

Abstract
Background:
Olfactory impairment is evident in Alzheimer’s disease (AD), however, its precise relationships with clinical biomarker measures of tau pathology and neuroinflammation are not well understood.
Objective:
To determine if odor identification performance measured with the University of Pennsylvania Smell Identification Test (UPSIT) is related to in vivo measures of tau pathology and neuroinflammation.
Methods:
Cognitively normal and cognitively impaired participants were selected from an established research cohort of adults aged 50 and older who underwent neuropsychological testing, brain MRI, and amyloid PET. Fifty-four participants were administered the UPSIT. Forty-one underwent 18F-MK-6240 PET (measuring tau pathology) and fifty-three underwent 11C-PBR28 PET (measuring TSPO, present in activated microglia). Twenty-three participants had lumbar puncture to measure CSF concentrations of total tau (t-tau), phosphorylated tau (p-tau) and β-amyloid (Aβ42).
Results:
Low UPSIT performance was associated with greater18F-MK-6240 binding in medial temporal cortex, hippocampus, middle/inferior temporal gyri, inferior parietal cortex and posterior cingulate cortex (p<0.05). Similar relationships were seen for 11C-PBR28. These relationships were primarily driven by amyloid-positive participants. Lower UPSIT performance was associated with greater CSF concentrations of t-tau and p-tau (p<0.05). Amyloid status and cognitive status exhibited independent effects on UPSIT performance (p<0.01).
Conclusions:
Olfactory identification deficits are related to extent of tau pathology and neuroinflammation, particularly in those with amyloid pathophysiology. The independent association of amyloid-positivity and cognitive impairment with odor identification suggests that low UPSIT performance may be a marker for AD pathophysiology in cognitive normal individuals, although impaired odor identification is associated with both AD and non-AD related neurodegeneration.
NCT Registration Numbers:
Keywords: Alzheimer disease, anosmia, tau proteins, olfaction, microglia
INTRODUCTION
Olfactory impairment is observed early in Alzheimer’s disease (AD) [1–4] and is thought to occur due to anatomical overlap of the regions involved in olfaction and early AD pathology. Olfactory bulb neurons project directly to limbic regions of the brain for olfactory processing [5, 6]. These regions, including the transentorhinal cortex and other medial temporal regions are known to be involved in early tau pathological changes of AD and correspond with Braak stages I- III [5–7]. Tau pathology in the olfactory bulb continues to increase with severity of AD, which provides a possible explanation for the progression of odor impairment that occurs with AD advancement [8].
Olfactory impairment observed in AD can be quantified with the University of Pennsylvania Smell Identification Test (UPSIT). UPSIT scores appear to correlate with measures of entorhinal cortex volume on MRI [9, 10]. Large community cohort studies have demonstrated that low UPSIT scores predict cognitive decline in cognitively normal elders and patients with mild cognitive impairment (MCI) [3, 11, 12]. These studies have also shown that UPSPIT performance is inversely related to performance on neuropsychological testing [12].
Several studies have investigated the relationships between UPSIT and in vivo measures of AD pathology, particularly amyloid. Some studies have demonstrated modest relationships between UPSIT performance and amyloid deposition on PET [9, 10]. Only one published study has examined the relationship between odor identification and PET measures of tau pathology, with results indicating that binding with the tau radioligand 18F-AV-1451 negatively correlated with UPSIT performance in cognitively normal adults, adults with subjective cognitive decline, and MCI patients [13]. However, that study did not include AD patients. One study evaluated the relationship between odor identification and CSF measures of tau pathology, demonstrating that low UPSIT performance was associated with elevated CSF tau [14].
Neuroinflammation is also associated with AD pathology and cognitive decline [15] and can be quantified using PET radioligands, such as 11C-PBR28, that bind the 18 kDa translocator protein (TSPO), a marker of immune activation. To our knowledge, no study has evaluated the relationship between odor identification and neuroinflammation.
We sought to determine the relationship between odor identification and neuroinflammation, measured by 11C-PBR28 PET. We further evaluated relationships between odor identification and tau pathology using PET imaging with 18F-MK-6240, a highly specific radioligand for phosphorylated tau, and CSF concentrations of total tau (t-tau) and phosphorylated tau (p-tau), and the relationship between odor identification and amyloid pathology using CSF concentrations of β-amyloid (Aβ42). We hypothesized that worse performance on odor identification testing would be associated with higher PET measures of neuroinflammation, and higher PET and CSF-biomarker measures of tau pathology, particularly in regions of early AD pathology, namely medial temporal lobe structures.
METHODS
Participant selection
Adults aged 50 years and older were recruited from Columbia University Irving Medical Center (CUIMC) Aging and Dementia clinic, the Columbia University Alzheimer’s Disease Research Center, other research cohorts at CUIMC or self-referral to establish the initial research cohort for a larger study (K23AG052633, PI = Kreisl). A subset of seventy-eight adults from the initial research cohort was considered for inclusion into this study. Study inclusion and exclusion criteria can be found in Supplementary Table 1.
All seventy-eight participants underwent an initial screening that included routine history and physical, neurological examination, routine laboratory tests, TSPO genotyping, neuropsychological evaluation and brain MRI. Screening measures were performed to exclude any participants with significant medical or psychiatric illness, cortical infarcts on brain MRI, and use of immunosuppressant medication.
TSPO genotyping of the rs6971 polymorphism was also performed. Participants homozygous for this polymorphism (low affinity binders) show negligible binding to 11C-PBR28 [16]. Those heterozygous for the polymorphism (mixed affinity binders) show reduced but reliable binding with 11C-PBR28; thus, TSPO genotype correction is required during statistical analysis to account for this heterogeneity in binding [17]. After screening, seventeen subjects were excluded from continuing participation in the study, including eight due to low affinity TSPO, three due to lab exclusions and six withdrawals (Supplementary Figure 1).
Neuropsychological evaluation including the Mini Mental State Examination, [18] Selective Reminding Test- Delayed Recall (SRT-DR), [19] Trail Making Test Parts A and B, and Category and Phonemic Fluency. These tests were selected to capture performance of specific cognitive domains, while the MMSE provided a global representation of cognition. The SRT-DR tested short-term memory, Trail Making Test Part A tested psychomotor functioning, Trail Making Test Part B tested executive functioning and Category and Phonemic Fluency tested language fluency. All cognitive test scores were transformed into z-scores using age-, sex- and education-adjusted normative data provided by the National Alzheimer’s Coordinating Center. All participants were assigned a Clinical Dementia Rating scale score (CDR) by a clinician based on history, examination, and neuropsychological test results. Only participants with a CDR score ≤1 (i.e. normal, mild cognitive impairment, or mild AD) were eligible, so this study could focus on pathological changes in early stages of AD, and to ensure that participants were able to complete study procedures.
Participants were defined as either cognitively normal or cognitively impaired based on history and cognitive examination. To qualify as cognitively impaired, participants had to have a primary memory complaint and meet clinical criteria for amnestic mild cognitive impairment (MCI) [20] or AD [21]. Participants who met clinical criteria for a non-AD neurodegenerative condition (e.g., dementia with Lewy bodies, vascular dementia, Parkinson’s disease, corticobasal degeneration, progressive supranuclear palsy, or frontotemporal dementia) were excluded. To qualify as cognitively normal, participants had to have no cognitive complaints and have absence of clinically significant cognitive impairment based on history and neuropsychological evaluation.
TSPO affinity determination
Blood samples were collected from all participants at the initial screening visit to utilize genomic DNA to genotype the rs6971 polymorphism using a TaqMan assay [16]. As mentioned under Participant Selection, eight participants from the original cohort (n = 78) were determined to be homozygous for the low affinity allele and were excluded from the remainder of the study (Supplementary Figure 1).
Amyloid PET Imaging
The sixty-one participants who met inclusion criteria after initial screening procedures had PET imaging with 18F-florbetaben (FBB) to determine amyloid status in a Siemens Biograph64 mCT/PET scanner at the CUIMC Kreitchman PET center (target dose: 8.1 mCi; 4x5 min frames), with a low-dose CT scan for attenuation correction. FBB images were acquired 50-70 minutes post-injection. All PET data were corrected for radioactive decay, attenuation of annihilation photons, scanner deadtime and normalization, and random and scatter events. Reconstructed FBB images were averaged to create a single static image for each participant. Amyloid status was determined by a binary visual read by an experienced neurologist (WCK), blinded to the participant diagnosis, according to established methods [22]. To validate the visual reads, we determined a SUVR cutoff of 1.27 for FBB as defined by the minimum among the visually amyloid-positive participants (Supplementary Figure 2). Using this cutoff, we found concordance in amyloid status determination between visual reads and use of SUVR in 58 of 61 participants (95.1%). The three discordant cases were then reviewed by a second trained and experienced reader (AJ), blind to diagnosis and the first reader’s interpretations, who agreed with the first reader on all thee visual interpretations. Therefore, we used the visual read results as the determinant for amyloid positivity or negativity. Studies have indicated that visual assessments perform similarly to SUVR cutoffs in interpreting amyloid status with FBB scans [23].
