

Abstract
Molecules that covalently bind macromolecular targets have found widespread applications as activity-based probes and as irreversibly binding drugs. However, the general reactivity of the electrophiles needed for covalent bond formation makes control of selectivity difficult. There is currently no rapid, unbiased screening method to identify new classes of covalent inhibitors from highly diverse pools of candidate molecules. Here we describe a phage display method to directly screen for ligands that bind to protein targets through covalent bond formation. This approach makes use of a reactive linker to form cyclic peptides on the phage surface while simultaneously introducing an electrophilic ‘warhead’ to covalently react with a nucleophile on the target. Using this approach, we identified cyclic peptides that irreversibly inhibit a cysteine protease and a serine hydrolase with nanomolar potency and exceptional specificity. This approach should enable rapid, unbiased screening to identify new classes of highly selective covalent inhibitors for diverse molecular targets.
Introduction
The efficacy of many drugs derives from their ability to bind a single target with high selectivity. While drug discovery efforts have historically focused on reversibly binding molecules, recent success with covalent inhibitors has highlighted their potential as therapeutic agents1, 2. Covalent drugs have the benefits of simplified pharmacokinetic properties and extended duration of therapeutic action compared to reversibly binding drugs. Covalent modifiers of enzyme active-site residues have also been used as activity-based probes (ABPs) to target and functionally characterize proteases and many other diverse families of enzymes3, 4. However, the discovery of covalent inhibitors that use reactive electrophiles to covalently bind nucleophilic residues (i.e. cysteine, serine and lysine) on proteins has been hindered by the challenge of controlling selectivity. While a small number of ABPs have been successfully designed to target a single enzyme, most react with a subset of related enzymes3, 4. This is largely due to the conserved active site regions in many enzyme family members and the use of small-molecule or peptide-like ABPs with limited surface area contacts between probe and target. This liability has been addressed by linking weakly reactive electrophiles to high-affinity, selective binding elements that limit off-target interactions1, 5. However, methods to identify sufficiently selective binding elements to pair with a reactive electrophile are often time consuming and unsuccessful. Thus, there is a need for a general approach to directly screen large numbers of ligands for their ability to bind proteins covalently and selectively.
Phage display is well suited to this application as it allows rapid generation of billions of peptides of exceptionally high diversity on the surface of phage particles, which can then be iteratively screened against molecular targets6, 7. However, conventional phage methods are limited in that only simple linear peptides made up of natural amino acid sequences can be screened, and the resulting hits are reversibly binding ligands. The approach described here overcomes these limitations. Inspired by previous work using chemical linkers to produce cyclic and bicyclic peptides on the phage surface8–11, we developed an approach that can be used both to form cyclic peptides and also to introduce a weak electrophile ‘warhead’ directly on the phage coat protein segment (Fig. 1). Cyclization helps to both rigidify the peptide scaffold for increased binding potency and selectivity and also to stabilize the resulting probes. These types of cyclized peptides also tend to adopt secondary structures that more closely mimic a folded protein and therefore can bind with a greater surface area of contacts compared to a linear peptide12, increasing the chances of identifying highly selective ligands.
Fig. 1. The phage display approach to screen for selective covalent inhibitors.
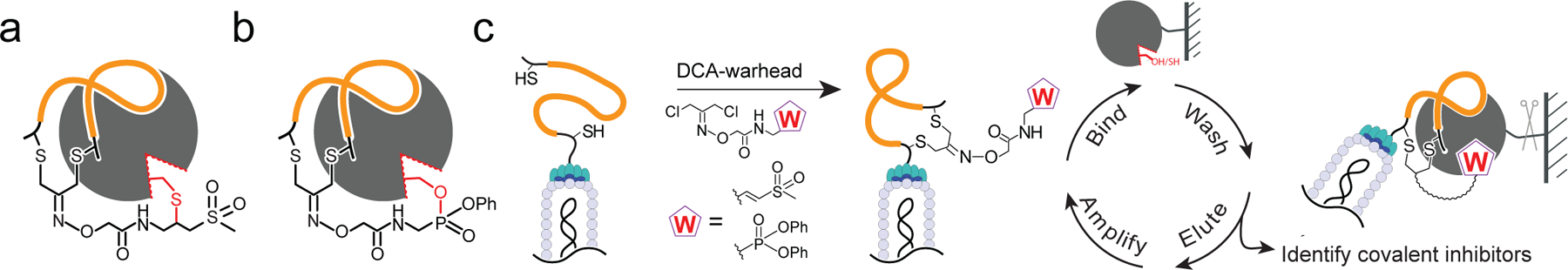
Schematic of the general strategy to covalently inhibit target cysteine and serine hydrolases by peptides (orange) cyclized by linkers containing a reactive a) vinyl sulfone or b) diphenyl phosphate ‘warhead’ that are presented to the reactive nucleophile (red) of targets. c) General workflow of the phage screening method to discover selective covalent inhibitors. The DCA-warhead linkers are used to chemically modify diverse peptides containing two cysteines to generate a cyclic peptide-warhead library on the phage surface. The resulting library can be screened through an iterative process in which phage that covalently bind the target are collected by affinity purification followed by washes to remove non-covalently bound phage and then elution by proteolytic digestion. The resulting released phage can then be amplified and subjected to the next round of screening or identified to unveil the sequence of binders.
Here we demonstrate the utility and general applicability of the screening approach using two mechanistically distinct enzyme targets. We chose the Tobacco Etch Virus (TEV) protease since it has well-defined primary sequence selectivity and no reported potent inhibitors13 and a recently identified serine hydrolase, fluorophosphonate-binding hydrolases F (FphF), from Staphylococcus aureus, which has poorly defined substrate specificity but is likely to be an important virulence factor14. Our screening efforts using a cyclization linker containing a cysteine reactive vinyl sulfone warhead for TEV (Fig. 1a) and a serine reactive diphenylphosphonate (DPP) warhead for FphF (Fig. 1b) identified a series of peptide sequences that irreversibly inhibited the targets. Further optimization of these sequences resulted in potent, irreversible inhibitors and a fluorescent probe that shows an exceptionally high degree of specificity over other highly related proteases. Therefore, phage screening has the potential to be a general method for rapid identification of highly selective and stable covalent binding molecules for diverse molecular targets.
Results
Design of the cyclization linker
Inspired by the use of chemical cyclization-linkers to generate phage-displayed monocyclic/bicyclic peptides8, 10, we set out to develop a linker that could be used to both drive the formation of constrained cyclic peptides while simultaneously introducing a reactive electrophile directly on the phage coat protein. The 1,3-dichloroacetone (DCA) linker is effective for cyclizing peptides in solution through reaction with two cysteines, as well as for directly modifying phage displayed peptides without causing toxicity11, 15. In addition, the ketone group of DCA allows convenient derivatization with alkoxy-amines/hydrazines to generate stable oxime/hydrazone linkages to introduce various functional groups16. For targeting the cysteine protease, TEV, we chose to use a vinyl sulfone (VS) electrophile due to its relatively weak reactivity and successful use for covalent inhibition of diverse cysteine protease targets17. To avoid any contribution of direct binding to the target protease by the linker itself, we used a glycine vinyl sulfone modified with an aminooxy group for attachment to the DCA linker. We also synthesized a similar analog containing a glycine diphenyl phosphonate (DPP)18 for use with serine hydrolase targets. We selected the DPP electrophile because it has been used in covalent inhibitors of serine hydrolase targets18. Both linkers were synthesized in one step by addition of the aminooxy linked glycine electrophile to the DCA linker (11 & 15, Supplementary Scheme 1 & 2).
The general screening workflow using the DCA linkers involves treating a library of phage expressing diverse peptides containing two cysteines on their pIII protein and then screening the resulting chemically modified phage for covalent binding to a labeled target (Fig. 1c). To confirm that the DCA-VS and DCA-DPP linkers form the desired cyclic product on phage, we tested their ability to cyclize a di-cysteine peptide C-X8-C (NACGSGSGSGCGC, A represents alanine, C represents cysteine, G represents glycine, and S represents serine), fused to the N1 and N2 ectodomains of the cysteine-free phage pIII protein20. After TCEP reduction, the peptide was incubated with either DCA-VS or DCA-DPP and the cyclization efficacies were monitored by mass spectrometry21. The results confirmed quantitative and specific cyclization by both linkers in a phage compatible buffer (Fig. 2a).
Fig. 2. Confirmation of selectivity and reactivity of the cyclization reactions and establishment of washing conditions for removal of non-covalently bound phage.
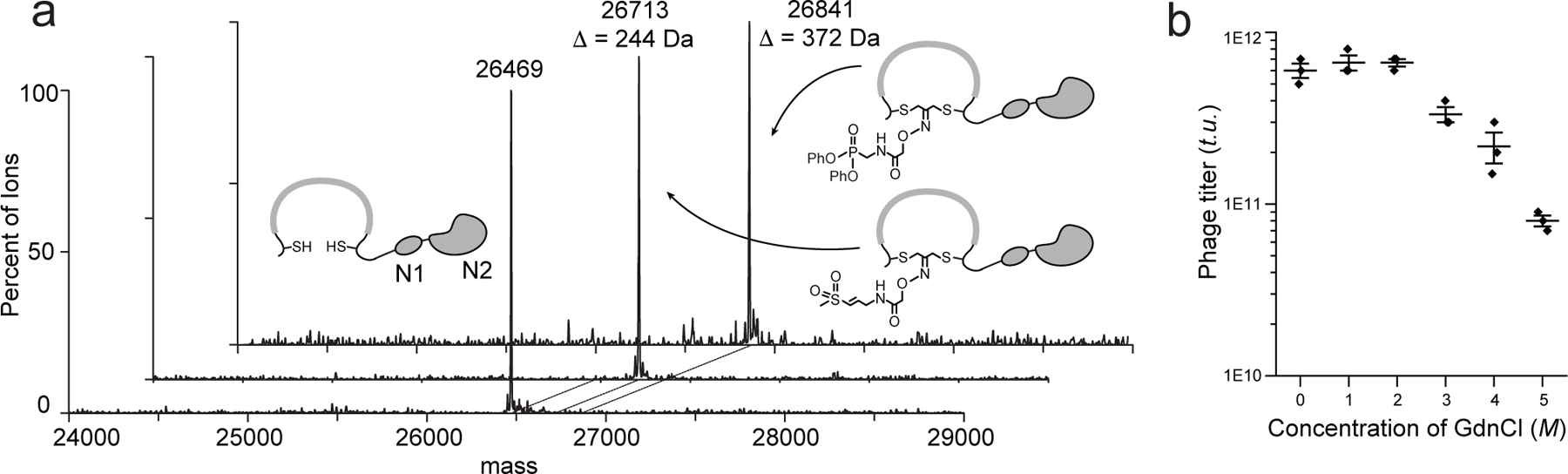
a) A test peptide containing two cysteine residues (NACGSGSGSGCGC) fused to the disulfide-free phage N1N2 domains is quantitatively modified to generate the desired cyclic peptide conjugates. The mass shift corresponds to the adduct of a single DCA-VS or DCA-DPP linker. b) Plot of phage titers for phage expressing a disulfide-free pIII protein treated with increasing concentrations of guanidine chloride (GdnCl) to determine the highest concentration that could be used in washing steps without causing toxicity to the phage. Assays were performed in triplicate, n= 3 biologically independent wells (mean + s.d. is depicted).
Design of target protease constructs and optimization of washing conditions
The covalent nature of binding of the phage to the target provides an opportunity to remove reversibly bound as well as non-specifically bound phage using stringent washing conditions. For releasing covalently bound phage, we engineered an orthogonal protease cut site either on the target protease or on the pIII coat protein of the phage. For the TEV protease, we engineered an expression construct containing an AviTag biotinylation tag attached to its N-terminus through a linker recognized by the Human Rhinovirus (HRV) 3C Protease (EVLFQGP)22. To improve the solubility and folding of the TEV protease, we also fused E. coli maltose-binding protein (MBP) through a TEV cleavable sequence (ENLYFQG) N-terminal to the AviTag23 (Supplementary Figure 1). For the FphF protein, we chose to use the native full-length protein expressed only with a N-terminal HisTag that could be removed after purification through a 3C protease cleavable site. Since this protein does not contain the AviTag, we performed chemical biotinylation of free lysine residues. Since this does not allow cleavage of the biotin tag, we constructed a phage library in which a TEV cleavage site was inserted between the peptide library and the pIII protein, to facilitate release of the covalently bound phage (see methods section). This approach is optimal for use with targets that do not tolerate a C or N termini tag, are hard to express, or that can be commercially purchased or purified from native sources (i.e. human targets isolated from cells and tissues). To identify the most optimal wash conditions to remove non-covalently bound phage without causing toxicity, we tested a range of concentrations of guanidine. We found that the infectivity of phage remains high even after treatment with 5 M guanidine chloride (Fig. 2b) and therefore used these washing conditions during each stage of the phage screening process.
Identification of covalent binding cyclic peptide probes that target TEV protease
For the phage panning experiments with TEV protease, we used a library of phage displaying peptides of eight random amino acids flanked by two cysteines (C-X8-C, C represents cysteine, X represents any canonical amino acid; Fig. 3a)24. The randomized DNA sequences encoding the peptide library were inserted into the phage DNA between the pelB signal peptide and the disulfide-free pIII protein of the fdg3p0ss phage vector20. The peptides present on the phage surface were reduced with TCEP and then modified with the DCA-VS linker to achieve the cyclic peptide vinyl sulfone (cpVS) phage library. To promote enrichment of stronger and faster binders upon increasing rounds of selection, we allowed phage and biotinylated TEV bound to streptavidin magnetic beads to incubate for 4 hours in the first round of selection, one hour in the second round, and a half hour in the third round. Measurement of the overall phage titer levels indicated a slight reduction in phage numbers compared to conventional phage screening, likely due to the stringent 5 M guanidine washes. However, we were able to achieve an approximately 55-fold enrichment in phage numbers through three rounds of panning, suggesting that the selection was successful. To release the selected phage, we incubated the resin with the 3C protease. Phage collected from the third round of selection were used to infect TG1 E. coli, that were then grown on plates to produce individual colonies for sequencing recovered phage plasmid DNA.
Fig. 3. Identification and optimization of covalent cyclic peptide inhibitors of TEV protease and FphF hydrolase.
