

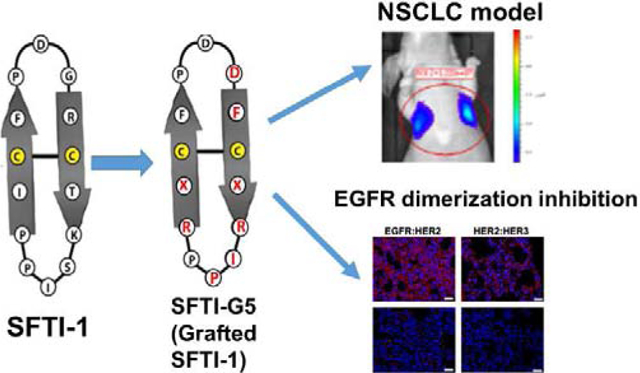
Abstract
Among the lung cancers, approximately 85% are histologically classified as non-small-cell lung cancer (NSCLC), a leading cause of cancer deaths worldwide. Epidermal growth factor receptors (EGFRs) are known to play a crucial role in lung cancer. HER2 overexpression is detected by immunohistochemistry in 2.4%–38% of NSCLC samples. EGFRs have been targeted with three generations of tyrosine kinase inhibitors (TKIs), and drug resistance has become a major issue; HER2 dimerization with EGFR also plays a major role in the development of resistance to TKI therapy. We have designed grafted peptides to bind to the HER2 extracellular domain (ECD) and inhibit protein-protein interactions of EGFR:HER2 and HER2:HER3. A sunflower trypsin inhibitor (SFTI-1) template was used to graft a peptidomimetic compound. Among several grafted peptides, SFTI-G5 exhibited antiproliferative activity in HER2-positive NSCLC cell lines such as Calu-3 cells with an IC50 value of 0.073 μM. SFTI-G5 was shown to bind to ECD of HER2 and inhibit EGFR:HER2 and HER2:HER3 dimerization and inhibit the phosphorylation of HER2 and downstream signaling proteins. As a proof-of-concept, the in vivo activity of SFTI-G5 was evaluated in two NSCLC mouse models. SFTI-G5 was able to inhibit tumor growth in both models. Furthermore, SFTI-G5 was shown to inhibit EGFR dimerization in tissue samples obtained from in vivo models. These grafted peptides can be used as novel dual inhibitors of EGFR dimerization in NSCLC.
Keywords: Grafted peptide, SFTI-1, EGFR, protein-protein interaction, NSCLC
Graphical Abstract
1. Introduction
The five-year relative survival rate for lung cancer patients is known to be around 18%, only a slight increase over what it was more than a decade ago, indicating that improvements in lung cancer therapy have been slow. Among lung cancers, non-small-cell lung cancer (NSCLC), a leading cause of cancer deaths worldwide1 and targeted therapy approaches are promising treatments for lung cancer2. Epidermal growth factor receptors (EGFRs) are known to play a crucial role in lung cancer. The receptor family consists of four members: HER1 or EGFR and HER2–4. These proteins have an extracellular domain (ECD)3, 4, a transmembrane helix, a cytoplasmic kinase domain, and a regulatory region. Ligand binding to EGFR or HER3 ECDs triggers a change in the conformation of the proteins, leading to their heterodimerization (Fig. 1) and, ultimately, to cell signaling5. In EGFR related NSCLC, EGFR and HER2 overexpression is observed. EGFR overexpression is evident and is observed in 40–89% of NSCLC6. However, HER2 overexpression has been reported at different frequencies in NSCLC patients with extremely wide ranges (2.4–38%)7, 8,9, 10. In HER2-related NSCLC, three mechanisms of HER2 activation have been described: a) HER2 protein overexpression, b) HER2 gene amplification, and c) HER2 gene mutations11, 12. These mechanisms also seem to have a role in the ability to develop resistance to targeted antibodies and tyrosine kinase inhibitors (TKIs). HER2 amplification is considered an alternative mechanism for the development of resistance to EGFR-targeted TKI therapy13. HER2 mutations and amplification are seen in 3% of lung cancers14. Thus, targeting HER2 and EGFR and their protein-protein interaction (PPI) or dimerization is important in NSCLC. The high co-expression of EGFR and HER2 in NSCLC patients was associated with a significantly shortened overall survival rate compared with that of cancer patients whose tumors had high levels of EGFR or HER2 alone15.
Fig. 1.
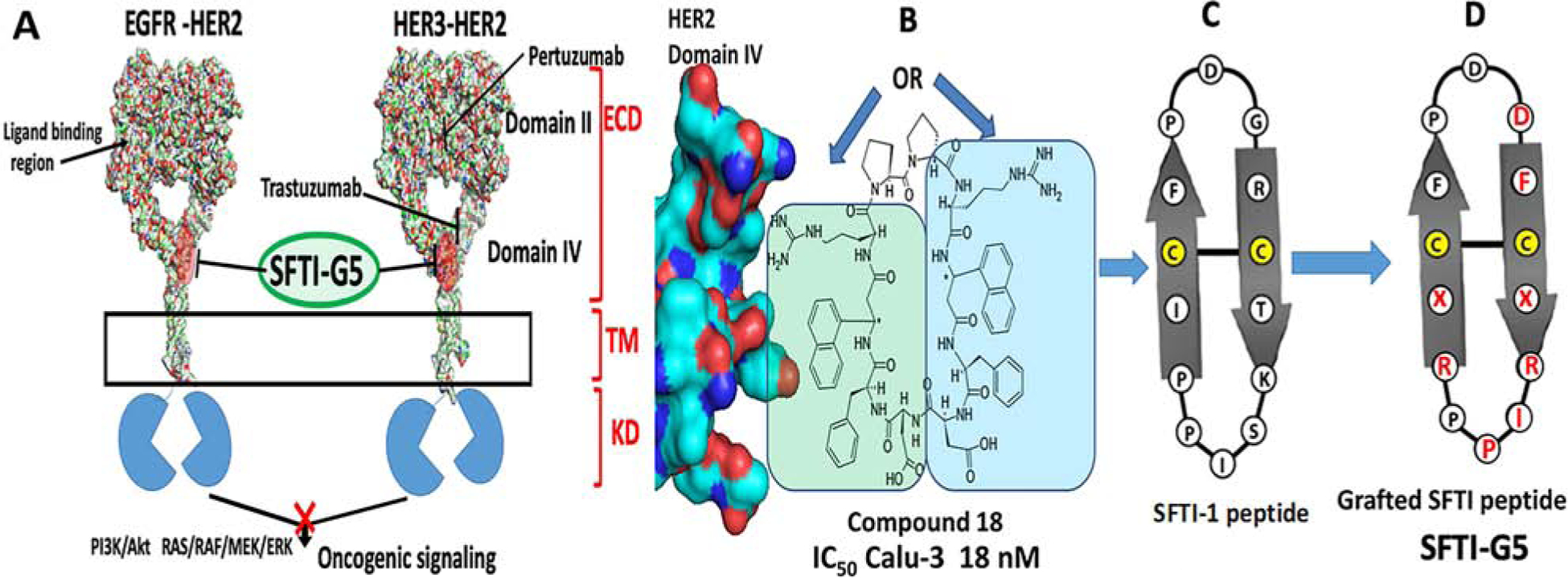
A) A schematic diagram of EGFR dimerization for downstream signaling in NSCLC showing the target site of SFTI-G5, a grafted peptide that targets EGFR:HER2 and HER3:HER2 dimerization. Design concept of grafted sunflower trypsin inhibitor (SFTI) peptide based on B) compound 18 that is known to bind to HER2 ECD domain IV and inhibit EGFR:HER2, HER2:HER3 dimerization. C) Sunflower trypsin inhibitor-1 (SFTI-1) peptide. D) Grafting of pharmacophore from compound 18 to SFTI-1 framework, resulting in a grafted peptide. Grafted residues are shown in red. X = 3-amino 3(1-napthyl) propionic acid (Anapa).
EGFR has been targeted with three generations of TKIs, and drug resistance has become a major issue16. EGFR-mutant cells can become resistant by acquiring the T790M mutation13, 17. Furthermore, mutant EGFRs, especially the L858R/T790M variant, have a propensity to heterodimerize with HER218. Thus, HER2 plays a significant role in mediating the sensitivity of EGFR-mutant lung tumors to anti-EGFR therapy. Taken together, these studies indicate that receptor signaling of EGFR, HER2, and HER3 is interdependent and emphasize the importance of inhibiting more than one HER family receptor for blockage of the signaling network of HER family receptors. Thus, inhibition of dimerization of EGFR:HER2 and HER2:HER3 signaling is necessary to achieve maximal anti-tumor responses in NSCLC therapy. Other approaches, such as immune checkpoint inhibitors, have been approved as immunotherapy for NSCLC but are limited to their expression in tissues19, 20. The use of monoclonal antibodies or a combination of antibodies with traditional chemotherapeutic agents or with kinase inhibitors has been widely seen in recent years. However, the antibodies that have been targeted to HER2 and EGFR proteins for the treatment of cancer have limitations in terms of stability, immunogenicity, size, cardiotoxicity, and cost21. Thus, there is a need for the development of novel molecules that are non-immunogenic and devoid of cardiotoxicity.
Extracellular domains of EGFRs homo- and heterodimerize by interacting with domain II and domain IV protein-protein surfaces (Fig. 1). Crystal structures of EGFR dimers suggest that domain IV interactions in the dimer are dominated by hydrophobic residues such as Leu, Trp, and Tyr22. Our interest is in targeting these with grafted peptides to modulate the cell signaling by inhibiting protein-protein interaction (PPI) “hotspots” in dimerization23, 24. Peptides are susceptible to enzymatic and chemical degradation25. To improve the stability of peptides, different approaches have been considered, such as N- and C-terminal modification, cyclization, side-chain modification, and chirality modification. The novel use of cyclotide-like multicyclic frameworks or sunflower trypsin inhibitor framework to graft an active peptide sequence is a more advanced method of stability improvement. Grafting of peptides onto cysteine-rich scaffolds is in high demand and has been successful in many studies26–28.
Sunflower trypsin inhibitor (SFTI-1), a cyclic peptide comprised of 14 amino acids found mainly in sunflower seeds. The structure consists of a cyclic backbone and is stabilized by a disulfide bridge. Due to its small size, extensive hydrogen-bonding network, and compact rigidity, it is one of the most widely employed molecular scaffolds for drug discovery29. In our previous work, we have shown that a peptidomimetic compound 18 binds specifically to ECD of HER2 and inhibits ECD heterodimerization of EGFRs, thereby preventing the downstream signaling by EGFR proteins (Table 1). Based on the design of compound 18, we have designed bicyclic peptides grafted onto a plant peptide, SFTI-129, to inhibit EGFR dimerization as a new class of grafted peptides. (Fig. 1B&C). Among the designed peptides, SFTI-G5, an SFTI-1 peptide grafted with a previously developed compound in our laboratory30, exhibited antiproliferative activity with an IC50 value of 0.073 μM in HER2-overexpressing lung cancer cell line Calu-3 (Fig. 1D) and was highly specific for HER2+ cancer cell lines. It also inhibited the dimerization of EGFR:HER2 and HER2:HER3 protein-protein interactions, thereby inhibiting the phosphorylation of HER2 via the Akt pathway. In addition, our results suggest that compound SFTI-G5 could suppress tumor growth in an animal model based on the in vivo study in mice.
Table 1.
