

Summary
Immunotherapy induces durable clinical responses in a fraction of patients with cancer. However, therapeutic resistance poses a major challenge to current immunotherapies. Here, we identify that expression of tumor stanniocalcin 1 (STC1) correlates with immunotherapy efficacy and is negatively associated with patient survival across diverse cancer types. Gain- and loss- of-function experiments demonstrate that tumor STC1 supports tumor progression and enables tumor resistance to checkpoint blockade in murine tumor models. Mechanistically, tumor STC1 interacts with calreticulin (CRT), an “eat-me” signal, and minimizes CRT membrane exposure - thereby abrogating membrane CRT-directed phagocytosis by antigen-presenting-cells (APCs), including macrophages and dendritic cells. Consequently, this impairs APC capacity of antigen presentation and T cell activation. Thus, tumor STC1 inhibits APC phagocytosis and contributes to tumor immune evasion and immunotherapy resistance. We suggest that STC1 is a previously unappreciated phagocytosis checkpoint and targeting STC1 and its interaction with CRT may sensitize to cancer immunotherapy.
Keywords: tumor, stanniocalcin 1, calreticulin, T-cell immunity, macrophages, dendritic cell, phagocytosis, checkpoint, PD-1, eat-me signal
Graphical Abstract
eTOC Blurb
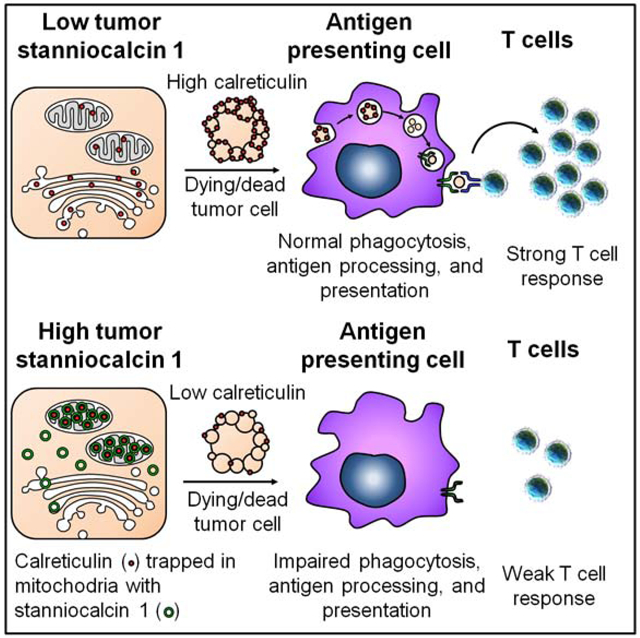
Lin et al. demonstrate tumor stanniocalcin-1 functions as an intracellular “eat-me” signal blocker by trapping calreticulin and impairs APC phagocytosis and T cell activation. Tumor stanniocalcin-1 negatively correlates with immunotherapy efficacy and patient survival.
Introduction
Immune checkpoint-based therapy manifests unprecedented success against cancer through revitalizing and boosting T-cell responses. However, the majority of patients with cancer do not respond to immune checkpoint therapy (Ribas and Wolchok, 2018; Topalian et al., 2015; Zou et al., 2016). A deeper understanding of complex interactions between different immune cell subsets and tumor cells in the tumor microenvironment (Zou, 2005) is crucial for dissecting intrinsic and adaptive immune resistance mechanisms. This will result in developing rationalized combinatorial therapeutic approaches and identifying potential novel targets. Recent studies demonstrate that tumor genetic and epigenetic mutations (Li et al., 2020; Mandal et al., 2019; Peng et al., 2015), mutations in interferon (IFN)γ and MHC signaling pathway (Gao et al., 2016; Sade-Feldman et al., 2017; Shin et al., 2017; Zaretsky et al., 2016), dysfunctional T-cell trafficking and cytotoxic T cell activity (Peng et al., 2015; Sheng et al., 2018; Spranger et al., 2015), abnormal metabolic networks (Bian et al., 2020; Buck et al., 2016; Karmaus et al., 2019; Li et al., 2018; Maj et al., 2017), and alterations in other biological pathways (Manguso et al., 2017; Pan et al., 2018; Patel et al., 2017) contribute to immunotherapy resistance. However, these mechanistic studies don’t explore a potential alteration of APC-mediated phagocytosis and its involvement in immunotherapy resistance.
To explore tumor intrinsic immune resistance mechanisms in the tumor microenvironment in patients with defined immune checkpoint therapy responsiveness, we have analyzed the transcriptomes of two data sets in patients with melanoma treated with checkpoint therapy (Hugo et al., 2016; Riaz et al., 2017). Our goals are to explore a potential correlation between specific unknown gene transcripts in tumor cells and patient therapeutic responses to both PD-1 (Hugo et al., 2016; Riaz et al., 2017) and CTLA-4 blockade (Van-Allen et al., 2015, Nathanson et al., 2017) in different patient cohorts, and subsequently elucidate how these gene(s) alters cancer immunity and immunotherapy efficacy. STC1 is reported as a hormone-like protein and may mediate multiple biological activities (Yeung et al., 2012). Nonetheless, its receptor and interaction partners, the mechanism(s) of action, and the potential importance of STC1 to tumor immunity remain unknown. Using several preclinical models and gain- and loss-of-function studies, we have demonstrated that tumor STC1 affected tumor immunity and impacted tumor response to immunotherapy.
APCs prime and activate tumor associated antigen (TAA)-specific T cells and are critical for defining the success of checkpoint therapy. This process is dependent on whether APCs, including macrophages and dendritic cells (DCs), could efficiently capture antigens from dead tumor cells via phagocytosis, present sufficient antigens to T cells, and activate T cells (Salmon et al., 2016). On this basis, we uncover that tumor STC1 interacted with CRT, an “eat-me” signal, trapped CRT in mitochondria area, and reduced membrane CRT. Consequently, membrane CRT-mediated phagocytosis by APCs is diminished, thereby resulting in impaired APC antigen presentation and T-cell activation. Thus, tumor STC1 contributes to tumor immune evasion and immunotherapy resistance. We suggest that tumor STC1 functions as an intracellular “eat-me” signal blocker and targeting STC1 and its interaction with CRT is a previously unknown anti-cancer immunotherapeutic approach to overcoming cancer checkpoint therapy resistance.
Results
STC1 correlates to cancer resistance to immunotherapy
To dissect immunotherapy resistant mechanism and define novel immunotherapeutic targets, we analyzed published data in patients with melanoma who have received immune checkpoint therapy (Hugo et al., 2016; Riaz et al., 2017). We first compared the transcriptomes of non-responders and responders to nivolumab therapy. Based on the threshold with > 2-fold change and P value < 0.1, we obtained the top 524 (Hugo et al., 2016) and 585 (Riaz et al., 2017) highly expressed genes in the non-responders in cohorts 1 (Hugo et al., 2016) and 2 (Riaz et al., 2017). We examined the overlap of these 2 cohorts and found 8 genes, including STC1 (Fig 1A). Then, we tested expression levels of the 8 genes in murine MC38 colon cancer cell line, B16-F10 melanoma cell line, and Lewis lung carcinoma cell line (LLC). We previously characterized that immune checkpoint therapy has high, intermediate, and low efficacy in these preclinical models, respectively (Lin et al., 2018). Among the 8 genes, we detected low, moderate, and high levels of Stc1 transcripts (Fig. S1A) and proteins (Fig. S1B) in MC38, B16-F10, and LLC cells, respectively. Hence, the levels of tumor Stc1 inversely correlate to different tumor sensitivities to immunotherapy in mouse models in vivo (Lin et al., 2018). The other 7 identified genes were minimally expressed in MC38, B16-F10, and LLC tumor cell lines (Fig. S1A). We validated the clinical relevance of STC1 in non-responders and responders in these 2 cohorts receiving immune checkpoint therapy. STC1 was highly expressed in the non-responders (Fig. 1B, C). High levels of STC1 correlated with low T-cell activation signature (Fig. S1C) (Wang et al., 2019b) and were associated with shorter overall survival in patients with melanoma treated with nivolumab (anti-PD-1) (Fig. 1D) or ipilimumab (anti-CTLA4) therapy (Fig. S1D, S1E) (Van-Allen et al., 2015, Nathanson et al., 2017). The results suggest a potential detrimental role of STC1 in tumor immunity and immunotherapy.
Figure 1. STC1 correlates to cancer resistance to immunotherapy.
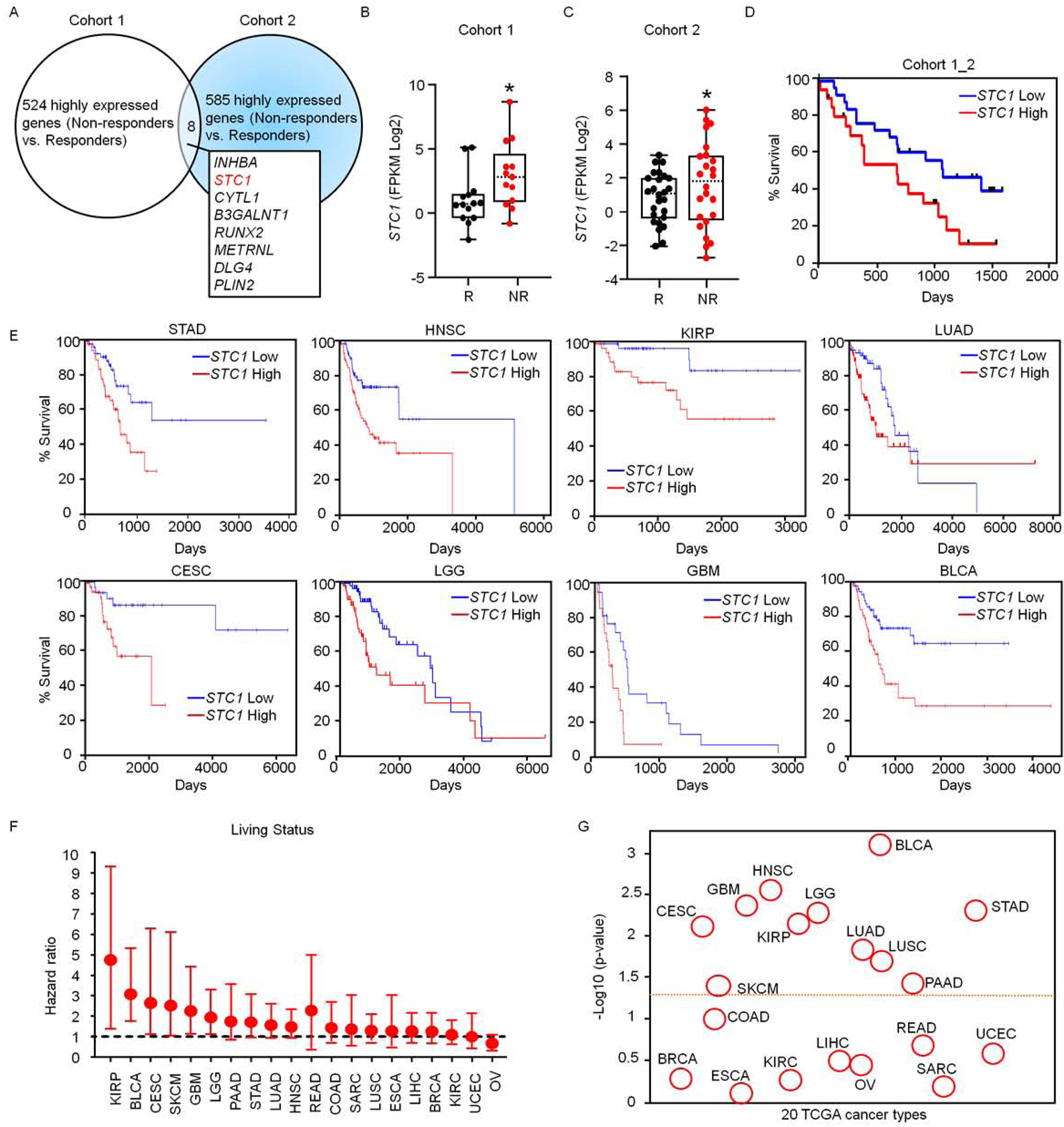
(A) Transcriptome analysis in patients treated with checkpoint blockade. Upregulated genes in non-responders treated with checkpoint blockade were determined in cohorts 1 (Hugo et al., 2016) and 2 (Riaz et al., 2017). The overlapping upregulated genes in 2 cohorts are shown. Cohort 1, n = 15 (responders), 13 (non-responders); Cohort 2, n = 26 (responders), 25 (non-responders).