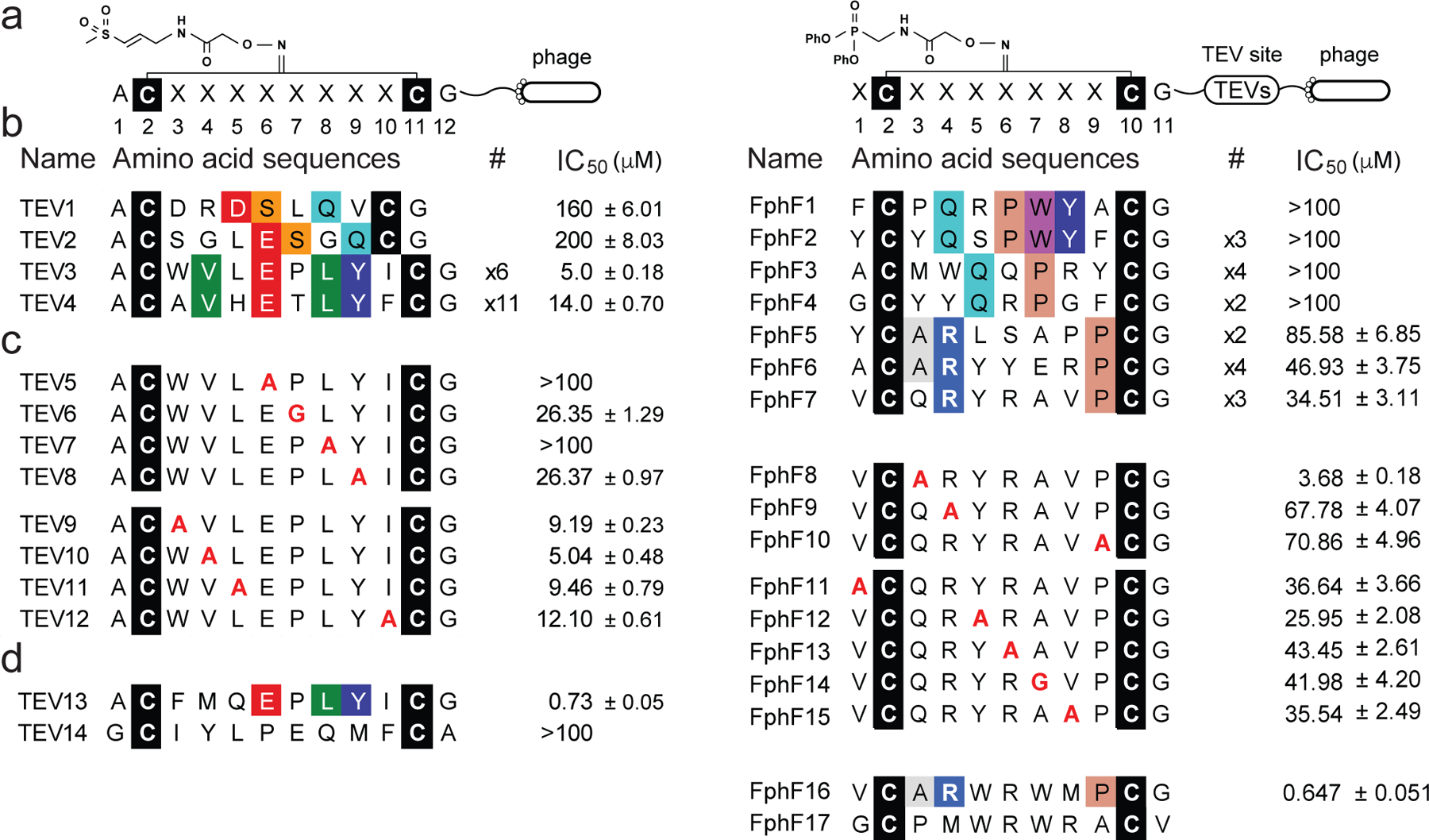
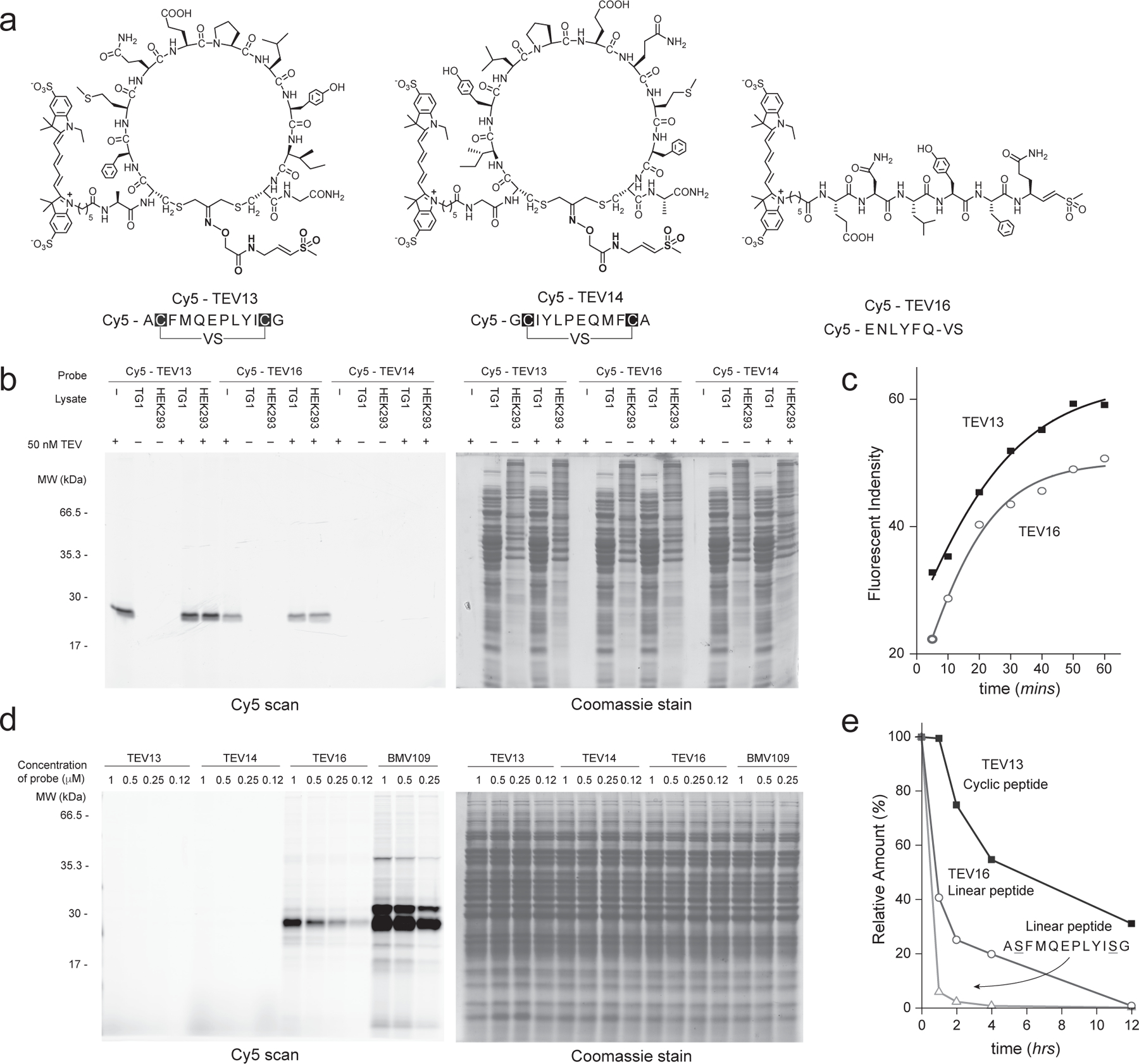
Schematic of the cyclic peptide phage library, generated by cyclizing linear peptides containing two cysteine residues separated by diversity positions (C-X8-C or X-C-X7-C, where C is cysteine, and X represents any canonical amino acid) using the cysteine reactive a) DCA-VS or e) DCA-DPP linker. The primary sequences of b) cpVS or f) cpDPP inhibitors selected after three rounds of panning against the TEV protease. The identified sequences are grouped according to similarity in sequences. Conserved amino acids are colored using the Shapely coloring rules. For peptides found more than once, the frequency in a pool of the sequenced clones is indicated (#). Each sequence was chemically synthesized, converted into a cyclic peptide containing the DCA-warhead linker and tested for potency against the recombinant TEV protease or FphF hydrolase using a fluorogenic substrate assay. The numbering for the amino acid positions is shown (top). c & g) Sequences of peptides used for structure activity relationship (SAR) studies to identify key binding residues. The top panel shows mutants in the conserved positions. The lower panel shows mutations of the position with non-conserved residues. The IC50 values for each peptide sequences when converted to the corresponding cpVS or cpDPP inhibitor are shown on the right. Sequence and IC50 values of the optimized d) cpVS TEV13 or h) cpDPP FphF16 constructed based on the results from the mutational analysis. The residues mutated are shown in red. The resulting average half maximal inhibitory concentrations (IC50) and standard deviations are shown, n= 3 biologically independent wells.
The sequencing results from 19 clones identified consensus peptide sequences that can be separated into two groups (Fig. 3b). The first group contained two peptides with a D/ESXQ sequence in the variable 7-residue stretch between the two cysteines (TEV1 and TEV2). The second group contained two peptides with a VXEXLY sequence in an 8-residue stretch between the two cysteines (TEV3 and TEV4). This sequence was isolated 17 times from the original 19 colonies. Both sets of sequences contained conserved residues found in the established TEV protease substrate binding sequence EXLYXQ. To validate the identified sequences, we synthesized all 4 consensus sequences as linear peptides and then produced the resulting cyclic peptides by reacting with the DCA-VS linker (Supplemental Figure 2). To test the inhibitory potency of the cyclized peptides for TEV, we used a quenched fluorogenic peptide (Cy5-ENLYFQGK(QSY21)-NH2) as a reporter for protease activity (Fig. 3b; Supplemental Figure 3). Inhibitors containing the D/ESXQ sequence (TEV1 & TEV2) were an order of magnitude less potent that the inhibitors containing the VXEXLY sequence (TEV3 & TEV4). The most potent inhibitor, TEV3, had an IC50 inhibition value of 5 μM, making it an ideal starting point for further optimization to achieve a high affinity TEV inhibitor. To determine which residues of the TEV3 sequence were most important for target binding, we replaced each residue at position 3–6 and 8–10 with alanine and used glycine to replace the proline at position 7 (Fig. 3c; Supplemental Figure 2). We found that the conserved glutamic acid at position 6 and leucine at position 8 were essential for binding (TEV5 & TEV7). Mutation of the proline at position 7 and tyrosine at position 9 resulted in a dramatic drop in activity (TEV6 & TEV8). Mutation of any of the other positions only slightly decreased inhibitor potency. We then performed optimization by keeping the essential residues (position 6–9), while substituting the remaining residues in the peptides with natural amino acids of different sizes, hydrophobicity and electrostatic properties (Supplementary Figure 4). For positions 3–5, phenylalanine at position 3, methionine at position 4 and glutamine at position 5 showed the most potent inhibition in each group. Modifications at position 10 did not improve potency so we fixed this position as the original isoleucine. Incorporation of the three preferred amino acids in positions 3–5 into a single peptide sequence resulted in TEV13, which was produced by efficient cyclization with the DCA-VS linker (Supplementary Figure 5). This inhibitor was the most potent TEV inhibitor synthesized, with nanomolar activity against the protease (IC50 of 730 nM with 1-hour preincubation; Fig. 3d) and a kinact/KI value of 6,500 ± 130 M−1s−1 (Supplementary Figure 6a).
Identification of covalent inhibitors with nanomolar potency against FphF
For screening of the serine hydrolase target, FphF, we performed similar steps as described for TEV protease except that we used a phage library containing a TEV-cleavable linker for release of the covalently bound phage. The library contained a random peptide sequence (X-C-X7-C, C represents cysteine, X represents any canonical amino acid; Fig. 3e), a short flexible gly/ser linker (GGSG) and the TEV protease recognition sequence (ENLYFQSG) inserted between the disulfide-free pIII protein and the pelB signal peptide of the fdg3p0ss phage vector20. The insertion of the TEV cleavage site sequence did not reduce the production yield or infectivity of the phage library.
We performed three rounds of panning against FphF using the same conditions applied to the TEV screens. Sequencing of 19 clones recovered from the third round of selections identified peptides with consensus sequences that can be separated into two groups (Fig. 3f). The first group contained four peptides with the sequence QXP at position 3–5 or 4–6 (FphF1 – FphF4). The second group contained three peptides with arginine in position 4 and proline in position 9 (FphF5 – FphF7). We synthesized all 7 peptides and cyclized with the DCA-DPP linker (Supplementary Figure 7). To test the inhibitory potency of these peptides, we used the recombinant FphF and a fluorogenic substrate, 4-methylumbelliferyl heptanoate (4-MU heptanoate). The first group of consensus peptides did not show any inhibition of the target at the highest concentration of 100 μM tested. However, the second group of peptides (FphF 5–7) showed promising initial activity in the micromolar range, with the most potent peptide, FphF7, inhibiting the enzyme with an IC50 inhibition value of around 35 μM. This sequence was therefore used as a starting point for further optimization.
To determine the most important residues of FphF7 for target binding, we replaced each of the variable residues with alanine (Fig. 3g) and found that alanine at position 3 (FphF8) greatly improved potency while replacement of arginine at position 4 and proline at position 9 resulted in a two-fold reduction of potency (FphF9 – FphF10). This relatively modest impact of alanine mutation at the two consensus residues may be due to the fact that the FphF lipid hydrolase is not expected to bind linear peptides substrates and thus the inhibitor relies on low affinity interactions between the target and multiple residues of the cyclic peptide. Mutations of other positions did not dramatically change inhibitory potency. We therefore performed optimization of the non-essential positions (position 5–8) using natural amino acids of different sizes, hydrophobicities and electrostatic properties, while keeping position 3 as the optimal alanine, 4 as arginine and 9 as a proline. The results indicated that tryptophan at positions 5 & 7, arginine at position 6 and methionine at position 8 showed the most potent inhibition in each group (Supplementary Figure 8). Incorporation of all four preferred amino acids into a single peptide sequence resulted in an inhibitor, FphF16, with nanomolar activity against FphF (IC50 of 879 nM with 1-hour pre-incubation, 95 % CI, 785 – 992 nM; Fig. 3h) and a kinact/KI value of 27,000 ± 840 M−1s−1 (Supplementary Figure 6b).
Evaluation of cyclic peptide probe and inhibitor specificity
To demonstrate the selectivity of the optimized inhibitors from the phage screening, we performed inhibition studies using the target proteases and closely related potential off-targets. We also synthesized a number of control molecules to further confirm the validity of the panning and optimization process. For the TEV target, we synthesized an isomer of TEV13 in which the order of the amino acid residues between the two cysteines was reversed to rule out inhibition by generic reactivity of the VS electrophile (TEV14; Fig. 4a). We also synthesized a second control containing the optimal TEV13 sequence cyclized with a linker that lacks the reactive VS electrophile (TEV15; Fig. 4a). As a final control we synthesized a linear hepta-peptide vinyl sulfone containing the optimal TEV protease substrate sequence ENLYFQ (TEV16; Fig. 4a). This linear peptide vinyl sulfone probe was synthesized directly on solid support by linking the P1 amino acid vinyl sulfone through the glutamine side chain to the resin as described previously25. Testing of the cyclic peptide controls for inhibition of recombinant TEV confirmed that the reversed sequence (TEV14) had no measurable activity even at concentrations as high as 200 μM and the cyclic peptide lacking the VS warhead (TEV15) showed only weak binding with less than 50% inhibition at concentrations over 200 μM (Fig. 4b). The potency of the optimized inhibitor TEV13 increased with increasing incubation time consistent with a covalent irreversible inhibition mechanism (Fig. 4c).
