

Abstract
Cas12a is an RNA-guided DNA endonuclease of the type V-A CRISPR-Cas system that has evolved convergently with the type II Cas9 protein. We previously showed that proline substitutions in the bridge helix (BH) impart target DNA cleavage selectivity in Streptococcus pyogenes (Spy) Cas9. Here, we examined a BH variant of Cas12a from Francisella novicida (FnoCas12aKD2P) to test mechanistic conservation. Our results show that for RNA-guided DNA cleavage (cis-activity), FnoCas12aKD2P accumulates nicked products while cleaving supercoiled DNA substrates with mismatches, with certain mismatch positions being more detrimental for linearization. FnoCas12aKD2P also possess reduced trans-single-stranded DNA cleavage activity. These results implicate the BH in substrate selectivity in both cis- and trans-cleavages and show its conserved role in target discrimination among Cas nucleases.
Keywords: adaptive immunity, Cas12a, Cas9, cis-DNA cleavage, Cpf1, CRISPR-Cas, DNA cleavage selectivity, gene editing, off-target, trans-DNA cleavage
CRISPR-Cas (Clustered Regularly Interspaced Short Palindromic Repeats and CRISPR-associated proteins) systems are adaptive immune systems present in bacteria and archaea [1–4]. The CRISPR array contains repeat-spacer units, and spacers are usually derived from foreign genetic elements [5–7]. The CRISPR arrays are transcribed into pre-CRISPR RNA and further processed into smaller, mature CRISPR RNAs (crRNA), which guide Cas nucleases to DNA and/or RNA targets to enable sequence-specific cleavage [2,8–10]. Over the past decade, the RNA-mediated DNA targeting, and cleavage activities possessed by Cas nucleases have been repurposed into powerful gene editing tools [11–16]. Based on their locus organization and functional mechanisms, CRISPR-Cas systems are classified into two classes that are further divided into six types and several subtypes [3,17]. Of these, type II and type V CRISPR-Cas systems are widely used for gene editing applications due to the simplicity of RNA-mediated DNA targeting offered by these systems, which can be attributed to their signature Cas nucleases, which can singly recognize, bind, and cleave target DNA in a sequence-specific manner [13,15,18,19].
Cas12a (previously known as Cpf1) is the signature protein of type V-A CRISPR-Cas systems. It is a single polypeptide comprised of multiple domains that are broadly divided into two structural lobes, the recognition (REC) lobe and the nuclease (NUC) lobe [20–26]. The two lobes are connected by an α-helix called bridge helix (BH) that is indispensable for the protein’s function [21]. Cas12a binds to crRNA, following which a 20-nucleotide (nt)-long guide region of the crRNA base pairs with complementary regions of a target DNA that is flanked by a protospacer adjacent motif (PAM) [21]. Recognition of the PAM and 17-nt of complementarity in the target DNA are required for a stable R-loop formation, where one of the strands of the DNA base pairs with the guide region of the crRNA [called the target strand (TS)] and the other strand is displaced [called the nontarget strand (NTS)] [27–29]. The double-stranded (ds) DNA cleavage is sequential, with NTS cleavage preceding that of TS. Both strands are cleaved by the endonuclease activity of the RuvC domain, even though cleavage of TS requires coordination of both the Nuc and RuvC domains [30–32]. The position of cleavage, as referenced by the PAM location, differs for each of the two strands, resulting in staggered DNA ends after cleavage [33]. In comparison, Cas9 uses two distinct domains for dsDNA cleavage, HNH for TS and RuvC for NTS cleavage. Several studies have shown that efficient cleavage by RuvC is dependent on HNH in Cas9, even though RuvC has demonstrated the ability to cleave NTS independent of HNH, albeit at a lower efficiency [34–36].
In addition to the RNA-guided DNA cleavage activity (also called as cis-cleavage), Cas12a possesses the ability to process its own crRNA, which has also been identified in several type V and type VI CRISPR-Cas systems [37,38]. Recent studies have identified the ability of Cas12a protein to cleave and degrade single-stranded (ss) DNA, nonsequence specifically (this activity is called trans-cleavage) [39,40]. The trans-cleavage is triggered by the binding of Cas12a-crRNA complex to a complementary DNA strand, referred to as an activator DNA [39,40]. The base pairing of crRNA guide region with the activator DNA induces conformational changes, opening the RuvC catalytic pocket to ssDNA [31,32,41]. Previous reports have shown that both ssDNA and dsDNA oligonucleotides (oligos) are effective activators, even though dsDNA has an additional requirement for a cognate PAM [39]. Cas12a can also cause RNA-independent degradation of ssDNA and non-sequence-specific nicking of ds plasmid in the presence of specific divalent metal ions [42,43].
One of the major problems associated with CRISPR-Cas-based gene editing is off-target DNA cleavage, where targets with partial complementarity to the crRNA are also cleaved, in addition to the on-target DNA, causing unwanted mutations during gene editing [44–47]. Even though Cas9 is most widely used for gene editing, it has been shown to cause considerable off-target effects [45–47]. Cas12a offers more advantages for gene editing due to its ability to cause staggered cleavage that is beneficial for homologous recombination, [48] the potential for multiplexing where several regions can be targeted simultaneously since Cas12a can process crRNA by itself, [49,50] and reduced off-target effects compared to Cas9 based on gene editing experiments [15,51,52]. However, Cas12a has not been used as widely as Cas9 in gene editing mainly due to limited in-depth mechanistic understanding when compared to Cas9.
We recently established that in Streptococcus pyogenes (Spy) Cas9, the BH plays a role in imparting selectivity in target DNA cleavage [53]. Substituting prolines in a region of the BH that adopts a loop conformation in the apo-SpyCas9 structure (PDB ID: 4CMP) [54] greatly reduced the cleavage of supercoiled DNAs containing PAM-proximal mismatches [53]. A recent work showed that other amino acid substitutions (Arg63Ala or Arg66Ala) in SpyCas9’s BH also created stringent Cas9 variants [55]. Interestingly, BH is conserved in several Cas9 orthologs and other Cas nucleases across different CRISPR systems including Cas12a [17]. Based on this, we hypothesized that amino acid substitutions in the BH of large multidomain Cas nucleases can alter selectivity in substrate cleavage.
In the present work, we analyzed the effect of BH perturbations of Cas12a on DNA cleavage. We substituted two amino acids, Lys969 and Asp970, in the BH of Francisella novicida (Fno) Cas12a with prolines (FnoCas12aKD2P). Our results showed that the variant protein, FnoCas12aKD2P, discriminates against DNA containing mismatches across several positions along the target DNA, causing a reduction in dsDNA breaks and an accumulation of nicked products while performing cis-cleavage. This feature establishes commonalities between Cas9 and Cas12a. We also observed that BH contributes to efficient cis-cleavage of different physical states of DNA substrates and that it is essential for trans-DNA cleavage, implicating BH in different aspects of DNA recognition and cleavage. Altogether, these results indicate that the role of BH in substrate discrimination is shared among BH-containing Cas nucleases and lays foundation for a common strategy where target selectivity of Cas nucleases can be tuned by manipulating BH residues.
Materials and methods
FnoCas12aKD2P construction and purification
The wild-type Fnocas12a gene cloned into a pET28m vector (His6 of pET28a replaced with His8–3C protease) with an N-terminal maltose-binding protein (MBP) tag for solubility and efficient purification (Table S1) was from our previous study [42]. Using this as a template, site-directed mutagenesis (SDM) was performed to introduce Lys969Pro and Asp970Pro substitutions to create FnoCas12aKD2P (Table S1) [56]. The purified PCR product (E.Z.N.A. kit) was treated with a KLD (kinase, ligase, and Dpn1) enzyme cocktail present in the Q5 site-directed mutagenesis kit and was transformed into Escherichia coli DH5α cells (NEB, Ipswich, MA, USA). Plasmids were isolated and sequenced to confirm the proline substitutions and the correctness of the rest of the coding region. The sequence-confirmed plasmid was transformed into Escherichia coli Rosetta strain 2 (DE3) cells. Both FnoCas12aWT and FnoCas12aKD2P were expressed and purified using a previously published protocol (Fig. S1) [42].
crRNA transcription and purification
The template strand for crRNA containing the T7 promoter region was ordered as an oligo DNA from Integrated DNA technologies (IDT) (Table S2). The complementary noncoding strand consisting of the T7 RNA polymerase promoter region was also ordered from IDT (Table S2). The coding and noncoding strands were annealed in a 1 : 1.5 molar ratio by heating at 95 °C for 2 min and slowly cooling to room temperature in the presence of 1X annealing buffer (10 mm Tris/HCl, pH 8, 50 mm NaCl). In vitro transcription reaction (200 μL) contained ~ 400 ng of annealed dsDNA transcription buffer [1X: 40 mm Tris/HCl, pH 8.0, 1 mm spermidine, 50 μg bovine serum albumin, 20 mm MgCl2, 5 mm dithiothreitol (DTT)], nucleotide triphosphates (9 mm GTP, 8 mm ATP, 8 mm CTP, and 8 mm UTP), 50 μg RNasin (Promega, Madison, WI, USA), 1 μg inorganic pyrophosphatase, and 40 μg T7 RNA polymerase. The reaction was carried out for 3 h at 37 °C. The transcription reaction was treated with DNaseI (NEB) (final: 0.01 mg/mL) in 1X DNase buffer (1X: 10 mm Tris, pH 7.5, 2.5 mm MgCl2 and 0.5 mm CaCl2) for 30 min at 37 °C to remove the template DNA. The products were ethanol-precipitated and further purified by extracting the RNA bands from a 12% urea (8 m)/poly-acrylamide gel. Aliquots of RNA were stored at −20 °C and were freshly annealed by heating at 95 °C for 2 min followed by slow cooling to room temperature using the 1X annealing buffer to allow proper secondary structure formation.
Construction of matched and mismatched plasmid DNA substrates
The strands corresponding to the matched DNA substrate were ordered from IDT, and it consisted of a 31-nt protospacer region, a 3-nt PAM, and ends resembling postcleav-age by BamHI and EcoRI restriction enzymes (Table S1 and Fig. S2). The DNA oligos were annealed, phosphorylated, and ligated to linearized and dephosphorylated pUC19. The corresponding clone was sequence-confirmed and was further used as a template to develop mismatched DNA substrates. Using SDM, single mismatches were introduced covering positions 1 to 22 of the protospacer region (Table S1, Fig. S2). The mismatch positions are numbered following the position downstream of the PAM with respect to NTS.
Supercoiled plasmid cleavage assays
The Cas12a proteins were diluted to the required concentration in 20 mm HEPES, pH 7.5, 150 mm KCl, 2 mm EDTA, 1 mm TCEP. The total reaction volume for all the experiments in this work was 10 μL, unless mentioned otherwise. This reaction volume of 10 μL includes 1X cleavage buffer (20 mm HEPES, pH 7.5, 150 mm KCl, 5% glycerol, and 0.5 mm DTT), 5 mm MgCl2, desired RNP, and 100 ng of plasmid DNA. The crRNA-Cas12a (RNP) complex at desired concentrations were preassembled in a 1.2:1 molar ratio in 1X cleavage buffer containing 5 mm MgCl2 by incubating at 37 °C for 10 min. The reaction was initiated by adding 100 ng (~5 nm) plasmid DNA and further incubated at 37 °C for different time points as required for the different analyses. The reactions were stopped by adding equal volume of 2X stop dye (1X: 100 mm EDTA, 2% SDS, 20% glycerol, and 0.08% orange G). The reactions were loaded on a 1% agarose gel, and products were resolved. Gels were poststained by ethidium bromide and imaged using a Bio-Rad GelDoc. The bands were quantified using ImageJ software [57]. Background correction was performed using baseline adjustment.
To quantify the reactions, the intensities [I] corresponding to nicked (N), linear (L), and supercoiled (SC) bands were designated respectively as IN, IL, and ISC. The background-corrected fractions of the cleavage products were calculated as:
| (1) |
| (2) |
where values with the ‘C’ subscript represent the intensities corresponding to the no enzyme control lane of each gel.
The fraction of total cleavage was then calculated as:
| (3) |
and the fraction of remaining supercoiled precursor (Frac[P]) was given by:
| (4) |
Standard deviation (SD) and standard error of mean (SEM) were calculated as reported previously [42].
Oligo DNA cleavage assays
A 50-nt oligo TS DNA and its complementary 50-nt strand (i.e., NTS), containing the protospacer and PAM, were independently labeled at their 5′ termini with 32P (Table S2). The labeled strands were further purified through Micro Bio-Spin columns (P-30) from Bio-Rad (Hercules, CA, USA). In this work, a 100% recovery of the labeled strands was assumed for calculations in downstream experiments. The strands were then annealed to excess of unlabeled complementary strand in a 1:1.2 ratio by heating at 95°C for 2 min followed by slow cooling to room temperature in the presence of 1X annealing buffer (10 mm Tris/HCl, pH 8, 50 mm NaCl). ~1 nm of duplex DNA was treated with varying concentrations of preformed RNP complexes (see section Supercoiled plasmid cleavage assays) ranging from 10 n to 500 nm in a reaction volume of 10 μL. The cleavage reactions were carried out at 37 °C. The cleavage reaction time was fixed at 45 min for concentration course, but varied in time-course measurements designed to measure the reaction rate. To stop the reaction at each desired time, EDTA (final of 11 mm), CaCl2 (14 mm), and Proteinase K (~1.8 μg/reaction, NEB) were added to the reaction bringing the reaction volume to ~ 14 μL, followed by incubation at 50 °C for 15 min. The reaction mixture was combined with equal volume of 2X RNA loading dye (1X concentration: 47.5% formamide, 9 mm EDTA, 1% SDS, and 0.0125% bromophenol blue dye), heated at 95 °C for 5 min, and loaded onto a prewarmed 16% urea/formamide/acrylamide gel. The gels were then exposed onto a phosphor imaging screen and imaged using a GE Typhoon FLA 7000 imager.
To quantify the reactions, the intensities of the precursor band and the product bands for either 32P-labeled TS (ITprecursor, ITproducts) or 32P-labeled NTS (INTprecursor, INTproducts) were measured. The fraction of precursor remaining was calculated as:
| (5) |
| (6) |
Linearized ds plasmid cleavage assays
Matched supercoiled plasmid DNA was digested with ScaI-HF (NEB) in Cutsmart buffer. The enzyme was inactivated by heating at 80 °C for 20 min, and the linearized DNA was purified using the E.Z.N.A Cycle Pure kit. For testing the effect of linear dsDNA on cis-cleavage, 25 nm of FnoCas12a was preincubated with 30 nm of crRNA in 1X cleavage buffer (section Supercoiled plasmid cleavage assays) for 10 min at 37 °C. 100 ng of linearized matched DNA was added, and the reactions were stopped at indicated time points using the 2X stop dye (section Supercoiled plasmid cleavage assays).
