

Abstract
Neurological disorders significantly outnumber diseases in other therapeutic areas. However, developing drugs for central nervous system (CNS) disorders remains the most challenging area in drug discovery, accompanied with the long timelines and high attrition rates. With the rapid growth of biomedical data enabled by advanced experimental technologies, artificial intelligence (AI) and machine learning (ML) have emerged as an indispensable tool to draw meaningful insights and improve decision making in drug discovery. Thanks to the advancements in AI and ML algorithms, now the AI/ML‐driven solutions have an unprecedented potential to accelerate the process of CNS drug discovery with better success rate. In this review, we comprehensively summarize AI/ML‐powered pharmaceutical discovery efforts and their implementations in the CNS area. After introducing the AI/ML models as well as the conceptualization and data preparation, we outline the applications of AI/ML technologies to several key procedures in drug discovery, including target identification, compound screening, hit/lead generation and optimization, drug response and synergy prediction, de novo drug design, and drug repurposing. We review the current state‐of‐the‐art of AI/ML‐guided CNS drug discovery, focusing on blood–brain barrier permeability prediction and implementation into therapeutic discovery for neurological diseases. Finally, we discuss the major challenges and limitations of current approaches and possible future directions that may provide resolutions to these difficulties.
Keywords: Alzheimer's, anesthesia, artificial intelligence, blood‐brain barrier, CNS, depression, disease subtyping, drug design, drug discovery, machine learning, neurological diseases, pain treatment, Parkinson's, schizophrenia, target identification
Abbreviations
- 3D
three‐dimensional
- ADME‐T
absorption, distribution, metabolism, and excretion—toxicity
- DTI
drug‐target interactions
- FDA
the US Food and Drug Administration
- HTS
high‐throughput screening
- PPI
protein–protein interactions
- QSAR
quantitative structure–activity relationship
- SAR
structure–activity relationship
- SMILES
simplified molecular input‐line entry system
1. INTRODUCTION
Disorders of the central nervous system (CNS) are responsible for multiple disease states of significant economic and social impact. Despite huge progress in our understanding of the structure and functions of the CNS, the development of new drugs for CNS disorders poses unique challenges. CNS drugs have lower success rates than other drug classes due to multiple factors, including an insufficient understanding of the pathophysiology of complex CNS conditions, poor target selection/engagement, lack of efficacy in early stages of development, and the presence of a blood–brain barrier (BBB). Such challenges have led to significantly longer development time for CNS drugs, which is, on average, 15–19 years to advance from discovery to regulatory approval. 1 The whole process of developing a new drug generates a lot of data. Over the past decades, the advances in “omics” technologies, high‐throughput screening (HTS), and chemical synthesis have led to a dramatic increase in the amount of available data on chemical activity 2 and functional genomics. 3 , 4 As a result, how to efficiently combine, correlate, and analyze existing large‐scale data has become a crucial problem for CNS drug discovery.
Artificial intelligence (AI) concepts such as machine learning (ML) have the potential to accelerate pharmaceutical research by extracting novel and important information from the vast amount of complex data generated from the drug discovery process. In recent years, AI/ML‐based methods have been widely applied to many therapeutic areas and achieved state‐of‐the‐art performance in addressing diverse problems in drug discovery. Such applications of AI/ML algorithms also have shown promise in the development of CNS therapeutics—the most challenging area in drug discovery. However, we have only just begun to explore the potential of these technologies for discovering novel therapeutics and repurposing old ones for CNS diseases. Therefore, this review will focus on AI/ML‐assisted drug discovery applications in this promising direction.
Here, we provide an overview of recent developments and applications in AI/ML‐assisted drug discovery, particularly for CNS diseases. This review is intended for biomedical researchers who are curious about the potential of AI/ML for advancing CNS drug discovery and consider AI‐based tools in their research. We first provide a broad overview of AI/ML approaches in drug discovery and then review AI/ML solutions to the issues in drug discovery specific for CNS diseases. We start with a brief introduction to AI algorithms and their input molecular descriptors and then summarize AI/ML‐based methods in various stages of drug discovery, including target identification and characterization, virtual screening, lead discovery, and physicochemical pharmacokinetic property prediction. We further review recent AI/ML applications in de novo design, predicting drug sensitivity and response, drug synergy prediction, and drug repurposing. For CNS diseases specific drug discovery, we focus on AI/ML solutions to key challenges such as BBB permeability and introduce AI/ML‐assisted applications to neurological diseases, including neurodevelopmental disorders, depression, Parkinson's disease, Alzheimer's disease, anesthesia and pain treatment. We conclude the review by highlighting challenges, limitations, and future directions of AI/ML‐aided drug discovery, especially for CNS diseases.
2. AI/ML APPLICATIONS IN DRUG DISCOVERY
AI/ML has been utilized at three different stages of early drug discovery process, including target identification, lead generation and optimization, and preclinical development (Figure 1). In target discovery, AI‐based approaches have been used to integrate heterogeneous data sets to identify patterns so as to understand molecular mechanisms underlying diseases and drug activities. For lead generation and optimization, AI/ML algorithms improve the scoring functions and quantitative structure–activity relationship (QSAR) models in virtual screening pipelines and support the automation and optimization of the de novo drug design processes. In preclinical development, AI/ML approaches are employed to generate predictive models of physicochemical properties by efficiently processing large amount of chemical data and further optimize absorption, distribution, metabolism, and excretion—toxicity (ADME‐T) profiles.
Figure 1.
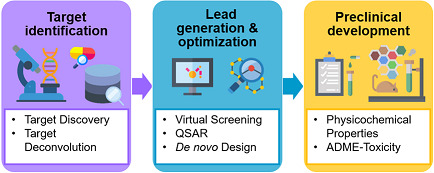
AI/ML applications in the drug discovery pipeline. AI/ML approaches provide a range of tools that can be applied in all the three stages of early drug discovery to improve decision making and speed up the process. ADME, absorption, distribution, metabolism, and excretion; AI, artificial intelligence; ML, machine learning; QSAR, quantitative structure–activity relationship
2.1. Overview of the AI/ML algorithms
To help the reader better understand AI/ML applications in CNS drug discovery, we provide a summary of AI‐based algorithms that are widely used in drug discovery. AI uses a large variety of models to build up intelligent systems, which can be classified by learning procedures. AI is frequently used to denote ML algorithms—yet they are not the same. So, it would be worth clarifying both terms at first. In this review, we follow the US Food and Drug Administration's (FDA) definition of AI. They describe AI as “the science and engineering of making intelligent machines”, while ML is “an artificial intelligence technique that can be used to design and train software algorithms to learn from and act on data”, 5 adding that all ML techniques are AI techniques, but not all AI techniques are ML techniques. Here, we provide brief definitions of the basic learning algorithms in Table 1, as these are most relevant in the context of drug discovery. AI‐related learning techniques are broadly categorized as supervised, unsupervised, semisupervised, active, reinforcement, transfer, and multitask learning. Different algorithms are used in those learning architectures to perform specific tasks such as classification or clustering. However, success with AI requires more than training an AI model. A robust AI workflow involves (i) formulating a problem, (ii) preparing data, (iii) extracting features, (iv) selecting training and testing data sets, (v) developing a model, (vi) training the model and testing its performance (cross‐validation), and (vii) applying the model to testing data sets and refining the model. Figure 2 displays the basics steps of building an AI architecture.
Table 1.
AI‐related learning techniques used in drug discovery
| Category of learning | Definition | |
|---|---|---|
| Supervised learning |
|
|
| Regression: Model finds outputs that are real variables | ||
| Classification: The model divides inputs into classes or groups | ||
| Algorithm | Task | Description |
| Naïve Bayes | Classification |
|
| Support vector machines | Classification |
|
| Random Forest | Classification/Regression |
|
| K‐nearest‐neighbors | Classification/Regression |
|
| Artificial neural networks | Classification/Regression |
|
| Deep neural network | Classification/Regression |
|
| Multiple regression | Regression |
|
| Unsupervised learning |
|
|
| Algorithm | Task | Description |
| K‐means clustering | Clustering |
|
| Fuzzy clustering | Clustering |
|
| Hierarchical clustering | Clustering |
|
| Principal component analysis | Dimensionality reduction |
|
| Independent component analysis | Dimensionality reduction |
|
| Autoencoders | Dimensionality reduction |
|
| Deep belief nets | Dimensionality reduction |
|
| Generative adversarial networks | Anomaly detection |
|
| Self‐organizing map | Dimensionality reduction |
|
| Semisupervised learning |
|
|
| Active learning |
|
|
| Reinforcement learning |
|
|
| Transfer learning |
|
|
| Multitask learning |
|
|
| Multiple kernel learning |
|
|
| Ensemble learning |
|
|
| End‐to‐end learning |
|
|
Note: The rows with gray backgrounds show the basic learning categories and their definition, while the rows following supervised and unsupervised learning parts display the different algorithms used in these categories.
Figure 2.
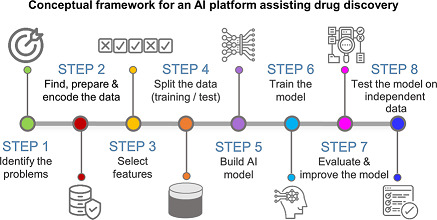
The basic steps of building an artificial intelligence (AI) platform for drug discovery. The process for developing an AI model as follows: (1) Define the problem appropriately (objective, desired outputs, etc.), (2) prepare the data (collection, exploration and profiling, formatting, and improving the quality), (3) transform raw data into features and select meaningful features (a.k.a. feature engineering), (4) split data into training and validation sets, (5) develop a model, (6) train the model with a fraction of the data, test its performance (cross‐validation) and tune its parameters with the validation set (7) evaluate model performance on the validation set and refine the model, and (8) evaluate the model on independent data not used for method development
2.2. Molecular descriptors and fingerprints for input data preparation
A key consideration in early drug discovery is to identify drug candidates with the desirable initial characteristics, which are then further developed into chemical structures with the desirable potency against the target molecule. Molecular descriptors and fingerprints are used for quantifying such physicochemical characteristics of both chemical entities and their biological target molecules. Molecular descriptors are experimentally quantified or theoretically characterized properties of a corresponding molecule that represent the physical, chemical, or topological characteristics, while molecular fingerprints are more complex descriptors that are encoded as binary bit strings. 6 , 7 Both molecular descriptors and fingerprints have crucial functions in ML‐based applications in drug discovery processes such as target molecule ranking, 8 , 9 similarity‐based compound search, 10 , 11 , 12 , 13 , 14 , 15 virtual screening, 16 , 17 QSAR analysis, 18 , 19 ADME‐T prediction of lead molecules. 20 , 21 , 22 , 23
There are various tools for molecular descriptor and fingerprint calculation, and each has a different set of features. Here, we explain the molecular descriptors (i.e., target protein descriptors and compound descriptors) and compound fingerprints, and provide the highly used programs for generating them (i.e., sequence‐based tools and structure‐based tools) in the Supporting Information. Additionally, Chuang et al. 24 comprehensively discussed how AI‐based methods (i.e., deep learning [DL]) could address limitations of molecular descriptors and fingerprints and thereby improve the predictive modeling of compound bioactivities.
2.3. AI/ML applications in target identification
A dominant approach to drug discovery is to design drug molecules that will reverse a disease course by modulating the activity of a target. 25 Drug development often begins with identification of a novel target whose modulation can lead to a therapeutic benefit with an acceptable safety margin. This is followed by validating the role of the selected target in disease in in vivo models and, ultimately, in clinical trials. Therefore, the ultimate success of a drug development project depends on early identification of promising drug targets.
A good drug target need be relevant to the disease phenotype as well as be suitable for therapeutic modulation (“druggable”). Biological and technological advances have continuously driven the generation of high‐throughput biomedical data, which present new opportunities for early identification of potential drug targets. However, the analysis of such large‐scale multidimensional biological data requires effective techniques that can produce accurate predictions for target identification. AI/ML has emerged as a powerful technology for analyzing the rapidly increasing multiomics data in the identification of potential therapeutic targets.
In literature, the “target identification” term is often used in two different contexts: Target discovery and target deconvolution. 26 The first is the discovery of a new disease target whose modulation would have therapeutic effects. The second is the identification of a target with a known active compound, which is also called “target fishing.” To avoid confusion, we will use context‐specific terms of target discovery and deconvolution rather than generic target identification.
2.3.1. Target discovery
Drug discovery begins with the identification of a novel target candidate that is followed by a target evaluation consisting of experimental target validation and theoretical assessment of its ability to bind small molecule drugs (druggability). 27 The target discovery process includes identification of targets that play a role in the disease pathophysiology, 28 assessment of druggability, and prioritization of candidate targets. However, because of the complex nature of human diseases, this process often requires more comprehensive approaches that integrate available heterogeneous data and information to understand the molecular mechanisms underlying disease phenotypes and identifying the patient‐specific changes. 29 To overcome such difficulties, researchers have applied AI/ML methods to predict “reliable” drug targets. The following sections demonstrate the AI/ML applications in different stages of the target discovery process (Figure 3).
Figure 3.
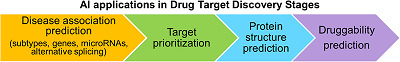
AI‐guided target discovery. AI/ML methods can efficiently analyze all available information to speed up the discovery of disease‐related drug targets. Specifically, AI/ML methods are utilized for disease subtyping, identification of disease driver genes and microRNAs, alternative splicing prediction, triaging of novel drug targets, modeling of three‐dimensional target structures, and druggability assessment. AI, artificial intelligence; ML, machine learning
2.3.1.1. Disease subtype prediction
In complex heterogeneous diseases, classifying patients into clinically and biologically homogenous subtypes is critical for understanding disease pathophysiology and developing appropriate subtype specific therapies. 30 Researchers have developed AI/ML algorithms that can integrate multiscale data to identify different etiological subtypes of complex diseases. For example, Shen et al. 31 developed iCluster, a joint latent variable model for integrative clustering analysis, which was applied to breast cancer and lung cancer and identified subtypes characterized by concordant DNA copy number changes and gene expression. 31 Yuan et al. 32 also integrated copy number variation and gene expression data by using a nonparametric Bayesian model and discovered prognostic subtypes in prostate cancer and breast cancer. 32 Zhang et al. 33 revealed the prognostic subtypes in neuroblastoma using DL‐based integration of multi‐Omics data and K‐means clustering analysis. Recently, Gao et al. 34 described a cancer classification method, deep cancer subtype classification (DeepCC), based on DL of functional spectra, which is a vector of gene set enrichment scores associating with biological functions for each patient sample. Overall, in recent years, AI/ML methods have been employed to analyze large‐scale genomic and other molecular profiling data in cancer for the identification of distinct, molecular disease subtypes. However, such AI‐based subtyping analysis have not been widely applied to other complex diseases. Implementation of robust and scalable AI/ML techniques for discovery of disease subtypes paves the way for developing more efficacious therapeutic strategies.
2.3.1.2. Prediction of disease driver genes
One of the most challenging tasks in target discovery is the prediction of disease‐causing genes from huge amount of genetic and functional genomic data. To predict these disease‐associated genes from multiomics data, researchers have employed various ML classifiers, 35 , 36 , 37 , 38 including Random Forest (RF)‐, 39 , 40 support vector machines (SVM)‐, 41 , 42 and decision tree (DT)‐based classifiers. 43 More detailed information about those applications can be found in the Supporting Information. Besides the ML‐methods using multiomics data, DriverML, 44 a supervised learning tool, identified cancer driver genes based on DNA sequence alterations from The cancer Genome Atlas (TCGA) data with superior performance over the other tools such as DriverDBv2 database. 45
In addition to ML classifiers, DL‐based methods have been implemented in more recently developed tools. For example, deepDriver 46 trained similarity networks and a convolutional neural network (CNN) on mutation data simultaneously to predict driver genes with better performance than the competing approaches when applied in breast cancer and colorectal cancer. In another example, Peng et al. 47 used deep neural network (DNN) to reduce the dimensionality of transcriptomics data to predict Parkinson's disease genes. This DNN‐based tool, namely, N2A‐SVM, consists of three steps, including extraction of vector representation of each gene in the protein–protein interaction (PPI) network, dimension reduction for the obtained vector with autoencoder, and prediction of the genes associated with Parkinson's disease using SVM.
Multitask learning has also been employed for the prediction of cancer driver genes. LOTUS, an ML‐based algorithm, predicts cancer driver genes in a pan‐cancer setting, as well as for specific cancer types, using a multitask learning strategy sharing information across cancer types. 48 For the readers who want to learn more about opportunities and challenges in predictive modeling for multiomics data sets, we suggest the review paper of Kim and Tagkopoulos. 49
Different from the tools using omics data sets, BeFree 50 was developed to extract relations between genes and diseases from text mining. This supervised learning approach utilized natural language processing (NLP) Kernel methods to identify gene–disease associations from the abstracts collected by Medline.
2.3.1.3. Prediction of disease‐associated microRNAs
The challenges in targeting disease proteins have shifted the focus in target selection to disease microRNAs (miRNAs), which are small noncoding RNAs that regulate gene expression by targeting messenger RNAs. 51 miRNAs are regarded as high‐potential drug targets due to their involvement in various diseases. 52 Therefore, considerable effort has been devoted in identifying relationships between miRNAs and diseases using ML‐based methods, such as the network based approach by Xu et al. 53 , 54 and RLSMDA. New strategies in miRNA target discovery have utilized neural networks (NN). Zeng et al. 55 developed a NN method, NNMDA to predict miRNA‐disease associations with the best performance among the existing algorithms. Application of NNMDA to lung neoplasm and breast neoplasm predicted novel disease‐related miRNAs. Very soon after that, Zheng et al. 56 published a new ML‐based method, MLMDA, which predicts miRNA–disease associations by integrating miRNA sequence, disease semantics, miRNA–disease association, and miRNA function but with slightly worse performance than NNMDA.
2.3.1.4. Prediction of alternative splicing
Alternative splicing (AS) plays a fundamental role in gene expression regulation and protein diversity by causing the generation of different transcripts from single genes. 57 Understanding the genetic variation in splicing signals is within the scope for AI/ML‐based models to discover therapeutic opportunities through novel targets. For splicing prediction and analysis, a web tool, AVISPA, 58 has been developed. For a given exon and its proximal sequence, AVISPA predicts if the exon is alternatively spliced and if it has associated regulatory elements by using a Bayesian NN classifier. However, the method by Leung et al. 59 outperformed the Bayesian NN approach for predicting AS by developing a DNN model inferred from mouse RNA‐Seq data that can predict splicing patterns in individual tissues and differences in splicing patterns across tissues. Later, Jha et al. 60 compared those two previous modeling approaches, Bayesian and Deep NN, and determined the confounding effects of data sets and target functions. On the basis of this knowledge, they developed a new target function for AS prediction with higher accuracy. For further improvement of the prediction, they developed a modeling framework that uses transfer learning to combine CLIP‐Seq, knockdown, and overexpression experiments. For enabling the usage of unlabeled data and the latent information, Stanescu et al. 61 applied semisupervised learning algorithms to AS prediction. Xiong et al. 62 built up a DL model trained to predict splicing from DNA sequence alone and successfully identified new autism‐linked genes.
2.3.1.5. Target prioritization
While increasing effort has been devoted to nominating novel drug targets involved in diseases, experimental validation of identified target candidates is an expensive and time‐consuming task. 63 Therefore, researchers have utilized AI/ML approaches to support the prioritization of the most promising target candidates for subsequent experiments. To identify and prioritize novel cancer drug targets, Jeon et al. 64 built an SVM classifier that uses features from various data types (DNA copy number, messenger RNA expression, mutation occurrence, and PPI) to prioritize drug targets specific for breast, pancreatic and ovarian cancers. To improve the disease gene prioritization process, Valentini et al. 65 combined different functional gene networks and applied a kernel‐based method to prioritize genes according to the disease MeSH terms. Then, Ferrero et al. 66 took advantage of the publicly available target–disease association data from the open targets platform training an NN classifier with semisupervised learning and predicted novel therapeutic targets. As another publicly available data source, Medline abstracts also have been benefited for developing prediction tools (i.e., DigSee 67 ) that identify disease–gene relationships and prioritize the genes based on evidence. Specifically, DigSee uses NLP to extract the relationship between diseases and genes and ranks the evidence sentences with a Bayesian classifier. Recently, Arabfard et al. 68 predicted and prioritized over 3,000 candidate age‐related human genes using three positive unlabeled learning algorithms, Naïve Bayes, Spy, and Rocchio‐SVM. They ranked the human genes according to their implication in aging based on binary gene features from 11 human biology databases. 68
2.3.1.6. Target protein structure prediction
AI/ML architectures have been applied in protein structure prediction over 30 years, and several groups have comprehensively reviewed those strategies. 69 , 70 , 71 , 72 , 73 Therefore, we will focus on recent applications in this field. Also, we provide a background of conventional protein structure prediction methods (i.e., template‐based and template‐free) for those who want to learn more about this field in the Supporting Information.
Since 1994, the Critical Assessment of protein Structure Prediction (CASP) competitions have been organized biannually for blind evaluation of the state‐of‐the‐art methods that predict three‐dimensional (3D) protein structures from protein sequences. There, each group submits structure predictions for each of the given protein sequences for which experimentally determined structures were sequestered. In December 2018, Google's AI firm DeepMind won the CASP13 competition with its latest AI system, AlphaFold. DeepMind's success generated significant interest in the protein folding community, where the researchers published several articles discussing the method. 74 , 75 , 76 , 77 AlphaFold determines the 3D shape of a protein from its amino acid sequence by merging two approaches: (i) Inferring physical contact in protein structure from residue covariation in protein sequence based on coevolution analysis of a multiple sequence alignment and (ii) identifying coevolutionary patterns in protein sequences as contact distributions by using DNNs and convert them into protein‐specific statistical energy potentials. AlphaFold system has achieved an unprecedented prediction accuracy among the ab initio methods. Although AlphaFold's performance represents a big leap in protein structure prediction, its accuracy still needs to be improved.
Inspired by AlphaFold as well as previous successful applications of DL to residue contact predictions, 78 researchers have developed different strategies to improve the protein structure prediction, including a deep residual network model, 79 a fragment library that is built using deep contextual learning techniques called DeepFragLib 80 and a community‐built, open‐source implementation of Alphafold (i.e., ProSPr). 81 The emergence of DL has suggested the rethinking of how to address the problem of protein structure and thereby, encourages the new approaches. RGN (recurrent geometric network) is an end‐to‐end differentiable model that takes a sequence of amino acids and position‐specific scoring matrices (a summary of residue propensities for mutation) as inputs and outputs a 3D structure. In contrast to the complexity of conventional structure prediction models, a trained RGN model is a single mathematical function that is evaluated once per prediction. Hence, a trained RGN makes predictions six to seven orders of magnitude faster than other methods. The same lab developed the RGN also published a data set to provide a standardized resource for training and assessing ML frameworks for predicting protein structures. The data set called ProteinNet integrates sequence, structure, and evolutionary information into preformatted input/output records. ProteinNet is available in a public repository, https://github.com/aqlaboratory/proteinnet.
Going beyond the structure prediction, researchers have employed the ML for the prediction of protein dynamics since target proteins are dynamic and sample multiple states. Ung et al. 82 used RF to classify pharmacologically relevant conformations of protein kinases. Using a 3D‐CNN, Okuno et al. 83 developed DEFMap, which extracts the dynamics information hidden in a given cryo‐EM density map. This approach allows us to grasp the dynamic changes associated with molecular recognition and the accompanying conformational selections from the cryo‐EM structure, which derive insights into the protein function as well.
The studies discussed above clearly demonstrate the utility of the AI/ML frameworks to make predictions of protein structural features from sequence alone. Rost et al. 84 comprehensively discussed how ML algorithms help to understand the effects of protein sequence variants on protein function and pathways. AI/ML algorithms are readily available for structural biologists to quickly estimate protein structures. Of course, the accuracy and speed of a framework will depend on the creativity in problem formulation, network design, and data storage. We can look forward to a rapid growth in the number of AI/ML applications in the prediction of protein structures.