Odor Identification Test Administration and Scoring
The 40-item UPSIT was administered by a trained technician on the same day as either the 18F-MK-6240 or 11C-PBR28 scan. For each of the 40 items on the UPSIT, the participants were provided with an odorant embedded in a microcapsule that could be scratched and smelled. They were instructed to choose from four distinct answer choices. The test was scored between 0 (no odors correctly identified) and 40 (all odors correctly identified). Because there is a 25% chance of guessing each odorant correctly, scores of 10 or below are consistent with anosmia and therefore were excluded from the analysis.
UPSIT was completed in 55 participants who had FBB PET. Four participants reported history of anosmia and did not have UPSIT performed. Two were unable to complete UPSIT. One participant who scored a 10 on the UPSIT was therefore excluded from analysis, leaving 54 participants with useable UPSIT data. Other known factors contributing to hyposmia such as smoking and current upper respiratory infection were considered, however, no participants were smokers or experiencing upper respiratory symptoms at the time of testing.
MRI Acquisition and Processing
T1-weighted MRI scans (160 slice 1 mm resolution, 256 x 200 voxel count) were acquired for all participants on a 3T Phillips Achieva MRI machine at CUIMC. Using PMOD 3.8 (PMOD Technologies), the T1 MR images were segmented and normalized to standard space. The Hammers-N30R83-1MM atlas was used to define regions of interest (ROIs), which were then consolidated into 10 volume-weighted ROIs. These ROIs included prefrontal cortex (middle frontal gyrus, superior/inferior frontal gyrus, posterior orbital gyrus); middle and inferior temporal gyri (medial part of anterior temporal lobe, lateral parts of anterior temporal lobe and middle and inferior temporal gyri); superior temporal gyrus (anterior part of superior temporal gyrus, posterior part of superior temporal gyrus); medial temporal cortex (amygdala, parahippocampal gyrus); posterior cingulate cortex; superior parietal lobule; inferior parietal lobule; lingual gyrus; striatum (caudate nucleus and putamen); and cerebellum. ROI volumes were reverse-warped to the participant’s native MRI space and manually corrected, if required. Left and right hippocampi were manually drawn on the native MRI by blinded investigators and the weighted-average volume was used as an ROI distinct from the remainder of the medial temporal cortex (i.e., the PMOD-derived amygdala and parahippocampal gyrus). The volume of each ROI was divided by total intracranial volume to adjust for differences in brain size.
Tau and Neuroinflammation PET Imaging
Forty-one participants underwent 18F-MK-6240 PET imaging to measure tau pathology (target dose: 5 mCi; 6x5 min frames). 18F-MK-6240 images were acquired 80-100 min post injection. Fifty-three participants underwent 11C-PBR28 PET imaging to measure TSPO (target dose: 20 mCi; 6x5 min frames). 11C-PBR28 PET images were acquired 60-90 min post-injection. 18F-MK-6240 and 11C-PBR28 PET imaging were performed on the same scanner as the FBB scans. Because it is a relatively novel radioligand, PET imaging with 18F-MK-6240 was not available at the initiation of this study. Therefore, not all fifty-four participants who completed the UPSIT were able to undergo 18F-MK-6240 PET imaging.
PET Image Processing
18F-MK-6240 and 11C-PBR28 PET images underwent the same processing steps. Reconstructed images were realigned and then corrected for participant movement with SPM12 (Wellcome Centre for Human Neuroimaging). The PNEURO tool in PMOD 3.8 was then used to coregister PET images into native MRI space and to perform correction for partial volume effects with the region-based voxelwise method [24]. The dynamic frames were then averaged to a single static image and the native MRI space ROIs defined above were applied to the averaged PET image. The concentration of radioactivity of each ROI was divided by the concentration of radioactivity of a reference region to generate standardized uptake value ratios (SUVRs). For 18F-MK-6240, inferior cerebellar gray matter was used as a reference region to avoid spill-over into the anterior lobe of the cerebellum from ventral temporal and occipital cortex [25]. For 11C-PBR28, the entire cerebellar gray matter was used as a “pseudo-reference” region, as previously validated [23, 26]. For 18F-MK-6240 and 11C-PBR28, both partial volume-corrected and uncorrected SUVRs were calculated.
CSF analysis
Lumbar puncture was optional for study participants. Twenty-three participants who had UPSIT also agreed to lumbar puncture and had CSF collected to measure concentrations of t-tau, p-tau (phosphorylated at threonine 181) and Aβ42.
Up to 15 cc of CSF was removed using a Sprotte 24G spinal needle, and placed in two 12 cc polypropylene tubes. All samples were centrifuged briefly, aliquoted using polypropylene pipettes within 30 minutes, and stored at −80°C. T-tau, p-tau (181), and Aβ42 concentrations were measured using the micro-bead-based multiplex immunoassay, the INNO-BIA AlzBio3 kit (Fujirebio, Ghent, Belgium), on the Luminex platform [27].
Statistical Analysis
Study participants were grouped based on amyloid status and CDR score into four groups: amyloid-negative controls (CDR = 0), amyloid-positive controls (CDR = 0), amyloid-positive patients (CDR = 0.5-1) and amyloid-negative patients (CDR = 0.5-1). For key characteristics, mean and standard deviations for continuous variables and frequencies for categorical variables were presented by each group. For continuous variables, the difference between groups was compared using analysis of variance (ANOVA) followed by post-hoc pairwise group difference tests, uncorrected for multiple comparisons. To assess the effect of amyloid status and cognitive status on UPSIT performance, we performed a 2-way ANOVA with factors amyloid status and cognitive status, controlling for age and sex. Partial eta squared () were calculated as effect size measures. Categorical demographic variables (e.g., sex, TSPO genotype) were tested for group differences with Chi-squared tests.
Partial correlation analyses evaluated the association between UPSIT total score and 11C-PBR28 binding, 18F-MK-6240 binding, CSF biomarkers, MMSE scores, and SRT-DR scores, covarying for age and sex (and TSPO genotype when applicable). The same partial correlation analyses were performed by amyloid status group (positive and negative) separately. For 11C-PBR28 binding and 18F-MK-6240 binding, partial correlation coefficients (rp) were computed in each ROI. The p-values of whole group association regarding 11C-PBR28 binding, 18F-MK-6240 binding, and CSF biomarkers were corrected for multiple comparisons controlling for false discovery rate using the Benjamini and Hochberg method [28]. Uncorrected p-values are also reported.
To assess the contributions of hippocampal volume, global amyloid burden, ROI-specific tau burden and ROI-specific neuroinflammatory burden to UPSIT performance, linear regression models were performed for all 11 ROIs and standardized coefficients were obtained as a measure of association. To consider how amyloidosis may modify the effect of tau and neuroinflammation on UPSIT, interaction terms between amyloid status and ROI-specific PET values were included in regression models. Effect sizes were calculated and reported as Cohen’s f 2 scores. All of the regression models controlled for age, sex, and TSPO genotype.
All statistical analyses were performed in R, version 3.6.0. Graphs were generated using GraphPad Prism 8. For visualization, residuals were calculated by regressing each variable on age and sex (and TSPO genotype when applicable).
Standard protocol approvals, registrations, and patient consents
This study was approved by the Columbia University Irving Medical Center Institutional Review Board. All participants (or their representative) provided informed consent according to the Declaration of Helsinki for participation in the study and for their health information to be used for research purposes.
Data availability
Anonymized data will be made available upon reasonable request to qualified investigators.
RESULTS
Participant Demographics
Fifty-four participants completed screening procedures and 18F-FBB PET scan and had UPSIT performed (23 amyloid-positive patients, 9 amyloid-negative patients, 6 amyloid-positive controls, 16 amyloid-negative controls) (Table 1). Forty-one of these participants (16 amyloid-positive patients, 8 amyloid-negative patients, 6 amyloid-positive controls, 11 amyloid-negative controls) underwent 18F-MK-6240 PET scan. Fifty-three of these participants (22 amyloid-positive patients, 9 amyloid-negative patients, 6 amyloid-positive controls, 16 amyloid-negative controls) underwent 11C-PBR28 PET scan. Twenty-three of these participants also underwent lumbar puncture (12 amyloid-positive patients, 3 amyloid-negative patients, 4 amyloid-positive controls, 4 amyloid-negative controls).
Table 1.