Fig. 4. TEV13 and FphF16 are potent and specific covalent inhibitors.
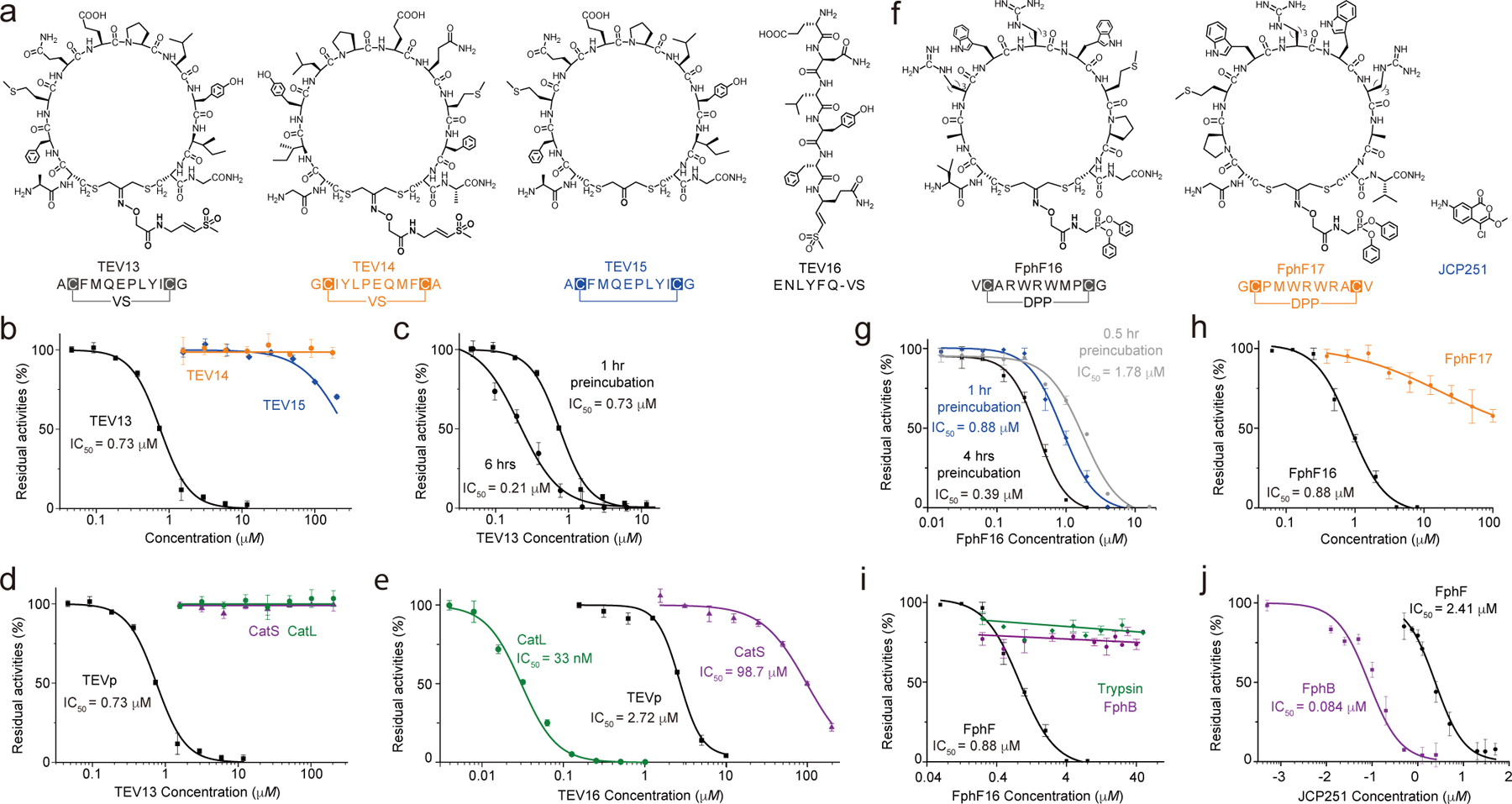
a) Structures of the optimized cpVS inhibitor TEV13 compared to the scrambled isomer, TEV14, the cyclic peptide lacking the VS electrophile in the linker, TEV15, and the linear peptide VS synthesized based on the conserved substrate specificity of TEV. b) Dose response inhibition studies of the cyclized peptides using recombinant TEV. Plots show residual enzyme activity over a range of inhibitor doses as measured by hydrolysis of a fluorogenic substrate. Enzyme was pre-treated with inhibitor at the indicated concentrations for 1 hour followed by addition of substrate and measurement of activity. IC50 values, when measurable are indicated. c) Time dependence of inhibition of TEV protease by TEV13. Recombinant enzyme was preincubated with a range of TEV13 concentrations for 1 hr or 6 hrs as indicated followed by measurement of residual activity using a fluorogenic substrate. Comparisons of the activity of (d) TEV13 and (e) TEV16 for TEV and the off-target cysteine proteases Cat S and Cat L. Inhibition studies were performed as in (b). (f) Structure of the optimized cpDPP inhibitor FphF16 compared to the sequence scrambled cpDPP, FphF17, and JCP251, a reported chloroisocoumarin inhibitor of the Fph hydrolases. g) Time dependence of inhibition of FphF hydrolase by FphF16. FphF16 was pre-incubated with the recombinant FphF hydrolase for 0.5 hr, 1 hr and 4 hrs, followed by the addition of a fluorogenic substrate, 4-methylumbelliferyl heptanoate. The residual enzyme activities represented by the fluorescent singles were plotted. h) Dose response of FphF16 and FphF17 to inhibit recombinant FphF hydrolase. IC50 values were determined as above. Comparisons of the activity of (i) FphF16 and (j) FphF17 for FphF hydrolase and the off-target FphB hydrolase and trypsin. Inhibition studies were performed as in (g). n= 3 biologically independent wells (mean + s.d. is depicted).
To determine the selectivity of TEV13 compared to the linear peptide TEV16, we tested both against other cysteine proteases. We chose sentrin-specific protease 1 (SENP1) because it has been successfully targeted with a linear peptide vinyl sulfone26 or the full length folded SUMO protein27. Neither the linear TEV16 nor the cyclic TEV13 was able to inhibit this protease at concentrations as high as 200 μM (Supplementary Figure 9). We then tested the inhibitors against two cysteine cathepsins (Cat S & Cat L) because both are relatively abundant and active proteases with broad substrate specificity profiles28 that have been effectively targeted with peptide vinyl sulfones29. We found that cyclic peptide TEV13 failed to inhibit cathepsin S or L even when preincubated for 1 hour 37 °C at concentrations over 200 μM (Fig. 4d). In contrast, the linear peptide VS TEV16 showed highly potent inhibition of cathepsin L with an IC50 value in the low nanomolar range and substantial inhibition of Cat S when used at high micromolar concentrations (Fig. 4e).
We then performed similar studies for the optimized FphF inhibitor, FphF16, to confirm selectivity. For controls, we used the reverse sequence cyclic peptide DPP (FphF17, Fig. 4f) and a small molecule chloroisocoumarin inhibitor (JCP251, Fig. 4f) that inhibits a highly related serine hydrolase, FphB, in the same family as FphF14. Inhibition of FphF by the optimized inhibitor Fph16 was time-dependent, consistent with a covalent mode of inhibition (Fig. 4g). The reverse sequence inhibitor, FphF17, showed no measurable inhibitory activity against FphF (Fig. 4h), confirming that the reactive DPP electrophile is not sufficient to promote inhibition of the target enzyme. To further confirm selectivity of the lead molecule, we tested it against several other serine hydrolase targets, including trypsin and FphB. Trypsin is an ideal off-target because is a highly active and promiscuous protease that can be inhibited by the DPP electrophile18. FphB is a stringent control for selectivity because the enzyme shares 52% sequence similarity to FphF and both enzymes have a highly conserved catalytic α/β hydrolase domain. The FphF16 inhibitor retained absolute specificity for FphF over both trypsin and FphB even when used at concentrations as high as 200 μM (Fig. 4i), while the FphB-selective inhibitor JCP251 showed potent activity for FphB but also low micromolar potency for FphF (Fig. 4j).
Generation of a selective ABP from phage selected inhibitors
One of the most effective ways to assess the selectivity of a covalent binding molecule is to make a tagged version for labeling complex mixtures containing off-target proteins and other reactive nucleophiles. We therefore conjugated a Cy5 fluorophore to the free N-terminus of TEV13, TEV14 and TEV16 to generate the corresponding fluorescent probes (Fig. 5a). We tested probe labeling of purified TEV protease in buffer as well as when added to total protein extracts from HEK293 and TG1 cells. The Cy5-TEV13 probe specifically labeled TEV protease both alone and when mixed with complex protein mixture while the corresponding reverse sequence probe, TEV14, showed no labeling, consistent with its lack of activity for TEV (Fig. 5b). The linear peptide VS probe Cy5-TEV16 effectively labeled TEV both in buffer and in total protein extracts. However, the intensity of TEV labeling was reduced compared to Cy5-TEV13 and the probe also showed an increased amount of background labeling in the lysates (Fig. 5c). Covalent binding of Cy5-TEV13 and Cy5-TEV16 was dependent on both concentration and incubation time (Supplementary Figure 10). Furthermore, the TEV labeling by Cy5-TEV13 could be blocked by preincubation with the TEV13 inhibitor lacking the Cy5 label (Supplementary Figure 11). To further assess selectivity of the probes, we performed labeling of RAW cell extracts with various concentrations of the probes at two pH values. We chose RAW cells because they have high levels of cysteine cathepsin expression30. The labeling results confirmed that the Cy5-TEV13 probe retained selectivity at both pH values with virtually no off-target background labeling even at the highest probe concentrations used. In contrast, the linear peptide Cy5-TEV16 labeled multiple cathepsins even at the lowest probe concentration and showed signs of labeling other off-target proteins indicated by increased background staining (Fig. 5d & Supplementary Figure 12). The TEV16 linear probe was as effective at labeling cathepsin L as the optimized cathepsin probe BMV10931.
Fig. 5. The Fluorescent cpABP Cy5-TEV13 specifically labels TEV protease in complex proteomic samples.
a) Chemical structures of the Cy5 fluorophore labeled ABP versions of the cyclic peptides TEV13, TEV14 and the linear peptide-VS TEV16. b) Samples of enzyme buffer (−) or lysates from TG1 or HEK293 cells (4 μg total protein) were spiked with purified recombinant TEV protease (15 ng) or buffer as indicated, followed by addition of the indicated Cy5-labeled probes. Samples were incubated for 1 hr at 37 °C and then SDS PAGE sample buffer was added followed by boiling. Samples were separated on SDS-PAGE gels followed by scanning for Cy5 fluorescence using a flatbed laser scanner (Left) and then stained with Coomassie brilliant blue (Right) to visualize total protein loading. c) Curves plotting the intensity of labeling of recombinant TEV protease (15 ng) in buffer over the range of indicated times. Samples were analyzed by SDS-PAGE analysis followed by scanning of the gel using a flatbed laser scanner to quantify the intensity of labeled protease at each time point. d) Images of SDS-PAGE gels containing samples of the indicated total cellular lysates labeled for 1 hr with the indicated concentrations of each Cy5-labeled probe. The probe BMV109 is a general ABP for cysteine cathepsins that was included for reference. The gels were processed as in (b). e) Plots of relative levels of each of the indicated cyclic and linear peptides after the indicated time of incubation in mouse plasma. Values were obtained by quantification of parent ions by mass spectrometry.
In addition to providing potentially improved target specificity by inducing protein-like folds, the cyclization of peptides also improves overall metabolic stability9. We therefore tested the serum stability of the optimal cyclic probe TEV13 and compared it to the linear peptide TEV16 as well as a linear version of the peptide portion of TEV13 in which the two cysteine residues are replaced with serine to prevent disulfide bond formation in the absence of the cyclization linker (Fig. 5e & Supplementary Figure 13). This data confirmed that both linear peptides were degraded within the first hour, while the cyclic peptide remained fully stable for 1 hour and then only partially metabolized over the next 12 hours.
Interactions between cyclic peptide and TEV protease
To better understand how the optimized cyclic peptide is able to induce selective binding to the TEV target, we sought to analyze this protein-ligand interaction using structural information. We applied molecular dynamics (MD) to simulate the interactions between TEV13 and TEV protease using the high-resolution structure of TEV protease bound to a 6-residue substrate peptide as a template (ENLYF; PDB ID: 1LVM, chain C). While the power of MD calculations for de novo prediction of ligand-protein interactions can be limited, there are a number of reasons why MD simulations are informative for predicting the interactions between TEV protease and TEV13. Firstly, the covalent linkage between TEV13 and TEV protease largely defines the orientation in which the peptide ligand can bind to the target protein, thus greatly narrowing the chemical space to explore and improving the probability of discovering relevant interactions. Secondly, the critical residues in the cyclic peptide portion of TEV13 are similar to the native TEV substrate sequence of EXLYXQ-S/G13. This allows us to generate a more reliable initial conformation of TEV13. To perform the MD simulations, we covalently linked the vinyl sulfone of TEV13 to the active site cysteine of TEV protease to generate input files that were loaded into the Assisted Model Building with Energy Refinement (AMBER) biomolecular simulation package32. The complex structure was subjected to MD simulation in explicit TIP3P solvent models with ff14SB and gaff parameter sets33, 34. After 40 ns of production simulation, the binding of TEV13 to TEV protease reached a constant and stable status (Supplementary video). The energy of the system remained smooth and the structure converged to a single low energy state for the complex (Supplementary Figure 14). This predicted structure shows TEV13 stably bound to the TEV protease in the primary substrate binding pocket (Fig. 6a). The side chains of residues Glu6, Leu8 and Tyr9 adopted a conformation similar to the native substrate peptide (Fig. 6b). In particular, Tyr9 in TEV13 matches the positioning of P3 tyrosine in the TEV substrate peptide, forming a backbone H-bond with Ser170 and interacting through a hydroxyl with residues of Asn174, T146 and H167 that form the S3 pocket of the TEV protease active site (Fig. 6c). Residue Leu8, which mimics the P4 leucine in the TEV substrate, interacts with Phe172, Val216, F225 and Ala169. Residue Glu6 inherited the role of P6 glutamate by forming several H-bonds with Tyr178, Asn176, Asn171 and His214 located at the lower end of the substrate recognition region. The MD results also indicated that Ile at position 10 contributed to the hydrophobic pocket formed by Val216 of TEV protease and Leu8 of TEV13. This could explain the preference of hydrophobic residues at the 10 position of the cyclic peptides. The flexible loop of the cyclic peptide in TEV13 composing residue 3–5 seems to interact weakly with the N-terminal β-sheet 214–217 of TEV protease, which could explain the relatively narrow SAR profile that we observed for residue 3–5. However, the optimized MD structure supports the contribution of cyclization to increase entropy to enhance binding of TEV13 in the active site. Overall, the MD simulations confirm that stable and low energy confirmations can be found in which the cyclic peptide of the TEV13 inhibitor positions the reactive vinyl sulfone for covalent modification by the active site cysteine residue.