(B-C) Expression of STC1 transcripts in Responders (R) and Non-Responders (NR) in cohort 1 (B) n = 14 (R), 13 (NR), p = 0.0287; and cohort 2 (C) n = 26 (R), 25 (NR), p = 0.0301. The dash line represents the median value, the bottom and top of the boxes are the 25th and 75th percentiles (interquartile range). Whiskers encompass 1.5 times the inter-quartile range.
(D) Association of STC1 expression levels with cancer patient survival analyzed on combined cohorts 1 and 2, STC1 high (n = 22) and low (n = 27) expression, p = 0.0385.
(E-G) Relationship of STC1 expression with cancer patient survival in 18 cancer types in TCGA data set. Results are shown as individual cancer survival curves with top 15% high and low STC1 expression (E); living status (F) Forest plot represents the adjust value of Cox proportional hazard ratio (HR) and 95% confidential interval (CI) of overall survival; and p-values (G) (STAD n = 56, p = 0.00475; HNSC n = 74, p = 0.00272; KIRP n = 42, p= 0.00698; LUAD n = 73, p = 0.0145; CESC n = 39, p = 0.00762; LGG n = 76, p = 0.00521; GBM n = 22, p = 0.00421; BLCA n = 60, p= 0.000765)
See also Figure S1.
We further analyzed the transcriptomes of STC1 in TCGA database (Uhlen et al., 2017). Among 20 cancer types, high levels of STC1 were broadly associated with poor patient survival in more than half of the cancer types, including bladder carcinoma (BLCA), stomach adenocarcinoma (STAD), head and neck squamous cell carcinoma (HNSC), renal papillary cell carcinoma (KIRP), lung squamous cell carcinoma (LUSC), lung adenocarcinoma (LUAD), glioblastoma (GBM), cervical squamous cell carcinoma (CESC), colorectal adenocarcinoma (COAD), and cutaneous melanoma (SKCM) (Fig. 1E–G, S1F). Notably, we did not observe any positive or negative associations of STC1 with cancer patient survival in several other cancer types (Fig. 1F–G). The results suggest that STC1 impacts human cancer outcomes in many cancer types.
Tumor STC1 is critical for intrinsic resistance to tumor immunity
Our aforementioned results (Fig. 1 and S1) suggest that murine MC38, B16-F10, and LLC tumor cells may be useful tools to explore the functional significance of tumor STC1 in tumor immunity and immunotherapy resistance in vivo. Based on different endogenous levels of STC1 in the three tumor cell lines (Fig. 1 and S1), we initially ectopically expressed Stc1 in MC38 (Stc1OE) cells and genetically knocked out Stc1 (Stc1−/−) in B16-F10 and LLC cells and confirmed altered expression by ELISA (Fig. S2A). We inoculated these cells into NOD.SCID γc deficient (NSG) mice and C57BL/6J mice. We found ectopic Stc1 expression accelerated MC38 tumor progression in C57BL/6J (immune competent) mice (Fig. 2A), but had no effect on tumor growth in NSG (immune deficient) mice (Fig. 2B). Accordingly, genetic knock out Stc1 resulted in slower B16-F10 and LLC tumor progression in C57BL/6J mice (Fig. 2C, D), but comparable tumor progression in NSG mice (Fig. 2E, F). To additionally validate these results, we knocked down Stc1 with short hairpin RNA (shRNA) in LLC tumor cells and conducted identical experiments. Again, knocking down Stc1 resulted in slower LLC tumor progression in C57BL/6J mice (Fig. 2G), but comparable tumor progression in NSG mice (Fig. 2H). These results indicate that tumor STC1 may modulate immune responses, supporting tumor progression.
Figure 2. Tumor STC1 is critical for intrinsic resistance to tumor immunity.
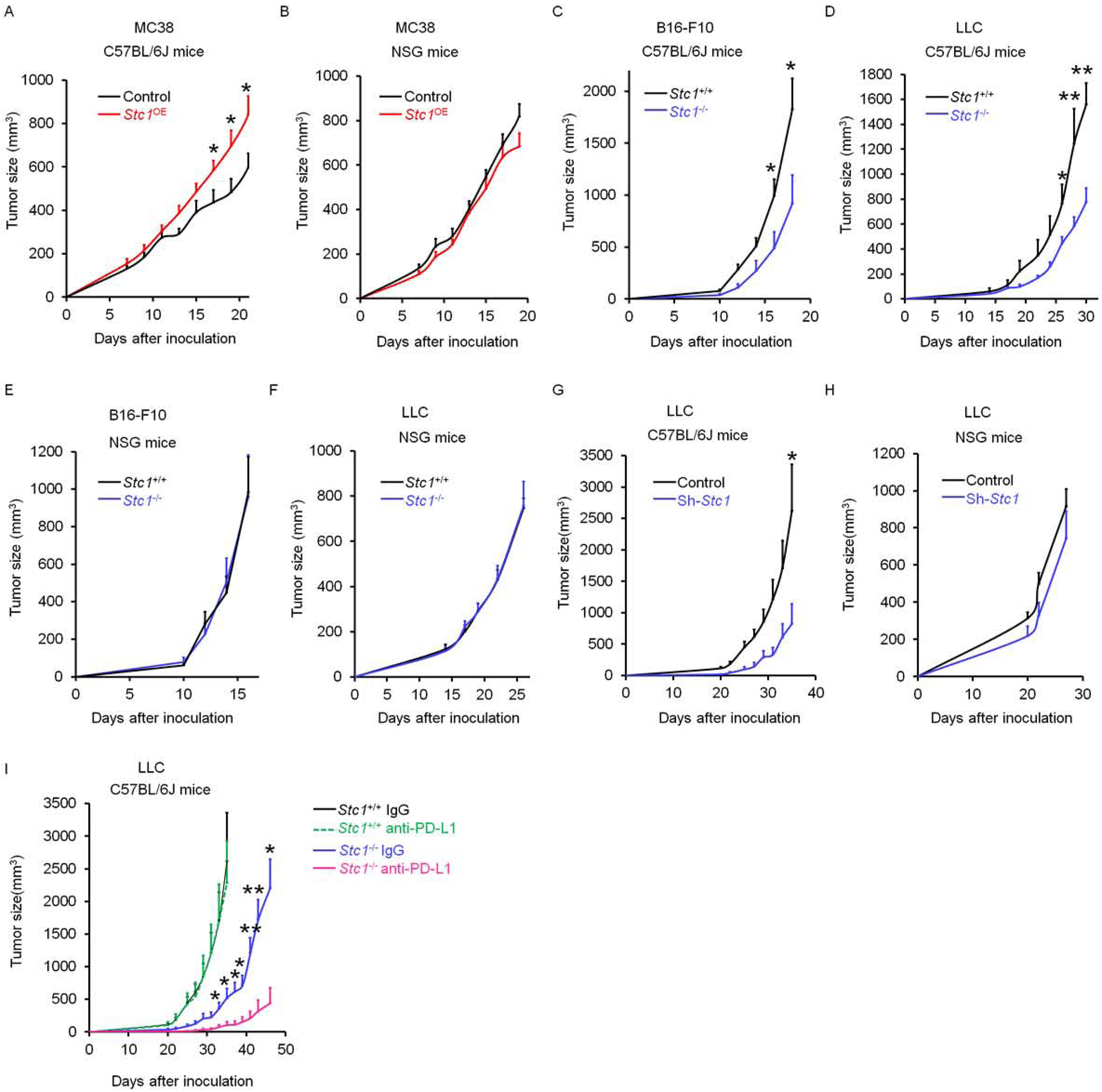
(A-B) Tumor growth curves of control MC38 and Stc1OE MC38 in (A) C57BL/6J mice and (B) NSG mice (n = 8).
(C-F) Tumor growth curves of Stc1+/+ and Stc1−/− LLC, and Stc1+/+ and Stc1−/− B16-F10 in (C, D) C57BL/6J mice and (E, F) NSG mice (n = 5–8).
(G-H) Tumor growth curves of control LLC and shStc1 LLC in (G) C57BL/6J mice and (H) NSG mice (n = 4–5).
(I) Tumor growth curves of Stc1+/+ and Stc1−/− LLC tumors in (I) C57BL/6J mice with anti-PD-L1 or isotype control antibodies treatments every 3 days starting day 3 (n = 4–7).
Data are shown as mean ± SEM, 2 tail t-test was used for two-way comparisons (A-I) (*p < 0.05, **p < 0.01).
See also Figure S2.
Given that LLC tumors are resistant to checkpoint blockade (Lin et al., 2018), we treated mice bearing Stc1−/− LLC tumor with anti-PD-L1 monoclonal antibody (mAb). As expected, mice bearing Stc1+/+ LLC tumors had no response to this therapy (Lin et al., 2018), whereas we observed a remarkable increase in the therapeutic efficacy of anti-PD-L1 in mice bearing Stc1−/−LLC tumors (Fig. 2I). In support of this, we inoculated Stc1−/− LLC (Fig. S2B) and Stc1OE B16-F10 (Fig. S2C) cells into Pdcd1−/− C57BL/6J mice. We observed a dramatic decrease in Stc1−/− LLC tumor growth (Fig. S2B) and a sharp increase in Stc1OE B16-F10 tumors (Fig. S2C) in these mice. Thus, tumor STC1 may function as an intrinsic resistant mechanism of spontaneous and immunotherapy-induced tumor immunity.
Tumor STC1 impairs anti-tumor CD8+ T cell responses
CD8+ T cells mediate anti-tumor immunity. Given that tumor STC1 promotes tumor progression in immune competent mice bearing multiple types of tumors (Fig. 2), we examined CD8+ T cell function in LLC tumor bearing mice (Fig. 2D). Flow cytometry analysis revealed an increase in Ki67+CD8+ T cells in tumor draining lymph nodes (Fig. 3A) and tumor tissues (Fig. 3B) in mice bearing Stc1−/− LLC tumors as compared to those bearing Stc1+/+ LLC tumors. In addition, the levels of tumor infiltrating IFNγ+, granzyme B+, and TNFα+ CD8+ T cells were higher in mice bearing Stc1−/− LLC tumors as compared to those bearing Stc1+/+ LLC tumors (Fig. 3C–D).
Figure 3. Tumor STC1 impairs anti-tumor CD8+ T cell responses.
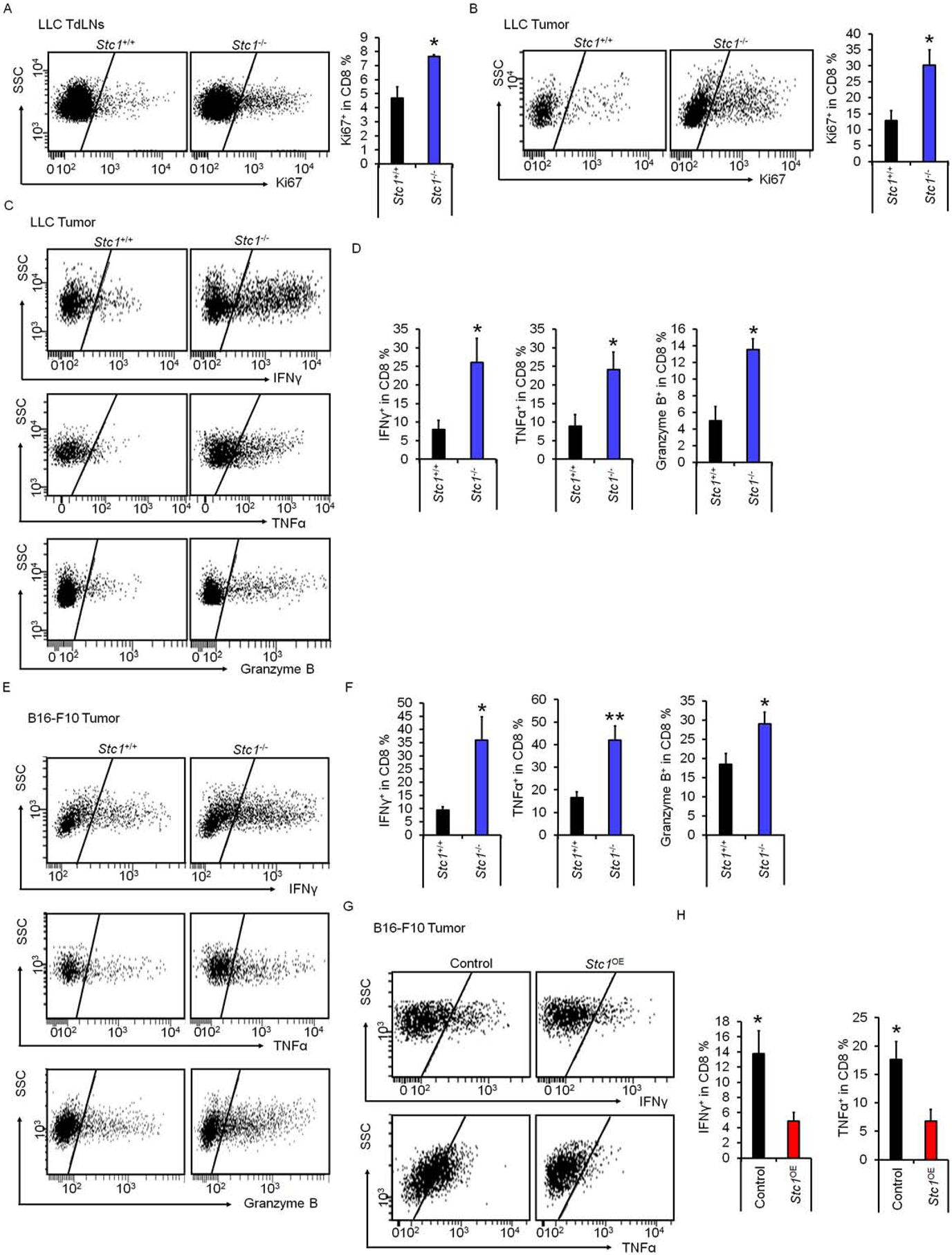
(A-B) Percentages of Ki67+CD8+ T cells in tumor drained lymph nodes (TdLNs) and tumor tissues in mice bearing Stc1+/+ and Stc1−/− LLC tumors determined by FACS.