2.3.1.7. Druggability
In target discovery, another crucial step is the evaluation of the target's druggability, “the likelihood of being able to modulate a target with a small‐molecule drug”. 85 In drug design, a selected target must have the biophysical properties that allow it to bind small molecules with drug‐like properties. ML‐based models usually estimate a target's druggability by using different features of it. As one of the earliest applications, SCREEN (Surface Cavity REcognition and EvaluatioN) webserver 86 was built based on an RF classifier trained on geometric, structural, and physicochemical features of drug‐binding and nondrug‐binding cavities on proteins. The classification process reveals that the most critical attributes to estimate druggability are the size and shape of the surface cavities of the protein. In the following studies, SVMs were applied to predict druggable targets based on various physicochemical properties from protein sequences. 87 , 88 Then, Costa et al. 89 constructed a DT‐based meta‐classifier by training on attributes including network topological features, tissue expression profile, and subcellular localization for each druggable and nondruggable gene. Later, Wang et al. 90 combined a biased SVM with a DL model, stacked autoencoders, to identify drug target proteins based on the sequence information of proteins. Recently, Kokh et al. 91 developed an ML tool for the druggability analysis of binding pocket variations during the protein movement. They used a logistic regression model and a CNN to identify potentially druggable protein conformations in trajectories from molecular dynamics simulations. On the contrary, Dezső and Ceccarelli 92 built up RF models for the druggability prediction of oncology drug targets to prioritize proteins according to their similarity to approved drug targets. More details on ML‐based tools designed to predict the druggability of targets can be found in the review from Kandoi et al. 93
2.3.2. Target deconvolution
Target deconvolution (a.k.a. target fishing) is an important step following the discovery of compounds that cause a desirable change in phenotype. Understanding the binding targets of phenotypic screen‐derived compounds can help design better analogs, find potential off‐targets, and thereby explain observed adverse events. However, existing experimental approaches for target deconvolution are labor, resource, and time‐intensive. Researchers have adapted computational approaches to target deconvolution problems to reduce the required sources for the experiments. Several studies implemented AI/ML algorithms into computational target deconvolution tools for higher predictive power. For example, Schneider and colleagues have widely applied self‐organizing maps (SOMs) to predict the macromolecular targets of compounds. 94 , 95 , 96 , 97 They preferred to use “fuzzy” molecular representations, such as pharmacophoric feature descriptors, since such fuzzy molecular representations demonstrated greater scaffold‐hopping potential than atomistic approaches in similarity searches. On the basis of the similarity of pharmacophoric features, their unsupervised SOM algorithm clustered the query molecules with unknown targets as well as drug‐like molecules with known targets. Hence, the trained SOM was able to transfer the knowledge of annotated drug targets to query molecules that are the nearest neighbors to known drugs. 94 They have applied this SOM approach to identify the macromolecular targets of de novo‐designed molecules, 95 complex natural products, 94 fragment‐like natural products, 96 and a natural anticancer compound. 97 Besides the SOM models, a multiple‐category Naïve Bayesian model was developed for the rapid identification of potential targets for compounds based on only chemical structure information, which is the connectivity fingerprints of compounds from 964 target classes in the WOMBAT (World Of Molecular BioAcTivity) chemogenomics database. 98 Moreover, a target‐fishing server named RF‐QSAR was built based on target SAR models that were created using an RF algorithm to rank candidate targets for a query compound. 99 A recent target identification tool, BANDIT, 100 uses a Bayesian approach to integrates six distinct data types—drug efficacies, posttreatment transcriptional responses, chemical structures, reported side effects, bioassay results, and known targets.
In the identification of the novel targets of drugs, there has been increasing interest in predicting drug–target interaction (DTI), given its relevance for side effect prediction and drug‐repositioning attempts. 101 The availability of heterogeneous biological data on known DTI has enabled the development of various AI/ML‐based strategies to exploit unknown DTI, 102 including ensemble learning, 103 , 104 , 105 , 106 tree‐ensemble learning, 107 active learning, 108 DL, 109 end‐to‐end DL, 110 and kernel‐based learning. 111 , 112 , 113 , 114 , 115 Such AI/ML‐enabled data integration strategies outperform the traditional methods in classifying both positive and negative interactions, 110 improved the quality of the predicted interactions, and expedited the identification of new DTI. 115
2.4. AI/ML applications in compound screening and lead discovery
To identify new compounds with potential interactions to target proteins, researchers commonly use HTS, an in vitro method that automatically tests large compound libraries towards a specific target. However, high cost and low hit rate of HTS have expedited the development of virtual screening (VS) alternatives, which enable cheaper and faster screening of larger compound libraries. 116 , 117 VS predicts the compounds that most likely to bind to a protein of interest using various approaches. Two broad categories of VS are structure‐based VS (SBVS) and ligand‐based VS (LBVS)—the former takes the structures of target proteins as input, 118 , 119 and the latter uses information on known inhibitors. 120 LBVS is basically “analoging” to some extent based on that similar molecules tend to exhibit similar properties, 121 and it also helps to build better pharmacophore models. SBVS and LBVS are often used synergistically: Leads from SBVS can be improved with LBVS, and data from improved yields can be used to refine models for SBVS. 122 For achieving better performance in VS workflows, AI/ML‐based methods have been utilized for both SBVS and LBVS. We will begin with the application of AI/ML methods in SBVS and continue with their applications in LBVS in the next section.
2.4.1. Structure‐based virtual screening
SBVS requires the 3D structure of a target protein to predict whether a compound is likely to bind the target. One widely used method to do this is molecular docking, which models the protein–ligand complex based on the estimated interaction energy. In recent years, ML methods have been employed in SBVS workflow to increase the robustness and accuracy of scoring functions (SFs), conformational sampling and ranking. Researchers have developed SFs using RF‐, 123 , 124 , 125 , 126 SVM‐, 127 , 128 and NN‐ 129 , 130 , 131 , 132 , 133 , 134 based learning algorithms and they outperformed the conventional SF predictions. 135 However, no ML‐based SF is superior to all the other approaches in all respects. 136 Indeed, the performance of an SF differs from target to target. 137 Therefore, researchers have developed ML‐based, target‐specific SFs to improve the efficiency of existing SFs for kinases, 138 , 139 , 140 , 141 histone methyltransferases, 142 cyclin‐dependent kinases and G protein‐coupled receptors (GPCRs), 137 and cytochrome P450 aromatase. 143
Moreover, such ML‐based models have been applied to post‐docking processes to improve the accuracy of molecular docking. For example, ML algorithms 142 , 144 , 145 , 146 , 147 , 148 improve pose/compound selection by automating the evaluation of docked ligands, which was done manually before. 149 Details about ML‐based scoring functions and AI/ML applications in the post‐docking stage can be found in the Supporting Information.
2.4.2. Ligand‐based virtual screening
When the 3D structure of a given target is available, SBVS approaches (i.e., molecular docking) can be employed. However, LBVS methods are the only option if the 3D structure of the target protein is not known. In contrast to the molecular docking that predicts the binding pose of ligands to the target protein using the protein structure, LBVS is based on the principle that ligands structurally similar to an active compound tend to have similar activity. 150 Hence, LBVS requires the information of known active compounds rather than the target protein structure. In drug discovery efforts, researchers often have a set of active compounds generated from testing molecules in biochemical or functional assays without knowing the target protein structure. In such cases, the LBVS approach can be utilized to find new ligands by assessing the structural similarity of candidate ligands to the known active compounds. The challenge is thereby to find an appropriate model for similarity that relates compound features to assay outcomes. In recent years, ML has emerged as an attractive approach to boost the predictive power of LBVS models. The specific aims of ML approaches include prediction of the active compounds against a particular target using models trained on input data sets, discrimination of drug modules from nondrug ones, and prioritization of compounds based on the probability of activity. For these purposes, researchers have used SVMs, Bayesian architectures, and artificial neural networks (ANNs) (Table S2). Further information regarding AI/ML applications in LBVS is available in some comprehensive review papers. 136 , 151 , 152
On the contrary, one of the most recent advances in AI/ML‐based LBSV was made by Stokes et al. 153 They successfully discovered new antibiotics by employing graph convolutional networks (GCN), whose outstanding performance over conventional ML models in predicting molecular properties was confirmed by two studies. 154 , 155 Using their GCN model, the authors performed a large‐scale screening and identified a promising new antibiotic, halicin. 153
In conclusion, the advances in selection and design of AI/ML algorithms for LBVS and the availability of large bioactivity data sets have enabled more accurate and faster selection of compounds that are predicted to be active against a particular target and will undergo further experimental assays eventually. Although traditional ML classifiers had been widely used in LBVS, recent successful applications have shown GCN's potential to become a popular approach for LBVS. 151
2.4.3. QSAR prediction
QSAR models are developed to identify a mathematical relationship between the physicochemical properties, which are represented by molecular descriptors, and biological activity of chemicals. These models play a prominent role in drug optimization, providing a preliminary in silico evaluation of essential attributes related to the activity, selectivity, and toxicity of candidate compounds. 156 , 157 , 158 By doing that, they significantly reduce the number of candidate compounds to be tested by in vivo experiments. QSAR models can be based on regression or classification models that depend on the underlying computational strategy. AI/ML approaches (i.e., RF, 159 , 160 SVM, 161 , 162 , 163 Naïve Bayesian, 164 , 165 , 166 , 167 , 168 , 169 , 170 , 171 , 172 , 173 and ANN 143 , 174 , 175 , 176 , 177 , 178 , 179 , 180 , 181 , 182 , 183 , 184 ) have been extensively employed in QSAR modeling (For the detailed discussion of the applications, see the Supporting Information). Notably, the RF algorithm is commonly used as a classification and regression tool 159 and considered to be the golden standard in QSAR studies. 185 Hence, the performance of new QSAR prediction tools often is compared with that of RF. Many RF‐based QSAR models have been developed, such as pQSAR, 186 a method for the soluble epoxide hydrolase, 187 and a model for Janus kinase 2. 188 When the predictive performance and interpretability of RF‐based QSAR models are compared to those of two widely used linear modeling approaches—SVMs and partial least‐squares, RF not only yields better predictive performance but also enables an amenable chemical and biological interpretation. 189
In the applications of NN to QSAR prediction, researchers use the data from a single assay using molecular descriptors as input to train an NN and record activities as training labels. However, the efficiency of those simple single‐task NN models depends on having sufficient training data in a single assay. To benefit from the data obtained from multiple assays, researchers aim to develop multitask QSAR models. Several groups constructed the multitask learning structures based on plain feed‐forward NN to avoid overfitting by learning multiple bioassays simultaneously. 190 , 191 , 192 , 193 , 194 , 195 , 196 Moreover, multitask QSAR models were also utilized for predicting the activity against multiple targets. 197 , 198 , 199
In 2012, a data science competition (www.kaggle.com/c/MerckActivity) was organized to find state‐of‐the‐art methods for QSAR. Using multitask DNNs, the winning team improved the prediction accuracy by 15% over the baseline RF method. 200 Since its introduction into the QSAR modeling, 159 RF has served as a “golden standard” and no QSAR methods other than DNNs outperform it. On the contrary, in the following DREAM challenges on predicting kinase‐drug‐binding, 201 the models based on DL algorithms did not perform better than the other learning algorithms. 202 In the next study, using the DNNs, Ma et al. 185 showed that DNNs could make better prospective predictions than RF, on large and diverse QSAR data sets. However, they could not propose a clear strategy for choosing between multitask and single‐task DNNs. Xu et al. 203 focused on demystifying multitask DNNs and explored why multitask DNNs perform significantly better or worse for some QSAR tasks. They found that multitask DNNs can boost the predictive performance if the assistant tasks have molecules in a training set with structures similar to those in the test set of the primary task and the activities between these similar molecules are correlated. Contrarily, if the assistant tasks do not include compounds structurally similar to those in the primary task test set, multitask DNNs show no improvement in prediction, regardless of correlated or uncorrelated activities. Recently, Zakharov et al. 204 combined multitask DNNs with consensus modeling to generate large‐scale QSAR models with improved prediction accuracy over the state‐of‐the‐art QSAR models.
Ensemble‐based ML approaches combining several basic models have also been used to overcome the weaknesses of individual learning models and thereby improve the overall performance of the QSAR predictors. There are various ensemble learning applications in QSAR predictions, including data sampling ensembles, method ensembles, and representation ensembles. Recently, Kwon et al. 205 proposed a model that is a combined ensemble of sampling, method, and representation with an end‐to‐end NN‐based individual classifier. Their ensemble model achieved better performance than the individual models in QSAR prediction.
2.5. AI/ML applications in prediction of physicochemical properties and ADME‐T
2.5.1. Prediction of physicochemical properties
Physicochemical properties indicate all aspects of drug action and profoundly affect the clinical success rates of drug candidates. A small molecule drug candidate must be sufficiently soluble and permeable to access its site of action and thereby engage its targets, with optimal safety profiles. Therefore, accurate prediction of the physicochemical characteristics can be beneficial for designing a new chemical entity with suitable pharmacokinetic and pharmacodynamic profiles. Researchers have adopted ML‐driven approaches to predict some key physicochemical properties, such as water solubility, membrane permeability, and lipophilicity. We provide a detailed description of each property and discuss the ML‐based techniques that specifically predict the water solubility, 206 , 207 , 208 , 209 , 210 membrane permeability, 211 , 212 , 213 and lipophilicity 214 , 215 , 216 , 217 , 218 , 219 in the Supporting Information. Although improved ML models have led to better prediction of molecular properties, the lack of standard criteria for performance evaluation has limited the progress. To address this, MoleculeNet, a benchmark collection for molecular ML was developed to serve as a unique resource for the scientific community to create advanced models for learning molecular properties. 154 To further support the comparison and development of novel models, MoleculeNet has implemented various ML algorithms. Benchmark results have shown that graph convolutional network (GCN) outperforms other traditional ML methods based on molecular fingerprints and descriptors to predict molecular properties. Recent studies have supported the superior performance of GCN. Applying GCN, Feinberg et al. 155 achieved an unprecedentedly high accuracy in predicting molecular physicochemical properties.
2.5.2. ADME‐T predictions
A successful drug development pathway must include the evaluation and optimization of pharmacokinetics, pharmacodynamics, and safety profiles of a candidate molecule. In early drug discovery, evaluation of the ADME‐T properties help researchers select good drug candidates for further development. ADME‐T properties are estimated to be responsible for half of all clinical failures. 220 In this context, in silico ADME‐T prediction models have received considerable progress over the past 40 years due to the availability of many compounds with known pharmacokinetic properties. 23 , 221 Prediction models usually try to build a direct relationship between a set of molecular descriptors and a given ADME‐T property. 222 These methods represent a compound by chemical descriptors as input features such as atom counts, surface areas, weight, van der Waals volume, partial charge information, and the presence or absence of a predefined substructure. The key substructures responsible for certain toxicity are structural alerts, of which detection in given small molecules could be used for toxicity prediction. 223 On the contrary, in these models, the toxicity properties of input compounds are HTS assay measurements of toxic effects that are highly relevant to human health, including nuclear receptor pathway assays (i.e., aryl hydrocarbon receptor, aromatase, androgen and estrogen receptor, PPAR‐gamma) and stress response pathway assays (i.e., ATAD5, antioxidant responsive element, heat shock factor response element, mitochondrial membrane potential, p53). 224 While the conventional approaches have yielded physiologically based pharmacokinetic and pharmacokinetic‐pharmacodynamic/quantitative systems pharmacology models, researchers have applied AI/ML algorithms to produce high‐quality models with improved accuracy and thus provide meaningful predictions of ADME‐T responses using chemical structure information. For predicting regulators of drug ADME‐T properties, the classification models—DT, K‐nearest‐neighbor (KNN), SVM, RF, and NN have been extensively used. Even beyond that, the introduction of DL models has led to further developments in this area. As a good example of recent advancements in AI. ML‐aided ADME‐T prediction, Alchemite 225 —a DL model—predicts ADME‐T properties by imputing heterogeneous drug discovery data, including multitarget biochemical activities, phenotypic activities in cell‐based assays, and ADME‐T endpoints.
Moreover, the introduction of capsule networks, a new class of DNN architectures, has remarkably improved the ADME‐T prediction. To predict the cardiotoxicity of drugs, Wang et al. 226 developed two capsule network architectures, including a convolution‐capsule network (Conv‐CapsNet) and a restricted Boltzmann machine‐capsule network (RBM‐CapsNet). Both models showed excellent performance with an accuracy of 91.8% for Conv‐CapsNet and 92.2% for RBM‐CapsNet. As the volume and chemotype coverage of the available ADME‐T databases are continually growing, we have witnessed a great progress in AI/ML‐guided ADME‐T prediction in recent years. Such advances in the field have been extensively reviewed. 136 , 227 , 228 , 229 , 230 , 231 , 232
2.6. AI/ML applications in de novo drug design
In de novo drug design, scientists generate novel chemical entities with desired chemical and biological characteristics from scratch, aiming to achieve particular efficacy and safety profiles in a cost‐ and time‐efficient manner. Advanced AI/ML‐based tools have enabled the automated generation of new chemical entities with suitable properties. As a result of such achievements, application of AI/ML to de novo discovery has become a popular topic over the last few years. Particularly, generative molecular design based on AI/ML has aroused considerable attention. In this section, we summarize the AI/ML algorithms utilized for de novo drug design with a focus on generative models. Those who want to learn more about this subject can check other comprehensive sources in the literature. 136 , 233 , 234
Traditional methods for generating novel chemical structures depend on the previously defined reaction or transformation rules, which bias the chemical space towards prior chemical knowledge. AI/ML‐based generative models are entirely data‐driven without relying on any explicit rules and can generate new molecules that are not present in a training set. Briefly, these generative models first learn from data, then create an abstract representation of the data, and finally use this representation to generate new data instances. 235 Thus, these generative models demonstrate all aspects of an artificially intelligent system (i.e., problem‐solving, learning from experience, and coping with new situations). 235
Recent de novo molecule‐generative models with an ML structure include adversarial autoencoders (AAE), 236 , 237 , 238 variational autoencoders (VAE), 239 , 240 and recurrent neural networks (RNN). 241 , 242 , 243 , 244 In generating novel molecules represented by simplified molecular input‐line entry system (SMILES) strings, RNN is a promising approach for learning from large sets of SMILES strings and generating ligands with similar activities to those of the training set templates, but with novel scaffolds. However, the percentage of valid SMILES, internal diversity, and the similarity of molecules to the training data set in the libraries generated by any given approach have been a matter of debate. To address these issues, Reinforcement learning (RL) has been embedded in ML architectures. 239 , 245 , 246 , 247 , 248 Introducing a task‐specific reward function, RL‐assisted models are able to produce chemically feasible and predominantly novel molecules with appropriate molecular properties. For the generation of novel small molecules with the desired characteristics, generative adversarial networks (GAN) also have been employed. For example, druGAN 237 (drug‐generative adversarial network) has been developed for producing new molecules with specific anticancer properties.
Another commonly used drug design approach is to generate new analogs/similar drugs of a given set of drugs. In such cases, the transfer learning models have been integrated into NN architectures to increase the prediction accuracy by taking knowledge acquired from training on a previous problem and applying them to a new but related problem. 249 , 250
In the generative drug design models above, many ML architectures use the SMILES as molecular representation. SMILES provides a linear representation, referred to as a SMILES string that can be translated into a graph and enables a straightforward application. However, it has one or more limitations: Generated SMILES may not represent a chemically feasible structure, and even a single character alteration in a SMILES representation can change the underlying molecular structure significantly. 251 To overcome its limitations, researchers proposed several solutions like converting SMILES strings into a new SMILES‐like syntax 252 or utilizing grammatical evaluation of the SMILES syntax. 253 Besides the SMILES string representation, molecular graphs have also been used to train ML‐based molecule generation algorithms. 254 In molecular graph generators, structures are directly represented as graphs in every step and substructures are inferred from the partially generated molecular graphs. 255 Examples of such ML models to design de novo molecules based on graph representation includes GANs 256 , 257 and VAEs. 258 , 259
In addition to the models mentioned above, some AI/ML‐driven de novo molecule design tools are distinguished by introducing novel approaches. An automated de novo molecular design tool, DINGOS, 260 has been developed to emulate the approach of a synthetic chemist. It assembles drug‐like new compounds through modular and synthetically feasible design schemes, considering the synthetic feasibility of each step. In brief, the DINGOS algorithm combines a rule‐based approach with an ML model trained on known successful synthetic routes, while the former ensures the synthesizability and the later provides a directed approach to limiting the output molecules to compounds with desirable similarity to the template. Another remarkable ML‐based generative approach is proposed by Méndez‐Lucio et al., 261 which bridges systems biology and molecular design. To our knowledge, it is the first AI/ML‐based drug design tool that combines transcriptomic and structural data. Conditioning a GAN architecture with compound‐induced transcriptomic data (i.e., L1000 data set), they can automatically design molecules that potentially produce the desired transcriptomic outcome. Their model allows the design of active‐like molecules for a desired target using just gene expression signature of target perturbation. However, the current version is not capable of generating compounds that can reverse disease‐related gene expression signatures. Also, its performance has not been evaluated in a real drug‐discovery setting yet.
Among all the studies of AI/ML‐based generative molecular design, maybe the most‐mentioned 262 one is published by Insilico Medicine, 263 showing how AI for generative chemistry can be used to drive rapid drug discovery. The goal of the study was to demonstrate that efficacious drugs can be developed in just 21 days for a new target. For this purpose, they have developed a generative tensorial reinforcement learning (GENTRL) model, which can be seen as an advanced version of their earlier algorithms on VAE 238 and GAN, 247 to design DDR1 kinase inhibitors. Notably, this study has two major limitations: First, DDR1 is considered to be the most promiscuous kinase 264 ; thus, developing compounds targeting this protein may be considered low hanging‐fruit. Second, the seemingly novel compound is highly similar to the widely used cancer drug ponatinib, indicating the limitation of the approach 265 in assessing truly novel scaffolds. Therefore, there is still room for improvement of AI/ML‐inferred small molecules to obtain a clinical candidate.
2.7. AI/ML applications in prediction of drug sensitivity and response
Personalized drug response prediction aims to improve the targeted therapy response in complex diseases like cancer. 266 However, the limited application of candidate drugs in clinical settings and the heterogeneity among cancer patients make it difficult to tailor therapy for each individual cancer patient. Personalized treatment design requires predictive methods that are capable of exploiting large, heterogeneous, and sparsely sampled data sets. Accurate AI/ML‐based models employing in vitro and in vivo data sets have the potential to improve the prediction of response of cancer cells to a given compound. There are various AI/ML models to predict drug sensitivity and anticancer drug response. In such efforts, elastic net regression, 267 , 268 , 269 ensemble‐based approaches, 270 , 271 transfer learning, 272 autoencoders, 266 , 273 , 274 , 275 and multitask learning approaches 276 , 277 , 278 have been widely used. The details about these AI/ML applications can be found in the Supporting Information.
2.8. AI/ML applications in prediction of drug–drug interactions
In the treatment of complex diseases such as neurological disorders, diabetes, cancer, or cardiovascular disease, drug combinations are highly utilized for medical intervention. Coadministration of drugs in the treatment aims to enhance efficacy, reduced toxicity, and prevent the emergence of resistance. Drug combinations are classified as synergistic, antagonistic, or additive. Drug synergy is the interaction of two or more drugs, causing the total effect of drugs to be greater than sum of individual effects of each drug. 279 If drugs act synergistically, lower doses of each drug could potentially be enough to provide the desired outcome allowing for less adverse effects. Opposite to synergism, the antagonistic combination means that the combined activity of the drugs is lower than the response of the individual agents. 280 Finally, a drug combination is considered to be additive when the response of each drug neither masks nor enhances the efficacy of others. 281 Although combinatorial therapy has advantages over monotherapy, developing a new drug combination regimen that can be transferred to the clinic is still challenging. So far, the effective drug combinations have been suggested based on either clinical experience or HTS of drug pairs at different concentrations on cell lines. However, the former involves the risk of harm to patients, and the latter is unfeasible to test the complete combinatorial space. 282 To accelerate conventional combinatorial therapy efforts, AI/ML algorithms have begun to be utilized for prioritizing the drug pairs and exploring the larger combinatorial space. Tonekaboni et al. 283 introduced some examples of various ML‐based prediction frameworks for drug–drug interactions. To avoid duplication, we overview the AI/ML applications in combinatorial therapy after that time, including the applications in cancer 284 , 285 , 286 , 287 , 288 and depression treatment, 289 antimalarial, 290 and antibiotic 291 discovery, along with the available AI/ML‐based tools to predict the synergistic effects of drug combinations 292 , 293 , 294 in the Supporting Information.
