

Abstract
Background:
Defective alleles within the PRF1 gene, encoding the pore-forming protein perforin, in combination with environmental factors, cause familial type 2 hemophagocytic lymphohistiocytosis (FHL2), a rare, severe autosomal recessive childhood disorder characterized by massive release of cytokines - cytokine storm.
Objective:
The aim of this study was to determine the function of hypomorph PRF1:p.A91V g.72360387 G>A on multiple sclerosis (MS) and type 1 diabetes (T1D).
Methods:
We cross-compare the association data for PRF1:p.A91V mutation derived from GWAS on adult MS and pediatric T1D in Sardinians. The novel association with T1D was replicated in metanalysis in 12,584 cases and 17,692 controls from Sardinia, UK and Scotland. To dissect this mutation function, we searched through the coincident association immunophenotypes in additional set of general population Sardinians.
Results:
We report that PRF1:p.A91V, is associated with increase of lymphocyte levels, especially within the cytotoxic memory T cells, at general population level with reduced interleukin 7 receptor expression on these cells. The minor allele increased risk of MS, in 2,903 cases and 2,880 controls from Sardinia p = 2.06×10−4, OR=1.29, replicating a previous finding, whereas it protects from T1D p = 1.04 × 10−5, OR= 0.82.
Conclusion:
Our results indicate opposing contributions of the cytotoxic T cell compartment to MS and T1D pathogenesis.
Keywords: perforin, type 1 diabetes, multiple sclerosis, hemophagocytic lymphohistiocytosis, cytokine storm, cytotoxic lymphocytes, inflammation
Introduction
Inter-individual variability in immune response is a cumulative result of both genetic and environmental factors and involves quantitative aspects, such as levels of immune cells, and qualitative features, such as the responses to antigens [1]. In particular, host-pathogen interactions prompt immune responses by activating immune cells to control pathogen clearance. For example, acute or chronic subclinical infections leave an imprint on levels and activation state of the subtypes of circulating immune cells [2]. Instead, genetic regulation leading to inter-individual differences in the basal levels of immune cells has been revealed by population-based studies, which found a number of association signals influencing levels of cellular and humoral immunophenotypes. Some of these signals coincide with associations for risk of autoimmunity, providing additional insights on pathological mechanisms [2,3]. Although of unknown relevance to clinical practice, those differences are observed in inflammation, immune related disorders including autoimmunity and cancer and in response to immunotherapy.
Cytotoxicity is the principal mechanism controlling infections and transformed cells, either directly through the perforin/granzyme or Fas/FasL pathways and secondarily by a release of cytokines [4]. Perforin, encoded by PRF1, is a pore-forming protein, stored in secretory vesicles and released by cytotoxic cells on contact with a target cell. It is critical for cytotoxicity of natural killer, CD8+, γδ+ and regulatory T lymphocytes [5].
While adaptive immunity and CD4+ T cell function have been extensively studied for both type 1 diabetes (T1D) and multiple sclerosis (MS), the role of innate immunity and CD8+ T cell function in autoimmunity remains less understood.
Recently, an Exome Chip - based meta-analysis of association statistics across 14 country-level strata found that the PRF1:p.A91V mutation confers predisposition to MS (pvalue = 1.04×10−10) [6]. This mutation has a dominant – negative function, presumably by disrupting pore formation when bound to unmutated molecules of perforin and slowing intracellular trafficking [7]. PRF1:p.A91V reduces protein expression of about 50% in homozygotes and significantly impairs function of cytotoxic T lymphocytes [7]. The same mutation is responsible for atypical late onset inherited, perforin deficiency syndrome – primary familial hemophagocytic lymphohistiocytosis 2 (FHL2) [7]. Usually, an infection primes the FHL2 syndrome early in childhood. However, PRF1:p.A91V atypical mutation causes FHL2 syndrome with variable age of diagnosis. The FHL2 pathology caused by this PRF1:p.A91V mutation includes massive cytokine release (cytokine storm), with IFN-γ leading a complex impact on immune-mediated disorders.
Perforin/granzyme pathway interruption leads to persistent inflammation, chronic antigen presentation and release of inflammation mediators. Primary and secondary lymphohistiocytosis have been associated with an ineffective immune responses to viral infection (Herpesviridae), susceptibility to inflammatory and autoimmune disorders, transplant rejection, neoplastic disease, and prolonged inflammation [8].
Here, we have used hematological and immunophenotyping data from the SardiNIA cohort to clarify the effect of PRF1:p.A91V in a general population. We also used case-control sample sets to further clarify the effects of this variant in autoimmunity. Our findings suggest that this PRF1:p.A91V mutation causes subclinical inflammation and defective cytotoxicity, with opposite effects on MS - and T1D - inherited risk.
Methods
Ethics
All participants gave informed consent to the study. The Sardinian autoimmunity study and SardiNIA studies were approved by the Sardinian local ethical committees: Comitato Etico di Azienda Sanitaria Locale 8, Lanusei (2009/0016600) and Comitato Etico di Azienda Sanitaria Locale 1, Sassari (2171/CE) and by the NIH Office of Human Subject Research as governed by Italian institutional review board approval. Experiments were conducted according to the principles expressed in the Declaration of Helsinki. Samples of venous blood were drawn after informed consent was obtained.