To quantify the reactions, the intensities of the precursor linear band and the product bands were measured. The intensities [I] corresponding to linear precursor (L), cleaved band 1 (B1), and cleaved band 2 (B2) were designated respectively as IL, IB1, and IB2. The background-corrected fractions of products were calculated as:
| (7) |
| (8) |
where values with the ‘C’ subscript represent the intensities corresponding to the no enzyme control lane of each gel.
The fraction of total cleavage was then calculated as:
| (9) |
and the fraction of remaining precursor (Frac[P]) was calculated using Eqn (4).
ss plasmid cleavage assays
Supercoiled ss plasmid
M13mp18 circular ssDNA was used for testing the effect of ss supercoiled plasmid DNA for cis-cleavage. The substrate and RNP concentration were same as Section Linearized ds plasmid cleavage assays.
Linearized ss plasmid
About 1.5 μg of M13mp18 was linearized using 70 units of EcoRI (NEB) enzyme for 90 min in IX NEB buffer 2.1 at 37 °C. The enzyme was heat-inactivated, and the DNA was purified using the E.Z.N.A Cycle Pure Kit. Note that ssDNA is not a preferred substrate for EcoRI, and control studies showed that the efficiency of M13mp18 DNA digestion ranged from 50% to 70% of linear form (details in Fig. S8). The digested M13mp18, containing a mixture of linear and circular forms, was treated with 25 nm RNP for FnoCas12aWT and different RNP concentrations for FnoCas12aKD2P in cleavage buffer listed in section Supercoiled plasmid cleavage assays. The reaction was stopped using 2X stop dye (section Supercoiled plasmid cleavage assays), and the products were resolved on a 1% agarose gel.
Determination of rate constants for precursor cleavage
Time-course measurements using linearized ds plasmid and oligo DNA substrates were fit [Origin (Pro) Version 2020b (Northampton, MA)] adequately to a single-exponential decay equation:
| (10) |
with y being the fraction of precursor of oligo-T strand (Eqn 5), NT strand (Eqn 6), or linearized plasmid (see Eqns 7–9 and Eqn 4), kobs being the reaction rate constants, and a being the total active fraction.
For cleavage of supercoiled plasmids, a double-exponential decay was required to properly fit the data:
| (11) |
with Frac[P] specified by Eqn (4), k1 and k2 being the reaction rate constants, and a1 and a2 being fraction of precursor that reacted respectively with the k1 and k2 rate constants. The total active fraction a = a1 + a2.
Electrophoretic mobility shift assay
The TS strands of matched or MM8 oligos (Table S2) were labeled with 32P on the 5′ termini. The labeled DNA strands were purified and annealed with the NTS oligo as described earlier (section Oligo DNA cleavage assays). ~1 nm of matched or MM8 TS-labeled DNA duplex was incubated with desired concentrations (5 nm to 250 nm) of preformed RNP in the presence of 1X cleavage buffer (section Supercoiled plasmid cleavage assays). No divalent metal was added in order to prevent DNA cleavage. After a 20-min incubation at room temperature, a glycerol dye was added to the mixture (final concentration: 14.3% glycerol, 0.0143 % bromophenol blue, and 0.0143% of xylene cyanol) and the sample was resolved on a 6% native acrylamide gel using 0.5X Tris borate (0.5X: 45 mm Tris/HCl, 45 mm boric acid) as the running buffer. The bands were visualized by exposing onto a phosphor imaging screen and imaged using a GE Typhoon FLA 7000 imager. The intensities of bands corresponding to the unbound DNA (Iunbound) and the ternary complexes (Icomplex) were measured. The fraction of bound DNA was calculated as:
| (12) |
The data were fit to:
| (13) |
with Kd being the dissociation constant between the RNP and the DNA.
trans-cleavage assays
trans-cleavage assays were tested on two different substrates: M13 ss circular DNA (NEB) and a 54-nt ss linear oligo DNA (Table S2). We also tested the effect of two different activators: a 20-nt-long ssDNA activator without a PAM and a 24-nt-long dsDNA activator that contained a PAM (Table S2). The forward and reverse strands of the ds activator (1 : 1 molar ratio) were annealed as previously mentioned (see section Oligo DNA cleavage assays). The concentrations of the activator DNA and the crRNA were the same in each reaction.
M13 trans-cleavage: For concentration titration, 100 ng (4.4 nm) of M13 ssDNA and 20-nt ssDNA or 24-nt dsDNA activator were incubated with preformed RNP at desired concentrations (5 mm to 500 mm) in the presence of 1X cleavage buffer (20 mm HEPES, pH 7.5, 150 mm KCl, 5% glycerol, and 0.5 mm DTT) and 5 mm MgCl2 for 60 min at 37 °C. The M13 ssDNA time-course reactions contained 25 nm Cas12a, 30 nm crRNA, 30 nm ssDNA activator, and 100 ng M13 ssDNA. The reactions were incubated for the indicated time points. All the reactions were stopped by adding an equal volume of 2X stop dye (1X concentration: 50 mm EDTA, 1% SDS, 10% glycerol, and 0.08% orange G). Reaction products were resolved on 1% agarose gels and poststained with ethidium bromide. Bands were quantified using the imagej [57].
To quantify the disappearance of M13 ssDNA, intensity of circular (ICr) band was quantified for control lane (ICr control) and reaction lane (ICr sample). The remaining in each lane circular (ICr) was calculated as:
| (14) |
Oligo trans-cleavage assay: A 54-nt oligo DNA (Table S2) was labeled at the 5′ termini with a 32P label and purified as described (section Oligo DNA cleavage assays). Approximately 1 nm of 32P-labeled ss oligo was incubated with 30 nm FnoCas12a, 36 nm crRNA, and 36 nm activator DNA (either ssDNA or dsDNA activator) in the presence of 1X cleavage buffer (section Supercoiled plasmid cleavage assays) and 10 mm MgCl2 for the indicated time points at 37 °C similar to a previous report [39]. The RNP was preformed, followed by the addition of activator and substrate to the reaction mix. The reactions were stopped by adding 2X RNA loading dye (1X concentration: 47.5% formamide, 9 mm EDTA, 1% SDS, and 0.0125% bromophenol blue) at the indicated time points. The samples were heated at 95 °C for 5 min and resolved using a prewarmed 16% urea/formamide/acrylamide gel. The gels were then exposed onto a phosphor screen and imaged using a GE Typhoon FLA 7000.
RNA-independent DNA cleavage assay
RNA-independent DNA cleavage assay was performed as previously described [42]. The different substrates tested include M13 ssDNA or pUC19 or a 60-nt ss oligo DNA (Table S2) that was labeled at its 5′ terminus with 32P.
Results
Analysis of interactions of FnoCas12a BH
To understand the functional role of BH in Cas12a, we analyzed interactions of the BH with crRNA/DNA and other surrounding domains in the FnoCas12a ternary complex structure (PDB ID: 5NFV) [20] (Fig. 1 and S3). The BH of FnoCas12a is shorter than that of SpyCas9 and interacts with crRNA and other regions of the protein through its N- and C-terminal amino acids (Fig. 1 and Table S3). The N terminus of FnoCas12a BH interacts with the pseudoknot region of the crRNA and its RuvC domain (Figs 1A, S3A, Table S3). The C-terminal of BH has multiple interactions with crRNA guide region and REC2 domain through residues 968–970 (Table S3). Arg968, a conserved arginine of BH, interacts with G11 and U12 of the guide region of crRNA (Fig. 1B and Table S3) [33]. Lys969 interacts with both guide region (A13, side chain interaction) and REC2 domain (K527, main chain interaction) (Fig. 1B, Table S3). Asp970 interacts with Lys524 of the REC2 domain (Fig. 1B and Table S3). Previous studies have shown that Trp971, the residue in the loop following BH, is highly conserved in Cas12a orthologs and its substitution drastically reduces protein activity (Fig. 1B) [21]. Trp971 has hydrophobic interactions with Tyr579 and Arg583 of REC2 domain and acts as a wedge, mediating the movement of the helices in the REC2 domain that are essential for conformational changes during transition from binary to ternary state (Figs 1B and S3B) [58].
Fig. 1.
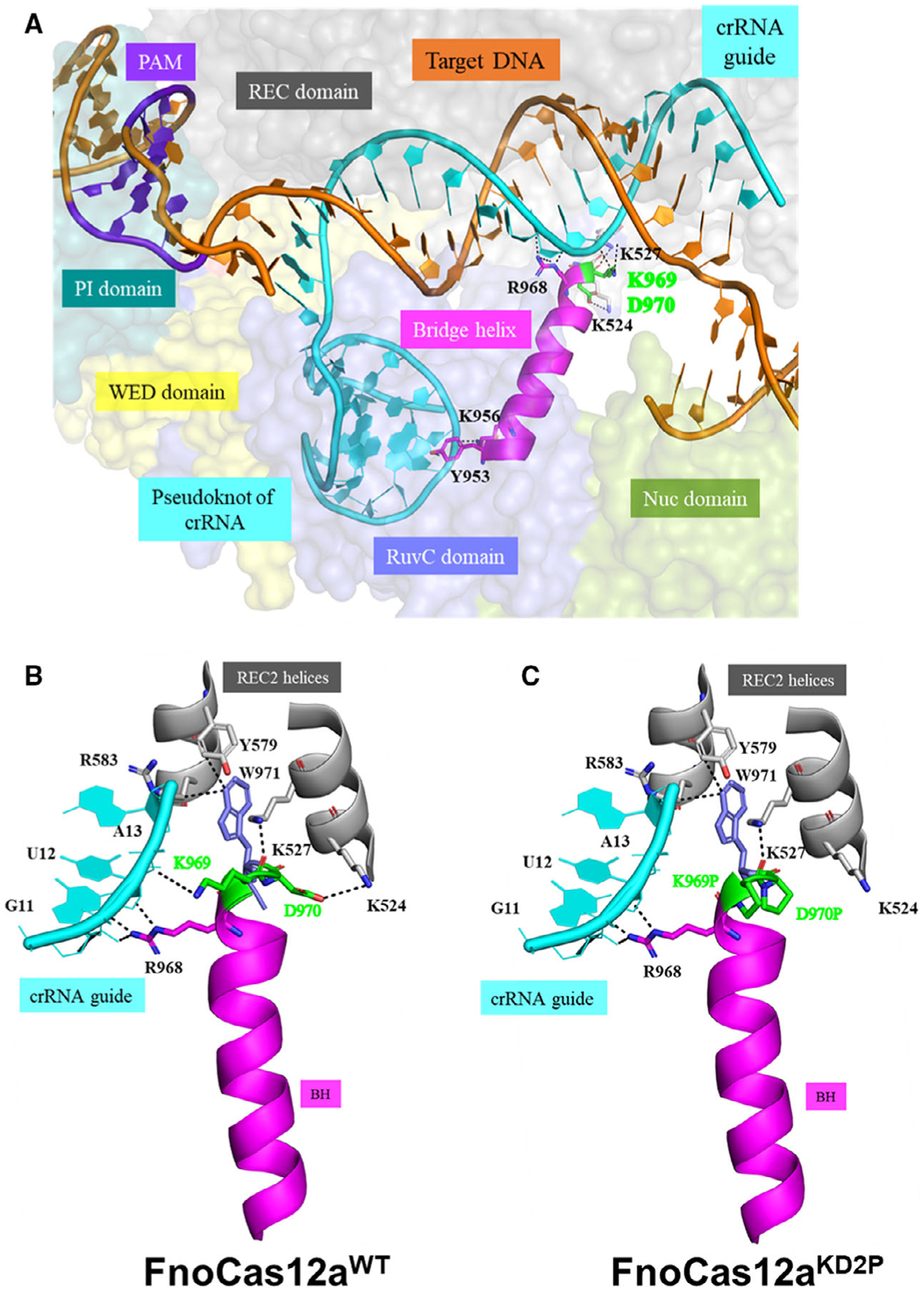
Interactions of FnoCas12a BH (residues Y953 to D970) with crRNA and REC domain. Crystal structure of FnoCas12a complexed to crRNA and dsDNA target (PDB ID: 5NFV) was used for this analysis [18]. (A) Representative figure showing orientation of BH and its interactions in FnoCas12a. The residues at the N-terminal end of BH interact with the pseudoknot of crRNA, while the residues at the C-terminal end interact with the guide region of crRNA and the residues K524 and K527 of the REC2 domain. The residues highlighted in green are substituted with prolines in the FnoCas12aKD2P variant. In this structure that represents pre-cleavage state, K969 is in helical conformation, while D970 is in a loop. (B) The zoomed-in figure shows the native interactions of residues K969, D970, and W971 with the crRNA guide and REC2 domain. W971 is present in the loop following the BH and is highly conserved in Cas12a orthologs. (C) Interactions of proline substitutions at positions 969 and 970. The view is the same as in (B). K969 and D970 were replaced by prolines in PyMOL, and the close interactions are shown. Figures were made using PyMOL [67].
Previous literature has shown a loop-to-helix transition at the C-terminal end of BH during transformation of the binary structure (Cas12a-crRNA, PDB: 5ID6 [22]) to the ternary state (Cas12-crRNA-DNA, PDB: 5XUS [58]) in a Cas12a ortholog from Lach-nospiraceae bacterium (LbCas12a). Similar observations can also be seen in the binary (PDB: 5NG6 [20]) and ternary (PDB: 5MGA [26]) structures of FnoCas12a. To characterize the effect of the loop-to-helix transition at the C-terminal end of BH, we introduced amino acids substitutions. Instead of changing Arg968 that is highly conserved across Cas12a orthologs [33], we chose to substitute amino acids Lys969 and Asp970 with proline (the variant is named FnoCas12aKD2P), as proline can not only impact helicity of a helix, enabling testing of loop-to-helix transition, but has also been shown to have less interactions with nucleic acids [59]. The substitutions cause the loss of the two side chain interactions, while maintaining the main chain interaction of the carbonyl oxygen of Pro969 with Lys527 of REC2 domain (analysis based on PDB ID: 5NFV [20], Fig. 1C). In addition, we hypothesize that the difference in helicity of BH due to the presence of two tandem prolines may affect the positioning of Typ971 that acts as a wedge in orchestrating the conformational changes.