Descriptive data for participant demographics based on amyloid and cognitive statusa
| Aβ (+) patients (n = 23) | Aβ (+) controls (n = 6) | Aβ (−) patients (n = 9) | Aβ (−) controls (n = 16) | F-statistic (continuous) / Chi-Squared (categorical) | P-value | |
|---|---|---|---|---|---|---|
| Age (years)b | 64.7 ± 8.6 | 71.3 ± 4.6 | 74.0 ± 8.0 | 67.8 ± 3.8 | 4.27 | 0.01 |
| Male/Female | 19/4 | 3/3 | 6/3 | 5/11 | 10.90 | 0.01 |
| Education (years) | 16.9 ± 2.4 | 15.0 ± 2.8 | 16.8 ± 3.5 | 15.8 ± 2.8 | 1.11 | 0.35 |
| MMSE scoreb, c, d | 23.6 ± 4.2 | 28.7 ± 2.1 | 27.6 ± 2.3 | 29.4 ± 0.8 | 13.15 | 0.02 |
| SRT-DR (z-score)c, d, e, f | −3.26 ± 0.65 | +0.26 ± 1.30 | −2.55 ± 0.69 | 0.74 ± 1.05 | 76.92 | <0.0001 |
| TSPO genotype (HAB/MAB) | 12/11 | 2/4 | 5/4 | 11/5 | 2.43 | 0.49 |
| % Hippocampal Volumec,f | 0.87 ± 0.16 | 1.00 ± 0.16 | 0.84 ± 0.18 | 1.05 ± 0.14 | 5.61 | <0.001 |
Thirteen participants did not undergo 18F-MK-6240 PET and 1 participant did not undergo 11C-PBR28 PET
Significant difference between Aβ (+) patients and Aβ (−) patients (p < 0.05)
Significant difference between Aβ (+) patients and Aβ (−) controls (p < 0.05)
Significant difference between Aβ (+) patients and Aβ (+) controls (p < 0.05)
Significant difference between Aβ (−) patients and Aβ (+) controls (p < 0.05)
Significant difference between Aβ (−) patients and Aβ (−) controls (p < 0.05)
Key: HAB, high affinity binder; MAB, mixed affinity binder; MMSE, Mini-Mental Status Examination; SRT-DR, Selective Reminding Test-Delayed Recall; TSPO, 18 kDa translocator protein.
Among all included participants who had UPSIT, amyloid-positive patients and amyloid-negative controls were younger than amyloid-positive controls and amyloid-negative patients (p < 0.01). We found no difference in years of education among participant groups. Amyloid-positive patients had lower MMSE scores than amyloid-negative controls, amyloid negative patients and amyloid positive controls (p’s < 0.01). Both amyloid-positive patients and amyloid-negative patients had smaller hippocampal volume, lower SRT-DR scores, and lower MMSE scores than the control groups (p < 0.01), suggesting that the impaired participants had hippocampal atrophy even when amyloid pathology was absent. There were more men in the amyloid-positive and amyloid-negative patient groups and more women among the amyloid-negative controls (χ2 (3, N = 54) = 10.9, p = 0.012), so statistical analysis accounted for sex as a co-variate.
UPSIT performance across study groups
We tested whether amyloid status and cognitive status were independently associated with UPSIT performance. We found that amyloid positivity (F1, 48= 9.15, p = 0.004) and cognitive impairment (F1, 48 = 8.66, p = 0.005) were each negatively associated with UPSIT score. These associations remained after controlling for age and sex. However, we found no interaction between amyloid status and cognitive status (F1, 47 = 0.776, p = 0.383). The of amyloid status and cognitive status were 0.103 and 0.163, respectively.
Amyloid-positive patients had lower UPSIT scores than amyloid-negative controls (p < 0.01, Fig 1). UPSIT performance of amyloid-negative patients did not differ significantly from amyloid-negative controls (p = 0.13) or amyloid-positive patients (p = 0.97). The of participant groups was 0.330.
Figure 1. UPSIT performance across study groups.
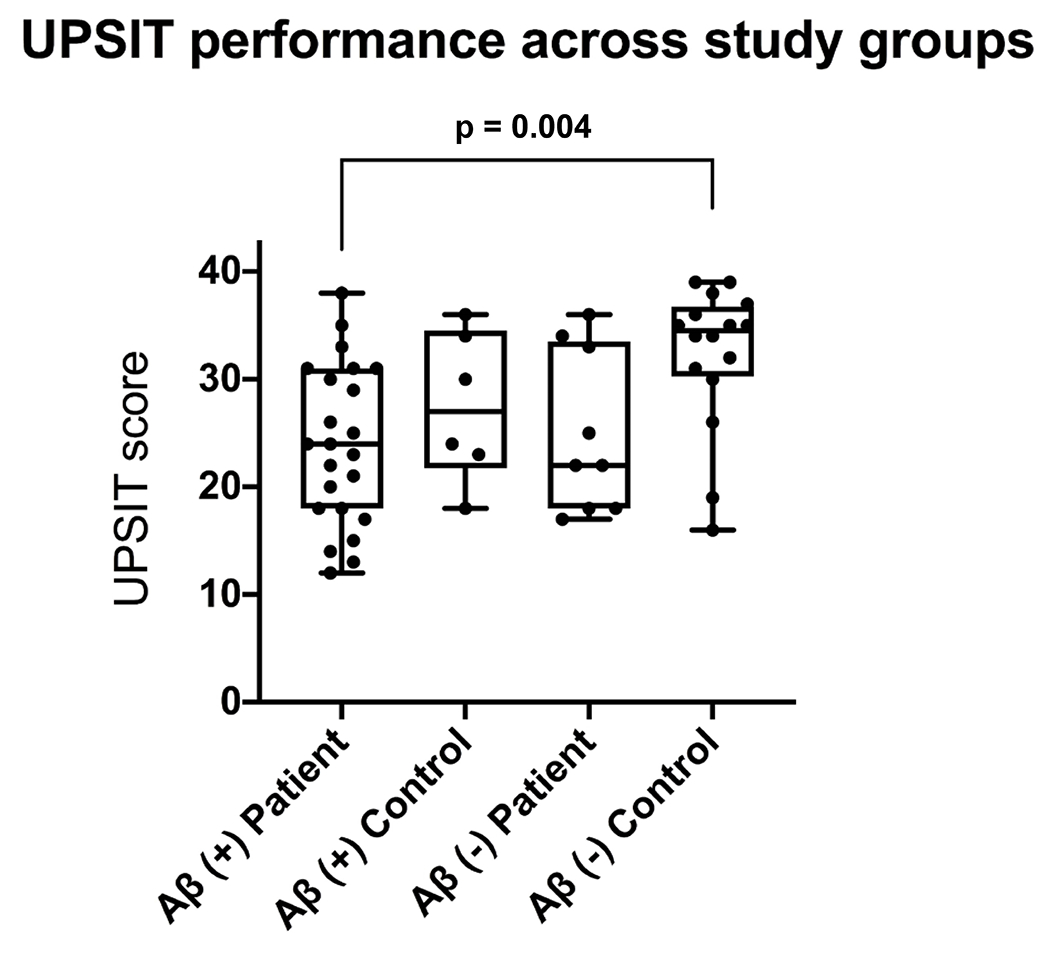
UPSIT scores across all four study groups. UPSIT scores were lower in amyloid-positive patients than amyloid-negative controls.
UPSIT performance and 18F-MK-6240 binding
For participants who underwent 18F-MK-6240 PET imaging (n= 41), we performed a partial correlation analysis between 18F-MK-6240 binding and UPSIT performance, correcting for age and sex. Using the partial volume corrected SUVR data, we found that 18F-MK-6240 binding was negatively associated with UPSIT performance in all ROIs except lingual gyrus when all participants were combined (r’s > −0.35, p’s < 0.05) (Fig 2, Table 2). Correlations between UPSIT performance and 18F-MK-6240 binding in all ROIs except for the lingual gyrus remained significant after multiple comparison correction (Table 2). When we stratified participants based on amyloid status, this significant negative partial correlation remained for amyloid-positive participants in medial temporal cortex (rp = −0.51, p = 0.02) and hippocampus (rp = −.53, p = 0.02). 18F-MK-6240 binding did not correlate with UPSIT performance in any regions when only amyloid-negative participants were included, not even at trend level (e.g., medial temporal cortex (rp = −0.27, p = 0.31), hippocampus (rp = −.17, p = 0.54)). Results from correlation analysis using PET data uncorrected for partial volume effects showed similar results (Supplementary Table 2).
Figure 2. Relationship between UPSIT score and 18F-MK-620 PET.