In addition to the synergistic effects, drug–drug interactions can induce unexpected adverse drug reactions. Such adverse reactions caused by drug–drug interactions could lead to death in some extreme cases. 295 Therefore, AI/ML‐based models have been developed to predict the risk of side effects due to drug–drug interactions. Applications of GCN, 296 DNN, 297 and ML architectures 298 showed promising results for predicting adverse drug reactions of drug combinations. Lee and Chen 299 extensively discussed the role of ML approaches in detection and classification of side effects caused by drug–drug interactions in their review of previous studies. In a recent study, Shankar et al. 300 predicted the adverse drug reactions of coadministered drug pairs using an ANN trained on transcriptomic data, compound chemical fingerprint, and Gene Ontologies. 300
2.9. AI/ML applications in drug repurposing
Drug development and trials in animals and humans is a time‐consuming and expensive process. In general, the whole process for developing a new FDA‐approved drug requires 10–17 years of period and the tremendous cost of $2.6 billion. 301 However, high expenditures for drug development has not been able to increase the rate of approved drugs. 302 Among the reasons for this limited approval rate, a key factor is the continued adherence to the classical “one gene, one drug, one disease” paradigm in the traditional drug development. 303 Since drug targets do not operate in isolation from the biochemical system, each DTI must be studied in a broader integrative context. 304 This approach provides new insights into “off‐target” effects (i.e., side effects), resistance to precision therapy, and drug mechanism of action that can inform drug‐repurposing efforts.
Drug repurposing, also known as drug repositioning, denotes the new indications of existing drugs and is an alternative over the de novo drug development. Although the unknown underlying complex biology and pharmacology has challenged the drug‐repurposing attempts, intelligent computer algorithms offer a strategy for detecting potential drug indications by integrating large‐scale heterogeneous data (i.e., genomic, transcriptomic, phenotypic, chemical, and bioactivity) from hundreds of approved drugs. Various specially designed AI/ML models have been proposed for detecting novel drug indications. Here, we classify the ML applications for drug repositioning into the following three categories: (i) Similarity‐based methods that employ different types of classifiers like logistic regression, 305 , 306 SVM, 307 , 308 , 309 RF, 310 , 311 KNN, 312 and CNN, 313 (ii) feature vector‐based methods that utilize supervised 314 , 315 , 316 , 317 , 318 and semisupervised 319 , 320 , 321 learning algorithms, and (iii) network‐based methods that mainly use semisupervised learning algorithms (e.g., Laplacian regularized least square, 322 , 323 , 324 label propagation, 325 random walk, 326 and RF 310 ). We provide an in‐depth discussion of these three classes of AI‐based drug repositioning applications in the Supporting Information. Particularly, in early 2020, researchers at MIT published a milestone paper using a DL approach to antibiotic discovery. 153 They trained the deep GCN model based on molecular features and predicted halicin as an antibacterial molecule from the Drug‐Repurposing Hub. Halicin showed a broad‐spectrum activity against drug‐resistant strains in mice. This is the first time an AI/ML‐assisted tool was used to identify thoroughly new types of antibiotic from scratch, without the need for any previous human assumptions.
3. AI/ML APPLICATIONS IN CNS DRUG DISCOVERY
CNS diseases are a group of neurological disorders that impose a significant economic and social impact. Development of new drugs for CNS diseases poses unique challenges compared to other diseases, including the complexity of brain anatomy and function, incomplete understanding of the biology of the complex nature of CNS diseases and the presence of BBB. In this section, we present an overview of AI/ML‐based approaches to meet challenges such as BBB permeability in CNS drug discovery (Figure 4).
Figure 4.
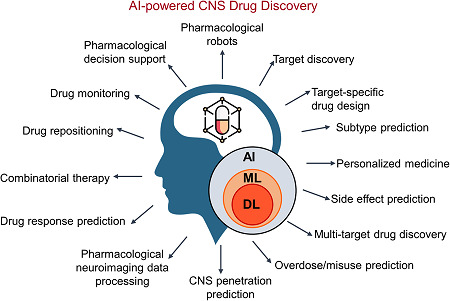
AI/ML‐enabled improvements in the treatment of CNS diseases. DL is a subset of ML, which is a subset of AI and their applications address a wide range of challenges in CNS drug discovery and development. The application fields portrayed here are discussed in the Section 3. AI, artificial intelligence; CNS, central nervous system; DL, deep learning; ML, machine learning
3.1. BBB permeability prediction
Despite significant progress in our understanding of CNS diseases, the development of novel therapies for CNS diseases faces some great challenges. In addition to the difficulties in CNS target identification, designing new molecules with the ability to penetrate the BBB is also a major obstacle. The role of the BBB is to protect the brain from variations in blood composition (e.g., hormones, amino acids, and potassium) and circulating pathogens. It consists of capillary endothelial cells that are lined by the basal lamina made from structural proteins (i.e., extracellular matrix proteins collagen and laminin), pericytes, astrocytic endfeet, and microglial cells. 327 This biologic membrane allows the uptake of water, glucose, and essential amino acids, the efflux of small molecules and nonessential amino acids from the brain to the blood and the passage of some molecules by passive diffusion. 328 While negligible penetration is desirable to minimize the brain side effects for peripheral drugs, high penetration is needed for CNS‐active drugs. To improve success rates in CNS drug discovery, the BBB permeability of drug candidates needs to be addressed early in the drug discovery process.
In recent years, AI‐based predictive models have been proposed to minimize the number of laborious, expensive, time‐consuming BBB permeability experiments that need be carried out in CNS drug discovery. For the construction of BBB permeability predictive models, researchers have employed various supervised learning approaches, such as SVM, 222 , 329 , 330 , 331 , 332 , 333 recursive partitioning (RP), 334 , 335 Gaussian process, 336 DT, 337 KNN, 338 linear discriminant analysis, 339 consensus classifier, 340 and ANN. 341 , 342 , 343 All of these methods were developed to process physical and chemical features, which mainly include molecular weight, hydrophilicity (ClogP), lipophilicity (ClogD), topological polar surface area, acidic and basic atoms numbers, hydrogen bond donors and acceptors, water‐accessible volume, flexibility (rotatable bonds), van der Waals volume, and ionization potential.
The predictive capability of all the methods mentioned above is limited to passive diffusional uptake and predominantly relies on few molecular descriptors. However, many molecules, for example, glucose and insulin, pass BBB via complex mechanisms that involve specific drug‐transporter/drug‐receptor interactions. 344 , 345 Hence, such mechanisms are hard to be described by simple physicochemical features of compounds. Moreover, achieving therapeutic drug concentrations in CNS may be limited by membrane transporters such as the ATP‐binding cassette and efflux transporter P‐glycoprotein (P‐gp), 346 which mediates efflux of drugs from the BBB. Although the primary role of these efflux transporters is limiting the brain entry of neurotoxins, they also limit the entry of many therapeutics and may contribute to CNS pharmacoresistance. 347 , 348 Therefore, prediction methods need to both overcome the limitations of physicochemical features and address the multiple mechanisms associated with the drugs that pass the barrier and sustain in the brain. For this purpose, Yuan et al. 333 developed an SVM model by combining physicochemical properties and molecular fingerprints: The former is related to passive diffusion while the latter is associated with specific interactions, such as uptake, efflux, and protein binding. When compared to other SVM‐based BBB permeability predictors, the improved accuracy of their model shows that integration of the physicochemical properties and fingerprints can yield better predictions. Actually, all the AI/ML‐based models we have mentioned so far have been trained only on molecular properties disregarding the other types of information related to the efficacy of CNS drugs.
Clinical trials of many drug candidates generate a large amount of phenotypic data in CNS, but the relationship between the CNS side‐effects of drugs and their BBB permeation has not been adequately captured. To bridge the knowledge gap, Gao et al. 349 developed a BBB permeability prediction tool utilizing drug clinical phenotypes (drug side effects and drug indications). Although they explored the BBB permeability prediction from a new angle by accounting for passive diffusion as well as putative contributions of active transport and other complex mechanisms, the accuracy of their SVM method still needs to be improved. In fact, the features based on physics and chemistry are different; hence, the relation between drug side effects and therapeutic effects is more abstract and deeper. 350 For this reason, classical classification algorithms are not able to efficiently explore the relationship between data and results. On the contrary, DL architectures have the ability to extract useful information from complex data structures with abstract relationships. Therefore, Miao et al. 350 built a DL model to predict the BBB permeability of drugs based on clinical features and achieved better performance than the other existing methods.
3.2. AI/ML applications in drug discovery for neurological disorders
3.2.1. AI/ML applications in drug discovery for neurodevelopmental disorders
Schizophrenia is arguably the most puzzling of psychiatric disorders. 351 As a neurodevelopmental disorder, 352 schizophrenia shows a lifetime prevalence of 0.30%–0.66%, 353 generally beginning before age 25 years and persisting throughout life, making it one of the leading factors of global disease burden. 354 Despite more than a century of research, its complex pathophysiology remains unknown, 355 and currently, there is no effective drug for schizophrenia. Therefore, there is a need for alternative strategies to develop innovative drug treatments for schizophrenia. 356 In recent years, AI/ML has seen as a promising technology to inform schizophrenia diagnosis, 355 , 357 detecting heterogeneity, 358 , 359 , 360 subtyping, 361 , 362 and treatment.
In drug discovery studies for schizophrenia, researchers have utilized AI/ML methods with various purposes, including drug target identification, 363 , 364 developing QSAR models, 365 predicting monitoring dosing compliance, 366 predicting GPCRs targeting compounds, 364 and drug repositioning. 367 Specifically, schizophrenia target genes were identified based on publicly available microarray data sets using an SVM‐RFE (recursive feature elimination)‐based feature selection, where the genes initially ranked by an SVM classifier and the signature was then identified by discarding the genes that were not differentially expressed. To detect optimal biomarkers of presynaptic dopamine overactivity, which may cause schizophrenia, an SVM classifier was used. 363 SVM classifiers were also used to predict QSAR models of the GABA (gamma aminobutyric acid) uptake inhibitor drugs, which can be beneficial in the treatment of schizophrenia. 365 Moreover, SVM outperformed the other ML methods in predicting the repositioning drugs for schizophrenia when trained on drug expression profiles. 367 On the contrary, for schizophrenia subtyping, an unsupervised learning approach, multi‐view clustering, was employed by combining transcriptomic data with clinical phenotypes. 368 Setting a good example of the beneficiary of AI/ML in clinical drug trials, a novel AI platform AiCure 366 on mobile devices was used to assess the dosing compliance in Phase 2 clinical trial in schizophrenia patients. It, simply, confirms the medication ingestion visually by using facial recognition and computer vision.
One of the major obstacles in developing AI/ML methods for schizophrenia drug discovery is data availability. 369 Publicly available, large‐scale, well‐structured information on neural phenotypes, genomics, and clinical stages are greatly lacking, which arouses questions for the generalizability of AI/ML algorithms across different data sets without performance loss. However, the availability of such integrative databases can encourage the development of AI/ML‐based methods to investigate personalized therapies by solving the disease heterogeneity.
Another neurodevelopmental disorder is autism spectrum disorder (ASD), which is characterized by deficits in social communication and social interaction and the presence of restricted, repetitive patterns in behaviors or interests. 370 ML methods have been utilized in ASD research for improving the diagnosis 371 and prognosis prediction. 371 Also, there are few ML applications in drug discovery for ASD. For example, ML‐based cluster analysis (i.e., affinity propagation and k‐medoids) of clinical data (i.e., signs and biomarkers) exhibited a good performance in drug response prediction of ASD patients. 372 Moreover, Bayesian ML models trained on HTS data revealed the potential repurposing of nicardipine or other dihydropyridine calcium channel antagonists for the treatment of Pitt Hopkins Syndrome, a rare genetic disorder that exhibits features of autistic spectrum disorders. 373 Recently, ML algorithms have been employed to predict the functional effects of variants in voltage‐gated sodium and calcium ion channels, which have been associated with ASD, schizophrenia and developmental encephalopathy. 374 Being trained on sequence‐ and structure‐based features, the ML model predicted the gain or loss of function effects of likely pathogenic missense variants in ion channels and the results were validated in exome‐wide data. On the contrary, the toxic compounds may trigger the recent increases in neurodevelopmental disorders among children. 375 To identify developmental neurotoxicants, researchers developed ML algorithms to predict the neurodevelopmental toxicity of compounds. 376 , 377
3.2.2. AI/ML applications in drug discovery for depression
AI/ML‐based methods have been utilized in psychiatric drug discovery, especially for pharmacological decision support. 367 , 378 , 379 In a depression study, researchers have developed a gradient boosting machine using the predictors identified by the elastic net to predict whether a patient will achieve symptomatic remission using an antidepressant, citalopram. 380 This model was also successfully applied to an escitalopram treatment group of an independent clinical trial. 378 In the next study of Chekroud et al., 381 they clustered the symptoms using an unsupervised learning approach (hierarchical clustering) and predict the responsiveness of each cluster to the treatment of different antidepressant drugs using the same model in the previous study. To provide decision support for clinicians to select the best drugs for a given cluster of symptoms, a web‐based application was designed. This AI‐based service is prospectively tested in hospital settings and thereby serve as a promising model for direct research translation. 382
On the contrary, the model of Chekroud et al. 380 has some limitations. The model only predicts whether a patient responds to a specific antidepressant without measuring the degree of antidepressant response. Since it was designed for only one antidepressant, the model is not capable of selecting the most effective drugs among various antidepressant candidates for patients. 383 To address these limitations, Chang et al. 383 developed an Antidepressant Response Prediction Network (ARPNet) model based on an NN architecture. Through the literature‐based and data‐driven feature selection process, ARPNet predicts the degree of antidepressant response, whether the patient will reach clinical remission from depression, and a patient's response to a combination of one or more antidepressants.
Electroencephalography (EEG) and functional magnetic resonance imaging (fMRI) data also have been employed in predicting drug responses to treatments of depression. Zhdanov et al. 384 used an SVM classifier to accurately predict the outcome of escitalopram treatment using patients' EEG data at the baseline and after the first 2 weeks of treatment. To identify a robust signature from resting‐state EEG that would predict response to antidepressants, Wu et al. 385 designed an end‐to‐end prediction algorithm with a latent space model. They applied their algorithm, Sparse EEG Latent SpacE Regression (SELSER), to data from an imaging‐coupled, placebo‐controlled antidepressant study and identified an EEG signature of patient's response to antidepressant treatment (i.e., sertraline). Ichikawa et al. 386 aimed to develop a melancholic depressive disorder biomarker to extract critically important functional connections (FCs) from fMRI data. By combining two ML algorithms (i.e., L1‐regularized sparse canonical correlation analysis and sparse logistic regression), they developed a classifier for melancholic depressive disorder and found out that antidepressants had a heterogeneous effect on the identified FCs of melancholic depressive disorder.
Although some of the recent AI/ML‐aided tools have been rapidly translated into the clinical trials, the AI/ML methods still are not used widely in clinical practice, while AI has been employed in psychiatric research over 20 years. 387 To close the gap between research and clinic, we need to improve the validity of diagnostic and prognostic labels, representability of the features, and generalizability of models. 388 As scientists continue to work to bridge the gap between research and clinic, it will be possible to provide efficient, personalized treatments based on a patient's unique characteristics. 389
3.2.3. AI/ML applications in drug discovery for Parkinson's disease
Parkinson's disease (PD) is the second most common age‐related neurodegenerative disorder, affecting over 1% of the population above the age of 60, increasing to 5% in individuals above 85 years of age. 390 PD is a prime example of a multifaceted disease, including a broad range of motor and non‐motor symptoms and possible contribution of genetic and environmental risk factors. 391 Currently, there is no treatment to prevent the progressive depletion of dopaminergic neurons in the substantia nigra that underlies the movement control and cognitive loss, which is manifested with tremors and memory loss. 392 , 393 Available drug treatments are based on the administration of levodopa (l‐dopa) and catechol‐O‐methyltransferase or monoamine oxidase B inhibitors, offering only symptomatic relief to the patients. 392
In PD research, previous AI applications have focused on diagnostic biomarker discovery in cerebrospinal fluid (CSF) and blood 394 , 395 , 396 , 397 and remote monitoring of treatment response by using electronic wearables. 398 , 399 , 400 , 401 , 402 On the contrary, recently, AI/ML has received little attention in PD drug discovery. Particularly, Shao et al. 403 initially built SVM models to quickly select the compounds containing indole–piperazine–pyrimidine scaffold among large chemical databases and subsequently identified novel compounds that simultaneously bind the two receptors—adenosine A2A receptor and dopamine D2 receptor—implicated in the PD pathophysiology. In another study, Sebastián‐Pérez 404 utilized several ML techniques to infer QSAR models for the identification of putative inhibitors of LRRK2 protein, a key genetic risk factor for familiar and sporadic PD. Moreover, AI‐based technologies have helped overcome the drug side effects in PD treatment. While l‐dopa has remained the cornerstone of PD therapy for reducing the symptoms associated with dopamine deficiency, almost half of PD patients treated with it eventually develop levodopa‐induced dyskinesia (LID), a side effect that causes abnormal involuntary movements. In a review paper, Johnston et al. 405 discussed the use of AI platforms to identify repurposing candidates for LID treatment and highlighted the potential of AI approaches by designing a drug repositioning case study. To identify novel repurposing candidates that may reduce LID, they utilized a literature mining approach based on an IBM Watson engine, where the semantic similarity and a “graph diffusion” algorithm were applied to score and rank each candidate drug.
Along with the identification of novel and repurposing candidates, AI/ML techniques have been applied to the development of in vitro and in vivo PD models for drug screening. Monzel et al. 406 created a human midbrain organoid model of PD as an in vitro toxicity assay and built an RF classifier to predict compounds' neurotoxic effect on organoids based on cellular features. To establish an efficient drug‐testing route, Hughes et al. 407 developed a zebrafish model of PD together with an AI/ML‐based method to classify movement disorders in this model using high‐resolution video captures. Encouraging results of all the studies discussed above, highlight the potential benefits of AI/ML applications for the discovery of efficient and multitargeting drugs against emerging targets in PD as well as the screening of the drug effects on PD models.
3.2.4. AI/ML applications in drug discovery for Alzheimer's disease
Increasing life expectancy has produced a dramatic rise in the prevalence, and thus impact, of aging‐related diseases. The most prevalent neurodegenerative disease in older adults is Alzheimer's disease (AD), characterized by insidious and progressive impairment of behavioral and cognitive functions, including memory. 408 The cause of AD is still unclear; however, generally accepted neuropathological hallmarks of AD include extracellular A‐beta plaques and intracellular neurofibrillary tangles, along with neuronal and synaptic loss and/or dysfunction. 409 Current drugs for AD target cholinergic and glutamatergic neurotransmission, thus improving symptoms, although they show limited benefits to most AD patients. 410 Therefore, new treatments are urgently needed to prevent or delay disease onset, slow its progression, or improve patients' symptoms. 411 However, drug development for AD has been extraordinarily difficult, with a failure rate of over 99% and no new drug approved since 2003. 411 AD drug failures are likely due to the lack of sufficient target engagement and toxicity, while drug discovery efforts mainly challenged by an incomplete understanding of AD pathogenesis, multifactorial etiology, and complex pathophysiology.
In recent years, AI/ML‐based models have become popular in AD research, mostly utilizing for AD diagnosis and prognosis in dealing with electronic health records and images. 412 On the contrary, AI/ML techniques have not been widely employed in AD drug discovery. However, there have been a few studies that show the potential benefits of AI/ML applications for the discovery of AD drugs. ML approaches have assisted the target identification and characterization in AD, which is the initial phase of drug discovery. For example, Cordax 413 (https://cordax.switchlab.org) is a novel structure‐based amyloid core sequence prediction method that implements ML to detect aggregation‐prone regions in proteins as well as to predict the structural topology, orientation and overall architecture of the resulting amyloid core. As an aggregation predictor, it uses structural information on amyloid cores currently available in the protein databank and translates structural compatibility and interaction energies into sequence aggregation propensity using logistic regression. Along with the characterization of amyloid fibrils, ML approaches have been utilized for identifying potential drug targets. HENA, 414 a heterogeneous network‐based data set for AD, integrates distinct data types (i.e., PPI, gene coexpression, epistasis, genome‐wide association study, gene expression in different brain regions, and positive selection data) through GCN to predict AD‐associated genes.
Researchers have built ML models—SVM, ANN, and RF—to predict the inhibitory effect of compounds against AD‐related proteins—histone deacetylase (HDAC), 415 acetylcholinesterase (AChE), 416 and S100 calcium‐binding protein A9 (S100A9), 417 respectively. Although these target‐specific models were successful for predicting the bioactive compounds, a high level of reliability is necessary for prioritizing compounds that are ultimately translated into assays. To generate hyper‐predictive ML models, Jamal et al. 418 have included dynamic properties of compounds and protein–ligand interactions. Extracting the dynamic descriptors from molecular dynamics simulations of caspase‐8 ligand complexes to train ANN and RF models, they predicted the active compounds against caspase‐8, which plays a key role in causing AD. The major challenge in developing such predictive models of inhibitor activity is the lack of data on true‐negative compound–protein interactions. To address this challenge, Miyazaki et al. 419 constructed a graph CNN model to explore compounds specifically targeting proteins without using the information on the true‐negative interaction and applied the model to identify inhibitors of BACE1 enzyme, a major target for AD.
Although these ML applications have advanced the discovery of single‐target inhibitors, the complex nature of AD requires the discovery of multitarget drugs to address the multiple pathways contributing to disease pathogenesis. Therefore, researchers have developed ML algorithms for predicting multitarget‐directed compounds against AD. Kleandrova et al. 420 designed seven molecules as triple target inhibitors of AD‐related proteins, namely GSK3B, HDAC1, and HDAC6 by combining perturbation theory and ML‐based on ANN. Using a new multitask QSAR model based on the linear discriminant analysis, Concu et al. 421 predicted the inhibitors of the two isoforms of the monoamine oxidase (MAO) enzymes, MAO‐A and MAO‐B, which are involved in the pathology of AD, PD, and other neuropsychiatric disorders. As epigenetic therapeutics for AD, HDAC inhibitors have shown promise; however, nonspecificity and nonselectivity are the major problems of current HDAC inhibitors. Therefore, Gupta et al. 422 combined VS and ML to classify the HDAC inhibitors and identified a novel compound that potentially inhibits all isoforms of class I and class IIb HDAC for AD therapy. In addition to these, Fang et al. 423 built 100 binary classifiers based on the naive Bayesian and RP algorithms to predict active small molecules against 25 key targets toward AD. Experimental validation of the predicted molecules yielded a compound that is a dual cholinesterase inhibitor and H3R antagonist. In their following study, 424 the system has been updated by assembling 204 binary classifiers towards 54 critical targets related to AD and the information of the classifiers was shared in a web server named AlzhCPI. Utilizing this classifier system, another group of researchers 425 has identified multiple targets of a traditional Chinese herbal medicine formula, Naodesheng, for application to AD. Natural products has continued to generate an increased interest as a mean of discovering novel bioactive compounds against AD. Grisoni et al. 426 proposed a VS protocol based on ML models to explore the bioactive synthetic mimetics of the natural product galantamine, which is the first natural product‐based AD drug approved by the FDA in 2001. 427 Using an ML‐based selection and target profiling program, they identified galantamine‐mimetic small molecules with multitarget activity on enzymes and receptors related to AD.
Besides the predictions of multitarget compounds based on their bioactivity against known drug targets in AD, Jamal et al. 428 predicted small molecules that show a high binding affinity for ML‐inferred possible therapeutic targets. Unlike previous studies that target known AD‐related proteins, they initially predicted the probable AD‐associated genes using ML classifiers that are trained on network, sequence and functional features. Then, they used a conventional VS tool to select the compounds that have high affinity for the majority of the predicted targets.
In addition to applications for identifying small molecules towards therapeutic targets for AD, ML techniques also have been utilized in drug repositioning efforts. For example, telmisartan has been associated with AD by a network‐based classification model. 310
AI/ML approaches have also been applied to drug response studies to treat AD patients in a more precise, personalized way. Hampel et al. 429 has built an AI/ML‐based precision medicine framework for identifying the genomic biomarkers of response to AD therapy. Specifically, they studied blarcamesine (ANAVEX2‐73), a selective sigma‐1 receptor agonist, in a Phase 2a trial, where they obtained the patients' whole‐exome and transcriptome data and recorded the measures of safety, clinical features, pharmacokinetics, and efficacy. They analyzed the relationship between the patient data and efficacy outcome measures using unsupervised formal concept analysis, which ultimately identified the biomarkers of drug response. On the contrary, Lu et al. 430 evaluated the therapeutic effects of Dengzhan Shengmai formula, a traditional Chinese medicine, on AD patients by analyzing the diffusion tensor imaging data with ML. Their ML classifier revealed significant white‐matter network alterations after treatment.