Patients population
Several cohorts have been used in this study. The first is the Sardinian autoimmunity case-control cohort of 2,903 multiple sclerosis (MS) and 1,558 Sardinian type 1 diabetes (T1D) patients [9,10]. They share the same set of 2,880 Sardinian controls that include blood donors.
Sardinian MS patients (female:male ratio 2.2:1) were diagnosed according to the McDonald criteria: 92.6% of them had a bout-onset disease course (mean age at diagnosis of 31.3+−10.55, range 5–88) [9]. Sardinian T1D patients (female:male ratio 0.8:1) have a mean age of onset 11.9, range 0.7–25. As a replication of T1D discovery, two cohorts were used: 5,854 cases and 7,336 controls from the UK and 5,172 cases and 7,474 controls from Scotland. The UK and Scotland T1D patients’ clinical descriptions were published before [11,12].
The SardiNIA general population cohort longitudinal study on ageing (6,921 volunteers, 3,985 of them female, aged 18–102), from the Lanusei valley area in Sardinia, Italy, has been previously described [13]. Volunteers participating in this study are generally healthy and were phenotyped for more than 1,000 quantitative traits. Details of phenotype and genotype assessments for these samples have been published previously [14]. For this study we used data of up to 6,521 individuals.
Genotyping, imputation and GWAS
The Sardinian T1D and MS GWAS case-control studies were performed using combined data sets where all samples were genotyped with ImmunoChip (Illumina) and subsets of the samples were genotyped with Genome-Wide Human SNP Array 6.0 (Affymetrix) and OmniExpress (Illumina). This combined approach after quality controls brought 883,557 SNPs subjected to the parallel GWAS studies on MS and T1D (Supplementary Figure.1). See Supplementary material for details for GWAS on MS and T1D using the integrated map (ImmunoChip, OmniExpress and Genome-Wide Human SNP Array 6.0).
To study PRF1:p.A91V mutation effect on phenotypes, the association results from the SardiNIA general population were used. The association results are based on 6,521 SardiNIA volunteers genotyped with Illumina arrays (OmniExpress, ImmunoChip, Cardio-MetaboChip and ExomeChip; 890,542 SNPs in total) [13]. Samples from both cohorts were imputed with Minimac3 on a Next Generation Sequencing based reference panel of 3,514 Sardinian individuals [13].
Immune phenotyping and data analysis in SardiNIA cohort
For this study we used the GWAS data on inflammatory parameters of 6,521 individuals and the immune phenotypes of 3,757 individuals derived from the extended flow cytometry (FACS) profiling [3,13] (Supplementary Figure 2). These individuals do not overlap with individuals participating in T1D and MS GWAS in Sardinia. For circulating immune cell types, FACS profiling fresh blood samples were used within 2 hours from the visit, as described previously in a study involving a smaller sample set [9].
The associations of 731 immune traits in the region of PRF1 gene were specifically assessed in the SardiNIA study. These traits include 118 absolute cell counts, 389 MFIs of surface antigens and 32 morphological parameters. The significant threshold has been calculated by correcting the nominal pvalue of 0.05 for these statistically independent 539 traits by applying a multiple independent test (Bonferroni correction) and reaching a significant threshold of 9.27×10−5. The remaining 192 relative counts, corresponding to percentage with respect to hierarchically higher cell population, are instead statistically dependent to the absolute cell count tested and were not considered for Bonferroni correction.
Colocalization analysis
Colocalization analysis were performed using R “coloc” package. We tested the colocalization between the summary statistics for the count of CD8dimCD28− cells in the SardiNIA cohort and the T1D summary statistics from the UK cohort. We used the recommended threshold for colocalization of 0.8.
Results
The p.A91V mutation in PRF1 confers predisposition to multiple sclerosis (MS) and protection from type 1 diabetes (T1D)
In an exploratory analysis to assess the statistical power to replicate the association of a recently described hypomorphic mutation PRF1:p.A91V (rs35947132) with MS, we observed that this functional variant is more common in Sardinians (9.4%) than in other European populations (average frequency 4.6% in the ExAc database), and far more than in African (0.6%) and South East Asian (0.4–0.01%) populations. Such allele frequency suggests that Sardinian population provides higher statistical power for single variant/hypothesis testing even for relatively small size effects compared to that observed in European populations.
We first assessed the known predisposing association of the minor allele A (p.Val91) of the rs35947132 mutation with MS in a Sardinian sample set of 2,903 patients and 2,880 controls (healthy blood donors) from across the island. We confirmed the predisposing effect of rs35947132 A allele on MS (p=2.06×10−4, OR=1.29) (Figure 1).
Figure 1. Association of the PRF1 p.Ala91Val (rs35947132) mutation with MS and T1D.