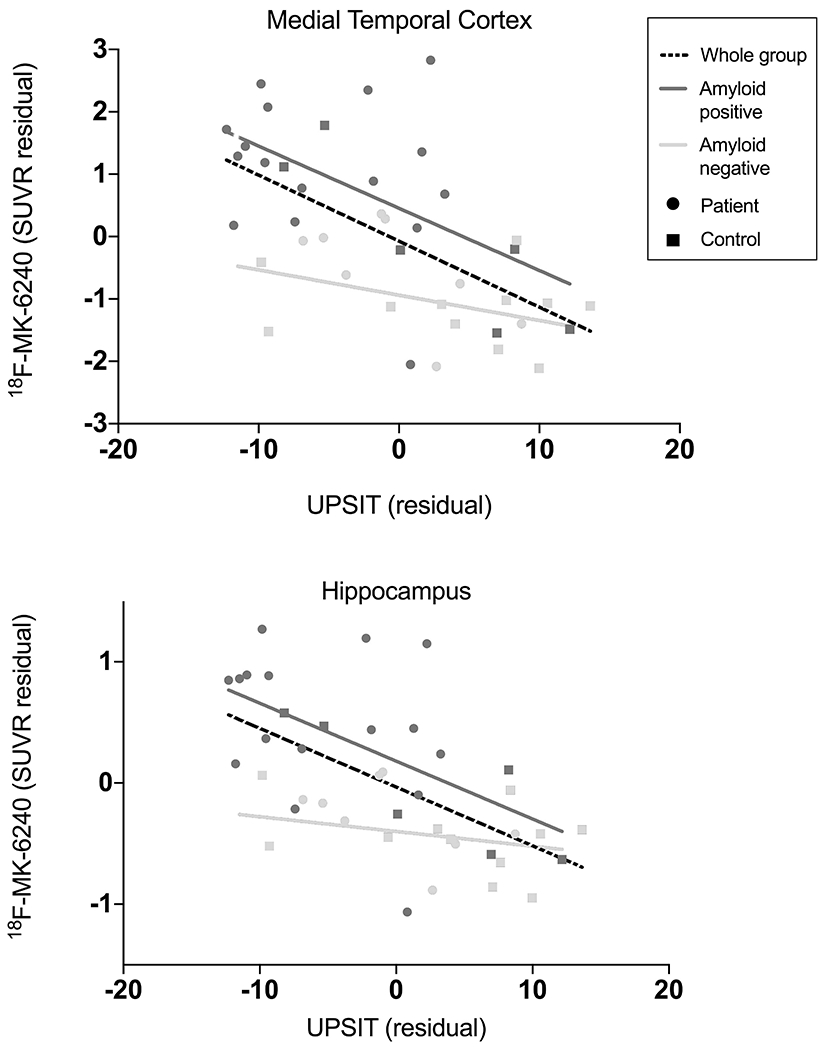
Lower UPSIT scores were associated with greater 18F-MK-6240 binding when all participants included in medial temporal cortex (r = −0.59, p < 0.01) and hippocampus (r = −0.60, p < 0.01). Correlations remained when only amyloid-positive participants were included (medial temporal cortex: r = −0.52, p = 0.02; hippocampus: r = −0.53, p = 0.02) but not when only amyloid-negative participants were included. Data corrected for age and sex.
Table 2.
Correlation analysis between UPSIT and partial volume corrected 18F-MK-6240 binding.
| All participants (n = 41) | Aβ (+) participants (n = 22) | Aβ (−) participants (n = 19) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Region of Interest | r | 95% CI | P-value | r | 95% CI | P-value | r | 95% CI | P-value |
| Pre-frontal Cortex | −0.44* | −0.66– −0.16 | 0.005 | −0.37 | −0.69– 0.06 | 0.12 | −0.32 | −0.68– 0.16 | 0.23 |
| Middle and Inferior Temporal Gyri | −0.48* | −0.69– −0.20 | 0.002 | −0.38 | −0.69– 0.05 | 0.11 | −0.24 | −0.63– 0.24 | 0.37 |
| Superior Temporal Cortex | −0.43* | −0.65– −0.14 | 0.006 | −0.32 | −0.65– 0.12 | 0.19 | −0.18 | −0.59– 0.30 | 0.51 |
| Medial Temporal Cortex Composite | −0.59* | −0.76– −0.35 | <0.001 | −0.52 | −0.77– −0.12 | 0.02 | −0.25 | −0.63– 0.23 | 0.36 |
| Posterior Cingulate Cortex | −0.35* | −0.60– −0.05 | 0.03 | −0.14 | −0.53– 0.30 | 0.56 | −0.14 | −0.56– 0.34 | 0.60 |
| Superior Parietal Cortex | −0.36* | −0.60– −0.06 | 0.02 | −0.18 | −0.56– 0.26 | 0.46 | −0.04 | −0.48– 0.42 | 0.89 |
| Inferior Parietal Cortex | −0.39* | −0.62– −0.09 | 0.02 | −0.20 | −0.57– 0.25 | 0.42 | −0.11 | −0.54– 0.37 | 0.69 |
| Striatum | −0.35* | −0.60– −0.05 | 0.03 | −0.08 | −0.48– 0.36 | 0.76 | −0.29 | −0.66– 0.18 | 0.27 |
| Hippocampus | −0.60* | −0.76– −0.36 | <0.001 | −0.53 | −0.78– −0.14 | 0.02 | −0.27 | −0.65– 0.21 | 0.31 |
| Lingual Gyrus | −0.21 | −0.49– 0.10 | 0.20 | 0.02 | −0.40– 0.44 | 0.92 | −0.02 | −0.47– 0.44 | 0.94 |
Survived multiple comparison correction.
UPSIT performance and 11C-PBR28 binding
For participants who underwent 11C-PBR28 PET imaging (n= 53), we performed a partial correlation analysis between 11C-PBR28 binding and UPSIT performance, correcting for age, sex and TSPO genotype. Using the partial volume corrected SUVR data, we found that 11C-PBR28 binding was negatively associated with UPSIT performance in the middle and inferior temporal gyri, medial temporal cortex, posterior cingulate cortex, inferior parietal cortex and hippocampus when all participants were combined (rp’s > −0.29, p’s < 0.05, Fig 3, Table 3). Correlations between UPSIT performance and 11C-PBR28 binding in the medial temporal cortex, posterior cingulate cortex, hippocampus and middle and inferior temporal gyri remained significant after multiple comparison correction (Table 3). When we stratified participants based on amyloid status, this negative partial correlation remained for amyloid-positive participants in the medial temporal cortex (rp = −0.74, p < 0.001) and the middle and inferior temporal gyri (rp = −0.48, p = 0.02). When only amyloid-negative participants were included, 11C-PBR28 binding did not correlate with UPSIT performance in any region except at trend level for the hippocampus (rp = −0.38, p = 0.08). Results from partial correlation analysis using imaging data uncorrected for partial volume effects showed similar results (Supplementary Table 3).
Figure 3. Relationship between UPSIT score and 11C-PBR28 PET.
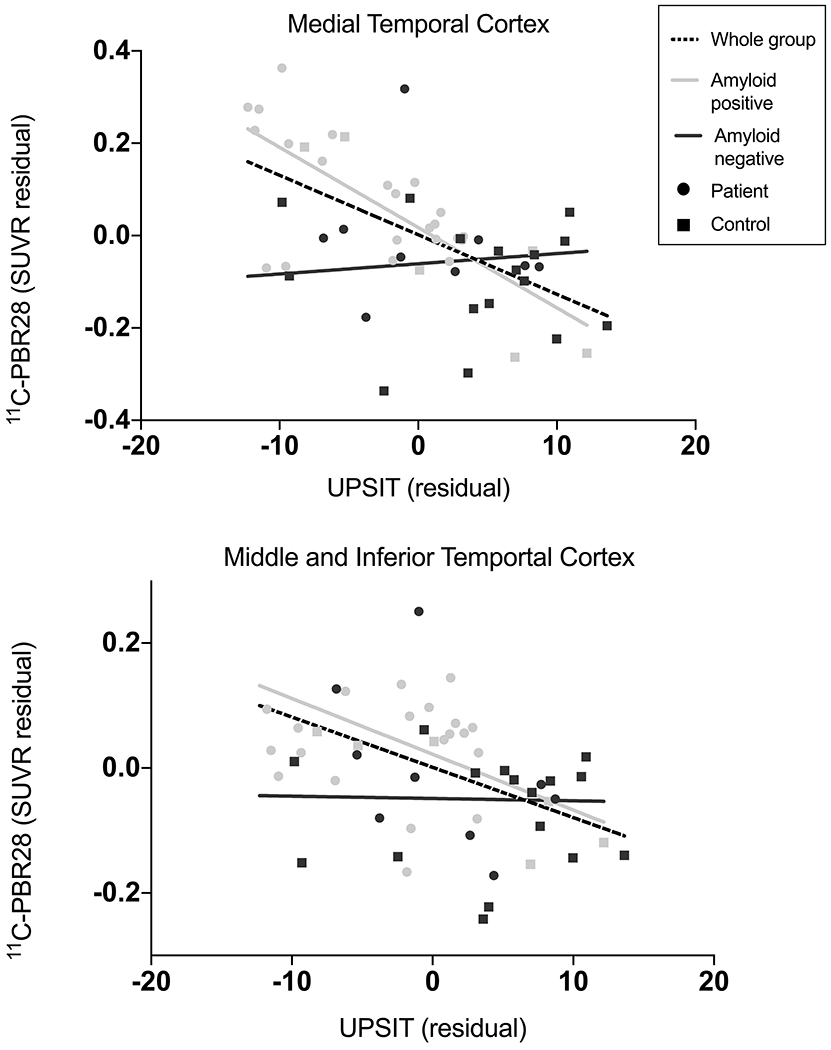
Lower UPSIT scores were associated with greater 11C-PBR28 binding when all participants were included in medial temporal cortex (r = −0.58, p < 0.01) and combined middle and inferior temporal gyri (r = −0.47, p < 0.01). Correlations remained when only amyloid-positive participants were included (medial temporal cortex: r = −0.74, p < 0.01; combined middle and inferior temporal gyri: r = −0.47, p = .02) but not when only amyloid-negative participants were included. Data corrected for age, sex, and TSPO genotype.
