-immune serum at the same dilution. After washing three times in PBS, sections were mounted on the slides and cover-slipped with Vectashield (Vector Laboratories, Burlingame, CA). Slides were then viewed using a Zeiss Confocal microscope (LSM 710) with Zen software (version 2012; Carl Zeiss, Jena, Germany). The number of immunopositive cells (NK1R) and fibers (5-HT, DβH, GAD67) in equally spaced serial sections were examined qualitatively and quantitatively in S2, VPL/VPM, and Sp5C. All microscopic images were captured in the same gain setting. An unbiased stereological method as described previously by us and others was used (Bernstein et al., 2011; Bernstein and O’Malley, 2013; Bose et al., 2005; Hou et al., 2014; West and Gundersen, 1990). Briefly, a standardized contour was used as the counting area following a previously published protocol (Lipton et al., 2008). IR-cells and fibers were quantified with Stereo Investigator software (MBF Bioscience, Williston, VT; Version 11.05) attached with a Zeiss Imager Z2 microscope (Carl Zeiss). The petrimetric (sine-wave) probe option was employed with a Merz radius of 20 μm, a grid size of 200 × 200 μm and a counting frame of 100 × 100 μm. An investigator blinded to the experimental condition quantified the IR-cells and fibers within each counting frame following a standard criteria previously reported (Lipton et al., 2008). Briefly, only IR-cells or fibers within the counting frame were included while those outside the frame or crossing over the frame were excluded as described previously (Lipton et al., 2008).
The fourth set of sections (B: −14.28 to B: −15.48) from spinal trigeminal nucleus was processed with conventional cresyl violet staining technique (Bose et al., 2012). Light microscopic qualitative evaluation was made to assess the morphology of the spinal trigeminal nucleus.
2.5. Statistics
Data were analyzed using GraphPad Prism 4 software (La Jolla, CA). A paired t-test was used to analyze the thermal nociception data comparing pre- and post-TBI conditions. Whereas, immunoshistochemistry data were analyzed using an unpaired t-test comparing data sets from TBI and age- and sex-matched control specimens. The potential for interaction between the altered expression of specific markers for 5-HT fibers, NK1R positive cells, and DβH fibers and behavioral scores for facial sensitization was tested using least squares linear regression of these respective data sets. The p values less than 0.05 were considered significant. For fiber density and immuno-positive cells, data are shown as corresponding mean ± SEM.
3. Results
3.1. Increased facial thermal sensitivity following TBI
Pre-injured rats (n = 8) were trained on the OPAD to assess their thermal sensitivity at neutral (32 °C) and two mildly noxious challenge temperatures (42 °C and 22 °C, hot and cold respectively). At two weeks following TBI, the ratio of lick per facial contact was significantly reduced at both challenge temperatures compared with the corresponding values recorded before injury (Fig. 1C and D). At 42 °C, the lick/contact ratio before injury was 28.00 and was observed to be 12.44 when tested two weeks following injury. The ratio observed following injury was found to be significantly decreased (paired t-test, t = 3.31, df = 7, and p = 0.0129). At cold noxious temperature, 22 °C, the lick/contact ratio was also decreased significantly in TBI animals compared to observations made during pre-injury normal recording sessions in a paired t-test (t = 2.48, df = 7, and p = 0.0423). However, at the neutral temperature (5 min–7 min in the ramp), the lick/face contact ratio was not observed to be different than ratios observed prior to injury (Fig. 1E) (t = 2.04, df = 7, and p = 0.097). These observations indicated that while the TBI animals had aversive responses to the challenge temperatures, they were not aversive to neutral temperature, indicating that there was facial thermal sensitization (allodynia) with unaltered reward-seeking behavior.
3.2. Histology of spinal trigeminal nucleus
Studies of cresyl violet stained tissues of the trigeminal nucleus revealed no detectable tissue damage (Fig. 2A–B). Other than a few large neurons (indicated by arrows), light microscopic qualitative studies of the stained TBI tissues revealed healthy neurons with normal cellular density without the presence of immune cell infiltrations.
Fig. 2.
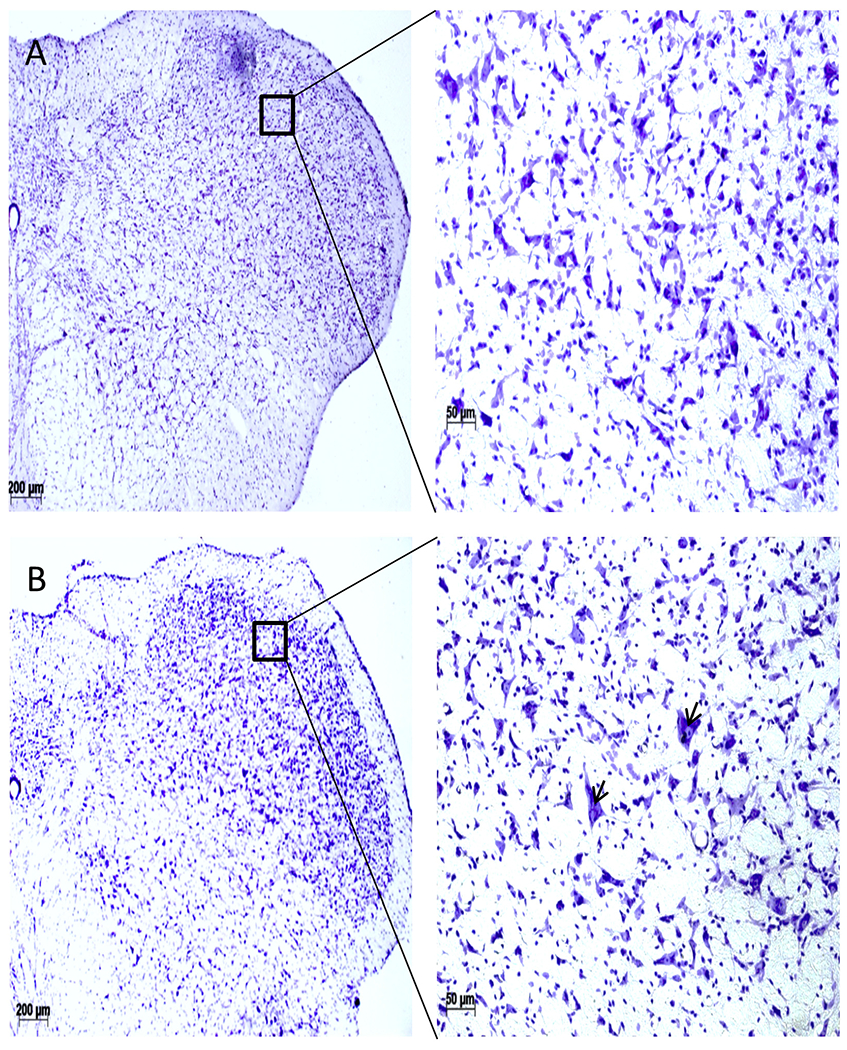
Cresyl-violet staining of the 5th cranial nucleus (SP5C), coronal sections (A, Normal; B, TBI). TBI SP5C area (B) did not show any detectable changes in the cellular morphology except appearance of a few largers cells (arrowhead). Scale bar = 100 μm.
3.3. Changes in 5-HT
Using an IR-expression marker for serotonin, the density of 5-HT fibers was compared between normal and cTBI animals following two weeks after TBI, in tissues from the brainstem, thalamic, and cortical levels of the trigeminal projection system.
3.3.1. SP5C
The density of 5-HT fibers in tissues from the SP5C region of the brainstem in cTBI and normal animals was 1744 ± 178.4 and 707.1 ± 90.26 respectively. These differences represented a significant (255%) increase of 5-HT fibers in the SP5C in cTBI animals (t-test, t = 3.56, df = 12, P < 0.05) (Fig. 5a–c), compared with the 5-HT fiber density observed in normal SP5C tissues.
Fig. 5.