BH variant of FnoCas12a cleaves matched DNA with a reduced efficiency
We performed in vitro cleavage assays using supercoiled plasmid substrates that carried a 31-nt-long protospacer and a flanking 3-nt-long PAM (5′-TTA-3′) (Fig. S2, Table S1). In this work, all activity assays involved the use of pre-incubated RNP complex, with RNA at a slightly higher molar excess than the protein. With a supercoiled plasmid DNA having complete complementarity with the guide region of the crRNA (matched DNA), FnoCas12aKD2P reached saturation levels of total cleavage (i.e., total DNA cleaved that includes both linear and nicked products) at 25 nm RNP (Fig. 2A). At higher RNP concentrations, total cleavage increased slightly only for FnoCas12aWT (Fig. 2A), and hence, an RNP concentration of 25 nm was selected for further experiments.
Fig. 2.
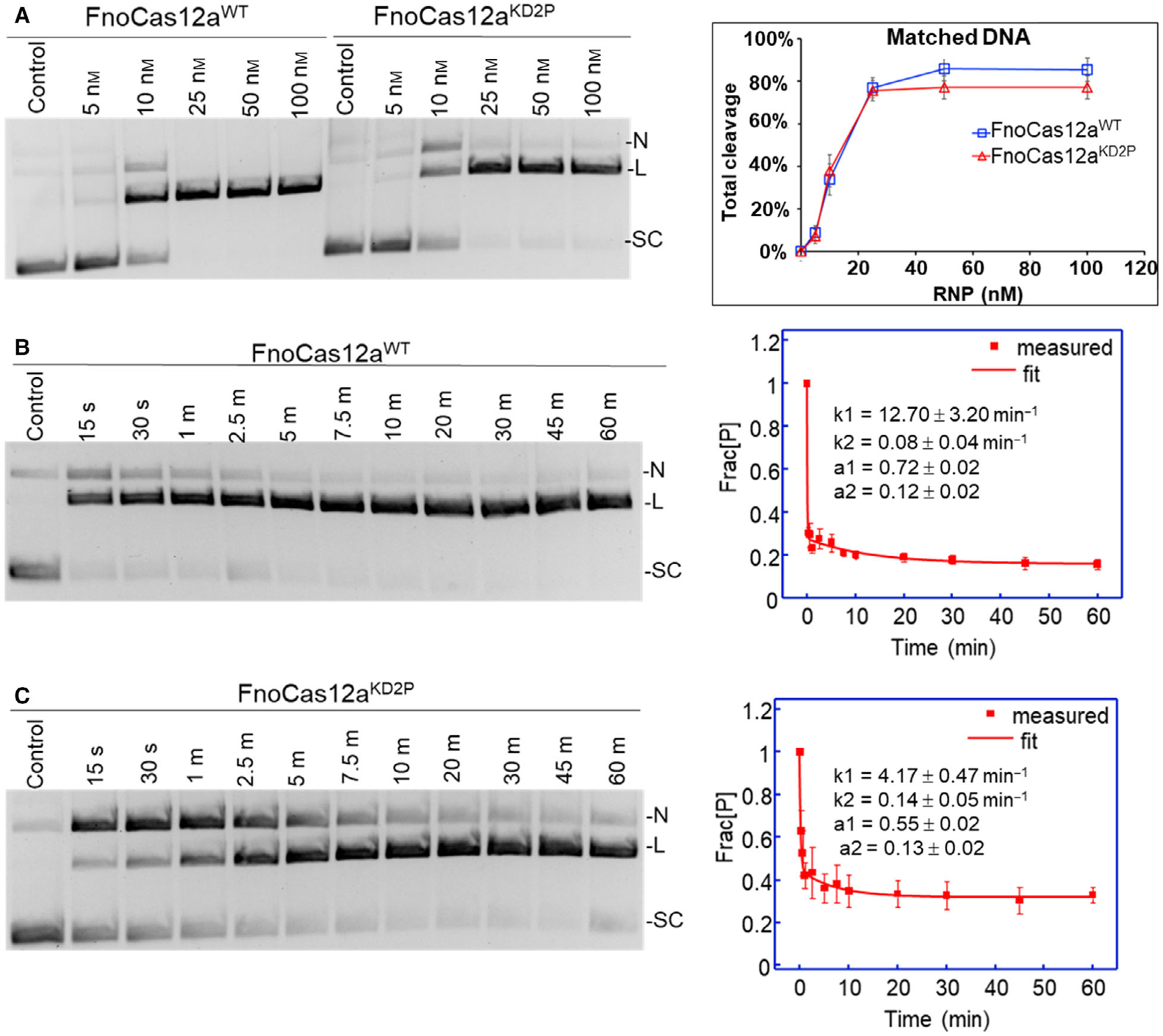
Effect of BH mutations on cis-cleavage of matched plasmid DNA substrate. (A) A representative gel showing cleavages of the matched DNA substrate with FnoCas12aWT and FnoCas12aKD2P at various RNP concentrations. The corresponding graph on the right shows total cleavage for FnoCas12aWT and FnoCas12aKD2P plotted against increasing RNP concentration. Total reaction time was 30 min. Data were obtained from six replications, and the error bars represent SEM. (B) A representative gel from time-course assays of cleavage by 25 nm RNP of FnoCas12aWT and the plot of the average fraction of precursor (Frac[P]) vs. time. (C) Representative gel from time-course assays of cleavage by 25 nm RNP of FnoCas12aKD2P and the plot of the average fraction of precursor (Frac[P]) vs. time. The error bars represent the SEM of three different replications. As shown, the data were fit to a double-exponential decay (Eqn 11). [N: nicked, L: linear, SC: supercoiled, s: sec and m: min].
While the total fraction of precursor cleavage at 30 min was comparable between FnoCas12aWT and FnoCas12aKD2P for the matched DNA (Fig. 2A), time-course analysis revealed a clearer deficiency in DNA cleavage rate for FnoCas12aKD2P (Fig. 2B,C, Table S4). For both FnoCas12aWT and FnoCas12aKD2P, the loss of supercoiled precursor can be fit to a double-exponential decay (Eqn 11, Materials and Methods), with the fast decay accounting for the majority of the population (Fig. 2B,C). For the fast decay, FnoCas12aKD2P showed a significant reduction in the rate constant (k1) as compared to FnoCas12aWT (4.17 ± 0.47 min−1 for FnoCas12aKD2P and 12.70 ± 3.20 min−1 for FnoCas12aWT), with a corresponding reduction in the fraction of populations (a1: 0.55 ± 0.02 for FnoCas12aKD2P vs. 0.72 ± 0.02 for FnoCas12aWT) (Table S4). For the slow decay, both the rate constant (k2) and fraction of population (a2) were comparable between FnoCas12aWT and FnoCas12aKD2P (Fig. 2B,C). However, the mechanistic origin of slow decay is rather unclear, as it may arise from ‘inactive’ RNP due to the preparation or minor ‘alternative’ RNP conformations. Furthermore, analyses validated the use of two-exponential fit (Eqn 11, Materials and Methods) instead of a one-exponential for analyzing the supercoiled plasmid DNA data (Fig. S4). Overall, the 3.0-fold reduction in k1 between FnoCas12aKD2P and FnoCas12aWT clearly indicates that proline substitutions in BH compromised the activity of FnoCas12aKD2P.
FnoCas12aKD2P exhibits selective nicking of supercoiled target DNA with a positional effect with respect to PAM
Our activity assays with a completely matched supercoiled target DNA showed that FnoCas12aKD2P has a lower efficiency to cleave on-target DNA, but proceeded to complete linearization of the substrate at longer time points. We proceeded to analyze the effect of BH modulations on cleavage of mismatch containing DNA. We created single mismatches across each position of the protospacer (Mismatch (MM)1–22) (Fig. S2) embedded in a supercoiled plasmid and assessed how the BH substitutions affect nicking and dsDNA cleavage activities of Cas12a.
The supercoiled plasmid DNA substrates were treated with 25 nm RNP complex at 37 °C for 15 min (Fig. S5A–C). Total cleavage, which measured the loss of the supercoiled precursor, was comparable for FnoCas12aWT and FnoCas12aKD2P across each mismatched position tested (Figs 3A, S6A, and S6B). Interestingly, there was a difference in the amount of nicking (N) and linearization (L) by FnoCas12aWT and FnoCas12aKD2P for each of these substrates (Figs 3B, S5, S6). In the case of FnoCas12aKD2P, even though nicked products are present across mismatches from 1 to 18 (MM1–MM18), positions MM12–17 are the most impacted with an accumulation of 61–77% nicked product in this region (Fig. S6B). The linearizing efficiency of FnoCas12aKD2P goes back to similar levels as with matched DNA for positions MM19–MM22, indicating a clear positional effect of mismatch cleavage efficiency (Figs 3, S5, S6). Interestingly, FnoCas12aWT also displays a similar pattern of accumulation of nicked products (3–19%) at mismatch positions MM12–MM17 (Fig. S6A) indicating higher sensitivity of Cas12a in these mismatch positions. To analyze whether longer incubations enable linearization of mismatches, we further performed cleavage assays with matched DNA, MM8, and MM12 substrates at various time points (Fig. S7). While FnoCas12aWT linearized a majority of the DNA within the first 30 min of the reaction across all the substrates, the nicked product accumulated at the 30 min time point by FnoCas12aKD2P for MM8 and MM12 was never converted to linear product even after 3 hours (Fig. S7).
Fig. 3.
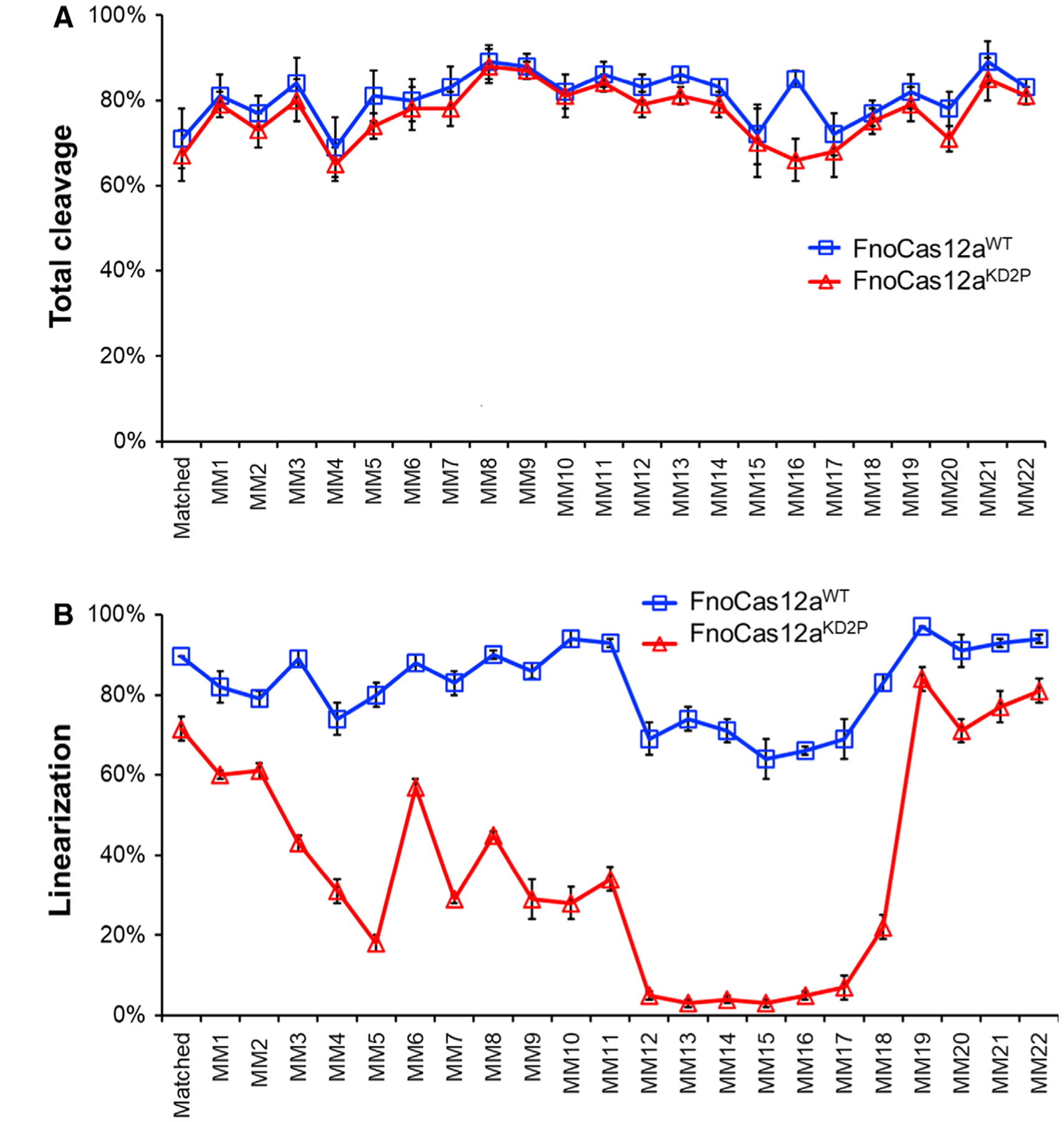
cis-cleavage of supercoiled target DNAs containing mismatches. (A) Graphs representing the total activity of FnoCas12aWT and FnoCas12aKD2P across all the substrates tested. (B) Graphs representing the linear activity of FnoCas12aWT and FnoCas12aKD2P across all the substrates tested. Data were obtained from three replications, and the error bars represent SEM. [MM: mismatch and the number indicate the mismatch position on the NTS with respect to PAM]. Note that the positions 12–18 lack linearization ability in the presence of an impaired BH.
The accumulation of nicked product by FnoCas12aKD2P across different positions of mismatched substrates indicates the inefficiency of FnoCas12aKD2P to cleave both strands of the DNA when mismatches are present (Figs 3B, S5, S6B). For comparison, FnoCas12aWT demonstrated comparable efficiencies to cleave mismatched DNA as that of the matched DNA (Fig. 3 B). Thus, substitutions in the BH of Cas12a significantly reduce dsDNA breaks in mismatched substrates, and the degree of such reduction varies depending on the specific positions of the target DNA.
