

Abstract
Calcific aortic valve disease (CAVD) is a condition causing stiffening of the aortic valve, impeding cardiac function and resulting in significant morbidity worldwide. CAVD is thought to be driven by the persistent activation of the predominant cell type in the valve, aortic valve interstitial cells (AVICs), into myofibroblasts, resulting in subsequent calcification and stenosis of the valve. Although much of the research into CAVD focuses on AVICs, the aortic valve endothelial cells (AVECs) have been shown to regulate AVICs and maintain tissue homeostasis. Exposed to distinct flow patterns during the cardiac cycle, the AVECs lining either side of the valve demonstrate crucial differences which could contribute to the preferential formation of calcific nodules on the aorta-facing (fibrosa) side of the valve. Cadherin-11 (CDH11) is a cell-cell adhesion protein which has been previously associated with AVIC myofibroblast activation, nodule formation, and CAVD in mice. In this study, we investigated the role of CDH11 in AVECs and examined side-specific differences. The aorta-facing or fibrosa endothelial cells (fibAVECs) express higher levels of CDH11 than the ventricle-facing or ventricularis endothelial cells (venAVECs). This increase in expression corresponds with increased contraction of a free-floating collagen gel compared to venAVECs. Additionally, co-culture of fibAVECs with AVICs demonstrated decreased contraction compared to an AVIC+AVIC control, but increased contraction compared to the venAVECs co-culture. This aligns with the known preferential formation of calcific nodules on the fibrosa. These results together indicate a potential role for CDH11 expression by AVECs in regulating AVIC contraction and subsequent calcification.
Keywords: Heart valve, cadherin-11, cell-cell mechanotransduction
INTRODUCTION
Calcific aortic valve disease (CAVD) is a condition characterized by the formation of calcific deposits on the aortic valve, resulting in impaired cardiac function and presenting in up to 25% of Americans over 65 (Bowler et al. 2018). This calcification predominantly occurs on the side of the leaflets facing the aorta, known as the fibrosa (the ventricle-facing side of the leaflet is known as the ventricularis) (Simmons et al. 2005). CAVD is primarily considered to be a fibroblast-driven disease orchestrated by the majority cell type populating the leaflets, the aortic valve interstitial cells (AVICs). However, the aortic valve endothelial cells (AVECs) lining either side of the leaflets, exposed to blood flow through the valve, are able to transduce mechanical signaling (Wang et al. 2013) and alter AVIC signaling via chemokine secretion (Richards et al. 2013). A hallmark of disease progression is the activation and differentiation of fibroblasts into myofibroblasts, increasing their proliferation, extracellular matrix deposition, and contraction (Hutcheson et al. 2013). Co-culture models have demonstrated that AVECs can inhibit this AVIC myofibroblast differentiation (Richards et al. 2013) and induce AVICs to maintain quiescence (Butcher and Nerem 2006). Under unidirectional flow conditions, AVECs exhibit anti-calcific gene expression profiles (Richards et al. 2013). Furthermore, endothelial dysfunction is often an early sign of valve disease (Farrar, Huntley, and Butcher 2015; Mahler et al. 2014; Richards et al. 2013). For these reasons, it is important to further incorporate AVECs into CAVD research in order to more fully understand their impact on AVIC signaling and disease progression.
The aorta-facing or fibrosa endothelial cells (fibAVECs) and the ventricle-facing or ventricularis endothelial cells (venAVECs) exist in very different hemodynamic environments and exhibit distinct gene expression profiles (Simmons et al. 2005). During the opening of the valve, venAVECs experience high shear stress unidirectional flow, whereas upon closure of the valve, fibAVECs are exposed to low recirculatory shear stress flow patterns (Mahler et al. 2014; Simmons et al. 2005). These distinct flow environments alter the ability of AVECs to inhibit myofibroblast differentiation and thus impact valve calcification. Anti-calcific gene expression profiles are found in AVECs exposed to unidirectional flow (Richards et al. 2013). Likewise, the fibrosa endothelium presents lower expression of anti-calcific genes (Simmons et al. 2005). Due to CAVD predominantly developing on the fibrosa where the endothelium is exposed to these recirculatory flow patterns, the differences in AVEC populations could be critical in further understanding contributions to disease initiation and progression.