(C-F) Percentages of IFNγ+, granzyme B+, and TNFα+CD8+ T cells in Stc1+/+ and Stc1−/− LLC tumor tissues (C, D) and B16-F10 tumor tissues (E, F) determined by FACS.
(G-H) Percentages of IFNγ+ and TNFα+ CD8 T cells in control and Stc1OE B16-F10 tumor tissues determined by FACS.
Data are shown as mean ± SEM, 2 tail T-test was used for two-way comparisons (n = 4–5, *p < 0.05; **p<0.01) (A-H).
See also Figure S3.
To validate these results, we examined mice bearing Stc1−/− and Stc1OE B16-F10 tumors (Fig. 2). Again, flow cytometry analysis revealed an increase in tumor infiltrating IFNγ+, granzyme B+, and TNFα+ CD8+ T cells in mice bearing Stc1−/− B16-F10 tumors as compared to those bearing Stc1+/+ B16-F10 tumors (Fig. 3E–F). In contrast, the levels of IFNγ+ and TNFα+ tumor infiltrating CD8+ T cells were lower in mice bearing Stc1OE B16-F10 tumors as compared to vector expressing B16-F10 tumors (Fig. 3G–H). To examine the effect of STC1 on tumor specific T cell response, we inoculated ovalbumin (OVA)-expressing MC38 cells expressing control vector or Stc1OE into C57BL/6J mice and performed IFNγ ELISOPT assays in tumor drained lymph node cells. As expected, in response to OVA-peptides, there were less IFNγ spots in mice bearing Stc1OE MC38 tumors as compared to mice bearing control vector expressing MC38 tumors (Fig. S3A). Thus, tumor STC1 dampens anti-tumor CD8+ T cell responses in vivo.
We next examined if recombinant STC1 and tumor-released STC1 directly inhibited T-cell activation in vitro. To this end, we stimulated splenocytes with anti-CD3 and anti-CD28 in the presence of recombinant STC1 or the cultured supernatants from tumor cells expressing different levels of STC1. We detected comparable levels of IFNγ, TNFα, and granzyme B expression in CD8+ T cells with or without recombinant STC1 (Fig. S3B). Furthermore, CD8+ T cells expressed similar levels of IFNγ, TNFα, and granzyme B in the presence of the supernatants from B16-F10 cells, Stc1OE B16-F10 cells, and Stc1−/− B16-F10 cells (Fig. S3C), or the supernatants from Stc1+/+ and Stc1−/− LLC cells (Fig. S3D). The data suggests that tumor-released STC1 may not directly regulate CD8+ T cell responses.
We next tested if intracellular STC1 in tumor cells effected T-cell activation. Despite the little understood mechanism, it has been reported that immunogenicity of dead cancer cells impacts tumor CD8+ T cell responses (Obeid et al., 2007; Zitvogel et al., 2008). To test whether tumor STC1 affects tumor immunogenicity, we loaded B16-F10 cells and Stc1OE B16-F10 cells with OVA (Theisen et al., 2018) and used ultraviolet irradiation to induce tumor cell death. We isolated splenic cells from OT-I transgenic mice, then, cultured these cells with different amount of dead OVA-expressing B16-F10 cells and Stc1OE B16-F10 cells (Fig. S3E). We detected a dose dependent increase in IFNγ production in the culture with dead OVA-B16-F10 cells, but not with dead OVA- Stc1OE B16-F10 cells (Fig. S3E). Apart from OT-I cells, there are APCs, including macrophages and myeloid dendritic cells (DCs), in splenic cells isolated from OT-I transgenic mice. The results suggest that APCs can capture OVA from dead B16-F10 tumor cells, present OVA to OT-I cells, and activate OT-I cells to express IFNγ. Meanwhile, as dead Stc1OE tumor cells triggered minimal OVA-specific CD8+ T cell responses, it suggests that STC1 in tumor cells may impair APC function. To additionally confirm this possibility, we activated C57BL/6J splenic T cells with anti-CD3 and anti-CD28 antibodies in the presence of dead B16-F10 cells and dead Stc1OE B16-F10 cells. We detected similar levels of IFNγ in co-cultures with dead B16-F10 tumor cells and dead Stc1OE B16-F10 tumor cells (Fig. S3F). Altogether, the results suggest that STC1 in tumor cells may not directly affect T cell activation, but rather potentially target APCs and reduces tumor immunity in an antigen presentation dependent manner.
STC1 abrogates tumor immunogenicity via targeting APCs
Damage-associated molecular patterns (DAMPs) alter APC phenotype and function in the course of antigen capturing and processing, including macrophage- and DC-mediated phagocytosis (Zitvogel et al., 2008). To assess a potential role of tumor STC1 in modulating APC function, we cultured bone-marrow-derived macrophages, CFSE-labeled OT-I cells, and dead OVA-B16-F10 cells or dead OVA- Stc1OE B16-F10 cells. We observed an increase in OT-I cell activation in a dose-dependent manner based on CFSE dilution in OT-I cells (Fig. S4A, B), and intracellular expression of granzyme B (Fig. S4C, D), IFNγ (Fig. S4E, F), and released IFNγ (Fig. S4G), followed by co-culture with dead B16-F10 cells. However, the magnitude of OT-I cell activation was reduced following co-culture with dead Stc1OE B16-F10 cells (Fig. S4G) or OVA-expressing dead Stc1OE MC38 cells (Fig. S4H) as shown by IFNγ production. We performed an identical experiment with Stc1+/+ and Stc1−/− LLC cells (Fig. 4A–F). In this setting, OT-I cell activation was superior in the presence of dead Stc1−/− LLC cells compared to dead Stc1+/+ LLC cells (Fig. 4A–F). We extended our studies from macrophages to myeloid DCs. In a similar setting (Fig. 4G–L), macrophages were replaced with DCs. Again, OT-I cell activation was superior in the presence of dead Stc1−/− LLC cells compared to dead Stc1+/+ LLC cells (Fig. 4G–L). Notably, OT-I cells were not activated in the absence of macrophages or DCs (Fig. 4G–K, S4A–E). Thus, tumor STC1 results in reduced T-cell activation in vitro via targeting APCs. We further tested this possibility in vivo. We established B16-F10 tumor in C57L/BL6 mice and conducted intratumoral injection of dead tumor cells from OVA-B16-F10 and OVA- Stc1OE B16-F10 cells. One day later, CSFE labeled OT-I cells were intravenously transferred into these tumor bearing mice. On day 3, we examined CFSE dilution in OT-I cells in tumor draining lymph nodes. We detected reduced percentages of OT-I cells in divisions 4 to 6 in mice receiving dead Stc1OE B16-F10 cells compared to those receiving dead B16-F10 cells, as shown by CFSE dilution (Fig. 4M–O). Altogether, tumor STC1 abrogates tumor immunogenicity via targeting APCs.
Figure 4. STC1 abrogates tumor immunogenicity via targeting APCs.
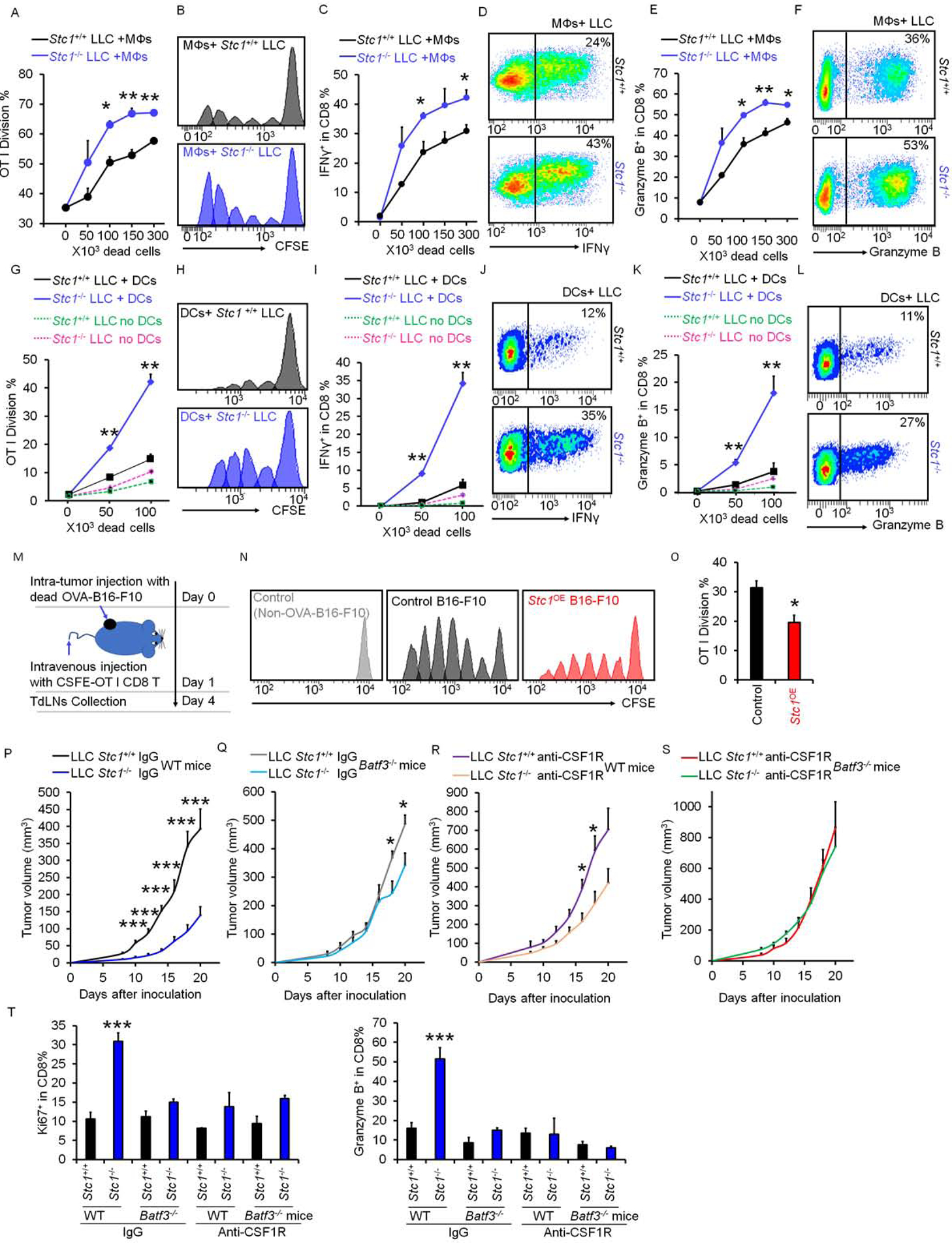
(A-L) Effect of Stc1 on OT-I cell activation in vitro. Stc1+/+ or Stc1−/− LLC cells were loaded with OVA and killed with UV-irradiation. CFSE-labeled OT-I cells were cultured with different numbers of dead LLC cells in the presence of macrophages (A-F) or DCs (G-L) for 3 days. CSFE dilution (A-B; G-H), IFNγ+ (C-D; I-J), and granzyme B+ (E-F; K-L) OT-I cells determined by FACS. Data are presented as mean ± SEM, 2 tail T-test was used for two-way comparisons (n = 3–5, *p < 0.05, **p < 0.01).