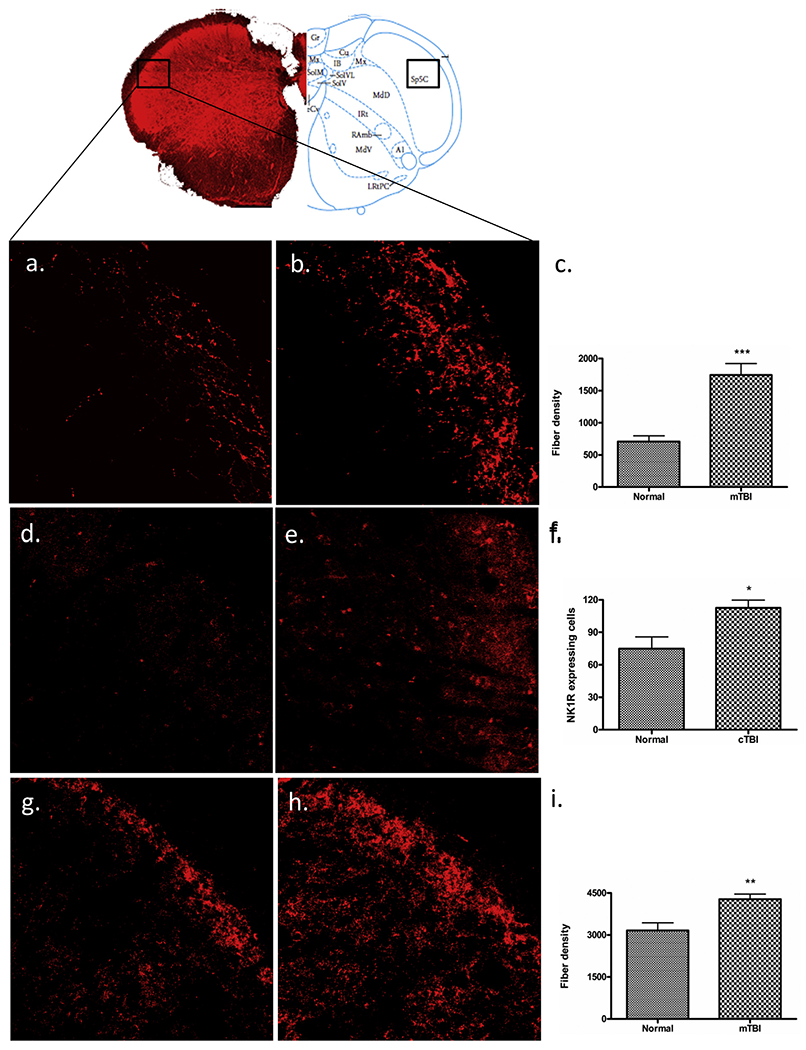
Immunofluorescent labelling of SP5C. Mild-TBI altered the immunoreactive expression of 5-HT (b), NK1R (e), and GAD67 (h) in the 5th cranial nerve nucleus caudalis (B: −15.0) according to the Paxinos atlas compare to the corresponding areas of control animals (a, d, g). Scale bar = 50 μm. A quantitative evaluation of fiber density for 5-HT (c), GAD67 (i) and NK1R expressing cells (f) showed significant elevation following m-TBI. “*”, P < 0.05, “**”, p < 0.005, and “***”, indicates P < 0.0005.
3.3.2. VPL/VPM
The density of 5-HT fibers in tissues from the VPL/VPM region of the thalamus in mTBI and normal animals was 1214 ± 25.17 and 530 ± 25.17 respectively. These differences represented a significant (219%) increase of 5-HT fibers in the VPL/VPM in cTBI animals (t-test, t = 5.19, df = 12, P < 0.005) (Fig. 4a–c), compared with the 5-HT fiber density observed in normal VPL/VPM tissues.
Fig. 4.
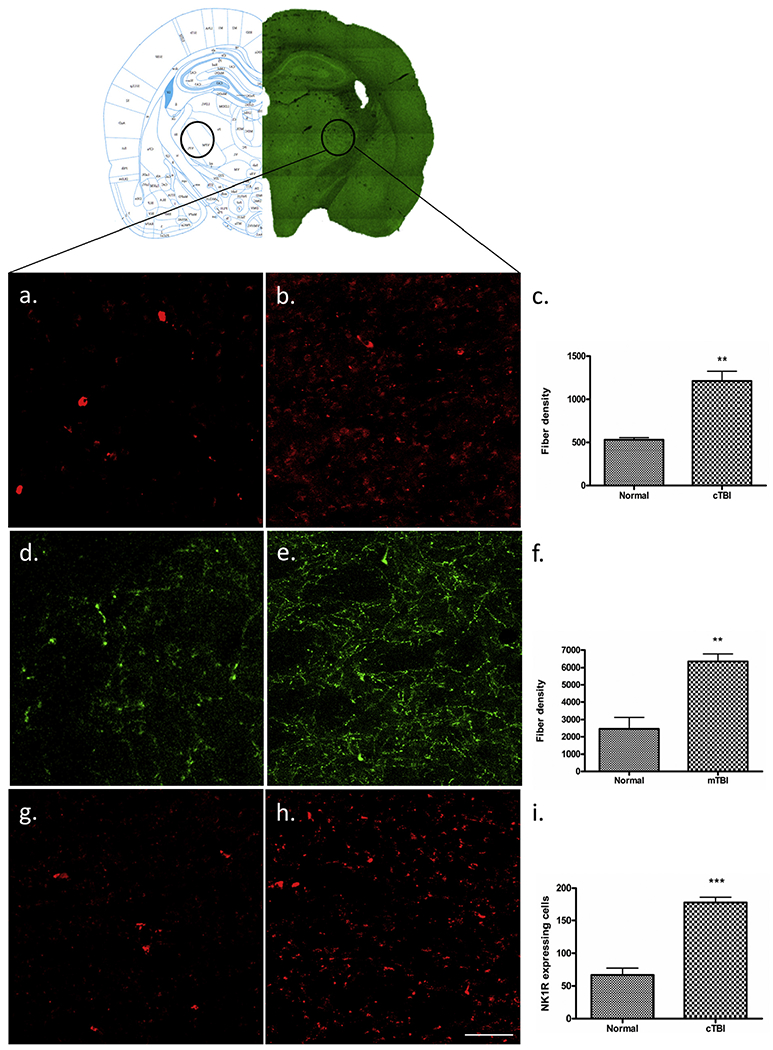
Immunofluorescent labelling of VPL and VPM. Mild-TBI altered the immunoreactive expression of 5-HT (b), DBH (e) and NK1R (h) in the VPL and VPM (B: −3.2) area of cortex according to the Paxinos atlas compare to the corresponding areas of control animals (a, d, g). Scale bar = 50 μm. A quantitative evaluation of fiber density for 5-HT (c), DβH (f) and NK1R expressing cells (i) showed significant elevation following m-TBI. “**”, indicates p < 0.005 and “***”, indicates P < 0.0005.
3.3.3. S1ULP/S2
The density of 5-HT fibers in tissues from the S1ULP/S2 regions of the cerebral cortex in cTBI and normal animals was 2496 ± 44.45 and 940.0 ± 47.61 respectively. These differences represented a significant (265%) increase of 5-HTfibers in the S1ULP/S2 in cTBI animals (t-test, t = 23.76, df = 12, P < 0.0005) (Fig. 3a–c), compared with the 5-HT fiber density observed in normal S1ULP/S2 tissues.
Fig. 3.