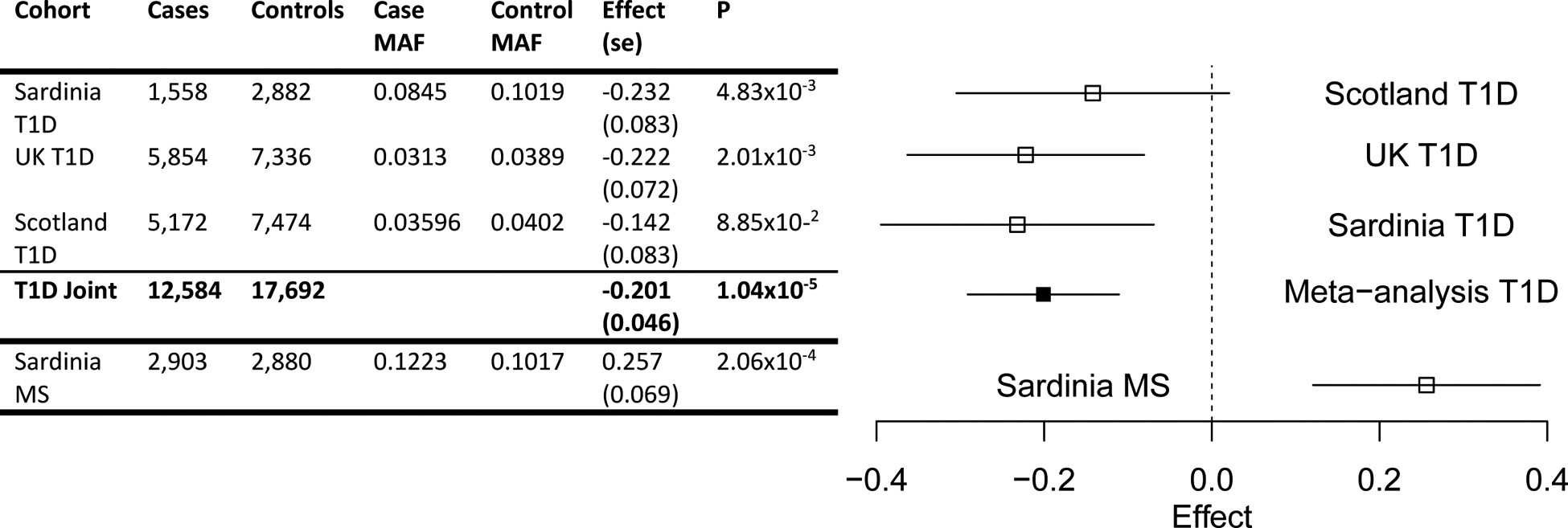
The forest plot (right) displays the distribution of the effects across study cohorts and the T1D meta-analysis result. The span of the horizontal lines corresponds to the 95% confidence interval of the log-odds ratio estimate.
Next we evaluated the role of (PRF1:p.A91V) on a T1D risk in a Sardinian cohort of 1,558 T1D patients and 3,323 controls. Notably, we found that the PRF1:p.A91V mutation confers protection from the disease (p = 4.8×10−3, OR=0.80). We then refined this finding in a meta-analysis using 12,584 T1D cases originating from Sardinia, the UK and Scotland, matched with 17,692 controls. PRF1:p.A91V mutation confers protection from T1D (p =1.04×10−5, OR=0.82) (Figure 1, Supplementary Material).
This protective effect in T1D of PRF1:p.A91V on T1D supports the recently described protection from another autoimmune disease, type 1 narcolepsy (T1N) [Ollila HM Biorxiv].
From these data, we infer that PRF1:p.A91V has opposite effects on the inherited risk for different autoimmune diseases.
PRF1:p.A91V driven expansion of cytotoxic CD8dim and CD8 dim CD28− producing subclinical inflammation state
In a search for cellular mechanisms underlying the distinct effects of PRF1:p.A91V on autoimmunity, we tested associations of this mutation in 731 FACS profiled cell immunophenotypes in up to 3,757 individuals from the SardiNIA general population cohort (Supplementary Figure 2).
We found that several CD8+ cytotoxic T lymphocyte subtypes were increased in individuals carrying PRF1:p.A91V (Table 1). Two T cell subpopulations, known to increase in inflammatory disorders, were expanded: CD8dimCD28− (p =5.06×10−6; effect = 0.20) and CD8dim (p =8.79×10−6, effect = 0.182) (Figure 2, Supplementary Figure 3A). This effect is in agreement with the known expansion of the CD8+CD28− T cells constrained by the size of the CD8+ T cell pool in relation to the CD4+ T cells [15]. Furthermore, PRF1:p.A91V is associated with reduced functionality of CD8+ T cells, as suggested by the reduced expression of CD127 (IL7R) in CD8+ T cells (p = 8.72×10−5; effect = −0.18) (Table 1).
Table 1. rs35947132 top associations across immunophenotyped traits.
Columns from left to right give the trait name, number of individuals (N), minor allele frequency (MAF), pvalue of association, the effect size expressed in standard deviation units (effect), its standard error (STERR). Traits shadowed in grey do not reach the significant threshold of association (p<9.27×10−5).