Sunflower trypsin inhibitor (SFTI) template-based grafted peptides designed based on compound 18. Single letter amino acid code was used for peptide sequence. Lower case letters refer to D amino acids and capital letters refer to L amino acids. Chirality of Anapa was indicated with R and S configuration in italics. Antiproliferative activity of peptides in cancer cell lines are given as IC50 in μM. Disulfide bonds between the cysteines are underlined. Replicates N=3, values were represented as ±S.E
| Code | Compound | IC50 (μM) | ||||
|---|---|---|---|---|---|---|
| BT-474 (HER2+) | MCF-7 (HER2−) | Calu-3 (HER2+) | A549 (HER2+) | MCF-10A | ||
| Compound 18* | Cyclo(PpR(R-X)FDDF(R-X)R) | 0.197±0.055 | >50 | 0.018 ± 0.013 | ND | 40 |
| SFTI-G1 | Cyclo(C(S-X)RDR(S-X)CFDSIDF) | 4.16 ± 0.18 | >50 | ND | ND | |
| SFTI-G2 | Cyclo(C(R-X)RPpR(R-X)CFF) | 5.23 ± 1.38 | >50 | ND | ND | |
| SFTI-G3 | Cyclo(C(R-X)RPpR(R-X)CFDDF) | 3.65±0.41 | >50 | ND | ND | |
| SFTI-G4 | Cyclo(C(S-X)-RPGR (S-X)CFDSIPPDF) | 7.22±1.03 | >30 | ND | ND | |
| SFTI-G5 | Cyclo(C(S-X)RIPPR(S-X)CFPDDF) | 0.280 ± 0.021 | >20 | 0.073±0.009 | 0.369±0.070 | >40 |
| SFTI-G6 | Cyclo(C(R-X)RIPPR(R-X)CFPDDF) | 2.94±1.05 | >50 | ND | ND | ND |
| Control | H2N-K(S-(3-amino-3(biphenyl)propionic acid)F-OH | >100 | >100 | >100 | >100 | |
| Control (SFTI-1) | Cyclo(GRCTKSIPPICFPD) | ND | ND | >100 | >100 | ND |
ND = not determined, X = Anapa, 3-amino napthyl propionic acid, a beta amino acid, * = Compound 18 is described in Kanthala et al. (2017) Oncotarget 8, 74244–74262.
2. Results and discussion
2.1. Design of cyclic grafted peptides
With the ultimate aim of improving the stability of the peptidomimetics for oral availability while maintaining or enhancing their potency, the peptidomimetic (compound 18)30 was grafted onto the SFTI-1 framework (Table 1) to generate new generation of grafted peptidomimetics. We used the disulfide-stabilized, antiparallel β-strand conformation of SFTI-1 (PDB ID: 1JBL)31 for our design. Since disulfide bond is known to play a major role in stability of SFTI peptides, we wanted to retain the disulfide bond in our design. We replaced the amino acids of SFTI-1 with Arg-Anapa-Phe-Asp, where Anapa is 3-amino-3-(1-naphthyl propionic acid). L-Pro-D-Pro in compound 18 was used to stabilize the structure. Since SFTI framework is known to be stable L-Pro-D-Pro sequence from compound 18 was not grafted onto the SFTI-1 framework. We compared the 3D structure of 18 and SFTI-1 by overlapping the backbone atoms. Since the number of amino acids in the two peptides are different and the binding of particular amino acid side chain function group to HER2 protein is not well established in 18, we had to design several possible grafted peptides. When we overlapped the 3D structure of 18 with the grafted peptides, orientation of Anapa with respect to the plane of the backbone was taken into consideration. To make the orientation of side chain of Anapa in the same direction of that observed in 18, we changed the chirality of Anapa to S or R (Table 1).
In addition, SFTI-1 has three proline amino acids, and some amino acids such as Asp, Phe, and Arg are present in both compound 18 and the SFTI-1 framework (Figs. 1 and 2). Thus, grafting was done with different possibilities without removing disulfide bonds in the SFTI-1 framework. For example, in SFTI-G1 (Fig. 2C and Table 1), Ile-Ser of the loop region, as well as Asp amino acids (in 18 and SFTI-1) and the disulfide bond from SFTI, were retained, and prolines were deleted. Arg-Anapa-Phe-Asp from 18 was grafted. In SFTI-G2 (Fig 2D), only a disulfide bridge was retained, and the entire SFTI-1 framework was replaced with Arg-Anapa-Phe-Pro from compound 18 to reduce the total number of amino acids in the SFTI-1 framework to ten amino acids. In SFTI-G3 (Fig. 2E), Arg-Anapa-Phe-Asp-Pro from 18 and the disulfide bond from the SFTI-1 framework were used. SFTI-G4 (Fig. 2F) was designed keeping eight amino acids of the SFTI-1 framework and introducing Arg-Anapa-Phe-Asp from 18. In SFTI-G5 and SFTI-G6 (Figs. 2G&H), the total number of fourteen amino acids in the SFTI-1 framework was maintained, including the Ile-Pro-Pro loop region, and the remaining amino acids were replaced by Arg-Anapa-Phe-Asp. To assess the role of chirality of a β-amino acid, namely, 3-amino-3-(1-naphthyl propionic acid) (Anapa), on biological activity, we synthesized compounds of both S-Anapa and R-Anapa. As a control a linear peptidomimetic with Anapa and SFTI-1 was used (Table 1).31, 29
Fig. 2.
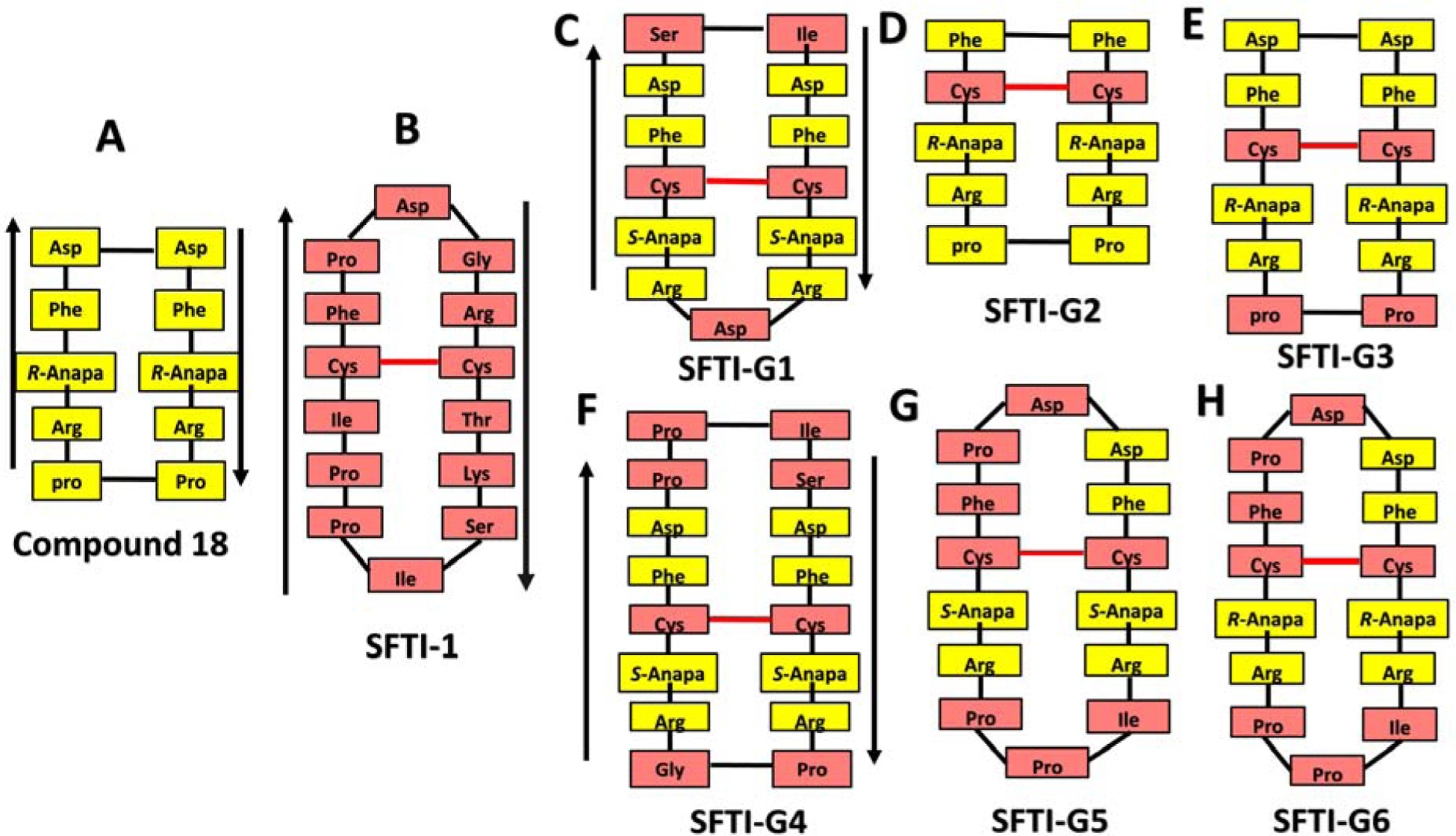
Design of grafted peptides. A) Amino acids and beta-amino acids from compound 18 were grafted onto B) SFTI-1framework to design different possible grafted peptides. C-H) SFTI-G1 to G6 peptides. Amino acids from compound 18 are highlighted in yellow and those from SFTI-1 are shown in pink. Anapa: 3-amino 3(1-napthyl) propionic acid. Direction of the peptide chain is shown with arrows. Disulfide bonds are indicated in red.
2.2. Peptide synthesis and characterization
The designed grafted peptides were synthesized and characterized by Fmoc solid-phase peptide synthesis as reported earlier32, 33. The sequences of the grafted cyclic peptides are shown in Table 1. (Supporting Information, Table S1, Figs. S1 to S7).
2.3. Antiproliferative activity of grafted cyclic peptides
Antiproliferative activity of the designed grafted peptides showed that one of the grafted peptides, SFTI-G5 exhibited antiproliferative activity with an IC50 value of 0.280 μM in HER2-overexpressing breast cancer cell line BT-474 and 0.073 μM in lung cancer Calu-3 cell line. In the A549 cell line, which is an EGFR wild-type and KRAS mutated NSCLC cell line34, the activity was found to be 0.369 μM. Furthermore, in the MCF-7 cell line, which is a HER2-negative cell line, the IC50 was found to be >20 μM (Table 1). The dose-response curves for SFTI-G5 in different cell lines are depicted in Supporting Information, Fig. S8 –S11. In the case of non-cancerous breast epithelial cell line MCF-10A, SFTI-G5 exhibited antiproliferative activity with an IC50 value of >40 μM, and in normal human lung fibroblast cell line (HLF) IC50 was >100 μM. Thus, we can conclude that the SFTI-G5 has high specificity toward HER2-overexpressing cancer cell lines. The activity of the designed grafted peptides was reduced slightly in all the HER2+ cell lines compared to the parent peptidomimetic 1830 (Table 1). This loss of activity may be due to altering positions of the vital amino acids in the SFTI-1 framework, which might be essential for interactions with the HER2 protein hot-spot region. We also evaluated the effect of the chirality of β-amino acid in SFTI-G5 on the antiproliferative activity. By altering the chirality of Anapa (from S-Anapa to R-Anapa) (SFTI-G6) (Table 1), there was a tenfold decrease in antiproliferative activity. This indicates the importance of the chirality of the Anapa group in the grafted peptide. As controls, SFTI-1 (Table 1) and a linear peptidomimetic was used. The antiproliferative activity of control peptide SFTI-1 was >100 μM in NSCLC cell lines A549 and Calu-3 indicating that grafting amino acids and Anapa on to SFTI-1 framework results in specificity towards HER2 positive cell lines. As SFTI-G5 exhibited the highest antiproliferative activity in HER2-overexpressing cancer cell lines among the grafted peptides designed, further studies related to the molecular mechanism and dimerization inhibition as well as in vivo studies were carried out on SFTI-G5. NCI-H1975 is a cell line that harbors the EGFR L858R/T790M double mutation35. Antiproliferative activity of SFTI-G5 in NCI-H1975 was IC50 =4.22 μM, which is much higher (low potency) than in HER2-overexpressing cancer cell lines, while the IC50 for erlotinib was 14 μM in NCI-H1975 cell lines36. We wanted to determine whether SFTI-G5 had any synergistic effect with erlotinib and lapatinib on different cancer cells when varying the concentration of SFTI-G5 and TKIs. The combination index (CI) for erlotinib and SFTI-G5 in the NCI-H1975 cell line was 0.76 for lapatinib and SFTI-G5 0.83 (Supporting Information Table S2).37, 38 In the MCF-7 cell line, there was no synergistic effect. These studies clearly suggest that SFTI-G5 has specificity towards HER2 positive NSCLC cell lines and have synergistic effect with TKIs in NSCLC cell lines that have resistance to EGFR TKI treatment.