3.2.5. AI/ML applications in anesthesia and pain treatment
The CNC drugs include general anesthetics and the analgesics, as well. In the past few years, we have witnessed the widespread use of autonomous and AI‐based recommender systems in therapeutic decision making in anesthesia and pain management. Especially, pharmacological robots have become an integral part of the anesthesia field, offering a personalized anesthetic drug dosage for maintaining patient homeostasis during general anesthesia and sedation. 431 These robots use complex ML algorithms based on patient data (e.g., EEG monitor, blood pressure, heart rate, etc.) and pharmacokinetic features of drugs to provide the optimal drug dosage. The role of pharmacological robots and even more intelligent autonomous systems (i.e., cognitive robot, which can recognize crucial clinical state that requires human intervention) in the anesthesia field has been comprehensively overviewed by Cédrick et al. 432 Besides the robotic systems, ML applications assisted the clinicians 433 to monitor the drug‐specific anesthetic states 434 , 435 , 436 and predict the adverse outcomes in anesthesia patients. 437 , 438 , 439
Similar to the anesthesia field, AI models have mainly utilized for clinical decision support in pain management. With the increasing amount of data collected by state‐of‐the‐art monitoring sensors and the Internet of Things, the AI‐assisted patient‐controlled analgesia has a great potential for personalized pain therapy. 440 The other clinical applications of AI systems in pain management include prediction of pain severity/modality and analgesic requirements, 441 , 442 , 443 individualized medicine decision support in analgesic treatment, 444 , 445 prediction of the effectiveness of the analgesics, 446 , 447 and prediction of medication overuse. 448 , 449 , 450 Besides the clinical applications, researchers have employed ML methods at the early stages of analgesic discovery, such as identifying novel genes and pathways associated with acute and chronic pain 451 and predicting inhibitors of a drug target for pain (i.e., NaV1.7 sodium channel). 452 To facilitate the prediction of novel multi‐target analgesics or drug combinations for pain treatment, researchers have established a comprehensive pain‐domain‐specific chemogenomics knowledgebase that includes the analgesics in current use, pain‐related targets with all available 3D structures, and the compounds reported for these target proteins. 453
4. CONCLUSIONS AND FUTURE DIRECTIONS
Given the complexity of neurological disorders, CNS drug development is still a long, expensive, inefficient, and challenging process with a low rate of new successful therapeutic discovery. To overcome the challenges of CNS drug discovery, researchers have utilized AI/ML‐based methods, which have played a promising role in all stages of drug discovery for a variety of diseases (Table 2). In general, AI/ML practices in pharmaceutical development have aroused great interest among researchers working in academia and industry. The number of start‐ups in this area has grown rapidly and reached 230 by June 2020. 454 Also, many pharmaceutical companies have invested in internal AI‐based research programs as well as in collaboration with AI start‐ups and academic institutions. 455 , 456 Recently, we have witnessed a massive collaborative effort by both academia and industry in response to the COVID‐19 outbreak. Labs and AI firms have shared their data and pipelines in open‐sourced platforms. For example, Google Deepmind has released the 3D structures of SARS‐CoV‐2 proteins that have been predicted by their AlphaFold system. Although AI‐enabled solutions have emerged as a crucial tool for transforming the process of therapeutic development, the use of AI technologies to improve CNS drug discovery is still at an early stage. Below, we discuss the limitations as well as the future directions to guide further advancement in this evolving field.
Table 2.
The promise of AI/ML‐based drug discovery strategies in CNS disorders
| Application field | Schizophrenia | Autism spectrum disorder | Depression | PD | AD | Anesthesia | Pain treatment |
|---|---|---|---|---|---|---|---|
| Diagnosis/prognosis | ✓ | ✓ | ✓ | ✓ | ✓ | ‐ | ✓ |
| Subtyping | ✓ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Heterogeneity detection | ✓ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Target identification | ✓ | ‐ | ‐ | ‐ | ✓ | ‐ | ✓ |
| Inhibitor discovery | ✓ | ✓ | ✓ | ✓ | ✓ | ‐ | ‐ |
| Multitarget drug discovery | ‐ | ‐ | ‐ | ‐ | ✓ | ‐ | ✓ |
| Drug repositioning | ✓ | ✓ | ✓ | ✓ | ✓ | ‐ | ‐ |
| Drug response | ‐ | ✓ | ✓ | ‐ | ‐ | ‐ | ‐ |
| Variant effect | ‐ | ✓ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Developmental neurotoxicants | ✓ | ✓ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Pharmacological decision support | ‐ | ‐ | ✓ | ‐ | ‐ | ✓ | ✓ |
| Drug response monitoring | ‐ | ‐ | ‐ | ✓ | ‐ | ✓ | ‐ |
| Adverse drug effects | ‐ | ‐ | ‐ | ✓ | ‐ | ✓ | ‐ |
| Drug screening | ‐ | ‐ | ‐ | ✓ | ‐ | ‐ | ‐ |
| Overdose and misuse | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ✓ |
Abbreviations: AD, Alzheimer's disease; AI, artificial intelligence; CNS, central nervous system; ML, machine learning; PD, Parkinson's disease.
The main bottleneck in applying AI/ML into CNS drug discovery is the lack of high‐quality, well‐annotated data sets to train effective algorithms. The data collected in the public databases are generally generated by different biological assays, methods, or conditions, which are not comparable. Also, multiple data sets on the same subject may contradict each other. Therefore, filtering the raw inputs to obtain high‐quality data is a crucial step before performing specific AI/ML tasks.
The “black box” nature of most next‐generation AI architectures an additional challenge in CNS drug discovery. The lack of interpretability of AI/ML‐generated results limits their applications. While this is not the case for simpler ML models (i.e., XGBoost, TensorFlow, Lasso, Ridge, Elastic Net), for more advanced ML models (i.e., DNN) the internal workings remain a mystery. Hence, researchers cannot explain how the model arrives at the result and understand the underlying biological mechanisms. This also makes it more difficult to troubleshoot these models when they unexpectedly fail. Therefore, there is a critical need to develop methods for decoding the black boxes of DL.
Also, the amount of the available data directly affects the performance of AI/ML models, since successful training of algorithms relies on suitably large amounts of training data. This is a particular challenge in nominating new targets or drugs for many neurological conditions for which no treatment reverses the disease state. When there are not sufficient training examples for performing a drug discovery task, transfer learning technology, which learns from one task and applies it to the other task, can offer a solution. However, in the long term, the most promising solution to overcome data scarcity would be for the scientific community to share their data. Such large‐scale sharing of data would make significant improvement in the CNS drug discovery process, with advances in hardware that lead to faster machines such as quantum computers in the near future.
A particular limitation for the AI/ML applications in CNS drug discovery is the unknown pathophysiology for many nervous system disorders, which makes target identification very challenging. To explore the complex disease mechanisms and define the right biological targets, we need better AI/ML tools that can pull information out of the data sets generated across the different biological layers (e.g., transcriptomics, proteomics, and metabolomics). Here, capsule networks, 457 a next‐generation AI architecture where CNNs are encapsulated in an interconnected module, can provide a solution. As the first application of capsule networks to drug discovery, capsule networks showed excellent performance to predict the cardiotoxicity of compounds, which highlights their unique potential in drug discovery efforts. 226 Because of the modular representation of the CNNs, capsule networks can learn from heterogeneous data sets by preserving the hierarchical aspects of the data itself. Considering the highly modular nature of CNS data sets with specified layers of genes, proteins, metabolites, capsule networks can analyze the changes in the functional organization and interplay of these layers upon the diseases.
Another critical issue in the application of AI/ML models into CNS drug discovery is the integration of different data types, including genotypic data from patients, multiomics data from drug treatments, and chemical data from bioactivity and toxicity assays. Considering the availability of various databases that include biological, structural, and chemical information, how to integrate these data to generate AI/ML models becomes a critical question in CNS drug discovery applications. Multitask learning, learning of different tasks jointly, can be suitable for these types of applications. Multitask NNs are capable of integrating data from many distinct sources. For example, a multitask architecture can predict the effects of a drug and its BBB permeability at the same time by learning from multiomics data sets, physicochemical properties, HTS, and bioactivity assays.
In recent years, we have seen the emergence of novel neuroimaging techniques such as pharmacological functional magnetic resonance imaging (pharmacoMRI) and pharmacologically induced functional ultrasound (pharmaco‐fUS), which provide in vivo functional data of specific effects of drugs on the brain. Although pharmacoMRI continues to play a useful role in neuropharmacology studies as a well‐established technique, 458 , 459 , 460 , 461 a variety of challenges (i.e., low sensitivity, the requirement for anesthesia, and blood oxygenation‐level dependent imaging) limit the preclinical use of it. A newer tool, pharmaco‐fUS enables brain activity imaging through the local monitoring of cerebral blood volume dynamics at an unprecedented spatiotemporal resolution without the bias of anesthesia. 462 , 463 Recent studies demonstrated fUS imaging's potential to characterize dynamic profiles of CNS drugs, including a drug combination of donepezil plus mefloquine for AD 464 and atomoxetine for attention‐deficit/hyperactivity disorder. 465 Moreover, Rabut et al. 466 adapted ML to analyze the rich data content provided by fUS connectivity imaging. Their ML model identified the “fingerprint” of drug‐induced brain connectivity changes in awake mice for scopolamine, a major preclinical drug to model AD. As evident from the previous applications, AI/ML methods hold the promise of characterization of treatment effects from novel neuroimaging data sets and thereby improving our understanding of the mechanism of action of drugs in the brain. Getting drugs across the BBB is an essential step to developing successful therapies to treat CNS disorders. However, it is often overlooked that BBB is not only a physical barrier for drug delivery to the CNS but also a complex, dynamic interface that might be affected by diseases. CNS disorders may result in dysfunction of BBB, such as its disruption or dysfunctions related to BBB transporters. To date, AI/ML‐based predictive algorithms have assumed that BBB is a static entity by neglecting the effects of CNS pathologies on it. Therefore, a prediction model for BBB penetrance that trained on data from non‐CNS diseases may not work for a CNS disease. To develop better prediction models for BBB permeability, we need to take into account disease‐related changes in the barrier. This also provides many unique opportunities for developing disease‐specific AI/ML tools in CNS drug discovery.
It is important to highlight that CNS drug discovery has a nondeterministic nature, where the neurological targets involve different pathways and their biological consequences are not the sums of the single functions, most drugs have diverse activities through multiple biological targets, and drug response is dependent on a range of factors (i.e., patient's genetic profile and drug's membrane permeability). Moreover, physiologic events are highly context‐specific: A receptor interaction may take place in the liver but not in the brain. AI/ML systems often fail to pick up such context‐specific nonlinear relationships and many other unknown contributing factors. As a result of incomplete domain representation, partial predictability in CNS drug discovery is inevitable. For example, an AI/ML algorithm may predict drug targets that neuroscientists know will likely have significant side effects in the brain or generate unsynthesizable molecules. Here, we need the human refinement process and hypothesis‐driven approach 467 to address many of these challenges to achieve better performance. Knowledge acquisition from the human experts to the AI systems can help the AI/ML system learn and thereby guarantee the best scientific results. In consequence, this mixture of machine and mind 468 will improve decision making as an essential component of the CNS drug discovery process.
Although AI/ML algorithms have already revolutionized other fields, the adoption of them to drug discovery is still at an eraly stage. Initially, AI/ML algorithms have been developed and practically used for certain areas such as image recognition, gaming, and internet search. Inspired by the successful applications in other disciplines, scientists have applied AI/ML algorithms to pharmaceutical research. And yet, we do not have any AI/ML algorithm that is developed specifically for a drug discovery problem. But this means that there should be many opportunities to develop innovative and novel algorithms in the field of therapeutic discovery. In this way, AI/ML methods will play an increasingly important role in not just the field of general pharmaceutical research but also CNS drug discovery.
In conclusion, we extensively review the latest AI/ML‐assisted drug discovery applications for the therapy of CNS diseases. These applications have been overgrowing in the past couple of years, fueled by the unprecedented success of AI/ML‐based approaches in different fields of science and technology. We envision that in the future, AI/ML will play more and more critical roles in CNS drug discovery towards personalized medicine, especially in the following areas: (1) patient subtyping, (2) identification of key disease drivers, (3) prediction of cell type‐specific drug response, (4) autonomous design of novel drugs, and (5) disease‐specific BBB permeability testing. Today there are structural constraints in data and algorithms that are limiting the role of AI/ML. Nonetheless, in the long run, ongoing and emerging developments in AI/ML approaches to neuropharmacology will enable us to develop more effective drugs for CNS diseases.
ACKNOWLEDGMENTS
This study was supported in parts by grants from the National Institutes of Health (NIH)/National Institute on Aging (U01AG046170, RF1AG054014, RF1AG057440, R01AG057907, U01AG052411, R01AG062355, U01AG058635, R01AG068030) and the Mount Sinai seed fund to Bin Zhang.
Biographies
Sezen Vatansever, MD, PhD, is currently a postdoctoral fellow in Dr. Bin Zhang's lab in the Department of Genetics and Genomic Sciences at the Icahn School of Medicine at Mount Sinai, New York. Sezen is a physician‐scientist, having earned her MD from Ankara University School of Medicine, Turkey, and her PhD in Biomedical Sciences and Engineering from Koc University Graduate School of Sciences and Engineering, Turkey. Her graduate studies focused on computer‐aided drug discovery approaches for the treatment of infectious diseases and cancer. Currently, she is identifying novel drug targets for Alzheimer's disease and translating them into new, small molecule drug therapies. She is applying artificial intelligence (AI) and other advanced bioinformatics methods to integrate large multiomics, chemical, and clinical data sets in the early stages of drug discovery.
Avner Schlessinger is an associate professor of Pharmacological Sciences at the Icahn School of Medicine at Mount Sinai in New York City, an associate director of Mt. Sinai Center for Therapeutics Discovery, and codirector of the Pharmacology, Discovery, and Therapeutics Training Area. Dr. Schlessinger graduated from Tel Aviv University with a BSc in Biology and Chemistry and completed his PhD from Columbia University in the Department of Biochemistry and Molecular Biophysics. As a PhD student, he developed programs predicting protein structure and function using various machine‐learning approaches such as artificial neural networks. Following his graduate studies, Dr. Schlessinger was an NIH NRSA postdoctoral fellow at the Department of Bioengineering and Therapeutic Sciences at UCSF, where he established methods for structure‐based drug design and used these approaches to rationally design tool compounds for membrane proteins. The overall goal of Dr. Schlessinger's lab is to improve and automate the drug discovery process by integrating approaches in computational chemistry and bioinformatics and applying these methods to characterize disease pathways. Dr. Schlessinger serves on the Editorial Boards of PLOS Computational Biology and Journal of General Physiology, as well as on the Advisory Board of various biotechnology companies and international consortia. He is also a cofounder of AIchemy, Inc, a startup company focused on the development of a machine learning‐based platform for kinase drug discovery.
Daniel Wacker is an Assistant Professor at the Icahn School of Medicine at Mount Sinai in the Departments of Pharmacological Sciences and Neuroscience. He studied chemistry and biochemistry for his Bachelor's and Master's degree at Ludwig Maximillian's University of Munich, Germany. After completing his thesis work at The Rockefeller University in New York, he completed his PhD at The Scripps Research Institute in La Jolla, California in 2013. Following his postdoctoral tenure at the University of North Carolina at Chapel Hill, Dr. Wacker became an Assistant Professor at the Icahn School of Medicine at Mount Sinai in New York in 2018. He has to date authored over 20 peer‐reviewed publications and has received several awards including a Sloan Research Fellowship in Neuroscience, an Edward Mallinckrodt, Jr Foundation grant, as well as a McKnight Scholar Award. His research focus is on the structure and function of G protein‐coupled receptors and neurotransmitter transporters, and Dr. Wacker has determined numerous first‐in‐class structures of serotonin, dopamine, and opioid receptors. He further investigates the molecular mechanisms of illicit and therapeutic drugs at receptors and transporters using a combination of structural, functional, and computational experiments. Using structure‐based drug discovery approaches that are based on his molecular studies, he has further developed several target‐selective small molecule compounds.
H. Ümit Kaniskan is an assistant professor in the Department of Pharmacological Sciences and assistant director of Mount Sinai Center for Therapeutics Discovery at the Icahn School of Medicine at Mount Sinai. He is earned his PhD in organic chemistry at Case Western Reserve University under the supervision of Dr. Philip Garner. During his doctoral study, he completed the formal total synthesis of Bioxalomycin β2 and Cyanocycline A. He then pursued his postdoctoral studies in Dr. Movassaghi's group at Massachusetts Institute of Technology (MIT), while working on the synthesis of Myrmicarin alkaloids. In January 2013, Dr. Kaniskan joined Dr. Jin's laboratory at the University of North Carolina at Chapel Hill and later at the Icahn School of Medicine at Mount Sinai as a postdoctoral researcher in the Department of Pharmacological Sciences. His research interests include the development of inhibitors of protein methyltransferases and biased ligands of G protein‐coupled receptors as well as targeted protein degradation, in efforts to discover innovative therapeutics for the treatment of human diseases including cancer and brain disorders.
Jian Jin is an internationally recognized medicinal chemist with more than 20 years of experience in small‐molecule drug discovery. He is currently the Mount Sinai Endowed Professor in Therapeutics Discovery, Professor in Departments of Pharmacological Sciences and Oncological Sciences, and the Director of the Mount Sinai Center for Therapeutics Discovery at Icahn School of Medicine at Mount Sinai. Dr. Jin's laboratory is a leader in discovering selective inhibitors of histone methyltransferases, novel degraders targeting oncogenic proteins, and biased ligands of G protein‐coupled receptors. Dr. Jian Jin received a Bachelor of Science degree in chemistry from the University of Science and Technology of China in 1991 and a PhD in organic chemistry from the Pennsylvania State University in 1997. After completing a postdoctoral training at the Ohio State University, Dr. Jin joined GlaxoSmithKline as a medicinal chemist in 1998 and had been a manager of medicinal chemistry from 2003 to 2008. In 2008, Dr. Jin joined the Division of Chemical Biology and Medicinal Chemistry at the University of North Carolina at Chapel Hill (UNC) as an associate professor. He had also served as an associate director of Medicinal Chemistry in the Center for Integrative Chemical Biology and Drug Discovery at UNC from 2008 to 2014. Dr. Jin was recruited to the Icahn School of Medicine at Mount Sinai as a professor with tenure in 2014. Dr. Jin has published close to 200 peer‐reviewed papers and delivered more than 100 invited talks. He is also an inventor of approximately 60 issued US patents and published international patent applications.
Ming‐Ming Zhou has a Dr. Harold and Golden Lamport Professorship in Physiology and Biophysics and Chairman of the Department of Pharmacological Sciences at the Icahn School of Medicine at Mount Sinai. He is also Co‐Director of Drug Discovery Institute and Professor of Oncological Sciences. His research interest is directed at better understanding of the basic principles that govern epigenetic regulation of gene transcription in human biology of health and diseases. The Zhou Lab was the first to discover the bromodomain as the lysine‐acetylated histone binding domain (“chromatin reader”) in gene transcription (Nature, 1999) and demonstrate druggability and therapeutic potential of modulating bromodomain/acetyl‐lysine binding in gene expression to treat a wide array of human diseases including cancer and inflammation, a concept that has had transformative impact in epigenetic drug discovery in the pharmaceutical industry. Dr. Zhou received his PhD degree in Chemistry from Purdue University and conducted postdoctoral study at Abbott Laboratories before he joined the faculty of Mount Sinai School of Medicine. Dr. Zhou serves on the Board of Directors at the New York Structural Biology Center and is a fellow of the American Association for the Advancement of Science.
Bin Zhang is currently a professor at the Department of Genetics and Genomic Sciences and the Director of the Mount Sinai Center for Transformative Disease Modeling, at the Icahn School of Medicine at Mount Sinai, New York, USA. His expertise lies in bioinformatics, systems biology, and artificial intelligence with applications to disease modeling and drug discovery. Over the past one and half decades, he has developed a series of influential gene network inference algorithms which have been extensively used for the identification of novel pathways and key gene targets, as well as the development of drugs for a variety of complex diseases such as cancer, atherosclerosis, Alzheimer's, obesity, and diabetes. His current research is focused on developing mechanistic molecular models of complex diseases by integrating large‐scale multiomics data and novel therapeutics for treating such diseases using artificial intelligence and deep machine learning approaches.
Vatansever S, Schlessinger A, Wacker D, et al. Artificial intelligence and machine learning‐aided drug discovery in central nervous system diseases: State‐of‐the‐arts and future directions. Med Res Rev. 2021;41:1427–1473. 10.1002/med.21764
REFERENCES
- 1. Mohs RC, Greig NH. Drug discovery and development: role of basic biological research. Alzheimers Dement (N Y). 2017;3(4):651‐657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Papadatos G, Gaulton A, Hersey A, Overington JP. Activity, assay and target data curation and quality in the ChEMBL database. J Comput Aided Mol Des. 2015;29(9):885‐896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wilson BJ, Nicholls SG. The Human Genome Project, and recent advances in personalized genomics. Risk Manag Healthc Policy. 2015;8:9‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. David L, Arús‐Pous J, Karlsson J, et al. Applications of deep‐learning in exploiting large‐scale and heterogeneous compound data in industrial pharmaceutical research. Front Pharmacol. 2019;10:1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Administration USFD. Artificial Intelligence and Machine Learning in Software as a Medical Device. https://www.fda.gov/medical-devices/software-medical-device-samd/artificial-intelligence-and-machine-learning-software-medical-device. Accessed October 26, 2020.