| TRAIT | N | MAF | PVALUE | EFFECT | STERR |
|---|---|---|---|---|---|
| CD28- CD8dim AC | 3408 | 0.094 | 5.06E-06 | 0.200 | 0.044 |
| CD8dim AC | 3652 | 0.094 | 8.79E-06 | 0.182 | 0.041 |
| EM CD8br AC | 3395 | 0.094 | 3.29E-05 | 0.184 | 0.044 |
| TCR gamma-delta AC | 3650 | 0.095 | 3.37E-05 | 0.172 | 0.041 |
| CD28- CD8dim %T cell | 3440 | 0.094 | 5.54E-05 | 0.176 | 0.044 |
| CD127 on CD28-CD8br | 2920 | 0.095 | 8.72E-05 | −0.182 | 0.046 |
| B cell AC | 3653 | 0.094 | 2.49E-04 | 0.151 | 0.041 |
| HLA DR+ CD8br AC | 3579 | 0.095 | 2.61E-04 | 0.152 | 0.042 |
| EM CD8br %T cell | 3427 | 0.094 | 3.61E-04 | 0.150 | 0.042 |
| Terminally Differentiated CD4-CD8- AC | 3395 | 0.094 | 4.73E-04 | 0.151 | 0.043 |
| CD28+ CD45RA+ CD8dim %CD8dim | 3440 | 0.094 | 5.15E-04 | −0.147 | 0.042 |
| CD8dim NKT AC | 3652 | 0.094 | 6.32E-04 | 0.143 | 0.042 |
| CD28- CD8br AC | 3408 | 0.094 | 6.58E-04 | 0.147 | 0.043 |
| EM CD8br %CD8br | 3427 | 0.094 | 7.50E-04 | 0.124 | 0.037 |
| CD8dim %T cell | 3668 | 0.094 | 8.30E-04 | 0.140 | 0.042 |
| TCR gamma-delta %T cell | 3666 | 0.095 | 8.66E-04 | 0.141 | 0.042 |
Figure 2. Regional plot of genetic association of the PRF1 gene region with CD28-CD8dim absolute count phenotype.
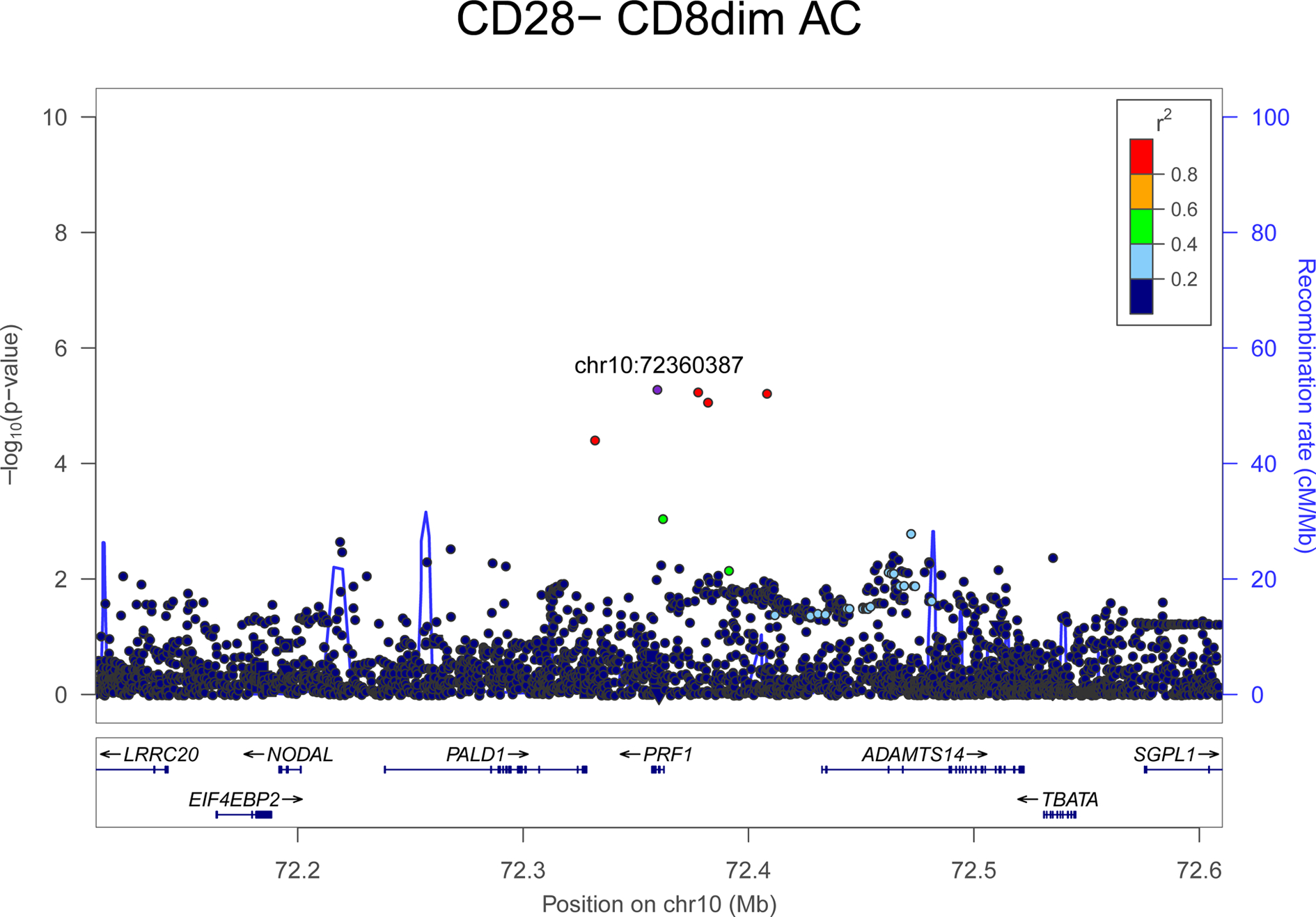
The association strength (-log10(pvalue); y axis) is plotted versus genomic positions (on the hg19/GRCh37 genomic build; x axis). SNPs are colored to reflect their LD with rs35947132 (p.A91V) (which is indicated with a purple dot).