2.4. Binding of SFTI-G5 to HER2 ECD
Compound 18 was shown to bind to the HER2 ECD and inhibit the PPI of EGFRs. To confirm that the grafted peptide, SFTI-G5, binds to the ECD of HER2, we performed peptide to protein binding studies using surface plasmon resonance (SPR)33. HER2 ECD protein (domains I-IV, 620 amino acids) or domain IV of ECD of HER2 was immobilized onto the CM5 sensor chip, and SFTI-G5 was injected over the immobilized chips at different concentrations. The SPR sensorgrams showed concentration-dependent binding of SFTI-G5 to HER2 ECD (Fig. 3A) as well as to domain IV (Supporting Information, Fig. S12A). Binding affinities were obtained by global fitting analysis of the titration curve to the 1:1 Langmuir interaction model, and the Kd value was calculated. The binding affinities (Kd values) of SFTI-G5 for HER2 ECD and domain IV of HER2 protein were found to be 4.87 × 10−7 M and 3.46 × 10−7 M, respectively. When control compounds (Table 1) was used as an analyte, no SPR response was recorded (Supporting Information, Fig. S12B & S13&14), indicating specific binding of SFTI-G5 to HER2 ECD.
Fig. 3.
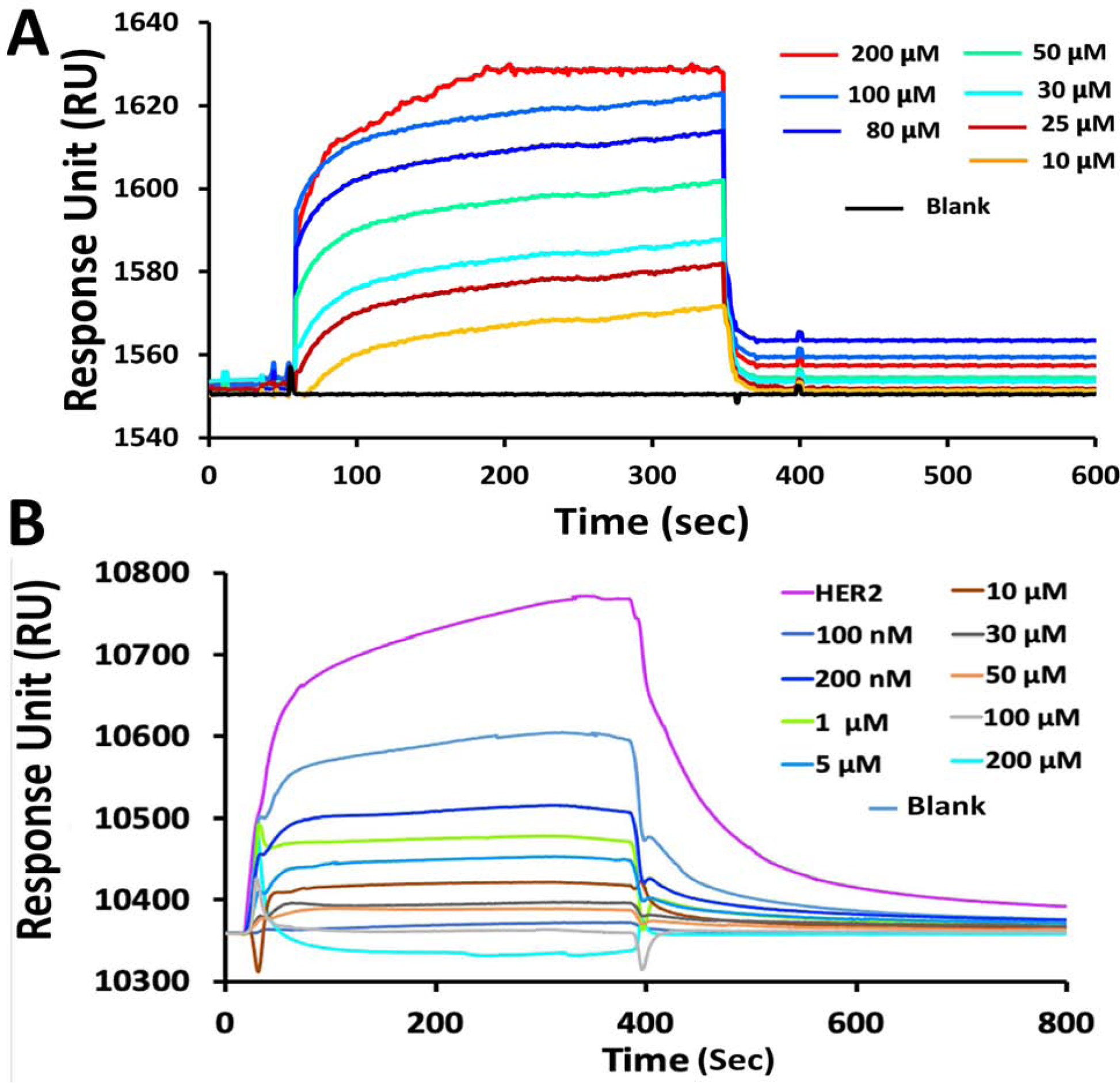
A) SPR analysis of binding of SFTI-G5 with HER2 protein. Binding to ECD of HER2 protein at different concentrations. Kinetics of association and dissociation are shown. B) SPR analysis of EGFR:HER2 dimerization and its inhibition by SFTI-G5. EGFR ECD was immobilized on CM5-chip and HER2 (200 nM) was added as analyte (violet). Different concentrations of SFTI-G5 along with constant concentration of HER2 ECD (200 nM) were added. Relative RU was decreased with increased concentration of SFTI-G5, indicating inhibition of protein-protein interaction of ECD of EGFR: HER2. Response to different concentrations of SFTI-G5 are shown in color for clarification.
Further, to confirm that SFTI-G5 inhibits the heterodimerization of EGFR-HER2 by binding to HER2, we performed a competitive binding study with SPR39. Here, EGFR protein was immobilized on CM5 chip, and varying amounts of SFTI-G5 (1–200 μM) were injected with a fixed concentration of HER2 protein (200 nM). When HER2 protein (200 nM) was injected alone, a response unit of 350 was recorded, which suggested protein-protein interactions of EGFR and HER2. As the concentration of SFTI-G5 increased in the presence of HER2 protein, we observed a decrease in the response unit (Fig. 3B), suggesting that SFTI-G5 competitively binds to the HER2 protein and prevents the dimerization of HER2 with EGFR. Hence, we can confirm that SFTI-G5 binds specifically to HER2 protein at the dimerization site of domain IV of HER2 and inhibits the dimerization of EGFR and HER2 protein.
2.5. Inhibition of EGFRs dimerization by proximity ligation assay (PLA)
Our aim is to inhibit the PPI of EGFR and, thus, modulate the cell signaling for cell growth. SFTI-G5 was designed to inhibit the EGFR heterodimers. To show that SFTI-G5 inhibits EGFR heterodimerization, we performed proximity ligation assay (PLA) in A549 and Calu-3 NSCLC cell lines. PLA is an established technique to quantify protein-protein interactions in cells40. A549/Calu-3 cells were incubated with different concentrations of SFTI-G5, and PLA assay was carried out using different antibodies (EGFR, HER2, and HER3). Cells without SFTI-G5 treatment were considered a positive control and cells not treated with the primary antibody but with a secondary antibody were regarded as another control.
Pertuzumab, an antibody that is known to bind to HER2 ECD domain II and inhibit the dimerization of EGFRs, was used as a positive control. In the absence of the SFTI-G5, we observed a high number of red fluorescence dots in the cells, indicating EGFR:HER2 dimerization in A459 cells (Fig. 4Aa Upper rows). Here, each red dot represents the dimerization of EGFR:HER2 protein. In the presence of SFTI-G5 at 0.5 μM and 1 μM concentrations, the number of red dots was significantly lower than in the positive control (Fig. 4A c & d). Our results indicate that SFTI-G5 inhibits heterodimerization in a concentration-dependent manner. We also found similar results when we probed for HER2:HER3 dimers with PLA probes (Fig. 4A e,f,g, & h). Similar results were observed when PLA assay was performed on Calu-3 cells with and without SFTI-G5 (Fig. 4A Lower two rows). Quantification of red fluorescence from PLA suggested that SFTI-G5 significantly inhibited the dimerization of HER2 compared to control in both cell lines (p < 0.05 for 0.5 μM and p < 0.01 for 1 μM SFTI-G5 concentration) (Fig. 4B). These results suggest that SFTI-G5 is a dual inhibitor of dimerization of ErbB heterodimers, EGFR:HER2 and HER2:HER3. Also, we compared SFTI-G5 against Pertuzumab, a HER2 dimerization inhibitor, for the inhibition of HER2 dimerization which showed similar result to Pertuzumab for inhibiting dimerization of HER2 (Supporting Information, Figure S15).
Fig. 4.
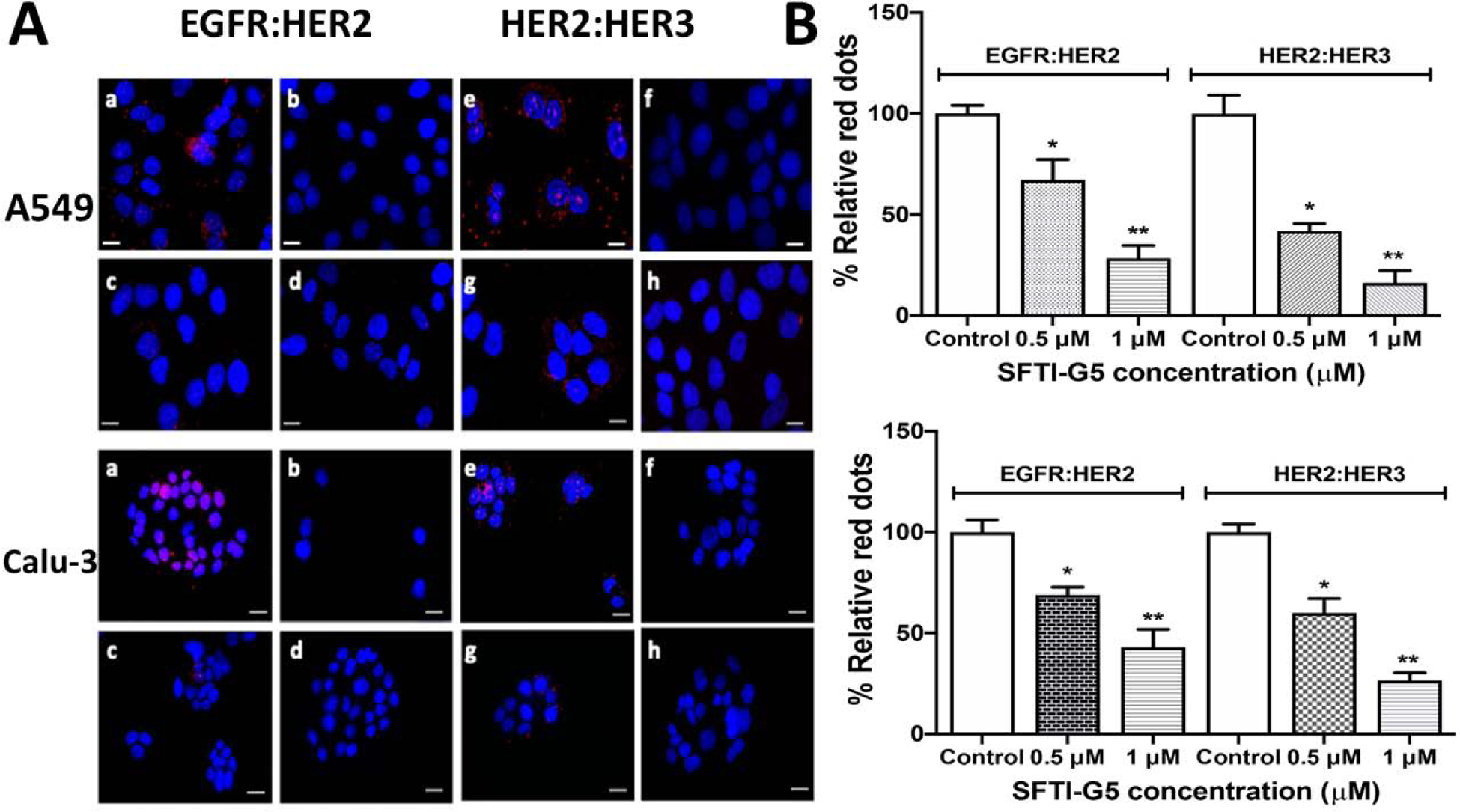
A) EGFR:HER2 and HER2:HER3 dimerization in A549 (upper two rows) and Calu-3 cells (lower two rows) and inhibition of dimerization by SFTI-G5 as shown by PLA assay. Upper rows: (a) EGFR:HER2 and (e) HER2:HER3 dimerization in A549 cells and its inhibition by SFTI-G5 at 0.5 and 1 μM (c & d, g & h). Controls (b&f) represents cells without the peptide and without one of the antibodies. Lower rows: (a) EGFR: HER2 and (e) HER2:HER3 dimerization in Calu-3 cells and its inhibition by SFTI-G5 at 0.5 and 1 μM (c & d, g & h). Controls (b&f) represent cells without the peptide and without one of the antibodies. Note the decrease in the number of red dots in the treatment group, suggesting the inhibition of dimerization. Nucleus was stained with DAPI B) Quantification of PLA assay fluorescence intensity. 3–6 different fields were chosen, and intensity of red fluorescence was measured using ImageJ software. Notice that, upon addition of SFTI-G5, red fluorescence was decreased, indicating inhibition of dimers. Scale 20 μm. Magnification 40×. *p < 0.05, ** p < 0.01. Mean ± SEM.