- 6. Todeschini R, Wiese M, Consonni V. Books—handbook of molecular descriptors. Angew Chem. 2001;40(10):1977. [Google Scholar]
- 7. Dong J, Cao D‐S, Miao H‐Y, et al. ChemDes: an integrated web‐based platform for molecular descriptor and fingerprint computation. J Cheminform. 2015;7(1):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cao DS, Zhou GH, Liu S, et al. Large‐scale prediction of human kinase‐inhibitor interactions using protein sequences and molecular topological structures. Anal Chim Acta. 2013;792:10‐18. [DOI] [PubMed] [Google Scholar]
- 9. Yee SW, Lin L, Merski M, et al. Prediction and validation of enzyme and transporter off‐targets for metformin. J Pharmacokinet Pharmacodyn. 2015;42(5):463‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Muegge I, Mukherjee P. An overview of molecular fingerprint similarity search in virtual screening. Expert Opin Drug Discovery. 2016;11(2):137‐148. [DOI] [PubMed] [Google Scholar]
- 11. Cereto‐Massague A, Ojeda MJ, Valls C, Mulero M, Garcia‐Vallve S, Pujadas G. Molecular fingerprint similarity search in virtual screening. Methods (San Diego, Calif). 2015;71:58‐63. [DOI] [PubMed] [Google Scholar]
- 12. Willett P. Similarity‐based virtual screening using 2D fingerprints. Drug Discovery Today. 2006;11(23‐24):1046‐1053. [DOI] [PubMed] [Google Scholar]
- 13. Heikamp K, Bajorat J. Fingerprint design and engineering strategies: rationalizing and improving similarity search performance. Future Med Chem. 2012;4(15):1945‐1959. [DOI] [PubMed] [Google Scholar]
- 14. Irwin JJ, Gaskins G, Sterling T, Mysinger MM, Keiser MJ. Predicted biological activity of purchasable chemical space. J Chem Inf Model. 2018;58(1):148‐164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Axen SD, Huang XP, Caceres EL, Gendelev L, Roth BL, Keiser MJ. A simple representation of three‐dimensional molecular structure. J Med Chem. 2017;60(17):7393‐7409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Geppert H, Vogt M, Bajorath J. Current trends in ligand‐based virtual screening: molecular representations, data mining methods, new application areas, and performance evaluation. J Chem Inf Model. 2010;50(2):205‐216. [DOI] [PubMed] [Google Scholar]
- 17. Berenger F, Voet A, Lee XY, Zhang KY. A rotation‐translation invariant molecular descriptor of partial charges and its use in ligand‐based virtual screening. J Cheminform. 2014;6:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roy K, Mitra I. Electrotopological state atom (E‐state) index in drug design, QSAR, property prediction and toxicity assessment. Curr Comput Aided Drug Des. 2012;8(2):135‐158. [DOI] [PubMed] [Google Scholar]
- 19. Cao D‐S, Xu Q‐S, Liang Y‐Z, Chen X, Li H‐D. Prediction of aqueous solubility of druglike organic compounds using partial least squares, back‐propagation network and support vector machine. J Chemom. 2010;24(9):584‐595. [Google Scholar]
- 20. Viswanadhan VN, Rajesh H, Balaji VN. Atom type preferences, structural diversity, and property profiles of known drugs, leads, and nondrugs: a comparative assessment. ACS Comb Sci. 2011;13(3):327‐336. [DOI] [PubMed] [Google Scholar]
- 21. Khan MT. Predictions of the ADMET properties of candidate drug molecules utilizing different QSAR/QSPR modelling approaches. Curr Drug Metab. 2010;11(4):285‐295. [DOI] [PubMed] [Google Scholar]
- 22. Cheng F, Li W, Zhou Y, et al. Correction to “admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties”. J Chem Inf Model. 2019;59:4959. [DOI] [PubMed] [Google Scholar]
- 23. Maltarollo VG, Gertrudes JC, Oliveira PR, Honorio KM. Applying machine learning techniques for ADME‐Tox prediction: a review. Expert Opin Drug Metab Toxicol. 2015;11(2):259‐271. [DOI] [PubMed] [Google Scholar]
- 24. Chuang KV, Gunsalus LM, Keiser MJ. Learning molecular representations for medicinal chemistry. J Med Chem. 2020;63:8705‐8722. [DOI] [PubMed] [Google Scholar]
- 25. Moffat JG, Vincent F, Lee JA, Eder J, Prunotto M. Opportunities and challenges in phenotypic drug discovery: an industry perspective. Nat Rev Drug Discovery. 2017;16(8):531‐543. [DOI] [PubMed] [Google Scholar]
- 26. Van den Broeck WMM. Chapter 3. Drug targets, target identification, validation, and screening. In: Wermuth CG, Aldous D, Raboisson P, Rognan D, eds. The Practice of Medicinal Chemistry. 4th Ed. San Diego, CA: Academic Press; 2015:45‐70. [Google Scholar]
- 27. Gashaw I, Ellinghaus P, Sommer A, Asadullah K. What makes a good drug target? Drug Discovery Today. 2011;16(23–24):1037‐1043. [DOI] [PubMed] [Google Scholar]
- 28. Gashaw I, Ellinghaus P, Sommer A, Asadullah K. What makes a good drug target? Drug Discovery Today. 2012;17(suppl):S24‐S30. [DOI] [PubMed] [Google Scholar]
- 29. Lindsay MA. Target discovery. Nat Rev Drug Discovery. 2003;2(10):831‐838. [DOI] [PubMed] [Google Scholar]
- 30. Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98(19):10869‐10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shen R, Olshen AB, Ladanyi M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics. 2009;25(22):2906‐2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yuan Y, Savage RS, Markowetz F. Patient‐specific data fusion defines prognostic cancer subtypes. PLOS Comput Biol. 2011;7(10):e1002227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang L, Lv C, Jin Y, et al. Deep learning‐based multi‐omics data integration reveals two prognostic subtypes in high‐risk neuroblastoma. Front Genet. 2018;9:477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gao F, Wang W, Tan M, et al. DeepCC: a novel deep learning‐based framework for cancer molecular subtype classification. Oncogenesis. 2019;8(9):44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Singh A, Shannon CP, Gautier B, et al. DIABLO: an integrative approach for identifying key molecular drivers from multi‐omics assays. Bioinformatics. 2019;35(17):3055‐3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tenenhaus A, Tenenhaus M. Regularized generalized canonical correlation analysis. Psychometrika. 2011;76(2):257‐284. [DOI] [PubMed] [Google Scholar]
- 37. Argelaguet R, Velten B, Arnol D, et al. Multi‐omics factor analysis—a framework for unsupervised integration of multi‐omics data sets. Mol Syst Biol. 2018;14(6):e8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lock EF, Hoadley KA, Marron JS, Nobel AB. Joint and individual variation explained (Jive) for integrated analysis of multiple data types. Ann Appl Stat. 2013;7(1):523‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wong WC, Kim D, Carter H, Diekhans M, Ryan MC, Karchin R. CHASM and SNVBox: toolkit for detecting biologically important single nucleotide mutations in cancer. Bioinformatics. 2011;27(15):2147‐2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Asif M, Martiniano H, Vicente AM, Couto FM. Identifying disease genes using machine learning and gene functional similarities, assessed through Gene Ontology. PLOS One. 2018;13(12):e0208626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mao Y, Chen H, Liang H, Meric‐Bernstam F, Mills GB, Chen K. CanDrA: cancer‐specific driver missense mutation annotation with optimized features. PLOS One. 2013;8(10):e77945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ghanat Bari M, Ung CY, Zhang C, Zhu S, Li H. Machine learning‐assisted network inference approach to identify a new class of genes that coordinate the functionality of cancer networks. Sci Rep. 2017;7(1):6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Botía JA, Guelfi S, Zhang D, et al. G2P: using machine learning to understand and predict genes causing rare neurological disorders. bioRxiv. 2018:288845. 10.1101/288845 [DOI] [Google Scholar]
- 44. Han Y, Yang J, Qian X, et al. DriverML: a machine learning algorithm for identifying driver genes in cancer sequencing studies. Nucleic Acids Res. 2019;47(8):e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chung IF, Chen CY, Su SC, et al. DriverDBv2: a database for human cancer driver gene research. Nucleic Acids Res. 2016;44(D1):D975‐D979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Luo P, Ding Y, Lei X, Wu FX. deepDriver: predicting cancer driver genes based on somatic mutations using deep convolutional neural networks. Front Genet. 2019;10:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Peng J, Guan J, Shang X. Predicting Parkinson's disease genes based on Node2vec and autoencoder. Front Genet. 2019;10:226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Collier O, Stoven V, Vert JP. LOTUS: a single‐ and multitask machine learning algorithm for the prediction of cancer driver genes. PLOS Comput Biol. 2019;15(9):e1007381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim M, Tagkopoulos I. Data integration and predictive modeling methods for multi‐omics datasets. Mol Omics. 2018;14(1):8‐25. [DOI] [PubMed] [Google Scholar]
- 50. Bravo A, Pinero J, Queralt‐Rosinach N, Rautschka M, Furlong LI. Extraction of relations between genes and diseases from text and large‐scale data analysis: implications for translational research. BMC Bioinformatics. 2015;16:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281‐297. [DOI] [PubMed] [Google Scholar]
- 52. Schmidt MF. miRNA targeting drugs: the next blockbusters? Methods Mol Biol (Clifton, NJ). 2017;1517:3‐22. [DOI] [PubMed] [Google Scholar]
- 53. Xu J, Li CX, Lv JY, et al. Prioritizing candidate disease miRNAs by topological features in the miRNA target‐dysregulated network: case study of prostate cancer. Mol Cancer Ther. 2011;10(10):1857‐1866. [DOI] [PubMed] [Google Scholar]
- 54. Xuan P, Han K, Guo M, et al. Prediction of microRNAs associated with human diseases based on weighted k most similar neighbors. PLOS One. 2013;8(8):e70204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zeng X, Wang W, Deng G, Bing J, Zou Q. Prediction of potential disease‐associated microRNAs by using neural networks. Mol Ther Nucleic Acids. 2019;16:566‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zheng K, You ZH, Wang L, Zhou Y, Li LP, Li ZW. MLMDA: a machine learning approach to predict and validate microRNA‐disease associations by integrating of heterogenous information sources. J Transl Med. 2019;17(1):260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Black DL. Mechanisms of alternative pre‐messenger RNA splicing. Annu Rev Biochem. 2003;72:291‐336. [DOI] [PubMed] [Google Scholar]
- 58. Barash Y, Vaquero‐Garcia J, González‐Vallinas J, et al. AVISPA: a web tool for the prediction and analysis of alternative splicing. Genome Biol. 2013;14(10):R114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Leung MK, Xiong HY, Lee LJ, Frey BJ. Deep learning of the tissue‐regulated splicing code. Bioinformatics. 2014;30(12):i121‐i129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jha A, Gazzara MR, Barash Y. Integrative deep models for alternative splicing. Bioinformatics. 2017;33(14):i274‐i282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stanescu A, Tangirala K, Caragea D. Predicting alternatively spliced exons using semi‐supervised learning. Int J Data Min Bioinform. 2016;14(1):1‐21. [Google Scholar]
- 62. Xiong HY, Alipanahi B, Lee LJ, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015;347(6218):1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Doncheva NT, Kacprowski T, Albrecht M. Recent approaches to the prioritization of candidate disease genes. Wiley Interdiscip Rev: Syst Biol Med. 2012;4(5):429‐442. [DOI] [PubMed] [Google Scholar]
- 64. Jeon J, Nim S, Teyra J, et al. A systematic approach to identify novel cancer drug targets using machine learning, inhibitor design and high‐throughput screening. Genome Med. 2014;6:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Valentini G, Paccanaro A, Caniza H, Romero AE, Re M. An extensive analysis of disease‐gene associations using network integration and fast kernel‐based gene prioritization methods. Artif Intell Med. 2014;61(2):63‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ferrero E, Dunham I, Sanseau P. In silico prediction of novel therapeutic targets using gene‐disease association data. J Transl Med. 2017;15(1):182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kim J, So S, Lee HJ, Park JC, Kim JJ, Lee H. DigSee: disease gene search engine with evidence sentences (version cancer). Nucleic Acids Res. 2013;41(Web Server issue):W510‐W517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Arabfard M, Ohadi M, Rezaei Tabar V, Delbari A, Kavousi K. Genome‐wide prediction and prioritization of human aging genes by data fusion: a machine learning approach. BMC Genomics. 2019;20(1):832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Holbrook SR, Muskal SM, Kim S‐H. Predicting protein structural features with Artificial neural networks. Artif Intell Mol Biol. 1993:161‐194. [Google Scholar]
- 70. Cheng J, Tegge AN, Baldi P. Machine learning methods for protein structure prediction. IEEE Rev Biomed Eng. 2008;1:41‐49. [DOI] [PubMed] [Google Scholar]
- 71. Kandathil SM, Greener JG, Jones DT. Recent developments in deep learning applied to protein structure prediction. Proteins. 2019;87(12):1179‐1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tsuchiya Y, Tomii K. Neural networks for protein structure and function prediction and dynamic analysis. Biophys Rev. 2020;12:569‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Torrisi M, Pollastri G, Le Q. Deep learning methods in protein structure prediction. Comput Struct Biotechnol J. 2020;18:1301‐1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. AlQuraishi M. AlphaFold at CASP13. Bioinformatics. 2019;35(22):4862‐4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wei G‐W. Protein structure prediction beyond AlphaFold. Nat Mach Intell. 2019;1(8):336‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Powell A. Science unfolding in time: a situated protein folding landscape in retrospect. OSF Preprints. 2019. 10.31219/osf.io/4e8j7 [DOI]
- 77. Zheng W, Li Y, Zhang C, Pearce R, Mortuza SM, Zhang Y. Deep‐learning contact‐map guided protein structure prediction in CASP13. Proteins. 2019;87(12):1149‐1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wang S, Sun S, Li Z, Zhang R, Xu J. Accurate de novo prediction of protein contact map by ultra‐deep learning model. PLOS Comput Biol. 2017;13(1):e1005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yang J, Anishchenko I, Park H, Peng Z, Ovchinnikov S, Baker D. Improved protein structure prediction using predicted interresidue orientations. Proc Natl Acad Sci. 2020;117(3):1496‐1503. 10.1073/pnas.1914677117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wang T, Qiao Y, Ding W, Mao W, Zhou Y, Gong H. Improved fragment sampling for ab initio protein structure prediction using deep neural networks. Nat Mach Intell. 2019;1(8):347‐355. [Google Scholar]
- 81. Billings WM, Hedelius B, Millecam T, Wingate D, Corte DD. ProSPr: democratized implementation of alphafold protein distance prediction network. bioRxiv. 2019:830273. 10.1101/830273 [DOI] [Google Scholar]
- 82. Ung PM, Rahman R, Schlessinger A. Redefining the protein kinase conformational space with machine learning. Cell Chem Biol. 2018;25(7):916‐924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Matsumoto S, Ishida S, Araki M, Kato T, Terayama K, Okuno Y. Extraction of protein dynamics information hidden in Cryo‐EM map using deep learning. bioRxiv. 2020. 10.1101/2020.02.17.951863 [DOI] [Google Scholar]
- 84. Rost B, Radivojac P, Bromberg Y. Protein function in precision medicine: deep understanding with machine learning. FEBS Lett. 2016;590(15):2327‐2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Owens J. Determining druggability. Nat Rev Drug Discovery. 2007;6(3):187. [Google Scholar]
- 86. Nayal M, Honig B. On the nature of cavities on protein surfaces: application to the identification of drug‐binding sites. Proteins. 2006;63(4):892‐906. [DOI] [PubMed] [Google Scholar]
- 87. Li Q, Lai L. Prediction of potential drug targets based on simple sequence properties. BMC Bioinformatics. 2007;8:353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bakheet TM, Doig AJ. Properties and identification of human protein drug targets. Bioinformatics. 2009;25(4):451‐457. [DOI] [PubMed] [Google Scholar]
- 89. Costa PR, Acencio ML, Lemke N. A machine learning approach for genome‐wide prediction of morbid and druggable human genes based on systems‐level data. BMC Genomics. 2010;11(suppl 5):S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wang Q, Feng Y, Huang J, Wang T, Cheng G. A novel framework for the identification of drug target proteins: combining stacked auto‐encoders with a biased support vector machine. PLOS One. 2017;12(4):e0176486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yuan JH, Han SB, Richter S, Wade RC, Kokh DB. Druggability assessment in TRAPP using machine learning approaches. J Chem Inf Model. 2020;60(3):1685‐1699. [DOI] [PubMed] [Google Scholar]
- 92. Dezső Z, Ceccarelli M. Machine learning prediction of oncology drug targets based on protein and network properties. BMC Bioinformatics. 2020;21(1):104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kandoi G, Acencio ML, Lemke N. Prediction of druggable proteins using machine learning and systems biology: a mini‐review. Front Physiol. 2015;6:366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Reker D, Perna AM, Rodrigues T, et al. Revealing the macromolecular targets of complex natural products. Nat Chem. 2014;6(12):1072‐1078. [DOI] [PubMed] [Google Scholar]
- 95. Reker D, Rodrigues T, Schneider P, Schneider G. Identifying the macromolecular targets of de novo‐designed chemical entities through self‐organizing map consensus. Proc Natl Acad Sci USA. 2014;111(11):4067‐4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Rodrigues T, Reker D, Kunze J, Schneider P, Schneider G. Revealing the macromolecular targets of fragment‐like natural products. Angew Chem Int Ed. 2015;54(36):10516‐10520. [DOI] [PubMed] [Google Scholar]
- 97. Schneider G, Reker D, Chen T, Hauenstein K, Schneider P, Altmann KH. Deorphaning the macromolecular targets of the natural anticancer compound doliculide. Angew Chem Int Ed. 2016;55(40):12408‐12411. [DOI] [PubMed] [Google Scholar]
- 98. NidhiGlick, M , Davies JW, Jenkins JL. Prediction of biological targets for compounds using multiple‐category Bayesian models trained on chemogenomics databases. J Chem Inf Model. 2006;46(3):1124‐1133. [DOI] [PubMed] [Google Scholar]
- 99. Lee K, Lee M, Kim D. Utilizing random Forest QSAR models with optimized parameters for target identification and its application to target‐fishing server. BMC Bioinformatics. 2017;18(suppl 16):567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Madhukar NS, Khade PK, Huang L, et al. A Bayesian machine learning approach for drug target identification using diverse data types. Nat Commun. 2019;10(1):5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. D'Souza S, Prema KV, Balaji S. Machine learning models for drug–target interactions: current knowledge and future directions. Drug Discovery Today. 2020;25(4):748‐756. 10.1016/j.drudis.2020.03.003 [DOI] [PubMed] [Google Scholar]
- 102. Anusuya S, Kesherwani M, Priya KV, et al. Drug‐target interactions: prediction methods and applications. Curr Protein Pept Sci. 2018;19(6):537‐561. [DOI] [PubMed] [Google Scholar]
- 103. Yuan Q, Gao J, Wu D, Zhang S, Mamitsuka H, Zhu S. DrugE‐Rank: improving drug‐target interaction prediction of new candidate drugs or targets by ensemble learning to rank. Bioinformatics. 2016;32(12):i18‐i27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ezzat A, Wu M, Li X, Kwoh CK. Computational prediction of drug‐target interactions via ensemble learning. Methods Mol Biol (Clifton, NJ). 2019;1903:239‐254. [DOI] [PubMed] [Google Scholar]
- 105. Ezzat A, Wu M, Li XL, Kwoh CK. Drug‐target interaction prediction using ensemble learning and dimensionality reduction. Methods (San Diego, Calif). 2017;129:81‐88. [DOI] [PubMed] [Google Scholar]
- 106. Rayhan F, Ahmed S, Shatabda S, et al. iDTI‐ESBoost: identification of drug target interaction using evolutionary and structural features with boosting. Sci Rep. 2017;7(1):17731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Pliakos K, Vens C. Drug‐target interaction prediction with tree‐ensemble learning and output space reconstruction. BMC Bioinformatics. 2020;21(1):49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Sharma A, Rani R. BE‐DTI': ensemble framework for drug target interaction prediction using dimensionality reduction and active learning. Comput Methods Programs Biomed. 2018;165:151‐162. [DOI] [PubMed] [Google Scholar]
- 109. Wan F, Hong L, Xiao A, Jiang T, Zeng J. NeoDTI: neural integration of neighbor information from a heterogeneous network for discovering new drug‐target interactions. Bioinformatics. 2019;35(1):104‐111. [DOI] [PubMed] [Google Scholar]
- 110. Monteiro NRC, Ribeiro B, Arrais J. Drug‐target interaction prediction: end‐to‐end deep learning approach. IEEE/ACM Trans Comput Biol Bioinform. 2020:1. [DOI] [PubMed] [Google Scholar]
- 111. Perlman L, Gottlieb A, Atias N, Ruppin E, Sharan R. Combining drug and gene similarity measures for drug‐target elucidation. J Comput Biol. 2011;18(2):133‐145. [DOI] [PubMed] [Google Scholar]
- 112. Wang YC, Zhang CH, Deng NY, Wang Y. Kernel‐based data fusion improves the drug‐protein interaction prediction. Comput Biol Chem. 2011;35(6):353‐362. [DOI] [PubMed] [Google Scholar]
- 113. Wang Y, Chen S, Deng N, Wang Y. Drug repositioning by kernel‐based integration of molecular structure, molecular activity, and phenotype data. PLOS One. 2013;8(11):e78518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Yamanishi Y, Araki M, Gutteridge A, Honda W, Kanehisa M. Prediction of drug‐target interaction networks from the integration of chemical and genomic spaces. Bioinformatics. 2008;24(13):i232‐i240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Nascimento AC, Prudencio RB, Costa IG. A multiple kernel learning algorithm for drug‐target interaction prediction. BMC Bioinformatics. 2016;17:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Ripphausen P, Nisius B, Peltason L, Bajorath J. Quo vadis, virtual screening? A comprehensive survey of prospective applications. J Med Chem. 2010;53(24):8461‐8467. [DOI] [PubMed] [Google Scholar]
- 117. Clark DE. What has virtual screening ever done for drug discovery? Expert Opin Drug Discovery. 2008;3(8):841‐851. [DOI] [PubMed] [Google Scholar]
- 118. Lionta E, Spyrou G, Vassilatis DK, Cournia Z. Structure‐based virtual screening for drug discovery: principles, applications and recent advances. Curr Top Med Chem. 2014;14(16):1923‐1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ghosh S, Nie A, An J, Huang Z. Structure‐based virtual screening of chemical libraries for drug discovery. Curr Opin Chem Biol. 2006;10(3):194‐202. [DOI] [PubMed] [Google Scholar]
- 120. Acharya C, Coop A, Polli JE, Mackerell AD Jr. Recent advances in ligand‐based drug design: relevance and utility of the conformationally sampled pharmacophore approach. Curr Comput Aided Drug Des. 2011;7(1):10‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Maldonado AG, Doucet JP, Petitjean M, Fan B‐T. Molecular similarity and diversity in chemoinformatics: from theory to applications. Mol Divers. 2006;10(1):39‐79. [DOI] [PubMed] [Google Scholar]
- 122. Keiser MJ, Irwin JJ, Shoichet BK. The chemical basis of pharmacology. Biochemistry. 2010;49(48):10267‐10276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Ballester PJ, Mitchell JB. A machine learning approach to predicting protein‐ligand binding affinity with applications to molecular docking. Bioinformatics. 2010;26(9):1169‐1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Zilian D, Sotriffer CA. SFCscore(RF): a random forest‐based scoring function for improved affinity prediction of protein‐ligand complexes. J Chem Inf Model. 2013;53(8):1923‐1933. [DOI] [PubMed] [Google Scholar]
- 125. Liu Q, Kwoh CK, Li J. Binding affinity prediction for protein‐ligand complexes based on beta contacts and B factor. J Chem Inf Model. 2013;53(11):3076‐3085. [DOI] [PubMed] [Google Scholar]
- 126. Li H, Leung KS, Ballester PJ, Wong MH. istar: a web platform for large‐scale protein‐ligand docking. PLOS One. 2014;9(1):e85678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Li GB, Yang LL, Wang WJ, Li LL, Yang SY. ID‐Score: a new empirical scoring function based on a comprehensive set of descriptors related to protein‐ligand interactions. J Chem Inf Model. 2013;53(3):592‐600. [DOI] [PubMed] [Google Scholar]
- 128. Ballester PJ. Machine learning scoring functions based on random forest and support vector regression. Paper presented at: IAPR International Conference on Pattern Recognition in Bioinformatics. Berlin, Heidelberg: Springer; 2012:14‐25. 10.1007/978-3-642-34123-6_2 [DOI]
- 129. Durrant JD, McCammon JA. NNScore: a neural‐network‐based scoring function for the characterization of protein‐ligand complexes. J Chem Inf Model. 2010;50(10):1865‐1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Ouyang X, Handoko SD, Kwoh CK. CScore: a simple yet effective scoring function for protein‐ligand binding affinity prediction using modified CMAC learning architecture. J Bioinform Comput Biol. 2011;9(suppl 1):1‐14. [DOI] [PubMed] [Google Scholar]