Fig. 6. Molecular dynamic simulations to predict the interactions between TEV13 and TEV protease.
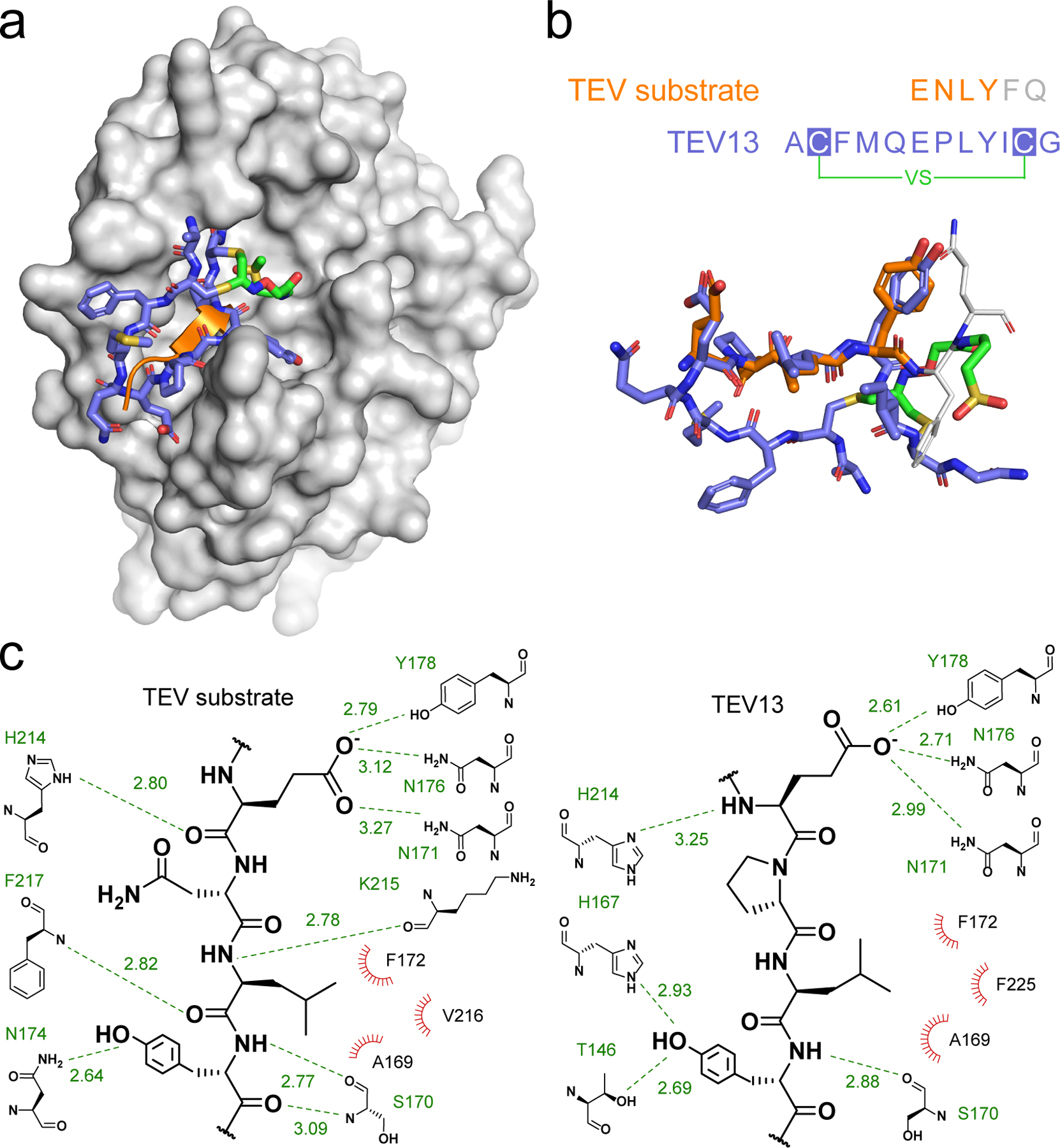
a) The MD minimized complex structure of TEV13 (colored in blue) bound to TEV protease (colored in gray). The native substrate of TEV protease is shown as an orange ribbon as found in the reported crystal structure (PDB ID: 1LVM). b) Structure of the MD optimized TEV-bound TEV13 layered onto the reported X-ray crystal structure of the native peptide substrate ENLYFQ. The key residues P3–6 that overlap with residues in TEV13 are shown in orange. c) Extended diagrammatic representation of interactions between key residues on the native substrate and TEV13 and the active site region of the TEV protease. H-bond interacting partners are shown with green dash lines with distance indicated in Å. Hydrophobic interactions are depicted as red radiations.
Discussion
Currently, the most common way to develop new covalent inhibitors involves starting with a potent and selective inhibitor of a target and identifying optimal locations for placement of a reactive functional group and reporter tag. This often requires prior access to a potent lead molecule and a significant amount of medicinal chemistry efforts. Even when such efforts are successful, in many cases the resulting compounds still lack absolute specificity for the target. In order to obtain selectivity over closely related enzymes, it is necessary to test a large diversity of complex ligand scaffolds. Here we demonstrate a phage display method that allows screening of libraries of potential covalent inhibitors that number in the billions, resulting in identification of sequences with high affinity and exceptional selectivity. We reasoned that cyclic peptides, with their rigid, protein-like structures, would have increased potential to direct selectivity of an ABP. Our results for the TEV protease and FphF support this hypothesis.
We chose TEV for our initial validation of the phage screening approach because it has a well-defined substrate specificity13 and there are currently no reported potent and selective inhibitors of this protease. We found that a vinyl sulfone inhibitor containing an optimal linear substrate recognition sequence (TEV16) could in fact inhibit TEV. However, this peptide inhibited the lysosomal cysteine cathepsin L substantially better than the intended target, highlighting the difficulty in generating highly selective covalent inhibitors using short linear peptides. On the other hand, our phage screening efforts identified cyclic peptide sequences that contain some residues that are part of the established substrate binding region of TEV, but lacked the key P1 glutamine residue that is essential for native substrate recongnition35. The resulting cyclic peptide VS compounds only inhibited TEV even though the reactive electrophile can effectively inhibit the other cysteine proteases. These data suggest we identified conformations that can present key anchor residues to the protease in the absence of key binding interactions of the native substrate and that these extended protein-like binding interactions drive highly selective target binding. Molecular dynamic simulation helped to clarify the binding mode and will direct future efforts to find optimal linkers for use with other enzyme targets.
We chose FphF as a second target for several reasons. First, this enzyme is a distinct enzyme class from TEV and uses a highly conserved α/β hydrolase domain for hydrolysis with an active site serine residue. Therefore, we could test the approach using a different enzyme/electrophile pair. Second, we have recently identified a family of Fph hydrolases in S. aureus that function as lipid esterases and at least one of the family members has a role in host colonization14. We have performed screening studies using the triazole urea scaffold and found it difficult to identify highly selective inhibitors due to the highly conserved nature of this family36. Finally, there is currently no evidence that the DPP electrophile can function as a covalent inhibitor of lipid esterases, thus allowing us to assess the potential of the cyclic peptide to induce covalent bond formation for even sub-optimal electrophiles. The result of these efforts is a compound that shows irreversible inhibition of the target hydrolase a high degree of specificity over the closely related FphB protein. We think this will be particularly valuable since Fph enzymes are expressed in biofilms of S. aureus and are therefore potentially valuable targets for imaging agents in vivo. For such imaging agents to be clinically valuable, they must have a high degree of target selectivity.
In summary, we believe that our phage screening approach could be used to find covalent binding ligands for virtually any biomolecule target (protein, glycan, nucleotide, lipid, etc.) that contains a slightly reactive nucleophile and the potential to interact with a macromolecular ligand. Our results suggest that the phage approach works well for proteases but It is likely that the same general linker could be used for screening against other targets that contain a reactive cysteine or serine residue. For example, there have been several recent reports of inhibitors that target cysteine residues in protein kinases2 and proteomic profiling studies have begun to catalog all potential reactive cysteine residues in a cell37. This work has been complemented by other global studies of lysine reactivity which potentially identifies targets that would be amenable to screening for covalent ligands using this approach38. Thus, the covalent phage screening approach could greatly enhance the application of selective covalent ligands in cell biology, imaging and drug development.
Methods
General synthesis methods
Chromatographic separations were performed by manual flash chromatography unless otherwise specified. Silica gel 60 (Merck 70–230 mesh) was used for manual column chromatography. Merck F-254 thickness (0.25 mm) commercial plates were used for analytical TLC to follow the progress of reactions. Unless otherwise specified, 1H NMR spectra and 13C NMR spectra were obtained on a Varian Mercury 400 MHz console connected with an Oxford NMR AS400 Actively Shielded magnet, or a 500 MHz Varian Inova spectrometer at room temperature. Chemical shifts for 1H or 13C are given in ppm (δ) relative to tetramethylsilane (TMS) as an internal standard. Mass spectra (m/z) of chemical compounds were recorded on a Thermo Finnigan LTQ mass spectrometer, waters Acquity UPLC SQD2 system or Agilent 1260 Infinity LC connected with an InfinityLab MS detector. Reverse phase HPLC purifications were performed in 1260 infinity HPLC equipped with a semi-prep C-18 column (Phenomenex, 5 μm C18 100 Å, 250 × 10 mm, LC Column) eluted over a linear gradient from 95% solvent A (H2O, 0.1% TFA) to 100% solvent B (ACN, 0.1% TFA).
Expression of biotinylated TEV protease with AviTag (Biotin-TEVp)
During expression, the TEV protease autocleaves the MBP moiety yielding a soluble TEV protease containing both a hexahistidine tag for affinity purification and the cleavable biotin tag for releasing covalently bound phage. The biotinylated TEV protease (Biotin-TEVp) expression vector pMAL-HisTag-AviTag-3Cs-TEV was built by inserting the AviTag sequence into the pRK793 vector39. The TEV protease catalytic domain and vector backbone were amplified by polymerase chain reaction (PCR) with primers TEV_forwards (5’-GTTTTATTCCAGGGTCCTAGTGGTGGCGGTGGAGAAAGCTTGTTTAAGGGGC-3’) and TEV_reverse (5’-ACCTTGAAAATAAAGATTTTCTCCCC-3’). The AviTag sequence was amplified from pAviTag N-His Kan Vector (Ludigen, #49041–1) with primers AviTag-5 (5’-GAGAAAATCTTTATTTTCAAGGT CATCATCACCACCATCACGG-3’) and AviTag-3 (5’-AGGACCCTGGAATAAAACTTCTAAAGAGCCACCGGTAGGAGGAG-3’). The two PCR products were then ligated with Gibson Assembly (New England Biolabs, E2611S). The sequence of the final plasmid was verified with sanger sequencing (McLab, south San Francisco). The plasmid was transformed into Biotin XCell F’ cells for expression. An over-night pre-culture in 5 mL 2YT media containing 100 μg/mL Carbenicillin was then used for inoculating 500 mL 2YT median containing 100 μg/mL Carbenicillin and 50 μM biotin on the second day. After culturing at 37 °C until cell density reached an OD600 of 0.4, 0.01 % arabinose and 1 mM IPTG was added to induce the expression of the BirA biotin transferase40 and the desired TEV protease. After further growing at 37 °C for another 4 hours, the cells of the expression culture were harvested by centrifugation and lysed in 50 mL 50 mM Tris 8.0, 100 mM KCl, 1 mM EDTA, 1 mM DTT, 0.01% Triton X100 buffer containing 10 mg lysozyme and 0.5 mg DNase with the help of sonication. After further incubation on ice for 30 mins, the insoluble cell debris was centrifuged at 10,000g for 20 mins. The clarified supernatant was pumped through a HisTrap™ HP 1mL column with a peristaltic pump (Rainin Dynamax RP-1) at a flow of 1 mL/min. After washing with 50 mL 50 mM Tris 8.0, 100 mM KCl, 1 mM EDTA, 1 mM DTT and 0.01 % Triton X100 buffer with 10 mM imidazole, the bound protein was eluted with an increasing imidazole gradient from 10 mM to 300 mM over 30 mins at a flow rate of 1 mL/min. The eluted protein fractions were combined and concentrated to 5 mL before being injected into a HiPrep 16/60 Sephacryl S-100 HR (GE healthcare) for purification. The purified fractions of TEV protease were analyzed by SDS-PAGE and fractions containing the enzyme were combined and stored in 50 mM Tris 8.0, 0.5 mM EDTA and 1 mM DTT at −80 °C.
TEV protease expression
The TEV protease conjugated with a N-terminus six Histidine tag was expressed in E. coli strain BL21(DE3) (Lucigen, #60401–1) using the cytoplasmic expression plasmid pDZ2087 (precious gift from Dr. David Waugh). BL21(DE3) harboring was used to inoculate 5 mL 2YT media with 100 μg/mL ampicillin and diluted with 500 mL 2YT media containing 100 μg/mL ampicillin for expression on the second day. The culture was shaken at 250 rpm at 37 °C until the density of the bacteria reached exponential phase (OD600 = 0.5) when 1 mM IPTG was added to induce the expression of TEV. After shaking at 30 °C and 250 rpm for 8 hours, the bacteria were harvested by spinning at 8000 rpm for 15min. The purification procedure was the same as the one used for purifying biotin-TEVp described above.
FphF expression & biotinylation
For the FphF protein which is challenging to express and whose function is poorly understood, we chose to use the native full-length protein (without tag) expressed only with a N-terminus HisTag that could be removed after purification through a 3C protease cleavable site. This construct produced soluble and catalytically active protein in E. coli and was therefore used for the phage panning experiments. Since this protein does not contain the AviTag, we performed chemical biotinylation of free lysine residues. Since this approach does not allow cleavage at the site of biotinylation, we constructed a phage library in which a TEV cleavage site was inserted between the N-terminus peptide library and the pIII protein, which allows efficient release of the covalently bound phage. This approach is a viable alternative to using a target protein that contains a cleavable tag and is optimal for use with targets that do not express well when modified at their C or N termini by the affinity tag. Furthermore, this approach can be used with targets that are hard to express and that can be commercially purchased or purified from native sources (i.e. human targets isolated from cells and tissues). In both cases, the presence of a cleavable linker enables stringent washing to remove reversibly bound phage and then subsequent release of the covalent bound phage by cleavage of the linker.