2.6. SFTI-G5 inhibits phosphorylation of HER2 kinase and modulates downstream signals
To confirm the inhibition of PPI of ECD of EGFRs by SFTI-G5 and its effects on intracellular signaling, Western blot analysis was performed on NSCLC cells. A549 that overexpress HER2 protein was incubated with SFTI-G5 and, after extraction of HER2 protein, the amount of phosphorylated protein was determined by Western blot (Fig. 5A). The phosphorylation of HER2 was measured by the p-HER2 monoclonal antibody. Quantitative analysis of Western blot indicated that SFTI-G5 significantly decreases the phosphorylation of HER2 kinase compared to control (without peptide treatment). Lapatinib41, an EGFR and HER2 kinase dual inhibitor were used as a positive control. The results suggest that the binding of SFTI-G5 to the ECD inhibits the phosphorylation of the intracellular kinase domain of HER2. However, there was no significant change in total HER2 protein levels (Fig. 5A). To understand whether SFTI-G5 inhibits any down-stream signaling molecules, we further evaluated the phosphorylation of Akt. As indicated in Fig. 5A, SFTI-G5 was able to inhibit the phosphorylation of Akt. Quantification of Western blot suggested that SFTI-G5 significantly inhibited the phosphorylation of Akt compared to control (Fig. 5B).
Fig. 5.

A) SFTI-G5 inhibits the phosphorylation of HER2 and Akt. Western blot analysis of phosphorylation of HER2 and Akt after treatment of vehicle, SFTI-G5 (1 μM), and Lapatinib (2 μM) for 40 h in A549 cells. The visualization of GAPDH was used to ensure equal sample loading in each lane. SFTI-G5 (1 μM) inhibits the phosphorylation of HER2 kinase and Akt compared to control in A549 cells as shown by Western blot. B) Quantification of Western blot images using densitometry. *p<0.05 ***p<0.001. Mean ± SEM.
2.7. Stability of the secondary structure of SFTI-G5
Due to its cyclic nature and stabilized disulfide bridge, the parent SFTI-1 molecule typically exhibits higher thermal stability than natural peptides, which are prone to conformational change with increase in temperature42. Sunflower trypsin inhibitor and other cyclic peptides with multiple disulfide bonded structures (cyclotides) exhibit exceptional thermal stability apart from chemical and enzymatic stability. Furthermore, sunflower trypsin inhibitor peptides do not exhibit one single major conformation in solution42−44. To evaluate whether grafted peptide of SFTI (changing the sequence of SFTI by grafting) exhibits thermal stability, we evaluated the thermal stability of SFTI-G5. The studies will also help to understand the stability of SFTI grafted peptides at different temperature in future formulation of such peptides under forced denatured/degradation conditions. We wanted to confirm whether, after the grafting, SFTI-G5 retains thermal stability. Circular dichroism spectroscopy was used to evaluate the thermal stability of grafted peptide SFTI-G5. There were no significant changes in the CD spectra of SFTI-G5 recorded at different temperatures (25–75 °C), which suggested that the overall secondary structure of the peptide remained the same at different temperatures (Supporting Information, Fig. 16A). To confirm this further, a freeze-dried samples of SFTI-G5 at different temperatures were analyzed via MALDI-TOF mass spectrometry (Supporting Information, Fig. S16B,C,D). Samples of SFTI-G5 from 25 to 70 °C showed intact molecular ion for the compound, suggesting that SFTI-G5 was stable at 70 °C. Thus, the CD and mass spectrometry results confirm that SFTI-G5 is stable against thermal denaturation.
To further evaluate the stability of the disulfide bond, the CD spectrum of SFTI-G5 was examined in the presence and absence of dithiothreitol (DTT)45. There was a slight change in CD spectrum of SFTI-G5 with the addition of DTT. Since the spectral changes observed may not be significant and CD spectra represents the average conformation of peptide in solution. DTT treated samples were subjected to mass spectrometry to evaluate whether disulfide bond was intact or not. The reduction was further confirmed by a shift in the molecular ion of SFTI-G5 upon the addition of DTT (m/z 1839 to 1841 after the addition of DTT, Supporting Information, Fig. S17).
2.8. In vivo studies
To obtain the proof-of-concept of SFTI-G5 dimerization inhibition and effect on NSCLC tumor growth in an animal model, we carried out in vivo studies on two NSCLC models46,47 namely xenograft model and an experimental metastasis model of lung cancer. In the xenograft model, compound was administered via intratumor injection and in metastasis model via tail vein intravenous injection.
2.8.1. Effect of SFTI-G5 on tumor growth in a xenograft model of lung cancer.
To evaluate whether the designed grafted peptide SFTI-G5 could reduce the progression of lung cancer tumor growth in an animal model, mice with xenograft tumors were treated with SFTI-G5 at 8 mg/kg thrice a week via intratumor injection just below the tumor. HER2 dimerization inhibitors such as pertuzumab and trastuzumab and pertuzumab combinations were also administered as control groups (Fig. 6A). Based on our hypothesis, SFTI-G5 is expected to inhibit HER2 heterodimerization in vivo, thus leading to delayed tumor growth progression. During the course of the experiment, the tumor size in the control xenograft group without any treatment continued to increase, reaching a diameter of ~9 mm in 28 days (Fig. 6B).
Fig. 6.
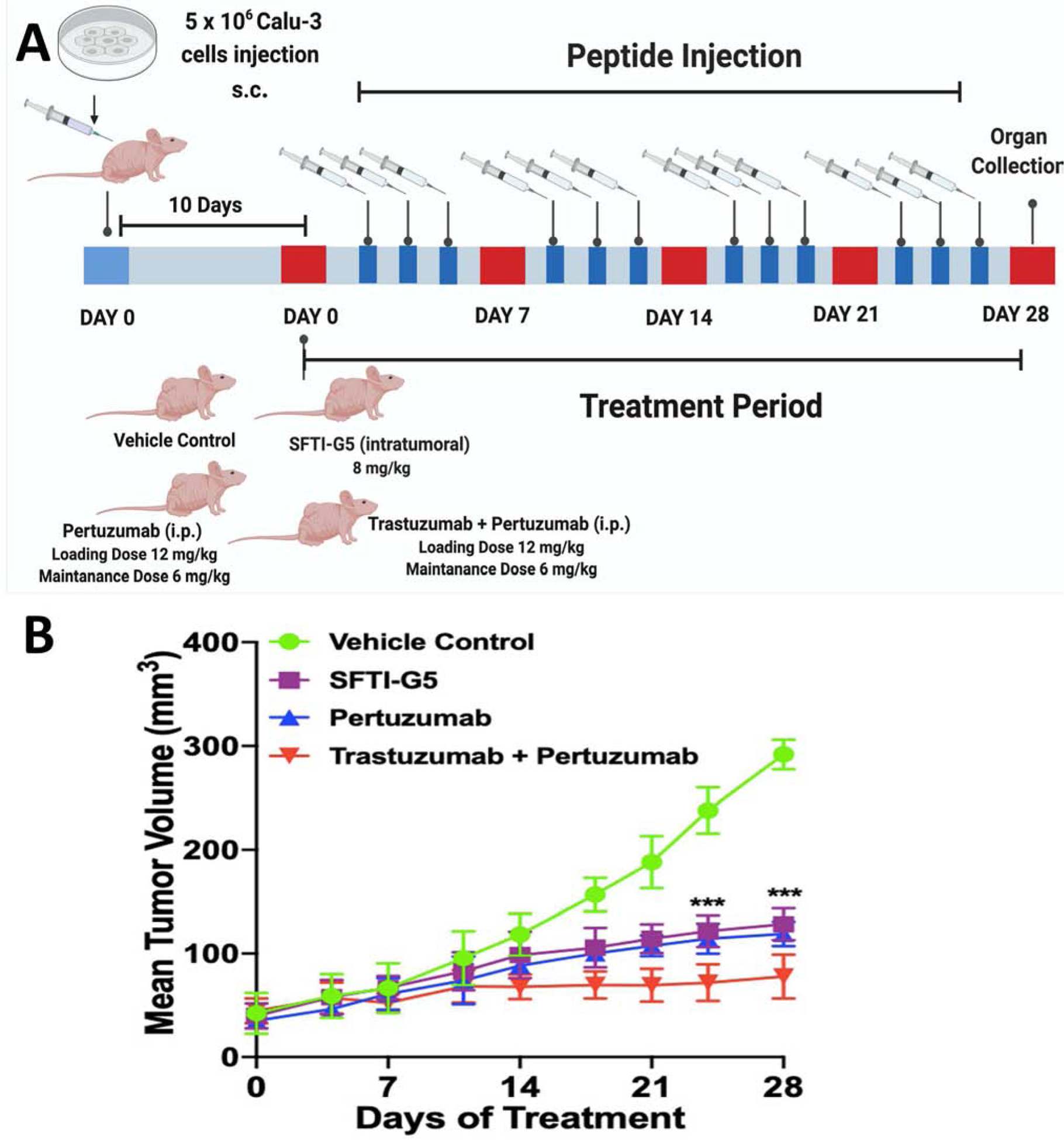
Xenograft lung cancer development and treatment by SFTI-G5. 5 × 106 Calu-3 cells suspended in serum-free medium were subcutaneously inoculated in left flank region of mice to induce lung cancer (Foxn1-nude). SFTI-G5 (8 mg/kg) was administered via intratumoral injection three times a week after 10 days of cell injection, Pertuzumab was injected with a loading dose of 12 mg/kg and then 6 mg/kg as maintenance dose intraperitonially once a week, trastuzumab and pertuzumab combination was injected at a loading dose of 12 mg/kg and then 6 mg/kg as maintenance dose once a week. A) Timeline of experiment; B) in vivo antitumorigenic activity of SFTI-G5; the tumor volume (V) was calculated as V = (L × W2)/2, where L was the length and W was the width of tumors. SFTI-G5(8 mg/kg) (square marks) delayed tumor growth in athymic nude mice significantly compared to the control group without any treatment (circles). For comparison, pertuzumab (up triangle, blue) and trastuzumab and pertuzumab combination (down triangle, red) also shown (with standard errors for n = 6). Statistical analysis indicated that there was a significant difference between control and SFTI-G5 (*** p<0.001 after 21 days.
On the other hand, SFTI-G5 showed a delay in tumor growth that was significant compared to control with p < 0.001 after 21 days (Fig. 6B). Based on the results of the two-tailed student t-test for statistical significance, treatment with pertuzumab also significantly reduced tumor growth progression compared to vehicle control and SFTI-G5. The combination treatment of trastuzumab and pertuzumab showed enhanced antitumor activity compared to both monotherapies. Further, the dosing schedule of SFTI-G5 as monotherapy was well-tolerated and had no significant effects on body weight (Supporting Information, Fig. S18) or clinical observations (not shown).