(M-O) Effect of Stc1 on OT-I cell activation in vivo. B16-F10 tumor bearing mice initially received intratumoral injection of dead tumor cells from OVA loaded-B16-F10 cells and OVA loaded-Stc1OE-B16-F10 cells, and were subsequently intravenously transfused with CSFE labeled OT-I cells (M). CFSE dilution and OT-I cell divisions in TdLNs (N, O) were determined by FACS. Data are presented as mean ± SEM, 2 tail T-test was used for two-way comparisons (n = 3, *p < 0.05).
(P-S) Effect of APCs on tumor growth curves. Stc1+/+ and Stc1−/− LLC cells were inoculated into Batf3+/+ or Batf3−/− mice. Starting 3 days before tumor inoculation, anti-CSF1R or isotype control antibodies were given every 3 days (n = 8). Data are shown as mean ± SEM, 2 tail t-test was used for two-way comparisons (P-R) (*p < 0.05, **p < 0.01).
(T) Percentages of Ki67+ and granzyme B+ CD8+ T cells in Stc1+/+ and Stc1−/− LLC tumor tissues from wild type or Batf3−/− mice under anti-CSF1R or isotype control antibody treatment (***p < 0.001, Stc1+/+ vs. Stc1−/− LLC tumor; p < 0.05, wild type vs Batf3−/− mice; p < 0.001, anti-CSF1R vs. isotype control antibodies).
See also Figure S4.
We next examined the in vivo impact of tumor STC1 on the immune responses mediated by different APC subsets, including DCs and macrophages. DC1s are genetically absent in Batf3−/− mice (Hildner et al., 2008) (Fig. S4I–M). Administration of anti-CSF1R mAb resulted in macrophage deletion (MacDonald et al., 2010, Yu et al. in press) (Fig. S4J–M). We inoculated Stc1+/+ and Stc1−/− LLC cells into Batf3−/− mice and wild type littermates (Batf3+/+) with or without anti-CSF1R mAb treatment. We observed that deletion of DC1s and macrophages alone partially, and simultaneous deletion of both DC1s and macrophages completely, abrogated the pro-tumor effect of STC1 as shown by tumor volume changes (Figure 4P–S). Accompanying with this, when compared to DC1 or macrophage deletion alone, simultaneous deletion of both DC1s and macrophages maximally reduced CD8+ T cell proliferation and function as shown by Ki67 and granzyme B expression (Fig. 4T). Thus, tumor STC1 may target both DCs and macrophages to abrogate tumor immunogenicity in tumor bearing hosts.
Tumor STC1 traps CRT to inhibit macrophage function
The phagocytosis of dead tumor cells is an initial step for APCs to capture, process, and present antigens to T cells (Houde et al., 2003; Joffre et al., 2012). To dissect the mechanism by which tumor STC1 downregulates tumor immunogenicity via targeting APCs, we hypothesized that tumor STC1 affected the nature of APC phagocytosis and in turn impaired APC-mediated antigen presentation and T-cell activation. To test this hypothesis, we cultured macrophages with fluorescently labeled dead B16-F10 cells or Stc1OE B16-F10 cells. We dynamically monitored fluorescent accumulation within macrophages. Compared to parental cells, there was less fluorescent uptake in macrophages cultured with dead B16-F10 Stc1OE cells throughout the experimental observation period (Fig. 5A). This data suggests that tumor STC1 inhibits engulfment and phagocytosis of dead tumor cells.
Figure 5. Tumor STC1 traps CRT to inhibit macrophage function.
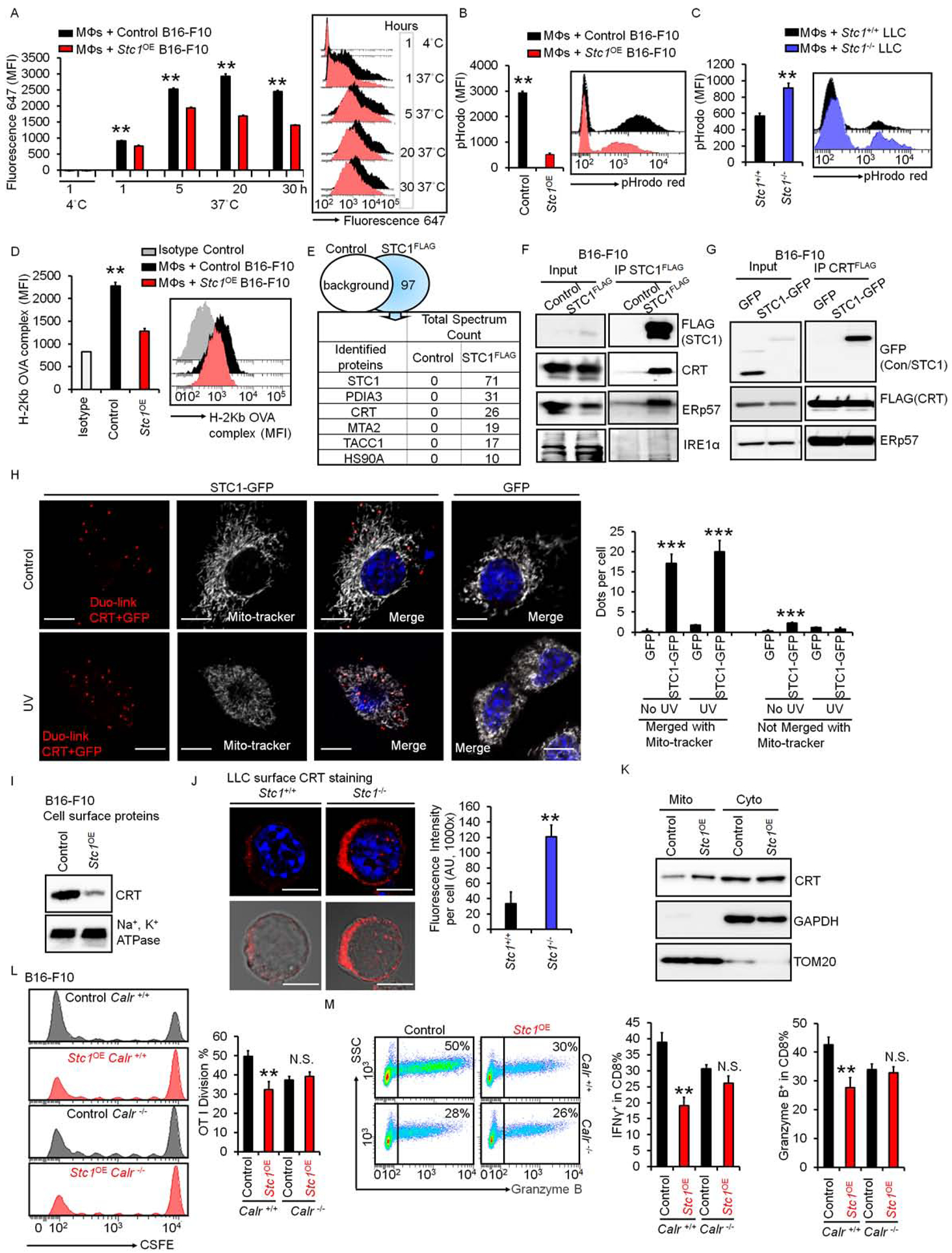
(A) Effect of Stc1 on macrophage-mediated phagocytosis. Macrophages were incubated with dead cells from fluor-647 labeled B16-F10 cells and fluor-647 labeled Stc1OE B16-F10 cells. Mean fluorescence intensity (MFI) of fluorescence 647 in macrophages, gated on CD11b+ cells, determined by FACS. Data are presented as mean ± SEM, 2 tail T-test was used for two-way comparisons (n = 6, *p < 0.05, **p < 0.01).
(B-C) Effect of Stc1 on macrophage-mediated bead up-taking. Macrophages were incubated with dead cells from B16-F10 cells and Stc1OE B16-F10 cells (B) or Stc1+/+ and Stc1+/+ LLC cells (C) for 20 hours. pHrodo™-SE labeled 3 μm latex beads were added for 1 hour. Red pHrodo signals in macrophages were determined by FACS. Data are shown as mean ± SEM (n = 4, *p < 0.05, **p < 0.01).
(D) Effect of Stc1 on antigen presentation. Macrophages were incubated with dead cells from OVA-loaded B16-F10 cells (control) and OVA-loaded Stc1OE B16-F10 cells for 48 hours. Surface OVA-binding-H2b complex expression (MFI) in macrophages were determined by FACS (n = 3, **p < 0.01).
(E) Mass spectrum showing STC1 interactive proteins. FLAG-IP was conducted in dead cells from STC1-FLAG expressing B16-F10 cells. Mass spectrum was subsequently performed in the FLAG-IP proteins. Control, cell lysates from B16-F10 cells without STC1-FLAG. Top 5 hits are shown.
(F) Interaction between endogenous CRT and STC1. Co-IP of STC1-FLAG was conducted with endogenous CRT, ERp57, and IRE1α in B16-F10 cells. One of 2 representative experiments is shown.
(G) Interaction between exogenous CRT and STC1. Co-IPs of CRT-FLAG with STC1-GFP and ERp57 were performed in cell lysates from UV-treated or non-treated B16-F10 cells. One of 2 representative experiments is shown.
(H) Duo-link showing the interactions (Red) of CRT and STC1-GFP, co-localizing with mito-tracker (white) in B16-F10 cells transfected with GFP or STC1-GFP with or without UV-treatment. Scale bars: 10 μm. Duo-link dots per cell merged or unmerged with mito-tracker were counted from over 20 images, mean ± SEM (n = 20, ***p < 0.001, STC1-GFP vs. GFP; # p <0.05, UV vs. No UV treatment).
(I) Membrane CRT in B16-F10 cells. UV-treated B16-F10 and Stc1OE B16-F10 cells were labeled with biotin. Western blot showed cell membrane CRT and Na+, K+-ATPase α1 in biotin-labeled proteins. One of 2 representative experiments is shown.
(J) Membrane CRT in UV-treated Stc1+/+ and Stc1−/− LLC cells. Confocal images showed membrane CRT expression. Scale bars: 10 μm. The intensity of CRT expression was analyzed through ImageJ software. Data are shown as mean ± SEM (n = 12, **p < 0.01).
(K) Western blots showing CRT distribution in mitochondria (TOM20) and cytosol (GAPDH) from UV-treated B16-F10 cells and Stc1OE B16-F10 cells. Mito, mitochondria; Cyto, cytosol. One of 2 experiments is shown.
(L-M) Effect of Stc1 on T cell activation in the context of Calr. Calr+/+ or Calr−/− vehicle control and Stc1OE B16-F10 cells were loaded with OVA and killed by UV-irradiation. CFSE-labeled OT-I cells were cultured with different numbers of dead B16-F10 cells with macrophages for 3–4 days. CSFE dilution (L), and granzyme B+ and IFNγ+ (M) OT-I cells were determined by FACS. Data are presented as mean ± SEM, 2 tail T-test was used for two-way comparisons (n = 3–5, **p < 0.01).
See also Figure S5.
We next examined the effect of tumor STC1 on the persistency of macrophage-mediated engulfment and phagocytosis (Wang et al., 2017). Similar to the above experiment (Fig. 5A), we incubated macrophages with dead B16-F10 and Stc1OE B16F10 cells for 20 hours, then pulsed pHrodo-SE beads for 20 minutes and chased for 40 minutes. pHrodo-SE dye is pH sensitive and increases fluorescence in acidic phagosomes (Savina et al., 2006). We monitored red fluorescence in macrophages and observed fewer beads in the phagosomes in macrophages cultured with dead B16-F10 Stc1OE cells than those cultured with dead B16-F10 cells (Fig. 5B). We performed similar experiments with dead Stc1−/− and Stc1+/+ LLC cells. In support of our results in B16-F10 cells, there were fewer beads in the phagosomes in macrophages cultured with dead Stc1+/+ LLC cells than dead Stc1−/− LLC cells (Fig. 5C). The data suggest that tumor STC1 potentially alters persistent macrophage-mediated phagocytosis. To additionally validate this possibility, we stained macrophages with LysoTracker™ Deep Red, a lysosome indicator dye. Confocal microscope revealed the beads within macrophages (Fig. S5A). The intensities of lysotracker-fluorescence reflected the maturation status of phagosomes. In macrophages incubated with dead Stc1OE B16-F10 cells, compared to those cultured with dead B16-F10 cells, we detected a decrease in lysotracker fluorescence intensity at the bead areas in macrophage phagosomes, suggesting a reduced phagosome maturation (Fig. S5A). Altogether, the results suggest that tumor STC1 negatively regulates the initiation and persistency of macrophage-mediated engulfment and phagocytosis of dead tumor cells.