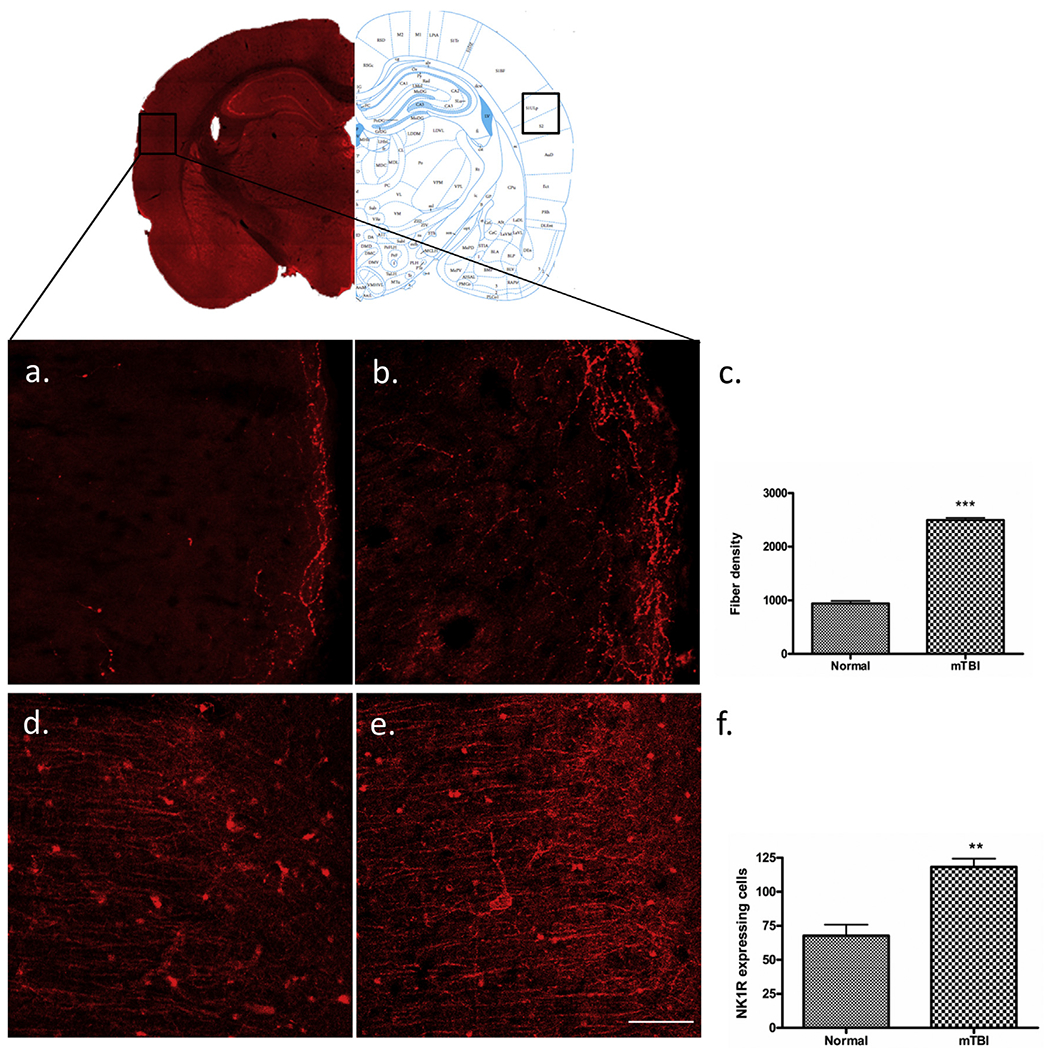
Immunofluorescent labelling of S1ULP and S2. Mild-TBI altered the immunoreactive expression of 5-HT (b) and NK1R (e) in secondary somatosensory cortex (B: −3.2) according to the Paxinos atlas compare to the corresponding areas of control animals (a, d). Scale bar = 50 μm. A quantitative evaluation of 5-HTfiber density (c) and NK1R expressing cells (f) showed significant elevation following m-TBI. “**”, p < 0.005, and “***”, indicates P < 0.0005.
3.4. Changes in NK1R
Using an IR-expression marker for the SP receptor NK-1, the immunoreactivity was compared between normal animals and that of cTBI animals following two weeks after TBI, in tissues from the brainstem, thalamic, and cortical levels of the trigeminal projection system.
3.4.1. SP5C
The number of NK1R expressing cells in tissues from the SP5C region of the brainstem in mTBI and normal animals was 112.5 ± 7.30 and 74.80 ± 10.99 respectively. These differences represented a significant (150.4%) increase of NK1R-expressing cells in the SP5C in cTBI animals (t-test, t = 2.98, df = 12, P < 0.05) (Fig. 5d–f), compared with the number of NK1R expressing cells observed in normal SP5C tissues.
3.4.2. VPL/VPM
The number of NK1R expressing cells in tissues from the VPL/VPM region of the thalamus in cTBI and normal animals was 177.8 ± 8.28 and 67.00 ± 9.85 respectively. These differences represented a significant (264.2%) increase in the NK1R-expressing cells in the VPL/VPM in cTBI animals (t = 6.59, df = 12, P < 0.0005) (Fig. 4g–i), compared with the number of NK1R expressing cells observed in normal VLP/VLM tissues.
3.4.3. S1ULP/S2
The number of NK1R expressing cells in tissues from the S1ULP/S2 regions of the cerebral cortex in cTBI and normal animals was 118.3 ± 6.142 and 67.67 ± 8.09 respectively. These differences represented a significant (173%) increase in the NK1R-expressing cells in the S1ULP/S2 in cTBI animals (t-test, t = 5.09, df = 12, P < 0.005) (Fig. 3d–f), compared with the number of NK1R expressing cells observed in normal S1ULP/S2 tissues.
3.5. Changes in DβH
DβH, a surrogate marker for norepinephrine (NE), IHC was used to identify and NE fibers in normal animals and in animals at two weeks following cTBI, in tissues from the brainstem, thalamic, and cortical levels of the trigeminal projection system. The mean density of DβH positive fiber from the VPL/VPM region of the thalamus in cTBI and normal tissues was 6340 ± 440.9 and 2450 ± 678.8 respectively. These differences represented a significant (258%) increase in the NE fiber expression in the VPL/VPM in cTBI animals (t-test, t = 5.04, df = 12, P < 0.005) (Fig. 4d–f), compared with the density of DβH fibers observed in normal VPL/VPM tissues. By contrast, no differences in NE fiber counts were observed in tissues from S1ULP/S2 (normal, 4781 ± 171.7vs. cTBI 4733 ± 568.7; t = 1.270, df = 12, P = 0.94) and SPC5 (normal, 2545 ± 514.0 vs. cTBI, 2415 ± 203.5; t = 0.24, df = 12, P = 0.82) (graphs not shown).
3.6. Changes in GAD67
As an index of GABA expression, GAD67 expression was studied in tissues from two weeks following TBI, in tissues from the brainstem, thalamic, and cortical levels of the trigeminal projection system. The density of GAD67 expressing fibers in tissues from the SP5C regions of the brainstem in cTBI and normal animals was 4275 ± 183.8 and 3164 ± 269.2 respectively. These differences represented a significant (135%) increase in GAD67 expressing fibers in the SP5C in cTBI animals (t = 3.287, df = 12, P < 0.005) (Fig. 5g–i). Compared with the number of GAD67 expressing fibers in normal SP5C tissues. In contrast, no significant differences were observed for GAD67 expressing fibers in tissues from cortical regions S1ULP/S2 (normal, 3331 ± 117.1 vs. cTBI 4571 ± 583.0; t = 2.09, df = 12, P = 0.11), as well as from tissues of the VPL/VPM thalamic nuclei (normal, 2334 ± 151.0 vs. cTBI 2582 ± 570.4; t = 0.42, df = 12, P = 0.69) (graphs not shown).
3.7. Correlation studies of orofacial sensitization and IHC data
Least squares linear regressions were performed to assess the conditional probability distribution of behavioral scores (set as scalar dependent variables) against IHC expression markers (set as independent variables). Simple linear regressions for each IHC factor is based upon the initial simplest assumption that the expression of each marker is not linked in a multivariate fashion.
3.7.1. 5-HT
The distributions for behavioral scores and 5-HT fiber density revealed linear relationships in SP5C, VPL/VPM, and S1ULP/S2, Fig. 6. Strong correlations of 0.719 (p = 0.038) and 0.766 (p = 0.0014) were observed for the regression of behavioral scores and 5-HT fiber counts in SP5C and VPL/VPM. However, the most powerful linear relationship revealed a correlation coefficient of 0.868 (p < 0.0001) in the S1ULP/S2 of corticle layer Fig. 6A, Table 1. Graphing the data in this fashion also emphasized the distinct separation of the data sets for normal vs TBI.
Fig. 6.