- 131. Cang Z, Wei GW. TopologyNet: topology based deep convolutional and multi‐task neural networks for biomolecular property predictions. PLOS Comput Biol. 2017;13(7):e1005690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Ragoza M, Hochuli J, Idrobo E, Sunseri J, Koes DR. Protein‐ligand scoring with convolutional neural networks. J Chem Inf Model. 2017;57(4):942‐957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Stepniewska‐Dziubinska MM, Zielenkiewicz P, Siedlecki P. Development and evaluation of a deep learning model for protein‐ligand binding affinity prediction. Bioinformatics. 2018;34(21):3666‐3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Ashtawy HM, Mahapatra NR. BgN‐Score and BsN‐Score: bagging and boosting based ensemble neural networks scoring functions for accurate binding affinity prediction of protein‐ligand complexes. BMC Bioinformatics. 2015;16(suppl 4):S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Ain QU, Aleksandrova A, Roessler FD, Ballester PJ. Machine‐learning scoring functions to improve structure‐based binding affinity prediction and virtual screening. Wiley Interdiscip Rev Comput Mol Sci. 2015;5(6):405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Yang X, Wang Y, Byrne R, Schneider G, Yang S. Concepts of artificial intelligence for computer‐assisted drug discovery. Chem Rev. 2019;119(18):10520‐10594. [DOI] [PubMed] [Google Scholar]
- 137. Wang D, Cui C, Ding X, et al. Improving the virtual screening ability of target‐specific scoring functions using deep learning methods. Front Pharmacol. 2019;10:924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Li L, Khanna M, Jo I, et al. Target‐specific support vector machine scoring in structure‐based virtual screening: computational validation, in vitro testing in kinases, and effects on lung cancer cell proliferation. J Chem Inf Model. 2011;51(4):755‐759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Xu D, Li L, Zhou D, Liu D, Hudmon A, Meroueh SO. Structure‐based target‐specific screening leads to small‐molecule CaMKII inhibitors. ChemMedChem. 2017;12(9):660‐677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Sun H, Pan P, Tian S, et al. Constructing and validating high‐performance MIEC‐SVM models in virtual screening for kinases: a better way for actives discovery. Sci Rep. 2016;6:24817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Berishvili VP, Voronkov AE, Radchenko EV, Palyulin VA. Machine learning classification models to improve the docking‐based screening: a case of PI3K‐tankyrase inhibitors. Mol Inf. 2018;37(11):e1800030. [DOI] [PubMed] [Google Scholar]
- 142. Yan Y, Wang W, Sun Z, Zhang JZH, Ji C. Protein‐ligand empirical interaction components for virtual screening. J Chem Inf Model. 2017;57(8):1793‐1806. [DOI] [PubMed] [Google Scholar]
- 143. Cherkasov A, Hilpert K, Jenssen H, et al. Use of artificial intelligence in the design of small peptide antibiotics effective against a broad spectrum of highly antibiotic‐resistant superbugs. ACS Chem Biol. 2009;4(1):65‐74. [DOI] [PubMed] [Google Scholar]
- 144. Kinnings SL, Liu N, Tonge PJ, Jackson RM, Xie L, Bourne PE. A machine learning‐based method to improve docking scoring functions and its application to drug repurposing. J Chem Inf Model. 2011;51(2):408‐419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Pereira JC, Caffarena ER, Dos Santos CN. Boosting docking‐based virtual screening with deep learning. J Chem Inf Model. 2016;56(12):2495‐2506. [DOI] [PubMed] [Google Scholar]
- 146. Leong MK, Syu RG, Ding YL, Weng CF. Prediction of N‐methyl‐d‐aspartate receptor GluN1‐ligand binding affinity by a novel SVM‐Pose/SVM‐Score combinatorial ensemble docking scheme. Sci Rep. 2017;7:40053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Li H, Leung KS, Wong MH, Ballester PJ. Improving Autodock Vina using Random Forest: the growing accuracy of binding affinity prediction by the effective exploitation of larger data sets. Mol Inf. 2015;34(2–3):115‐126. [DOI] [PubMed] [Google Scholar]
- 148. Arciniega M, Lange OF. Improvement of virtual screening results by docking data feature analysis. J Chem Inf Model. 2014;54(5):1401‐1411. [DOI] [PubMed] [Google Scholar]
- 149. Waszkowycz B. Towards improving compound selection in structure‐based virtual screening. Drug Discovery Today. 2008;13(5–6):219‐226. [DOI] [PubMed] [Google Scholar]
- 150. Johnson MA, Maggiora GM. Concepts and Applications of Molecular Similarity. Wiley; 1990. [Google Scholar]
- 151. Carpenter KA, Cohen DS, Jarrell JT, Huang X. Deep learning and virtual drug screening. Future Med Chem. 2018;10:2557‐2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Melville JL, Burke EK, Hirst JD. Machine learning in virtual screening. Comb Chem High Throughput Screen. 2009;12(4):332‐343. [DOI] [PubMed] [Google Scholar]
- 153. Stokes JM, Yang K, Swanson K, et al. A deep learning approach to antibiotic discovery. Cell. 2020;180(4):688‐702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Wu Z, Ramsundar B, Feinberg EN, et al. MoleculeNet: a benchmark for molecular machine learning. Chem Sci. 2018;9(2):513‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Feinberg EN, Joshi E, Pande VS, Cheng AC. Improvement in ADMET prediction with multitask deep featurization. J Med Chem. 2020;63(16):8835‐8848. [DOI] [PubMed] [Google Scholar]
- 156. Wang T, Wu MB, Lin JP, Yang LR. Quantitative structure‐activity relationship: promising advances in drug discovery platforms. Expert Opin Drug Discovery. 2015;10(12):1283‐1300. [DOI] [PubMed] [Google Scholar]
- 157. Kumar R, Chaudhary K, Singh Chauhan J, et al. An in silico platform for predicting, screening and designing of antihypertensive peptides. Sci Rep. 2015;5:12512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Briard JG, Fernandez M, de Luna P, Woo TK, Ben RN. QSAR Accelerated discovery of potent ice recrystallization inhibitors. Sci Rep. 2016;6:26403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Svetnik V, Liaw A, Tong C, Culberson JC, Sheridan RP, Feuston BP. Random forest: a classification and regression tool for compound classification and QSAR modeling. J Chem Inf Comput Sci. 2003;43(6):1947‐1958. [DOI] [PubMed] [Google Scholar]
- 160. Zakharov AV, Varlamova EV, Lagunin AA, et al. QSAR modeling and prediction of drug‐drug interactions. Mol Pharmaceutics. 2016;13(2):545‐556. [DOI] [PubMed] [Google Scholar]
- 161. Fang X, Bagui S, Bagui S. Improving virtual screening predictive accuracy of Human kallikrein 5 inhibitors using machine learning models. Comput Biol Chem. 2017;69:110‐119. [DOI] [PubMed] [Google Scholar]
- 162. Chen JJF, Visco DP Jr. Developing an in silico pipeline for faster drug candidate discovery: virtual high throughput screening with the signature molecular descriptor using support vector machine models. Chem Eng Sci. 2017;159:31‐42. [Google Scholar]
- 163. Chen JJF, Visco DP Jr. Identifying novel factor XIIa inhibitors with PCA‐GA‐SVM developed vHTS models. Eur J Med Chem. 2017;140:31‐41. [DOI] [PubMed] [Google Scholar]
- 164. Xia X, Maliski EG, Gallant P, Rogers D. Classification of kinase inhibitors using a Bayesian model. J Med Chem. 2004;47(18):4463‐4470. [DOI] [PubMed] [Google Scholar]
- 165. Bender A, Mussa HY, Glen RC. Screening for dihydrofolate reductase inhibitors using MOLPRINT 2D, a fast fragment‐based method employing the Naive Bayesian classifier: limitations of the descriptor and the importance of balanced chemistry in training and test sets. J Biomol Screen. 2005;10(7):658‐666. [DOI] [PubMed] [Google Scholar]
- 166. Prathipati P, Ma NL, Keller TH. Global Bayesian models for the prioritization of antitubercular agents. J Chem Inf Model. 2008;48(12):2362‐2370. [DOI] [PubMed] [Google Scholar]
- 167. Ekins S, Reynolds RC, Kim H, et al. Bayesian models leveraging bioactivity and cytotoxicity information for drug discovery. Chem Biol. 2013;20(3):370‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Vijayan RS, Bera I, Prabu M, Saha S, Ghoshal N. Combinatorial library enumeration and lead hopping using comparative interaction fingerprint analysis and classical 2D QSAR methods for seeking novel GABA(A) alpha(3) modulators. J Chem Inf Model. 2009;49(11):2498‐2511. [DOI] [PubMed] [Google Scholar]
- 169. Liu L, Lu J, Lu Y, et al. Novel Bayesian classification models for predicting compounds blocking hERG potassium channels. Acta Pharmacol Sin. 2014;35(8):1093‐1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Singh N, Chaudhury S, Liu R, AbdulHameed MDM, Tawa G, Wallqvist A. QSAR classification model for antibacterial compounds and its use in virtual screening. J Chem Inf Model. 2012;52(10):2559‐2569. [DOI] [PubMed] [Google Scholar]
- 171. Renault N, Laurent X, Farce A, et al. Virtual screening of CB(2) receptor agonists from bayesian network and high‐throughput docking: structural insights into agonist‐modulated GPCR features. Chem Biol Drug Des. 2013;81(4):442‐454. [DOI] [PubMed] [Google Scholar]
- 172. AbdulHameed MD, Ippolito DL, Wallqvist A. Predicting rat and human pregnane X receptor activators using Bayesian classification models. Chem Res Toxicol. 2016;29(10):1729‐1740. [DOI] [PubMed] [Google Scholar]
- 173. Shi H, Tian S, Li Y, et al. Absorption, distribution, metabolism, excretion, and toxicity evaluation in drug discovery. 14. Prediction of human pregnane X receptor activators by using naive bayesian classification technique. Chem Res Toxicol. 2015;28(1):116‐125. [DOI] [PubMed] [Google Scholar]
- 174. Murcia‐Soler M, Pérez‐Giménez F, García‐March FJ, et al. Artificial neural networks and linear discriminant analysis: a valuable combination in the selection of new antibacterial compounds. J Chem Inf Comput Sci. 2004;44(3):1031‐1041. [DOI] [PubMed] [Google Scholar]
- 175. Douali L, Villemin D, Cherqaoui D. Neural networks: accurate nonlinear QSAR model for HEPT derivatives. J Chem Inf Comput Sci. 2003;43(4):1200‐1207. [DOI] [PubMed] [Google Scholar]
- 176. Sabet R, Fassihi A, Hemmateenejad B, Saghaei L, Miri R, Gholami M. Computer‐aided design of novel antibacterial 3‐hydroxypyridine‐4‐ones: application of QSAR methods based on the MOLMAP approach. J Comput Aided Mol Des. 2012;26(3):349‐361. [DOI] [PubMed] [Google Scholar]
- 177. Fjell CD, Jenssen H, Hilpert K, et al. Identification of novel antibacterial peptides by chemoinformatics and machine learning. J Med Chem. 2009;52(7):2006‐2015. [DOI] [PubMed] [Google Scholar]
- 178. Torrent M, Andreu D, Nogues VM, Boix E. Connecting peptide physicochemical and antimicrobial properties by a rational prediction model. PLOS One. 2011;6(2):e16968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Sardari S, Kohanzad H, Ghavami G. Artificial neural network modeling of antimycobacterial chemical space to introduce efficient descriptors employed for drug design. Chemometr Intell Lab. 2014;130:151‐158. [Google Scholar]
- 180. Khatri N, Lather V, Madan A. Diverse classification models for anti‐hepatitis C virus activity of thiourea derivatives. Chemometr Intell Lab. 2015;140:13‐21. [Google Scholar]
- 181. Hu L, Chen G, Chau RM. A neural networks‐based drug discovery approach and its application for designing aldose reductase inhibitors. J Mol Graph Model. 2006;24(4):244‐253. [DOI] [PubMed] [Google Scholar]
- 182. Patra JC, Chua BH. Artificial neural network‐based drug design for diabetes mellitus using flavonoids. J Comput Chem. 2011;32(4):555‐567. [DOI] [PubMed] [Google Scholar]
- 183. Myint KZ, Wang L, Tong Q, Xie XQ. Molecular fingerprint‐based artificial neural networks QSAR for ligand biological activity predictions. Mol Pharmaceutics. 2012;9(10):2912‐2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Geanes AR, Cho HP, Nance KD, et al. Ligand‐based virtual screen for the discovery of novel M5 inhibitor chemotypes. Bioorg Med Chem Lett. 2016;26(18):4487‐4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Ma J, Sheridan RP, Liaw A, Dahl GE, Svetnik V. Deep neural nets as a method for quantitative structure‐activity relationships. J Chem Inf Model. 2015;55(2):263‐274. [DOI] [PubMed] [Google Scholar]
- 186. Martin EJ, Polyakov VR, Tian L, Perez RC. Profile‐QSAR 2.0: kinase virtual screening accuracy comparable to four‐concentration IC50s for realistically novel compounds. J Chem Inf Model. 2017;57(8):2077‐2088. [DOI] [PubMed] [Google Scholar]
- 187. Shamsara J. A random forest model to predict the activity of a large set of soluble epoxide hydrolase inhibitors solely based on a set of simple fragmental descriptors. Comb Chem High Throughput Screen. 2019;22:555‐569. [DOI] [PubMed] [Google Scholar]
- 188. Simeon S, Jongkon N. Construction of quantitative structure activity relationship (QSAR) models to predict potency of structurally diverse Janus kinase 2 inhibitors. Molecules (Basel, Switzerland). 2019;24(23):4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Marchese Robinson RL, Palczewska A, Palczewski J, Kidley N. Comparison of the predictive performance and interpretability of Random Forest and Linear Models on Benchmark Data Sets. J Chem Inf Model. 2017;57(8):1773‐1792. [DOI] [PubMed] [Google Scholar]
- 190. Speck‐Planche A, Kleandrova VV, Cordeiro MN. New insights toward the discovery of antibacterial agents: multi‐tasking QSBER model for the simultaneous prediction of anti‐tuberculosis activity and toxicological profiles of drugs. Eur J Pharm Sci. 2013;48(4–5):812‐818. [DOI] [PubMed] [Google Scholar]
- 191. Tenorio‐Borroto E, Peñuelas Rivas CG, Vásquez Chagoyán JC, et al. ANN multiplexing model of drugs effect on macrophages; theoretical and flow cytometry study on the cytotoxicity of the anti‐microbial drug G1 in spleen. Bioorg Med Chem. 2012;20(20):6181‐6194. [DOI] [PubMed] [Google Scholar]
- 192. Tenorio‐Borroto E, Peñuelas‐Rivas CG, Vásquez‐Chagoyán JC, et al. Model for high‐throughput screening of drug immunotoxicity—study of the anti‐microbial G1 over peritoneal macrophages using flow cytometry. Eur J Med Chem. 2014;72:206‐220. [DOI] [PubMed] [Google Scholar]
- 193. Speck‐Planche A, Cordeiro MN. Simultaneous modeling of antimycobacterial activities and ADMET profiles: a chemoinformatic approach to medicinal chemistry. Curr Top Med Chem. 2013;13(14):1656‐1665. [DOI] [PubMed] [Google Scholar]
- 194. Speck‐Planche A, Cordeiro MN. Simultaneous virtual prediction of anti‐Escherichia coli activities and ADMET profiles: a chemoinformatic complementary approach for high‐throughput screening. ACS Comb Sci. 2014;16(2):78‐84. [DOI] [PubMed] [Google Scholar]
- 195. Kleandrova VV, Ruso JM, Speck‐Planche A, Dias Soeiro Cordeiro MN. Enabling the discovery and virtual screening of potent and safe antimicrobial peptides. Simultaneous prediction of antibacterial activity and cytotoxicity. ACS Comb Sci. 2016;18(8):490‐498. [DOI] [PubMed] [Google Scholar]
- 196. Speck‐Planche A, Dias Soeiro Cordeiro MN. Speeding up early drug discovery in antiviral research: a fragment‐based in silico approach for the design of virtual anti‐hepatitis C leads. ACS Comb Sci. 2017;19(8):501‐512. [DOI] [PubMed] [Google Scholar]
- 197. Vina D, Uriarte E, Orallo F, Gonzalez‐Diaz H. Alignment‐free prediction of a drug‐target complex network based on parameters of drug connectivity and protein sequence of receptors. Mol Pharmaceutics. 2009;6(3):825‐835. [DOI] [PubMed] [Google Scholar]
- 198. Speck‐Planche A, Kleandrova VV, Luan F, Cordeiro MN. Chemoinformatics in multi‐target drug discovery for anti‐cancer therapy: in silico design of potent and versatile anti‐brain tumor agents. Anticancer Agents Med Chem. 2012;12(6):678‐685. [DOI] [PubMed] [Google Scholar]
- 199. Speck‐Planche A, Cordeiro M. Fragment‐based in silico modeling of multi‐target inhibitors against breast cancer‐related proteins. Mol Divers. 2017;21(3):511‐523. [DOI] [PubMed] [Google Scholar]
- 200. Dahl GE, Jaitly N, Salakhutdinov R Multi‐task neural networks for QSAR predictions. arXiv preprint arXiv:14061231. 2014.
- 201. Schlessinger A, Abagyan R, Carlson HA, Dang KK, Guinney J, Cagan RL. Multi‐targeting drug community challenge. Cell Chemical Biology. 2017;24(12):1434‐1435. [DOI] [PubMed] [Google Scholar]
- 202. Cichonska A, Ravikumar B, Allaway RJ, et al. Crowdsourced mapping extends the target space of kinase inhibitors. bioRxiv. 2020. 10.1101/2019.12.31.891812 [DOI] [Google Scholar]
- 203. Xu Y, Ma J, Liaw A, Sheridan RP, Svetnik V. Demystifying multitask deep neural networks for quantitative structure‐activity relationships. J Chem Inf Model. 2017;57(10):2490‐2504. [DOI] [PubMed] [Google Scholar]
- 204. Zakharov AV, Zhao T, Nguyen DT, et al. Novel consensus architecture to improve performance of large‐scale multitask deep learning QSAR Models. J Chem Inf Model. 2019;59(11):4613‐4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Kwon S, Bae H, Jo J, Yoon S. Comprehensive ensemble in QSAR prediction for drug discovery. BMC Bioinformatics. 2019;20(1):521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Cheng T, Li Q, Wang Y, Bryant SH. Binary classification of aqueous solubility using support vector machines with reduction and recombination feature selection. J Chem Inf Model. 2011;51(2):229‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Lind P, Maltseva T. Support vector machines for the estimation of aqueous solubility. J Chem Inf Comput Sci. 2003;43(6):1855‐1859. [DOI] [PubMed] [Google Scholar]
- 208. Lusci A, Pollastri G, Baldi P. Deep architectures and deep learning in chemoinformatics: the prediction of aqueous solubility for drug‐like molecules. J Chem Inf Model. 2013;53(7):1563‐1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Coley CW, Barzilay R, Green WH, Jaakkola TS, Jensen KF. Convolutional embedding of attributed molecular graphs for physical property prediction. J Chem Inf Model. 2017;57(8):1757‐1772. [DOI] [PubMed] [Google Scholar]
- 210. Boobier S, Osbourn A, Mitchell JBO. Can human experts predict solubility better than computers? J Cheminform. 2017;9(1):63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Shaik B, Gupta R, Louis B, Agrawal VK. Prediction of permeability of drug‐like compounds across polydimethylsiloxane membranes by machine learning methods. J Pharm Investig. 2015;45(5):461‐473. [Google Scholar]
- 212. Chi CT, Lee MH, Weng CF, Leong MK. In silico prediction of PAMPA effective permeability using a two‐QSAR approach. Int J Mol Sci. 2019;20(13):3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Brocke SA, Degen A, MacKerell AD, Dutagaci B, Feig M. Prediction of membrane permeation of drug molecules by combining an implicit membrane model with machine learning. J Chem Inf Model. 2019;59(3):1147‐1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Cheng T, Zhao Y, Li X, et al. Computation of octanol‐water partition coefficients by guiding an additive model with knowledge. J Chem Inf Model. 2007;47(6):2140‐2148. [DOI] [PubMed] [Google Scholar]
- 215. Zhang H, Xiang ML, Ma CY, et al. Three‐class classification models of logS and logP derived by using GA‐CG‐SVM approach. Mol Divers. 2009;13(2):261‐268. [DOI] [PubMed] [Google Scholar]
- 216. Liao Q, Yao J, Yuan S. SVM approach for predicting LogP. Mol Divers. 2006;10(3):301‐309. [DOI] [PubMed] [Google Scholar]
- 217. Tetko IV, Poda GI. Application of ALOGPS 2.1 to predict log D distribution coefficient for Pfizer proprietary compounds. J Med Chem. 2004;47(23):5601‐5604. [DOI] [PubMed] [Google Scholar]
- 218. Tetko IV, Tanchuk VY, Villa AE. Prediction of n‐octanol/water partition coefficients from PHYSPROP database using artificial neural networks and E‐state indices. J Chem Inf Comput Sci. 2001;41(5):1407‐1421. [DOI] [PubMed] [Google Scholar]
- 219. Bruneau P, McElroy NR. logD7.4 modeling using Bayesian regularized neural networks. Assessment and correction of the errors of prediction. J Chem Inf Model. 2006;46(3):1379‐1387. [DOI] [PubMed] [Google Scholar]
- 220. Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discovery. 2004;3(8):711‐716. [DOI] [PubMed] [Google Scholar]
- 221. Bhhatarai B, Walters WP, Hop C, Lanza G, Ekins S. Opportunities and challenges using artificial intelligence in ADME/Tox. Nat Mater. 2019;18(5):418‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Shen J, Cheng F, Xu Y, Li W, Tang Y. Estimation of ADME properties with substructure pattern recognition. J Chem Inf Model. 2010;50(6):1034‐1041. [DOI] [PubMed] [Google Scholar]
- 223. Yang H, Sun L, Li W, Liu G, Tang Y. In silico prediction of chemical toxicity for drug design using machine learning methods and structural alerts. Front Chem. 2018;6:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Huang R, Xia M, Nguyen D‐T, et al. Tox21Challenge to build predictive models of nuclear receptor and stress response pathways as mediated by exposure to environmental chemicals and drugs. Front Environ Sci. 2016;3(85). 10.3389/fenvs.2015.00085 [DOI] [Google Scholar]
- 225. Irwin BWJ, Levell JR, Whitehead TM, Segall MD, Conduit GJ. Practical applications of deep learning to impute heterogeneous drug discovery data. J Chem Inf Model. 2020;60(6):2848‐2857. [DOI] [PubMed] [Google Scholar]
- 226. Wang Y, Huang L, Jiang S, et al. Capsule networks showed excellent performance in the classification of hERG blockers/nonblockers. Front Pharmacol. 2019;10:1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Tao L, Zhang P, Qin C, et al. Recent progresses in the exploration of machine learning methods as in‐silico ADME prediction tools. Adv Drug Deliv Rev. 2015;86:83‐100. [DOI] [PubMed] [Google Scholar]
- 228. Bhhatarai B, Walters WP, Hop CECA, Lanza G, Ekins S. Opportunities and challenges using artificial intelligence in ADME/Tox. Nat Mater. 2019;18(5):418‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Zang Q, Mansouri K, Williams AJ, et al. In silico prediction of physicochemical properties of environmental chemicals using molecular fingerprints and machine learning. J Chem Inf Model. 2017;57(1):36‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Alqahtani S. In silico ADME‐Tox modeling: progress and prospects. Expert Opin Drug Metab Toxicol. 2017;13(11):1147‐1158. [DOI] [PubMed] [Google Scholar]
- 231. Wenzel J, Matter H, Schmidt F. Predictive multitask deep neural network models for ADME‐Tox properties: learning from large data sets. J Chem Inf Model. 2019;59(3):1253‐1268. [DOI] [PubMed] [Google Scholar]
- 232. Göller AH, Kuhnke L, Montanari F, et al. Bayer's in silico ADMET platform: a journey of machine learning over the past two decades. Drug Discovery Today. 2020;25:1702‐1709. [DOI] [PubMed] [Google Scholar]
- 233. Schneider G. Future de novo drug design. Mol Inf. 2014;33(6‐7):397‐402. [DOI] [PubMed] [Google Scholar]
- 234. Struble TJ, Alvarez JC, Brown SP, et al. Current and future roles of artificial intelligence in medicinal chemistry synthesis. J Med Chem. 2020;63:8667‐8682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Schneider G. Generative models for artificially‐intelligent molecular design. Mol Inf. 2018;37(1‐2):1880131. [DOI] [PubMed] [Google Scholar]
- 236. Kadurin A, Aliper A, Kazennov A, et al. The cornucopia of meaningful leads: applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget. 2017;8(7):10883‐10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Kadurin A, Nikolenko S, Khrabrov K, Aliper A, Zhavoronkov A. druGAN: an advanced generative adversarial autoencoder model for de novo generation of new molecules with desired molecular properties in silico. Mol Pharmaceutics. 2017;14(9):3098‐3104. [DOI] [PubMed] [Google Scholar]
- 238. Polykovskiy D, Zhebrak A, Vetrov D, et al. Entangled conditional adversarial autoencoder for de novo drug discovery. Mol Pharmaceutics. 2018;15(10):4398‐4405. [DOI] [PubMed] [Google Scholar]
- 239. Gómez‐Bombarelli R, Wei JN, Duvenaud D, et al. Automatic chemical design using a data‐driven continuous representation of molecules. ACS Cent Sci. 2018;4(2):268‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Lim J, Ryu S, Kim JW, Kim WY. Molecular generative model based on conditional variational autoencoder for de novo molecular design. J Cheminform. 2018;10(1):31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Olivecrona M, Blaschke T, Engkvist O, Chen H. Molecular de‐novo design through deep reinforcement learning. J Cheminform. 2017;9(1):48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Segler MHS, Kogej T, Tyrchan C, Waller MP. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent Sci. 2018;4(1):120‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Yuan W, Jiang D, Nambiar DK, et al. Chemical space mimicry for drug discovery. J Chem Inf Model. 2017;57(4):875‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Merk D, Grisoni F, Friedrich L, Schneider G. Tuning artificial intelligence on the de novo design of natural‐product‐inspired retinoid X receptor modulators. Communications Chemistry. 2018;1(1):68. [Google Scholar]
- 245. Popova M, Isayev O, Tropsha A. Deep reinforcement learning for de novo drug design. Sci Adv. 2018;4(7):eaap7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Henderson P, Islam R, Bachman P, Pineau J, Precup D, Meger D. Deep reinforcement learning that matters. Paper presented at: Thirty‐Second AAAI Conference on Artificial Intelligence, 2018.