Substitutions in the BH of FnoCas12a impact both TS and NTS cleavages
Our results with supercoiled plasmids showed accumulation of nicked products when FnoCas12aKD2P cleaves both matched and mismatch containing plasmids, indicating that cleavage of one of the DNA strands is compromised in the BH variant. To identify which strand cleavage is impacted, we performed cleavage assays using a 50 base pair (bp) linear dsDNA oligo substrate that contained a 31-nt-long protospacer region and a cognate PAM (Table S2). For the matched oligo substrate, time-course assays were performed by incubating 1 nm matched duplex DNA with 25 nm of RNP complex (Fig. 4). FnoCas12aWT cleaved around 87% of TS and NTS at 1 min, and the cleavage rate constants (kobs) were estimated to be 2.03 ± 0.14 min−1 for TS and 2.90 ± 0.36 min−1 for NTS (Figs. 4A–C). The slightly faster kobs for NTS is consistent with previous observations that NTS cleavage precedes TS cleavage in wild-type Cas12a [31,32].
Fig. 4.
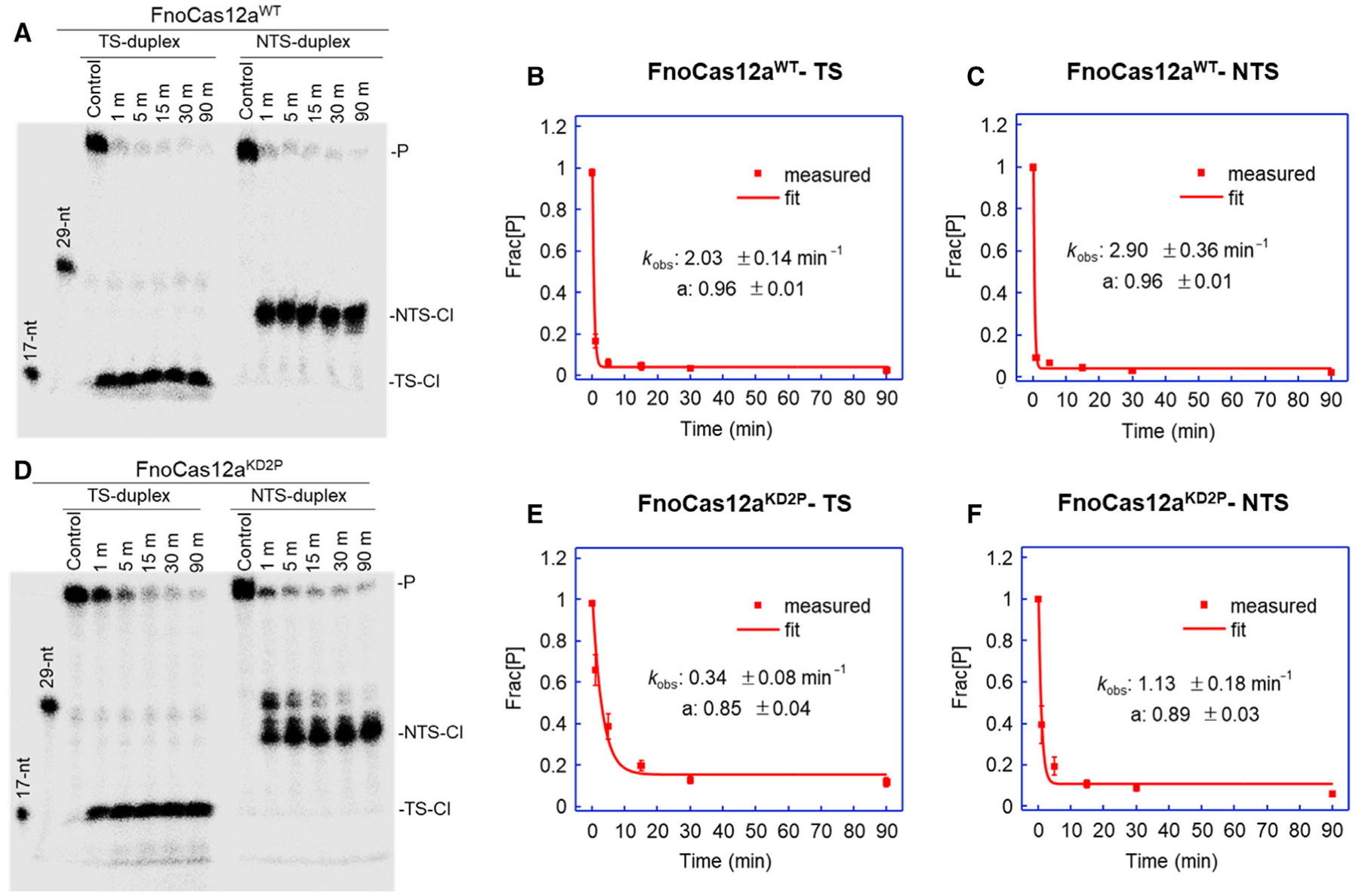
Kinetic analyses of matched oligo DNA cleavage. (A) A representative gel for the time-course assay with matched oligo DNA duplex and FnoCas12aWT. Fitting of the disappearance of matched TS oligo precursor (B) and matched NTS oligo precursor (C) by FnoCas12aWT. (D) A representative gel for the time-course assay with matched oligo DNA duplex and FnoCas12aKD2P. Fitting of the disappearance of matched TS oligo precursor (E) and matched NTS oligo precursor (F) by FnoCas12aKD2P. In each panel, the average fraction of precursor (Frac[P]) is plotted vs. time, with the error bars representing the SEM of four replications. The data were fit to a single-exponential decay (Eqn 10). [P: precursor; TS: target strand; NTS: nontarget strand; TS-Cl: TS cleavage products; NTS-Cl: NTS cleavage products; nt: nucleotides].
FnoCas12aKD2P cleaved around 32% TS and 61% NTS of matched duplex oligo at 1 min and the kobs was determined to be 0.34 ± 0.08 min−1 for TS and 1.13 ± 0.18 min−1 for NTS (Figs. 4D–F). NTS cleavage was faster than TS, but cleavage of both TS and NTS was slower compared with FnoCas12aWT (Table S4). Compared with that of FnoCas12aWT, kobs was reduced by 6-fold and 3-fold, respectively, for TS and NTS cleavages in FnoCas12aKD2P (Table S4). These results suggest that the BH variation in FnoCas12aKD2P negatively impacts TS cleavage more than NTS cleavage. Consequently, this suggests the accumulation of nicked population by FnoCas12aKD2P in the plasmid cleavage assay (Fig. 2) is likely due to the impaired TS cleavage.
Furthermore, cleavage of a 50 bp linear dsDNA oligo substrate containing an MM8 mismatch was analyzed at increasing RNP concentrations (Fig. 5A,B). FnoCas12aWT cleaved MM8 oligo to a lower extent when compared to the matched oligo at the different RNP concentrations tested (Fig. 5C). Interestingly, our results indicate that the NTS cleavage is more efficient than that of TS, even in the case of cleavage of MM8 oligo by FnoCas12aWT (Fig. 5C). On the other hand, FnoCas12aKD2P did not show any apparent cleavage under the experimental conditions that were tested (Fig. 5B). Importantly, binding analysis showed similar dissociation constants (Kd) between FnoCas12aWT and FnoCas12aKD2P with either matched or MM8 oligo (Kd, ranging from 17 to 32 nm, Fig. 6), indicating that deficiency in the cleavage of MM8 does not stem from weaker binding by FnoCas12aKD2P.
Fig. 5.
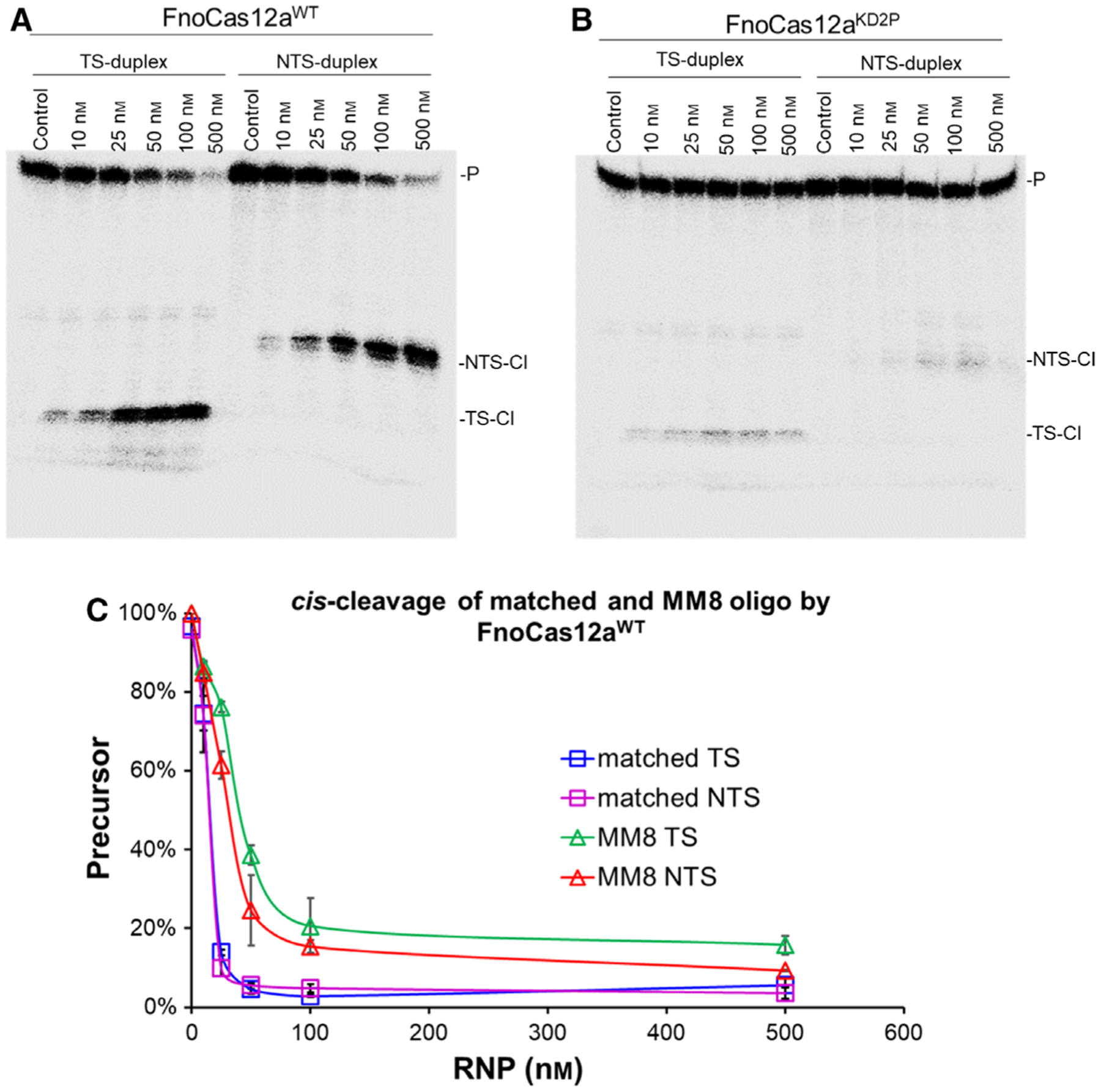
cis-cleavage of MM8 oligo DNA substrate. An RNP concentration course for cleavage of MM8 oligo dsDNA by FnoCas12aWT (A) and FnoCas12aKD2P (B). Reaction time was 45 min. [P: precursor; TS: target strand; NTS: nontarget strand; TS-Cl: TS cleavage products; NTS-Cl: NTS cleavage products]. Representative gels from two replications. (C) Graph showing the disappearance of TS and NTS precursors of matched and MM8 oligo DNA for FnoCas12aWT.
Fig. 6.
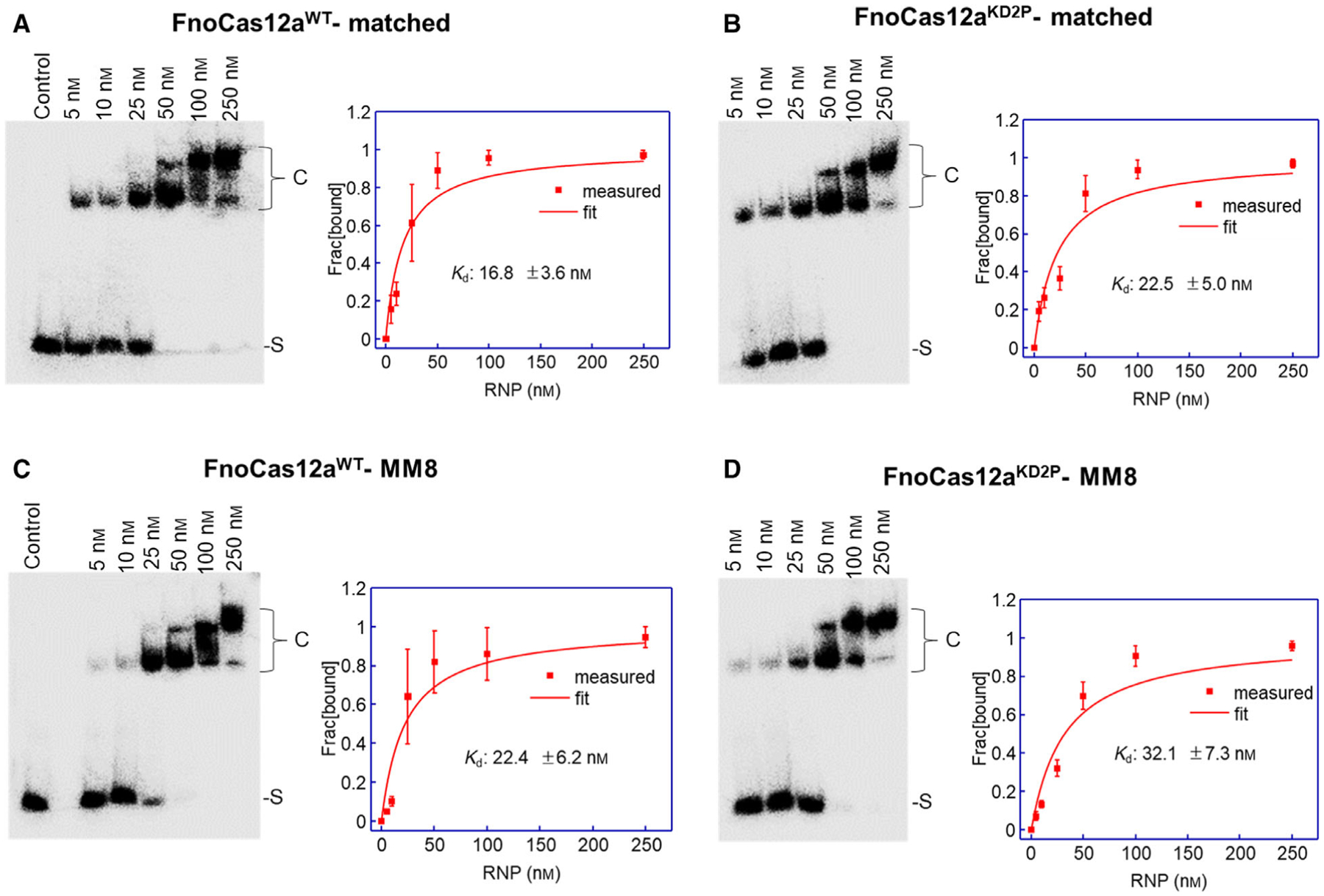
Electrophoretic mobility shift assay of matched and MM8 oligo DNA with FnoCas12aWT and FnoCas12aKD2P. In each panel, a representative gel is shown on the left (only the TS of the duplex is labeled for visualization), and a fitting of average fraction of bound DNA for Kd determination is shown on the right. Data were obtained from three replications, and the error bars represent SEM. (A) FnoCas12aWT binding to matched oligo DNA. (B) FnoCas12aKD2P binding to matched oligo DNA. (C) FnoCas12aWT binding to MM8 oligo DNA. (D) FnoCas12aKD2P binding to MM8 oligo DNA. [S: unbound substrate; C: ternary complex].