Cadherin-11 (CDH11) is a cell-cell adhesion protein which functions via homophilic extracellular bonds and anchoring to the actin cytoskeleton via catenins (Cavallaro and Christofori 2004). CDH11 is involved in migration and differentiation (Bowen et al. 2015), often associated with a more invasive phenotype (Cavallaro and Christofori 2004), and implicated in a range of fibrotic and inflammatory diseases, including pulmonary fibrosis (Schneider et al. 2012), scleroderma (Wu et al. 2014), rheumatoid arthritis (Kyung Chang, Gu, and Brenner 2010; Lee et al. 2007), and CAVD (Clark et al. 2017; Hutcheson et al. 2013). Diseased human valves with CAVD show enrichment of CDH11 (Sung et al. 2016). Additionally, AVICs treated with transforming growth factor-β (TGF-β), a known inducer of myofibroblast differentiation and a key initiator in valve calcification (Fisher, Chen, and Merryman 2013), demonstrate significant CDH11 upregulation(Chen et al. 2015; Hutcheson et al. 2013; Wang, Leinwand, and Anseth 2014). Previous studies have described the necessity of CDH11 expression by AVICs for nodule formation in vitro (Hutcheson et al. 2013; Sung et al. 2016). Interestingly, CDH11−/− murine AVICs demonstrate lower contraction in vitro, despite increased α-smooth muscle actin (α-SMA), a common indicator of contractile ability that is significantly upregulated during myofibroblast differentiation (Bowler et al. 2018). Furthermore, targeting CDH11 reduced aortic valve stenosis in a mouse model of CAVD (Clark et al. 2017). Despite this body of research on CDH11 in myofibroblasts and fibrotic tissue, little is known about the role of CDH11 in AVECs. CDH11 is known to be mechanically regulated, showing decreased expression in AVECs under unidirectional flow conditions (Butcher et al. 2006), such as those found in the environment of the venAVECs. However, it is unknown whether there is a side-specific difference in AVEC CDH11 expression or whether that has an impact on AVEC signaling and subsequent AVIC behavior. Our objective in this study was to determine if side-specific AVECs demonstrated differences in CDH11 expression and if that difference resulted in altered contraction profiles either in single culture or in co-culture with AVICs.
MATERIALS AND METHODS
AVEC isolation
Aortic valves were isolated from pig hearts obtained at a local abattoir (Hampton Meats, Hopkinsville, KY). Valves were transported in PBS on ice, and cells were isolated within 8 hours of dissection. Side-specific endothelial cell isolation was done as described previously by Gould, et al (Gould and Butcher 2010). Briefly, aortic valves were digested in collagenase II, placed in a dish with the fibrosa side face up, and a cotton swab was used to gently release the endothelial cells from the surface. Cells were plated in endothelial media (Endothelial Growth Media Bullet Kit, Lonza, #CC-3162) on tissue culture-treated plastic coated with 1% gelatin. The leaflets were flipped, and this process was repeated for the ventricularis side. Colonies were purified via clonal expansion according to Cheung et al (Cheung, Young, and Simmons 2008). Briefly, cells were lifted and plated in a 96-well plate at a final concentration of 0.3 cells/well, resulting in approximately one-third of the wells containing a cell and giving a high probability of colonies originating from a single cell. After two weeks, thriving colonies of endothelial morphology were passaged and grown until confirmation of phenotype. After phenotype confirmation, lines arising from several different wells were combined in order to prevent the final cell line from being completely homogenous.
Immunostaining and western blots
Immunostaining was performed using the following antibodies: CDH11, Thermo Fisher Scientific, #71–7600, 1:50 dilution; α-SMA, Cy3-conjugated, Millipore Sigma, C6198, 1:300 dilution; Prolong™ Gold Antifade Mountant with DAPI, P36931. Western blotting was performed using the following antibodies: CDH11, Cell Signaling Technologies, #4442BF, 1:4000 dilution; α-SMA, Abcam, ab5694, 1:1000 dilution; α-Tubulin, Vanderbilt MCBR Core, 1:1000 dilution.
Gel contraction assays
Gel contraction assays were performed using a free-floating collagen gel. For single-cell assays, 100,000 cells were seeded onto the top of the gel. For co-culture assays, 50,000 AVICs were embedded within the gel during polymerization, and 50,000 AVICs, fibAVECs, or venAVICs were seeded on top of the gel post-polymerization. Media was changed every 48 hours. Gels were allowed to contract for up to 96 hours, and images were taken periodically for quantification. ImageJ was used to quantify the fraction of contraction.
