

Abstract
A key factor for developing gene therapy strategies for neurological disorders is the availability of suitable vectors. Currently, the most advanced are adeno-associated vectors that, while being safe and ensuring long-lasting transgene expression, have a very limited cargo capacity. In contrast, herpes simplex virus-based amplicon vectors can host huge amounts of foreign DNA, but concerns exist about their safety and ability to express transgenes long-term. We aimed at modulating and prolonging amplicon-induced transgene expression kinetics in vivo using different promoters and preventing transgene silencing. To pursue the latter, we deleted bacterial DNA sequences derived from vector construction and shielded the transgene cassette using AT-rich and insulator-like sequences (SAm technology). We employed luciferase and GFP as reporter genes. To determine transgene expression kinetics, we injected vectors in the hippocampus of mice that were longitudinally scanned for bioluminescence for 6 months. To evaluate safety, we analyzed multiple markers of damage and performed patch clamp electrophysiology experiments. All vectors proved safe, and we managed to modulate the duration of transgene expression, up to obtaining a stable, long-lasting expression using the SAm technology. Therefore, these amplicon vectors represent a flexible, efficient, and safe tool for gene delivery in the brain.
Keywords: herpes simplex virus, amplicon vector, Central Nervous System, hippocampus
Graphical abstract
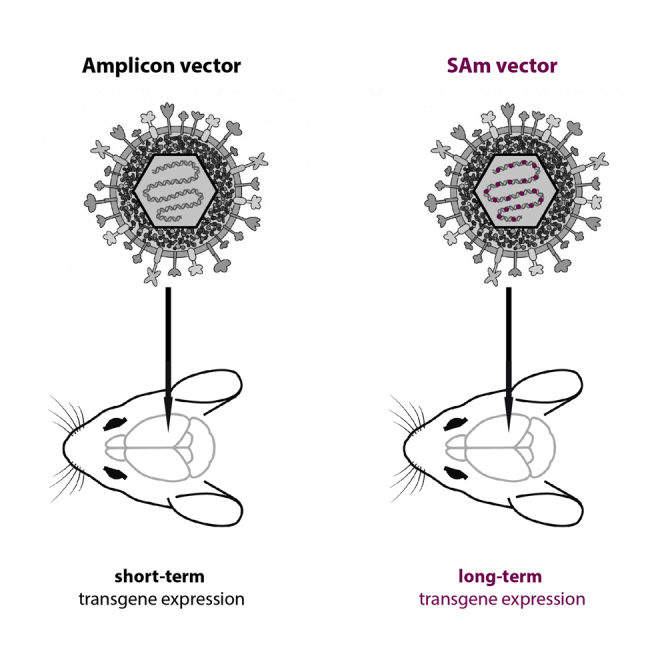
By shielding the transgene cassette using AT-rich and insulator-like sequences, we developed a new generation of amplicon vectors that offer the opportunity for efficient, safe, and (in contrast with “traditional” amplicon vectors) prolonged and stable gene expression in neuronal cells.
Introduction
Disorders affecting the central nervous system (CNS) entail important unmet medical needs. Their pathogenic mechanisms are not yet completely understood, and therapeutic approaches, when available, are not satisfactorily effective and safe. By delivering genes into defined target cells, gene therapy may represent not only a tool for studying the mechanisms of complex neurological diseases1 but also a new therapeutic option.2 To pursue these goals, one major hurdle to overcome is the development of an efficient tool for gene transfer.