The full-length gene of FphF was cloned into a modified pET28a vector carrying a 3C protease cleavage site following the N-terminus 6Histadine tag. After transforming the sequence verified plasmid, FphF was expressed in BL21(DE3) E.coli. grown in 1L LB media and induced with 0.2mM IPTG at 18°C overnight. Following cell lysing, the cytosol fraction of expression host was recovered and mixed with Ni affinity resin for separating crude FphF protein. After washing with purification buffer (50mM Tris pH 8.0, 300mM NaCl, 10mM Imidazole, 10% (v/v) glycerol, 10% (w/v) Sucrose), the tagged FphF protein was eluted from the affinity resin with elution buffer (50mM Tris pH 8.0, 300mM NaCl, 300mM Imidazole, 10% (v/v) glycerol, 10% (w/v) Sucrose). 50ug/mL 3C protease and 2mM DTT were added to the elution buffer to remove the N-terminus His tag at 4°C overnight. The final FphF was obtained by purifying the protein with ionic exchange resource Q column and Superdex 75 columns sequentially and was stored in 50 mM HEPES pH 7.5, 100 mM NaCl buffer. For the biotinylation, FphF (100 uL, 2mg/mL) was incubated with EZ-link Sulfo-NHS-LC-biotin (5 uL, 28mM in DMSO; Pierce) for 2 h on ice. Excess of the biotinylation reagent was removed by a PD-10 desalting column and eluted with 50 mM HEPES pH 7.5, 100 mM NaCl buffer. The biotinylation efficiency was verified by incubating the protein with magnetic streptavidin and neutravidin beads respectively and analyzing the bound and unbound protein fraction by SDS-PAGE.
Peptide-D1-D2 fusion protein expression
A previously reported plasmid41 (pET28b-C7C-D1D2) was used for expressing a peptide (ACGSGSGSGCG) containing two cysteine residues fused to the phage D1D2 protein (ACGSGSGSGCG-D1D2). The expression and purification procedures are similar to the expression procedure described above except the following changes were made: BL21(DE3) cells were used for expression at 30 °C for 14 hours. The final protein was eluted with the buffer R (20 mM NH4HCO3, 5 mM EDTA, pH 8.0 with 1 mM TCEP).
Modification of the peptide-D1-D2 fusion protein
Excess TCEP was removed by exchanging the protein buffer to buffer R without TCEP using a PD-10 column (GE Healthcare). The concentration of the protein was determined using a Nanodrop spectrometer based on absorption at 280 nm. The DCA linker (300 μM) was added to 2 μM fusion protein in buffer R with 20 % (v/v) acetonitrile and incubated at 30 °C for 2 hours or 42 °C for 1 hour before being analyzed using a Waters ACQUITY UPLC equipped with a C8 column and SQ Detector 2. The acquired mass of the protein was deconvoluted using MassLynx software.
DCA-VS linker synthesis
For scheme of synthesis of DCA-VS (11) see Extended Data Figure 1.
tert-butyl (2,3-dihydroxypropyl)carbamate (2)
Triethylamine (4.11 mL, 29.5 mmol) was added dropwise to a solution of 1-aminopropane-2,3-diol (23.2 g, 255 mmol) in methanol at room temperature, followed by slow addition of a solution of di-tert-butyl dicarbonate (66.8 g, 306 mmol) in dichloromethane (200mL). The reaction mixture was further stirred at room temperature for 3 hours. After removing the solvent, the oil-like residue was purified using a silica column and eluted with dichloromethane/methanol to give 40 g of the desired product as an oil (82% yield). 1H NMR (500 MHz, Chloroform-d) δ 5.02 (sb, 1H), 3.89 – 3.72 (m, 1H), 3.68 – 3.54 (m, 2H), 3.46 – 3.17 (m, 2H), 2.81 (sb, 2H), 1.47 (s, 9H).
tert-butyl (2-oxoethyl)carbamate (3)
Sodium periodate (53.78 g, 251 mmol) was added in portions to a tert-butyl 2,3-dihydroxypropylcarbamate (40 g, 209 mmol) solution in water (300 mL). After stirring at room temperature for 2 hours, the resulting solids were removed by filtration, and the aqueous fraction was extracted 6 times with dichloromethane. The combined organic fractions were washed with brine and dried over magnesium sulfate. After filtration, the organic fraction was dried by rotary evaporation and used directly for the next synthesis step (crude yield 85%). 1H NMR (500 MHz, Chloroform-d) δ 9.68 (s, 1H), 5.21 (sb, 1H), 4.10 (d, J = 5.1 Hz, 2H), 1.48 (s, 9H).
diethyl ((methylsulfonyl)methyl)phosphonate (5)
To a solution of diethyl (methylthio)methylphosphonate (10 g, 50.5 mmol) in acetic acid (34.6 mL, 605 mmol) was slowly added 50% hydrogen peroxide in water (7.4 mL, 260 mmol). Following completion of addition, the reaction mixture was heated to 70 °C for 30 min. After removing the solvent by rotary evaporation, the oily residue was diluted with water and extracted six times with dichloromethane. The organic fraction was further washed with saturated sodium bicarbonate in water, brine and dried over anhydrous magnesium sulfate. After filtration, the organic fraction was further dried by rotary evaporation. The resulting white solid was recrystallized in a mixture of ethyl acetate / hexane to give 9.3 g of a colorless crystalline solid (yield 80%). 1H NMR (500 MHz, Chloroform-d) δ 4.57 – 4.11 (m, 4H), 3.62 (d, J = 16.4 Hz, 2H), 3.24 (s, 3H), 1.41 (t, J = 7.1 Hz, 6H).
tert-butyl (E)-(3-(methylsulfonyl)allyl)carbamate (6)
In dry flask under argon, methylmagnesium bromide solution (3M; 13 mL, 39 mmol) in diethyl ether was added slowly through a syringe to a solution of diethyl ((methylsulfonyl)methyl)phosphonate (8.95 g, 38.9 mmol) in dry tetrahydrofuran (100 mL). After stirring at room temperature for 15 mins, tert-butyl (2-oxoethyl)carbamate (6.8 g, 42.8 mmol) solution in dry tetrahydrofuran (100 mL) was slowly added. After addition, the solution was further refluxed for 2.5 hours. To stop the reaction, 20 mL saturated ammonium chloride solution was added. The product was extracted by washing the aqueous layer with three times with 40 mL diethyl ether. After drying with magnesium sulfate, the organic fraction was concentrated and the crude product purified using a silica column. The desired product was eluted with Hexane and ethyl acetate at a ratio of 3:1 (6.6 g; yield 72 %). 1H NMR (500 MHz, Chloroform-d) δ 6.94 (dt, J = 15.7, 4.8 Hz, 1H), 5.97 (dt, J = 15.8, 1.9 Hz, 1H), 4.73 (s, 1H), 3.95 (t, J = 5.6 Hz, 2H), 3.76 (s, 3H), 1.48 (s, 9H). 13C NMR (126 MHz, Chloroform-d) δ 166.55, 145.26, 120.71, 79.73, 60.37, 51.58, 41.28, 28.30.
tert-butyl (E)-(2-((3-(methylsulfonyl)allyl)amino)-2-oxoethoxy)carbamate (9)
A solution of tert-butyl (E)-(3-(methylsulfonyl)allyl)carbamate (80 mg, 0.34 mmol) in 50 % trifluoroacetic acid with dichloromethane was stirred at room temperature for 1 hour before the solvent was removed under vacuum and toluene was added twice to co-evaporate the TFA. The remaining oily residue was triturated with diethyl ether to produce a brownish solid (50 mg, 0.216 mmol). Then 2-(((tert-butoxycarbonyl)amino)oxy)acetic acid (50 mg, 0.262 mmol) was added followed by N,N’-Diisopropylcarbodiimide (27.2 mg, 0.216 mmol) and N,N-Diisopropylethylamine (38 uL, 0.218 mmol). After stirring at room temperature overnight, the insoluble solids were filtered off and the dichloromethane fraction was purified directly by silica chromatography to produce 42 mg of product, for a 63% yield. 1H NMR (500 MHz, Chloroform-d) δ 8.68 (sb, 1H), 7.81 (s, 1H), 6.92 (dt, J = 15.2, 4.3 Hz, 1H), 6.55 (dt, J = 15.2, 2.0 Hz, 1H), 4.37 (s, 2H), 4.18 (ddd, J = 6.2, 4.3, 2.0 Hz, 2H), 2.93 (s, 3H), 1.47 (s, 9H). 13C NMR (126 MHz, Chloroform-d) δ 169.32, 158.18, 143.91, 129.88, 105.00, 83.69, 42.84, 38.88, 28.06.
(E)-2-(((1,3-dichloropropan-2-ylidene)amino)oxy)-N-(3-(methylsulfonyl)allyl)acetamide (DCA-VS) (11)
Tert-butyl (E)-(2-((3-(methylsulfonyl)allyl)amino)-2-oxoethoxy)carbamate (42 mg, 0.136 mmol) was placed in 1 mL of a 50 % trifluoroacetic acid solution in dichloromethane at room temperature for 1 hour. The solvent was removed under vacuum. Toluene was added and dried twice to remove the residual trifluoroacetic acid. The resulting alkoxyl amine was mixed with 1,3-Dichloroacetone (25 mg, 0.198 mmol ) in 1 mL of dimethylformamide. The mixture was stirred at room temperature overnight. After removing dimethylformamide, the residue was purified by silica chromatography and the desired product was eluted with 3% methanol in dichloromethane to produce 26 mg product for a 60 % yield. 1H NMR (400 MHz, Chloroform-d) δ 8.01 (s, 1H), 6.90 (dt, J = 15.2, 4.4 Hz, 1H), 6.46 (d, J = 15.2 Hz, 1H), 4.71 (s, 2H), 4.35 (s, 2H), 4.27 (s, 2H), 4.19 (ddd, J = 6.4, 4.5, 1.9 Hz, 2H), 2.94 (s, 3H). 13C NMR (101 MHz, cdcl3) δ 168.98, 154.30, 143.64, 130.22, 73.65, 42.84, 41.78, 38.84, 32.79. MS (ESI): calcd for C9H15Cl2N2O4S+: 317.0; found: 317.0.
DCA-DPP Linker Synthesis
For scheme of synthesis of DCA-DPP (15) see Extended Data Figure 2.
benzyl ((diphenoxyphosphoryl)methyl)carbamate (13)
A mixture of benzyl carbamate (6.04 g, 40 mmol),) acetic anhydride (5.18 g, 51 mmol), and paraformaldehyde (1.22 g, 40 mmol) in acetic acid (40 mL) was heated to 70 °C for 3 h. Then triphenyl phosphite (12.4 g, 40 mmol) was added and the mixture was heated at 120 °C for 2 h. The volatile components were removed in vacuo and the residue was dissolved in methanol and left overnight at −20 °C. The precipitate was isolated via filtration, washed with cold methanol, and dried at room temperature to afford 13 as a white solid (46 %). 1H NMR (400 MHz, Chloroform-d) δ 7.48–7.02 (m, 15H), 5.46 (s, 1H, NH), 5.13 (s, 2H), 4.04–3.92 (m, 2H). MS (ESI): calcd for C21H21NO5P+: 398.1; found: 398.1.
tert-butyl (2-(((diphenoxyphosphoryl)methyl)amino)-2-oxoethoxy)carbamate (14)
benzyl ((diphenoxyphosphoryl)methyl)carbamate (13, 2.22 g, 5.60 mmol) was treated with 33 % HBr/acetic acid (10 mL) and the reaction mixture was stirred for 2 h. After 2 h, the reaction mixture was concentrated, and the residue was dissolved in diethyl ether and left overnight at – 20 °C. The resulting white precipitate was filtered, washed with cold diethyl ether and dried at room temperature. Directly to this was added (Boc-aminooxy)acetic acid (1.13 g, 6.71 mmol), N,N’-Diisopropylcarbodiimide (865 μL, 5.60 mmol) and N,N-Diisopropylethylamine (1.95 mL, 11.20 mmol) in 20 mL DMF and the reaction mixture was stirred overnight at room temperature. The reaction mixture was concentrated, and the residue purified by flash silica chromatography using a gradient of 10%−100% EtOAc in hexane. After concentration of pure fractions 14 was obtained as a white solid in 67% yield. 1H NMR (400 MHz, DMSO) δ 7.48 – 7.35 (m, 4H), 7.31 – 7.15 (m, 6H), 4.30 (d, J = 1.3 Hz, 2H), 4.09 (dd, J = 10.9, 6.0 Hz, 2H), 1.40 (s, 9H). MS (ESI): calcd for C20H25N2O7P+: 436.1; found: 436.1.
diphenyl ((2-(((1,3-dichloropropan-2-ylidene)amino)oxy)acetamido)methyl)phosphonate (15)
tert-butyl (2-(((diphenoxyphosphoryl)methyl)amino)-2-oxoethoxy)carbamate (14, 1 g, 2.30 mmol) was dissolved in 10 mL of 50 % trifluoroacetic acid in DCM and stirred at room temperature for 1 h. The reaction mixture was concentrated and the resulting alkoxyl amine was mixed with 1,3-Dichloroacetone (0.43 g, 3.45 mmol) in 1 mL of DMF overnight at room temperature. The reaction mixture was concentrated, and the residue was purified by flash silica chromatography using a gradient of 0%−10% MeOH in DCM. After concentration of pure fractions 15 was obtained as a white solid in 72 % yield. 1H NMR (400 MHz, CDCl3) δ 7.39 – 7.32 (m, 4H), 7.25 – 7.17 (m, 6H), 6.70 (t, J = 4.3 Hz, 1H), 4.71 (d, J = 1.4 Hz, 2H), 4.33 (s, 2H), 4.20 (s, 2H), 4.16 (s, 1H). 13C NMR (101 MHz, CDCl3) δ 168.71 (s), 168.65 (s), 154.19 (s), 149.91 (s), 149.83 (s), 129.93 (s), 125.68 (s), 120.56 (s), 120.49 (s), 73.64 (s), 41.70 (s), 35.55 (s), 33.97 (s), 32.61 (s). 31P NMR (162 MHz, CDCl3) δ 14.71 (s). MS (ESI): calcd for C18H19Cl2N2O5P+: 444.0; found: 444.0.
Gln-Vinyl Sulfone Warhead Synthesis
For scheme of synthesis of Gln-Vinyl Sulfone peptides see Extended Data Figure 3.