RESULTS
Clonal expansion ensured purity of isolated AVEC populations
The side-specific endothelial cells were first clonally expanded prior to experimentation to ensure both that the populations were free from contaminating AVICs isolated along with the AVECs and that the endothelial cells did not undergo endothelial-to-mesenchymal transformation. Clonal expansion resulted in purified AVEC populations. Immunostaining was performed using CD31 as an endothelial marker and α-SMA as a marker of AVIC contamination. Another sample from the line of venAVECs was allowed to grow without clonal expansion and used as a control for comparison. Figure 1 illustrates the confirmation of endothelial phenotype with minimal presence of other cell types. Both the clonally expanded venAVEC and fibAVEC lines show strong CD31 expression with minimal to no α-SMA expression. Without clonal expansion, the venAVEC line was overtaken with fibroblasts, demonstrating minimal CD31 expression and an abundance of α-SMA fibers.
Figure 1.
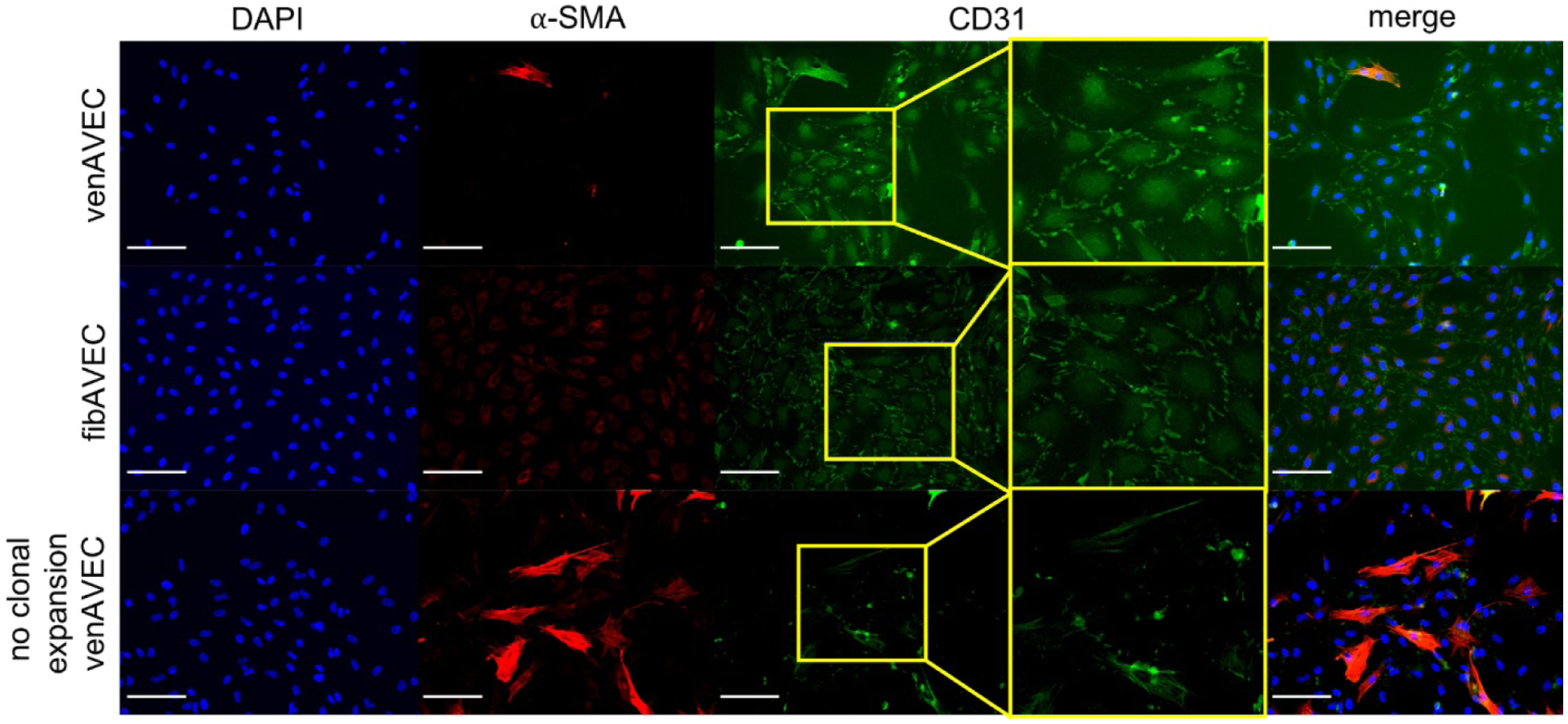
Both clonally expanded AVEC lines (venAVEC and fibAVEC) were stained using CD31 as an endothelial marker and α-SMA as a fibroblast marker, confirming endothelial phenotype. A line of non-clonally expanded venAVECs was used as a control. Scale bar is 100 μm.