Table 3.
Correlation analysis between UPSIT and partial volume corrected 11C-PBR28 binding.
| All participants (n = 53) | Aβ (+) participants (n = 28) | Aβ (−) participants (n = 25) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Region of Interest | r | 95% CI | P-value | r | 95% CI | P-value | r | 95% CI | P-value |
| Pre-frontal Cortex | −0.22 | −0.46– 0.05 | 0.12 | −0.19 | −0.53– 0.20 | 0.36 | 0.12 | −0.28– 0.50 | 0.58 |
| Middle and Inferior Temporal Gyri | −0.47* | −0.66– −0.23 | <0.001 | −0.47 | −0.72– −0.12 | 0.02 | −0.09 | −0.47– 0.031 | 0.68 |
| Superior Temporal Cortex | −0.20 | −0.45– 0.07 | 0.16 | −0.15 | −0.50– 0.24 | 0.48 | 0.16 | −0.25– 0.52 | 0.49 |
| Medial Temporal Cortex Composite | −0.58* | −0.74– −0.37 | <0.001 | −0.74 | −0.87– −0.50 | <0.001 | −0.06 | −0.45– 0.34 | 0.77 |
| Posterior Cingulate Cortex | −0.34* | −0.56– −0.08 | 0.01 | −0.15 | −0.49– 0.24 | 0.48 | −0.18 | −0.54– 0.23 | 0.43 |
| Superior Parietal Cortex | −0.25 | −0.49– 0.02 | 0.08 | 0.00 | −0.34– 0.38 | 0.98 | 0.10 | −0.31– 0.47 | 0.67 |
| Inferior Parietal Cortex | −0.29 | −0.52– −0.02 | 0.04 | −0.15 | −0.50– 0.23 | 0.46 | 0.10 | −0.31– 0.47 | 0.67 |
| Striatum | −0.09 | −0.36– 0.18 | 0.51 | −0.07 | −0.44– 0.31 | 0.72 | −0.15 | −0.51– 0.26 | 0.50 |
| Hippocampus | −0.35* | −0.57– −0.08 | 0.01 | −0.31 | −0.61– 0.08 | 0.15 | −0.38 | −0.67– 0.02 | 0.08 |
| Lingual Gyrus | −0.20 | −0.45– 0.07 | 0.16 | −0.02 | −0.39– 0.36 | 0.94 | 0.06 | −0.34– 0.44 | 0.79 |
Survived multiple comparison correction
UPSIT performance and CSF biomarkers
For participants who underwent lumbar puncture (n=23), we performed a partial correlation analysis between CSF biomarkers burden and UPSIT performance. We found that UPSIT performance was negatively associated with CSF concentrations of t-tau (rp = −0.52, p = 0.02) and p-tau (rp = −0.53, p = 0.012) when all participants were combined (Fig 4, Table 4). We did not observe a significant negative association between UPSIT performance and CSF concentrations of Aβ42 (rp = −0.12, p = 0.60). We did not observe significant associations between UPSIT performance and CSF t-tau: Aβ42 ratios (rp = −0.32, p = 0.17) or between UPSIT performance and p-tau (181): Aβ42 ratios (rp = −0.38, p = 0.10). Correlations between UPSIT performance and CSF measures of t-tau and p-tau remained significant after multiple comparison correction (Table 4). Due to the smaller sample size of participants who underwent lumbar puncture, we did not stratify participants based on amyloid status for subgroup evaluation.
Figure 4. Relationship between UPSIT score and CSF concentrations of total tau and phosphorylated tau.
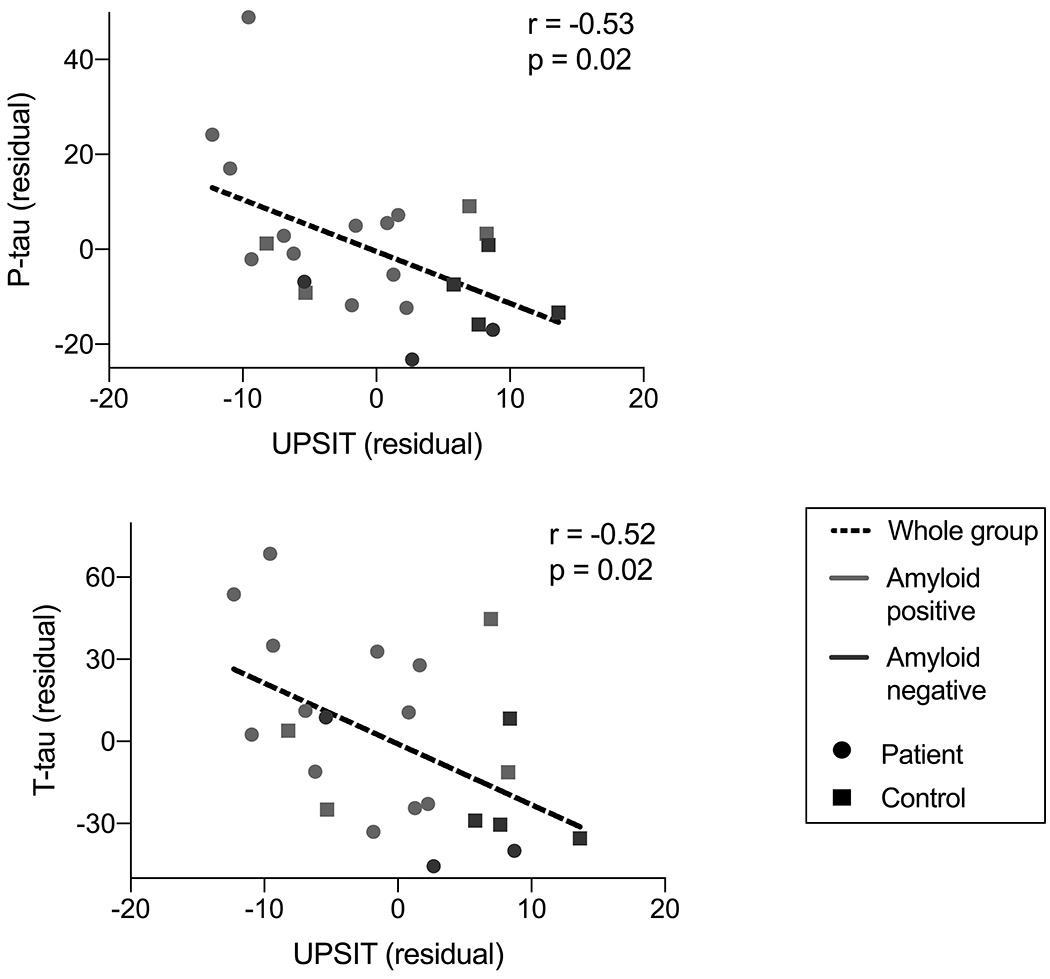
Lower UPSIT scores were associated with greater CSF concentrations of phosphorylated tau (p-tau, r = −0.53, p = .02) and total tau (t-tau, r = −0.52, p = .02), after controlling for age and sex.
Table 4.
Correlation analysis between UPSIT and CSF measures
| CSF Biomarker | r (whole Group) | 95% CI | P-value |
|---|---|---|---|
| T-tau | −0.52 | −0.77– −0.14 | 0.02* |
| P-tau | −0.53 | −0.78– −0.16 | 0.02* |
| Aβ42 | 0.14 | −0.30– 0.51 | 0.60 |
| T-tau: Aβ42 | −0.32 | −0.65- 0.11 | 0.17 |
| P-tau: Aβ42 | −0.38 | −0.69- 0.04 | 0.10 |
Survived multiple comparison correction
UPSIT performance, hippocampal volume, and cognition
Performance on the UPSIT positively correlated with hippocampal volume, such that lower UPSIT scores were associated with smaller hippocampal volumes, when all participants were included (rp = 0.53, p < 0.001) and when only amyloid-positive participants were included (rp = 0.69, p < 0.001, Fig 5A). UPSIT performance positively correlated with MMSE scores (rp = 0.42, p < 0.001) and z-scores for performance on the SRT-DR (rp = 0.65, p < 0.001), such that lower UPSIT scores were associated with worse cognitive performance, when all participants were included (Fig 5B). The partial correlation between UPSIT and SRT-DR performance remained significant when only amyloid-positive participants were included (rp = 0.68, p < 0.001, Fig 5C).
Figure 5. Relationship among UPSIT score and hippocampal volume, Mini Mental State Exam (MMSE) score and Selective Reminding Test – Delayed Recall (SRT-DR) score.