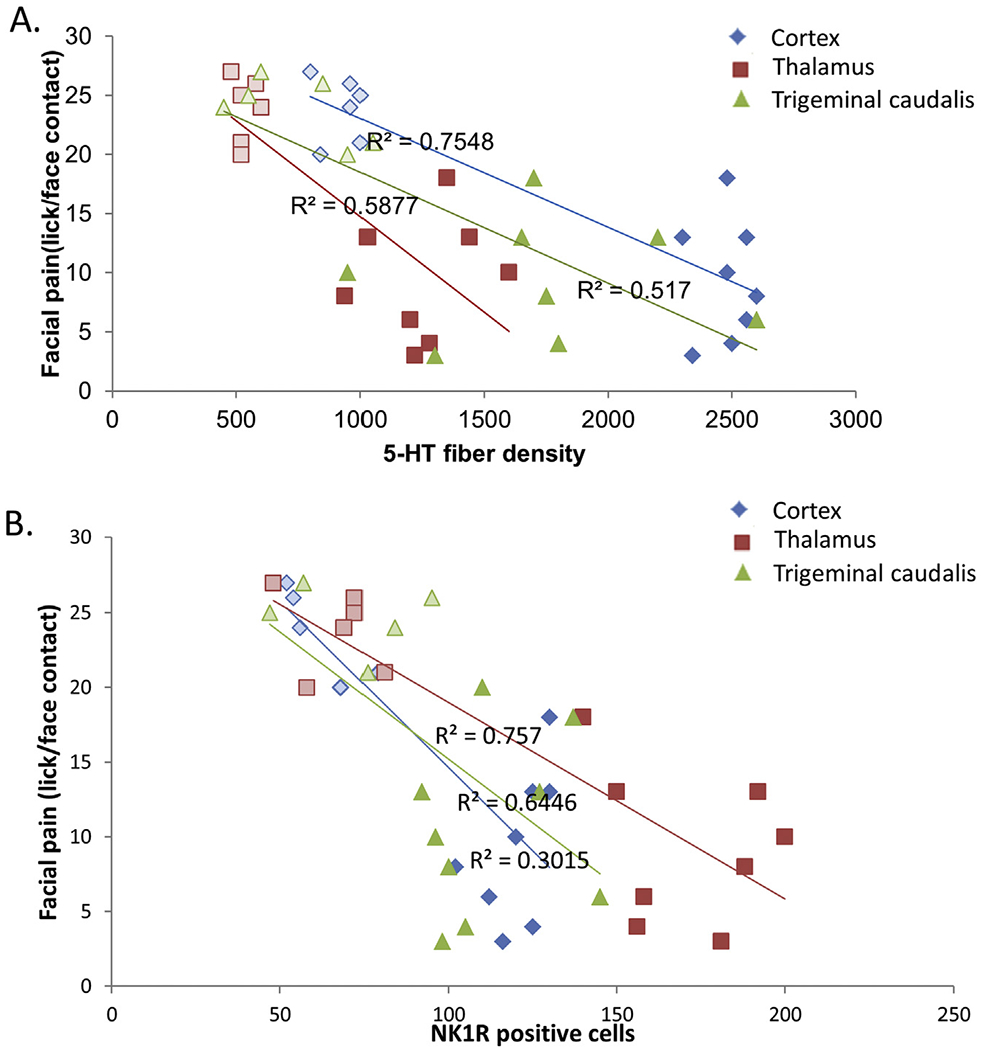
Representative correlative changes in immunohistochemistry and orofacial allodynia. The changes in different neurotransmitters, (A) 5-HT, (B) NK1R, were compared with the changes with the orofacial sensitivity in OPAD system in different regions of the brain, for sensory cortex (S1ULP and S2), thalamic areas (VPL and VPM) and 5th cranial nucleus of caudal region (SP5C). Open and solid blocks represent Normal and TBI animals respectively.
Table 1:
Statistical values of correlation experiment.
| Neurotransmitters | 95% Confidence interval | p-value | Correlation coefficients (r) | |
|---|---|---|---|---|
| Cortex | 5-HT | −0.01251 to −0.005907 | <0.0001 | 0.868** |
| NK1R | −0.3267 to −0.1187 | 0.0005 | 0.802** | |
| DBH | −0.001246 to 0.003994 | 0.2755 | 0.313 | |
| GAD67 | −0.006340 to 0.0005117 | 0.0885 | 0.471 | |
| Thalamus | 5-HT | −0.02471 to 0.007657 | 0.0014 | 0.766** |
| NK1R | −0.1782 to −0.08455 | <0.0001 | 0.870** | |
| DBH | −0.004211 to −0.001661 | 0.0003 | 0.822** | |
| GAD67 | −0.005404 to 0.006346 | 0.8630 | 0.053 | |
| Nucleus Caudalis | 5-HT | −0.01509 to −0.003678 | 0.0038 | 0.719** |
| NK1R | −0.3359 to −0.02382 | 0.0273 | 0.587* | |
| DBH | −0.005585 to 0.005547 | 0.9943 | 0.002 | |
| GAD67 | −0.01284 to −0.003217 | 0.0037 | 0.742** |
indicate significant correlation with p < 0.005
indicate significant correlation with P < 0.05.
3.7.2. NK1R
The distributions for behavioral scores and NK1R positive cells revealed linear relationships in SP5C, VPL/VPM, and S1ULP/S2. The strongest correlations were observed in the VPL/VPM (r 0.870) (p < 0.0001) and S1ULP/S2 (r = 0.805, p = 0.0005).
3.7.3. DβH
No significant correlations for the distribution of behavioral scores and DβH fiber density were observed in SP5C and the S1ULP/S2. However, a strong correlation (r = 0.822, p = 0.0003) was observed between the distribution of behavioral scores and DβH fiber density in the VPL/VPM.
3.7.4. GAD67
A strong correlation (r = 0.742, p = 0.0037) was also observed between the distribution of behavioral score and GAD67 fiber density in the SP5C.
4. Discussion
PTH pain is the most frequent and persistent symptom after mild TBI (Theeler et al., 2013). cTBIs often involve acceleration/deceleration of the brain that induces shear of brain gray, white matter, and vasculature. The primary, secondary, and chronically progressing distributed injuries of the white matter, e.g., diffuse axonal injury (DAI), are considered to have the greatest impact on disability (Blennow et al., 2012; Buki and Povlishock, 2006). Laboratory models of cTBI have been used to gain insights into TBI-induced disabilities and their pathophysiology especially for motor, cognitive and anxiety dysfunction (Bose et al., 2013). However, TBI-induced alterations in sensitization aimed at increasing the understanding of the neurobiology of TBI-induced pain syndromes have not been documented in this model. It has been reported (Hoffman et al., 2011; Jensen and Thulstrup, 2001) that females develop posttraumatic headache 2.6 times more frequently than males. Accordingly, these studies were performed in female rats to evaluate alterations in sensory sensitivity in this model of cTBI. These studies benefitted from the use of a sensitive tool (OPAD) and technique (operant conditioning) to assess thermosensitivity in the orofacial region (Vierck and Yezierski, 2015). Our results suggest that mild cTBI induces an alteration in thermosensitivity in the orofacial region of these rats. The significant change in behavioral responses to the challenge temperatures revealed alteration in thermal-sensitivity to both hot and cold modalities. Moreover, the IHC studies of tissues from these animals indicated significant changes in the expression of several key neuromodulators/neurotransmitters compared with tissues from normal animals. Changes such as these have been proposed to underlie the observed alterations in thermosensitivity in previous studies of chronic pain (Coderre et al., 1993; Ofek and Defrin, 2007). Accordingly, we conclude that injury-induced changes in key neuromodulators/neurotransmitters resulted in a significant increase in facial sensitivity following cTBI. The OPAD paradigm’s detection of sensitization is based upon the observations that application of a stimulus that is perceived as noxious commonly triggers avoidance and escape behavior; these behavioral responses have been argued to be motivated by the conscious phenomenon of pain (Chery-Croze, 1983; Vierck and Yezierski, 2015). Among the different natural stimuli, thermal stimulation has been widely used to evaluate changes in sensory perception.
This study tested the thermal sensitivity of the face in normal and post-TBI animals using an operant behavior that required facial contact with a thermal stimulus to permit a goal behavior (access to a lick spout). Based upon previous human and animals sensory studies the temperatures used in the present studies (32 °C, 42 °C, and 22 °C) represent neutral and mildly hot and cold nociceptive stimuli, respectively. The challenge temperatures (42 °C and 22 °C) were chosen based upon a consensus that these represent threshold levels for eliciting heat and cold nociceptive inputs (Defrin et al., 2002; Wasner and Brock, 2008). We observed that while the challenge temperatures did not block lick/facial contact in the normal animals, lick/facial contact was significantly reduced in the TBI animals. Although significantly increased avoidance behaviors were observed at the two challenge temperatures, no differences in goal behavior were observed at the neutral temperature. These avoidance behaviors were indicative of a facial thermal-hypersensitivity following a weight drop-induced closed head traumatic brain injury. These changes in behavior were correlated with changes in the immuno-expression of selected receptors and neuromodulators throughout the trigeminal, thalamic, and cortical components of the facial sensory projection system compared with studies in tissues from age- and sex-matched normal animals. In particular, significant differences in the IHC expression of NK1 receptors, DβH (marker for NA), serotonin, and GAD67 were observed in the post-TBI animals.