We further questioned if the negative role of tumor STC1 on macrophage phagocytosis is involved in impaired antigen presentation. We cultured macrophages with dead tumor cells from vector expressing OVA-B16-F10 cells and Stc1OE OVA-B16-F10 cells. We detected lower levels of OVA-MHC-I binding-complexes on macrophages cultured with dead Stc1OE B16-F10 cells compared to those cultured with dead B16-F10 cells (Figure 5D). Our previous experiments demonstrated a negative role of tumor STC1 in antigen-specific T-cell activation (Figure 4). Altogether, the data suggest that tumor STC1 targets macrophage phagocytosis, resulting in a reduced antigen presentation and T-cell activation.
We next investigated how tumor STC1 affected macrophage phagocytosis. Given recombinant STC1 had no direct effect on APC-mediated T-cell activation, we hypothesized that tumor STC1 may indirectly regulate macrophage phagocytosis through an interaction partner. To identify the potential partners of STC1 at the protein level, we performed mass spectrometry (MS) on B16-F10 tumor cells stably expressing FLAG-tagged STC1. B16-F10 tumor cells served as controls (Figure 5E). We detected 97 specific bindings with STC1 (Table S1). Among them, the top 5 binding proteins were PDIA3 (protein disulfide isomerase associated 3, also named as ERp57), MTA2 (metastasis-associated gene family, member 2), CRT, TACC1 (transforming, acidic coiled-coil containing protein 1), and HSP90a (heat shock protein 90, alpha) (Fig. 5E). Among these 5 proteins, membrane CRT has been reported to facilitate APC phagocytosis (Obeid et al., 2007). Thus, we explored a potential role of CRT in macrophage phagocytosis-regulated by tumor STC1. We initially validated the binding of STC1 to CRT. We transfected B16-F10 cells with STC1-FLAG and immunoprecipitated with STC1-FLAG. STC1-FLAG immunoprecipitation (IP) revealed that endogenous CRT and ERp57, but not IRE1a, interacted with STC1 (Fig. 5F). Indeed, the interaction between ERp57 and CRT has been previously reported in mouse CT26 cells (Panaretakis et al., 2008). We ectopically expressed CRT-FLAG and STC1-GFP in B16-F10 cells. Following IP with CRT-FLAG, the immunoblot of STC1-GFP confirmed an interaction between CRT and STC1 (Fig. 5G). Additionally, STC1-GFP did not affect the interaction between CRT and ERp57 (Fig. 5G). Then, we transfected B16-F10 cells with GFP-labeled STC1, and examined the intracellular localizations of STC1 and CRT. Confocal microscopic studies showed that STC1 was co-localized with apoptosis-inducing-factor (AIF), a mitochondria marker (Fig. S5B). This suggests that STC1 is localized in the mitochondrial area. CRT can be localized in the areas of endoplasmic reticulum (ER) (Panaretakis et al., 2008) and mitochondria (Shan et al., 2014). We found a co-localization of CRT with mito-tracker in B16-F10 cells (Fig. S5C). As expected, STC1 was also localized with mito-tracker in B16-F10 cells. Interestingly, we observeda co-localization of STC1 and CRT in mitochondria in B16-F10 cells (Fig. S5D). Next we used Duolink™ technology and additionally validated the co-localization of STC1 and CRT in the mitochondrial area (Fig. 5H). In addition to the mitochondrial marker in the microscopic studies, we also employed the Co-IP experiments to further test the interaction of STC1 and CRT with a specific mitochondrial protein. Based on STC1 mitochondrial localization (Fig. S5B) and STC1 binding protein profile (Table S1), we selected SDHB, a mitochondrial protein in the Co-IP experiments. As expected, following CRT-FLAG IP, we found CRT interacted with SDHB (Fig. S5E). The data suggest that tumor STC1 interacts with and traps CRT in the mitochondrial area.
We assessed whether tumor STC1 affected the levels of CRT in different organelles. Western blot showed that over expression of STC1 did not alter the levels of CRT in whole cell lysates as compared to controls (Fig. S5F). However, we detected lower levels of CRT in cell membrane in Stc1OE B16-F10 cells than B16-F10 cells (Fig. 5I) or in Stc1OE MC38 cells than vehicle control cells (Fig. S5G). As assessed by confocal microscope, we detected lower levels of membrane CRT in Stc1+/+ LLC cells than Stc1−/− LLC cells (Fig. 5J). Furthermore, we treated B16-F10 cells with oxaliplatin to induce tumor cell death. Again, there were lower levels of CRT in cell membrane in Stc1OE B16-F10 cells than B16-F10 cells (Fig. S5H), whereas the expression levels of CD47 were comparable on Stc1OE and B16-F10 cells (Fig. S5I). Then, we assessed whether tumor STC1 affected the distribution of CRT in different organelles. We enriched lysosomes and ER from UV irradiated Stc1OE and vector control B16-F10 cells through ultracentrifugation with different Opti-prep gradients. In Stc1OE tumors and control tumors, we detected similar amounts of CRT in lysosome-enriched compartments as shown by LAMP1, and similar amounts of CRT in ER-enriched compartments as shown by Bip expression (Figure S5J). However, there were higher amounts of CRT in mitochondria fraction in Stc1OE tumors compared to control tumors (Figure 5K). The data confirm that tumor STC1 interacts with CRT, traps CRT in mitochondria, and reduces membrane levels of CRT.
Membrane CRT functions as an “eat-me” signal and facilitates APC phagocytosis (Obeid et al., 2007). To test if CRT is essential for the effect of STC1 on macrophage phagocytosis, we genetically knocked out CRT in B16-F10 cells and Stc1OE B16-F10 cells. We cultured macrophages with dead cells from Calr+/+ or Calr−/− B16-F10 cells, and Calr+/+ or Calr−/− Stc1OE B16-F10 cells, and added pHrodo-SE labeled latex beads to macrophages. Consistent with previous results (Fig. 5B), there were fewer beads in phagosomes in macrophages incubated with B16-F10 Calr+/+ Stc1OE cells than B16-F10 Calr+/+ control cells. Genetic deletion of tumor CRT led to a comparable reduction of beads into phagosomes in macrophages incubated with Calr−/− Stc1OE B16-F10 cells and Calr−/− B16-F10 cells (Fig. S5K). To explore a role of CRT in CD8+ T cell response in the context of STC1, we cultured OT-I cells with macrophages in the presence of OVA-loaded dead tumor cells, including Calr+/+ and Calr−/− B16-F10 cells, and Calr+/+ and Calr−/− Stc1OE B16-F10 cells. Consistent with previous results (Fig. S4A–G), OT-I cells were less activated in the presence of Stc1OE B16-F10 cells, while Calr deficiency abolished the negative role of tumor STC1 in T cell proliferation and activation as shown by CFSE dilution and expression of granzyme B and IFNγ (Figure 5L–M). Thus, tumor STC1 regulates macrophage function via interacting with CRT, thereby reducing membrane CRT.
Finally, to gain a global understanding of the impact of tumor STC1 on APC function, we performed an RNA-sequencing study on macrophages engulfed tumor cells with or without Stc1 overexpression (GEO: GSE161813). We found expression of multiple gene signatures was decreased in macrophages engulfed Stc1OE tumors compared to control macrophages, including several gene sets closely related to actin cytoskeletal modeling (such as myosin filament, microtubule bundle formation, dynein complex, and synaptic vesicle membrane genes), calcium channel genes, chloride channel genes, and APC-co-stimulation and maturation molecules (Figure S5L–M). This may provide a potential explanation as to why STC1-tumors mediate a long-lasting impact on macrophage function, including phagocytosis and T cell activation.
Discussion
As the majority of patients with cancer are not responsive to immune checkpoint therapy, recent studies have extensively explored different layers of immune resistance mechanisms (Kalbasi and Ribas, 2020; Nagarsheth et al., 2017; O’Sullivan et al., 2019; Ribas and Wolchok, 2018). We have focused our studies on the cross-talk between immune cell subsets and tumor cells in the tumor microenvironment, with the goal of gaining a comprehensive understanding on the nature of tumor immune responses induced by immunotherapy. In this current work, we have discovered that tumor STC1 reversely correlates with checkpoint therapy efficacy in patients with cancer and is associated with poor patient survival across multiple cancer types.
Despite its unknown receptor and/or partner(s), and undefined mode of action, STC1 has been reported to be a hormone like glycoprotein and may play a role in wound healing, inflammation, and carcinogenesis (Liu et al., 2010; Wang et al., 2009; Yeung et al., 2012). Several studies have detected elevated levels of STC1 in cancers, such as breast cancer, colorectal cancer, and hepatic carcinoma (Tamura et al., 2011, Chan et al., 2017). Unexpectedly, we have found that STC1 is an immune regulatory molecule in the context of tumor immunity. We have functionally validated the relationship between STC1 and tumor immunity in multiple murine tumor models. Hence, our work has identified that tumor STC1 functions as a previously unappreciated intrinsic resistance mechanism in tumor immunity and immunotherapy.
In spite of an obvious immune suppressive role of tumor STC1 in vivo in multiple tumor bearing models and its negative impact on immunotherapy efficacy in patients with cancer, it has been challenging to dissect how STC1 impairs tumor immunity and endows immunotherapy resistance. We have discerned that tumor cells release STC1, and different levels of tumor cell released STC1 proportionally correlate with different sensitivities of tumor cells to checkpoint therapy in vivo (Lin et al., 2018). Thus, we hypothesized that STC1, similar to TGF-β or IL-10, mediates a direct immune suppressive effect on T cells. Unfortunately, regardless of antigen specific and polyclonal TCR-mediated stimulation, it appears that tumor cell-released and recombinant STC1 has failed to directly suppress T cell activation. Interestingly, when we stimulated T cells in the presence of APCs, along with Stc1+/+ or STC1 overexpressing dead tumor cells, but not Stc1−/−dead tumor cells, we observed a consistent inhibition of T cell activation. Notably, no free tumor STC1 is available in this T cell culture system. This result prompts us to speculate that tumor cell-associated STC1, but not tumor released STC1, affects APC function and alters APC-directed T cell activation.
To explore this possibility, we initially examined a potential role of tumor cell STC1 in APC-mediated phagocytosis. Phagocytosis is an early step in the course of APC-mediated antigen capturing, processing, and presentation (Joffre et al., 2012). Indeed, we have demonstrated that tumor cell STC1 negatively affects APC-mediated phagocytosis, accompanied by reduced antigen presentation. As it is unknown from current literature if STC1 alters APC function, and our experiments show no impact of tumor released and recombinant STC1 on antigen presentation, we have presumed that tumor cell-associated STC1 may interact with and regulate unknown molecule(s), which may be involved in controlling APC phagocytosis. In line with this possibility, our proteomic profiling has identified a close interaction between STC1 and CRT in tumor cells. Furthermore, the interaction of STC1 and CRT is largely co-localized in the mitochondria area; high expression of STC1 traps and retains CRT in the mitochondria area, minimizes membrane CRT levels, and results in reduced APC phagocytosis. In this regard, CRT, as a chaperone, may bind to newly synthesized glycoproteins to facilitate macrophage-mediated phagocytosis (Feng et al., 2018). Therefore, it is unsurprising for us to observe an interaction of CRT with STC1, a glycoprotein.
Previous reports have elucidated that tumor cells, particularly dead tumor cells, can express “don’t eat-me” signals, (such as CD47 and CD24), to avoid APC-mediated phagocytosis (Barkal et al., 2019; Feng et al., 2019; Majeti et al., 2009; Wang et al., 2019a). Cell membrane CRT is a key “eat-me” signal, secondary to phosphatidylserine (Gardai et al., 2005). Macrophages fail to capture and engulf CRT-deficient dead cells, even in the presence of normal phosphatidylserine activity (Gardai et al., 2005). In fact, CRT signal counterbalances the CD47–SIRPα axis, a “don’t eat-me” signal pathway, and functions as a pro-phagocytic signal for CD47-blockade-mediated phagocytosis (Chao et al., 2010). Recent study has shown that CRT surface exposure is crucial to determine immunogenicity versus non-immunogenicity of dead tumor cells. Along this line, some chemotherapeutic drugs, such as mitoxantrone and oxaliplatin, are able to induce membrane CRT, -thereby leading to immunogenic tumor cell death via membrane CRT-directed APC phagocytosis (Obeid et al., 2007). In an extensive search of novel checkpoints for cancer immunotherapy, several recent studies reveal that the CD47–SIRPα, CD24–Siglec-10, and Siglec-15 function as potential phagocytosis checkpoints, becoming potential novel targets for cancer immunotherapy (Barkal et al., 2019; Feng et al., 2019; Majeti et al., 2009; Wang et al., 2019a). In line with these important discoveries (Barkal et al., 2019; Feng et al., 2019; Majeti et al., 2009; Wang et al., 2019a), our work is a novel expansion in this active research area, indicating that STC1 may function as an intracellular “eat-me” signal blocker and could be a previously unappreciated phagocytosis checkpoint. Along a similar vein, small molecular inhibitors have been designed to target the interaction between P53 and MDM2 (Chène, 2003). Thus, targeting the interaction between STC1 and CRT may be an approach to sensitize cancer immunotherapy.