Fibrosa AVECs demonstrated higher expression of CDH11 compared to ventricularis AVECs
Previous studies have established distinct gene expression profiles between venAVECs and fibAVECs (Simmons et al. 2005). Due to prior work implicating CDH11 expression by AVICs in nodule formation and in valve calcification(Hutcheson et al. 2013; Sung et al. 2016), we sought to examine possible differences in CDH11 expression by AVECs. Both AVEC lines were grown on 1% gelatin-coated tissue culture plastic, and cell lysates were analyzed via Western blot. We found that fibAVECs expressed approximately three times as much CDH11 compared to the venAVECs (Figure 2A). Interestingly, the fibAVECs also had slightly higher expression of CDH11 compared to AVICs. Neither AVEC line expressed α-SMA, and we observed no differences in eNOS expression (Figure 2A). Immunostaining of both venAVECs and fibAVECs confirmed the Western blot results, illustrating higher levels of CDH11 in the fibrosa-side endothelial cells compared to the ventricularis-side (Figure 2B).
Figure 2.
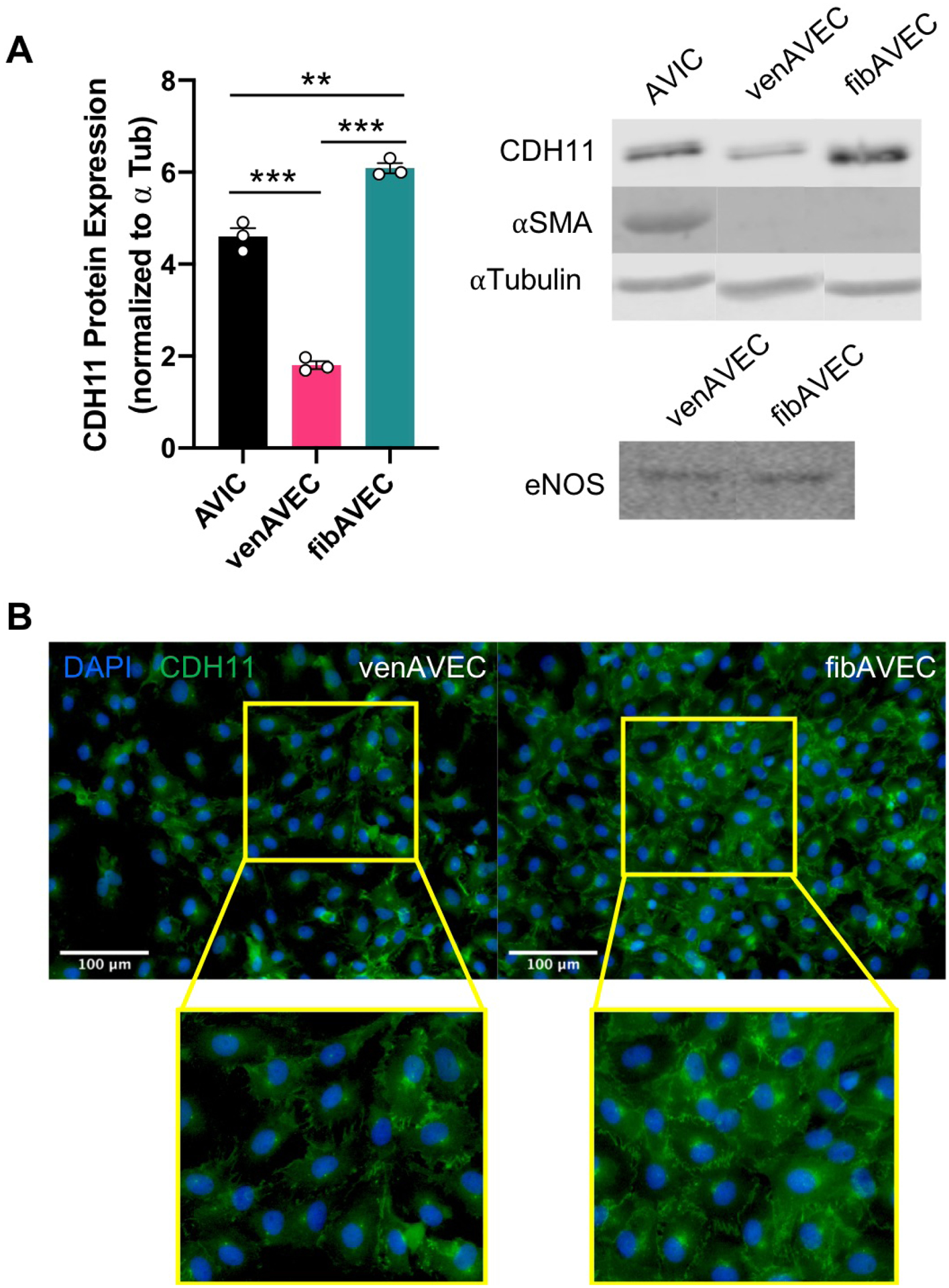
(A) FibAVECs express higher levels of CDH11 as measured by Western blot compared to both venAVECs and AVICs, while venAVECs express substantially less CDH11 than both. Neither of the AVEC lines demonstrated any α-SMA. There were no changes in eNOS expression between the two AVEC lines. (B) Immunostaining also illustrated higher expression of CDH11 by fibAVECs compared to venAVECs.