Together, our results show that BH plays a role in the coordinated cleavages of NTS and TS in FnoCas12a. Further experiments are needed to elucidate the exact mechanism of coordinated cleavage. Comparison of the rates for cleaving supercoiled and oligo DNA substrates indicates preferences of Cas12a for different types of DNA and a possible role of BH in supporting cleavage of such substrates (Table S4).
Superhelicity and strandedness of substrate DNA are sensed by FnoCas12a BH
Previous research has shown that Cas9 exhibits preferences toward different physical states of DNA substrates [60]. We analyzed whether FnoCas12a’s BH plays a role in the cleavage of DNA with different characteristics, such as superhelicity and strandedness. To test the effect of superhelicity, we measured the efficiency of FnoCas12aWT and FnoCas12aKD2P to cleave linearized matched DNA plasmid. Time-course measurements were carried out using 25 nm RNP (Fig. 7A,B), and the loss of the linearized substrate was adequately fit to a one-exponential decay (Eqn 10). The kobs was determined to be 3.12 ± 0.24 min−1 for FnoCas12aWT and 0.35 ± 0.03 min−1 for FnoCas12aKD2P, resulting in a 9-fold reduction in the rate of cleavage of linear substrate by FnoCas12aKD2P (Fig. 7 A,B, Table S4). Even though both FnoCas12aWT and FnoCas12aKD2P had a slower cleavage rate on linearized plasmid compared with that of supercoiled substrate, the reduction was more drastic in FnoCas12aKD2P (Figs 2, 7, Table S4).
Fig. 7.
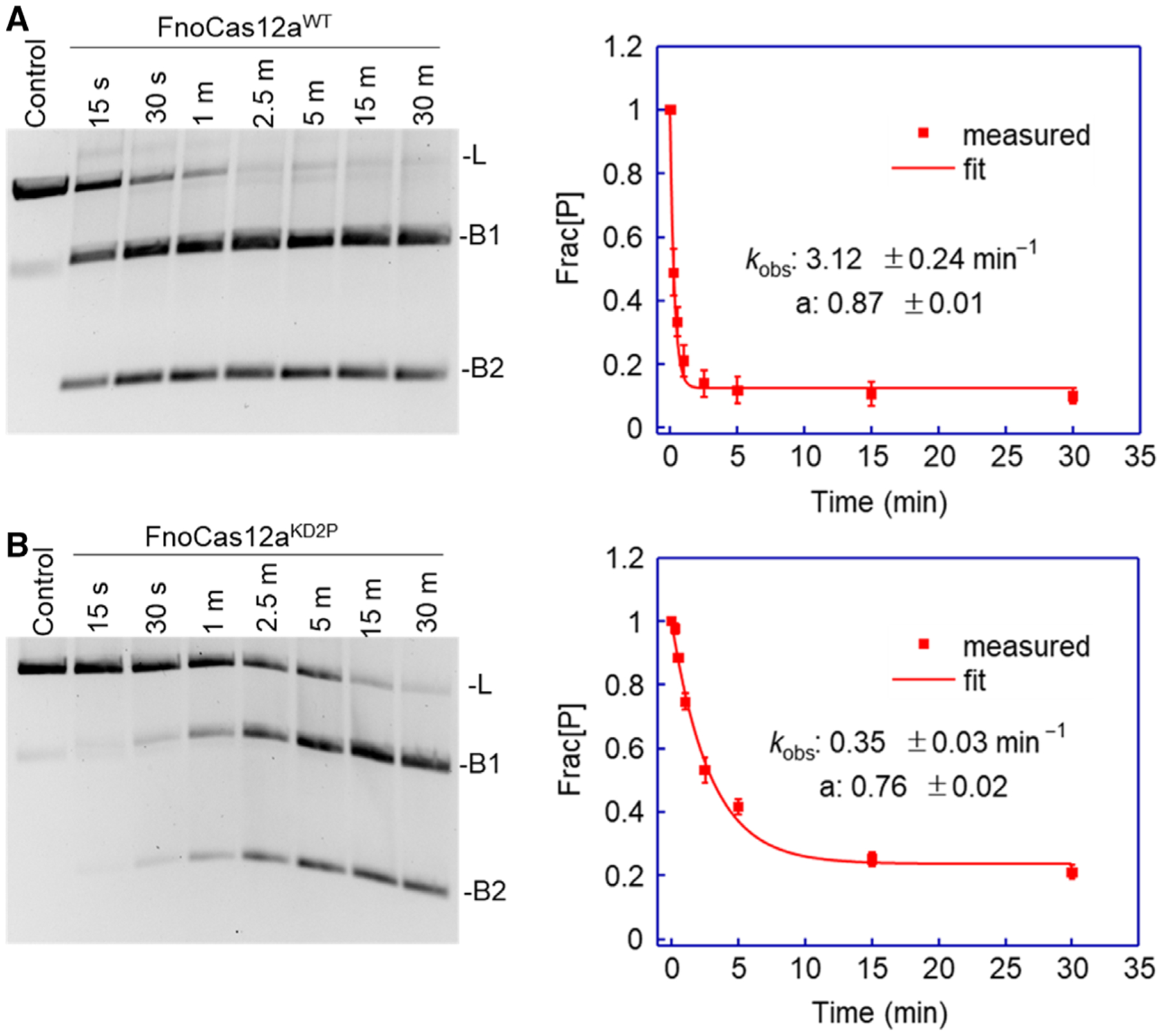
cis-cleavage of linearized matched ds plasmid by FnoCas12aWT and FnoCas12aKD2P. (A) A representative gel showing the time-dependent cleavage of linear DNA by 25 nm RNP of FnoCas12aWT and the plot of the average fraction of precursor (Frac[P]) vs. time. (B) A representative gel showing the time-dependent cleavage of linear DNA by 25 nm RNP of FnoCas12aKD2P RNP and the plot of the average fraction of precursor (Frac[P]) vs. time. Data were obtained from three replications, and the error bars represent SEM. [L: linear, B1: cleaved band 1; B2: cleaved band 2; s: sec and m: min].
To test the effect of strandedness of DNA on cis-cleavage efficiency, circular and linearized M13 mp18 ssDNA were used as substrates (Fig. S8). FnoCas12aWT at 25 nm RNP was able to completely degrade the circular M13 ssDNA (Fig. S8B), although the process took approximately an hour, which was slower than that observed with the supercoiled dsDNA substrate under identical conditions (Fig. 2B). It is interesting to note that in addition to linearization of ssDNA by cis-cleavage, the substrate is being further degraded, due to the trans-cleavage activity exhibited by Cas12a proteins (discussed later). FnoCas12aKD2P had a significantly lower extent of cleavage of circular M13 DNA as compared to FnoCas12aWT, for both cis- and the associated trans-activities (Fig. S8B).
To test the effect of superhelicity on ssDNA, we linearized M13 ssDNA and performed cis-cleavage. FnoCas12aWT displayed a lower efficiency to cleave linear M13 ssDNA compared with that of circular M13 DNA. This observation is based on the amount of substrate left after 60 min of incubation with 25 nm RNP complex (0% for circular M13 vs. ~ 40% for linear M13, Fig. S8B, S8C). FnoCas12aKD2P displayed further impairment as observed in reactions with even increasing RNP concentrations (Fig. S8C). Thus, Cas12a does not prefer ssDNA as a substrate, and superhelical ssDNA can be tolerated by the wild-type protein. BH appears to play a role in supporting ssDNA cleavage since both circular and linear M13 ssDNA were cleaved to a much lower extent by FnoCas12aKD2P.
In conclusion, our results clearly establish that FnoCas12a prefers dsDNA substrates over ssDNA, and supercoiled DNA over linear DNA (Figs 2, 4, 7, S8). With each of these DNA substrate states, FnoCas12aKD2P shows reduced cleavage activity when compared to that of FnoCas12aWT, with a varied degree of reduction depending on the physical state of the DNA (Figs 2, 4, 7, S8). These results implicate that BH plays a role in supporting cleavage of different physical states of DNA.
FnoCas12a BH perturbations affect trans-cleavage activity
We proceeded to analyze whether BH perturbations affect the trans-ssDNA cleavage efficiency of FnoCas12a. A PAM-less 20-nt ssDNA activator that is completely complementary to the guide region of the crRNA was used to initiate trans-cleavage (Table S2). With FnoCas12aWT, 25 nm of RNP activator completely degraded an M13 ss circular DNA substrate by trans-cleavage at 37°C in 60 min, with a 10 nm FnoCas12aWT RNP required for initiating trans-cleavage. (Fig. 8A). In the case of FnoCas12aKD2P, a minimum RNP concentration of 25 nm was required for initiating trans-cleavage, with ~ 90% of M13 circular ssDNA left uncut at 60 min (Fig. 8A). At higher concentrations of RNP, FnoCas12aKD2P degraded M13 ssDNA to completion (Fig. 8A). This indicates that the BH substitution in FnoCas12a reduces the efficiency of trans-M13 cleavage.
Fig. 8.
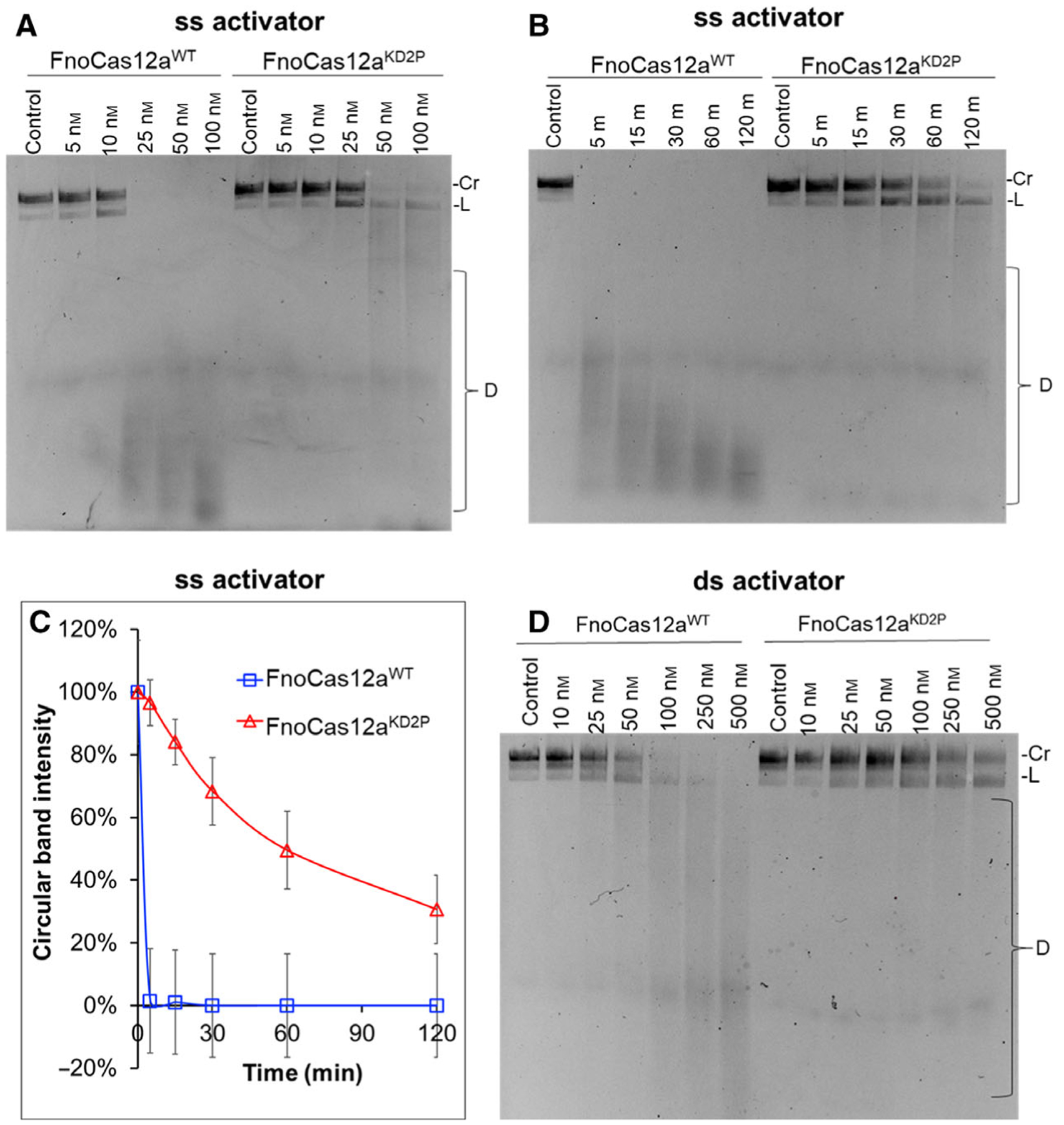
trans-cleavage of FnoCas12aWT and FnoCas12aKD2P on circular M13 ssDNA. (A) Gel showing trans-cleavage of circular M13 ssDNA with increasing RNP and ssDNA activator concentrations. Total reaction time was 60 min. Representative gel from four replications. (B) Time course of trans-cleavage of circular M13 ssDNA by 25 nm RNP-ssDNA activator. Representative gel from three replications. (C) Graph tracking the disappearance of circular form of M13 ssDNA over time. The error bars represent the SEM of three replications. (D) Gel showing trans-cleavage of circular M13 ssDNA with increasing RNP and dsDNA activator concentrations. Total reaction time was 60 min. Representative gel from three replications. [Cr: circular; L: linear; D: degradation; m: min].