- 247. Putin E, Asadulaev A, Ivanenkov Y, et al. Reinforced adversarial neural computer for de novo molecular design. J Chem Inf Model. 2018;58(6):1194‐1204. [DOI] [PubMed] [Google Scholar]
- 248. Sanchez‐Lengeling B, Outeiral C, Guimaraes GL, Aspuru‐Guzik A. Optimizing distributions over molecular space. An objective‐reinforced generative adversarial network for inverse‐design chemistry (ORGANIC). ChemRxiv. 2017. 10.26434/chemrxiv.5309668.v3 [DOI] [Google Scholar]
- 249. Gupta A, Muller AT, Huisman BJH, Fuchs JA, Schneider P, Schneider G. Generative recurrent networks for de novo drug design. Mol Inf. 2018;37(1–2):1700111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Awale M, Sirockin F, Stiefl N, Reymond JL. Drug analogs from fragment‐based long short‐term memory generative neural networks. J Chem Inf Model. 2019;59(4):1347‐1356. [DOI] [PubMed] [Google Scholar]
- 251. Liu B, Ramsundar B, Kawthekar P, et al. Retrosynthetic reaction prediction using neural sequence‐to‐sequence models. ACS Cent Sci. 2017;3(10):1103‐1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Noel OB, Andrew D. DeepSMILES: an adaptation of SMILES for use in machine‐learning of chemical structures. ChemRxiv. 2018. 10.26434/chemrxiv.7097960.v1 [DOI] [Google Scholar]
- 253. Yoshikawa N, Terayama K, Sumita M, Homma T, Oono K, Tsuda KJCL. Population‐based de novo molecule generation, using grammatical evolution. Chem Lett. 2018;47(11):1431‐1434. [Google Scholar]
- 254. Pope PE, Kolouri S, Rostami M, Martin CE, Hoffmann H. Discovering molecular functional groups using graph convolutional neural networks. ArXiv. 2018. abs/1812.00265. [Google Scholar]
- 255. You J, Liu B, Ying R, Pande V, Leskovec J. Graph convolutional policy network for goal‐directed molecular graph generation. In: Proceedings of the 32nd International Conference on Neural Information Processing Systems. Montréal, Canada; 2018:6412‐6422.
- 256. De Cao N, Kipf T. MolGAN: an implicit generative model for small molecular graphs. ArXiv Preprint. 2018;arXiv:180511973.
- 257. Maziarka Ł, Pocha A, Kaczmarczyk J, Rataj K, Danel T, Warchoł M. Mol‐CycleGAN: a generative model for molecular optimization. J Cheminform. 2020;12(1):1‐18. 10.1186/s13321-019-0404-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Liu Q, Allamanis M, Brockschmidt M, Gaunt A. Constrained graph variational autoencoders for molecule design. In: Proceedings of the 32nd International Conference on Neural Information Processing Systems. Montréal, Canada; 2018:7806‐7815.
- 259. Skalic M, Jiménez J, Sabbadin D, de Fabritiis G. Shape‐based generative modeling for de novo drug design. J Chem Inf Model. 2019;59(3):1205‐1214. [DOI] [PubMed] [Google Scholar]
- 260. Button A, Merk D, Hiss JA, Schneider G. Automated de novo molecular design by hybrid machine intelligence and rule‐driven chemical synthesis. Nat Mach Intell. 2019;1(7):307‐315. [Google Scholar]
- 261. Méndez‐Lucio O, Baillif B, Clevert D‐A, Rouquié D, Wichard J. De novo generation of hit‐like molecules from gene expression signatures using artificial intelligence. Nat Commun. 2020;11(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Overview of attention for article published in Nature Biotechnology, September 2019. Altmetric. https://www.altmetric.com/details/65797801. Accessed October 2, 2020. [DOI] [PubMed]
- 263. Zhavoronkov A, Ivanenkov YA, Aliper A, et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nature Biotechnol. 2019;37(9):1038‐1040. [DOI] [PubMed] [Google Scholar]
- 264. Hanson SM, Georghiou G, Thakur MK, et al. What makes a kinase promiscuous for inhibitors? Cell Chem Biol. 2019;26(3):390‐399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Walters WP, Murcko M. Assessing the impact of generative AI on medicinal chemistry. Nature Biotechnol. 2020;38(2):143‐145. [DOI] [PubMed] [Google Scholar]
- 266. Rampasek L, Hidru D, Smirnov P, Haibe‐Kains B, Goldenberg A. Dr.VAE: improving drug response prediction via modeling of drug perturbation effects. Bioinformatics. 2019;35(19):3743‐3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Geeleher P, Cox NJ, Huang RS. Clinical drug response can be predicted using baseline gene expression levels and in vitro drug sensitivity in cell lines. Genome Biol. 2014;15(3):R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Ding Z, Zu S, Gu J. Evaluating the molecule‐based prediction of clinical drug responses in cancer. Bioinformatics. 2016;32(19):2891‐2895. [DOI] [PubMed] [Google Scholar]
- 269. Iorio F, Knijnenburg TA, Vis DJ, et al. A landscape of pharmacogenomic interactions in cancer. Cell. 2016;166(3):740‐754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Tan M, Ozgul OF, Bardak B, Eksioglu I, Sabuncuoglu S. Drug response prediction by ensemble learning and drug‐induced gene expression signatures. Genomics. 2019;111(5):1078‐1088. [DOI] [PubMed] [Google Scholar]
- 271. Graim K, Friedl V, Houlahan KE, Stuart JM. PLATYPUS: a multiple‐view learning predictive framework for cancer drug sensitivity prediction. Pac Symp Biocomput. 2019;24:136‐147. [PMC free article] [PubMed] [Google Scholar]
- 272. Sharifi‐Noghabi H, Zolotareva O, Collins CC, Ester M. MOLI: multi‐omics late integration with deep neural networks for drug response prediction. Bioinformatics. 2019;35(14):i501‐i509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Dincer AB, Celik S, Hiranuma N, Lee S‐I. DeepProfile: deep learning of cancer molecular profiles for precision medicine. bioRxiv. 2018:278739. 10.1101/278739 [DOI] [Google Scholar]
- 274. Li M, Wang Y, Zheng R, et al. DeepDSC: a deep learning method to predict drug sensitivity of cancer cell lines. IEEE/ACM Trans Comput Biol Bioinform. 2019:1. 10.1109/tcbb.2019.2919581 [DOI] [PubMed] [Google Scholar]
- 275. Ding MQ, Chen L, Cooper GF, Young JD, Lu X. Precision oncology beyond targeted therapy: combining omics data with machine learning matches the majority of cancer cells to effective therapeutics. Mol Cancer Res. 2018;16(2):269‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Iwata M, Yuan L, Zhao Q, et al. Predicting drug‐induced transcriptome responses of a wide range of human cell lines by a novel tensor‐train decomposition algorithm. Bioinformatics. 2019;35(14):i191‐i199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Tan M. Prediction of anti‐cancer drug response by kernelized multi‐task learning. Artif Intell Med. 2016;73:70‐77. [DOI] [PubMed] [Google Scholar]
- 278. Ammad‐Ud‐Din M, Khan SA, Wennerberg K, Aittokallio T. Systematic identification of feature combinations for predicting drug response with Bayesian multi‐view multi‐task linear regression. Bioinformatics. 2017;33(14):i359‐i368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Greco WR, Faessel H, Levasseur L. The search for cytotoxic synergy between anticancer agents: a case of Dorothy and the ruby slippers? J Natl Cancer Inst. 1996;88(11):699‐700. [DOI] [PubMed] [Google Scholar]
- 280. Roell KR, Reif DM, Motsinger‐Reif AA. An Introduction to terminology and methodology of chemical synergy‐perspectives from across disciplines. Front Pharmacol. 2017;8:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Gibbs BK, Sourbier C. Detecting the potential pharmacological synergy of drug combination by viability assays in vitro. Methods Mol Biol (Clifton, NJ). 2018;1709:129‐137. [DOI] [PubMed] [Google Scholar]
- 282. Mayr A, Klambauer G, Unterthiner T, Hochreiter S. DeepTox: toxicity prediction using deep learning. Front Environ Sci. 2016;3(80). 10.3389/fenvs.2015.00080 [DOI] [Google Scholar]
- 283. Madani Tonekaboni SA, Soltan Ghoraie L, Manem VSK, Haibe‐Kains B. Predictive approaches for drug combination discovery in cancer. Brief Bioinform. 2018;19(2):263‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Preuer K, Lewis RPI, Hochreiter S, Bender A, Bulusu KC, Klambauer G. DeepSynergy: predicting anti‐cancer drug synergy with deep learning. Bioinformatics. 2018;34(9):1538‐1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Malyutina A, Majumder MM, Wang W, Pessia A, Heckman CA, Tang J. Drug combination sensitivity scoring facilitates the discovery of synergistic and efficacious drug combinations in cancer. PLOS Comput Biol. 2019;15(5):e1006752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Sidorov P, Naulaerts S, Ariey‐Bonnet J, Pasquier E, Ballester PJ. Predicting synergism of cancer drug combinations using NCI‐ALMANAC data. Front Chem. 2019;7:509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Celebi R, Bear Don't Walk O 4th, Movva R, Alpsoy S, Dumontier M. In‐silico prediction of synergistic anti‐cancer drug combinations using multi‐omics data. Sci Rep. 2019;9(1):8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Singh H, Rana PS, Singh U. Prediction of drug synergy in cancer using ensemble‐based machine learning techniques. Mod Phys Lett B. 2018;32(11):1850132. [Google Scholar]
- 289. Gallagher PJ, Castro V, Fava M, et al. Antidepressant response in patients with major depression exposed to NSAIDs: a pharmacovigilance study. Am J Psychiatry. 2012;169(10):1065‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Mason DJ, Eastman RT, Lewis RPI, Stott IP, Guha R, Bender A. Using machine learning to predict synergistic antimalarial compound combinations with novel structures. Front Pharmacol. 2018;9:1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Mason DJ, Stott I, Ashenden S, et al. Prediction of antibiotic interactions using descriptors derived from molecular structure. J Med Chem. 2017;60(9):3902‐3912. [DOI] [PubMed] [Google Scholar]
- 292. Ianevski A, Giri AK, Gautam P, et al. Prediction of drug combination effects with a minimal set of experiments. Nat Mach Intell. 2019;1(12):568‐577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Ding P, Yin R, Luo J, Kwoh CK. Ensemble prediction of synergistic drug combinations incorporating biological, chemical, pharmacological, and network knowledge. IEEE J Biomed Health Inform. 2019;23(3):1336‐1345. [DOI] [PubMed] [Google Scholar]
- 294. Zhang C, Yan G. Synergistic drug combinations prediction by integrating pharmacological data. Synth Syst Biotechnol. 2019;4(1):67‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Vilar S, Harpaz R, Uriarte E, Santana L, Rabadan R, Friedman C. Drug‐drug interaction through molecular structure similarity analysis. J Am Med Inform Assoc. 2012;19(6):1066‐1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Zitnik M, Agrawal M, Leskovec J. Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics. 2018;34(13):i457‐i466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Ryu JY, Kim HU, Lee SY. Deep learning improves prediction of drug–drug and drug–food interactions. Proc Natl Acad Sci. 2018;115(18):E4304‐E4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Zheng Y, Peng H, Zhang X, Zhao Z, Yin J, Li J. Predicting adverse drug reactions of combined medication from heterogeneous pharmacologic databases. BMC Bioinformatics. 2018;19(19):517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Lee CY, Chen YP. Prediction of drug adverse events using deep learning in pharmaceutical discovery. Brief Bioinform. 2020. 10.1093/bib/bbaa040 [DOI] [PubMed] [Google Scholar]
- 300. Shankar S, Bhandari I, Okou DT, Srinivasa G, Athri P. Predicting adverse drug reactions of two‐drug combinations using structural and transcriptomic drug representations to train an artificial neural network. Chem Biol Drug Des. 2020:cbdd.13802 [DOI] [PubMed] [Google Scholar]
- 301. Avorn J. The $2.6 billion pill—methodologic and policy considerations. N Engl J Med. 2015;372(20):1877‐1879. [DOI] [PubMed] [Google Scholar]
- 302. Mullard A. FDA drug approvals. Nat Rev Drug Discovery. 2016;2017 16(2):73‐76. [DOI] [PubMed] [Google Scholar]
- 303. Tan SY, Grimes S. Paul Ehrlich (1854–1915): man with the magic bullet. Singapore Med J. 2010;51(11):842‐843. [PubMed] [Google Scholar]
- 304. Greene JA, Loscalzo J. Putting the patient back together—social medicine, network medicine, and the limits of reductionism. N Engl J Med. 2017;377(25):2493‐2499. [DOI] [PubMed] [Google Scholar]
- 305. Gottlieb A, Stein GY, Ruppin E, Sharan R. PREDICT: a method for inferring novel drug indications with application to personalized medicine. Mol Syst Biol. 2011;7:496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Liu Z, Guo F, Gu J, et al. Similarity‐based prediction for anatomical therapeutic chemical classification of drugs by integrating multiple data sources. Bioinformatics. 2015;31(11):1788‐1795. [DOI] [PubMed] [Google Scholar]
- 307. Napolitano F, Zhao Y, Moreira VM, et al. Drug repositioning: a machine‐learning approach through data integration. J Cheminform. 2013;5(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Masoudi‐Sobhanzadeh Y, Omidi Y, Amanlou M, Masoudi‐Nejad A. DrugR+: a comprehensive relational database for drug repurposing, combination therapy, and replacement therapy. Comput Biol Med. 2019;109:254‐262. [DOI] [PubMed] [Google Scholar]
- 309. Ravikumar B, Timonen S, Alam Z, Parri E, Wennerberg K, Aittokallio T. Chemogenomic analysis of the druggable kinome and its application to repositioning and lead identification studies. Cell Chem Biol. 2019;26(11):1608‐1622. [DOI] [PubMed] [Google Scholar]
- 310. Oh M, Ahn J, Yoon Y. A network‐based classification model for deriving novel drug‐disease associations and assessing their molecular actions. PLOS One. 2014;9(10):e111668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Zheng Y, Peng H, Zhang X, Zhao Z, Gao X, Li J. Old drug repositioning and new drug discovery through similarity learning from drug‐target joint feature spaces. BMC Bioinformatics. 2019;20(suppl 23):605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Zhang P, Agarwal P, Obradovic Z. Computational Drug Repositioning by Ranking and Integrating Multiple Data Sources. Berlin, Heidelberg, Germany: Springer Verlag; 2013. [Google Scholar]
- 313. Xuan P, Zhao L, Zhang T, Ye Y, Zhang Y. Inferring drug‐related diseases based on convolutional neural network and gated recurrent unit. Molecules (Basel, Switzerland). 2019;24(15):2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Aliper A, Plis S, Artemov A, Ulloa A, Mamoshina P, Zhavoronkov A. Deep learning applications for predicting pharmacological properties of drugs and drug repurposing using transcriptomic data. Mol Pharmaceutics. 2016;13(7):2524‐2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Wu G, Liu J, Yue X. Prediction of drug‐disease associations based on ensemble meta paths and singular value decomposition. BMC Bioinformatics. 2019;20(suppl 3):134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Zhao D, Wang J, Sang S, Lin H, Wen J, Yang C. Relation path feature embedding based convolutional neural network method for drug discovery. BMC Med Inform Decis Mak. 2019;19(suppl 2):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Jiang HJ, Huang YA, You ZH. Predicting drug‐disease associations via using Gaussian interaction profile and Kernel‐based autoencoder. BioMed Res Int. 2019;2019:1‐11. 10.1155/2019/2426958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Lee M, Kim H, Joe H, Kim H‐G. Multi‐channel PINN: investigating scalable and transferable neural networks for drug discovery. J Cheminform. 2019;11(1):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Wang R, Li S, Cheng L, Wong MH, Leung KS. Predicting associations among drugs, targets and diseases by tensor decomposition for drug repositioning. BMC Bioinformatics. 2019;20(suppl 26):628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Bahi M, Batouche M. Drug‐Target Interaction Prediction in Drug Repositioning Based on Deep Semi‐Supervised Learning. Cham, Switzerland: Springer; 2018. [Google Scholar]
- 321. deAndres‐Galiana EJ, Bea G, Fernandez‐Martinez JL, Saligan LN. Analysis of defective pathways and drug repositioning in Multiple Sclerosis via machine learning approaches. Comput Biol Med. 2019;115:103492. [DOI] [PubMed] [Google Scholar]
- 322. Xia Z, Wu LY, Zhou X, Wong ST. Semi‐supervised drug‐protein interaction prediction from heterogeneous biological spaces. BMC Syst Biol. 2010;4(suppl 2):S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Chen H, Zhang Z. A semi‐supervised method for drug‐target interaction prediction with consistency in networks. PLOS One. 2013;8(5):e62975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Nam Y, Kim M, Chang H‐S, Shin H. Drug repurposing with network reinforcement. BMC Bioinformatics. 2019;20(13):383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Yan XY, Zhang SW, Zhang SY. Prediction of drug‐target interaction by label propagation with mutual interaction information derived from heterogeneous network. Mol BioSyst. 2016;12(2):520‐531. [DOI] [PubMed] [Google Scholar]
- 326. Zeng X, Zhu S, Liu X, Zhou Y, Nussinov R, Cheng F. deepDR: a network‐based deep learning approach to in silico drug repositioning. Bioinformatics. 2019;35(24):5191‐5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Feustel SM, Meissner M, Liesenfeld O. Toxoplasma gondii and the blood‐brain barrier. Virulence. 2012;3(2):182‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Ballabh P, Braun A, Nedergaard M. The blood‐brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16(1):1‐13. [DOI] [PubMed] [Google Scholar]
- 329. Doniger S, Hofmann T, Yeh J. Predicting CNS permeability of drug molecules: comparison of neural network and support vector machine algorithms. J Comput Biol. 2002;9(6):849‐864. [DOI] [PubMed] [Google Scholar]
- 330. Martins IF, Teixeira AL, Pinheiro L, Falcao AO. A Bayesian approach to in silico blood‐brain barrier penetration modeling. J Chem Inf Model. 2012;52(6):1686‐1697. [DOI] [PubMed] [Google Scholar]
- 331. Majumdar S, Basak SC, Lungu CN, Diudea MV, Grunwald GD. Finding needles in a haystack: determining key molecular descriptors associated with the blood‐brain barrier entry of chemical compounds using machine learning. Mol Inf. 2019;38:1800164. [DOI] [PubMed] [Google Scholar]
- 332. Golmohammadi H, Dashtbozorgi Z, Acree WE Jr. Quantitative structure‐activity relationship prediction of blood‐to‐brain partitioning behavior using support vector machine. Eur J Pharm Sci. 2012;47(2):421‐429. [DOI] [PubMed] [Google Scholar]
- 333. Yuan Y, Zheng F, Zhan CG. Improved prediction of blood‐brain barrier permeability through machine learning with combined use of molecular property‐based descriptors and fingerprints. AAPS J. 2018;20(3):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. Mente SR, Lombardo F. A recursive‐partitioning model for blood‐brain barrier permeation. J Comput Aided Mol Des. 2005;19(7):465‐481. [DOI] [PubMed] [Google Scholar]
- 335. Zhao YH, Abraham MH, Ibrahim A, et al. Predicting penetration across the blood‐brain barrier from simple descriptors and fragmentation schemes. J Chem Inf Model. 2007;47(1):170‐175. [DOI] [PubMed] [Google Scholar]
- 336. Obrezanova O, Csanyi G, Gola JM, Segall MD. Gaussian processes: a method for automatic QSAR modeling of ADME properties. J Chem Inf Model. 2007;47(5):1847‐1857. [DOI] [PubMed] [Google Scholar]
- 337. Andres C, Hutter MC. CNS permeability of drugs predicted by a decision tree. QSAR Comb Sci. 2006;25(4):305‐309. [Google Scholar]
- 338. Zhang L, Zhu H, Oprea TI, Golbraikh A, Tropsha A. QSAR modeling of the blood‐brain barrier permeability for diverse organic compounds. Pharm Res. 2008;25(8):1902‐1914. [DOI] [PubMed] [Google Scholar]
- 339. Vilar S, Chakrabarti M, Costanzi S. Prediction of passive blood‐brain partitioning: straightforward and effective classification models based on in silico derived physicochemical descriptors. J Mol Graph Model. 2010;28(8):899‐903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340. Wang Z, Yang H, Wu Z, et al. In silico prediction of blood‐brain barrier permeability of compounds by machine learning and resampling methods. ChemMedChem. 2018;13(20):2189‐2201. [DOI] [PubMed] [Google Scholar]
- 341. Garg P, Verma J. In silico prediction of blood brain barrier permeability: an Artificial Neural Network model. J Chem Inf Model. 2006;46(1):289‐297. [DOI] [PubMed] [Google Scholar]
- 342. Garg P, Dhakne R, Belekar V. Role of breast cancer resistance protein (BCRP) as active efflux transporter on blood‐brain barrier (BBB) permeability. Mol Divers. 2015;19(1):163‐172. [DOI] [PubMed] [Google Scholar]
- 343. Lingineni K, Belekar V, Tangadpalliwar SR, Garg P. The role of multidrug resistance protein (MRP‐1) as an active efflux transporter on blood‐brain barrier (BBB) permeability. Mol Divers. 2017;21(2):355‐365. [DOI] [PubMed] [Google Scholar]
- 344. Geier EG, Schlessinger A, Fan H, et al. Structure‐based ligand discovery for the large‐neutral amino acid transporter 1, LAT‐1. Proc Natl Acad Sci USA. 2013;110(14):5480‐5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Dolghih E, Jacobson MP. Predicting efflux ratios and blood‐brain barrier penetration from chemical structure: combining passive permeability with active efflux by P‐glycoprotein. ACS Chem Neurosci. 2013;4(2):361‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346. Hindle SJ, Munji RN, Dolghih E, et al. Evolutionarily conserved roles for blood‐brain barrier xenobiotic transporters in endogenous steroid partitioning and behavior. Cell Rep. 2017;21(5):1304‐1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347. Miller DS. Regulation of ABC transporters blood‐brain barrier: the good, the bad, and the ugly. Adv Cancer Res. 2015;125:43‐70. [DOI] [PubMed] [Google Scholar]
- 348. Qosa H, Miller DS, Pasinelli P, Trotti D. Regulation of ABC efflux transporters at blood‐brain barrier in health and neurological disorders. Brain Res. 2015;1628(pt B):298‐316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349. Gao Z, Chen Y, Cai X, Xu R. Predict drug permeability to blood‐brain‐barrier from clinical phenotypes: drug side effects and drug indications. Bioinformatics. 2017;33(6):901‐908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Miao R, Xia LY, Chen HH, Huang HH, Liang Y. Improved classification of blood‐brain‐barrier drugs using deep learning. Sci Rep. 2019;9(1):8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Sullivan PF. Puzzling over schizophrenia: schizophrenia as a pathway disease. Nat Med. 2012;18(2):210‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Stachowiak MK, Kucinski A, Curl R, et al. Schizophrenia: a neurodevelopmental disorder—Integrative genomic hypothesis and therapeutic implications from a transgenic mouse model. Schizophrenia Research. 2013;143(2):367‐376. [DOI] [PubMed] [Google Scholar]
- 353. McGrath J, Saha S, Chant D, Welham J. Schizophrenia: a concise overview of incidence, prevalence, and mortality. Epidemiol Rev. 2008;30:67‐76. [DOI] [PubMed] [Google Scholar]
- 354. Mathers C. The global burden of disease: 2004 update. World Health Organization; 2008. [Google Scholar]
- 355. Kambeitz J, Kambeitz‐Ilankovic L, Leucht S, et al. Detecting neuroimaging biomarkers for schizophrenia: a meta‐analysis of multivariate pattern recognition studies. Neuropsychopharmacology. 2015;40(7):1742‐1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. Hyman SE. Revolution stalled. Sci Transl Med. 2012;4(155):155cm111. [DOI] [PubMed] [Google Scholar]
- 357. Palaniyappan L, Deshpande G, Lanka P, et al. Effective connectivity within a triple network brain system discriminates schizophrenia spectrum disorders from psychotic bipolar disorder at the single‐subject level. Schizophr Res. 2019;214:24‐33. [DOI] [PubMed] [Google Scholar]
- 358. Schnack HG. Improving individual predictions: machine learning approaches for detecting and attacking heterogeneity in schizophrenia (and other psychiatric diseases). Schizophr Res. 2019;214:34‐42. [DOI] [PubMed] [Google Scholar]
- 359. Honnorat N, Dong A, Meisenzahl‐Lechner E, Koutsouleris N, Davatzikos C. Neuroanatomical heterogeneity of schizophrenia revealed by semi‐supervised machine learning methods. Schizophr Res. 2019;214:43‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Tandon N, Tandon R. Using machine learning to explain the heterogeneity of schizophrenia. Realizing the promise and avoiding the hype. Schizophr Res. 2019;214:70‐75. [DOI] [PubMed] [Google Scholar]
- 361. Talpalaru A, Bhagwat N, Devenyi GA, Lepage M, Chakravarty MM. Identifying schizophrenia subgroups using clustering and supervised learning. Schizophr Res. 2019;214:51‐59. [DOI] [PubMed] [Google Scholar]