** indicates p<0.01; *** indicates p<0.001
Fibrosa AVECs exhibit higher contraction of a free-floating collagen gel both in single culture and in co-culture with AVICs when compared to ventricularis AVECs
In order to determine any differences in contraction between the side-specific endothelial cells, we performed a gel contraction assay. Cells were seeded onto a free-floating collagen gel and allowed to contract for up to 96 hours in order to measure the degree of contraction. Figure 3A illustrates the culture conditions for the single and co-culture models. For single cultures (Figure 3B), gels were seeded with either venAVECs, fibAVECs, or AVICs. Predictably, the AVICs contracted sooner and to a greater degree than either of the endothelial lines. However, although neither of the AVEC cell lines express detectable α-SMA, a typical marker of contractile ability, the fibAVECs began to contract earlier at 36 hours and sustained that contraction until the experiment ended at 96 hours.
Figure 3.
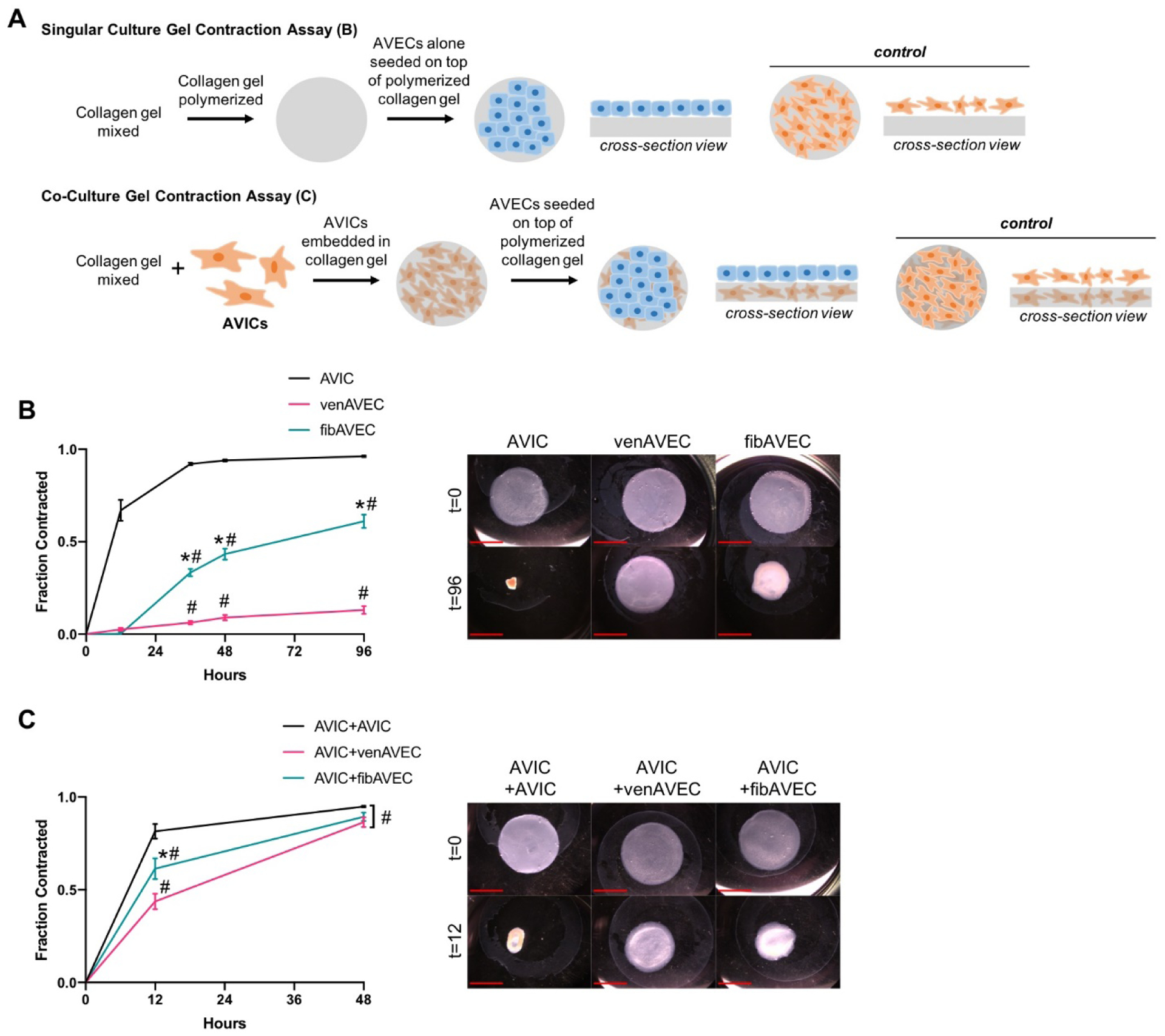
(A) An illustration of the set-up for both the single and co-culture gel contraction assays. (B) FibAVECs contracted a free-floating collagen gel significantly more than venAVECs over a 96 hour period. Each point represents n=3. (C) In a co-culture with AVICs, fibAVECs contracted a free-floating collagen gel less than an AVIC-AVIC co-culture at 12 hours, but significantly more than an AVIC-venAVEC co-culture. Each point for AVIC+venAVEC and AVIC+fibAVEC represents n=6, and for AVIC+AVIC, each point represents n=5. Scale bar is 1 mm.
* indicates difference of p<0.05 between fibAVEC and venAVEC at the same timepoint; # indicates difference of p<0.05 from AVIC at the same timepoint
Additionally, the collagen gels were also used to perform co-culture contraction experiments (Figure 3C), with one cell type embedded within the gel (AVICs) and one seeded on top (AVECs), representative of the aortic valve structure. One group contained AVICs both embedded and seeded on top of the gel to be used as a control. After 12 hours, both the AVIC+AVEC co-culture collagen gels had contracted less than the AVIC+AVIC control. The AVIC+fibAVEC gels contracted significantly more than the AVIC+venAVEC gels, aligning with the difference in contraction seen in the single culture model.
DISCUSSION
Previous research has examined the role of CDH11 in the activation, proliferation, and contraction of myofibroblasts, as well as its role in the formation of AVIC calcific nodules characteristic of CAVD (Hutcheson et al. 2013; Wang et al. 2014). However, despite the known ability of AVECs to inhibit myofibroblast activation and otherwise affect calcification (Richards et al. 2013), little is known about the role of CDH11 in AVECs and that impact on AVIC disease pathology. The results from this study both show significant CDH11 expression by AVECs and demonstrate a previously unreported difference in CDH11 expression between the AVECs of the fibrosa and ventricularis sides of the aortic valve. This is of importance due to the side-specific