We decided to test whether longer incubation times at 25 nm RNP would enhance trans-cleavage by FnoCas12aKD2P by varying incubation times from 5 min to 2 h (Fig. 8B). FnoCas12aWT degraded 99% of M13 circular ssDNA within 5 min, the first time point measured; while FnoCas12aKD2P cleaved only around 70% of the circular form of M13 ssDNA after 2 h (Fig. 8B, C). FnoCas12aKD2P also showed a significant accumulation of linear M13 ssDNA product in contrast to the complete degradation observed for FnoCas12aWT (Fig. 8B). This suggests that substitution in the BH decreases the ability of FnoCas12aKD2P to processively cleave ssDNA.
The trans-cleavage activity was also tested using a 24-nt PAM containing dsDNA activator (Table S2). With FnoCas12aWT, trans-cleavage activity appeared at RNP activator concentrations higher than 50 nm, with nearly complete cleavage observed only at concentrations higher than 100 nm RNP (Fig. 8D). Interestingly, no significant trans-cleavage was observed in case of FnoCas12aKD2P even at the highest concentration tested (500 nm) (Fig. 8D).
Since results from the cis-cleavage assay show that DNA substrate lengths and superhelicity (plasmid vs. oligo) can affect the cleavage efficiency, we tested the trans-cleavage activity on a 54-nt-long 32P-labeled linear ssDNA substrate that bears no complementarity with the crRNA or the activator DNA (Fig. S9). Both ssDNA and dsDNA activators (Figs S9A, S9B) were tested to analyze the efficiency of oligo trans-cleavage. The results showed that FnoCas12aWT possessed efficient trans-cleavage with both ssDNA and dsDNA activators similar to what was observed for a different ortholog (LbCas12a) in a previous study (Fig. 9A,B) [39]. Interestingly, under identical reaction conditions, FnoCas12aKD2P-crRNA- activator showed no trans-cleavage activity on the ss oligo substrate either with the ssDNA or with dsDNA activator (Fig. 9A,B). The dsDNA activator produced an intermediate product compared with that of ssDNA activator in the case of FnoCas12aWT. Together, our data suggest that perturbations in the BH of FnoCas12a not only affect cis-cleavage, but also impact the trans-cleavage activity.
Fig. 9.
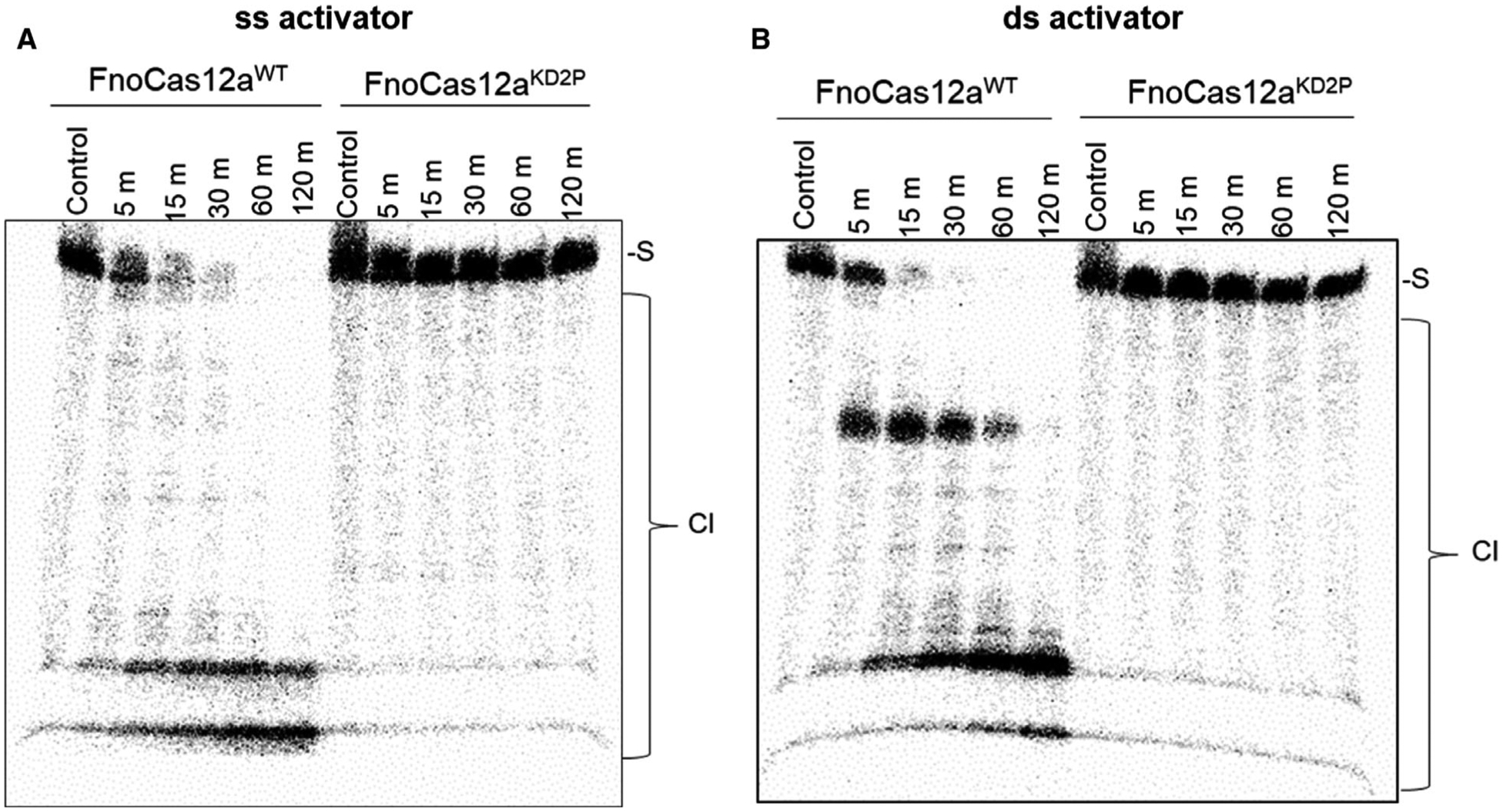
trans-cleavage of oligo DNA. Time-course assay showing trans-oligo cleavage with a 20-nt ssDNA activator (A) and 24-nt dsDNA activator (B). While FnoCas12aWT exhibits trans-cleavage with both ssDNA and dsDNA activators, there is an intermediate product being accumulated before complete degradation in the case of dsDNA activator. FnoCas12aKD2P did not show any visible cleavage even at the end of 2 h of incubation. Both panels had three independent replications. [S: ss oligo DNA substrate; Cl: cleavage products; m: min].
RNA-independent DNA cleavage activity is insensitive to BH modulations
To test the ability of FnoCas12aKD2P to cause RNA-independent DNA cleavage, we incubated M13 circular ssDNA and supercoiled pUC19 DNA with FnoCas12aWT and FnoCas12aKD2P proteins in the presence of 10 mm Mn2+ for 30 min at 37 °C [42]. The FnoCas12aKD2P protein completely degraded M13 ssDNA and nicked pUC19 DNA to the same extent as FnoCas12aWT (Fig. 10A,B). Following this, we tested the RNA-independent activity on a 60-mer ss oligo DNA substrate. With oligo DNA, FnoCas12aKD2P had a reduced efficiency for RNA-independent DNA cleavage compared with FnoCas12aWT (Fig. 10C), indicating that preferences for DNA physical states are more prominent in FnoCas12aKD2P compared with FnoCas12aWT. These results show that features of BH and its interactions with crRNA impact Cas12a activities that depend on its interactions with crRNA (cis- and trans-cleavages), but do not contribute to the RNA-independent DNA cleavage. The reduction in RNA-independent DNA cleavage by FnoCas12aKD2P on oligo substrate is most probably due to the reduced preference for oligo DNA, similar to what was observed for cis-oligo cleavage (Figs 2, 4, Table S4).
Fig. 10.
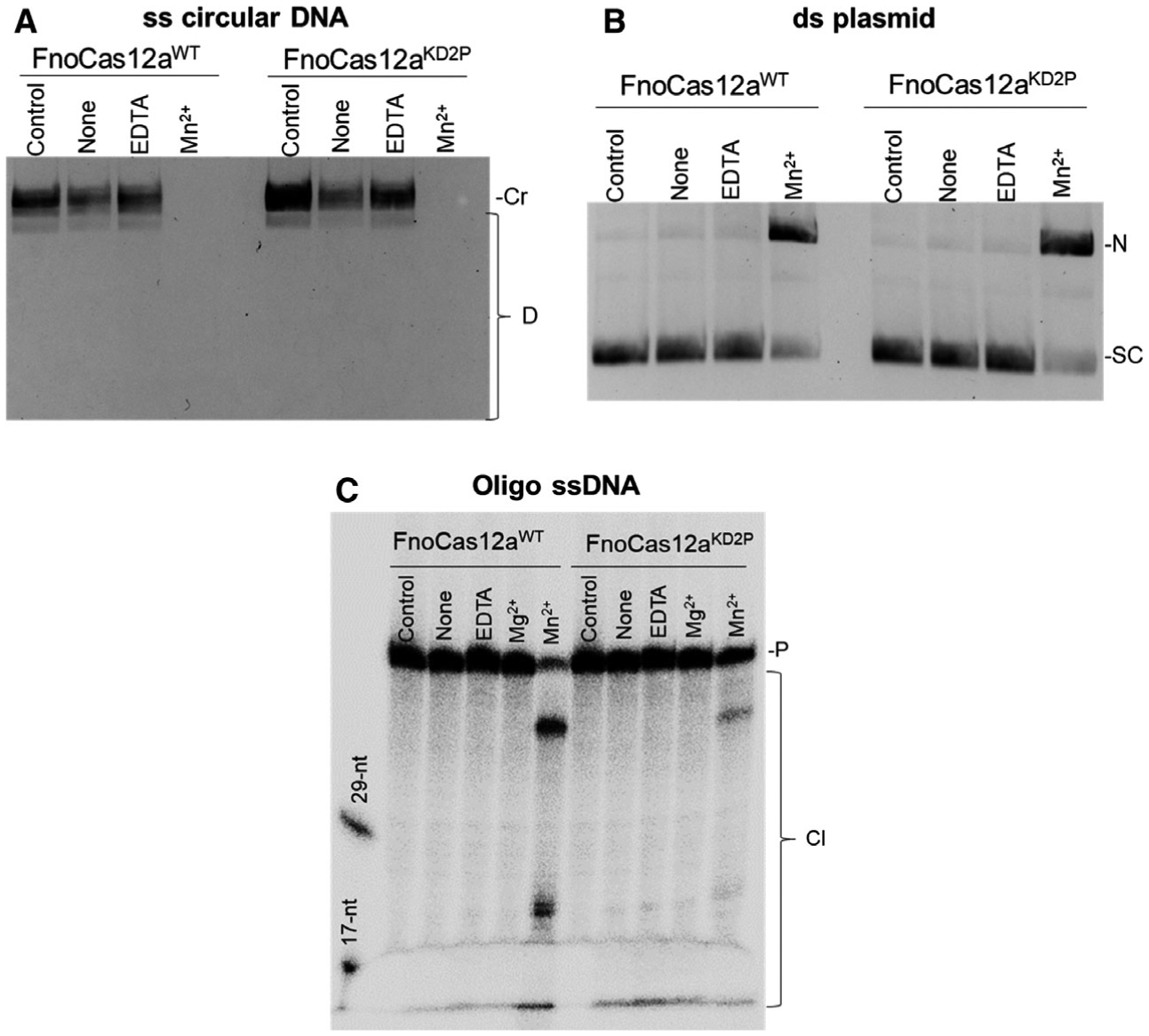
RNA-independent DNA cleavage activity of FnoCas12aWT and FnoCas12aKD2P. (A) Cleavage of M13 circular ssDNA by FnoCas12aWT and FnoCas12aKD2P in the absence of crRNA. Representative gel from three replications. (B) Cleavage of supercoiled pUC19 by FnoCas12aWT and FnoCas12aKD2P in the absence of crRNA. Representative gel from three replications is shown. Total reaction time was 30 min for (A) and (B). (C) Cleavage of ss oligo DNA (60-mer) by FnoCas12aWT and FnoCas12aKD2P in the absence of crRNA. Representative gel from three replications. Total reaction time was 1 h. [None: no added metal, Cr: circular, D: degradation, N: nicked, SC: supercoiled, P: precursor, Cl: cleavage; nt: nucleotides].
Discussion
Cas12a BH contributes to the efficiency and selectivity of target DNA cleavage
With matched DNA protospacers, data obtained from supercoiled plasmid (Fig. 2) indicate that FnoCas12aKD2P cleaved at a slower rate than FnoCas12aWT, but can completely cleave the precursor when given sufficient time. This reveals the importance of BH in efficient DNA cleavage. In our studies, when supercoiled plasmid DNA containing mismatches were treated with FnoCas12aKD2P, it resulted in a mixture of linear and nicked products. Although the total cleavage with FnoCas12aKD2P averaged between 65% and 88%, there was a significant variation in the ratio of nicked and linear products (Figs S5, S6). Specifically, mismatches at positions 12–17 showed significant accumulation of nicked products (61–77%), while the corresponding values for FnoCas12aWT were 3–19% (Fig. S6). These results indicate that FnoCas12aKD2P is compromised in cleaving both strands of the DNA efficiently with mismatches along the first 18 positions of the target DNA and that the effect is more pronounced in an area covering the middle of the R-loop.
To understand the positional effect, we analyzed the crystal structure (PDB: 5NFV) [20] and observed several interactions across this region of R-loop, which could contribute to this effect: (i) interactions of Arg968 and Lys969 with residues G11 to A13 of the guide region of crRNA (Fig. 1B), and (ii) interactions of the amino acids in REC2 helices (Val576, Tyr579, Asn580, and Arg583) with nt 12–15 of crRNA and the corresponding complementary region of TS (Table S5). It should be noted that the highly conserved Trp971 that acts as a wedge to move REC2 domain in response to DNA binding is very closely located to Val576, Tyr579, Asn580, and Arg583 (Figs 1B, S3B, Table S3 and S5). The proline substitutions in our study can potentially impair these interactions. For example, Lys969Pro removes interaction with A13 nucleotide of crRNA guide (Fig. 1B,C). As mentioned previously, the C-terminal end has been observed in loop and helical conformations in several crystal structures (PDB: 5NFV, 5MGA, 5B43, 5ID6, 6I1L, 6I1K, and 5XUS) [20–22,26,31,58]. It is possible that the proline substitutions that impact the helicity of the BH may affect the positioning of Trp971 in FnoCas12aKD2P. We believe that a combination of these factors contributes to the position-dependent accumulation of nicked product on mismatch substrates covering MM12–MM17. The reduction in the accumulation of nicked product past the 18th position (MM19–MM22) (Figs 3B, S5, S6) in FnoCas12aKD2P further supports these observations.