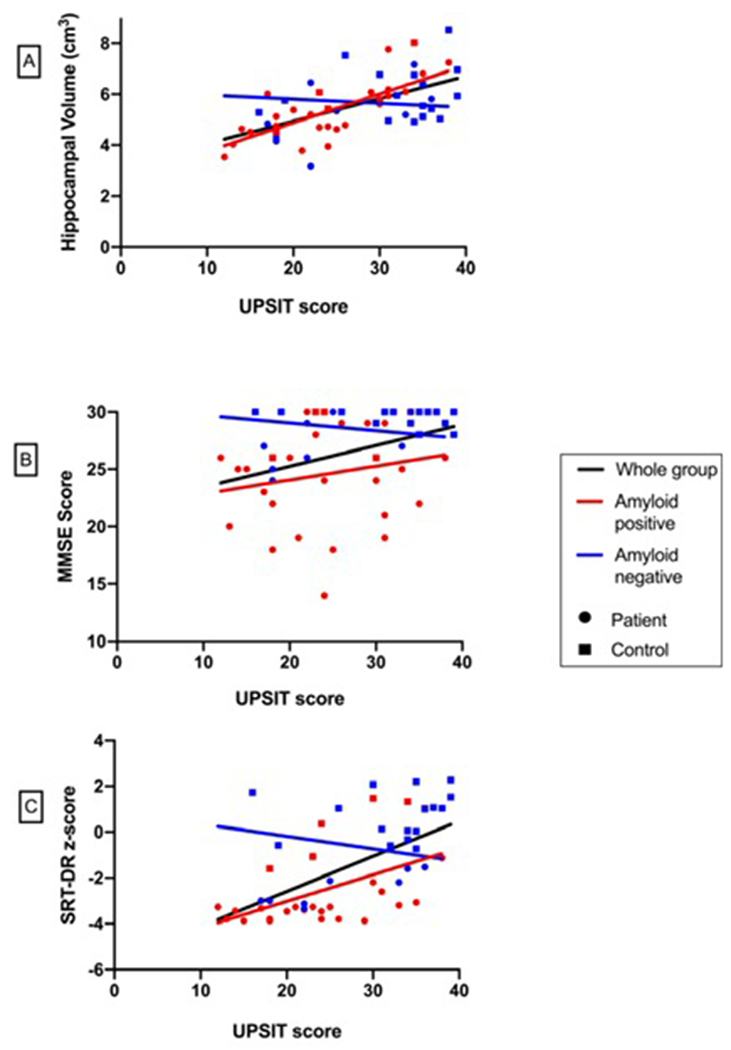
Positive correlations were observed between UPSIT performance and (A) hippocampal volume (r = 0.53, p< 0.001), (B) MMSE (r = 0.42, p< 0.001) and (C) SRT-DR performance (r = 0.65, p< 0.001) when all participants were included. Positive correlations between UPSIT performance and hippocampal volume (r= 0.69, p< 0.001) and UPSIT and SRT-DR performance (r = 0.68, p< 0.001) remained when only amyloid-positive participants were included.
Linear Regression Models of UPSIT Performance
Across the linear regression models to determine whether global amyloid burden, tau burden, neuroinflammatory burden or hippocampal volume exhibited the greatest association with UPSIT, hippocampal volume consistently demonstrated the strongest relationship with UPSIT in all 11 ROIs (p’s < 0.0001). Additionally, ROI-specific neuroinflammatory burden measured by 11C-PBR28 PET exhibited significant associations with UPSIT in the medial temporal cortex, hippocampus and middle/inferior temporal gyri, using both partial volume corrected and uncorrected data (p’s < 0.05) (Supplementary Table 4).
In the regression models considering effect modification by amyloidosis on 18F-MK-6240 binding and 11C-PBR28 binding, none of the interaction terms were significant. We observed small to medium effect sizes in the medial temporal lobe for 11C-PBR28 binding (f 2= 0.042) and 18F-MK-6240 binding (f 2= 0.025).
DISCUSSION
We demonstrated that olfactory identification is negatively associated with progression along the Alzheimer’s disease clinical continuum, such that amyloid-positive patients had lower UPSIT scores than amyloid-negative controls, and that UPSIT score positively correlated with cognitive performance and hippocampal volume. We also found that UPSIT score negatively correlated with PET and CSF measures of tau pathology and neuroinflammation. Taken together, these results suggest that odor identification worsens with AD progression in a manner that may be related to both tau and neuroinflammatory burden.
When we considered the amyloid-positive group separately, we found inverse relationships between olfactory identification ability and both tau pathology and neuroinflammation in medial temporal regions (hippocampus and the combined amygdala/parahippocampal gyrus). Our results suggest that decreased ability to identify odors may reflect the burden of tau-mediated neurodegeneration in these regions, which are among the first to show tau pathology and correspond to Braak stages I-III [5–7]. This topographical specificity is notable because these regions, which are affected early in AD, receive afferent input from primary neurons originating in the olfactory bulb [5].
Our results build on early findings demonstrating relationships between UPSIT performance and AD. In one sample of cognitively normal adults, poorer performance on the 12-item Brief Smell Identification (B-SIT) was associated with a 50% increased risk of developing MCI over the following five years and exhibited predictive value for developing dementia [12]. Lower UPSIT scores are associated with smaller hippocampal and entorhinal volumes in cognitively normal elders, particularly in those with high amyloid burden on PET [9, 10]. Another PET study found that lower odor identification scores in a shorter version of the UPSIT were modestly associated with greater neocortical amyloid binding in cognitively normal, MCI and AD patients when combined, but not when MCI participants were considered independently, suggesting that olfactory impairment is not directly related to amyloid burden alone [29]. Additionally, UPSIT performance prior to death predicted neurofibrillary tangle burden in the CA1/subiculum of the hippocampus in AD patients [30]. Our finding of a negative relationship between UPSIT score and CSF concentrations of tau, but not Aβ42 is in agreement with prior studies showing that low performance on the B-SIT and UPSIT have been associated with increased CSF tau but not with measurement of CSF Aβ42 [14, 31]. These results suggest that UPSIT may provide more insight into burden of tau pathology in early AD than amyloid pathology. While a prior study showed associations between low UPSIT score and increased ratios of CSF t-tau and p-tau (181) to Aβ42 we did not observe a relationship between odor identification and these CSF ratio measurements in our sample [31]. This discrepancy may be due to lower sample size in our study; however, since we saw no association between UPSIT and CSF Aβ42 alone, the Aβ42 concentrations may have introduced variance into our t-tau: Aβ42 and p-tau: Aβ42 ratios, explaining why UPSIT correlated with t-tau and p-tau alone but not with the ratio values.
To our knowledge, the only prior study comparing odor identification and tau pathology in vivo using PET imaging used flortaucipir (18F-AV-1451) and likewise found an inverse relationship between UPSIT score and tau binding in temporal and parietal cortices in cognitively normal adults and patients with subjective cognitive decline. In our study, we extended these results to include clinically affected AD patients and patients who are amyloid-negative but exhibit evidence of hippocampal neurodegeneration and AD patterns of cognitive impairment. In addition, we used 18F-MK-6240, an improved tau radioligand with less off-target binding in basal ganglia and choroid plexus than 18F- AV-1451, and confirmed our imaging findings by demonstrating correlations between UPSIT score and CSF concentrations of t-tau and p-tau [25, 32].
We also showed that there is a strong relationship between UPSIT performance and PET measures of neuroinflammation. While the staging of neuroinflammation is poorly understood, one meta-analysis including results from a range of TSPO radioligands, including 11C-PBR28, reported that the difference in microglial activation measured on PET imaging between AD patients and healthy controls existed in several cortical areas, but was greatest in the middle and inferior temporal gyri and the parahippocampal gyrus [31]. The same relationships were observed in MCI, albeit more modestly. Interestingly, these regions were among those that exhibited the strongest inverse relationships with UPSIT performance in our present study, suggesting that these regions experience early neuroinflammation in AD. Further, the inverse relationships between UPSIT performance and PET measures of neuroinflammation were observed in nearly identical brain regions as PET measures of tau pathology. These results support the possibility of a topographical overlap between neuroinflammation and tau deposition in early neurodegeneration, and align with results of prior PET studies showing colocalization of TSPO and tau binding, which is largely driven by amyloid-positive individuals [33, 34]. These findings raise the question of whether tau and inflammation are independent processes in AD pathogenesis. In our earlier study, we showed that, among amyloid-positive participants, earliest increases in tau pathology were found in medial temporal regions, while increases in TSPO were first found in neocortical regions [33, 34]. Therefore, even though neuroinflammation and tau pathology are likely related to each another, they may have distinct spatial patterns early in the AD continuum and may therefore independently contribute to olfactory impairment. Longitudinal studies in cognitively normal older adults could help identify the downstream effects of these distinct early spatial patterns of pathology, and clarify the temporal relationships between olfactory impairment and pathological changes in amyloid, tau, and neuroinflammation.
Our current study selected a subset of participants from a pre-established research cohort. In a prior study of the larger cohort, both 18F-MK-6240 and 11C-PBR28 binding were greater in amyloid-positive than in amyloid-negative participants, specifically in neocortical regions for 11C-PBR28 and in the medial temporal lobe for 18F-MK-6240 [34]. In our sub-sample, we observed similar regional patterns of increased 11C-PBR28 and 18F-MK-6240 binding in association with lower UPSIT scores, suggesting that odor identification impairment may be mechanistically linked to inflammation and tau pathology, and not just a nonspecific measure of neurodegeneration.