4.1. 5-HT
lt is important to note that significant increases in 5-HT expression were observed at all three levels (SP5C, thalamus, and cortex) of the TSS. In the context of sensitization, these serial changes provide the potential for serial amplification of sensory transmission at each respective level. The relative expression of 5-HT in the trigeminal nucleus was of particular interest due to the significant role that 5-HT plays in inhibition/facilitation of pain transmission (Aicher et al., 2012; Cai et al., 2014; Chai et al., 2012). It is known that 5-HT acts as a pain transmitter via 5-HT2 and 5-HT3 receptors. It has been shown that 5-HT acts directly as a hyperalgesic agent in rats through 5 HT1A receptors (Taiwo and Levine, 1992; Takeda et al., 2012). The 5-HT1A receptor is present in both pre-and post-synaptic locations in different regions of the brain and spinal cord. Especially the pre-synaptic receptor possesses autoreceptor function that has been reported to modulate pain in spinal and midbrain level (Bervoets et al., 1990; Millan et al., 1996; Song et al., 2007). It is known that activation of 5-HT projections from the rostroventral medulla to caudal trigeminal nucleus can significantly increase orofacial pain sensitivity (Cai et al., 2014; Chai et al., 2012). Recently it was demonstrated that secondary hyperalgesia in persistent orofacial pain after nerve injury involves a supraspinal serotonin mechanism where descending 5-HT from the rostral ventromedial medulla and 5HT3 receptors work together to develop central sensitization (Okubo et al., 2013). While previous studies have clearly demonstrated significant loss of noradrenergic (NA) fibers in the locus coerules after a closed head injury in a rat (Bose et al., 2013), and an acute widespread reduction in endogenous NE has been reported after TBI (Dunn-Meynell et al., 1994), by contrast, little is known regarding changes in 5-HT following TBI. The significant increase of 5-HT expression throughout the trigeminal pathway correlated with significant increases in facial sensitivity, suggests a role for 5-HT in the enhanced thermal hypersensitivity in the orofacial region after TBI. Our data of increased 5-HT expression after TBI are in agreement with similar previous studies which proposed that 5-HT facilitates transition of acute pain to chronic orofacial pain by developing central sensitization.
4.2. NK1
Although neuropathological changes have been reported for chief sensory nucleus following fluid percussion TBI (Miremami et al., 2014), the present study is the first to report changes in the caudal trigeminal nucleus following experimental close head TBI. Changes in NK1R expression were of particular interest because it is well known that activation of the tachykinin receptor (NK1) by SP elicits multiple intracellular messengers that influence cellular excitability, such as an increase in activity burst rate due to increased NMDA current as well as a decreased n-type Calcium-dependent potassium channel activation related decrease in the membrane after hyperpolarization (Perez et al., 2013). SP has a preferential affinity for NK1R and acts directly in the processing of noxious information in the spinal cord (Abbadie et al., 1997; Lagraize et al., 2010; Mansikka et al., 2000). SP also plays a key role in producing heat hyperalgesia via the activation of TRPV1 receptor (Holzer, 2004; Zhang et al., 2007). In terms of orofacial pain the peripheral SP/NK1 system has been reported to play a significant role in the development of hyperalgesia and increased pain experience (Awawdeh et al., 2002; Bowles et al., 2003; Denadai-Souza et al., 2009; Rodd and Boissonade, 2000; Takeda et al., 2005). Our data indicate a robust increase of expression of NK1R in areas of the sensory cortex, thalamic region, and spinal trigeminal nucleus related to orofacial pain processing. It is known that SP binds to the NK1R which causes long lasting membrane depolarization. This, in turn, stimulates the C-fiber evoked potential and ultimately triggers central sensitization (Dougherty and Willis, 1991; Latremoliere and Woolf, 2009). While other key factors in TBI-induced central sensitization (e.g. glutamate, CGRP, and BDNF) remain to be investigated, the current studies indicate that TBI-induced upregulation of NK1R could play an important part in TBI-related hypersensitivity.
4.3. DβH
The current studies of TBI-induced changes in NE expression were prompted by our recent findings of significant injury of the locus coeruleus (LC) following TBI (Bose et al., 2013) and the long standing knowledge of the importance of LC originating NE projections in the regulation of pain transmission in general (Jones, 1991) and, particularly, in the trigeminal system (Couto et al., 2006; Sasa et al., 1979, 1974, 1986; Tsuruoka et al., 2003). More recently, it was reported that absence of NA results in a SP/NK1-mediated chronic hyperalgesia particularly to thermal stimuli (Jasmin et al., 2002). These findings indicated that a loss of NA-mediated inhibitory control of sensory transmission in the SP5C induced a trigeminal sensitization and associated allodynia of the trigeminal exteroceptive dermatome. Accordingly, we were surprised that although decreased, significant changes were not observed in DβH expression in the SP5C nucleus or in the cerebral cortex. However, the current studies utilizing DβH expression to assess changes in NA, revealed significant increases in fiber counts in VPL/VPM. This increased in DβH expression in the context of sensitization appears to be a pardox, given the known importance of central role of NA in the inhibitory regulation of sensory transmission. On the other hand, it has also long been known that norepinephrine exerts potent and lasting ionic effects on thalamic neurons. This is in part due to alpha-1 adrenergic suppression of a resting leak potassium current and a beta-adrenergic mediated enhancement of the hyperpolarizing activated cationic current; collectively, these noradrenergic effects facilitate the switch of these thalamic neurons to a state of increased excitability and responsiveness (Hirata et al., 2006; McCormick and Wang, 1991; Samuels and Szabadi, 2008). These thalamic influences, acting in conjunction with increased excitability of the SP5C, and increased 5-HT mediated excitability could accordingly, amplify thalamocortical transmission. Finally, the increased expression of 5-HT at the cortical level completes a pattern of TBI-induced sensitization due to TBI-induced changes in multiple factors at multiple levels in trigeminal sensory transmission.
4.4. GAD67
We also noticed an increased expression of GAD67, a key synthetic enzyme for producing GABA. The increased expression was significant in the SP5C nucleus. This observation seems paradoxical in the setting of sensitization, although there are other considerations (Egea et al., 2012; Viggiano et al., 2004). It has long been known that serotonin and GABA are colocalized in neurons of the rostral ventral medulla (Millhorn et al., 1988; Stamp and Semba, 1995). Accordingly, it is consistent that an increased expression of 5-HT fibers would also account for an increased GAD67 expression. A natural possibility for the role of the increased GAD67 is the production of GABA for inhibition of noxious pain. On the other hand, unlike GAD65, GAD67 produces GABA for neuron activities unrelated to neurotransmission such as synaptogenesis and protection from injury (Pinal and Tobin, 1998). Another possibility is that in the setting of chronic pain, the increased expression of GABA may not necessarily induce more inhibitory control. The GABAergic transmission depends on the cation chloride co-transporters. These transporters are often involved in the central mechanism of inflammatory or neuropathic pain at the spinal level. Peripheral pain has been shown to alter K+-Cl− co-transporter 2 (KCC2) in neuropathic pain following spinal cord injury (Boulenguez et al., 2010; Cramer et al., 2008) in rat and in the formalin induced orofacial pain in mouse (Wu et al., 2009). The inhibition of KCC2 expression has been shown to directly cause a positive shift in the reversal potential of the GABAA response that establishes a depolarizing effect of GABA, which is excitatory (Rivera et al., 1999). Thus, such a down regulation of cation chloride could affect the inhibitory action of GABAergic transmission and promote the central sensitization in TBI-induced orofacial pain and hyperalgesia. (Lee et al., 2011).
Collectively, these findings indicate that orofacial thermal hypersensitivity following cTBI was correlated with significant changes in neuromodulatory amines and receptors that have been reported to be integral to nociceptive transmission and processing throughout the investigated sensory projection system.
Moreover, the IHC studies indicate significant changes in the expression of several key neuromodulators/neurotransmitters. It is a hypothesis that such changes could underlie the observed alterations in thermosensitivity (Coderre et al., 1993; Ofek and Defrin, 2007). We concluded that such changes in these neuromodulators/neurotransmitters could potentially trigger facial allodynia following cTBI. In-summary, acute TBI causes a significant neuromodulatory changes in the cranial nerve nucleus V without detectable changes in the cellular morphology. In addition, remarkable neuromodulatory changes were observed in cortical and thalamic areas related to orofacial pain processing. Accordingly, these observations suggest potentially multiple targets for intervention to addresses changes as an effective strategy to control orofacial sensitization and headache after TBI. The existing treatment of using Non-steroidal anti-inflammatory drugs (NSAIDS), calcium channel blockers, beta-adrenergic blockers, anticonvulsants, and antidepressants possess contraindications, and often manage post-TBI headache non-selectively (Arciniegas et al., 2002). Findings in the neuromodulatory changes related to TBI will be more beneficial toward the specific intervention in post TBI headache and pain.