In the UK Biobank dataset (UKB Neale v2 2018) the PRF1:A91V variant is in strong linkage disequilibrium (r2=0.98) with the rs142239370 variant that was found associated with higher lymphocyte count (p = 3.20×10−19, effect = 0.40) [16] (Figure 3). The lymphocyte count, however, in the SardiNIA cohort only marginally increase in the presence of p.Val91 (p = 3.5×10−3; effect = 0.12). This finding likely reflects an effect of CD8+ cytotoxic T lymphocytes, which - even if diluted in the total lymphocyte count - was still detectable on lymphocyte count because of the large sample size in the UK Biobank. Indeed, the contribution of PRF1:p.A91V to predisposition to the persistent subclinical inflammatory state is supported by the increase of both total lymphocyte counts and specific CD8+ subtypes (from our more refined cytofluorimetric analysis).
Figure 3.
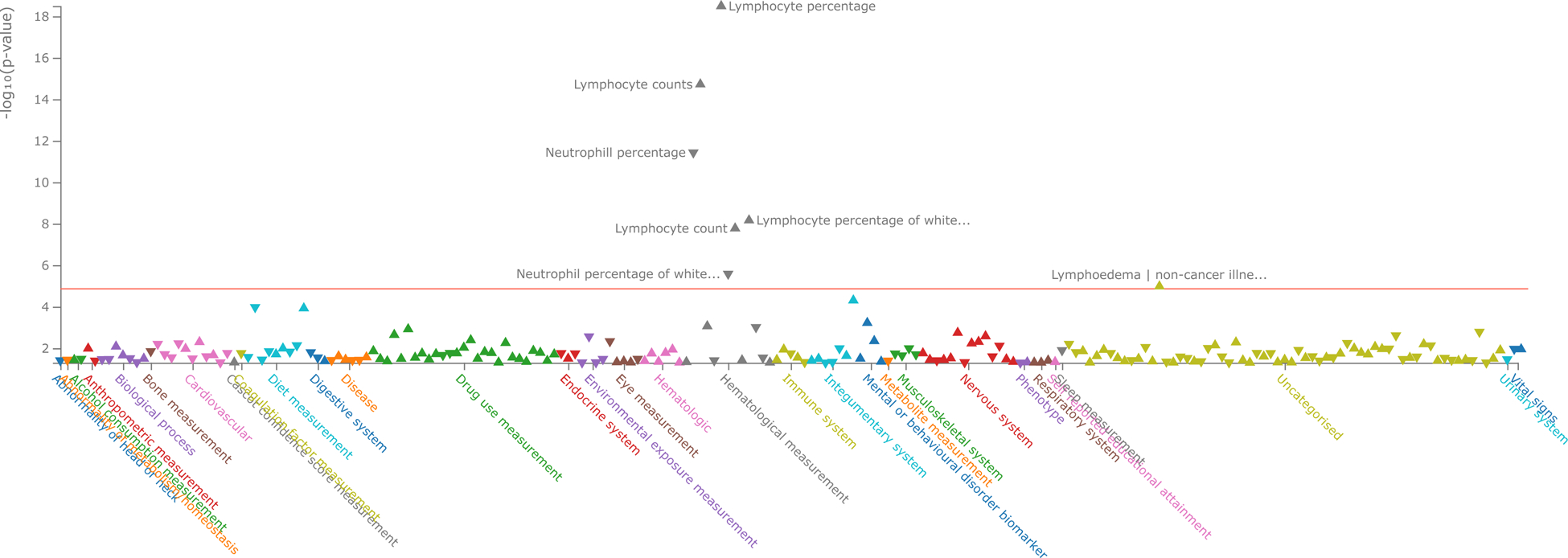
PheWAS data of traits associated with rs35947132 (PRF1) variant in the UK Biobank and GWAS Catalog summary statistics (p<0.05) according to Open Targets Genetics.
Moreover, we found that the colocalization probability between the two associations -the count of CD8dim CD28− cells in the SardiNIA cohort and the T1D summary statistics from the UK cohort profile is 0.97.
Immune memory is potentiated in PRF1:p.A91V individuals
Consistent with the chronic inflammatory process, we observed that PRF1:p.Val91 expands the CD8bright T effector memory pool (EM, p = 3.29×10−5, effect = 0.18) (Table1, Supplementary Figure 3B). CD8brightT EM cells are considered primarily responsible for cytotoxic action against pathogens, because they increase during immunosenescence and chronic inflammation and appear to be long-lived cytokine producers [17]. The CD8bright T EM persistent expansion has been described in individuals after cytomegalovirus infection. This phenomenon is known as CD8+ T cell inflation, in which the cells expand after infection and do not constrain to the small pool of memory cells; instead they remain expanded.
Moreover, we observed two independent associations for HLA-DR CD8+ T cell subpopulation (Supplementary Figure 4A, B).
γδ+ TCR cells are augmented in individuals carrying PRF1:p.A91V mutation
In agreement with our observation of the increase of all cytotoxic cell levels, the γδ+ subpopulation was expanded in individuals carrying PRF1:p.A91V (p = 3.37×10−5; effect = 0.17) (Table 1, Supplementary Figure 3C). γδ+ cells are unconventional cells that constitute 1–5% of total T cells circulating in healthy adults, but they are a major component in intestinal intraepithelial lymphocytes [18]. These cells rapidly produce cytokines and have potent cytotoxic activity.