nature of the clinical disease progression, with preferential calcification on the fibrosa (Simmons et al. 2005). Additionally, this lines up with what is known about CDH11 mechanotransduction. Previous studies have shown that CDH11 expression is decreased in AVECs under unidirectional flow (Butcher et al. 2006). The venAVECs, exposed to high shear stress unidirectional flow in the valve, correspondingly exhibited lower expression of CDH11. Of clinical relevance, healthy human aortic valves show higher expression of CDH11 along the endothelium, while diseased samples show CDH11 distributed more evenly through the interstitium (Hutcheson et al. 2013), indicating that CDH11 expression by the endothelium could play a role earlier in disease initiation. Given the established link between CDH11 expression and CAVD, this finding could have implications in further understanding the disease pathology.
The gel contraction results demonstrate an innate difference between the fibrosa and ventricularis AVECs in vitro. Although the single cultures of AVECs lacked any α-SMA expression, there was still observable gel contraction and a disparity between the venAVECs and fibAVECs. This difference in gel contraction may likely be a result of the difference in CDH11 expression between the fibAVECs and venAVECs. Murine CDH11−/− AVICs contract less in culture despite an upregulation of α-SMA (Bowler et al. 2018), opposing conventional understanding of contractile ability and underlining the importance of CDH11 in contraction. However, it is also possible that this difference in gel contraction observed is not due to the cells contracting, but due to the cells exerting traction forces on the collagen gel. It is important to note that these expression differences in CDH11 observed here were measured in two-dimensional culture, and the three-dimensional nature of the gels could have affected protein expression. Additional studies could gather pertinent information by quantifying protein expression in the contracted gels. Likewise, although these VEC populations remained endothelial in phenotype during static culture, the possibility that they underwent endothelial to mesenchymal transition (EndMT) in the softer environment of the collagen gel cannot be ruled out. Higher rates of EndMT could be an explanation for the higher gel contraction observed in the fibAVECs. Although these results cannot conclusively link the CDH11 disparity between venAVECs and fibAVECs to their contractile differences, it is reasonable to consider CDH11 a likely perpetrator due to the previously established relationship between CDH11 expression and contraction both in other cell types, most notably AVICs (Bowler et al. 2018), and in CAVD (Hutcheson et al. 2013). These results illustrate another side-specific difference between AVECs and reveal another possible pathway through which AVEC signaling could impact AVIC function during disease progression. Future studies can draw stronger conclusions by utilizing a method of genetic knockdown of CDH11 in both cell types in the same assay.