4.5. Probability distribution of behavioral scores and IHC expression
The linear regression statistical studies revealed highly significant linear relationships between the distributions for behavioral scores for facial sensitization and scores for immuno-expression markers of 5-HT fiber density, NK1R positive cells, and DβH fiber density. These correlations indicate that the probability distribution of behavioral markers of facial sensitization were significantly correlated with numerical scoring of markers for 5-HT fibers, NK1R positive cells, and DβH fibers at all three levels (SP5C, thalamus, and cortex) of the TSS. Accordingly, it is reasonable to propose that the sensitization behaviors observed in the TBI animals were, in part, correlated with TBI-induced plasticity of sensory transmission at all three levels of the TSS.
5. Conclusion
In conclusion, this study provides evidence of a TBI-induced alterations in facial sensory sensitivity using operant behavioral responses, which are considered to be the most accurate markers for alterations in sensitivity (Vierck and Yezierski, 2015). This presumptive sensitization was correlated with changes in several key neuromodulators in three levels of the TSS, particularly 5-HT, and the receptor (NK1R). Currently, there are important questions regarding the etiology of TBI-induced headache/pain. It has been suggested that PTH is derived from central neural injuries producing dysfunctional pain modulation (Defrin et al., 2014). In this regard, our data revealed a strong linear correlation between increased immunomarker expression (particularly for 5-HT) and increased sensitization. On the other hand, recent studies have suggested that PTH could be ascribed to vascular injuries at the base of the calvarium and meningeal blood vessels. Since SP5C receives sensory innervation from meningeal blood vessel (in addition to all three branches of the trigeminal nerve), imbalance in neurotransmitter levels in this nucleus may result in an alteration to the vascular properties and may contribute headache. Collectively these results suggest that TBI-induced a broad spectrum of changes in the neurotransmission that could trigger sensory abnormalities related to facial allodynia and PTH. Accordingly, therapies aimed at peripheral nociceptors may not effectively address sensitization stemming from significant plasticity throughout the TSS. Moreover, single pharmacological therapy aiming at one of the neuromodulators may not be appropriate to treat TBI-induced allodynia. Thus, the findings of the present study reveal important new information regarding the neurobiology of TBI-induced facial sensory sensitization. However, further studies are needed testing specific agonist or antagonists to augment or reverse the behavioral outcome which may provide additional understanding of the role of each transmitter or receptor in TBI induced altered sensitization and/or to develop future therapies.
Acknowledgement
This work was supported by Merit Review Award # B6570R, B78071, B1005-R, from the United States (U.S.) Department of Veterans Affairs Rehabilitation Research and Development Service.
Footnotes
Conflict of interest statement
All authors have no conflicts of interest to declare.
References
- Abbadie C, Trafton J, Liu H, Mantyh PW, Basbaum AI, 1997. Inflammation increases the distribution of dorsal horn neurons that internalize the neurokinin-1 receptor in response to noxious and non-noxious stimulation. J. Neurosci. Off. J. Soc. Neurosci. 17 (20), 8049–8060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aicher SA, Hermes SM, Whittier KL, Hegarty DM, 2012. Descending projections from the rostral ventromedial medulla (RVM) to trigeminal and spinal dorsal horns are morphologically and neurochemically distinct. J. Chem. Neuroanat. 43 (2), 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alwis DS, Yan EB, Morganti-Kossmann M-C, Rajan R, 2012. Sensory cortex underpinnings of traumatic brain injury deficits. PLoS One 7 (12), e52169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson EM, Mills R, Nolan TA, Jenkins AC, Mustafa G, Lloyd C, Caudle RM, Neubert JK, 2013. Use of the operant orofacial pain assessment device (OPAD) to measure changes in nociceptive behavior. J. Vis. Exp. JoVE 76 e50336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arciniegas DB, Held K, Wagner P, 2002. Cognitive impairment following traumatic brain injury. Curr. Treat. Options Neurol. 4 (1), 43–57. [DOI] [PubMed] [Google Scholar]
- Awawdeh LA, Lundy FT, Linden GJ, Shaw C, Kennedy JG, Lamey PJ, 2002. Quantitative analysis of substance P, neurokinin A and calcitonin gene-related peptide in gingival crevicular fluid associated with painful human teeth. Eur. J. Oral Sci. 110 (3), 185–191. [DOI] [PubMed] [Google Scholar]
- Baandrup L, Jensen R, 2005. Chronic post-traumatic headache – a clinical analysis in relation to the International Headache Classification 2nd edition. Cephalalgia 25 (2), 132–138. [DOI] [PubMed] [Google Scholar]
- Bernstein AI, Garrison SP, Zambetti GP, O’Malley KL, 2011. 6-OHDA generated ROS induces DNA damage and p53- and PUMA-dependent cell death. Mol. Neurodegener. 6 (1), 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein AI, O’Malley KL, 2013. MPP+-induces PUMA- and p53-dependent, but ATF3-independent cell death. Toxicol. Lett. 219 (2), 93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bervoets K, Millan MJ, Colpaert FC, 1990. Agonist action at 5-HT1C receptors facilitates 5-HT1A receptor-mediated spontaneous tail-flicks in the rat. Eur. J. Pharmacol. 191 (2), 185–195. [DOI] [PubMed] [Google Scholar]
- Blennow K, Hardy J, Zetterberg H, 2012. The neuropathology and neurobiology of traumatic brain injury. Neuron 76 (5), 886–899. [DOI] [PubMed] [Google Scholar]
- Bose P, Hou J, Nelson R, Nissim N, Parmer R, Keener J, Wacnik PW, Thompson FJ, 2013. Effects of acute intrathecal baclofen in an animal model of TBI-induced spasticity, cognitive, and balance disabilities. J. Neurotrauma 30 (13), 1177–1191. [DOI] [PubMed] [Google Scholar]
- Bose P, Parmer R, Reier PJ, Thompson FJ, 2005. Morphological changes of the soleus motoneuron pool in chronic midthoracic contused rats. Exp. Neurol. 191 (1), 13–23. [DOI] [PubMed] [Google Scholar]
- Bose PK, Hou J, Parmer R, Reier PJ, Thompson FJ, 2012. Altered patterns of reflex excitability, balance, and locomotion following spinal cord injury and locomotor training. Front. Physiol. 3, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulenguez P, Liabeuf S, Bos R, Bras H, Jean-Xavier C, Brocard C, Stil A, Darbon P, Cattaert D, Delpire E, Marsala M, Vinay L, 2010. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat. Med. 16 (3), 302–307. [DOI] [PubMed] [Google Scholar]
- Bowles WR, Withrow JC, Lepinski AM, Hargreaves KM, 2003. Tissue levels of immunoreactive substance P are increased in patients with irreversible pulpitis. J. Endod. 29 (4), 265–267. [DOI] [PubMed] [Google Scholar]
- Buki A, Povlishock JT, 2006. All roads lead to disconnection?–traumatic axonal injury revisited. Acta Neurochir. 148 (2), 181–193 discussion 193–184. [DOI] [PubMed] [Google Scholar]
- Burstein R, Jakubowski M, Garcia-Nicas E, Kainz V, Bajwa Z, Hargreaves R, Becerra L, Borsook D, 2010. Thalamic sensitization transforms localized pain into widespread allodynia. Ann. Neurol. 68 (1), 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai YQ, Wang W, Hou YY, Pan ZZ, 2014. Optogenetic activation of brainstem serotonergic neurons induces persistent pain sensitization. Mol. Pain 10, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai B, Guo W, Wei F, Dubner R, Ren K, 2012. Trigeminal-rostral ventromedial medulla circuitry is involved in orofacial hyperalgesia contralateral to tissue injury. Mol. Pain 8, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chery-Croze S, 1983. Painful sensation induced by a thermal cutaneous stimulus. Pain 17 (2), 109–137. [DOI] [PubMed] [Google Scholar]