Our linear regression models demonstrated that hippocampal volume showed the strongest association with UPSIT when accounting for global amyloid burden, ROI-specific tau burden and ROI-specific neuroinflammatory burden. Neuroinflammatory burden in the medial temporal cortex, hippocampus and middle/inferior temporal gyri also exhibited associations with UPSIT when accounting for other variables. Although we are unable to determine causation, if any, in this cross-sectional model, it is worth noting these relationships. The largest estimates across models for neuroinflammatory burden were in ROIs that exhibited the strongest partial correlations with UPSIT performance, namely Braak I-III regions.
We also found that amyloid status and cognitive status are independently associated with UPSIT performance. That amyloid-positivity is associated with lower UPSIT score is consistent with prior studies showing that lower performance on odor identification predicts decline in cognitively normal elderly and UPSIT scores correlate with amyloid deposition on PET [35, 36]. Therefore, UPSIT may be useful as a selection tool to identify cognitively normal elders more likely to be amyloid-positive for preventative clinical trials. That impaired cognition is associated with lower UPSIT score independent of amyloid status is not surprising, given that impaired odor identification has also been reported in amyloid negative dementias, or non-AD dementias such as dementia with Lewy bodies, Huntington’s disease, and frontotemporal dementia [37]. Notably, amyloid-negative patients did not have lower UPSIT scores than amyloid-negative controls, although this may relate to our modest sample size. That amyloid-positive patients had the lowest UPSIT scores in our cohort may reflect greater overall pathology in this group than in the amyloid-negative participants. We don’t have histopathological confirmation in the amyloid-negative patients; however, given the overall small hippocampal volumes and AD-like patterns of impairment in this group, they may represent hippocampal sclerosis/TDP-43 pathology, argyrophilic grain disease, or other AD mimics that may have more indolent clinical trajectories than patients with biomarker evidence of AD pathophysiology [38]. Importantly, none of the amyloid-negative patients had clinical or radiographic (on MRI) signs or symptoms indicative of non-AD dementias such as frontotemporal dementia, dementia with Lewy bodies, vascular dementia, progressive supranuclear palsy, or corticobasal syndrome).
While the positive correlations in amyloid-positive but not amyloid negative participants suggests these relationships are moderated in part by amyloidosis, we additionally performed an interaction regression model to see if the association between UPSIT score and either 11C-PBR28 binding or 18F-MK-6240 binding differ by amyloid status. In the 47 models, none of the interaction terms reached significance; however, we may have been underpowered for this particular analysis. We saw small-to-medium effect sizes in the interactions with the medial temporal cortex for both 11C-PBR28 and 18F-MK-6240 binding. Therefore, a larger study is warranted to better characterize how the relationships among olfactory identification, inflammation, and tau are influenced by amyloid status.
Our conclusions are limited by our sample size. We did not observe any significant relationships within the amyloid-negative subgroups alone and many of the overall relationships observed were driven by amyloid-positive patients. We cannot say, however, that olfactory identification is not related to tau or neuroinflammation in amyloid-negative participants, only that we failed to find such a relationship and that presumably tau, neuroinflammation, and impaired odor identification are mediated at least in part by amyloid. Sample size was particularly limiting for our CSF analysis, as only 23 participants in our cohort elected to have lumbar puncture performed, and therefore we did not evaluate amyloid-positive and amyloid-negative groups separately. Further, our sample size did not permit stratification of these relationships by sex given that there were more male participants in both the amyloid-positive and amyloid-negative patient groups and more female participants overall in the control group. However, sex was considered as a biological covariate in statistical analysis. We did not evaluate relationships between olfactory impairment and performance on neuropsychological testing batteries beyond the MMSE and SRT-DR. However, the relationship between UPSIT and cognitive performance was more comprehensively investigated in a larger community cohort of over 1000 participants, with results demonstrating that UPSIT significantly correlated with neuropsychological measures of memory, fluency and executive functioning [12]. While these results do not imply causation due to the limitations of a cross-sectional observational study, our results indicate that there appear to be significant associations between olfactory impairment, tau pathology and neuroinflammation that could be further investigated with a larger sample size. 18F-MK-6240 is still an early tau radioligand with an off-target binding profile that is not yet fully understood. Early studies, however, suggest that the radioligand has adequate sensitivity for detecting tau pathology [32, 39].
In conclusion, while reduced olfactory identification ability has previously been linked to cognitive decline and amyloid deposition, we have demonstrated that UPSIT performance is also related to other contributors of AD pathophysiology. Therefore, the UPSIT appears to serve broader utility beyond being a marker of disease severity, but rather an inexpensive, non-invasive screening tool that may provide insight into the burden of tau pathology and neuroinflammation. Based on our results and the literature, the UPSIT could also be considered for use as an initial screening tool to identify participants in at-risk populations who may be more likely to test positive of PET for amyloid, tau or other in vivo measures of AD pathology, saving time and cost in clinical trials involving preventative treatments.
Supplementary Material
ACKNOWLEDGEMENTS
18F-Florbetaben was supplied by Life Molecular Imaging. 18F-MK-6240 was supplied by Cerveau Technologies. TSPO genotyping was performed by Regina Santella, PhD and the Columbia University Biomarkers Shared Resource. We wish to acknowledge the contributions of the staff and faculty of the Irving Institute for Clinical Research, the MRI Center and the David A. Gardner PET Imaging Center at the Columbia University Irving Medical Center.
FUNDING
This research was funded by National Institute of Aging grants K23AG052633, R01AG026158, R01AG062578 and R56AG034189. Support for this study came from NIA grant T35AG044303 and the Columbia University Alzheimer’s Disease Research Center (P50AG008702). Data collection and sharing for this project was supported by the Washington Heights-Inwood Columbia Aging Project (WHICAP, P01AG07232, R01AG037212, RF1AG054023). We acknowledge the WHICAP study participants and the WHICAP research and support staff for their contributions to this study.
Footnotes
CONFLICTS OF INTEREST
Julia Klein- Reports no disclosures
Xinyu Yan- Reports no disclosures
Aubrey Johnson- Reports no disclosures
Zeljko Tomljanovic- Reports no disclosures
James Zou- Reports no disclosures
Krista Polly- Reports no disclosures
Lawrence Honig- Consultant: Cortexyme, Eisai, Medscape, Prevail; Research Grants: Abbvie, Alector, Biogen, Genentech, Eli Lilly, Roche
Adam M. Brickman- Consultant: Regeneron Pharmaceuticals, Cognition Therapeutics; Equity: Mars Holding Limited
Yaakov Stern- Reports no disclosures
D.P Devanand- Consultant: Acadia, BXcel, Corium, Genentech, Grifols
Seonjoo Lee- Reports no disclosures
William C. Kreisl- Consultant for Cerveau Technologies. However, Cerveau was not involved in the design or execution of this study or in the interpretation of the results.