In summary, we have found that tumor STC1 dampens tumor immunity and immunotherapy via diminishing membrane exposure of CRT, -thereby impairing membrane CRT-directed APC phagocytosis and T cell activation. Given that STC1 counteracts the effect of CRT, we suggest that tumor STC1 behaves as a previously unknown intracellular “eat-me” signal blocker and functions as a potential phagocytosis checkpoint for cancer immunotherapy.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for materials should be directed to the Lead Contact: Weiping Zou (wzou@med.umich.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
RNA seq raw data and files have been deposited to GEO: GSE161813. The MS files in mzIdentML format have been deposited to the ProteomeXchange consortium: PXD023251.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Mouse melanoma cell line B16-F10 and lung cancer cell line LLC were purchased from ATCC. Mouse colon cancer cell line MC38 (Lin et al., 2018; Tanikawa et al., 2012) were previously reported. All cell lines were regularly examined for mycoplasma contamination.
Animals
All animal work was approved by the Institutional Animal Care and Use Committee at the University of Michigan. Mice of both sexes, between the ages of 6–10 weeks of age were used for the study. NOD.SCID γc deficient (NSG) mice, wild type C57BL/6J mice, and OT-I TCR transgenic mice, Batf3−/− (Batf3tm1Kmm/J) mice were obtained from The Jackson Laboratory. Pdcd1−/− mice were originally from Dr. Tasuku Honjo (Kyoto University) (Nishimura et al., 1998). All mice are maintained under pathogen-free conditions.
METHOD DETAILS
In vivo experiments
For MC38, B16-F10, and LLC tumor models, 106 tumor cells were subcutaneously injected on the right flank of male mice. Tumor diameters were measured using calipers. Tumor volume was calculated. Anti-PD-L1 and IgG1 isotype antibodies were given intraperitoneally at a dose of 100 μg per mouse on day 3 after tumor cells inoculation, then every 3 days for the duration of the experiment.
For the vivo macrophage depletion experiments, wild type C57BL/6J mice or Batf3−/− mice were treated with anti-CSF1R as described previously (MacDonald et al., 2010). Mice were pre-treated with 800 μg anti-CSF1R (clone AFS98, BioXCell) or IgG control (clone 2A3, BioXCell) 4 days before tumor inoculation, followed with 400 μg anti-CSF1R or IgG control every 3 days sustained throughout tumor progression.
Flow cytometry analysis (FACS)
Single cell suspensions were prepared from fresh mouse tumor tissues or tumor draining lymph nodes. Cells were stained with specific antibodies against mouse CD45 (30-F11), CD90 (53–2.1), and CD8 (53–6.7) to define CD45+CD90+CD8+ T cells. T cell cytokine expression was determined by intracellular staining; antibodies against mouse IFNγ (XMG1.2), TNFα (MP6-XT22), granzyme B (GB11), Ki67 (SolA15) from eBioscience or BD Biosciences were used. Macrophages were stained with anti-CD45 (30-F11) and CD11b (M1/70). All flow samples were acquired through LSR Fortessa (BD) and data were analyzed with DIVA software (BD Biosciences).
In vitro splenocytes activation
Splenocytes (106/ml) from C57BL/6J mice were activated with anti-CD3 (eBioscience, 5 μg/ml) and anti-CD28 (eBioscience, 2.5 μg/ml) or other indicated concentration in the presence of recombinant mouse STC1 (Creative Biomart Inc., 100 ng/ml) or tumor cell culture supernatants. The cells were subject to surface marker and intracellular cytokine staining and analyzed by FACS. In a different setting, splenocytes from OT-I TCR transgenic mice were cultured with OVA loaded dead tumor cells for 3 days. Supernatants from these cells were collected for IFNγ detection with ELISA (R&D, DY485).
Bone marrow-derived macrophages (MΦs) and dendritic cells (DCs)
Bone marrow was obtained from the hind legs of mice. Erythrocytes were lysed with Red Blood Cell Lysis Buffer (Sigma Aldrich). Bone marrow-derived macrophages were generated from bone marrow cells with M-CSF (20 ng/ml), culture medium was half changed every 3 days. On days 7 to 9, macrophages were collected for further experimentation (Lin et al., 2018). DCs were generated from bone marrow cells with FLT3L (100 ng/ml) in IMDM, 10%FBS (Theisen et al., 2018). On days 8–10, floating cells were enriched using XCR1-PE (Biolegend, ZET) antibody and PE Beads from Miltenyl Biotec.
CRISPR gene targeting
Gene targeting by CRISPR/Cas9 was accomplished by transfection with the guide sequence targeting mouse Stc1 and mouse Calr (Synthego) together with Cas9 Nuclease (NEB #M0646). Successful Stc1 targeting was determined by sequencing the cell clones and detecting STC1 in the culture of each clone. Multiple Stc1 deficient clones were pooled. Successful Calr knockout cell clones were determined by Western blot. Multiple Calr deficient clones were pooled. Guide sequence targeting mouse Stc1: 1# CUU GUCUCUUCCAGCUGAAG; 2# AGGCAGCGAACCACUUCAGC; 3# AGCCCCGCAGCCAACCU GCA. Guide sequence targeting mouse Calr: 1# UUUGGAUUCGACCCAGCGGU; 2# UU CGACCCAGCGGUUGGUCC; 3# CAGAUGCCUGGACCAACCGC.
Stc1 overexpression
MC38 or B16-F10 cells were transfected with lentivirus encoding Stc1 (MR203105L1, Origene™ Technologies) or scrambled control. After transfection, the transfected cells were selected by puromycin for 7 days, tested for Stc1 expression, and cultured for the in vivo experiments. Stc1OE and scrambled control cell lines were further constructed on B16-F10 sub cell line, which was pooled with Stc1 knockout cell clones and used for experiments in Figure 4 to 5.
Antigen presentation and T cell activation assay
For detecting surface OVA-peptide SIINFEKL presentation on H-2Kb (MHC-I), macrophages or DCs were cultured with dead tumor-OVA cells for 48 hours and stained with antibodies against OVA-peptide-MHC-I binding epitope (eBioscience™, 25-D1.16) and CD11b (BD Biosciences, M1/70). Cells were analyzed by FACS. For T cell activation assay (Theisen et al., 2018), tumor cells were osmotically loaded with 10 mg/ml ovalbumin (OVA, Sigma Aldrich), then irradiated with UV in 10 mm dishes. 4 ×104 macrophages or DCs were co-cultured with 2 × 105 CFSE-labeled OT-I cells and dead tumor cells at indicated number in flat 96 well plates. After 3 days, cells were collected and analyzed for CFSE dilution and cytokine expression in OT-I cells.
Phagocytosis assay
We have established 3 complementary and confirmatory experiments to examine the role of tumor STC1 in macrophage-mediated phagocytosis. In the first set of experiments, we used flow cytometry analysis to kinetically test the impact of tumor STC1 on macrophage-mediated phagocytosis. 2 ×105 macrophages were incubated with 106 AlexaFluor647 labeled dead tumor cells from Stc1+/+, Stc1−/−, and Stc1OE tumor cells. By gating on CD11b, macrophages were kinetically sampled to determine the mean fluorescence intensity (MFI) of AlexaFluor647 in different time points by FACS.
In the second set of experiments, we used flow cytometry analysis in combination with pH sensitive pHrodo™-SE (Invitrogen™, P36600) labeled beads to assess the effect of tumor STC1 on macrophage phagosome maturation. As pHrodo-SE dye is sensitive to acidic condition and appears brightly red fluoresce in acidic phagosomes (Savina et al., 2006), these beads enable accurate monitoring whether the beads were in mature acidic lysosome compartments in macrophages. Similar to the first experiments, macrophages were incubated with different dead tumor cells for 20 hours. Then, pHrodo™-SE labeled 3 μm latex beads (Sigma Aldrich) were pulsed for 20 minutes. Macrophages were extensively washed in cold PBS and chased for 40 minutes (Savina et al., 2006). By gating on CD11b, the MFI of red pHrodo fluorescence in macrophages was analyzed by FACS.
In the third set of experiments, we used confocal microscopic analysis in combination with bead-uptake and lysosome labeling to validate the effect of tumor STC1 on macrophage phagosome maturation. Similar to the second experiments, macrophages were incubated with different dead tumor cells for 20 hours. Then, 3 μm latex beads (Sigma Aldrich) were pulsed for 20 minutes. Macrophages were extensively washed in cold PBS and chased for 40 minutes. Lysotracker dye was added for the last 30 minutes of chasing time. Macrophages were extensively washed and fixed with paraffin. Immunofluorescence images were acquired with Nikon A1 inverted confocal microscope. Fluorescence intensity of Lysotracker dye was determined by FIJI-ImageJ software at the bead areas in individual macrophages with single bead per cell (Savina et al., 2006).
Real time PCR
Total RNA was extracted using TRIzol™ LS Reagent (Invitrogen™, 10296010) according to the manufacturer’s instructions. cDNA synthesis was performed with 0.5~1 μg of total RNA using RevertAid RT Reverse Transcription Kit (Invitrogen™, K1691). mRNA levels were measured with gene-specific primers using the SYBR™ Green PCR Master Mix (Invitrogen™, 4368702). The results were normalized to GAPDH. The primers are shown as follows:
mouse Stc1 forward: AGGAGGACTGCTACAGCAAGCT
mouse Stc1 reverse: TCCAGAAGGCTTCGGACAAGTC
mouse Inhba forward: TGCTGCTCAAGTGCCAATAC
mouse Inhba reverse: AGCAAAAGTCGTGTGGTTGC
mouse Cytl1 forward: TTCAGAGCCTGAGGATTCCTGT
mouse Cytl1 reverse: CTTCCGCACTCTGTCCTTCA
mouse Runx2 forward: TCGCCTCACAAACAACCACA
mouse Runx2 reverse: CTGCTTGCAGCCTTAAATATTCCT
mouse Metrnl forward: TAAGACTGTTGGTGCGGGAC
mouse Metrnl reverse: GCCTCGGACAACAAAGTCAC
mouse Dlg4 forward: GATGAAGACACGCCCCCTCT
mouse Dlg4 reverse: CTGCAACTCATATCCTGGGGCTT
mouse Plin2 forward: GCTCTCCTGTTAGGCGTCTC
mouse Plin2 reverse: AACAATCTCGGACGTTGGCT
mouse Gapdh forward: CATCACTGCCACCCAGAAGACTG
mouse Gapdh reverse: ATGCCAGTGAGCTTCCCGTTCAG
ELISA and ELISPOTS detection
ELISA:
Culture mediums were collected for STC1 (R&D, DY2958) or IFNγ (R&D, DY485) detection following the kit manufacturers’ instructions.
IFNγ ELISPOTS:
Cells collected from tumor drained lymph nodes from MC38-OVA tumor bearing mice. 250,000 cells were cultured with or without OVA peptide for 48 hours and processed with detection antibody and following the kit manufacturer’s instructions (R&D, EL485).
Immune blotting and cell surface protein detection
For immunoblot analysis, whole-cell lysates were prepared in RIPA lysis buffer (Thermo Scientific™, 89900) containing Halt™ Protease Inhibitor Cocktail (Thermo Scientific™, 78429). The protein concentrations were determined by BCA Protein Assay Kits (Pierce, 23227). Cell surface protein was prepared using Cell Surface Protein Isolation Kit (Thermo Fisher, 89881) and assessed via immunoblot. 20–60 μg protein samples were loaded into SDS-PAGE and transferred to polyvinylidene difluoride membrane. Immune blotting antibodies included anti-CRT (D3E6), anti-ERp57 (G117), anti-IRE1α (14C10), anti-GAPDH (D16H11), anti-Na+, K+-ATPase (3010), anti-BiP (C50B12), anti-LAMP1 (C54H11), and anti-GFP (4B10) from Cell Signaling Technology, Inc, or anti-CD9 antibody (EPR2949) from Abcam.