PRF1:p.Val91 allele does not predispose to augmented inflammation
Further searching for the clinical and immune phenotypes of PRF1:p.A91V, we characterized a subgroup of 60 homozygous individuals from the SardiNIA cohort.
The course of FHL2 syndrome is clinically characterized by the spleen enlargement, recurrent fever and pulmonary complications that are the most frequent symptoms of organ damage. These, PRF1:p.A91V homozygote subjects did not exhibit significant differences compared to the rest of the cohort in the size of their spleens or a history of frequent complicated infections. Similarly, the physiological levels of hematological traits (hemoglobin levels, thrombocytopenia, neutropenia) and the inflammatory markers (ferritin, transferrin, triglycerides, fibrinogen) are consistent with the absence of symptomatic inflammation. Similarly, we observed no increased in soluble IL2RA, the biomarker used for detection of the FHL2 course (unpublished data).
Discussion
To enhance our knowledge on the immunological and clinical effects of the PRF1:p.A91V mutation, and more in general of PRF1 variation, we used a systems biology approach based on the characterization of this variant in large sample sets of individuals with different autoimmune diseases, coupled with extensive immunophenotyping of well-characterized phenotypically individuals from the general population of Sardinia. We discovered that PRF1:p.A91V contributes to the regulation of distinct immune subtypes, notably augmenting the memory cytotoxic T cell compartment: CD8dim, CD8dimCD28−, TCR γδ+, and effector-memory CD8bright T cell pool expansion. These effects are likely to have a role in the spectrum of clinical manifestations associated with this variant.
Similar to some pathogens, the PRF1:p.A91V mutation resets basal immune status, possibly predisposing to persistent subclinical, asymptomatic inflammation observed at a population level, which in turn contributes to MS but conversely protects from T1D (Supplementary Figure 4).
We have shown that increased level of total circulating lymphocytes observed in the UK Biobank data-set for PRF1:p.A91V are caused in particular, by increased cytotoxic cell subpopulations, probably due to a feedback mechanism to compensate their lower perforin-mediated cytotoxic function.
The differences in immune cell subtypes that we find in PRF1:p.A91V individuals are typical of subclinical inflammation characteristic of chronic inflammatory and autoimmune states. Indeed, the increase in cytotoxic CD8+CD28− T cells has been reported in almost every chronic inflammatory disease as well as in senescence, Herpesviriadae infections (CMV, EBV), HIV, tumor microenvironment, organ transplantation and autoimmunity [17,19,20].
Physiologically, CD8+ T cells exposed to antigens, such as viruses, proliferate and expand but then constrain to memory cells. However, certain infections cause long persistent expansion of the CD8+ T cell pool [19], contributing to different basal thresholds of activation of immune system. For instance, after infection with Herpesviriadae, through the mechanism known as CD8+ T cell inflation, T cell populations specific for certain epitopes do not contract but accumulate as effector-memory cells [21]. Thus, Herpesviriadae produces continuous inflammation, similar to the effect of PRF1:p.A91V in our data – an accumulation of CD8dimCD28− and long-lived CD8bright EM T cells, that are known to rapidly respond to the pathogens and express perforin [22].
The memory T cell pool results from the clonal expansion of antigen-experienced lymphocytes that accumulate over the lifetime of an individual. These cells carry large amounts of perforin, produce IFN-gamma, IL-4 and IL-5 following antigen stimulation, and are characterized by rapid effector function [23].
The CD8+ T cells act in peripheral non-lymphoid tissues against pathogens [22], suggesting a lower threshold of immune activation in PRF1:p.A91V individuals.
Inefficient cytotoxicity could provoke multiple effects: i) accumulation of cell subpopulations that normally are removed and lead to continuous antigenic stimulation; ii) the accumulation of memory cells and lymphocytosis that results in to immune system exhaustion; iii) continuous release of cytokines and mediators of inflammation; and iv) possible exposure to local antigens released from persisting cellular debris at the site of inflammation.
Genetic predisposition of the partially penetrant PRF1:p.A91V mutation to augmented cytotoxic T cell levels observed in our study, could, when reinforced by synergistic environmental factors, explain exaggerated immune system activation in adult familial hemophagocytic lymphohistiocytosis type 2 (FHL2) by unbalancing an endogenous immune feedback loop critical for immune homeostasis. That mechanism involves perforin-dependent elimination of antigen presenting dendritic cells by CD8+ T cells and has a dominant influence on the magnitude of T cell activation after viral infection [24].
Expanded CD8+ T cells are the leading cell subpopulation in organ specific, cell-mediated autoimmune disorders such as the pancreas beta cells in T1D [25]. CD8+ T cells mainly mediate pancreas damage in vitro and allogenic islet graft in vivo through the perforin/granzyme and Fas/FasL mechanisms. Local secretion of IFN-γ by effector T cells induces additional T, B cells and innate cells to migrate into the islets [21]. Accordingly, perforin deficiency diminishes the equivalent of T1D diabetes in the NOD mouse model [26]. Consistent with those observations, we found that partial perforin deficiency protects against human T1D but, because PRF1:p.A91V is a milder mutation, it still provides about half of perforin expression, contributing to modest protective effects. In accord with these findings, anti-PD-1 and PD-L1 antibodies used to potentiate exhausted CD8+ T cells in NOD mice and in human studies also result in rapid onset of autoimmune diabetes [27].