- 362. Mothi SS, Sudarshan M, Tandon N, et al. Machine learning improved classification of psychoses using clinical and biological stratification: update from the bipolar‐schizophrenia network for intermediate phenotypes (B‐SNIP). Schizophr Res. 2019;214:60‐69. [DOI] [PubMed] [Google Scholar]
- 363. Hsu KC, Wang FS. Model‐based optimization approaches for precision medicine: a case study in presynaptic dopamine overactivity. PLOS One. 2017;12(6):e0179575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364. Yang QX, Wang YX, Li FC, et al. Identification of the gene signature reflecting schizophrenia's etiology by constructing artificial intelligence‐based method of enhanced reproducibility. CNS Neurosci Ther. 2019;25(9):1054‐1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Marunnan SM, Pulikkal BP, Jabamalairaj A, et al. Development of MLR and SVM aided QSAR models to identify common SAR of GABA uptake herbal inhibitors used in the treatment of schizophrenia. Curr Neuropharmacol. 2017;15(8):1085‐1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Bain EE, Shafner L, Walling DP, et al. Use of a novel artificial intelligence platform on mobile devices to assess dosing compliance in a phase 2 clinical trial in subjects with schizophrenia. JMIR Mhealth Uhealth. 2017;5(2):e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367. Zhao K, So HC. Drug Repositioning for schizophrenia and depression/anxiety disorders: a machine learning approach leveraging expression. Data. IEEE J Biomed Health Inform. 2019;23(3):1304‐1315. [DOI] [PubMed] [Google Scholar]
- 368. Yin L, Chau CKL, Sham PC, So HC. Integrating clinical data and imputed transcriptome from GWAS to uncover complex disease subtypes: applications in psychiatry and cardiology. Am J Hum Genet. 2019;105(6):1193‐1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369. Chakravarty MM. Guest editorial: Special issue on machine learning in schizophrenia. Schizophr Res. 2019;214:1‐2. [DOI] [PubMed] [Google Scholar]
- 370. Fitzpatrick SE, Srivorakiat L, Wink LK, Pedapati EV, Erickson CA. Aggression in autism spectrum disorder: presentation and treatment options. Neuropsychiatr Dis Treat. 2016;12:1525‐1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Anzulewicz A, Sobota K, Delafield‐Butt JT. Toward the autism motor signature: gesture patterns during smart tablet gameplay identify children with autism. Sci Rep. 2016;6:31107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372. Obara T, Ishikuro M, Tamiya G, et al. Potential identification of vitamin B6 responsiveness in autism spectrum disorder utilizing phenotype variables and machine learning methods. Sci Rep. 2018;8(1):14840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373. Ekins S, Gerlach J, Zorn KM, Antonio BM, Lin Z, Gerlach A. Repurposing approved drugs as inhibitors of Kv7.1 and Nav1.8 to treat Pitt Hopkins Syndrome. Pharm Res. 2019;36(9):137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Heyne HO, Baez‐Nieto D, Iqbal S, et al. Predicting functional effects of missense variants in voltage‐gated sodium and calcium channels. Sci Transl Med. 2020;12(556):eaay6848. [DOI] [PubMed] [Google Scholar]
- 375. Grandjean P, Landrigan PJ. Neurobehavioural effects of developmental toxicity. The Lancet Neurology. 2014;13(3):330‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376. Frank CL, Brown JP, Wallace K, Mundy WR, Shafer TJ. From the cover: developmental neurotoxicants disrupt activity in cortical networks on microelectrode arrays: results of screening 86 compounds during neural network formation. Toxicol Sci. 2017;160(1):121‐135. [DOI] [PubMed] [Google Scholar]
- 377. Liu SH, Bobb JF, Claus Henn B, et al. Bayesian varying coefficient kernel machine regression to assess neurodevelopmental trajectories associated with exposure to complex mixtures. Stat Med. 2018;37(30):4680‐4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378. Tian S, Sun Y, Shao J, et al. Predicting escitalopram monotherapy response in depression: the role of anterior cingulate cortex. Hum Brain Mapp. 2019;41(5):1249‐1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Koutsouleris N, Kahn RS, Chekroud AM, et al. Multisite prediction of 4‐week and 52‐week treatment outcomes in patients with first‐episode psychosis: a machine learning approach. Lancet Psychiatry. 2016;3(10):935‐946. [DOI] [PubMed] [Google Scholar]
- 380. Chekroud AM, Zotti RJ, Shehzad Z, et al. Cross‐trial prediction of treatment outcome in depression: a machine learning approach. Lancet Psychiatry. 2016;3(3):243‐250. [DOI] [PubMed] [Google Scholar]
- 381. Chekroud AM, Gueorguieva R, Krumholz HM, Trivedi MH, Krystal JH, McCarthy G. Reevaluating the efficacy and predictability of antidepressant treatments: a symptom clustering approach. JAMA Psychiatry. 2017;74(4):370‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382. Chekroud AM, Koutsouleris N. The perilous path from publication to practice. Mol Psychiatry. 2018;23(1):24‐25. [DOI] [PubMed] [Google Scholar]
- 383. Chang B, Choi Y, Jeon M, et al. ARPNet: antidepressant response prediction network for major depressive disorder. Genes (Basel). 2019;10:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384. Zhdanov A, Atluri S, Wong W, et al. Use of machine learning for predicting escitalopram treatment outcome from electroencephalography recordings in adult patients with depression. JAMA Netw Open. 2020;3(1):e1918377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385. Wu W, Zhang Y, Jiang J, et al. An electroencephalographic signature predicts antidepressant response in major depression. Nat Biotechnol. 2020;38(4):439‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386. Ichikawa N, Lisi G, Yahata N, et al. Primary functional brain connections associated with melancholic major depressive disorder and modulation by antidepressants. Sci Rep. 2020;10(1):3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Bzdok D, Meyer‐Lindenberg A. Machine learning for precision psychiatry: opportunities and challenges. Biol Psychiatry Cogn Neurosci Neuroimaging. 2018;3(3):223‐230. [DOI] [PubMed] [Google Scholar]
- 388. Dwyer DB, Falkai P, Koutsouleris N. Machine learning approaches for clinical psychology and psychiatry. Annu Rev Clin Psychol. 2018;14(1):91‐118. [DOI] [PubMed] [Google Scholar]
- 389. Graham S, Depp C, Lee EE, et al. Artificial intelligence for mental health and mental illnesses: an overview. Curr Psychiatry Rep. 2019;21(11):116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390. Reeve A, Simcox E, Turnbull D. Ageing and Parkinson's disease: why is advancing age the biggest risk factor? Ageing Res Rev. 2014;14:19‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391. Glaab E. Computational systems biology approaches for Parkinson's disease. Cell Tissue Res. 2018;373(1):91‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392. Pinto M, Fernandes C, Martins E, et al. Boosting drug discovery for Parkinson's: enhancement of the delivery of a monoamine oxidase‐B inhibitor by brain‐targeted PEGylated polycaprolactone‐based nanoparticles. Pharmaceutics. 2019;11(7):331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393. Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296(5575):1991‐1995. [DOI] [PubMed] [Google Scholar]
- 394. Varadi C, Nehez K, Hornyak O, Viskolcz B, Bones J. Serum N‐glycosylation in Parkinson's disease: a novel approach for potential alterations. Molecules (Basel, Switzerland). 2019;24(12):2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395. Ishigami N, Tokuda T, Ikegawa M, et al. Cerebrospinal fluid proteomic patterns discriminate Parkinson's disease and multiple system atrophy. Mov Disord. 2012;27(7):851‐857. [DOI] [PubMed] [Google Scholar]
- 396. Potashkin JA, Santiago JA, Ravina BM, Watts A, Leontovich AA. Biosignatures for Parkinson's disease and atypical parkinsonian disorders patients. PLOS One. 2012;7(8):e43595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397. Abdi F, Quinn JF, Jankovic J, et al. Detection of biomarkers with a multiplex quantitative proteomic platform in cerebrospinal fluid of patients with neurodegenerative disorders. J Alzheimers Dis. 2006;9(3):293‐348. [DOI] [PubMed] [Google Scholar]
- 398. Matarazzo M, Arroyo‐Gallego T, Montero P, et al. Remote Monitoring of treatment response in Parkinson's disease: the habit of typing on a computer. Mov Disord. 2019;34(10):1488‐1495. [DOI] [PubMed] [Google Scholar]
- 399. Hssayeni MD, Burack MA, Ghoraani B. Automatic assessment of medication states of patients with Parkinson's disease using wearable sensors. Conf Proc IEEE Eng Med Biol Soc. 2016;2016:6082‐6085. [DOI] [PubMed] [Google Scholar]
- 400. Nguyen V, Kunz H, Taylor P, Acosta D. Insights into pharmacotherapy management for Parkinson's disease patients using wearables activity data. Stud Health Technol Inform. 2018;247:156‐160. [PubMed] [Google Scholar]
- 401. Hssayeni MD, Burack MA, Jimenez‐Shahed J, Ghoraani B. Assessment of response to medication in individuals with Parkinson's disease. Med Eng Phys. 2019;67:33‐43. [DOI] [PubMed] [Google Scholar]
- 402. Asakawa T, Sugiyama K, Nozaki T, et al. Can the latest computerized technologies revolutionize conventional assessment tools and therapies for a neurological disease? The example of Parkinson's disease. Neurol Med Chir (Tokyo). 2019;59(3):69‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403. Shao YM, Ma X, Paira P, et al. Discovery of indolylpiperazinylpyrimidines with dual‐target profiles at adenosine A2A and dopamine D2 receptors for Parkinson's disease treatment. PLOS One. 2018;13(1):e0188212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404. Sebastian‐Perez V, Martinez MJ, Gil C, Campillo NE, Martinez A, Ponzoni I. QSAR modelling to identify LRRK2 inhibitors for Parkinson's disease. J Integr Bioinform. 2019;16(1):20180063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405. Johnston TH, Lacoste AMB, Visanji NP, Lang AE, Fox SH, Brotchie JM. Repurposing drugs to treat l‐DOPA‐induced dyskinesia in Parkinson's disease. Neuropharmacology. 2019;147:11‐27. [DOI] [PubMed] [Google Scholar]
- 406. Monzel AS, Hemmer K, Kaoma T, et al. Machine learning‐assisted neurotoxicity prediction in human midbrain organoids. Parkinsonism Rel Disord. 2020;75:105‐109. [DOI] [PubMed] [Google Scholar]
- 407. Hughes GL, Lones MA, Bedder M, Currie PD, Smith SL, Pownall ME. Machine learning discriminates a movement disorder in a zebrafish model of Parkinson's disease. Dis Model Mech. 2020;13(10):dmm045815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408. Goedert M, Spillantini MG. A century of Alzheimer's disease. Science. 2006;314(5800):777‐781. [DOI] [PubMed] [Google Scholar]
- 409. Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M. Alzheimer's disease: clinical trials and drug development. Lancet Neurol. 2010;9(7):702‐716. [DOI] [PubMed] [Google Scholar]
- 410. Misra S, Medhi B. Drug development status for Alzheimer's disease: present scenario. Neurol Sci. 2013;34(6):831‐839. [DOI] [PubMed] [Google Scholar]
- 411. Cummings JL, Morstorf T, Zhong K. Alzheimer's disease drug‐development pipeline: few candidates, frequent failures. Alzheimer's Res Ther. 2014;6(4):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412. Khan S, Barve KH, Kumar MS. Recent advancements in pathogenesis, diagnostics and treatment of Alzheimer's disease. Curr Neuropharmacol. 2020;18:1106‐1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413. Louros N, Orlando G, de Vleeschouwer M, Rousseau F, Schymkowitz J. Structure‐based machine‐guided mapping of amyloid sequence space reveals uncharted sequence clusters with higher solubilities. Nat Commun. 2020;11(1):3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414. Sügis E, Dauvillier J, Leontjeva A, et al. HENA, heterogeneous network‐based data set for Alzheimer's disease. Sci Data. 2019;6(1):151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415. Hung TC, Lee WY, Chen KB, Chan YC, Lee CC, Chen CY. In silico investigation of traditional Chinese medicine compounds to inhibit human histone deacetylase 2 for patients with Alzheimer's disease. BioMed Res Int. 2014;2014:769867. 10.1155/2014/769867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416. Cavas L, Topcam G, Gundogdu‐Hizliates C, Ergun Y. Neural network modeling of AChE inhibition by new carbazole‐bearing oxazolones. Interdiscip Sci. 2019;11(1):95‐107. [DOI] [PubMed] [Google Scholar]
- 417. Lee J, Kumar S, Lee SY, Park SJ, Kim MH. Development of predictive models for identifying potential S100A9 inhibitors based on machine learning methods. Front Chem. 2019;7:779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418. Jamal S, Grover A, Grover S. Machine learning from molecular dynamics trajectories to predict caspase‐8 inhibitors against Alzheimer's disease. Front Pharmacol. 2019;10:780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419. Miyazaki Y, Ono N, Huang M, Altaf‐Ul‐Amin M, Kanaya S. Comprehensive exploration of target‐specific ligands using a graph convolution neural network. Mol Inf. 2020;39(1–2):e1900095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420. Kleandrova VV, Speck‐Planche A. PTML modeling for Alzheimer's disease: design and prediction of virtual multi‐target inhibitors of GSK3B, HDAC1, and HDAC6. Curr Top Med Chem. 2020;20(19):1661‐1676. [DOI] [PubMed] [Google Scholar]
- 421. Riccardo C, Michael G‐D, Maria Natália DSC. Developing a multi‐target model to predict the activity of monoamine oxidase A and B drugs. Curr Top Med Chem. 2020;20(18):1593‐1600. [DOI] [PubMed] [Google Scholar]
- 422. Gupta R, Ambasta RK, Kumar P. Identification of novel class I and class IIb histone deacetylase inhibitor for Alzheimer's disease therapeutics. Life Sci. 2020;256:117912. [DOI] [PubMed] [Google Scholar]
- 423. Fang J, Li Y, Liu R, et al. Discovery of multitarget‐directed ligands against Alzheimer's disease through systematic prediction of chemical‐protein interactions. J Chem Inf Model. 2015;55(1):149‐164. [DOI] [PubMed] [Google Scholar]
- 424. Fang J, Wang L, Li Y, et al. AlzhCPI: a knowledge base for predicting chemical‐protein interactions towards Alzheimer's disease. PLOS One. 2017;12(5):e0178347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425. Pang XC, Kang D, Fang JS, et al. Network pharmacology‐based analysis of Chinese herbal Naodesheng formula for application to Alzheimer's disease. Chin J Nat Med. 2018;16(1):53‐62. [DOI] [PubMed] [Google Scholar]
- 426. Grisoni F, Merk D, Friedrich L, Schneider G. Design of natural‐product‐inspired multitarget ligands by machine learning. ChemMedChem. 2019;14(12):1129‐1134. [DOI] [PubMed] [Google Scholar]
- 427. Thompson CA. FDA approves galantamine for Alzheimer's disease. Am J Health Syst Pharm. 2001;58(8):649. [DOI] [PubMed] [Google Scholar]
- 428. Jamal S, Goyal S, Shanker A, Grover A. Integrating network, sequence and functional features using machine learning approaches towards identification of novel Alzheimer genes. BMC Genomics. 2016;17(1):807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429. Hampel H, Williams C, Etcheto A, et al. A precision medicine framework using artificial intelligence for the identification and confirmation of genomic biomarkers of response to an Alzheimer's disease therapy: analysis of the blarcamesine (ANAVEX2‐73) Phase 2a clinical study. Alzheimers Dement (NY). 2020;6(1):e12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430. Lu H, Zhang J, Liang Y, et al. Network topology and machine learning analyses reveal microstructural white matter changes underlying Chinese medicine Dengzhan Shengmai treatment on patients with vascular cognitive impairment. Pharmacol Res. 2020;156:104773. [DOI] [PubMed] [Google Scholar]
- 431. Hemmerling TM, Taddei R, Wehbe M, Morse J, Cyr S, Zaouter C. Robotic anesthesia—a vision for the future of anesthesia. Transl Med UniSa. 2011;1:1‐20. [PMC free article] [PubMed] [Google Scholar]
- 432. Zaouter C, Joosten A, Rinehart J, Struys MMRF, Hemmerling TM. Autonomous systems in anesthesia: where do we stand in 2020? A narrative review. Anesth Analg. 2020;130(5):1120‐1132. [DOI] [PubMed] [Google Scholar]
- 433. Char DS, Burgart A. Machine‐learning implementation in clinical anesthesia: opportunities and challenges. Anesth Analg. 2020;130(6):1709‐1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434. Ramaswamy SM, Weerink MAS, Struys MMRF, Nagaraj SB. Dexmedetomidine‐induced deep sedation mimics non‐rapid eye movement stage 3 sleep: large‐scale validation using machine learning. Sleep. 2020. 10.1093/sleep/zsaa167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435. Kashkooli K, Polk SL, Hahm EY, et al. Improved tracking of sevoflurane anesthetic states with drug‐specific machine learning models. J Neural Eng. 2020;17(4):046020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436. Belur Nagaraj S, Ramaswamy SM, Weerink MAS, Struys M. Predicting deep hypnotic state from sleep brain rhythms using deep learning: a data‐repurposing approach. Anesth Analg. 2020;130(5):1211‐1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437. Kang AR, Lee J, Jung W, et al. Development of a prediction model for hypotension after induction of anesthesia using machine learning. PLOS One. 2020;15(4):e0231172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438. Ermer SC, Farney RJ, Johnson KB, Orr JA, Egan TD, Brewer LM. An automated algorithm incorporating poincare analysis can quantify the severity of opioid‐induced ataxic breathing. Anesth Analg. 2020;130(5):1147‐1156. [DOI] [PubMed] [Google Scholar]
- 439. Moghadam MC, Abad EMK, Bagherzadeh N, Ramsingh D, Li GP, Kain ZN. A machine‐learning approach to predicting hypotensive events in ICU settings. Comput Biol Med. 2020;118:103626. [DOI] [PubMed] [Google Scholar]
- 440. Wang R, Wang S, Duan N, Wang Q. From patient‐controlled analgesia to artificial intelligence‐assisted patient‐controlled analgesia: practices and perspectives. Front Med. 2020;7(145). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441. Johnson A, Yang F, Gollarahalli S, et al. Use of mobile health apps and wearable technology to assess changes and predict pain during treatment of acute pain in sickle cell disease: feasibility study. JMIR Mhealth Uhealth. 2019;7(12):e13671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442. Nair AA, Velagapudi MA, Lang JA, et al. Machine learning approach to predict postoperative opioid requirements in ambulatory surgery patients. PLOS One. 2020;15(7):e0236833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443. Walter S, Al‐Hamadi A, Gruss S, Frisch S, Traue HC, Werner P. Multimodal recognition of pain intensity and pain modality with machine learning. Schmerz. 2020;34(5):400‐409. [DOI] [PubMed] [Google Scholar]
- 444. Gudin J, Mavroudi S, Korfiati A, Theofilatos K, Dietze D, Hurwitz P. Reducing opioid prescriptions by identifying responders on topical analgesic treatment using an individualized medicine and predictive analytics approach. J Pain Res. 2020;13:1255‐1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445. Lopez‐Martinez D, Eschenfeldt P, Ostvar S, Ingram M, Hur C, Picard R. Deep reinforcement learning for optimal critical care pain management with morphine using dueling double‐deep Q networks. Annu Int Conf IEEE Eng Med Biol Soc. 2019;2019:3960‐3963. [DOI] [PubMed] [Google Scholar]
- 446. Parthipan A, Banerjee I, Humphreys K, et al. Predicting inadequate postoperative pain management in depressed patients: a machine learning approach. PLOS One. 2019;14(2):e0210575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447. Wei M, Liao Y, Liu J, et al. EEG beta‐band spectral entropy can predict the effect of drug treatment on pain in patients with herpes zoster [published online ahead of print July 14, 2020]. J Clin Neurophysiol. 2020. 10.1097/WNP.0000000000000758 [DOI] [PubMed] [Google Scholar]
- 448. Ferroni P, Zanzotto FM, Scarpato N, et al. Machine learning approach to predict medication overuse in migraine patients. Comput Struct Biotechnol J. 2020;18:1487‐1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449. Lee S, Wei S, White V, Bain P, Baker C, Li J. Classification of opioid usage through semi‐supervised learning for total joint replacement patients. IEEE J Biomed Health Inform, 2020:1. [DOI] [PubMed] [Google Scholar]
- 450. Zhang Y, Fatemi P, Medress Z, et al. A predictive‐modeling‐based screening tool for prolonged opioid use after surgical management of low back and lower extremity pain. Spine J. 2020;20(8):1184‐1195. [DOI] [PubMed] [Google Scholar]
- 451. Chidambaran V, Ashton M, Martin LJ, Jegga AG. Systems biology‐based approaches to summarize and identify novel genes and pathways associated with acute and chronic postsurgical pain. J Clin Anesth. 2020;62:109738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452. Kong W, Tu X, Huang W, Yang Y, Xie Z, Huang Z. Prediction and optimization of NaV1.7 sodium channel inhibitors based on machine learning and simulated annealing. J Chem Inf Model. 2020;60(6):2739‐2753. [DOI] [PubMed] [Google Scholar]
- 453. Feng Z, Chen M, Shen M, Liang T, Chen H, Xie XQ. Pain‐CKB, a pain‐domain‐specific chemogenomics knowledgebase for target identification and systems pharmacology research. J Chem Inf Model. 2020;60:4429‐4435. [DOI] [PubMed] [Google Scholar]
- 454.Startups using artificial intelligence in drug discovery. https://blog.benchsci.com/startups-using-artificial-intelligence-in-drug-discovery
- 455. Chan HCS, Shan H, Dahoun T, Vogel H, Yuan S. Advancing drug discovery via artificial intelligence. Trends Pharmacol Sci. 2019;40(8):592‐604. [DOI] [PubMed] [Google Scholar]
- 456. Mak K‐K, Pichika MR. Artificial intelligence in drug development: present status and future prospects. Drug Discovery Today. 2019;24(3):773‐780. [DOI] [PubMed] [Google Scholar]
- 457. Hinton GE, Krizhevsky A, Wang SD. Transforming Auto‐Encoders. Berlin, Heidelberg, Germany: Springer; 2011. [Google Scholar]
- 458. Becker G, Bolbos R, Costes N, Redoute J, Newman‐Tancredi A, Zimmer L. Selective serotonin 5‐HT1A receptor biased agonists elicit distinct brain activation patterns: a pharmacoMRI study. Sci Rep. 2016;6:26633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459. Downey D, Dutta A, McKie S, et al. Comparing the actions of lanicemine and ketamine in depression: key role of the anterior cingulate. Eur Neuropsychopharmacol. 2016;26(6):994‐1003. [DOI] [PubMed] [Google Scholar]
- 460. von Pföstl V, Li J, Zaldivar D, et al. Effects of lactate on the early visual cortex of non‐human primates, investigated by pharmaco‐MRI and neurochemical analysis. Neuroimage. 2012;61(1):98‐105. [DOI] [PubMed] [Google Scholar]
- 461. Carmichael O, Schwarz AJ, Chatham CH, et al. The role of fMRI in drug development. Drug Discovery Today. 2018;23(2):333‐348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462. Urban A, Golgher L, Brunner C, et al. Understanding the neurovascular unit at multiple scales: advantages and limitations of multi‐photon and functional ultrasound imaging. Adv Drug Deliv Rev. 2017;119:73‐100. [DOI] [PubMed] [Google Scholar]
- 463. Macé E, Montaldo G, Cohen I, Baulac M, Fink M, Tanter M. Functional ultrasound imaging of the brain. Nat Methods. 2011;8(8):662‐664. [DOI] [PubMed] [Google Scholar]
- 464. Vidal B, Droguerre M, Valdebenito M, et al. Pharmaco‐fUS for characterizing drugs for Alzheimer's disease—the case of THN201, a drug combination of donepezil plus mefloquine. Front Neurosci. 2020;14:835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465. Vidal B, Droguerre M, Venet L, et al. Functional ultrasound imaging to study brain dynamics: application of pharmaco‐fUS to atomoxetine. Neuropharmacology. 2020;179:108273. [DOI] [PubMed] [Google Scholar]
- 466. Rabut C, Ferrier J, Bertolo A, et al. Pharmaco‐fUS: quantification of pharmacologically‐induced dynamic changes in brain perfusion and connectivity by functional ultrasound imaging in awake mice. Neuroimage. 2020;222:117231. [DOI] [PubMed] [Google Scholar]
- 467. Chuang KV, Keiser MJ. Adversarial controls for scientific machine learning. ACS Chem Biol. 2018;13(10):2819‐2821. [DOI] [PubMed] [Google Scholar]
- 468. Schneider G. Mind and machine in drug design. Nat Mach Intell. 2019;1(3):128‐130. [Google Scholar]