The co-culture of both AVEC lines with AVICs demonstrated decreased contraction compared to AVICs alone, exhibiting another beneficial effect of AVEC co-culture. However, the co-culture of fibAVECs+AVICs exhibited higher contraction at 12 hours compared to venAVECs+AVICs. Although studies have reported preferential nodule formation on the fibrosa of porcine valves cultured ex vivo (Richards et al. 2013) in addition to lower expression of anti-calcific genes by the fibrosa (Simmons et al. 2005), a difference in contraction had not been previously observed. This is similar with what is observed in vivo, with the fibrosa side of the valve demonstrating the majority of the calcification. Given previous findings illustrating the importance of CDH11 expression by AVICs in nodule formation (Hutcheson et al. 2013) and the predisposition for calcification on the fibrosa side of the leaflet (Simmons et al. 2005), these results are relevant in further understanding of the disease pathology of CAVD. Further study will be crucial in understanding to what extent CDH11 is directly involved in the contractile differences observed between the two different types of AVECs.
ACKNOWLEDGEMENTS
We thank Dr. Joseph Chen for his assistance in isolating the porcine cell lines.
SOURCES OF FUNDING
This work was supported by the National Science Foundation (DGE-1445197), American Heart Association (18PRE34070125), and by the National Institutes of Health (HL135790).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST STATEMENT
The authors have no conflicts of interest.
REFERENCES
- Bowen Caitlin J., Zhou Jingjing, Sung Derek C., and Butcher Jonathan T.. 2015. “Cadherin-11 Coordinates Cellular Migration and Extracellular Matrix Remodeling during Aortic Valve Maturation.” Developmental Biology 407(1):145–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowler Meghan A., Bersi Matthew R., Ryzhova Larisa M., Jerrell Rachel J., Parekh Aron, and Merryman W. David. 2018. “Cadherin-11 as a Regulator of Valve Myofibroblast Mechanobiology.” American Journal of Physiology-Heart and Circulatory Physiology 315(6):H1614–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher Jonathan T. and Nerem Robert M.. 2006. “Valvular Endothelial Cells Regulate the Phenotype of Interstitial Cells in Co-Culture: Effects of Steady Shear Stress.” Tissue Engineering 12(4):905–15. [DOI] [PubMed] [Google Scholar]
- Butcher Jonathan T., Tressel Sarah, Johnson Tiffany, Turner Debi, Sorescu George, Jo Hanjoong, and Nerem Robert M.. 2006. “Transcriptional Profiles of Valvular and Vascular Endothelial Cells Reveal Phenotypic Differences: Influence of Shear Stress.” Arteriosclerosis, Thrombosis, and Vascular Biology 26(1):69–77. [DOI] [PubMed] [Google Scholar]
- Cavallaro Ugo and Christofori Gerhard. 2004. “Cell Adhesion and Signalling by Cadherins and Ig-CAMs in Cancer.” Nature Reviews Cancer 4(2):118–32. [DOI] [PubMed] [Google Scholar]
- Chen Joseph, Ryzhova Larisa M., Sewell-Loftin MK, Brown Christopher B., Huppert Stacey S., Baldwin H. Scott, and Merryman W. David. 2015. “Notch1 Mutation Leads to Valvular Calcification through Enhanced Myofibroblast Mechanotransduction.” Arteriosclerosis, Thrombosis, and Vascular Biology 35(7):1597–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung Wing-Yee, Young Edmond W. K., and Simmons Craig A.. 2008. Techniques for Isolating and Purifying Porcine Aortic Valve Endothelial Cells. [PubMed]