- Coderre TJ, Katz J, Vaccarino AL, Melzack R, 1993. Contribution of central neuroplasticity to pathological pain: review of clinical and experimental evidence. Pain 52 (3), 259–285. [DOI] [PubMed] [Google Scholar]
- Cook GA, Hawley JS, 2014. A review of mild traumatic brain injury diagnostics: current perspectives, limitations, and emerging technology. Mil. Med. 179 (10), 1083–1089. [DOI] [PubMed] [Google Scholar]
- Couto LB, Moroni CR, dos Reis Ferreira CM, Elias-Filho DH, Parada CA, Pela IR, Coimbra NC, 2006. Descriptive and functional neuroanatomy of locus coeruleus-noradrenaline-containing neurons involvement in bradykinin-induced antinociception on principal sensory trigeminal nucleus. J. Chem. Neuroanat. 32 (1), 28–45. [DOI] [PubMed] [Google Scholar]
- Cramer S, Baggott C, Cain J, Tilghman J, Allcock B, Miranpuri G, Rajpal S, Sun D, Resnick D, 2008. The role of cation-dependent chloride transporters in neuropathic pain following spinal cord injury. Mol. Pain 4 (1), 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defrin R, Ohry A, Blumen N, Urca G, 2002. Sensory determinants of thermal pain. Brain J. Neurol 125 (Pt 3), 501–510. [DOI] [PubMed] [Google Scholar]
- Defrin R, Riabinin M, Feingold Y, Schreiber S, Pick CG, 2014. Deficient pain modulatory systems in patients with mild traumatic brain and chronic Post-Traumatic headache: implications for its mechanism. J. Neurotrauma 32 (1), 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denadai-Souza A, Camargo LdL., Ribela MTCP, Keeble JE, Costa SKP, Muscará MN, 2009. Participation of peripheral tachykinin NK1 receptors in the carrageenan-induced inflammation of the rat temporomandibular joint. Eur. J. Pain 13 (8), 812–819. [DOI] [PubMed] [Google Scholar]
- Dougherty PM, Willis WD, 1991. Enhancement of spinothalamic neuron responses to chemical and mechanical stimuli following combined microiontophoretic application of N-methyl-D-aspartic acid and substance P. Pain 47 (1), 85–93. [DOI] [PubMed] [Google Scholar]
- Dunn-Meynell A, Pan S, Levin BE, 1994. Focal traumatic brain injury causes widespread reductions in rat brain norepinephrine turnover from 6 to 24 h. Brain Res. 660 (1), 88–95. [DOI] [PubMed] [Google Scholar]
- Egea J, Malmierca E, Rosa AO, del Barrio L, Negredo P, Nunez A, Lopez MG, 2012. Participation of calbindin-D28K in nociception: results from calbindin-D28K knockout mice. Pflugers Archiv. Eur. J. Physiol. 463 (3), 449–458. [DOI] [PubMed] [Google Scholar]
- Elliott MB, Oshinsky ML, Amenta PS, Awe OO, Jallo JI, 2012. Nociceptive neuropeptide increases and periorbital allodynia in a model of traumatic brain injury. Headache 52 (6), 966–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul MXL, Wald MM, Coronado VG, 2010. Traumatic brain injury in the United States: emergency department visits, hospitalizations, and deaths. In: NCfIPaC Centers for Disease Control and Prevention Editor. [Google Scholar]
- Foda MA, Marmarou A, 1994. A new model of diffuse brain injury in rats. Part II: morphological characterization. J. Neurosurg. 80 (2), 301–313. [DOI] [PubMed] [Google Scholar]
- Guven H, Cilliler AE, Comoglu SS, 2013. Cutaneous allodynia in patients with episodic migraine. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 34 (8), 1397–1402. [DOI] [PubMed] [Google Scholar]
- Haas DC, 1996. Chronic post-traumatic headaches classified and compared with natural headaches. Cephalalgia Int. J. Headache 16 (7), 486–493. [DOI] [PubMed] [Google Scholar]
- Haggman-Henrikson B, Gronqvist J, Eriksson PO, 2011. Frequent jaw-face pain in chronic whiplash-associated disorders. Swed. Dent. J. 35 (3), 123–131. [PubMed] [Google Scholar]
- Hirata A, Aguilar J, Castro-Alamancos MA, 2006. Noradrenergic activation amplifies bottom-up and top-down signal-to-noise ratios in sensory thalamus. J. Neurosci. Off. J. Soc. Neurosci. 26 (16), 4426–4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman JM, Lucas S, Dikmen S, Braden CA, Brown AW, Brunner R, Diaz-Arrastia R, Walker WC, Watanabe TK, Bell KR, 2011. Natural history of headache after traumatic brain injury. J. Neurotrauma 28 (9), 1719–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer P, 2004. TRPV1 and the gut: from a tasty receptor for a painful vanilloid to a key player in hyperalgesia. Eur. J. Pharmacol. 500 (1–3), 231–241. [DOI] [PubMed] [Google Scholar]
- Hou J, Nelson R, Nissim N, Parmer R, Thompson FJ, Bose P, 2014. Effect of combined treadmill training and magnetic stimulation on spasticity and gait impairments after cervical spinal cord injury. J. Neurotrauma 31 (12), 1088–1106. [DOI] [PubMed] [Google Scholar]
- Jasmin L, Tien D, Weinshenker D, Palmiter RD, Green PG, Janni G, Ohara PT, 2002. The NK1 receptor mediates both the hyperalgesia and the resistance to morphine in mice lacking noradrenaline. Proc. Natl. Acad. Sci. U. S. A 99 (2), 1029–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen OK, Thulstrup AM, 2001. Gender differences of post-traumatic headache and other post-commotio symptoms. A follow-up study after a period of 9–12 months. Ugeskrift laeger 163 (37), 5029–5033. [PubMed] [Google Scholar]
- Jones BE, 1991. Noradrenergic locus coeruleus neurons: their distant connections and their relationship to neighboring (including cholinergic and GABAergic) neurons of the central gray and reticular formation. Prog. Brain Res. 88, 15–30. [DOI] [PubMed] [Google Scholar]
- Lagraize SC, Guo W, Yang K, Wei F, Ren K, Dubner R, 2010. Spinal cord mechanisms mediating behavioral hyperalgesia induced by neurokinin-1 tachykinin receptor activation in the rostral ventromedial medulla. Neuroscience 171 (4), 1341–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ, 2009. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J. Pain Off. J. Am. Pain Soc. 10 (9), 895–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov NE, 2012. The Neurochemical Anatomy of Trigeminal Primary Afferent Neurons. INTECH Open Access Publisher. [Google Scholar]
- Lee M, Schwab C, McGeer PL, 2011. Astrocytes are GABAergic cells that modulate microglial activity. Glia 59 (1), 152–165. [DOI] [PubMed] [Google Scholar]
- Lipton JW, Tolod EG, Thompson VB, Pei L, Paumier KL, Terpstra BT, Lynch KA, Collier TJ, Sortwell CE, 2008. 3,4-Methylenedioxy-N-metham-phetamine (ecstasy) promotes the survival of fetal dopamine neurons in culture. Neuropharmacology 55 (5), 851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas S, Hoffman JM, Bell KR, Walker W, Dikmen S, 2012. Characterization of headache after traumatic brain injury. Cephalalgia Int. J. Headache 32 (8), 600–606. [DOI] [PubMed] [Google Scholar]
- MacGregor AJ, Dougherty AL, Tang JJ, Galarneau MR, 2013. Postconcussive symptom reporting among US combat veterans with mild traumatic brain injury from operation Iraqi freedom. J. Head Trauma Rehabil. 28 (1), 59–67. [DOI] [PubMed] [Google Scholar]
- Mansikka H, Sheth RN, DeVries C, Lee H, Winchurch R, Raja SN, 2000. Nerve injury-induced mechanical but not thermal hyperalgesia is attenuated in neurokinin-1 receptor knockout mice. Exp. Neurol. 162 (2), 343–349. [DOI] [PubMed] [Google Scholar]
- Marmarou A, Foda MA, van den Brink W, Campbell J, Kita H, Demetriadou K, 1994. A new model of diffuse brain injury in rats. Part I: pathophysiology and biomechanics. J. Neurosurg. 80 (2), 291–300. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Wang Z, 1991. Serotonin and noradrenaline excite GABAergic neurones of the guinea-pig and cat nucleus reticularis thalami. J. Physiol. 442, 235–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ, Seguin L, Honore P, Girardon S, Bervoets K, 1996. Pro- and antinociceptive actions of serotonin (5-HT)1A agonists and antagonists in rodents: relationship to algesiometric paradigm. Behav. Brain Res. 73 (1–2), 69–77. [DOI] [PubMed] [Google Scholar]