Pancreatic inflammation induced by certain viral infections is also protective in mouse models of T1D by expansion of antiviral cytotoxic T lymphocytes involved in insulitis [28]. Consistent with PRF1:pA91V protection against T1D, human cytomegalovirus infection has been shown to be protective against T1D in a small pediatric cohort [29].
In line with our observation that perforin deficiency predisposes to MS, a recent study demonstrated that clonally expanded CD8+ T cells in blood and central nervous system in MS are antigen nonspecific regulatory populations [30]. They inhibit disease by suppressing the proliferation of myelin oligodendrocyte glycoprotein (MOG)-specific CD4+ T cells. Furthermore, CD8+ T cells derived from perforin knockout mice immunized with myelin oligodendrocyte glycoprotein MOG and surrogate peptides fail in their suppressive function [6,30]. We observed that the increased cell levels to compensate for perforin deficiency also affects the levels of HLA-DR CD8+ T - cells considered by some authors to have suppressive activity and may represent a regulatory T cell subpopulation (Supplementary Figure 5A, B).
The pathogenic function in MS has, in contrast, been attributed to clonally expanded γδ+ T cells with an IL-17 phenotype [30]. That population of cells is important in early host defense against bacteria, protozoa and fungi and expands in PRF1:p.A91V individuals. In a mouse model, ablation of these cells after vaccination impaired protective and effector CD8+ T cell responses and diminished activation of CD8+ T cells in the periphery, liver, and spleen [31]. These results provide a rationale for use of that subpopulation of cells for therapeutic manipulation.
Supplementary Material
Funding
The authors thank all volunteers from Sardinian study on autoimmunity and SardiNIA cohorts. We thank to all the retired members of the Sardinian study on autoimmunity, in particular Paolo Pusceddu MD, Gianna Costa, Antonietta Zedda MD, Miriam Soro MD, Anna Rosa Mureddu MD. We thank to Mario Pani MD (Centro Trasfusionale AOBrotzu) and to all nurses from different Sardinian hospitals for enrolment assistance. To members of HPC group of CRS4: Chris Jones, Carlo Podda, Marco Moro and Michele Muggiri.
Sardinia Autonomus Region and its regional agency for research and development in Sardinia - Sardegna Ricerche and local Lanusei government for continues support.
That study was supported by grants (2011/R/13 and 2015/R/09 to Prof. Cucca) from Italian Foundation for Multiple Sclerosis; This work was supported in part by the Intramural Research Program of the NIH, National Institute on Aging (N01-AG-1–2109 and HHSN271201100005C, to Prof. Cucca); a grant (633964, to Prof. Cucca from the Horizon 2020 Research and Innovation Program of the European Union; a grant (U1301.2015/AI.1157.BE Prat.2015–1651, to Prof. Cucca) from Fondazione di Sardegna.
The T1D UK genotype data were generated using funding from the JDRF (9–2011- 253 and 5-SRA-2015–130-A-N) and Wellcome (091157 and 107212) to the Diabetes and Inflammation Laboratory, University of Oxford, as well as funding from the Wellcome Trust Case Control Consortium (076113) and support from the NIHR Oxford Biomedical Research Centre.
Footnotes
Declaration of Conflicting Interests
The authors declare no conflicting interests.
Supplementary material
Supplementary information to this article can be found online at:
References
- [1].Brodin P, Davis MM. Human immune system variation. Nat Rev Immunol 2017;17:21–9. 10.1038/nri.2016.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Patin E, Hasan M, Bergstedt J, Rouilly V, Libri V, Urrutia A, et al. Natural variation in the parameters of innate immune cells is preferentially driven by genetic factors. Nat Immunol 2018;19:302–14. 10.1038/s41590-018-0049-7. [DOI] [PubMed] [Google Scholar]
- [3].Orrù V, Steri M, Sole G, Sidore C, Virdis F, Dei M, et al. Genetic variants regulating immune cell levels in health and disease. Cell 2013;155:242–56. 10.1016/j.cell.2013.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol 2015;15:388–400. 10.1038/nri3839. [DOI] [PubMed] [Google Scholar]
- [5].Voskoboinik I, Dunstone MA, Baran K, Whisstock JC, Trapani JA. Perforin: structure, function, and role in human immunopathology. Immunol Rev 2010;235:35–54. 10.1111/j.0105-2896.2010.00896.x. [DOI] [PubMed] [Google Scholar]
- [6].International Multiple Sclerosis Genetics Consortium. Electronic address: chris.cotsapas@yale.edu, International Multiple Sclerosis Genetics Consortium. Low-Frequency and Rare-Coding Variation Contributes to Multiple Sclerosis Risk. Cell 2018;175:1679–1687.e7. 10.1016/j.cell.2018.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Voskoboinik I, Sutton VR, Ciccone A, House CM, Chia J, Darcy PK, et al. Perforin activity and immune homeostasis: the common A91V polymorphism in perforin results in both presynaptic and postsynaptic defects in function. Blood 2007;110:1184–90. 10.1182/blood-2007-02-072850. [DOI] [PubMed] [Google Scholar]