Among different types of viral and non-viral vectors that can be used to vehicle genes into specific brain areas or cells, three viral vectors are most commonly used in preclinical and clinical studies: those based on adeno-associated virus (AAV),3, 4, 5 lentivirus (LV),6, 7, 8 or herpes simplex virus 1 (HSV-1).9,10 AAV vectors are the most advanced, as they already reached successful results in clinical studies, for example for the treatment of spinal muscular dystrophy,11 that eventually led to US Food and Drug Administration (FDA) and European Medicines Agency (EMA) approval for marketing. However, both AAV and, even if to a lesser extent, LV vectors cannot carry large amounts of DNA, a prerequisite for many research and therapeutic strategies. This is instead the main advantage of HSV-1-based vectors, in particular of amplicon vectors, in which nearly all viral DNA (up to 150 Kb) can be substituted by a concatemer of multiple copies of transgenes.12
In principle, amplicon vectors may be considered the safest among HSV vectors, because their genome does not carry viral genes. Unfortunately, they are contaminated by helper viruses during production.13 However, important manufacturing breakthrough, like amplicon vector improved packaging system and helper-free amplicon vector production, may overcome this problem.13, 14, 15, 16 Another issue is that amplicon vectors provide only temporary expression in non-dividing and dividing cells, as the transgene(s) expression is rapidly silenced and the vector DNA remains in episomal form and is therefore lost during mitosis.17 The latter issue inevitably limits the applicability of these vectors for a stable gene transfer in replicating cell populations, but may not constitute a substantial problem for terminal elements like neurons. As for the former issue, i.e., silencing of transgene expression, gene-regulating strategies may be developed based on insulators, promoters, or enhancers.
The aim of this study was to develop safe amplicon vectors with different kinetics of transgene expression. This goal was pursued by (1) comparing different promoters and titers (doses) of vectors; (2) deleting bacterial DNA sequences and incorporating in the genome sequences with high content of adenine and thymine residues (AT-rich) and insulator-like sequences, to shield promoter methylation and thereby prevent silencing of transgene expression; and (3) test vector safety at histological and electrophysiological level.
Specifically, we systematically compared expression of two reporter genes (luciferase and green fluorescent protein, GFP) when driven by 3 different promoters: the immediate-early (IE) 4/5 promoter, known to induce only transient gene expression,18 a constitutively active enhanced cytomegalovirus/chicken-β-actin based promoter (ECBA) and the neuron-specific enhanced human synapsin 1 (ESyn) promoter,19 known to drive a long-lasting expression of transgenes in the CNS.20,21
In addition, we combined the ESyn promoter with an improved amplicon technology (“steady” amplicons, SAm) that uses cis-regulatory elements to attempt obtaining sustained levels of transgene expression in time. Most undesired silencing processes of transgene expression are caused by methylations of cytosines followed by guanine residues (CpG), which trigger chromatin condensation spreading to the downstream promoter. This phenomenon has been reported for both integrated and episomal transgenes.22 More recently, the extragenic spacer lenght between the 5' and 3' ends of the expression cassette, rather than CpG methylation, has been shown to play the key role in episomal transgene silencing in vivo.23 To prevent such potential reduction in transgene expression, all amplicons used in this study were produced removing all the bacterial backbone. In addition, because insulators proved capable to shield transgenes from the effects of flanking viral sequences,24, 25, 26, 27 we inserted insulators-like and AT-rich DNA sequences in the SAm backbone.
Results
Time course of luciferase transgene signal in the mouse hippocampus
We developed and tested the following portfolio of amplicon vectors: vAm-IE4/5-LiG2 (LiG standing for Luciferase-IRES-GFP), vAm-ECBA-LiG2, vAm-ESyn-LiG2, and vSAm-ESyn-LiG2, based on the amplicon plasmids schematized in Figure 1. The first three amplicon vectors differ for the promoter, which drives the expression of the reporter genes GFP and luciferase. We chose to compare an IE promoter constitutively active in most cell types and known to induce solely a transient gene expression18 and the constitutive ECBA and the neuron-specific ESyn promoters, known to drive a long-lasting expression of transgenes in the CNS.20,21 Finally, we combined the ESyn promoter with an improved amplicon technology.
Figure 1.