To confirm whether SFTI-G5 inhibited the heterodimerization, fixed tumor sections from SFTI-G5-treated, trastuzumab- and pertuzumab-treated, and vehicle control groups were incubated with primary and secondary antibodies of HER2 and EGFR, and PLA assay was carried out. Samples from tumors treated with SFTI-G5 exhibited markedly diminished signals from dimerization as indicated by a relative decrease in red fluorescence compared to the control tumor sample, indicating a decrease in the heterodimerization of EGFR:HER2 in vivo (Fig. 7A). The results suggest that inhibition of dimerization inhibits the signaling for cell growth and correlates with the reduction of tumor growth progression, as observed in the antitumor study.
Fig. 7.
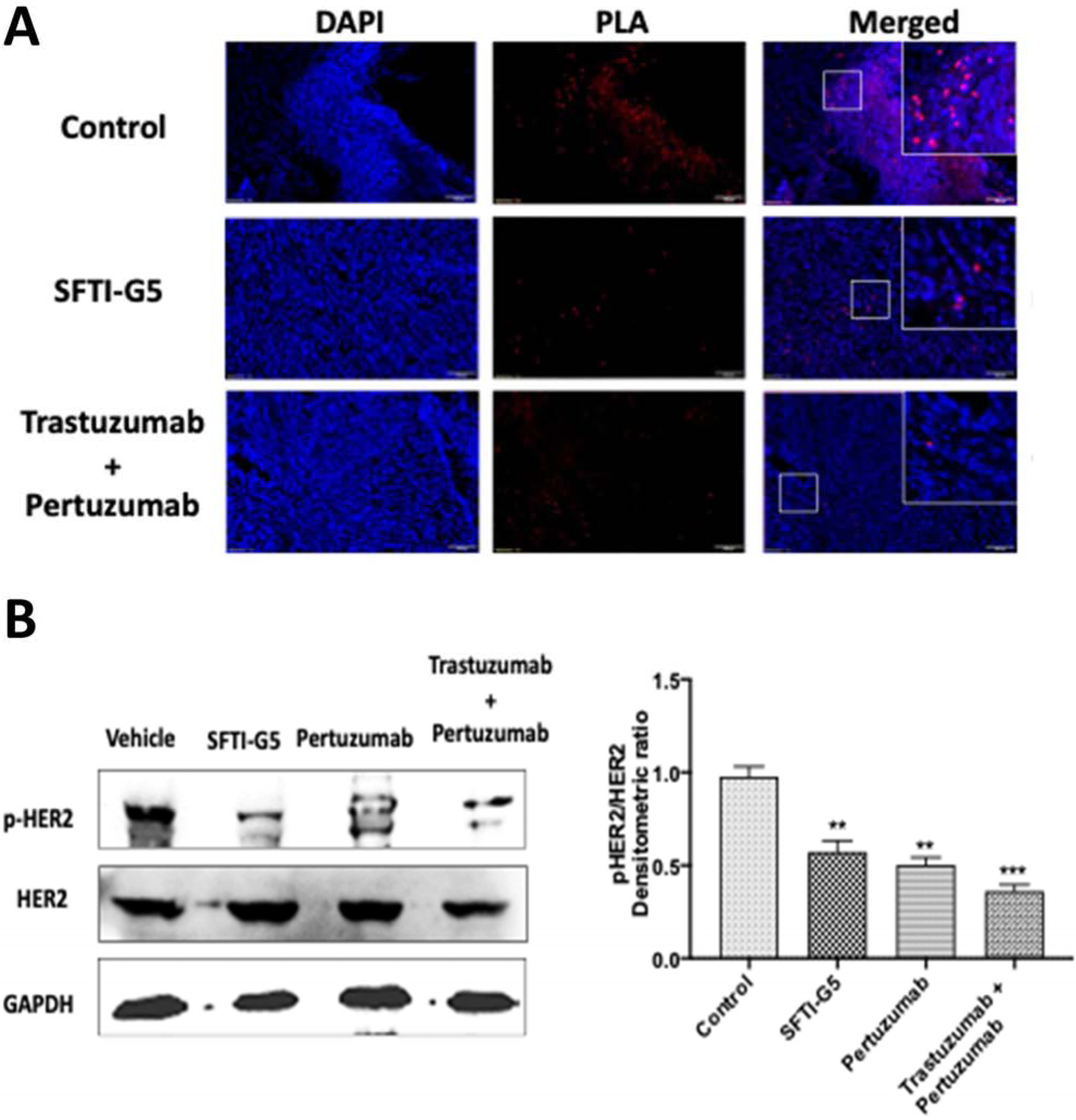
A) PPI inhibition assessed by PLA on tumor tissue sample. EGFR:HER2 PPI and its inhibition in vehicle control, SFTI-G5-treated and trastuzumab- and pertuzumab-treated mice groups. Magnification 10×, scale 100 μm, SFTI-G5 as well as trastuzumab+pertuzumab treated samples showed decrease in the number of red fluorescence dots indicating PPI inhibition. B) Western blot analysis of tumor sections of control group, mice treated with SFTI-G5 group, pertuzumab group, and trastuzumab+pertuzumab combination group. Tumor sections were homogenized, and protein was extracted and subjected to Western blot. SFTI-G5 showed a decrease in phosphorylation of HER2 kinase. Statistical analysis showed that there was significant reduction of phosphorylation HER2 protein in SFTI-G5 group. (** p<0.01) Mean ± SEM.
To evaluate the phosphorylation of HER2 protein, Western blot analysis was performed on frozen tumor samples. Results from Western blot analysis of the tumor samples showed significant inhibition of HER2 phosphorylation by SFTI-G5 compared to vehicle treatment (Fig. 7B). Thus, SFTI-G5 binds to the ECD and inhibits the phosphorylation of HER2.
2.8.2. Effect of SFTI-G5 on tumor growth in an experimental metastasis model of lung cancer.
In the experimental metastasis model, the tumor can grow in particular organs following intravascular injection. Before injecting to the animals, the Bioware® Brite A549 Red-FLuc luciferase transfected cells (PerkinElmer) were first evaluated in vitro for their bioluminescence efficiency. The reason for selecting A549 cells is that these are derived from NSCLC and are known to have KRAS mutation and EGFR wild-type48. Using whole-body bioluminescence imaging, the growth of the tumor was monitored once a week. After two weeks of injection of cells, mice with lung tumors in were divided into two groups and injected intravenously (tail vein) with either vehicle or SFTI-G5 (6 mg/kg) in 100 μL prepared in PBS twice a week. This dosage was based on our previously reported work on HER2+ breast cancer using peptidomimetics30, 49. All mice were imaged weekly with bioluminescence imaging. It has been reported that the photon flux from the tumor is directly proportional to the number of light-emitting cells that express luciferase, and that the signal can be measured to monitor tumor growth and development50. As shown in Fig. 8A, after two weeks, the rate of tumor growth, indicated by total flux (p/s), decreased in mice treated with SFTI-G5 (6 mg/kg) compared with mice injected with vehicle. There was a significant difference in the tumor growth rate between control and treated groups (Fig. 8AB). Statistical analysis was done using the two-tailed student t-test. Mice were sacrificed on day 35, and excised lungs and other organs were individually analyzed.
Fig. 8.
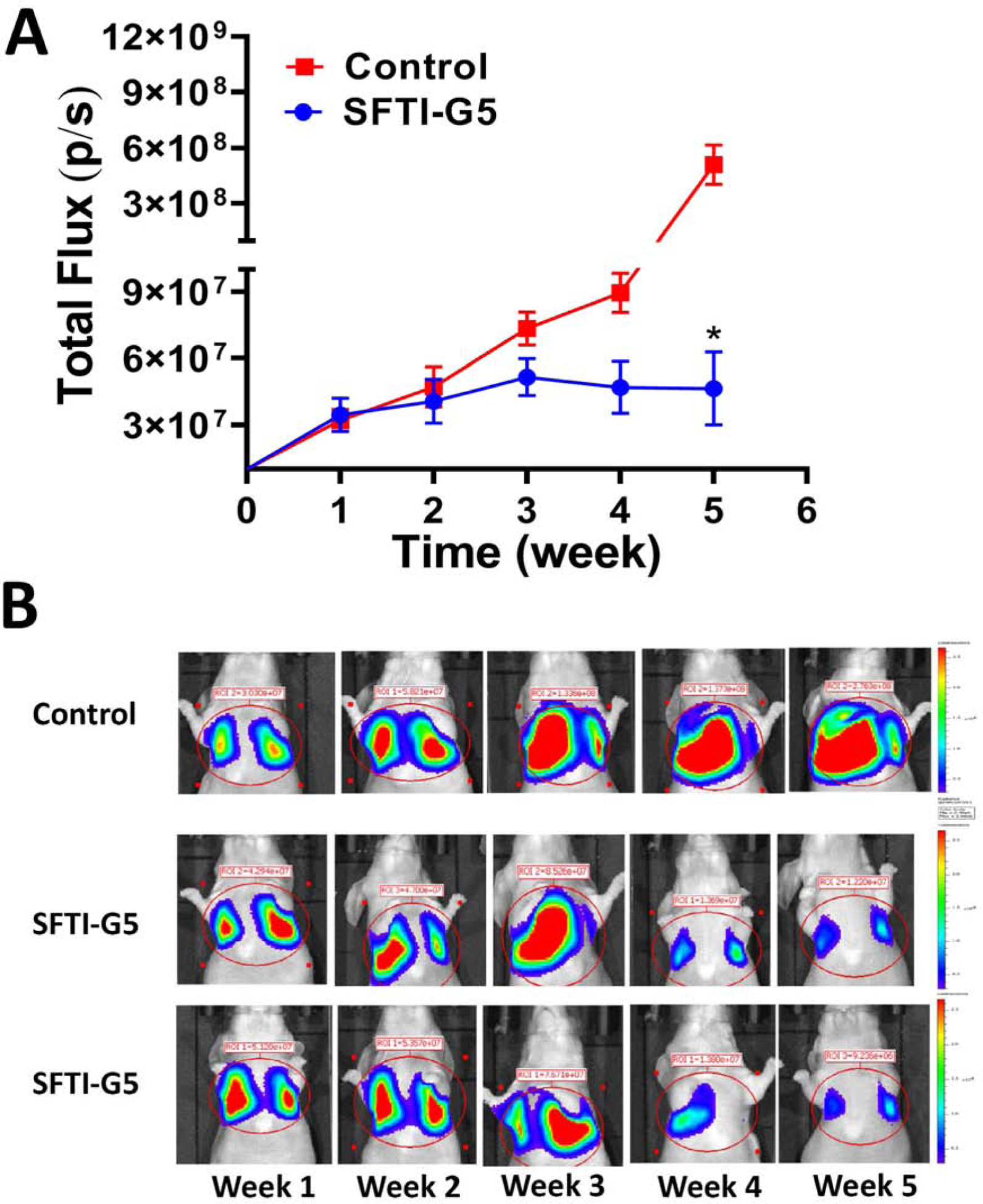
A) Lung cancer development and treatment by SFTI-G5. A549 Red-FLuc Luciferase transfected cells were injected to induce lung cancer in mice (Foxn1-nude). One week after injection of cells, mice were monitored for lung cancer development using luciferin imaging. SFTI-G5 was administered via IV twice a week after 2 weeks. Relative luminescence intensity (total photon flux/sec) plotted for 4 animals and controls. B) Representative images: In the control group (Row 1) lung cancer tumor growth increased in 5 weeks. In animals that were treated with SFTI-G5 (Rows 2 and 3) via IV injection (6 mg/kg, 100 μL), tumor growth was reduced compared to the control group as shown by reduction in bioluminescence of tumor in weeks 4 and 5.
To gain further understanding of the therapeutic effects of treatments on lung tumor, hematoxylin & eosin (H&E)-stained cross-sections of the lungs with tumors were studied. H&E staining of histological sections of the lungs from mice without treatment (vehicle control) showed the presence of enlarged, hyperchromatic nuclei and abundant eosinophilic cytoplasm (Fig. 9A), while in the SFTI-G5-treated group there were less.
Fig. 9.