- Millhorn DE, Hokfelt T, Seroogy K, Verhofstad AA, 1988. Extent of colocalization of serotonin and GABA in neurons of the ventral medulla oblongata in rat. Brain Res. 461 (1), 169–174. [DOI] [PubMed] [Google Scholar]
- Miremami JD, Talauliker PM, Harrison JL, Lifshitz J, 2014. Neuropathology in sensory, but not motor, brainstem nuclei of the rat whisker circuit after diffuse brain injury. Somatosens. Mot. Res. 31 (3), 127–135. [DOI] [PubMed] [Google Scholar]
- Morales DM, Marklund N, Lebold D, Thompson HJ, Pitkanen A, Maxwell WL, Longhi L, Laurer H, Maegele M, Neugebauer E, Graham DI, Stocchetti N, McIntosh TK, 2005. Experimental models of traumatic brain injury: do we really need to build a better mousetrap? Neuroscience 136 (4), 971–989. [DOI] [PubMed] [Google Scholar]
- Murphy NP, Mills RH, Caudle RM, Neubert JK, 2014. Operant assays for assessing pain in preclinical rodent models: highlights from an orofacial assay. Curr. Top. Behav. Neurosci. 20, 121–145. [DOI] [PubMed] [Google Scholar]
- Mustafa G, Anderson EM, Bokrand-Donatelli Y, Neubert JK, Caudle RM, 2013. Anti-nociceptive effect of a conjugate of substance P and light chain of botulinum neurotoxin type A. Pain 154 (11), 2547–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nampiaparampil DE, 2008. Prevalence of chronic pain after traumatic brain injury: a systematic review. JAMA 300 (6), 711–719. [DOI] [PubMed] [Google Scholar]
- Neubert JK, Widmer CG, Malphurs W, Rossi HL, Vierck CJ Jr., Caudle RM, 2005. Use of a novel thermal operant behavioral assay for characterization of orofacial pain sensitivity. Pain 116 (3), 386–395. [DOI] [PubMed] [Google Scholar]
- Ofek H, Defrin R, 2007. The characteristics of chronic central pain after traumatic brain injury. Pain 131 (3), 330–340. [DOI] [PubMed] [Google Scholar]
- Okubo M, Castro A, Guo W, Zou S, Ren K, Wei F, Keller A, Dubner R, 2013. Transition to persistent orofacial pain after nerve injury involves supraspinal serotonin mechanisms. J. Neurosci. Off. J. Soc. Neurosci. 33 (12), 5152–5161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez CT, Hill RH, Grillner S, 2013. Modulation of calcium currents and membrane properties by substance P in the lamprey spinal cord. J. Neurophysiol. 110(2), 286–296. [DOI] [PubMed] [Google Scholar]
- Pinal CS, Tobin AJ, 1998. Uniqueness and redundancy in GABA production. Perspect. Dev. Neurobiol. 5 (2–3), 109–118. [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K, 1999. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397 (6716), 251–255. [DOI] [PubMed] [Google Scholar]
- Rodd HD, Boissonade FM, 2000. Substance P expression in human tooth pulp in relation to caries and pain experience. Eur. J. Oral Sci. 108 (6), 467–474. [DOI] [PubMed] [Google Scholar]
- Samuels ER, Szabadi E, 2008. Functional neuroanatomy of the noradrenergic locus coeruleus: its roles in the regulation of arousal and autonomic function part II: physiological and pharmacological manipulations and pathological alterations of locus coeruleus activity in humans. Curr. Neuropharmacol. 6 (3), 254–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasa M, Fujimoto S, Igarashi S, Munekiyo K, Takaori S, 1979. Microiontophoretic studies on noradrenergic inhibition from locus coeruleus of spinal trigeminal nucleus neurons. J. Pharmacol. Exp. Ther. 210 (3), 311–315. [PubMed] [Google Scholar]
- Sasa M, Munekiyo K, Ikeda H, Takaori S, 1974. Noradrenaline-mediated inhibition by locus coeruleus of spinal trigeminal neurons. Brain Res. 80 (3), 443–460. [DOI] [PubMed] [Google Scholar]
- Sasa M, Ohno Y, Ito J, Kashii S, Utsumi S, Takaori S, 1986. Beta-receptor involvement in locus coeruleus-induced inhibition of spinal trigeminal nucleus neurons: microiontophoretic and HRP studies. Brain Res. 377 (2), 337–343. [DOI] [PubMed] [Google Scholar]
- Smith DH, Johnson VE, Stewart W, 2013. Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat. Rev. Neurol. 9 (4), 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B, Chen W, Marvizon JC, 2007. Inhibition of opioid release in the rat spinal cord by serotonin 5-HT(1A) receptors. Brain Res. 1158, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamp JA, Semba K, 1995. Extent of colocalization of serotonin and GABA in the neurons of the rat raphe nuclei. Brain Res. 677 (1), 39–49. [DOI] [PubMed] [Google Scholar]
- Sterling M, Jull G, Vicenzino B, Kenardy J, 2003. Sensory hypersensitivity occurs soon after whiplash injury and is associated with poor recovery. Pain 104 (3), 509–517. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Levine JD, 1992. Serotonin is a directly-acting hyperalgesic agent in the rat. Neuroscience 48 (2), 485–490. [DOI] [PubMed] [Google Scholar]
- Takeda M, Takahashi M, Matsumoto S, 2012. Suppression of neurokinin-1 receptor in trigeminal ganglia attenuates central sensitization following inflammation. J. Peripher. Nerv. Syst. JPNS 17 (2), 169–181. [DOI] [PubMed] [Google Scholar]
- Takeda M, Tanimoto T, Nasu M, Ikeda M, Kadoi J, Matsumoto S, 2005. Activation of NK1 receptor of trigeminal root ganglion via substance P paracrine mechanism contributes to the mechanical allodynia in the temporomandibular joint inflammation in rats. Pain 116 (3), 375–385. [DOI] [PubMed] [Google Scholar]
- Teodoro FC, Tronco Junior MF, Zampronio AR, Martini AC, Rae GA, Chichorro JG, 2013. Peripheral substance P and neurokinin-1 receptors have a role in inflammatory and neuropathic orofacial pain models. Neuropeptides 47(3), 199–206. [DOI] [PubMed] [Google Scholar]
- Theeler B, Lucas S, Riechers RG 2nd, Ruff RL, 2013. Post-traumatic headaches in civilians and military personnel: a comparative, clinical review. Headache 53 (6), 881–900. [DOI] [PubMed] [Google Scholar]
- Tsuruoka M, Matsutani K, Maeda M, Inoue T, 2003. Coeruleotrigeminal inhibition of nociceptive processing in the rat trigeminal subnucleus caudalis. Brain Res. 993 (1–2), 146–153. [DOI] [PubMed] [Google Scholar]
- Vierck CJ, Yezierski RP, 2015. Comparison of operant escape and reflex tests of nociceptive sensitivity. Neurosci. Biobehav. Rev. 51, 223–242. [DOI] [PubMed] [Google Scholar]
- Viggiano A, Monda M, Chiefari M, Aurilio C, De Luca B, 2004. Evidence that GABAergic neurons in the spinal trigeminal nucleus are involved in the transmission of inflammatory pain in the rat: a microdialysis and pharmacological study. Eur. J. Pharmacol. 496 (1–3), 87–92. [DOI] [PubMed] [Google Scholar]
- Wasner GL, Brock JA, 2008. Determinants of thermal pain thresholds in normal subjects. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 119 (10), 2389–2395. [DOI] [PubMed] [Google Scholar]
- West MJ, Gundersen HJ, 1990. Unbiased stereological estimation of the number of neurons in the human hippocampus. J. Comp. Neurol. 296 (1), 1–22. [DOI] [PubMed] [Google Scholar]
- Wu L-A, Huang J, Wang W, Wang W, Wang X-J, Wu S-X, 2009. Down-regulation of K+-Cl− co-transporter 2 in mouse medullary dorsal horn contributes to the formalin-induced inflammatory orofacial pain. Neurosci. Lett. 457 (1), 36–40. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Mahmood A, Chopp M, 2009. Emerging treatments for traumatic brain injury. Expert Opin. Emerg. drugs 14 (1), 67–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Cang CL, Kawasaki Y, Liang LL, Zhang YQ, Ji RR, Zhao ZQ, 2007. Neurokinin-1 receptor enhances TRPV1 activity in primary sensory neurons via PKCepsilon: a novel pathway for heat hyperalgesia. J. Neurosci. Off. J. Soc. Neurosci. 27 (44), 12067–12077. [DOI] [PMC free article] [PubMed] [Google Scholar]