Fmoc-Glu(OtBu)-dimethyl hydroxyl (Weinreb) amide (16):
Fmoc-Glu(OtBu)-OH (10.34 g, 24.3 mmol), HOBt (3.6g, 26.7 mmol) and dicyclohexylcarbodiimide (DCC; 5.5g, 26.7 mmol) were dissolved in 50 mL DMF. After stirring for 30 minutes at room temperature a solid formed and was removed by filtration. N,O-dimethyl hydroxyl amine (2.84g, 29.2 mmol) was added to the filtered reaction mixture along with triethyl amine (2.94g, 29.2 mmol). The reaction was stirred for an additional 12 hours and then concentrated. The crude oil was then dissolved in ethyl acetate and washed with three portions each of saturated sodium bicarbonate, 0.1N HCl and brine. The organic layer was dried over magnesium sulfate and concentrated to dryness. This crude preparation was used in subsequent reactions without further purification. 1H NMR (400 MHz, Chloroform-d) δ 7.76 (d, J = 7.4 Hz, 2H), 7.64 – 7.56 (m, 2H), 7.40 (t, J = 7.4 Hz, 2H), 7.36 – 7.27 (m, 2H), 5.60 (d, J = 8.8 Hz, 1H), 4.36 (d, J = 7.3 Hz, 2H), 4.21 (t, J = 7.2 Hz, 1H), 3.78 (s, 3H), 3.21 (s, 3H), 2.38 – 2.29 (m, 2H), 2.16 – 1.81 (m, 2H), 1.44 (s, 9H).
Fmoc-Glu(OtBu)-H (17):
Crude 16 (11 g, 23.5 mmol) was dissolved in anhydrous ethyl ether and stirred on ice under a positive argon flow. Lithium aluminum hydride (0.891 g, 23.5 mmol) was slowly added leading to gas evolution. The reaction was stirred for an additional 20 minutes and then quenched by the addition of potassium hydrogen sulfate (6.67g, 49 mmol). The quenched reaction was stirred on ice for 20 minutes and then at room temperature for an additional 30 minutes. The mixture was extracted three times with ethyl acetate and the combined organics washed with three portions of 0.1M HCl, saturated sodium carbonate and brine. The organic phase was concentrated to an oil. The product was purified by flash chromatography in hexanes:ethyl acetate (2:1, v:v). Yield (1 g, 10.4%). 1H NMR (400 MHz, Chloroform-d) δ 9.59 (s, 1H), 7.77 (d, J = 7.8 Hz, 2H), 7.60 (d, J = 7.5 Hz, 2H), 7.41 (t, J = 7.5 Hz, 2H), 7.32 (t, J = 7.5 Hz, 2H), 5.61 – 5.51 (m, 1H), 4.50 – 4.38 (m, 2H), 4.22 (t, 1H), 2.44 – 2.28 (m, 2H), 1.98 – 1.85 (m, 2H), 1.44 (s, 9H).
Fmoc-Glu(OtBu)-vinyl sulfone (18):
Diethyl (methylthiomethyl) phosphonate was oxidized to the corresponding phosphonate sulfone using hydrogen peroxide in acetic acid as described above for compound 5. Diethyl phosphonate sulfone (96 mg, 0.417 mmol) was dissolved in anhydrous THF under argon. Methylmagnesium bromide solution (3 M; 139 μL, 0.417 mmol) in diethyl ether was added and the reaction was stirred for 30 minutes at room temperature. Pure 17 (170 mg, 0.416 mmol) was dissolved in anhydrous THF under argon and added to the stirring reaction by syringe. After 30 minutes of stirring at room temperature the reaction was quenched with water and the aqueous phase was washed three times with dichloromethane. The organic phases were combined, dried over MgSO4 and evaporated to dryness. The crude oil was purified by flash chromatography over silica gel in hexane/ethyl acetate (1.5:1 v:v). Yield (168 mg, 8.7 mmol, 34.6%). 1H NMR (500 MHz, Chloroform-d) δ 7.81 (d, J = 7.5 Hz, 2H), 7.62 (s, 2H), 7.48 – 7.41 (m, 2H), 7.40 – 7.34 (m, 2H), 6.84 (dd, J = 14.6, 4.9 Hz, 1H), 6.46 (d, J = 15.0 Hz, 1H), 5.26 (d, J = 8.0 Hz, 1H), 4.49 (d, 2H), 4.23 (t, J = 6.7 Hz, 1H), 2.96 (s, 3H), 2.46 – 2.28 (m, 2H), 2.04 – 1.83 (m, 2H), 1.48 (s, 9H).
Fmoc-Glu(Rink amide resin)-vinyl sulfone (19):
Pure 18 (17 mg, 35 μmol) was dissolved in methylene chloride (0.5 ml) and an equal volume of anhydrous trifluoracetic acid was added. After 1 h of stirring at room temperature an equal volume of toluene was added and the reaction mixture was concentrated by rotary evaporation. The crude product was then coupled to Rink amide resin (60 mg, 0.35 mmol/g load, 21 μmol) in DMF (1 mL) using HATU (13.3 mg, 35 μmol) and DIEA (11 μL, 105 μmol) for 2 hours at room temperature. The resin was washed with DMF and peptide chain extension was achieved with standard solid phase peptide synthesis.
Construction of a phage peptide library with a TEV protease cutting site
The insertion of the TEV cleavage site sequence did not reduce the production yield or infectivity of the phage library (data not shown). The phage library was created by inserting DNA sequences encoding the semi-random peptide sequences (Ala-(Xaa)-Cys-(Xaa)7-Cys, Xaa is any colonial amino acid), the linker Gly-Gly-Ser-Gly, TEV protease cleavable peptide ENLYFQSG and the disulfide free domains D1 and D2 into the phage vector fdg3p0ss vector20, resulting at the generation of a library with 8E8 diversity. The insert was stepwise created in two consecutive PCR reactions. First, the genes of the TEV protease peptide andD1 and D2 of the phage pIII protein were PCR amplified with the two primers SecPCRba (5’- TGT GGC GGT TCT GGC GCT C −3’) and sfi2notfo (5’- CCA TGG CCC CCG AGG CCG CGG CCG CAT TGA CAG G −3’) using the vector fdg3p0ss21 as a template. Second, the DNA encoding the random peptides was appended in a PCR reaction using primers of C7C_lib_F (5’-TATGCGGCCCAGCCGGCCATGGCANNKTGTNNKNNKNNKNNKNNKNNKNNKTGTGGCGGTTCTGGCGCTC-3’) and sfi2notfo. The PCR products were digested with SfiI and ligated into a SfiI-digested phage vector. After verifying the efficiency of ligation, the library plasmids were electroporated into E. coli TG1 cells and incubated for 1 hour in 2YT at 37°C. The achieved phage library was then amplified by plating on large (20 cm diameter) chloramphenicol (30 mg/ml) 2YT plates. After growing at 37°C overnight for 16 hours, colonies were scraped off the plates with 2YT media, supplemented with 10% v/v glycerol and stored at −80°C.
Phage panning
Phage harboring the cysteine rich peptide library (CX8C, X is any 20 canonical amino acid, C is cysteine), a generous gift from Dr. Christian Heinis (EPFL, Lausanne), were used for selecting peptides against the TEV protease. The randomized DNA sequences encoding the peptide library were inserted into the phage DNA between the pelB signal peptide and the disulfide-free pIII protein of the fdg3p0ss phage vector20. The phage peptide library bearing a TEV protease cleavable site was used for selection of peptides against FphF hydrolase. To overcome the low infectivity of the disulfide-free phage strain used in our screens, we used a large volume of 2YT rich media (1 L) for the production of phage. The phage peptide libraries in TG1 E.coli bacteria (Lucigen, # 60502–1) were thawed from stock and used for inoculating 2YT media containing 30 μg/mL chloramphenicol. Phage were produced at 30 °C with shaking at 250 rpm for 16 hours. On the second day, the media rich with secreted phage was separated from the host bacteria by spinning at 8,500 rpm for 30 mins and the supernatant was mixed with 250 mL 20 % PEG 6000, 2.5 M sodium chloride solution. After being incubated on ice for another hour, the precipitated phage was spun at 9,000 rpm for 45 min. The resulting phage pellet was then dissolved in 10 mL buffer R (20 mM ammonium bicarbonate pH 8.0, 5 mM EDTA) and reduced with 1 mM TCEP at 42 °C for 1 hour. After removing the excess TCEP by filtration, 16 mL buffer R was added and 4 mL linker (DCA-VS; 1.5 mM) in acetonitrile was added to modify the phage at 30 °C for 2 hours. Then 5 mL 20 % PEG 6000, 2.5 M sodium chloride solution was added to precipitate the phage and the recovered phage pellets were resuspended in TEV Buffer (50 mM Tris 8.0, 100 mM potassium chloride, 1 mM EDTA and 1 mM DDT) for selections against TEV protease or FphF buffer (50 mM HEPES pH 7.5, 100 mM NaCl) for selections against FphF hydrolase, both supplemented with 1 % BSA and 0.1 % TX100 for blocking at room temperature for 0.5 hour. Biotin-TEV protease or FphF-biotin (10 μg) was incubated with 50 μL of hydrophilic streptavidin magnetic beads (New England Biolabs, S1421S) for 0.5 h and washed 5 times with TEV buffer supplemented with 1 % BSA and 0.1% TX100. After being blocked in the same buffer for 0.5 h, the phage library was mixed with the magnetic beads and incubated at room temperature for different amounts of time, depending on the round of screening being performed. After washing 10 times with PBS buffer, the magnetic beads were incubated with 100 μL guanidine chloride in PBS for 5 mins and then each eluted with 10 μM TEV protease in 300 μL TEV buffer (50 mM Tris 8.0, 0.5 mM EDTA and 1 mM DTT) buffer at 34 °C for 1 hour.to release the covalently bound phage from the solid support. The released phage were used to infect 45 mL TG1 bacteria at OD600 of 0.4 for 1.5 hours in a 37 °C incubator without shaking. Afterwards the TG1 cells were pelleted at 3,000 rpm for 15 min and plated on two 15 cm 2YT agar plate with 30 μg/mL chloramphenicol antibiotic for each protease. The agar plate was then incubated at 37 °C overnight to allow the infected TG1 cells to expand. On the second day, TG1 cells were scrapped off the agar plates with 5 mL 2YT median, mixed with 5 mL 50 % glycerol and stored at −80 °C. This glycerol stock was then used for preparing plasmids for sequencing or generating phage for the next round of phage panning.
Phage infectivity measurement
To the volume of 1 mL PBS containing 0, 1, 2, 3, 4, 5 and 6 M guanidine chloride, 5 μL of phage stock solution was added and incubated at room temperature for 5 mins. To the volume of 1 mL of TEV buffer containing 0–25 ng/mL TEV protease, 5 μL of phage stock solution was added and incubated at room temperature for 0.5 hour. Each solution was serially diluted 12 times by taking 20 μL samples and adding to 180 μL of 2YT medium. Each dilution (20 μL) was added to 180 μL exponentially growing TG1 cells (OD600 = 0.4) and incubated for 90 minutes at 37 °C. 10 μL of the infected TG1 cells were then spotted onto 2YT agar plates containing 30 μg/ml chloramphenicol. The number of colonies was counted the next day and the number of infective phage was calculated.
Peptide modification
All peptides were prepared using standard solid-phase peptide synthesis on a Syro II automated parallel peptide synthesizer (Biotage, Charlotte, NC USA) using standard 9-fluorenylmethoxycarbonyl (FMOC) chemistry protocols and Rink Amide AM resin support (0.02 mmol scale). Fmoc groups were removed using 300 μL of a 20% (v/v) solution of piperidine in DMF and amino acid coupling was carried out using a double coupling strategy with a 4-fold excess of Fmoc-amino acids (0.5 M solution in DMF). Coupling was achieved using a 1/1/1.5 ratio of amino acid/HBTU/DIEA in DMF. The deprotection and coupling times were 5 and 30 minutes, respectively. Six times 600 μL DMF washes were performed between deprotection and coupling steps. The peptide-loaded resin was treated with cleavage cocktail K (90/2.5/2.5/2.5/2.5 (v/v) mixture of TFA/thioanisole/water/phenol/EDT) for 2 h, which simultaneously cleaved the peptide from the resin and removed all side-chain protecting groups. The cleaved peptides were subsequently isolated by precipitation in cold diethyl ether followed by centrifugation. The precipitated peptides were resuspended in diethyl ether and centrifuged (3 times). Finally, the peptides were dissolved in degassed MilliQ water and lyophilized42. To perform peptide cyclization, 10 mg of the crude peptide was dissolved in 9 mL degassed solvent mixture of 20 % acetonitrile and 80 % water resulting in a peptide concentration of about 1 mM, followed by addition of 3.5 mg DCA-VS or DCA-DPP linker in 0.5 mL acetonitrile and 0.5 mL 1 M NH4HCO3 in water. After incubation at 30 °C for 1 hour, the reaction mixture was quenched with 50 μL TFA and lyophilized to dryness. For purification, the modified peptides were dissolved in 400 μL water and injected onto a 1260 infinity HPLC (Agilent technologies, USA) equipped with a semi-prep C-18 column (Phenomenex, 5 μm C18 100 Å, 250 × 10 mm, LC Column), and separated over a linear gradient from 95 % solvent A (H2O, 0.1% TFA) to 100% solvent B (ACN, 0.1% TFA) in 15 mins. The mass of peptides was measured using a Bruker Maldi Microflex LRF, and HPLC fractions were lyophilized and resulting solids dissolved in DMSO to a stock concentration of 10 mM and stored at −20 °C.
Fluorogenic substrate synthesis
The TEV substrate peptide (ENLYFQGK, 20 μmol) was synthesize by standard Fmoc based solid phase peptide synthesis on rink amide resin (CHEM-IMPEX INT’L INC., #02900, 0.33 mmol/g load) as described above. After deprotecting the Fmoc from the N-terminus of the synthesized peptide, Cy5 acid (14 mg, 22 μmol), HATU (12 mg, 31.5 μmol) and DIPEA (18 μL, 103 μmol) were added to the washed resin and incubated for 2 hours at room temperature. After releasing and deprotecting the peptide with cleavage cocktail K, the Cy5 labeled peptide was purified by HPLC and lyophilized to dryness. MS (ESI) negative: calculated for M-H+: 1620.7; found: 1620.4. Cy5-ENLYFQGK-NH2 (10 mg, 6.2 μmol), QSY21 (7 mg, 7.5 μmol) and DIPEA (6.5 μL, 37.4 μmol) were mixed in 1 mL DMSO and incubated at room temperature overnight. The reaction mixture was injected into a RP-HPLC connected with a semi-prep C18 column to purify the Cy5-ENLYFQGK(QSY21)-NH2 as described above. MS (ESI) negative: calculated for M-2H+: 1221.4; found: 1221.4.
TEV inhibition assay
The inhibitory activity of cyclic peptides was determined by incubating TEV protease (50 nM) with different concentrations of inhibitor and quantifying residual activity using the fluorogenic substrate synthesized as described above (50 μM, Cy5-ENLYFQGK(QSY21)-NH2). Peptide inhibitors and TEV protease were pre-incubated at 30 °C for 1 hour, and then the enzymatic assays were performed at 30 °C in buffer containing 50 mM Tris-Cl, pH 8.0, 100 mM KCl, 1 mM EDTA 1 mM DTT, 1 % DMSO, 1 % BSA and 0.01% TX100. The TEV activity was measured by monitoring the change in fluorescence intensity during one hour using a Cytation 3 Multi-Mode Reader (excitation at 640 nm, emission recorded at 670 nm; BioTek Instruments, Inc, Winooski, VT, USA). Calculations were done with OriginPro 2019 software (OriginLab Corporation). Sigmoidal curves were fitted to the data using the following dose response equation: y = A1 + (A2-A1)/1 + 10(logx0-x)p wherein x = peptide concentration, y = % activity of reaction without peptide, A1 = 100%, A2 = 0%, p = 1. IC50 values were derived from the fitted curve.