Co-immunoprecipitation (Co-IP)
Cell pellets were lysed with coimmunoprecipitation buffer (10 mM Tris/Cl, pH 7.5, 250 mM NaCl, 0.5 mM EDTA, 0.5 % Nonidet™ P40 Substitute, and 0.15 % Triton™ X-100) and IP Magnetic Agarose overnight at 4°C with shaking, and were washed with Co-IP buffer. Interaction complexes were competitively eluted using 3x DYKDDDDK Peptide (Pierce™, A36806), 4X Bolt™ LDS Sample Buffer (Invitrogen™, B0008) was added, and then boiled for 10 minutes. The samples were subjected to SDS-PAGE and detected with immune blotting.
Mass spectrometry (MS) analysis
Whole-cell lysates were prepared from UV-irradiated, FLAG-tagged STC1 or control vector expressing B16-F10 tumor cells. The cell lysates were subjected to IP with anti-FLAG Magnetic Agarose (Pierce, A36797) overnight at 4°C. Interaction complexes were separated from the beads by competitive elution using 3x DYKDDDDK Peptide (Pierce™, A36806). The samples were analyzed using LC/MS/MS on Orbitrap Velos mass spectrometer. Product ion data were searched against the NCBI protein database using the Mascot and X-Tandem search engines. Mascot output files were parsed into the Scaffold program (www.proteomesoftware.com) for filtering to assess false discovery rates and allow only correct protein identifications.
Immunofluorescence analysis and imaging
UV-irradiated tumor cells were fixed with 4% paraformaldehyde. For surface CRT detection, cells were blocked with 5% goat serum without permeabilization, stained with primary antibody anti-CRT (1:100, CST, D3E6), and followed by secondary antibodies conjugated with Alexa Fluro594 (1:1000, Thermo Scientific, A32740). For intracellular protein detection, fixed cells were permeabilized in methanol for 20 minutes, blocked with 5% goat serum, and stained with primary antibodies anti-GFP (1:400, CST, 4B10), anti- CRT (1:100, CST, D3E6), and anti-AIF (1:400, CST, D39D2), followed by secondary antibodies conjugated with Alexa Fluro488 (1:1000, Thermo Scientific, A32723) and Alexa Fluro594 (1:1000, Thermo Scientific, A32740). In some cases, cells were pre-labeled with Mito-Tracker™ Deep Red FM (Invitrogen™, M22426) before fixation. The cell nuclei were stained with DAPI. Slides were mounted with ProLong™ Gold Antifade (Invitrogen™, P36931). Fluorescence images were obtained by using a laser scanning confocal imaging system (Nikon A1 inverted High Sensitivity Confocal).
Subcellular fractionation studies
B16-F10 cells grown in 15 cm plates to near confluence were treated with UV and cultured for other 3 h, then used for next experiments.
Lysosome and ER enrichment:
The cells were homogenized in ice-cold isolation buffer (250 mM sucrose, 5 mM Hepes, pH 7.4) supplemented with protease inhibitors using a Dounce homogenizer, then subjected to centrifugation for 10 minutes at 900 × g to remove nuclei fraction. The supernatant was then centrifuged for 1 hour at 100,000 × g to separate cytosol from the post-nuclear particulate fraction pellets. The pellets were resuspended in 0.5 ml of isolation medium. A discontinuous gradient was prepared using 25, 20, 15, and 10% Opti-prep™. The resuspended pellet was overlaid onto the discontinuous gradient and centrifuged at 100,000 × g for 3 hours at 4 °C. Fractions were collected from the top of the gradient. Lysosome and ER enrichment was determined by Western blotting with lysosome and ER markers.
Mitochondria isolation:
The crude mitochondria were enriched by using a Mitochondria isolation kit (Thermo Scientific™, 89874). Then, the crude mitochondria were further purified through centrifuged at 100,000 × g for 1 hour on 25, 20, 15, and 10% Opti-prep™ gradients. Purified mitochondria were determined by Western blotting with mitochondrial marker.
RNA-sequencing (seq)
Macrophages were loaded with Stc1OE or control B16F-F10 corpses at 1 to 5 ratios. After culture for 20 hours, unbound tumor corpses were washed away. Total RNA was isolated using the Direct-zol RNA Miniprep Plus kit (Zymo Research) and mRNA libraries were prepared using the Illumina TruSeq technology. Libraries were then sequenced using an Illumina NextSeq sequencer at 150 bp, paired end reads with approximately 30 million reads per sample. Four independent experiments were sequenced for each condition. RNA-seq Fastq data were mapped to the reference genome mm10 using Hisat2 (Kim et al., 2015). The reads were counted with HTseq (Anders et al., 2015), and the differential expression between experimental groups was quantified using DESeq2 (Love et al., 2014). Gene categories were chosen according to the similarity of primary function or GSEA reference. Differential gene expression was calculated with DESeq2; significance was assigned with adjusted P value per algorithm DESeq2 as < 0.05. RNA seq data can be found at GSE161813.
QUANTIFICATION AND STATISTICAL ANALYSIS
Bioinformatics analysis
Transcriptomic analysis was conducted in two cohorts of patients with melanoma treated with immunotherapy (Hugo et al., 2016; Riaz et al., 2017). Statistical methods were chosen according to the original data sources: Unpaired, nonparametric t-test was used to compare the responders(R), including patients with complete response (CR), partial response (PR), stable disease (SD), and non-responders (NR), as well as patients with progressive disease (PD). T cell activation gene signature was calculated as previously reported (Wang et al., 2019b).
Statistical methods and software
Wilcoxon rank-sum and 2 tailed t tests were used to compare two independent groups. Survival functions were estimated by the Kaplan-Meier methods and compared using the log-rank test. All analyses were done using GraphPad Prism. P < 0.05 was considered significant. Sample size was determined on the basis of animal experimental trials and in consideration of previous publications on similar experiments to allow for confident statistical analysis. Unless noted, samples were independent biological replicates.
Supplementary Material
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| V500 Rat Anti-Mouse CD45 (30-F11) | BD Bioscience | Cat# 561487 |
| FITC Rat Anti-Mouse CD90.2 (53–2.1) | BD Bioscience | Cat# 553003 |
| Alexa Fluor® 700 Rat Anti-Mouse CD8a (53–6.7) | BD Bioscience | Cat# 557959 |
| BV786 Rat Anti-Mouse IFNγ (XMG1.2) | BD Bioscience | Cat# 563773 |
| PE-Cy™7 Rat Anti-Mouse TNFα (MP6-XT22) | BD Bioscience | Cat# 557644 |
| PE Mouse Anti-Human Granzyme B (GB11) | BD Bioscience | Cat# 561142 |
| BV786 Rat Anti-Mouse CD11b (M1/70) | BD Bioscience | Cat# 740861 |
| Armenian hamster Anti-Mouse CD3e (145–2C11) | Thermo Fisher | Cat# 14-0031-82; RRID: AB 467048 |
| Syrian hamster Anti-Mouse CD28 (37.51) | Thermo Fisher | Cat# 14-0281-82; RRID: AB 466413 |
| PE Mouse anti-mouse/rat XCR1 Antibody (ZET) | BioLegend | Cat# 148204; RRID: AB 2563842 |
| PE Mouse anti-OVA257–264 (SIINFEKL) peptide bound to H-2Kb (eBio25-D1.16) | Thermo Fisher | Cat# 12-5743-82; RRID: AB 925771 |
| Rabbit monoclonal Anti-Calreticulin (D3E6) | Cell Signaling Technology | Cat# 12238; RRID: AB 2688013 |
| Rabbit monoclonal Anti-ERp57 (G117) | Cell Signaling Technology | Cat# #2881; RRID: AB 2160840 |
| Rabbit monoclonal Anti-IRE1α (14C10) | Cell Signaling Technology | Cat# 3294 |
| Rabbit monoclonal Anti-GAPDH (D16H11) | Cell Signaling Technology | Cat# 5174; RRID: AB 11129865 |
| Rabbit monoclonal Anti -Na+,K+-ATPase (3010) | Cell Signaling Technology | Cat# 3010 |
| Rabbit monoclonal Anti-BiP (C50B12) | Cell Signaling Technology | Cat# 3177; RRID: AB 2119845 |
| Rabbit monoclonal Anti-LAMP1 (C54H11) | Cell Signaling Technology | Cat# 3243; RRID: AB 2134478 |
| Mouse Anti-GFP (4B10) | Cell Signaling Technology | Cat# 2955; RRID: AB_1196614 |
| Rabbit monoclonal Anti-AIF (D39D2) XP® mAb | Cell Signaling Technology | Cat# 5318 |
| Rabbit monoclonal Anti-CD9 (EPR2949) | Abcam | Cat# ab92726 |
| InVivoMab Anti-mouse CSF1R (CD115) antibody (AFS98) | Bio X Cell | Cat# BE0213; RRID: AB 2687699 |
| InVivoMAb rat IgG2a isotype control (2A3) | Bio X Cell | Cat# BE0089 |
| InVivoPlus Anti-mouse PD-L1 (10F.9G2) | Bio X Cell | Cat# BE0101, RRID: AB_10949073 |
| InVivoPlus rat IgG2b isotype control (LTF-2) | Bio X Cell | Cat# BP0090 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Mouse STC1 | Creative Biomart Inc. | Cat# STC1–16117M |
| Mouse Recombinant M-CSF | Stem Cell Technologies | Cat# 78059 |
| Recombinant Mouse Flt-3 Ligand/FLT3L Protein | R&D Systems | Cat# 427-FL-025 |
| EnGen® Spy Cas9 NLS | NEB | Cat# M0646T |
| Ovalbumin Peptide (257–264) chicken | Sigma Aldrich | Cat# S7951 |
| Ovalbumin | Sigma Aldrich | Cat# A5503 |
| 3x DYKDDDDK Peptide | Thermo Fisher | Cat# A36806 |
| Critical Commercial Assays | ||
| Mouse IFN-gamma DuoSet ELISA | R&D Systems | Cat# DY485 |
| Mouse IFN-gamma ELISpot Kit | R&D Systems | Cat# EL485 |
| Human STC1 DuoSet ELISA | R&D Systems | Cat# DY2958 |
| Anti-PE MicroBeads | Miltenyi Biotec | Cat# 130-048-801 |
| pHrodo™ Red, SE | Thermo Fisher | Cat# P36600 |
| Pierce™ Cell Surface Protein Isolation Kit | Thermo Fisher | Cat# 89881 |
| Anti-FLAG Magnetic Agarose | Thermo Fisher | Cat# A36797 |
| Mito-Tracker™ Deep Red FM | Thermo Fisher | Cat# M22426 |
| Opti-prep™ Density Gradient Medium | Sigma Aldrich | Cat# D1556 |
| Mitochondria isolation kit (crude) | Thermo Fisher | Cat# 89874 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE161813 |
| IP-based mass spectrometry | This paper | PXD023251 |
| Experimental Models: Cell Lines | ||
| Mouse cell line: B16-F10 | ATCC | CRL-6475 |
| Mouse cell line: LLC | ATCC | CRL-1642 |
| Mouse cell line: MC38 | (Tanikawa et al., 2012) | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: NOD.SCID γc deficient (NSG) | The Jackson Laboratory | JAX: 005557 |
| Mouse: C57BL/6J | The Jackson Laboratory | JAX: 000664 |
| Mouse: OT-I TCR transgenic mice | The Jackson Laboratory | JAX: 003831 |
| Mouse: Pdcd1−/− mice | (Nishimura et al., 1998) | N/A |
| Mouse: Batf3−/− (Batf3tm1Kmm/J) | The Jackson Laboratory | JAX: 013755 |
| Oligonucleotides | ||
| sgRNA targeting sequence: Mouse Stc1 #1: CUUGUCUCUUCCAGCUGAAG | Synthego | N/A |
| sgRNA targeting sequence: Mouse Stc1 #2: AGGCAGCGAACCACUUCAGC | Synthego | N/A |
| sgRNA targeting sequence: Mouse Stc1 #3: AGCCCCGCAGCCAACCUGCA | Synthego | N/A |
| sgRNA targeting sequence: Mouse Calr #1: UUUGGAUUCGACCCAGCGGU | Synthego | N/A |
| sgRNA targeting sequence: Mouse Calr #2: UUCGACCCAGCGGUUGGUCC | Synthego | N/A |
| sgRNA targeting sequence: Mouse Calr #3: CAGAUGCCUGGACCAACCGC | Synthego | N/A |
| Primer: mouse Stc1 forward: AGGAGGACTGCTACAGCAAGCT | IDT | N/A |
| Primer: mouse Stc1 reverse: TCCAGAAGGCTTCGGACAAGTC | IDT | N/A |
| Primer: mouse Inhba forward: TGCTGCTCAAGTGCCAATAC | IDT | N/A |
| Primer: mouse Inhba reverse: AGCAAAAGTCGTGTGGTTGC | IDT | N/A |
| Primer: mouse Cytl1 forward: TTCAGAGCCTGAGGATTCCTGT | IDT | N/A |
| Primer: mouse Cytl1 reverse: CTTCCGCACTCTGTCCTTCA | IDT | N/A |
| Primer: mouse Runx2 forward: TCGCCTCACAAACAACCACA | IDT | N/A |
| Primer: mouse Runx2 reverse: CTGCTTGCAGCCTTAAATATTCCT | IDT | N/A |
| Primer: mouse Metrnl forward: TAAGACTGTTGGTGCGGGAC | IDT | N/A |
| Primer: mouse Metrnl reverse: GCCTCGGACAACAAAGTCAC | IDT | N/A |
| Primer: mouse Dlg4 forward: GATGAAGACACGCCCCCTCT | IDT | N/A |
| Primer: mouse Dlg4 reverse: CTGCAACTCATATCCTGGGGCTT | IDT | N/A |
| Primer: mouse Plin2 forward: GCTCTCCTGTTAGGCGTCTC | IDT | N/A |
| Primer: mouse Plin2 reverse: AACAATCTCGGACGTTGGCT | IDT | N/A |
| Primer: mouse Gapdh forward: CATCACTGCCACCCAGAAGACTG | IDT | N/A |
| Primer: mouse Gapdh reverse: ATGCCAGTGAGCTTCCCGTTCAG | IDT | N/A |
| Recombinant DNA | ||
| Stc1 (NM_009285) Mouse Tagged ORF Clone | Origene™ Technologies | Cat# MR203105L1 |
| Software and Algorithms | ||
| BD FACSDiva™ Software | BD Bioscience | https://www.bdbiosciences.com/en-us/instruments/research-instruments/research-software/flow-cytometry-acquisition/facsdiva-software |
| Graphpad Prism 6.0 software | GraphPad Software, Inc. | http://www.graphpad.com/scientific-software/prism/ |
| Hisat2 | (Kim et al., 2015) | |
| HTSeq | (Anders et al., 2015) | |
| DESeq2 | (Love et al., 2014) | |
Highlights.