REFERENCES:
- [1].Doty RL, Reyes PF, Gregor T (1987) Presence of both odor identification and detection deficits in Alzheimer’s disease. Brain Res Bull 18, 597–600. [DOI] [PubMed] [Google Scholar]
- [2].Murphy C, Gilmore MM, Seery CS, Salmon DP, Lasker BR (1990) Olfactory thresholds are associated with degree of dementia in Alzheimer’s disease. Neurobiol Aging 11, 465–469. [DOI] [PubMed] [Google Scholar]
- [3].Devanand DP, Michaels-Marston KS, Liu X, Pelton GH, Padilla M, Marder K, Bell K, Stern Y, Mayeux R (2000) Olfactory deficits in patients with mild cognitive impairment predict Alzheimer’s disease at follow-up. Am J Psychiatry 157, 1399–1405. [DOI] [PubMed] [Google Scholar]
- [4].Djordjevic J, Jones-Gotman M, De Sousa K, Chertkow H (2008) Olfaction in patients with mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging 29, 693–706. [DOI] [PubMed] [Google Scholar]
- [5].Masurkar AV, Devanand DP (2014) Olfactory Dysfunction in the Elderly: Basic Circuitry and Alterations with Normal Aging and Alzheimer’s Disease. Curr Geriatr Rep 3, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Laske C, Sohrabi HR, Frost SM, Lopez-de-Ipina K, Garrard P, Buscema M, Dauwels J, Soekadar SR, Mueller S, Linnemann C, Bridenbaugh SA, Kanagasingam Y, Martins RN, O’Bryant SE (2015) Innovative diagnostic tools for early detection of Alzheimer’s disease. Alzheimers Dement 11, 561–578. [DOI] [PubMed] [Google Scholar]
- [7].Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82, 239–259. [DOI] [PubMed] [Google Scholar]
- [8].Attems J, Lintner F, Jellinger KA (2005) Olfactory involvement in aging and Alzheimer’s disease: an autopsy study. J Alzheimers Dis 7, 149–157; discussion 173–180. [DOI] [PubMed] [Google Scholar]
- [9].Vassilaki M, Christianson TJ, Mielke MM, Geda YE, Kremers WK, Machulda MM, Knopman DS, Petersen RC, Lowe VJ, Jack CR Jr., Roberts RO (2017) Neuroimaging biomarkers and impaired olfaction in cognitively normal individuals. Ann Neurol 81, 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Growdon ME, Schultz AP, Dagley AS, Amariglio RE, Hedden T, Rentz DM, Johnson KA, Sperling RA, Albers MW, Marshall GA (2015) Odor identification and Alzheimer disease biomarkers in clinically normal elderly. Neurology 84, 2153–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Devanand DP, Tabert MH, Cuasay K, Manly JJ, Schupf N, Brickman AM, Andrews H, Brown TR, DeCarli C, Mayeux R (2010) Olfactory identification deficits and MCI in a multi-ethnic elderly community sample. Neurobiol Aging 31, 1593–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Devanand DP, Lee S, Manly J, Andrews H, Schupf N, Masurkar A, Stern Y, Mayeux R, Doty RL (2015) Olfactory identification deficits and increased mortality in the community. Ann Neurol 78, 401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Risacher SL, Tallman EF, West JD, Yoder KK, Hutchins GD, Fletcher JW, Gao S, Kareken DA, Farlow MR, Apostolova LG, Saykin AJ (2017) Olfactory identification in subjective cognitive decline and mild cognitive impairment: Association with tau but not amyloid positron emission tomography. Alzheimers Dement (Amst) 9, 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Reijs BLR, Ramakers I, Elias-Sonnenschein L, Teunissen CE, Koel-Simmelink M, Tsolaki M, Wahlund LO, Waldemar G, Hausner L, Johannsen P, Vanderstichele H, Verhey F, Devanand DP, Visser PJ (2017) Relation of Odor Identification with Alzheimer’s Disease Markers in Cerebrospinal Fluid and Cognition. J Alzheimers Dis 60, 1025–1034. [DOI] [PubMed] [Google Scholar]
- [15].McGeer EG, McGeer PL (2003) Inflammatory processes in Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry 27, 741–749. [DOI] [PubMed] [Google Scholar]
- [16].Owen DR, Yeo AJ, Gunn RN, Song K, Wadsworth G, Lewis A, Rhodes C, Pulford DJ, Bennacef I, Parker CA, StJean PL, Cardon LR, Mooser VE, Matthews PM, Rabiner EA, Rubio JP (2012) An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab 32, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kreisl WC, Jenko KJ, Hines CS, Lyoo CH, Corona W, Morse CL, Zoghbi SS, Hyde T, Kleinman JE, Pike VW, McMahon FJ, Innis RB, Biomarkers Consortium PETRPT (2013) A genetic polymorphism for translocator protein 18 kDa affects both in vitro and in vivo radioligand binding in human brain to this putative biomarker of neuroinflammation. J Cereb Blood Flow Metab 33, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Folstein MF, Robins LN, Helzer JE (1983) The Mini-Mental State Examination. Arch Gen Psychiatry 40, 812. [DOI] [PubMed] [Google Scholar]
- [19].Ruff RM, Light RH, Quayhagen M (1989) Selective Reminding Tests: a normative study of verbal learning in adults. J Clin Exp Neuropsychol 11, 539–550. [DOI] [PubMed] [Google Scholar]
- [20].Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Carrillo MC, Thies B, Phelps CH (2011) The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr., Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH (2011) The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bullich S, Villemagne VL, Catafau AM, Jovalekic A, Koglin N, Rowe CC, De Santi S (2017) Optimal Reference Region to Measure Longitudinal Amyloid-beta Change with (18)F-Florbetaben PET. J Nucl Med 58, 1300–1306. [DOI] [PubMed] [Google Scholar]
- [23].Bullich S, Seibyl J, Catafau AM, Jovalekic A, Koglin N, Barthel H, Sabri O, De Santi S (2017) Optimized classification of (18)F-Florbetaben PET scans as positive and negative using an SUVR quantitative approach and comparison to visual assessment. Neuroimage Clin 15, 325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Thomas BA, Erlandsson K, Modat M, Thurfjell L, Vandenberghe R, Ourselin S, Hutton BF (2011) The importance of appropriate partial volume correction for PET quantification in Alzheimer’s disease. Eur J Nucl Med Mol Imaging 38, 1104–1119. [DOI] [PubMed] [Google Scholar]
- [25].Betthauser TJ, Cody KA, Zammit MD, Murali D, Converse AK, Barnhart TE, Stone CK, Rowley HA, Johnson SC, Christian BT (2019) In Vivo Characterization and Quantification of Neurofibrillary Tau PET Radioligand (18)F-MK-6240 in Humans from Alzheimer Disease Dementia to Young Controls. J Nucl Med 60, 93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lyoo CH, Ikawa M, Liow JS, Zoghbi SS, Morse CL, Pike VW, Fujita M, Innis RB, Kreisl WC (2015) Cerebellum Can Serve As a Pseudo-Reference Region in Alzheimer Disease to Detect Neuroinflammation Measured with PET Radioligand Binding to Translocator Protein. J Nucl Med 56, 701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Vanderstichele H, De Meyer G, Andreasen N, Kostanjevecki V, Wallin A, Olsson A, Blennow K, Vanmechelen E (2005) Amino-truncated beta-amyloid42 peptides in cerebrospinal fluid and prediction of progression of mild cognitive impairment. Clin Chem 51, 1650–1660. [DOI] [PubMed] [Google Scholar]
- [28].Benjamini Y, Hochberg Y (1995) Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B-Statistical Methodology 57, 289–300. [Google Scholar]
- [29].Bahar-Fuchs A, Chetelat G, Villemagne VL, Moss S, Pike K, Masters CL, Rowe C, Savage G (2010) Olfactory deficits and amyloid-beta burden in Alzheimer’s disease, mild cognitive impairment, and healthy aging: a PiB PET study. J Alzheimers Dis 22, 1081–1087. [DOI] [PubMed] [Google Scholar]
- [30].Wilson RS, Arnold SE, Schneider JA, Tang Y, Bennett DA (2007) The relationship between cerebral Alzheimer’s disease pathology and odour identification in old age. J Neurol Neurosurg Psychiatry 78, 30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bradburn S, Murgatroyd C, Ray N (2019) Neuroinflammation in mild cognitive impairment and Alzheimer’s disease: A meta-analysis. Ageing Res Rev 50, 1–8. [DOI] [PubMed] [Google Scholar]
- [32].Hostetler ED, Walji AM, Zeng Z, Miller P, Bennacef I, Salinas C, Connolly B, Gantert L, Haley H, Holahan M, Purcell M, Riffel K, Lohith TG, Coleman P, Soriano A, Ogawa A, Xu S, Zhang X, Joshi E, Della Rocca J, Hesk D, Schenk DJ, Evelhoch JL (2016) Preclinical Characterization of 18F-MK-6240, a Promising PET Tracer for In Vivo Quantification of Human Neurofibrillary Tangles. J Nucl Med 57, 1599–1606. [DOI] [PubMed] [Google Scholar]
- [33].Dani M, Wood M, Mizoguchi R, Fan Z, Walker Z, Morgan R, Hinz R, Biju M, Kuruvilla T, Brooks DJ, Edison P (2018) Microglial activation correlates in vivo with both tau and amyloid in Alzheimer’s disease. Brain 141, 2740–2754. [DOI] [PubMed] [Google Scholar]
- [34].Zou J, Tao S, Johnson A, Tomljanovic Z, Polly K, Klein J, Razlighi QR, Brickman AM, Lee S, Stern Y, Kreisl WC (2019) Microglial activation, but not tau pathology, is independently associated with amyloid positivity and memory impairment. Neurobiol Aging 85, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Schubert CR, Carmichael LL, Murphy C, Klein BE, Klein R, Cruickshanks KJ (2008) Olfaction and the 5-year incidence of cognitive impairment in an epidemiological study of older adults. J Am Geriatr Soc 56, 1517–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kreisl WC, Jin P, Lee S, Dayan ER, Vallabhajosula S, Pelton G, Luchsinger JA, Pradhaban G, Devanand DP (2018) Odor Identification Ability Predicts PET Amyloid Status and Memory Decline in Older Adults. J Alzheimers Dis 62, 1759–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Benarroch EE (2010) Olfactory system: functional organization and involvement in neurodegenerative disease. Neurology 75, 1104–1109. [DOI] [PubMed] [Google Scholar]
- [38].Murray ME, Cannon A, Graff-Radford NR, Liesinger AM, Rutherford NJ, Ross OA, Duara R, Carrasquillo MM, Rademakers R, Dickson DW (2014) Differential clinicopathologic and genetic features of late-onset amnestic dementias. Acta Neuropathol 128, 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lohith TG, Bennacef I, Vandenberghe R, Vandenbulcke M, Salinas CA, Declercq R, Reynders T, Telan-Choing NF, Riffel K, Celen S, Serdons K, Bormans G, Tsai K, Walji A, Hostetler ED, Evelhoch JL, Van Laere K, Forman M, Stoch A, Sur C, Struyk A (2019) Brain Imaging of Alzheimer Dementia Patients and Elderly Controls with (18)F-MK-6240, a PET Tracer Targeting Neurofibrillary Tangles. J Nucl Med 60, 107–114. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Anonymized data will be made available upon reasonable request to qualified investigators.