- Clark Cynthia R., Bowler Meghan A., Snider J. Caleb, and Merryman W. David. 2017. “Targeting Cadherin-11 Prevents Notch1-Mediated Calcific Aortic Valve Disease.” Circulation 135(24):2448–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar Emily J., Huntley Geoffrey D., and Butcher Jonathan. 2015. “Endothelial-Derived Oxidative Stress Drives Myofibroblastic Activation and Calcification of the Aortic Valve.” PLoS ONE 10(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher Charles I., Chen Joseph, and Merryman W. David. 2013. “Calcific Nodule Morphogenesis by Heart Valve Interstitial Cells Is Strain Dependent.” Biomechanics and Modeling in Mechanobiology 12(1):5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould Russell A. and Butcher Jonathan T.. 2010. “Isolation of Valvular Endothelial Cells.” Journal of Visualized Experiments (46):e2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutcheson Joshua D., Chen Joseph, Sewell-Loftin MK, Ryzhova Larisa M., Fisher Charles I., Su Yan Ru, and Merryman W. David. 2013. “Cadherin-11 Regulates Cell-Cell Tension Necessary for Calcific Nodule Formation by Valvular Myofibroblasts.” Arteriosclerosis, Thrombosis, and Vascular Biology 33(1):114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sook Kyung Chang, Gu Zhizhan, and Brenner Michael B.. 2010. “Fibroblast-like Synoviocytes in Inflammatory Arthritis Pathology: The Emerging Role of Cadherin-11.” Immunological Reviews 233(1):256–66. [DOI] [PubMed] [Google Scholar]
- Lee David M., Kiener Hans P., Agarwal Sandeep K., Noss Erika H., Watts Gerald F. M., Chisaka Osamu, Takeichi Masatoshi, and Brenner Michael B.. 2007. “Cadherin-11 in Synovial Lining Formation and Pathology in Arthritis.” Science 315(5814):1006–10. [DOI] [PubMed] [Google Scholar]
- Mahler Gretchen J., Frendl Christopher M., Cao Qingfeng, and Butcher Jonathan T.. 2014. “Effects of Shear Stress Pattern and Magnitude on Mesenchymal Transformation and Invasion of Aortic Valve Endothelial Cells.” Biotechnology and Bioengineering 111(11):2326–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards Jennifer, El-Hamamsy Ismail, Chen Si, Sarang Zubair, Sarathchandra Padmini, Yacoub Magdi H., Chester Adrian H., and Butcher Jonathan T.. 2013. “Side-Specific Endothelial-Dependent Regulation of Aortic Valve Calcification: Interplay of Hemodynamics and Nitric Oxide Signaling.” American Journal of Pathology 182(5):1922–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider Daniel J., Wu Minghua, Le Thuy T., Cho Seo-Hee, Brenner Michael B., Blackburn Michael R., and Agarwal Sandeep K.. 2012. “Cadherin-11 Contributes to Pulmonary Fibrosis: Potential Role in TGF-β Production and Epithelial to Mesenchymal Transition.” The FASEB Journal 26(2):503–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons Craig A., Grant Gregory R., Manduchi Elisabetta, and Davies Peter F.. 2005. “Spatial Heterogeneity of Endothelial Phenotypes Correlates with Side-Specific Vulnerability to Calcification in Normal Porcine Aortic Valves.” Circulation Research 96(7):792–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung Derek C., Bowen Caitlin J., Vaidya Kiran A., Zhou Jingjing, Chapurin Nikita, Recknagel Andrew, Zhou Bin, Chen Jonathan, Kotlikoff Michael, and Butcher Jonathan T.. 2016. “Cadherin-11 Overexpression Induces Extracellular Matrix Remodeling and Calcification in Mature Aortic Valves.” Arteriosclerosis, Thrombosis, and Vascular Biology 36(8):1627–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Chong, Baker Brendon M., Chen Christopher S., and Schwartz Martin Alexander. 2013. “Endothelial Cell Sensing of Flow Direction.” Arteriosclerosis, Thrombosis, and Vascular Biology 33(9):2130–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Huan, Leinwand Leslie A., and Anseth Kristi S.. 2014. “Roles of Transforming Growth Factor-B1 and OB-Cadherin in Porcine Cardiac Valve Myofibroblast Differentiation.” FASEB Journal 28(10):4551–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Minghua, Pedroza Mesias, Lafyatis Robert, George Anuh Teresa, Mayes Maureen D., Assassi Shervin, Tan Filemon K., Brenner Michael B., and Agarwal Sandeep K.. 2014. “Identification of Cadherin 11 as a Mediator of Dermal Fibrosis and Possible Role in Systemic Sclerosis.” Arthritis and Rheumatology 66(4):1010–21. [DOI] [PMC free article] [PubMed] [Google Scholar]