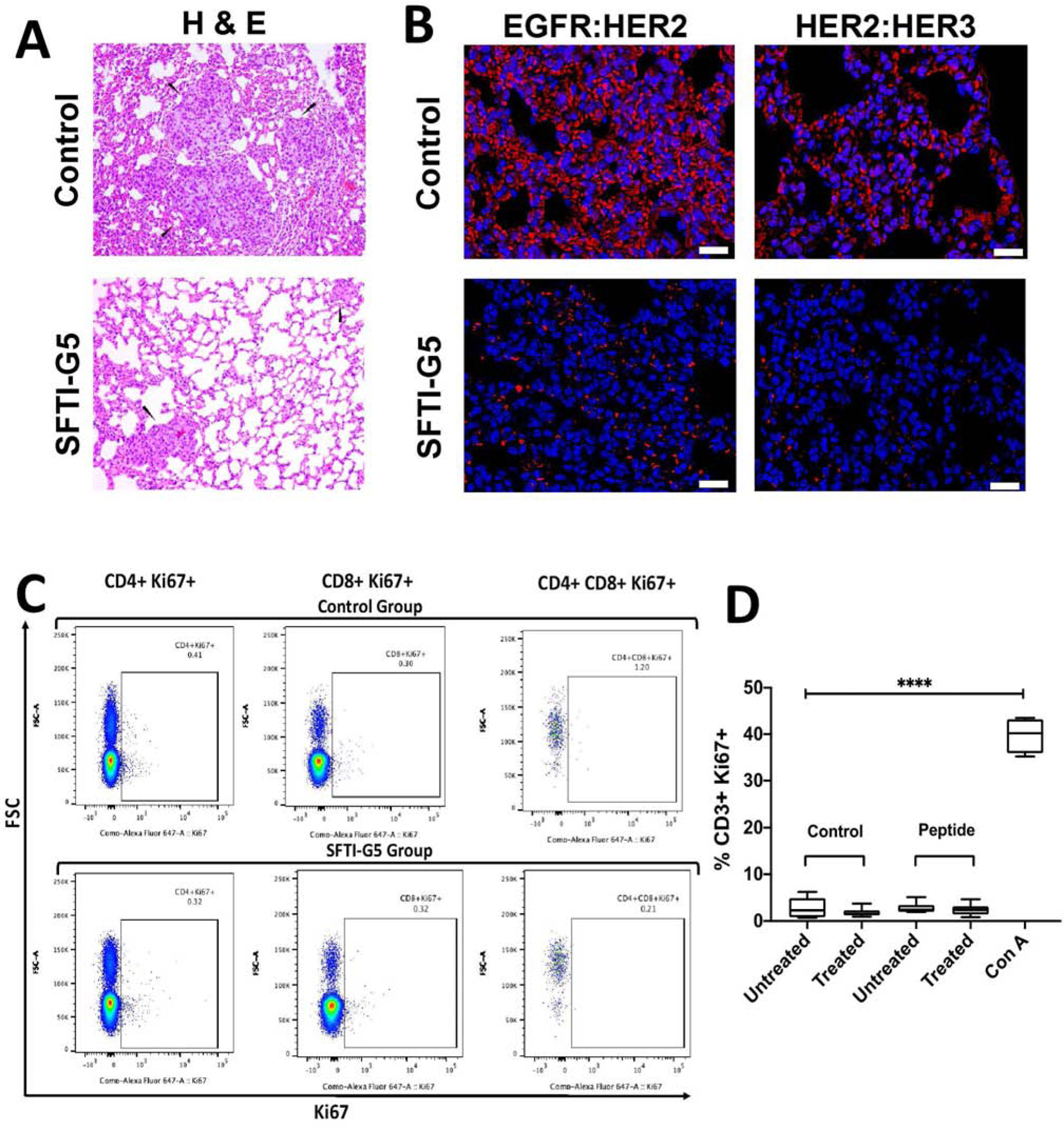
A) Histopathology of tumor samples from in vivo study of lung cancer. Mice without treatment (vehicle control) showed the presence of enlarged, hyperchromatic nuclei and abundant eosinophilic cytoplasm, while in the SFTI-G5-treated group there were less. B) PPI inhibition assessed by PLA on lung tumor tissue. EGFR:HER2 and HER2:HER3 PPI and its inhibition by SFTI-G5 treated mice. Note the decrease in red fluorescence in treated mice lung cancer tissue due to inhibition of PPI by SFTI-G5. Magnification 40×. Scale bar 20 μm. C) Immunogenicity of SFTI-G5 in vivo. BALB/C mice were primed with SFTI-G5 (6 mg/kg IV) on day 0 and day 10, and splenocytes were isolated on day 13 from untreated and treated groups. Splenocytes were incubated with peptide (SFTI-G5 at 5 μM) or control media for 48 hours before flow cytometry. Splenocytes treated with ConA (5μg/ml) served as a positive control. Representative dot plots are shown, and D) the %CD3+Ki67+ of CD3+ cells are quantified across groups. (n = 2–3, **** p < 0.0001)
2.9. Inhibition of EGFR dimerization by SFTI-G5
Tissue sections of lung tumor samples were further evaluated by proximity ligation assay for EGFR dimerization inhibition by SFTI-G5 in vivo51. Tissue sections from the vehicle control group showed high red fluorescence, indicating EGFR:HER2 and HER2:HER3 dimerization. Sections of tissues that were treated with SFTI-G5 showed a reduction in red fluorescence, indicating inhibition of EGFR:HER2 and HER2:HER3 dimerization (Fig. 9B). Thus, SFTI-G5 inhibits both dimers in the in vivo NSCLC model. The results suggest that inhibition of the ECD of HER2 inhibits signaling for cell growth and, hence, reduction in tumor growth in the animal model of cancer.
2.10. SFTI-G5 is not immunogenic in mice
To investigate whether the grafted peptide SFTI-G5 is immunogenic and initiates an antigen-specific immune response after administration, we performed a flow cytometry-based T-lymphocyte (T-cell) proliferation assay in a mouse model52. We evaluated the Ki-67 expressions in T cells of both control and the SFTI-G5 primed group using different T-cell markers (CD3, CD4, and CD8). Ki-67 is a proliferation marker of nuclear protein and has been used to measure specific T-cell responses53. Here, BALB/c mice were primed with SFTI-G5 (6 mg/kg), followed by a booster dose on the 10th day. Animals were sacrificed on the 13th day, and splenocytes were harvested and cultured in vitro from both untreated and treated mice, either in the presence or absence of the peptide. After 48 h incubation, the cells were stained for flow cytometry. The expression of Ki-67 in CD3+ subsets (CD4+, CD8+, and CD 4+CD8+) of SFTI-G5-primed mice was not significantly different between cells exposed to ex vivo peptide compared to those exposed to control media (Fig. 9C). Positive control T cells treated with Concanavalin A (5 μg/ml) expressed significantly greater Ki67 compared to all other treatment groups (p < 0.0001) (Fig. 9D) (Supporting Information S19).
3. Conclusions
We successfully synthesized a series of grafted cyclic peptides by grafting potent peptidomimetics (compound 18) onto the SFTI-1 framework without altering the disulfide bridge. Among these grafted peptides, SFTI-G5 was found to have higher potency than others, as confirmed from the antiproliferative assay. Further, SPR results showed that SFTI-G5 binds to the ECD of HER2 protein, specifically Domain IV of HER2 protein. PLA and Western blot results showed that the grafted cyclic peptide inhibits the dimerization of EGFRs and ultimately reduces the downstream signaling. In vivo studies suggested that SFTI-G5 reduces tumor growth significantly compared to control. Stability studies indicated that the compound is retains its secondary structure even at high temperatures.
4. Experimental
4.1. Materials
Anti-EGFR, anti-phospho-EGFR, anti-phospho-HER2, anti-HER2, anti-phopho-akt, anti-akt monoclonal antibodies were purchased from Cell Signaling Technology. Anti-GAPDH monoclonal antibody (sc-365052) was purchased from Santacruz Biotechnology (Dallas, TX, USA). Cell-titer Glo assay kit was purchased from Promega (Madision, MI, USA). HER2 protein and EGFR protein for surface plasmon resonance study was obtained from Abcam and Leinco Technologies Inc, respectively. 1× RBC lysis buffer (420301, Biolegend, San Diego, CA); Concanavalin A (ConA; C5275, Sigma-Aldrich, Saint Louis, MO, USA); viability dye (LIVE/DEAD™ Fixable Near-IR Dead Cell Stain Kit, L34975, Invitrogen, Eugene, OR, USA); Fixation/Permeabilization solution (00-5123-43 and 00-5223-56, eBioscience, Carlsbad, CA, USA); CD3-PerCP-eFlour710 (17A2, Invitrogen); CD4-FITC (GK 1.5, BD Biosciences); CD8a-BV650 (53–6.7, BD Horizon); and Ki67-AF647(B56, BD Pharmingen) for peptide immunogenicity study were purchased. Proximity ligation assay was performed using the Duolink® II assay kit (Sigma Aldrich). Primary antibodies for EGFR, HER2 and HER3 were from Enzo Life Sciences.
4.2. Peptide synthesis
All the grafted peptides (Table 1) were synthesized via Fmoc based solid-phase peptide synthesis method described previously33. (Details are provided in in supporting information).
4.3. Cell viability assay
CellTiter Glo® Assay kit (Promega Corporation, Madison, WI)54 was performed to access the antiproliferative activity of cyclic peptides on different HER2 over expressive cancer cell lines (BT-474, Calu-3, and A549), HER2 negative cell line MCF-7, nontumorigenic cell line MCF-10A and normal human lung fibroblast cell line (HLF). Briefly, 1 × 104 were coated in each well of 96 well plate and incubated overnight at 37 °C and 5% CO2. Then the cells were treated with different concentrations of grafted peptides (200 μm−0.01 μM) along with 1% SDS and 1% DMSO as negative and positive controls respectively. After 72 h incubation, cells were washed with PBS, and CellTiter-Glo detection reagent was added. Luminescence measurements were obtained using a plate reader (Biotek Synergy, Winooski, VT). IC50 values were calculated using GraphPad Prism software (La Jolla, CA) from three independent experiments for each cell line.
To evaluate the synergistic effect of SFTI-G5 with erlotinib, first a dose response curve was generated for erlotinib, and SFTI-G5 alone in EGFR mutated NCI-H1975 cells. Then cells were treated with different concentrations of SFTI-G5 (0.005–10 μM) along with fixed concentration of erlotinib (10 μM). Similarly, cells were treated with different concentrations of erlotinib (0.005–10 μM) with fixed concentration of SFTI-G5 (3 μM). Combination data were analysed, and results showed as combination index (CI) values according to the median-effect principle, where CI <1, =1, and >1 indicate synergism, additive effect, and antagonism, respectively37, 38. CI values calculated as follows: CI = [CAx/ICxA + CBx/ICxB], where ICxA and ICxB are the concentrations of individual combination ingredients, Erlotinib and SFTI-G5 inducing 50% cell growth inhibition (IC50); CAx and CBx are the concentrations of combination ingredients that induce 50% cell growth inhibition when used combined as determined by non-linear regression curve fit analysis. Similar studies were conducted with lapatinib (Table S2).
4.4. Surface Plasmon Resonance (SPR) binding assay
To analyze the binding affinity of SFTI-G5 to HER2 protein, SPR (Reichert Technologies, SR700DC, Buffalo, NY) instrument was used55. The HER2 extracellular domain protein was immobilized on a CM5 SPR sensor chip (Reichert Technologies, NY), in pH 4.5 acetate buffer at a rate of 10 μL/min using standard amine coupling procedure. Various concentrations of SFTI-G5 (0.5–200 μM) prepared in PBS buffer containing Tween-20 (Sigma-Aldrich, USA), were injected on immobilized protein at a flow rate of 25 μL/min at 25 °C and binding kinetics were analyzed with 1:1 Langmuir binding model using Tracedrawer software. Binding studies of SFTI-G5 also performed using HER2 extracellular domain IV. Peptides that does not exhibit antiproliferative activity (IC50 >50 μM) against HER2 positive cell line (Table S1) was used as a control for the experiment. All the solutions were freshly prepared and filtered (0.22 μm pore size).
To confirm the inhibition of protein-protein interactions by SFTI-G5 in vitro, a competitive SPR binding study was performed39. For this study, EGFR protein (Leinco Technologies Inc, MO) with EGF (Leinco Technologies Inc, MO) was immobilized on a CM5 sensor chip as described above. A fixed concentration of 200 nM HER2 protein was incubated with various concentrations of SFTI-G5 for 15 minutes in HEPES buffer. Then the mixture was injected over the immobilized chip as described above. The surface was regenerated after each injection with glycine pH 2.2.