It was also interesting to note that upon longer incubations, FnoCas12aWT showed complete linearization of all the substrates that were tested, while accumulated nicked products were not linearized in the case of FnoCas12aKD2P. This emphasizes the importance of BH interactions with the guide region on mismatch sensitivity and its possible role in contributing to reaction pathways due to trapping of reaction intermediates in FnoCas12aKD2P (Fig. S7). Our results implicate that BH is critical in imparting dsDNA breaks in Cas12a. Nicks created in an in vivo gene editing setting will be repaired back to the native genomic content by repair enzymes, while dsDNA breaks can create indels during the repair process [61]. Thus, accumulation of nicked products in response to BH variation has wide range of implications. Similarities between target selectivity by two distinct BH variants of two different Cas proteins, belonging to two different CRISPR types, indicate conservations in the mechanism of BH-mediated activation and target DNA cleavage by Cas proteins.
Previous studies have shown that in several RNA-binding proteins that possess an Arg-rich BH, the BHs are disordered and unstructured in the absence of RNA [62]. Binding of specific RNA induces conformational changes in the protein especially in the Arg-rich BH, which are found to eventually impact protein function [63]. Substitution of specific amino acid residues in the BH of Cas9 was shown to contribute toward mismatch DNA cleavage sensitivity [53,55]. Our results with FnoCas12a further strengthen the role of BH in supporting the wild-type protein to cleave mismatch containing DNA, as demonstrated by activity assays that showed tolerance of wild-type protein against mismatches in DNA. This ability to cleave mismatched DNA that is imparted by BH may be advantageous for bacteria, since a less-stringent Cas nuclease can provide protection against mutated intruders, offering an enhanced immune protection.
BH is essential to support cis-cleavage of different types of DNA substrates
Our analysis of different DNA substrates showed that for cis-cleavage, Cas12a inherently has preferences for DNA substrates with different physical states. Those preferences vary between FnoCas12aWT and FnoCas12aKD2P indicating that BH plays a role in supporting cleavage of DNA substrates in different physical states. In the case of FnoCas12aWT, both helicity and strandedness contribute to efficient DNA cleavage, while length of the dsDNA did not change the efficiency. In FnoCas12aKD2P, all the factors tested, superhelicity, strandedness, and length, contribute to DNA cleavage. It is interesting to notice the 12-fold reduction in cleaving long linearized dsDNA compared with a 4-fold reduction in short oligo dsDNA in FnoCas12aKD2P. This may suggest a role of BH in supporting unwinding of the dsDNA during R-loop formation, as it may be more efficient to unwind a short oligo DNA compared to a fully base-paired long dsDNA. Interestingly, a comparison between both the proteins also shows the highest impairment occurs for FnoCas12aKD2P on linearized dsDNA, further supporting this observation (Table S4). Together, our results may suggest that unwinding of DNA to form an R-loop is more efficient in negatively supercoiled DNA containing single-stranded regions, compared with tightly base-paired linear dsDNA. Cas12a’s discretion against ssDNA substrate indicates that the additional stability of the complex provided by the NTS in the ternary complex may also contribute to the efficiency of cleavage. Interestingly, a wild-type BH allows Cas12a to accommodate several of these substrates to perform cleavage at variable efficiencies, while this ability is highly reduced in the presence of an impaired BH.
BH may play a role in coordinating cis-TS and NTS cleavages
The matched and mismatched plasmid cis-cleavage assays showed the accumulation of nicked product suggesting proline substitutions in the BH affects cleavage of one of the DNA strands (Figs 2 and 3). Previous literature has shown that the cleavage of NTS precedes TS cleavage [31,32]. We tracked the strand preference using oligo cleavage assays and showed a faster rate of appearance of NTS cleavage products over TS cleavage in FnoCas12aKD2P. This indicates that the order of sequential cleavage did not change in the variant (Fig. 4). Interestingly, upon comparison of both the proteins, there is a 6-fold reduction in the observed rate for TS cleavage and a 3-fold reduction in NTS cleavage by FnoCas12aKD2P (Fig. 4, Table S4). These results implicate the role of BH in coordinating the cleavage of both the DNA strands, especially the TS. Together, our data suggest that the accumulation of nicked products in FnoCas12aKD2P may be the result of inefficient cleavage of TS.
It was previously established that the movement of REC2 domain away from the RuvC pocket is a pre-requisite for both cis- and trans-cleavages [31]. Recent molecular dynamics simulation studies showed that upon initial cleavage of NTS at the RuvC catalytic site, the REC2 and Nuc domains move closer to each other, enabling the approach of TS closer to RuvC site for cleavage [64]. Even though cleavage of TS is through the RuvC domain, based on the crystal structures of Cas12b, it is speculated that a combined path created by residues from both RuvC and Nuc domains is essential for TS cleavage [31,65]. These observations indicate that proline substitutions in the BH may impact the movement of REC2, thus hindering cleavage of TS.
Cleavage assays with mismatched MM8 oligo dsDNA resulted in negligible cleavage by FnoCas12aKD2P for either strand compared with near-complete cleavage by FnoCas12aWT (Fig. 5A,B), despite there being no apparent deficiency in DNA binding (Fig. 6). This further supports the notion that BH is essential for Cas12a to tolerate mismatches and activate conformational changes essential for DNA cleavage. The proline substitutions in FnoCas12aKD2P does not impact DNA binding, but rather may be impacting BH-mediated conformational changes essential for DNA cleavage.
Overall, in both Cas9 and Cas12a, studies have shown that BH interacts with the crRNA and the RNA-DNA hybrid [53,55]. Proline substitutions in the BH (i.e., FnoCas12aKD2P in this work and SpyCas92Pro reported in our previous work [53]) may affect these interactions, which in turn appears to perturb the communications that are essential for interdependent cleavage of TS and NTS of target DNA. Such perturbations become much more significant with certain mismatch positions, resulting in reduced cleavage activities on mismatched DNAs.
BH regulates trans-cleavage activity of Cas12a but not RNA-independent DNA cleavage
Cas12a proteins also possess a unique ability to cleave ssDNA (circular and oligo) in trans, after activation by RNA-guided DNA cleavage [31,39,40], and this property has been repurposed into developing specific tools for viral detection [39,66]. Our results show a clearly reduced trans-cleavage activity for FnoCas12aKD2P when compared to FnoCas12aWT (Figs 8 and 9).
Current understanding indicates that RNA-guided DNA binding or cleavage exposes Cas12a’s RuvC nuclease domain, subsequently enabling ssDNA cleavage in a sequence-independent manner [31,32]. The crystal structures depicting the pre- (PDB: 5NFV [20]) and post-cleavage (PDB: 5MGA [26]) ternary states of FnoCas12a show differences in the conformation of Lys969 and Asp970, the two amino acids that were substituted to proline in the present study. In the pre-cleavage state, Lys969 is in the helical form and Asp970 is in the loop, while in the post-cleavage structure, both the amino acids are in a helix. The post-cleavage state will have an unblocked RuvC active site following the release of PAM-distal DNA product from cis-cleavage and can enable trans-cleavage [26,31]. In FnoCas12aKD2P, the BH mutations likely affect the conformational changes needed to initiate or maintain this state of RuvC for trans-cleavage.
Our results also showed that dsDNA activator was less effective in inducing trans-cleavage in FnoCas12a compared with that of a ssDNA activator (Fig. 8). In addition, an intermediate product was formed during trans-cleavage of oligo ssDNA with a dsDNA activator (Fig. 9B). One possible explanation for this is the differences in the conformational states of Lys969 and Asp970 in the ternary structures of FnoCas12a with ssDNA activator (PDB: 6I1L) and dsDNA activator (PDB: 6I1K) [31]. The conformation of BH in the ssDNA activator-ternary complex is similar to that of post-cleavage state (PDB: 5MGA) with both amino acids in the helical conformation, while in the dsDNA activator-ternary state, both these amino acids are in a loop conformation [26,31]. This indicates that dsDNA activator might need to induce additional conformational changes to effectively activate FnoCas12a for trans-cleavage. Further detailed experiments are needed to identify how BH contributes to trans-cleavage activity and what are the effects of different activators (ss vs. ds) in promoting trans-cleavage.
While BH modulations of FnoCas12a cause an overall reduction in both cis- and trans-DNA cleavage activities, FnoCas12aKD2P maintains comparable amounts of RNA-independent DNA cleavage in the presence of Mn2+ on plasmid substrates (Fig. 10A,B). The possible explanation for this is that BH modulations are impairing only crRNA-mediated protein interactions, and since RNA-independent DNA cleavage does not require these interactions, it stays intact. The efficiency of RNA-independent DNA cleavage by FnoCas12aKD2P is reduced considerably on an oligo DNA, which may be due to the reduced preference toward oligo DNA as was observed in oligo ds cis-cleavage (Figs 4, 10C, Table S4).
The in vitro data presented here show that a strong parallel can be drawn between SpyCas9 and FnoCas12a, regarding a similar mechanistic role for the BH in imparting target DNA selectivity and coordination of TS and NTS cleavages. Our previous work with SpyCas92Pro has established that in vitro DNA cleavage selectivity translates to lower off-target rates in cell-based gene editing [53]. Another study on SpyCas9 BH variations utilizing Arg63 and Arg66 also has shown lower off-targets for gene editing using BH variants [55]. These previous results suggest a possibility of Cas12a BH variants to elicit lower off-target effects in gene editing, though further studies are required to conclusively establish this.
In summary, the studies presented here on FnoCas12aKD2P build upon our prior work on SpyCas9 [53], and together establish a framework indicating that BH modulations can increase the stringency of target DNA cleavage in Cas proteins. This aids in further deciphering the DNA recognition and cleavage mechanisms of various BH-containing Cas proteins.
Supplementary Material
Table S1. Primers used in the study.
Table S2. DNA substrates used in the study.
Table S3. List of interactions of FnoCas12a BH with crRNA and different protein domains.
Table S4. The compilation of the rate constants calculated for different physical states of dsDNA substrates in the study and respective fold changes for FnoCas12aWT and FnoCas12aKD2P.
Table S5. List of interactions of crRNA and DNA bound to FnoCas12a 2.
Fig. S1. A 10% SDS gel showing the purity of the FnoCas12aWT and FnoCas12aKD2P proteins after a three-step purification protocol.
Fig. S2. Illustration of R-loop formation with matched DNA.
Fig. S3. Interactions of BH with crRNA and REC2 domain.
Fig. S4. One-exponential fit for the loss of supercoiled plasmid DNA precursor.
Fig. S5. (A-C) Representative gel images comparing the activities of FnoCas12aWT and FnoCas12aKD2P on different mismatch-containing plasmid substrates.
Fig. S6. Tables depicting the values of nicked, linear and total cleavage for (A) FnoCas12aWT and (B) FnoCas12aKD2P.
Fig. S7. Gels showing the effect of longer incubation times on cleavage of matched DNA, MM8 DNA and MM12 DNA by (A) FnoCas12aWT and (B) FnoCas12aKD2P.
Fig. S8. (A) Gels showing the efficiency of linearization of M13 ssDNA on a native (left) and an alkaline (right) gel. The alkaline gel, which can give molecular weight corresponding to ssDNA shows a linearized M13 ssDNA at 7 kilobases (kb) and the uncut circular M13 above the linear band. Our preparations gave 50–70% linearization of M13 circular ssDNA with EcoRI. (B) Gel showing the cis-cleavage of circular M13 ssDNA by FnoCas12aWT and FnoCas12aKD2P. At 25 nM RNP, FnoCas12aWT completely degrades M13 DNA by a combinational effect of cis- and trans-activities, while FnoCas12aKD2P shows reduced cleavge abilities even after 2 hours. (C) Efficiency of cis-cleavage by FnoCas12a on linear M13 ssDNA. FnoCas12aWT degraded the linear M13 ssDNA with a moderate efficiency compared to that of FnoCas12KD2P, which did not cleave even at the highest concentration tested. Reactions with linear M13 ssDNA were for 60 min. Representative gels from two replications. [Cr: circular, L: linear, D: degradation, m: min].
Fig. S9. Constructs used to test trans-cleavage of ss oligonucleotide DNA.
Acknowledgements
We thank the OU Protein Production & Characterization Core (PPC Core) facility for protein purification services and instrument support. The OU PPC core is supported by an Institutional Development Award (IDeA) grant from the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) [grant number P20GM103640]. We thank Mason Van Orden for critical reading of the manuscript. Work reported here was supported in part by grants from the National Science Foundation [grant number MCB-1716423, RR], Oklahoma Center for the Advancement of Science and Technology (OCAST) award [grant number HR20-103, RR], National Institutes of Health [grant number R01GM124413, PZQ], and in part by a grant from the Research Council of the University of Oklahoma Norman Campus to RR.
Abbreviations
- bp
base pair
- BH
bridge helix
- CRISPR-Cas
Clustered Regularly Interspaced Short Palindromic Repeats and CRISPR-associated proteins
- ds
double-stranded
- Fno
Francisella novicida
- MBP
maltose-binding protein
- MM
mismatch
- nt
nucleotides
- NTS
nontarget strand
- PAM
protospacer adjacent motif
- ss
single-stranded
- oligos
oligonucleotides
- Spy
Streptococcus pyogenes
- TS
target strand
Footnotes
Conflict of interest
A patent application (US20200332275A1) has been filed based on this work. The authors declare no other conflicts of interest with regard to this manuscript.