- [8].Nathan C, Ding A. Nonresolving inflammation. Cell 2010;140:871–82. 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- [9].Steri M, Orrù V, Idda ML, Pitzalis M, Pala M, Zara I, et al. Overexpression of the Cytokine BAFF and Autoimmunity Risk. N Engl J Med 2017;376:1615–26. 10.1056/NEJMoa1610528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sanna S, Pitzalis M, Zoledziewska M, Zara I, Sidore C, Murru R, et al. Variants within the immunoregulatory CBLB gene are associated with multiple sclerosis. Nat Genet 2010;42:495–7. 10.1038/ng.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 2009;41:703–7. 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Akbar T, McGurnaghan S, Palmer CNA, Livingstone SJ, Petrie J, Chalmers J, et al. Cohort Profile: Scottish Diabetes Research Network Type 1 Bioresource Study (SDRNT1BIO). Int J Epidemiol 2017;46:796–796i. 10.1093/ije/dyw152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sidore C, Busonero F, Maschio A, Porcu E, Naitza S, Zoledziewska M, et al. Genome sequencing elucidates Sardinian genetic architecture and augments association analyses for lipid and blood inflammatory markers. Nat Genet 2015;47:1272–81. 10.1038/ng.3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pilia G, Chen W-M, Scuteri A, Orrú M, Albai G, Dei M, et al. Heritability of cardiovascular and personality traits in 6,148 Sardinians. PLoS Genet 2006;2:e132. 10.1371/journal.pgen.0020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Clementi M, Forabosco P, Amadori A, Zamarchi R, De Silvestro G, Di Gianantonio E, et al. CD4 and CD8 T lymphocyte inheritance. Evidence for major autosomal recessive genes. Hum Genet 1999;105:337–42. [DOI] [PubMed] [Google Scholar]
- [16].Astle WJ, Elding H, Jiang T, Allen D, Ruklisa D, Mann AL, et al. The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell 2016;167:1415–1429.e19. 10.1016/j.cell.2016.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Arosa FA, Esgalhado AJ, Padrão CA, Cardoso EM. Divide, Conquer, and Sense: CD8+CD28- T Cells in Perspective. Front Immunol 2016;7:665. 10.3389/fimmu.2016.00665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Carding SR, Egan PJ. Gammadelta T cells: functional plasticity and heterogeneity. Nat Rev Immunol 2002;2:336–45. 10.1038/nri797. [DOI] [PubMed] [Google Scholar]
- [19].Hooper M, Kallas EG, Coffin D, Campbell D, Evans TG, Looney RJ. Cytomegalovirus seropositivity is associated with the expansion of CD4+CD28- and CD8+CD28- T cells in rheumatoid arthritis. J Rheumatol 1999;26:1452–7. [PubMed] [Google Scholar]
- [20].Chen X, Liu Q, Xiang AP. CD8+CD28- T cells: not only age-related cells but a subset of regulatory T cells. Cell Mol Immunol 2018;15:734–6. 10.1038/cmi.2017.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].O’Hara GA, Welten SPM, Klenerman P, Arens R. Memory T cell inflation: understanding cause and effect. Trends Immunol 2012;33:84–90. 10.1016/j.it.2011.11.005. [DOI] [PubMed] [Google Scholar]
- [22].Park CO, Kupper TS. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med 2015;21:688–97. 10.1038/nm.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol 2004;22:745–63. 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- [24].Terrell CE, Jordan MB. Perforin deficiency impairs a critical immunoregulatory loop involving murine CD8(+) T cells and dendritic cells. Blood 2013;121:5184–91. 10.1182/blood-2013-04-495309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Skulina C, Schmidt S, Dornmair K, Babbe H, Roers A, Rajewsky K, et al. Multiple sclerosis: brain-infiltrating CD8+ T cells persist as clonal expansions in the cerebrospinal fluid and blood. Proc Natl Acad Sci U S A 2004;101:2428–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Thomas HE, Trapani JA, Kay TWH. The role of perforin and granzymes in diabetes. Cell Death Differ 2010;17:577–85. 10.1038/cdd.2009.165. [DOI] [PubMed] [Google Scholar]
- [27].Kotwal A, Haddox C, Block M, Kudva YC. Immune checkpoint inhibitors: an emerging cause of insulin-dependent diabetes. BMJ Open Diabetes Res Care 2019;7:e000591. 10.1136/bmjdrc-2018-000591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Christoffersson G, Chodaczek G, Ratliff SS, Coppieters K, von Herrath MG. Suppression of diabetes by accumulation of non-islet-specific CD8+ effector T cells in pancreatic islets. Sci Immunol 2018;3. 10.1126/sciimmunol.aam6533. [DOI] [PubMed] [Google Scholar]
- [29].Ekman I, Vuorinen T, Knip M, Veijola R, Toppari J, Hyöty H, et al. Early childhood CMV infection may decelerate the progression to clinical type 1 diabetes. Pediatr Diabetes 2019;20:73–7. 10.1111/pedi.12788. [DOI] [PubMed] [Google Scholar]
- [30].Saligrama N, Zhao F, Sikora MJ, Serratelli WS, Fernandes RA, Louis DM, et al. Opposing T cell responses in experimental autoimmune encephalomyelitis. Nature 2019;572:481–7. 10.1038/s41586-019-1467-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zupan B, Liu B, Taki F, Toth JG, Toth M. Maternal Brain TNF-α Programs Innate Fear in the Offspring. Curr Biol CB 2017;27:3859–3863.e3. 10.1016/j.cub.2017.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.