Synthesis of SENP1 substrate Ac-QTGG-AFC
POCl3 (400 μL, 4.29 mmol) was slowly added to a solution of 7-amino-4-trifluoromethylcoumarin (AFC, 500 mg, 2.18 mmol) and Boc-Gly-OH (381 mg, 2.18 mmol) in pyridine (10 mL) at −15 °C under argon and the resulting mixture was further stirred at −15 °C for 0.5 h. The reaction was then quenched with water and extracted with ethyl acetate. The organic phase was combined and washed with 0.1 M HCl, water, and brine. After drying over magnesium sulfate, the organic phase was concentrated And the resulting crude material was purified by silica column to afford Boc-G-AFC as white solid (630 mg, 74% yield). After Boc deprotection with a mixture of TFA:DCM (1:1, 10mL) at room temperature for 1 hour, the reaction mixture was dried and injected into a CombiFlash Companion Flash Chromatography System equipped with a RediSep® Rf Reversed-phase C18 column (Teledyne, Thousand Oaks, CA, USA) for purification. H2N-G-AFC was isolated as a white solid (550 mg, 88%). MS (ESI): calcd for M+H+: 287.2; found: 287.1.
Ac-Q(trt)T(t-Bu)G-OH was synthesized on chlorotrityl resin as described above and cleaved with 20% hexafluoroisopropanol in DCM 3×5 mins to keep the peptide fully protected. MS (ESI): calcd for M+H+: 645.3; found: 644.9. Ac-Q(trt)T(t-Bu)G-OH, H2N-G-AFC, DIC and DIPEA was mixed in DCM and stirred at room temperature overnight. The next morning, the generated white solid was removed by filtration and the reaction mixture was dried under vacuum and deprotected with 1 mL TFA:TIPS:water (95:2.3:2.5) for 2 hours at room temperature. After removing the solvent, the residue was dissolved in DMSO and purified by HPLC. Fractions with the correct MS were combined and lyophilized to give a white solid. MS (ESI): calcd for M+H+: 615.2; found: 615.3.
FphF inhibitory activity test
FphF peptides were tested in a similar manner to TEV peptides with the following changes:: All assays were performed in FphF buffer (20 mM HEPES pH 7.5, 50 mM NaCl + 0.01%TX100 + 5% DMSO) with final concentrations of 1 nM FphF and 20 μM 4-methylumbelliferyl heptanoate. FphF without any inhibitor was used as a positive control, and substrate in buffer without enzyme was taken as negative control. Peptides were pre-incubated for 1 h at 37°C before residual FphF activity was determined.
Cat S, Cal L, SENP1, FphB and trypsin inhibition
The inhibitory activity of peptides against cathepsin S (Cat S), cathepsin L (Cat L), sentrin-specific protease 1 (SENP1)43 fluorophosphonate-binding hydrolases B (FphB) and trypsin, was determined by pre-incubating peptide inhibitors with proteases and quantifying residual activity with a fluorogenic substrate in the same procedure as described for determining TEV inhibition activities. For Cat S and Cat L inhibition, a final concentration of 2–200 μM peptide was pre-incubated with 2.5 nM Cat S or Cat L enzyme at 37 °C for 1 hour before 100 μM Z-FR-AMC substrate was added in 100 mM NaAc, 2.5mM EDTA, 2.5mM DTT, 0.01% Tween20, 1% DMSO buffer. Monitoring the generation of free AMC due to hydrolysis of fluorogenic substrate was achieved by reading fluorescent signal excited at 380 nm and emission at 460 nm. For SENP1 inhibition, final concentrations of 2–200 μM of peptide was pre-incubated with 500 nM SENP1 enzyme at 37 °C for 1 hour before 100 μM Ac-QTGG-AFC substrate was added in 50 mM Tris 7.5, 100 mM NaCl, 1 mM EDTA, 1 mM DTT, 5% DMSO, 0.01% Tween20 buffer. Monitoring the generation of free AMC due to the hydrolysis of fluorogenic substrate was achieved by reading fluorescent signals excited at 400 nm and emission at 505 nm. For the FphB inhibition assay, a final concentration of 0.5–50 μM peptide was pre-incubated with 200 nM FphB enzyme at 37 °C for 1 hour before 20 μM 4-methylumbelliferyl butyrate substrate was added in PBS buffer. Monitoring the generation of free 4-methylumbelliferone (4-MU) due to hydrolysis of fluorogenic substrate was achieved by reading fluorescent signals excited at 380 nm and emission at 460 nm. For the trypsin inhibition assay, a final concentration of 0.5–50 μM peptide was pre-incubated with 20 μg/mL trypsin enzyme at 37 °C for 1 hour before 50 ng FITC-Casein Substrate was added in 100 mM Tris 8.5 buffer. Monitoring the generation of free FITC due to hydrolysis of fluorogenic substrate was achieved by reading the fluorescent signal excited at 490 nm and emission at 525 nm. The slope of the developing curve reflected the residual activity of enzymes after covalent inhibition. The IC50 values were calculated as described above.
Determination of kinact/KI
Under pseudo-first order conditions using the same buffers as outlined above, a fixed amount of TEV protease (50 nM) or FphF (100 nM) was incubated with TEV13 or FphF16 respectively, at different concentrations in the presence of the appropriate substrate (50 μM, Cy5-ENLYFQGK(QSY21)-NH2 for TEV and 20 μM 4-methylumbelliferyl heptanoate for FphF). The pseudo first-order rate constant, kobs, was calculated for each inhibitor concentration using non-linear regression analysis in GraphPad Prism 5. The obtained values were plotted against inhibitor concentration to afford the apparent second order rate constant kinact/KI.
Cy5 labeled TEV13 peptide
Cy5-NHS (1 mg, 1.35 μmol) and DIPEA (1 μL, 5.75 μmol) was added to a solution of the TEV13 peptide (10 mM, 50 μL) in DMSO. The reaction mixture was incubated at room temperature overnight and injected into the HPLC for purification as described above. The fractions with the desired product were combined and lyophilized to dryness. MS (MALDI): calcd for M+H+: 2276.8; found: 2276.5.
Labeling with Cy5-TEV13-DCA-VS
HEK293 cells from ATCC were cultured in DMEM medium (Gibco, #11995–073) supplemented with antibiotics (Gibco, #15140148) and 10% FBS (GeneMate, #S-1200–500), and no mycoplasma test was performed. One 10 cm petri dish of HEK293 cells or TG1 E.coli grown in 5 mL 2YT media were individually resuspended in 1 mL TEV buffer (50 mM Tris 8, 100 mM KCl, 1 mM EDTA and 1 mM DTT) with 0.2% Triton X100. The cells were transferred to 2.0 mL O-ring tubes and mixed with 100 μL 0.1mm glass beads (Biospec Products, Bartlesville, OK, USA). Tubes were then placed in a prechilled aluminum vial rack and cell samples were lysed in a Mini-BeadBeater-96 (Biospec Prod) three times (30 s with 2 min resting on ice in between). After centrifugation at 13,000Xg, at 4 °C for 5 min, 50 μL of the cleared supernatant was mixed with 50 μL TEV buffer with/without 100 nM TEV protease for labeling with 1 μM Cy5-TEV13-DCA-VS at 30 °C for 1 hour. Then SDS PAGE loading buffer (40 μL) was added and the mixtures were heated to 95 °C for 5 mins. Samples from each condition (15 μL) were separated by 12% SDS-PAGE. Cy5 fluorescence was detected by scanning on a Typhoon 9410 variable mode imager (GE Healthcare, λex=633 nm, 670 BP30 filter)44. The fluorescent intensity of labeled proteins was analyzed with LI-COR Image Studio Lite software, and plotted with OriginPro 2019 software (OriginLab Corporation). Afterwards, the gel was stained with Coomassie blue to visualize total protein loads.
Ellman’s test of free cysteine in TEV13
Free cysteine solutions (0.5 μL of 2, 4 6 8 and 10 mM) in buffer D (0.1 M sodium phosphate pH 8.0, 1 mM EDTA) were mixed with 9.5 μL buffer D and 10 μL DTNB (80 μg/mL) and incubated at room temperature for 15 mins. Three replicates of each condition were obtained. Then the absorbance at 412 nm was measured in a UV spectrometer to generate a standard curve. Cyclic peptide TEV13 was dissolved in DMSO to achieve a stock concentration of 10 mM. The stock solution (0.5 μL) was mixed with 9.5 μL buffer D and 10 μL DTNB (80 μg/mL) and incubated at room temperature for 15 mins. Then the absorbance at 412 nm was measured in a UV spectrometer and fit to the standard curve generated with free cysteine45.
Competition of TEV13 for labeling by Cy5-TEV13
TEV13 (0.64 μL of 2 mM stock in DMSO) was added to 40 μL of TEV buffer and then 20 μL was taken to perform another 6 2X dilutions. TEV buffer was used as a zero standard. Then 20 μL of 400 nM TEV protease and 40 μL of cell lysates were added to each well and the samples were incubated at 30 °C for 1 hour. Then 0.8 μL of 0.1 mM Cy5-TEV13 fluorescent probe was added to each condition and incubated at 30 °C for another hour before 30 μL 4X SDS PAGE sample buffer was added to each well to quench the reaction. After boiling the samples for 5 mins, 15 μL of the samples from each condition was analyzed on a 12 % SDS PAGE gel. Cy5 fluorescence was measured by scanning the gel with a Typhoon 9410 variable mode imager (GE Healthcare, λex=633 nm, 670 BP30 filter)46. The fluorescent intensity of the protein bands at the expected size was analyzed using LI-COR Image Studio Lite software and plotted with OriginPro 2019 software (OriginLab Corporation). Afterwards, the gel was stained with Coomassie blue to visualize the total protein load.
Plasma stability of TEV13
TEV13 (10 μL of 2 mM stock in DMSO) was added to 290 μl mouse serum (final peptide concentration was 66.7 μM in 300 μL final volume in Balb C Mouse Serum (Innovative Research)) and the mixture was incubated at 37 °C in a water bath. After 0, 1, 2, 4 and 12 hours, 50 μL samples were taken, mixed with 100 μL analytical grade ethanol and kept at −20 °C for at least 1 hour to precipitate the proteins and lipids in plasma. Then the precipitants were centrifuged in an Eppendorf 5417 centrifuge at 14k rpm at 4 °C and the supernatant containing the peptides was mixed with 300 μL water before being lyophilized to dryness. Finally, the samples were resuspended in 55 μL water and 15 μL was characterized by analytical reversed-phase high performance liquid chromatography (RP-HPLC) on an Agilent HPLC system (1100 series) equipped with a Waters Agilent ZORBAX 300SB-C3 reverse phage column. The elution linear gradient was 0 – 95% v/v solvent B over 14 minutes at a flow rate of 1 mL/min (A: 99.9% v/v H2O and 0.1% v/v TFA; B: 99.9% v/v ACN and 0.1% v/v TFA). The elutes were detected at a wavelength of 220 nm.
HPLC analysis of peptide purity
Peptide stock solutions (1 μL of 3 mM) were injected into an Agilent 1260 Infinity HPLC system equipped with a Poroshell 120 EC-C18 2.7 μm (Agilent #699975–902) analytical column, and run with a linear gradient of a mobile phase composed of eluent A (99.9 % v/v H2O, 0.1% v/v TFA) and eluent B (99.9% v/v acetonitrile, 0.1% v/v TFA) from 5% to 95% over 6 minutes at a flow rate of 0.6 ml/min. The absorbance at a wavelength of 220 nm was used to generate plots of peptide purity.
Amber MD simulation
Homology models of TEV protease with TEV13 residues Glu6, Pro7, Leu8 and Tyr9 bound in the standard mechanism to the S6, S5, S4 and S3 sub-sites were built using X-ray cocrystal structure data sets using 1LVM (available at protein data bank) as a template. Non-essential water molecules and ions were deleted in the PDB files and the peptide ligands truncated to retain only the P6-P3 (ENLY) residues. The remaining residues in the peptide chain were added manually in randomly extended conformation. The initial structure of the DCA-VS linker was minimized with the Gaussian 16 software package and parameterized with GAFF. Tleap was used for generating molecular dynamic simulation input files, and the linkages between DCA-VS and cysteines residues in TEV13 were manually specified. Two individual models were built when the linkage between vinyl sulfone and active site cysteine was generated in R and S conformations. Force field parameters of DCA-VS and surrounding atoms were taken from GAFF and partial charges were calculated with HF/6–31G* RESP charge method. All structures were first minimized by holding the conformation of TEV13 Glu6, Pro7, Leu8 and Tyr9 residues and TEV protease, followed by whole system minimization. Then the system was heated to 300 K before equilibration on the whole system. The production equilibration was run at 300 K with constant pressure and periodic boundary for 40 ns. Langevin dynamics was used to control the temperature using a collision frequency of 1.0 /ps. Then the final structures were analyzed, the lowest energy structure is presented47. All the calculations were performed by Assisted Model Building with Energy Refinement (AMBER, code available at http://ambermd.org/) biomolecular simulation package compiled in the sherlock High-Performance Computing (HPC) cluster provide by Stanford University and the Stanford Research Computing Center.
Preparation of RAW cell extracts and labeling with Cy5 probes
RAW 264.7 cells (ATCC® TIB-71™) were cultured in DMEM (Gibco, #11995–073) supplemented with 10% animal serum (GeneMate, #S-1200–500), 100 units/mL penicillin and 100 μg/mL streptomycin (Gibco, #15140148). Cells were cultured in a 5% CO2 humidified incubator at 37˚C on 10 cm culture dishes (corning, # 430167), and no mycoplasma test was performed. At around 90% confluency, cells were scraped off into 1 mL 100 mM NaAc pH 5.5, 2.5 mM EDTA, 2 mM DTT buffer or 1 mL PBS pH 7.4 buffer supplemented with 2 mM DTT followed by lysis by ultrasound sonication. After centrifugation at 18,000xg at 4 °C for 15 mins, the supernatant was taken and aliquoted into 60 μL fractions. The probes (0.6 μL) were added to the lysates to achieve final probe concentrations of 1 μM, 0.5 μM, 0.25 μM, and 0.125 μM and incubated at 37 °C for 1 hour. After adding 15 μL SDS-PAGE sample buffer and boiling, 15 μL of each labeling mixture was analyzed on a 12 % SDS PAGE gel. Cy5 fluorescence was measured by scanning of the gel on a Typhoon 9410 variable mode imager (GE Healthcare, λex=633 nm, 670 BP30 filter). The fluorescent intensity of the labeled proteins was analyzed using LI-COR Image Studio Lite software, and plotted with OriginPro 2019 software (OriginLab Corporation). Afterwards, the gel was stained with Coomassie blue to visualize the total proteins.