Tumor STC1 negatively correlates with immunotherapy efficacy and patient survival
STC1 interacts with CRT and traps CRT in mitochondria
STC1 impairs APC phagocytosis and T cell activation via trapping CRT
STC1 functions as an “eat-me” signal blocker and may be a phagocytosis checkpoint
Acknowledgements:
We thank Drs. Malini Raghavan and Joel Swanson for their scientific input. This work was supported in part by research grants from the NIH/NCI grants for WZ (CA248430, CA217648, CA123088, CA099985, CA193136, and CA152470) and the NIH through the University of Michigan Rogel Cancer Center Support Grant (P30CA46592).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no conflict of interest.
References:
- Anders S, Pyl PT, & Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics, 31(2), 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, Krishnan V, Hatakeyama J, Dorigo O, Barkal LJ, et al. (2019). CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 572, 392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian Y, Li W, Kremer DM, Sajjakulnukit P, Li S, Crespo J, Nwosu ZC, Zhang L, Czerwonka A, Pawłowska A, Xia H, Li J, Liao P, Yu J, et al. (2020). Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, Qiu J, Kretz O, Braas D, van der Windt GJ, et al. (2016). Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 166, 63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KK, Leung CO, Wong CC, Ho DW, Chok KS, Lai CL, Ng IO, and Lo RC (2017). Secretory Stanniocalcin 1 promotes metastasis of hepatocellular carcinoma through activation of JNK signaling pathway. Cancer Lett 403, 330–338. [DOI] [PubMed] [Google Scholar]
- Chao MP, Jaiswal S, Weissman TR, Alizadeh A, Gentles A, Volkmer J, Weiskopf K, Willingham S, Raveh T, Park C, et al. (2010). Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med 2, 63ra94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chène P (2003). Inhibiting the p53-MDM2 interaction: an important target for cancer therapy. Nature reviews. Cancer, 3, 102–109. [DOI] [PubMed] [Google Scholar]
- Feng M, Jiang W, Kim B, Zhang CC, Fu Y, and Weissman IL (2019). Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer 19, 568–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng M, Marjon KD, Zhu F, Weissman-Tsukamoto R, Levett A, Sullivan K, Kao KS, Markovic M, Bump PA, Jackson HM, et al. (2018). Programmed cell removal by calreticulin in tissue homeostasis and cancer. Nat Commun 9, 3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. (2016). Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 167, 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardai SJ,K,M, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, and Henson PM (2005). Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 123, 321–334. [DOI] [PubMed] [Google Scholar]
- Hildner K, Edelson BT, Purtha WE, et al. (2008) Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322,1097–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houde M, Bertholet S, Gagnon E, Brunet S, Goyette G, Laplante A, Princiotta MF, Thibault P, Sacks D, and Desjardins M (2003). Phagosomes are competent organelles for antigen cross-presentation. Nature 425, 402–406. [DOI] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. (2016). Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 165, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joffre OP, Segura E, Savina A, and Amigorena S (2012). Cross-presentation by dendritic cells. Nat Rev Immunol 12, 557–569. [DOI] [PubMed] [Google Scholar]
- Kalbasi A, and Ribas A (2020). Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol 20, 25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmaus PWF, Chen X, Lim SA, Herrada AA, Nguyen TM, Xu B, Dhungana Y, Rankin S, Chen W, Rosencrance C, et al. (2019). Metabolic heterogeneity underlies reciprocal fates of T(H)17 cell stemness and plasticity. Nature 565, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, & Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nature methods, 12(4), 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wang W, Zhang Y, Cieślik M, Guo J, Tan M, Green MD, Lin H, Li W, Wei S, et al. (2020). Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy. J Clin Invest, 134402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, Wei S, Zhao L, Vatan L, Wen B, et al. (2018). Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab 28, 87–103.e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Wei S, Hurt EM, Green MD, Zhao L, Vatan L, Szeliga W, Herbst R, Harms PW, Fecher LA, et al. (2018). Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J Clin Invest 128, 805–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Yang G, Chang B, Mercado-Uribe I, Huang M, Zheng J, Bast RC, Lin SH, and Liu J (2010). Stanniocalcin 1 and ovarian tumorigenesis. J Natl Cancer Inst 102, 812–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, & Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology, 15(12), 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald KP, Palmer JS, Cronau S, Seppanen E, Olver S, et al. (2010). An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood, 116, 3955–3963. [DOI] [PubMed] [Google Scholar]
- Maj T, Wang W, Crespo J, Zhang H, Wei S, Zhao L, Vatan L, Shao I, Szeliga W, Lyssiotis C, et al. (2017). Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol 18, 1332–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KDJ, Van-Rooijen N, and Weissman IL (2009). CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 138, 286–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal R, Samstein RM, Lee KW, Havel JJ, Wang H, Krishna C, Sabio EY, Makarov V, Kuo F, Blecua P, et al. (2019). Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 364, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, Collins NB, Bi K, LaFleur MW, Juneja VR, et al. (2017). In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarsheth N, Wicha MS, and Zou W (2017). Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol 17, 559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanson T, Ahuja A, Rubinsteyn A, Aksoy BA, Hellmann MD, Miao D, Van Allen E, Merghoub T, Wolchok JD, Snyder A, et al. (2017). Somatic Mutations and Neoepitope Homology in Melanomas Treated with CTLA-4 Blockade. Cancer immunology research 5, 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura H, Minato N, Nakano T, and Honjo T (1998). Immunological studies on PD-1 deficient mice: implication of PD-1 as a negative regulator for B cell responses. Int Immunol 10, 1563–1572. [DOI] [PubMed] [Google Scholar]
- O’Sullivan D, Sanin DE, Pearce EJ, and Pearce EI (2019). Metabolic interventions in the immune response to cancer. Nat Rev Immunol 19, 324–335. [DOI] [PubMed] [Google Scholar]
- Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, et al. (2007). Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 13, 54–61. [DOI] [PubMed] [Google Scholar]
- Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, Tsoucas D, Qiu X, Lim K, Rao P, et al. (2018). A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaretakis T, Joza N, Modjtahedi N, Modjtahedi N, Tesniere A, Tesniere A, Vitale I, Vitale I, Durchschlag M, Durchschlag M, Fimia GM, et al. (2008). The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ 15, 1499–1509. [DOI] [PubMed] [Google Scholar]
- Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, Gartner JJ, Jia L, Steinberg SM, Yamamoto TN, et al. (2017). Identification of essential genes for cancer immunotherapy. Nature 548, 537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, Sun Y, Zhao E, Vatan L, Szeliga W, et al. (2015). Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, Martín-Algarra S, Mandal R, Sharfman WH, et al. (2017). Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 171, 934–949.e916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, and Wolchok JD (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, Bjorgaard SL, Hammond MR, Vitzthum H, Blackmon SM, et al. (2017). Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun 8, 1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, et al. (2016). Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 44, 924–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savina A, Jancic C, Hugues S, Guermonprez PI, Vargas P, Moura IC, Lennon-Duménil AM, Seabra MC, Raposo G, and Amigorena S (2006). NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 126, 205–218. [DOI] [PubMed] [Google Scholar]
- Shan H, Wei JF, Zhang M, Lin L, Yan R, Zhu Y, and Zhang R (2014). Calreticulin is localized at mitochondria of rat cardiomyocytes and affected by furazolidone. Mol Cell Biochem 397, 125–130. [DOI] [PubMed] [Google Scholar]
- Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, Li Y, Chen H, Yang H, Hsu PH, et al. (2018). LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 174, 549–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, Grasso CS, Hugo W, Sandoval S, Torrejon DY, et al. (2017). Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov 7, 188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Bao R, and Gajewski TF (2015). Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235. [DOI] [PubMed] [Google Scholar]
- Tamura S, Oshima T, Yoshihara K, Kanazawa A, Yamada T, Inagaki D, Sato T, Yamamoto N, Shiozawa M, Morinaga S, et al. (2011). Clinical significance of STC1 gene expression in patients with colorectal cancer. Anticancer Res 31, 325–329. [PubMed] [Google Scholar]
- Tanikawa T, Wilke CM, Kryczek I, Chen GY, Kao J, Núñez G, and Zou W (2012). Interleukin-10 ablation promotes tumor development, growth, and metastasis. Cancer Res 72, 420–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theisen DJ, Davidson JT, Briseño CG, Gargaro M, Lauron EJ, Wang Q, Desai P, Durai V, Bagadia P, Brickner JR, et al. (2018). WDFY4 is required for crosspresentation in response to viral and tumor antigens. Science 362, 694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Drake CG, and Pardoll DM (2015). Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27, 450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, et al. (2017). A pathology atlas of the human cancer transcriptome. Science 357, eaan2507. [DOI] [PubMed] [Google Scholar]
- Van-Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen M, Goldinger SM, Utikal J, et al. (2015). Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Sun J, Liu LN, Flies DB, Nie X, Toki M, Zhang J, Song C, Zarr M, Zhou X, et al. (2019a). Siglec-15 as an immune suppressor and potential target for normalization cancer immunotherapy. Nat Med 25, 656–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, et al. (2019b). CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Huang L, Abdelrahim M, Cai Q, Truong A, Bick R, Poindexter B, and Sheikh-Hamad D (2009). Stanniocalcin-1 suppresses superoxide generation in macrophages through induction of mitochondrial UCP2. J Leukoc Biol 86, 981–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Subramanian M, Yurdagul A Jr., Barbosa-Lorenzi VC, Cai B, de Juan-Sanz J, Ryan TA, Nomura M, Maxfield FR, and Tabas I (2017). Mitochondrial Fission Promotes the Continued Clearance of Apoptotic Cells by Macrophages. Cell 171, 331–345.e322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung BH, Ay. L, and Wong CK (2012). Evolution and roles of stanniocalcin. Mol Cell Endocrinol 349, 272–280. [DOI] [PubMed] [Google Scholar]
- Yu J, Green MD, Li S, Sun Y, Journey S, et al. (in press). Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T-cell elimination. Nat Med, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. (2016). Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med 375, 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L, Apetoh L, Ghiringhelli F, and Kroemer G (2008). Immunological aspects of cancer chemotherapy. Nat Rev Immunol 8, 59–73. [DOI] [PubMed] [Google Scholar]
- Zou W (2005). Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 5, 263–274. [DOI] [PubMed] [Google Scholar]
- Zou W, Wolchok JD, and Chen L (2016). PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med 8, 328rv324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA seq raw data and files have been deposited to GEO: GSE161813. The MS files in mzIdentML format have been deposited to the ProteomeXchange consortium: PXD023251.