Supporting information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
References
- 1.Mojica FJ, Diez-Villasenor C, Garcia-Martinez J and Almendros C (2009) Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155, 733–740. [DOI] [PubMed] [Google Scholar]
- 2.Gasiunas G, Barrangou R, Horvath P and Siksnys V (2012) Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A 109, E2579–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns SJJ, Charpentier E, Cheng D, Haft DH, Horvath P et al. (2020) Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol 18, 67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA and Horvath P (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. [DOI] [PubMed] [Google Scholar]
- 5.Pourcel C, Salvignol G and Vergnaud G (2005) CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151, 653–663. [DOI] [PubMed] [Google Scholar]
- 6.Mohanraju P, Makarova KS, Zetsche B, Zhang F, Koonin EV and van der Oost J (2016) Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science 353, aad5147. [DOI] [PubMed] [Google Scholar]
- 7.Bolotin A, Quinquis B, Sorokin A and Ehrlich SD (2005) Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151, 2551–2561. [DOI] [PubMed] [Google Scholar]
- 8.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA and Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF et al. (2011) Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol 9, 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sapranauskas R, Gasiunas G, Fremaux C, Barrangou R, Horvath P and Siksnys V (2011) The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res 39, 9275–9282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fellmann C, Gowen BG, Lin PC, Doudna JA and Corn JE (2017) Cornerstones of CRISPR-Cas in drug discovery and therapy. Nat Rev Drug Discov 16, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI and Liu DR (2017) Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knott GJ and Doudna JA (2018) CRISPR-Cas guides the future of genetic engineering. Science 361, 866–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin RM, Ikeda K, Cromer MK, Uchida N, Nishimura T, Romano R, Tong AJ, Lemgart VT, Camarena J, Pavel-Dinu M et al. (2019) Highly efficient and marker-free genome editing of human pluripotent stem cells by CRISPR-Cas9 RNP and AAV6 donor-mediated homologous recombination. Cell Stem Cell 24, 821–828 e825. [DOI] [PubMed] [Google Scholar]
- 15.Kim D, Kim J, Hur JK, Been KW, Yoon SH and Kim JS (2016) Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat Biotechnol 34, 863–868. [DOI] [PubMed] [Google Scholar]
- 16.Gifford CA, Ranade SS, Samarakoon R, Salunga HT, de Soysa TY, Huang Y, Zhou P, Elfenbein A, Wyman SK, Bui YK et al. (2019) Oligogenic inheritance of a human heart disease involving a genetic modifier. Science 364, 865–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shmakov S, Smargon A, Scott D, Cox D, Pyzocha N, Yan W, Abudayyeh OO, Gootenberg JS, Makarova KS, Wolf YI et al. (2017) Diversity and evolution of class 2 CRISPR-Cas systems. Nat Rev Microbiol 15, 169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adli M (2018) The CRISPR tool kit for genome editing and beyond. Nat Commun 9, 1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jinek M, East A, Cheng A, Lin S, Ma E and Doudna J (2013) RNA-programmed genome editing in human cells. Elife 2, e00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swarts DC, van der Oost J and Jinek M (2017) Structural basis for guide RNA processing and seed-dependent DNA targeting by CRISPR-Cas12a. Mol Cell 66, 221–233 e224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamano T, Nishimasu H, Zetsche B, Hirano H, Slaymaker IM, Li Y, Fedorova I, Nakane T, Makarova KS, Koonin EV et al. (2016) Crystal structure of Cpf1 in complex with guide RNA and target DNA. Cell 165, 949–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dong D, Ren K, Qiu X, Zheng J, Guo M, Guan X, Liu H, Li N, Zhang B, Yang D et al. (2016) The crystal structure of Cpf1 in complex with CRISPR RNA. Nature 532, 522–526. [DOI] [PubMed] [Google Scholar]
- 23.Stella S, Alcon P and Montoya G (2017) Class 2 CRISPR-Cas RNA-guided endonucleases: Swiss Army knives of genome editing. Nat Struct Mol Biol 24, 882–892. [DOI] [PubMed] [Google Scholar]
- 24.Murugan K, Babu K, Sundaresan R, Rajan R and Sashital DG (2017) The revolution continues: Newly discovered systems expand the CRISPR-Cas toolkit. Mol. Cell 68, 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao P, Yang H, Rajashankar KR, Huang Z and Patel DJ (2016) Type V CRISPR-Cas Cpf1 endonuclease employs a unique mechanism for crRNA-mediated target DNA recognition. Cell Res 26, 901–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stella S, Alcon P and Montoya G (2017) Structure of the Cpf1 endonuclease R-loop complex after target DNA cleavage. Nature 546, 559–563. [DOI] [PubMed] [Google Scholar]
- 27.Singh D, Mallon J, Poddar A, Wang Y, Tippana R, Yang O, Bailey S and Ha T (2018) Real-time observation of DNA target interrogation and product release by the RNA-guided endonuclease CRISPR Cpf1 (Cas12a). Proc Natl Acad Sci U S A 115, 5444–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeon Y, Choi YH, Jang Y, Yu J, Goo J, Lee G, Jeong YK, Lee SH, Kim IS, Kim JS et al. (2018) Direct observation of DNA target searching and cleavage by CRISPR-Cas12a. Nat Commun 9, 2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang L, Sun R, Yang M, Peng S, Cheng Y and Chen C (2019) Conformational dynamics and cleavage sites of Cas12a are modulated by complementarity between crRNA and DNA. iScience 19, 492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swarts DC and Jinek M (2018) Cas9 versus Cas12a/Cpf1: structure-function comparisons and implications for genome editing. Wiley Interdis Rev 9, e1481. [DOI] [PubMed] [Google Scholar]
- 31.Swarts DC and Jinek M (2019) Mechanistic insights into the cis- and trans-acting DNase activities of Cas12a. Mol. Cell 73, 589–600 e584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stella S, Mesa P, Thomsen J, Paul B, Alcon P, Jensen SB, Saligram B, Moses ME, Hatzakis NS and Montoya G (2018) Conformational activation promotes CRISPR-Cas12a catalysis and resetting of the endonuclease activity. Cell 175, 1856–1871 e1821. [DOI] [PubMed] [Google Scholar]
- 33.Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A et al. (2015) Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sternberg SH, LaFrance B, Kaplan M and Doudna JA (2015) Conformational control of DNA target cleavage by CRISPR-Cas9. Nature 527, 110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raper AT, Stephenson AA and Suo Z (2018) Functional insights revealed by the kinetic mechanism of CRISPR/Cas9. J Am Chem Soc 140, 2971–2984. [DOI] [PubMed] [Google Scholar]
- 36.Yamada M, Watanabe Y, Gootenberg JS, Hirano H, Ran FA, Nakane T, Ishitani R, Zhang F, Nishimasu H and Nureki O (2017) Crystal structure of the minimal Cas9 from Campylobacter jejuni reveals the molecular diversity in the CRISPR-Cas9 systems. Mol Cell 65, 1109–1121 e1103. [DOI] [PubMed] [Google Scholar]
- 37.Fonfara I, Richter H, Bratovic M, Le Rhun A and Charpentier E (2016) The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 532, 517–521. [DOI] [PubMed] [Google Scholar]
- 38.East-Seletsky A, O’Connell MR, Knight SC, Burstein D, Cate JH, Tjian R and Doudna JA (2016) Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen JS, Ma E, Harrington LB, Da Costa M, Tian X, Palefsky JM and Doudna JA (2018) CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 360, 436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li SY, Cheng QX, Liu JK, Nie XQ, Zhao GP and Wang J (2018) CRISPR-Cas12a has both cis- and trans-cleavage activities on single-stranded DNA. Cell Res 28, 491–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swarts DC (2019) Making the cut(s): how Cas12a cleaves target and non-target DNA. Biochem Soc Trans 47, 1499–1510. [DOI] [PubMed] [Google Scholar]
- 42.Sundaresan R, Parameshwaran HP, Yogesha SD, Keilbarth MW and Rajan R (2017) RNA-independent DNA cleavage activities of Cas9 and Cas12a. Cell Rep 21, 3728–3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li B, Yan J, Zhang Y, Li W, Zeng C, Zhao W, Hou X, Zhang C and Dong Y (2020) CRISPR-Cas12a possesses unconventional DNase activity that can be inactivated by synthetic oligonucleotides. Mol Ther Nucleic Acids 19, 1043–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kosicki M, Tomberg K and Bradley A (2018) Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol 36, 765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shin HY, Wang C, Lee HK, Yoo KH, Zeng X, Kuhns T, Yang CM, Mohr T, Liu C and Hennighausen L (2017) CRISPR/Cas9 targeting events cause complex deletions and insertions at 17 sites in the mouse genome. Nat Commun 8, 15464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK and Sander JD (2013) High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31, 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang XH, Tee LY, Wang XG, Huang QS and Yang SH (2015) Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther Nucleic Acids 4, e264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li P, Zhang L, Li Z, Xu C, Du X and Wu S (2020) Cas12a mediates efficient and precise endogenous gene tagging via MITI: microhomology-dependent targeted integrations. Cell Mol Life Sci 77, 3875–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Breinig M, Schweitzer AY, Herianto AM, Revia S, Schaefer L, Wendler L, Cobos Galvez A and Tschaharganeh DF (2019) Multiplexed orthogonal genome editing and transcriptional activation by Cas12a. Nat Methods 16, 51–54. [DOI] [PubMed] [Google Scholar]
- 50.Zhong G, Wang H, Li Y, Tran MH and Farzan M (2017) Cpf1 proteins excise CRISPR RNAs from mRNA transcripts in mammalian cells. Nat Chem Biol 13, 839–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kleinstiver BP, Tsai SQ, Prew MS, Nguyen NT, Welch MM, Lopez JM, McCaw ZR, Aryee MJ and Joung JK (2016) Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat Biotechnol 34, 869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim Y, Cheong SA, Lee JG, Lee SW, Lee MS, Baek IJ and Sung YH (2016) Generation of knockout mice by Cpf1-mediated gene targeting. Nat Biotechnol 34, 808–810. [DOI] [PubMed] [Google Scholar]
- 53.Babu K, Amrani N, Jiang W, Yogesha SD, Nguyen R, Qin PZ and Rajan R (2019) Bridge helix of Cas9 modulates target DNA cleavage and mismatch tolerance. Biochemistry 58, 1905–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, Anders C, Hauer M, Zhou K, Lin S et al. (2014) Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343, 1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bratovič M, Fonfara I, Chylinski K, Gálvez EJC, Sullivan TJ, Boerno S, Timmermann B, Boettcher M and Charpentier E (2020) Bridge helix arginines play a critical role in Cas9 sensitivity to mismatches. Nat Chem Biol 16, 587–595. [DOI] [PubMed] [Google Scholar]
- 56.Bachman J (2013) Site-directed mutagenesis. Methods Enzymol 529, 241–248. [DOI] [PubMed] [Google Scholar]
- 57.Schneider CA, Rasband WS and Eliceiri KW (2012) NIH image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamano T, Zetsche B, Ishitani R, Zhang F, Nishimasu H and Nureki O (2017) Structural basis for the canonical and non-canonical PAM recognition by CRISPR-Cpf1. Mol Cell 67, 633–645.e633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hoffman MM, Khrapov MA, Cox JC, Yao J, Tong L and Ellington AD (2004) AANT: the amino acid-nucleotide interaction database. Nucleic Acids Res 32, D174–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tsui TKM, Hand TH, Duboy EC and Li H (2017) The impact of DNA topology and guide length on target selection by a cytosine-specific Cas9. ACS Synth Biol 6, 1103–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vriend LE and Krawczyk PM (2017) Nick-initiated homologous recombination: protecting the genome, one strand at a time. DNA Repair (Amst) 50, 1–13. [DOI] [PubMed] [Google Scholar]
- 62.Calnan BJ, Biancalana S, Hudson D and Frankel AD (1991) Analysis of arginine-rich peptides from the HIV Tat protein reveals unusual features of RNA-protein recognition. Genes Dev 5, 201–210. [DOI] [PubMed] [Google Scholar]
- 63.Casu F, Duggan BM and Hennig M (2013) The arginine-rich RNA-binding motif of HIV-1 Rev is intrinsically disordered and folds upon RRE binding. Biophys J 105, 1004–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saha A, Arantes PR, Hsu RV, Narkhede YB, Jinek M and Palermo G (2020) Molecular dynamics reveals a DNA-induced dynamic switch triggering activation of CRISPR-Cas12a. J Chem Inf Model 60, 6427–6437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang H, Gao P, Rajashankar KR and Patel DJ (2016) PAM-dependent target DNA recognition and cleavage by C2c1 CRISPR-Cas endonuclease. Cell 167, 1814–1828.e1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gootenberg JS, Abudayyeh OO, Kellner MJ, Joung J, Collins JJ and Zhang F (2018) Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 360, 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.DeLano WL (2002) Pymol: An open-source molecular graphics tool. CCP4 Newslett Prot Crystallog 82–92. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers used in the study.
Table S2. DNA substrates used in the study.
Table S3. List of interactions of FnoCas12a BH with crRNA and different protein domains.
Table S4. The compilation of the rate constants calculated for different physical states of dsDNA substrates in the study and respective fold changes for FnoCas12aWT and FnoCas12aKD2P.
Table S5. List of interactions of crRNA and DNA bound to FnoCas12a 2.
Fig. S1. A 10% SDS gel showing the purity of the FnoCas12aWT and FnoCas12aKD2P proteins after a three-step purification protocol.
Fig. S2. Illustration of R-loop formation with matched DNA.
Fig. S3. Interactions of BH with crRNA and REC2 domain.
Fig. S4. One-exponential fit for the loss of supercoiled plasmid DNA precursor.
Fig. S5. (A-C) Representative gel images comparing the activities of FnoCas12aWT and FnoCas12aKD2P on different mismatch-containing plasmid substrates.
Fig. S6. Tables depicting the values of nicked, linear and total cleavage for (A) FnoCas12aWT and (B) FnoCas12aKD2P.
Fig. S7. Gels showing the effect of longer incubation times on cleavage of matched DNA, MM8 DNA and MM12 DNA by (A) FnoCas12aWT and (B) FnoCas12aKD2P.
Fig. S8. (A) Gels showing the efficiency of linearization of M13 ssDNA on a native (left) and an alkaline (right) gel. The alkaline gel, which can give molecular weight corresponding to ssDNA shows a linearized M13 ssDNA at 7 kilobases (kb) and the uncut circular M13 above the linear band. Our preparations gave 50–70% linearization of M13 circular ssDNA with EcoRI. (B) Gel showing the cis-cleavage of circular M13 ssDNA by FnoCas12aWT and FnoCas12aKD2P. At 25 nM RNP, FnoCas12aWT completely degrades M13 DNA by a combinational effect of cis- and trans-activities, while FnoCas12aKD2P shows reduced cleavge abilities even after 2 hours. (C) Efficiency of cis-cleavage by FnoCas12a on linear M13 ssDNA. FnoCas12aWT degraded the linear M13 ssDNA with a moderate efficiency compared to that of FnoCas12KD2P, which did not cleave even at the highest concentration tested. Reactions with linear M13 ssDNA were for 60 min. Representative gels from two replications. [Cr: circular, L: linear, D: degradation, m: min].
Fig. S9. Constructs used to test trans-cleavage of ss oligonucleotide DNA.