Probe concentration and incubation time dependent labeling
To four tubes of 200 μL 100 nM TEV protease in 50 mM Tris-Cl, pH 8.0, 100 mM KCl, 1 mM EDTA 1 mM DTT and 0.01% Tween X100 buffer was added 2 μL of 100 μM, 50 μM, 25 μM, 12.5 μM peptide inhibitors in DMSO to achieve final peptide inhibitor concentrations of 1 μM, 0.5 μM, 0.25 μM and 0.125 μM. The reaction tubes were immediately transferred to a 30 °C incubator, and after 5, 10, 20, 30, 40, 50, 60 mins, 20 μL of the reaction mixture from each condition was mixed with 10 μL SDS-PAGE sample buffer and boiled. Samples from each condition (15 μL) were resolved on a 12 % SDS PAGE gel. Cy5 fluorescence was measured by scanning of the gel on a Typhoon 9410 variable mode imager (GE Healthcare, λex=633 nm, 670 BP30 filter). The fluorescent intensity of the labeled proteins was analyzed using the LI-COR Image Studio Lite software, and plotted with OriginPro 2019 software (OriginLab Corporation).
Extended Data
Extended Data Fig. 1.
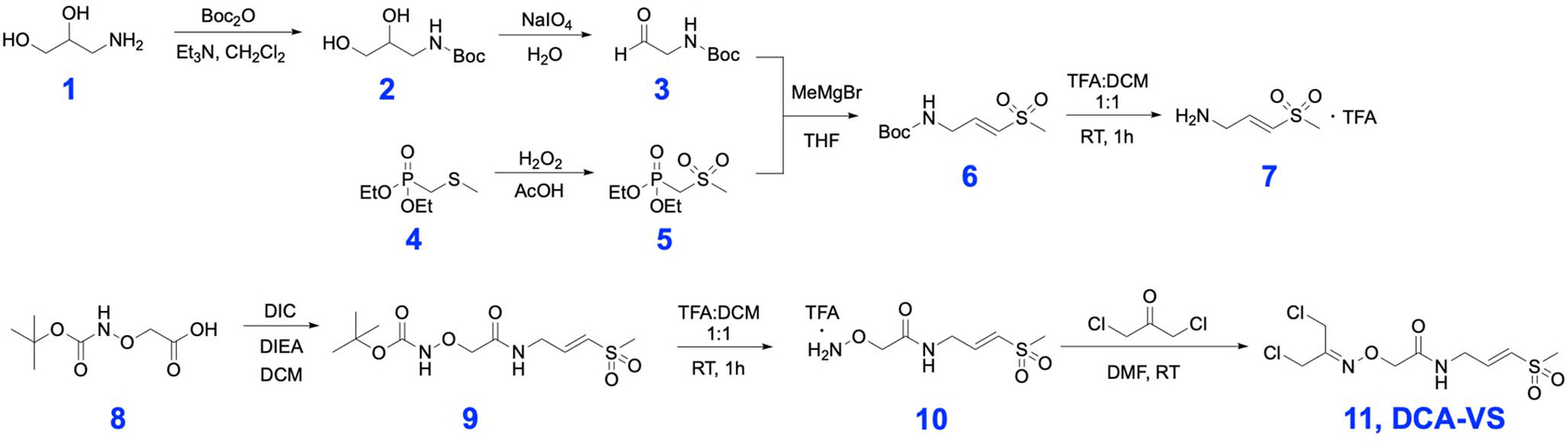
Strategy for the synthesis of DCA-VS, derivatizing a vinyl sulfone cysteine reactive warhead with 1,3-Dichloroacetone.
Extended Data Fig. 2.
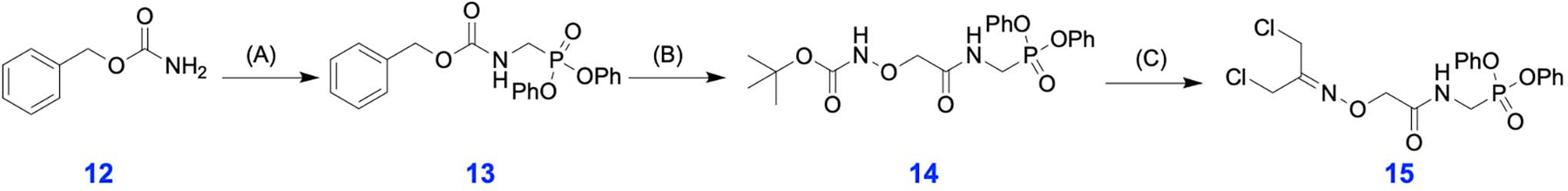
Strategy for the synthesis of DCA-DPP, derivatizing a diphenylphosphonate serine reactive warhead with 1,3-Dichloroacetone. (A) Acetic Anhydride, paraformaldehyde, triphenyl phosphite, acetic acid, 5 h, 120 °C, 46 % (B) i. HBr in acetic acid ii. (Boc-aminooxy)acetic acid, DCC, DIPEA, DMF, RT, O/N, 67 % (C) i. TFA in DCM, RT, 1 h ii. Dichloroacetone, DMF, RT, O/N, 72 %.
Extended Data Fig. 3.
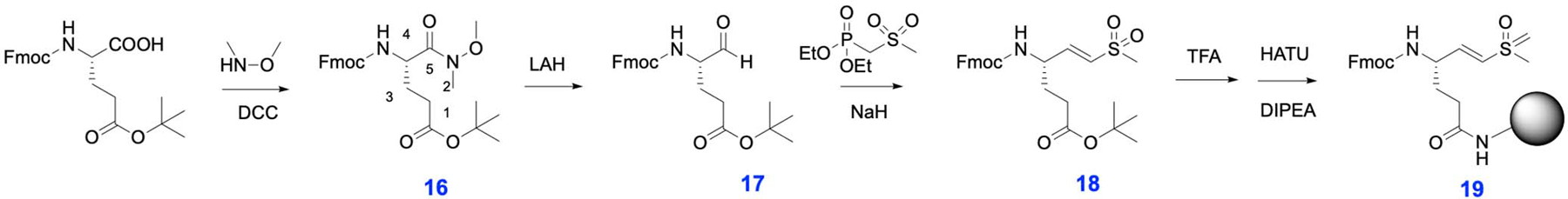
Strategy for synthesis of the Fmoc-Gln-vinyl sulfone warhead.
Supplementary Material
Acknowledgements
The authors thank the Vincent Coates Foundation Mass Spectrometry Laboratory at Stanford University for providing technical assistance with mass spectrometry. The authors also thank Dr. David Waugh (NCI, Frederick, MD) for providing the TEV expression construct pDZ2087 and Dr. Christian Heinis (EPFL, Lausanne, Switzerland) for providing the phage library. The work was supported by The Swiss National Science Foundation Postdoc Mobility fellowship P2ELP3_155323 P300PB_164725 (to S.C.) and by funding from The National Institutes of Health grants R01 EB026285 and R01 EB026285 02S1 (to M.B.).
Footnotes
Competing financial interests
The authors declare no competing financial interests.
Data availability
All data presented in this manuscript are available from the corresponding author upon reasonable request. The TEV protease expressing plasmid sequence is available at GenBank with accession number MN480436. Characterization of cyclization linker and probes are available in the Supplementary Information.
Reporting Summary
Additional information regarding statistics, data availability, study design and cell lines can be found in the Reporting Summary.
References
- 1.Singh J, Petter RC, Baillie TA & Whitty A The resurgence of covalent drugs. Nat Rev Drug Discov 10, 307–317 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Ghosh AK, Samanta I, Mondal A & Liu WR Covalent Inhibition in Drug Discovery. ChemMedChem 14, 889–906 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cravatt BF, Wright AT & Kozarich JW Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu Rev Biochem 77, 383–414 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Sanman LE & Bogyo M Activity-based profiling of proteases. Annu Rev Biochem 83, 249–273 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Cohen MS, Zhang C, Shokat KM & Taunton J Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science 308, 1318–1321 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith GP Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 228, 1315–1317 (1985). [DOI] [PubMed] [Google Scholar]
- 7.Matthews DJ & Wells JA Substrate phage: selection of protease substrates by monovalent phage display. Science 260, 1113–1117 (1993). [DOI] [PubMed] [Google Scholar]
- 8.Heinis C, Rutherford T, Freund S & Winter G Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat Chem Biol 5, 502–507 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Chen S, Bertoldo D, Angelini A, Pojer F & Heinis C Peptide ligands stabilized by small molecules. Angew Chem Int Ed Engl 53, 1602–1606 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Jafari MR et al. Discovery of light-responsive ligands through screening of a light-responsive genetically encoded library. ACS Chem Biol 9, 443–450 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Ng S & Derda R Phage-displayed macrocyclic glycopeptide libraries. Org Biomol Chem 14, 5539–5545 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Cardote TA & Ciulli A Cyclic and Macrocyclic Peptides as Chemical Tools To Recognise Protein Surfaces and Probe Protein-Protein Interactions. ChemMedChem 11, 787–794 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cesaratto F, Burrone OR & Petris G Tobacco Etch Virus protease: A shortcut across biotechnologies. J Biotechnol 231, 239–249 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Lentz CS et al. Identification of a S. aureus virulence factor by activity-based protein profiling (ABPP). Nat Chem Biol 14, 609–617 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Assem N, Ferreira DJ, Wolan DW & Dawson PE Acetone-Linked Peptides: A Convergent Approach for Peptide Macrocyclization and Labeling. Angew Chem Int Ed Engl 54, 8665–8668 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larsen D et al. Exceptionally rapid oxime and hydrazone formation promoted by catalytic amine buffers with low toxicity. Chem Sci 9, 5252–5259 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palmer JT, Rasnick D, Klaus JL & Bromme D Vinyl sulfones as mechanism-based cysteine protease inhibitors. J Med Chem 38, 3193–3196 (1995). [DOI] [PubMed] [Google Scholar]
- 18.Oleksyszyn J, Boduszek B, Kam CM & Powers JC Novel amidine-containing peptidyl phosphonates as irreversible inhibitors for blood coagulation and related serine proteases. J Med Chem 37, 226–231 (1994). [DOI] [PubMed] [Google Scholar]
- 19.Timmerman P, Beld J, Puijk WC & Meloen RH Rapid and quantitative cyclization of multiple peptide loops onto synthetic scaffolds for structural mimicry of protein surfaces. Chembiochem 6, 821–824 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Kather I, Bippes CA & Schmid FX A stable disulfide-free gene-3-protein of phage fd generated by in vitro evolution. J Mol Biol 354, 666–678 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Chen S, Morales-Sanfrutos J, Angelini A, Cutting B & Heinis C Structurally diverse cyclisation linkers impose different backbone conformations in bicyclic peptides. Chembiochem 13, 1032–1038 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Hornsby M et al. A High Through-put Platform for Recombinant Antibodies to Folded Proteins. Mol Cell Proteomics 14, 2833–2847 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raran-Kurussi S, Cherry S, Zhang D & Waugh DS Removal of Affinity Tags with TEV Protease. Methods Mol Biol 1586, 221–230 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rebollo IR, Angelini A & Heinis C Phage display libraries of differently sized bicyclic peptides. MedChemComm 4, 145–150 (2013). [Google Scholar]
- 25.Nazif T & Bogyo M Global analysis of proteasomal substrate specificity using positional-scanning libraries of covalent inhibitors. Proc Natl Acad Sci U S A 98, 2967–2972 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Albrow VE et al. Development of small molecule inhibitors and probes of human SUMO deconjugating proteases. Chem Biol 18, 722–732 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hemelaar J et al. Specific and covalent targeting of conjugating and deconjugating enzymes of ubiquitin-like proteins. Mol Cell Biol 24, 84–95 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choe Y et al. Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J Biol Chem 281, 12824–12832 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Bromme D, Klaus JL, Okamoto K, Rasnick D & Palmer JT Peptidyl vinyl sulphones: a new class of potent and selective cysteine protease inhibitors: S2P2 specificity of human cathepsin O2 in comparison with cathepsins S and L. Biochem J 315 ( Pt 1), 85–89 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanman LE, van der Linden WA, Verdoes M & Bogyo M Bifunctional Probes of Cathepsin Protease Activity and pH Reveal Alterations in Endolysosomal pH during Bacterial Infection. Cell Chem Biol 23, 793–804 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verdoes M et al. Improved quenched fluorescent probe for imaging of cysteine cathepsin activity. J Am Chem Soc 135, 14726–14730 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salomon-Ferrer R, Case DA & Walker RC An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev.-Comput. Mol. Sci 3, 198–210 (2013). [Google Scholar]
- 33.Maier JA et al. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J Chem Theory Comput 11, 3696–3713 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J, Wang W, Kollman PA & Case DA Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model 25, 247–260 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Phan J et al. Structural basis for the substrate specificity of tobacco etch virus protease. J Biol Chem 277, 50564–50572 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Chen L, Keller LJ, Cordasco E, Bogyo M & Lentz CS Fluorescent Triazole Urea Activity-Based Probes for the Single-Cell Phenotypic Characterization of Staphylococcus aureus. Angew Chem Int Ed Engl 58, 5643–5647 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weerapana E et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hacker SM et al. Global profiling of lysine reactivity and ligandability in the human proteome. Nat Chem 9, 1181–1190 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only References:
- 39.Kapust RB et al. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Engineering, Design and Selection 14, 993–1000 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Shen A et al. Simplified, enhanced protein purification using an inducible, autoprocessing enzyme tag. PLoS One 4, e8119 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bellotto S, Chen S, Rentero Rebollo I, Wegner HA & Heinis C Phage Selection of Photoswitchable Peptide Ligands. Journal of the American Chemical Society 136, 5880–5883 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Chen S et al. Improving binding affinity and stability of peptide ligands by substituting glycines with D-amino acids. Chembiochem 14, 1316–1322 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Mikolajczyk J et al. Small ubiquitin-related modifier (SUMO)-specific proteases: profiling the specificities and activities of human SENPs. J Biol Chem 282, 26217–26224 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Li H et al. Structure- and function-based design of Plasmodium-selective proteasome inhibitors. Nature 530, 233–236 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen S et al. Bicyclic peptide ligands pulled out of cysteine-rich peptide libraries. J Am Chem Soc 135, 6562–6569 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Lentz CS et al. Design of Selective Substrates and Activity-Based Probes for Hydrolase Important for Pathogenesis 1 (HIP1) from Mycobacterium tuberculosis. ACS Infect Dis 2, 807–815 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen S et al. Dithiol amino acids can structurally shape and enhance the ligand-binding properties of polypeptides. Nat Chem 6, 1009–1016 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.