4.5. Western blot analysis
To evaluate the role of SFTI-G5 in phosphorylation of kinase domain, western blot analysis was performed. 1 × 106 A549 cells were treated with SFTI-G5 and lapatinib (positive control) at 1 μM and 2 μM concentrations respectively. Cells without any treatment was taken as the negative control. Cells were treated for 40 h and the cell lysate was collected using cell lysis buffer containing protease inhibitor and phosphatase inhibitor. The protein concentration in each sample was obtained using BCA protein assay reagent kit (Thermo Fisher scientific). 40 μg of protein from each sample was loaded on 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels and western blot analysis was carried out as described previously30. Different primary antibodies against phosphorylated HER2 (phospho-Tyr1248; #2247), total HER2 (#2242S), Akt (#9272), p-Akt (Ser473; #9271, all from Cell Signaling Technology, were used at 1:1000 dilutions. After addition of corresponding horseradish peroxidase-conjugated secondary antibodies (anti-mouse or anti-rabbit from Cell Signaling) were used against each primary antibody, and the images were captured using C-Digit Blot Scanner (LI-COR Biotechnology, Lincoln, NE). Experiments were repeated three times and a representative western blot image was used for the final presentation. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used to ensure equal sample loading in each lane. Data are from triplicates and are presented as the mean ± standard error of the mean (SEM). To evaluate the significance, statistical analysis was performed by two tailed student t-test using Prism Graph Pad (GraphPad Software, San Diego). P< 0.05 was considered statistically significant.
4.6. Proximity ligation assay (PLA)
To determine the inhibition of EGFR: HER2 and HER2: HER3 protein-protein interaction PLA assay was performed as described previously30, 40, 55. Approximately 1 × 104 cells (A549 and Calu-3) were incubated in 8-well slide plates and then treated with different concentrations of SFTI-G5 (0.5 μM and 1 μM) and controls (no compound as a positive control and negative control where one of the primary antibodies was omitted) for 48 h and then fixed using cold methanol. Cells were incubated with primary antibodies (EGFR, HER2, and HER3) overnight at 4 °C and washed, and secondary antibodies with PLA probes (positive and negative) were added. Images were taken with Olympus BX63 fitted with deconvolution optics using DAPI, FITC, and Texas Red filters at 40x magnification and processed by using CellSens dimension software. Images were obtained at 40X. PLA dots were quantified using ImageJ software (NIH). Pertuzumab was used as positive control (Genentech, MTA).
4.7. Thermal stability
Circular dichroism spectrometer JASCO-815 (Jasco Inc., Easton, MD) was used to observe the peptide secondary structure stability as a function of temperature33. SFTI-G5 was dissolved in acetonitrile to a concentration of 15 μM. The ellipticity of the SFTI-G5 solution was measured at temperatures between 25°C to 75°C with an increment of 5°C. Mass of the SFTI-G5 was confirmed at 25°C, 50°C, 70°C, and also at the end of the experiment.
4.8. In-vivo activity in an NSCLC xenograft model
The aim of this study was to evaluate the anti-cancer activity of the grafted peptide in a xenograft model. Athymic nude mice (Foxn1nu/Foxn1+, 6–7 weeks) were purchased from Envigo Laboratories and maintained in the ULM College of Pharmacy vivarium. All animals were handled in strict accordance with good animal practice as defined by NIH guidelines, and all animal work was approved by the IACUC, University of Louisiana at Monroe (ULM). Mice were kept for 1 week to acclimatize the local environment. Nearly 5 × 106 Calu-3 cells suspended in serum-free EMEM medium (50 μL) were injected into the left flank of the mice. Calu-3 cell lines overexpress HER2 protein. Animals were monitored daily, and body weight was measured every week. Ten days after the injections, visible tumors were developed. Tumor size was assessed twice weekly by measuring tumor diameter with a caliper; tumor volume was calculated using the formula V = [(length × width2)/2]. Once the tumors reached 3 mm in diameter, mice were randomly divided into four groups of six animals (3 male and 3 female) and categorized as a positive control, SFTI-G5, Pertuzumab, and Trastuzumab and Pertuzumab, respectively. The total treatment period for each group was 28 days. Treatment groups received compounds dissolved in saline, and the intratumor injections were given at a dose of 8 mg/kg three times a week. This dosage was based on the results obtained from the pharmacokinetic analysis of SFTI-G5 and our previous work on HER2 positive cancer treatment using peptidomimetics. Control group was given intratumor saline injections three times a week, Pertuzumab was administered at 12 mg/kg as loading dose and 6 mg/kg as maintenance dose once a week intraperitonially, and Trastuzumab and Pertuzumab was administered at 12 mg/kg (6 mg/kg each) as loading dose and 6 mg/kg (3 mg/kg each) as maintenance dose once a week intraperitonially. Mice were sacrificed when tumors reached 10 mm diameter in size. The final tumor volume in the control group was used as a marker to end the experiments. Tumor growth curves for all the groups were constructed and compared using statistical analysis. Two tumors per group were removed surgically and maintained at −80 °C for immunoblot analysis, while the remaining three tumors were fixed and maintained in paraffin blocks. Microsections of tumors (5 μm) were performed, stained with hematoxylin and eosin (H & E), and analyzed by a pathologist for necrosis. All data are presented as the mean ± SEM. Statistical differences were evaluated student two-tailed t-test on data from different groups and the criterion for statistical significance was p<0.05. The doses of SFTI-G5 was based on our previous work on compound 1830, 56.
4.9. Anti-tumor efficacy in experimental metastasis in-vivo model
All animals were handled according to the approved protocol from IACUC at the University of Louisiana at Monroe. Athymic nude mice (Foxn1nu/Foxn1+, female, 4–5 weeks) were purchased from Envigo Laboratories (Indianapolis, IN) and maintained in the ULM college of pharmacy vivarium. After acclimatization of animals to the local environment for a week, nearly 4.5 × 106 Luciferase transfected A549-Red-FLuc Bioware® Brite Cell Line (Perkin Elmer) in 100 μL PBS were injected into mice via tail vein (i.v.). Mice were monitored for 1 week prior to bioluminescence imaging. Bioluminescence was measured for each animal by imaging under anesthesia using an IVIS Lumina series III (Perkin Elmer) instrument after injecting with D-luciferin (XenoLight D-Luciferin - K+ Salt Bioluminescent Substrate, PerkinElmer) at a dose of 150 mg/kg per animal in PBS intraperitonially. The photons emitted from luciferase-expressing cells within the animal body and transmitted through the tissue were quantified using Living Image software program (PerkinElmer) according to manufacturer’s instructions. After 2 weeks of tumor inoculation, mice were randomly assigned to two groups (n=4). These groups contain negative control (vehicle control), and a treatment group (SFTI-G5). The mice were treated for 3 weeks. SFTI-G5 was administered at a dose of 6 mg/kg while the control group received saline injections twice a week intravenously via tail vein. Animals were imaged and bioluminescence were recorded once a week to monitor the progression of lung cancer. The animal’s health status was monitored daily for weight loss or any signs of altered motor ability while in their cages. At the end of the study, mice were sacrificed according to the approved IACUC protocol. Lung tumors along with other organs (liver, kidney, heart, and pancreas) from all animals were harvested and embedded in paraffin for PLA and IHC analysis. All data are presented as the mean ± SEM. Statistical differences were evaluated different methods such as student two –tailed t-test and the criterion for statistical significance was p < 0.05.
4.10. Immunohistochemistry studies and PLA on tumor tissue sections
For Immunohistochemistry study, formalin fixed paraffin embedded (FFPE) 5 μm cross-sections slides were de-paraffinized by incubation in xylene (soaking twice for 10 minute each), rehydrated with decreasing concentrations of ethanol30. Antigen retrieval was performed by treatment with fresh sodium citrate buffer with 0.05% Tween 20 at pH 6.0, for 20 minutes at 100°C, and sections were later stained with H&E and evaluated for necrosis.
Proximity ligation assay on tissue samples was carried out as described above30, 49. After the antigen retrieval on the tissue section of slides is done as discussed above, endogenous peroxidase was blocked with 3% H2O2 in methanol and the tissue sections were incubated with blocking solution containing 3% BSA/3% normal goat serum (NGS)/0.4% Triton X-100 in PBS for 60 min at RT, followed by overnight incubation at 4°C with primary antibodies. The tumor sections were then used to analyze inhibition of EGFR: HER2 as well as HER2:HER3 dimerization using secondary antibodies with PLA probes similar to in-vitro PLA methods as discussed previously. Then the slides were visualized under the microscope (Olympus BX63 fitted with deconvolution optics), and images were taken at 10X and 40X by using CellSens Dimension software.
4.11. Peptide immunogenicity study
The immunogenicity of peptide SFTI-G5 was determined by using BALB/c mice (5–6 weeks, female). Mice were bred at the LSU Division of Laboratory Animal Medicine (DLAM) facility and maintained under spcific pathogen free conditions. All procedures were approved by the LSU IACUC committee (#19–064). Twelve mice were divided into two groups: untreated and treated. The untreated group was given PBS and the treated group was given 6 mg/kg of SFTI-G5 peptide in (PBS) intravenously via tail vein on day 0 and day 10. On day 13, all animals were sacrificed and splenocytes were isolated. Splenocytes were incubated with 1x red blood cell (RBC) lysis buffer (420301, Biolegend, San Diego, CA) to remove the RBCs for 3 min on ice. At least 2 × 106 cells were plated in each well of a 24-well plate and incubated in RPMI media supplemented with 10% FBS at 37 °C. Each group were further divided into two subgroups: control and peptide. Splenocytes from untreated and treated mice were exposed to either control media or 2 μM of SFTI-G5 peptide. As a positive control, some splenocytes were treated with Concanavalin A (ConA; 5 μg/mL; C5275, Sigma-Aldrich, MO, USA).
After 48 hours of incubations, the cells were stained with different antibodies for flow cytometric analysis as described previously57. Briefly, the cells were washed in PBS, and stained with viability dye (LIVE/DEAD™ Fixable Near-IR Dead Cell Stain Kit, L34975, Invitrogen, Eugene, OR, USA) at a dilution of 1:1000 in Dulbecco’s phosphate buffered saline (DPBS; Gibco, Carlsbad, CA, USA) for 30 min in a total volume of 100 μL. Samples were further washed again in PBS before staining with a cocktail of surface antibodies or isotype control diluted in staining buffer (3% fetal bovine serum (FBS, heat-inactivated; Omega Scientific, Tarzana, CA, USA) and 1mM of EDTA (Thermo Fisher Scientific, Carlsbad, CA, USA) in DPBS) at 1:100 dilutions for 20 min. The following antibodies were used for this study: CD3-PerCP-eFlour710 (17A2), Invitrogen; CD4-FITC (GK 1.5), BD Biosciences; CD8a-BV650 (53–6.7), BD Horizon; as surface antibodies. Samples were next washed in staining buffer. Cells were then incubated in 250 μL of Fixation/Permeabilization solution (00-5123-43 and 00-5223-56, eBioscience, Carlsbad, CA, USA) for 20 min. A cocktail of intracellular antibody Ki67-AF647(B56), BD Pharmingen diluted in permeabilization buffer (1:100) was then incubated for 30 min. Finally, all samples were washed again in permeabilization buffer and then suspended in 1% paraformaldehyde before undergoing flow cytometry. All antibody-staining steps were performed in a total volume of 50 μL per sample. All the incubation steps were performed at 4 °C.
Controls included a fluorescence minus one (FMO) or staining with an isotype control for antibodies that did not produce a bimodal distribution of cell staining. Flow cytometry was performed on a Becton Dickinson LSRFortessa X-20 flow cytometer, with blue (488nm), violet (405nm) and red (633nm) lasers. Compensation settings were conducted using BD CompBeads (BD Biosciences). Flow cytometry data was analyzed using FlowJo software (Version 10; TreeStar Inc.). The gating strategy used is illustrated in Fig. S16. The percentage of CD3+ cells expressing Ki67 was compared between groups.
4.12. Statistical analysis
Values are expressed as mean ± standard error mean (S.E.M.) and analyzed using statistical package for GraphPad Prism software version 8 (San Diego, CA) using two tailed student t-test; p < 0.05 were considered statistically significant.
Supplementary Material
Highlights.
A grafted peptide was designed to inhibit protein-protein interactions (PPI) of EGFRs
The peptide binds to HER2 protein extracellular domain IV and inhibits PPI of EGFRs
It was shown to inhibit the phosphorylation of HER2 kinase in vitro and in vivo
The grafted peptide was able to inhibit tumor growth of NSCLC in mice model
Stable grafted peptides can be used as therapeutic agents for NSCLC
Acknowledgements
Authors would like to thank core facilities at Louisiana State University Baton Rouge and at University of Louisiana Monroe for providing high-performance computer (HPC) at LSU, Baton Rouge via the Louisiana Optical Network Initiative (LONI), Microscopy facility and Mass spectrometry facility. Trastuzumab and pertuzumab were generously provided by Genentech Inc. under MTA. Sitanshu S. Singh was also supported by the Louisiana Biomedical Research Network summer research program. Authors would also like to thank Dr. David Craik and Thomas Durek for their useful suggestions for using the SFTI-1 template for grafting. Dr. Ted Gauthier, LSU Ag Center, Louisiana State University, Baton Rouge for help with synthesis, and Dr. Konstantin G Kousoulas, LSU School of Veterinary Medicines, Baton Rouge for graciously providing antibodies for the immunogenicity study. This work was supported by funding from NCI under Grant 1R15CA188225-01A1 (SDJ). This publication is subject to the NIH Public Access Policy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Appendix A. Supplementary data: HPLC and mass and NMR spectra of compounds, dose-response curves, SPR analysis of SFTI-G5 Binding to HER2 Domain IV, proximity ligation assay, HER2 expression in various cancer cell lines using Flow cytometry, Thermal stability data of SFTI-G5 in various temperatures are available.
Declaration of interests
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
Data related to this work was submitted as a US patent application No. 16/970,198.
References
- 1.Milano MT, Strawderman RL, Venigalla S, Ng K and Travis LB, J Thorac Oncol, 2014, 9, 1081–1090. [DOI] [PubMed] [Google Scholar]
- 2.Rothschild SI, Cancers (Basel), 2015, 7, 930–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F and Gianni L, Nature reviews. Clinical oncology, 2012, 9, 16–32. [DOI] [PubMed] [Google Scholar]
- 4.Tebbutt N, Pedersen MW and Johns TG, Nature reviews. Cancer, 2013, 13, 663–673. [DOI] [PubMed] [Google Scholar]
- 5.Wang SE, Narasanna A, Perez-Torres M, Xiang B, Wu FY, Yang S, Carpenter G, Gazdar AF, Muthuswamy SK and Arteaga CL, Cancer Cell, 2006, 10, 25–38. [DOI] [PubMed] [Google Scholar]
- 6.Al Olayan A, Al Hussaini H and Jazieh AR, J Infect Public Health, 2012, 5 Suppl 1, S50–60. [DOI] [PubMed] [Google Scholar]
- 7.Mazieres J, Peters S, Lepage B, Cortot AB, Barlesi F, Beau-Faller M, Besse B, Blons H, Mansuet-Lupo A, Urban T, Moro-Sibilot D, Dansin E, Chouaid C, Wislez M, Diebold J, Felip E, Rouquette I, Milia JD and Gautschi O, J Clin Oncol, 2013, 31, 1997–2003. [DOI] [PubMed] [Google Scholar]
- 8.Ricciardi GR, Russo A, Franchina T, Ferraro G, Zanghi M, Picone A, Scimone A and Adamo V, J Thorac Oncol, 2014, 9, 1750–1762. [DOI] [PubMed] [Google Scholar]
- 9.Zhao J and Xia Y, JCO Precision Oncology, 2020, 4, 411–425. [DOI] [PubMed] [Google Scholar]
- 10.Yoshizawa A, Sumiyoshi S, Sonobe M, Kobayashi M, Uehara T, Fujimoto M, Tsuruyama T, Date H and Haga H, Lung Cancer, 2014, 85, 373–378. [DOI] [PubMed] [Google Scholar]
- 11.Peters S and Zimmermann S, Transl Lung Cancer Res, 2014, 3, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mar N, Vredenburgh JJ and Wasser JS, Lung Cancer, 2015, 87, 220–225. [DOI] [PubMed] [Google Scholar]
- 13.Chong CR and Janne PA, Nat Med, 2013, 19, 1389–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li BT, Ross DS, Aisner DL, Chaft JE, Hsu M, Kako SL, Kris MG, Varella-Garcia M and Arcila ME, J Thorac Oncol, 2016, 11, 414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brabender J, Danenberg KD, Metzger R, Schneider PM, Park J, Salonga D, Holscher AH and Danenberg PV, Clin Cancer Res, 2001, 7, 1850–1855. [PubMed] [Google Scholar]
- 16.Camidge DR, Pao W and Sequist LV, Nat Rev Clin Oncol, 2014, 11, 473–481. [DOI] [PubMed] [Google Scholar]
- 17.Pao W and Chmielecki J, Nat. Rev. Cancer, 2010, 10, 760–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takezawa K, Pirazzoli V, Arcila ME, Nebhan CA, Song X, de Stanchina E, Ohashi K, Janjigian YY, Spitzler PJ, Melnick MA, Riely GJ, Kris MG, Miller VA, Ladanyi M, Politi K and Pao W, Cancer Discov, 2012, 2, 922–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cho JH, Immune Netw, 2017, 17, 378–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okita R, Maeda A, Shimizu K, Nojima Y, Saisho S and Nakata M, Cancer Immunol Immunother, 2017, 66, 865–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansel TT, Kropshofer H, Singer T, Mitchell JA and George AJ, Nature reviews. Drug discovery, 2010, 9, 325–338. [DOI] [PubMed] [Google Scholar]
- 22.Lu C, Mi LZ, Grey MJ, Zhu J, Graef E, Yokoyama S and Springer TA, Mol Cell Biol, 2010, 30, 5432–5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sperandio O, Reynes CH, Camproux AC and Villoutreix BO, Drug Discov Today, 2010, 15, 220–229. [DOI] [PubMed] [Google Scholar]
- 24.Reichmann D, Rahat O, Cohen M, Neuvirth H and Schreiber G, Curr Opin Struct Biol, 2007, 17, 67–76. [DOI] [PubMed] [Google Scholar]
- 25.Bruno BJ, Miller GD and Lim CS, Ther Deliv, 2013, 4, 1443–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ji Y, Majumder S, Millard M, Borra R, Bi T, Elnagar AY, Neamati N, Shekhtman A and Camarero JA, J Am Chem Soc, 2013, 135, 11623–11633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li P, Jiang S, Lee SL, Lin CY, Johnson MD, Dickson RB, Michejda CJ and Roller PP, J Med Chem, 2007, 50, 5976–5983. [DOI] [PubMed] [Google Scholar]
- 28.Chan LY, Gunasekera S, Henriques ST, Worth NF, Le SJ, Clark RJ, Campbell JH, Craik DJ and Daly NL, Blood, 2011, 118, 6709–6717. [DOI] [PubMed] [Google Scholar]
- 29.Lesner A, Legowska A, Wysocka M and Rolka K, Curr Pharm Des, 2011, 17, 4308–4317. [DOI] [PubMed] [Google Scholar]
- 30.Kanthala SP, Liu YY, Singh S, Sable R, Pallerla S and Jois SD, Oncotarget, 2017, 8, 74244–74262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Korsinczky ML, Schirra HJ, Rosengren KJ, West J, Condie BA, Otvos L, Anderson MA and Craik DJ, J Mol Biol, 2001, 311, 579–591. [DOI] [PubMed] [Google Scholar]
- 32.Cheneval O, Schroeder CI, Durek T, Walsh P, Huang YH, Liras S, Price DA and Craik DJ, J Org Chem, 2014, 79, 5538–5544. [DOI] [PubMed] [Google Scholar]
- 33.Sable R, Durek T, Taneja V, Craik DJ, Pallerla S, Gauthier T and Jois S, ACS Chem Biol, 2016, 11, 2366–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang S, Yu X, Fan Y, Shi X and Jin Y, J Cancer, 2018, 9, 2930–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walter AO, Sjin RT, Haringsma HJ, Ohashi K, Sun J, Lee K, Dubrovskiy A, Labenski M, Zhu Z, Wang Z, Sheets M, St Martin T, Karp R, van Kalken D, Chaturvedi P, Niu D, Nacht M, Petter RC, Westlin W, Lin K, Jaw-Tsai S, Raponi M, Van Dyke T, Etter J, Weaver Z, Pao W, Singh J, Simmons AD, Harding TC and Allen A, Cancer Discov, 2013, 3, 1404–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tallarida RJ, Genes Cancer, 2011, 2, 1003–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang L, Jiang Y and Chen Y, Sci Rep, 2017, 7, 40752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chou TC, Pharmacol Rev, 2006, 58, 621–681. [DOI] [PubMed] [Google Scholar]
- 39.Li Q, Quan L, Lyu J, He Z, Wang X, Meng J, Zhao Z, Zhu L, Liu X and Li H, Oncotarget, 2016, 7, 64967–64976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sable R, Jambunathan N, Singh S, Pallerla S, Kousoulas KG and Jois S, Biotechniques, 2018, 65, 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Diaz R, Nguewa PA, Parrondo R, Perez-Stable C, Manrique I, Redrado M, Catena R, Collantes M, Penuelas I, Diaz-Gonzalez JA and Calvo A, BMC Cancer, 2010, 10, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Craik DJ, Swedberg JE, Mylne JS and Cemazar M, Expert Opin Drug Discov, 2012, 7, 179–194. [DOI] [PubMed] [Google Scholar]
- 43.Brauer AB, Domingo GJ, Cooke RM, Matthews SJ and Leatherbarrow RJ, Biochemistry, 2002, 41, 10608–10615. [DOI] [PubMed] [Google Scholar]
- 44.Tischler M, Nasu D, Empting M, Schmelz S, Heinz DW, Rottmann P, Kolmar H, Buntkowsky G, Tietze D and Avrutina O, Angew Chem Int Ed Engl, 2012, 51, 3708–3712. [DOI] [PubMed] [Google Scholar]
- 45.Lopes de Almeida JP and Saldanha C, Clin Hemorheol Microcirc, 2010, 46, 51–56. [DOI] [PubMed] [Google Scholar]
- 46.Price JE, Methods Mol Biol, 2014, 1070, 223–233. [DOI] [PubMed] [Google Scholar]
- 47.Wu S, Kasim V, Kano MR, Tanaka S, Ohba S, Miura Y, Miyata K, Liu X, Matsuhashi A, Chung UI, Yang L, Kataoka K, Nishiyama N and Miyagishi M, Cancer Res, 2013, 73, 1787–1799. [DOI] [PubMed] [Google Scholar]
- 48.Del Re M, Rofi E, Restante G, Crucitta S, Arrigoni E, Fogli S, Di Maio M, Petrini I and Danesi R, Oncotarget, 2018, 9, 6630–6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kanthala S, Banappagari S, Gokhale A, Liu YY, Xin G, Zhao Y and Jois S, Chem Biol Drug Des, 2015, 85, 702–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Justilien V and Fields AP, Curr Protoc Pharmacol, 2013, 62, Unit 14 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Toki MI, Carvajal-Hausdorf DE, Altan M, McLaughlin J, Henick B, Schalper KA, Syrigos KN and Rimm DL, J Thorac Oncol, 2016, 11, 1901–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arbelaez CA, Estrada J, Gessner MA, Glaus C, Morales AB, Mohn D, Phee H, Lipford JR and Johnston JA, NPJ Vaccines, 2020, 5, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Soares A, Govender L, Hughes J, Mavakla W, de Kock M, Barnard C, Pienaar B, Janse van Rensburg E, Jacobs G, Khomba G, Stone L, Abel B, Scriba TJ and Hanekom WA, J Immunol Methods, 2010, 362, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tolliday N, Curr Protoc Chem Biol, 2010, 2, 153–161. [DOI] [PubMed] [Google Scholar]
- 55.Naik H, Gauthier T, Singh S and Jois S, Bioorg Med Chem Lett, 2018, 28, 3506–3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shrestha L, Singh SS, Parajuli P, Dahal A, Mattheolabakis G, Meyer S, Bhattacharjee J and Jois SD, Journal of Cancer, 2020, 11, 5982–5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Withers SS, Moore PF, Chang H, Choi JW, McSorley SJ, Kent MS, Monjazeb AM, Canter RJ, Murphy WJ, Sparger EE and Rebhun RB, Dev Comp Immunol, 2018, 87, 64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.