Structure of the plasmids used for construction of amplicon vectors
Genes encoding two reporter products, firefly luciferase (Luc) and green fluorescence protein (GFP), are linked with an internal ribosomal entry site (IRES) element, and the GFP gene is followed by a poly(A) tail. (A–D) Different promotor sequences were placed ahead of Luc to regulate transcription: the IE4/5 promoter in the pAm-IE4/5-luciferase-GFP plasmid (A); the cytomegalovirus enhancer chicken-β-actin (ECBA) promoter in the pAm-ECBA-luciferase-GFP plasmid (B); the neuron-specific enhanced synapsin (ESyn) promoter in the pAm-ESyn-luciferase-GFP plasmid (C), and the pSAm-ESyn-luciferase-GFP plasmid (D). The size of each plasmid is reported in the respective scheme. Based on these sizes and considering that each plasmid will produce a concatemer closely matching the size of the HSV genome (about 150 kb), it can be estimated that pAm-IE4/5-LiG2 will be represented 20 times in the respective amplicon vector, pAm-ECBA-LiG2 16 times, pAm-ESyn-LiG2 17 times, and pSAm-ESyn-LiG2 16 times. “Steady” amplicon (SAm) sequences, i.e., insulator sequences of about 0.5 Kb and an approximately 2 Kb AT-rich sequence, were cloned in the pSAm-ESyn-luciferase-GFP plasmid (D). Amplicons vectors were assembled from these plasmids as described in the Materials and methods.
For all amplicon vectors, we carried out longitudinal in vivo studies to evaluate the transgenes expression. Specifically, we examined the following: first, the bioluminescence (BLI) level induced by luciferase expression, acquired using an IVIS 2D Lumina in vivo imaging system (PerkinElmer); second, the number of GFP-positive cells detectable around the site of injection. All amplicon vectors were tested at low titer (1 × 108 TU/mL) and two of them also at high titer (1 × 1010 TU/mL).
To assess whether the different amplicon vectors express the luciferase gene in vivo and to characterize the expression kinetics of each vector, we injected mice into the dorsal hippocampus with 1 μL of the amplicon vectors and BLI scanning was performed at different time points (Figure 2). Control (i.e., sham operated) mice received the vehicle (UltraMEM). All animals were scanned before vector inoculation, under basal conditions, and then regularly at defined time-points from 1 day post-infection (dpi) to 6 months post-infection (mpi). As expected, no BLI signal was detectable before vectors inoculation (−1 dpi) or in animals injected with vehicle (data not shown). On the contrary, a clear BLI signal was detected in the hippocampal region of all vector-injected animals. Representative images of the BLI signal emitted from the inoculation site are shown for two amplicon vectors (vAm-ESyn-LiG2, vSAm-ESyn-LiG2) in Figure 2.
Figure 2.

Bioluminescence (BLI) imaging experimental plan
(Top) Experimental flow chart illustrating the time points when BLI imaging was longitudinally performed. (Bottom) Representative BLI images of the head of mice injected with the two vectors in which transgenes are driven by the ESyn promoter (vAm-ESyn-LiG2, vSAm-ESyn-LiG2). Animals were anesthetized and BLI scanned 15–20 min after i.p. injection of D-luciferin (150 mg/kg).
All vectors produced a rapid expression of luciferase, peaking at our earliest time point (1 dpi). This early peak was in the range of 106 p/s for all low titer vectors (Figures 3A–3C). Two-log higher titers (i.e., two-log higher doses) of the vAm-IE4/5-LiG2 and vAm-ECBA-LiG2 vectors produced significantly higher peaks, indicating that the effects are dose-dependent (Figures 3A, 3B, and 3D). However, the response was about 2 logs higher for vAm-IE4/5-LiG2 and only about 1 log higher vAm-ECBA-LiG2 (Figures 3A, 3B, and 3D). The levels of expression declined very rapidly with vAm-IE4/5-LiG2, dropping by more than 3 logs within a week even at the higher dose (for this reason, we did not continue BLI scanning after 14 dpi in this group). In contrast, luciferase expression declined much more slowly when driven by the ECBA or the ESyn promoter, such that it remained above 105 p/s for up to 6 months with the high titer vAm-ECBA-LiG2 vector (Figures 3B, 3C, and 3E). Apart from these differences in transgene levels, the kinetics of expression were similar in low and high titer vectors. More interestingly, the novel vector backbone proved efficient in maintaining high levels of transgene expression in time. In fact, vSAm-ESyn-LiG2-induced luciferase expression remained high (around 106 p/s, i.e., >1 log higher than with vAm-ESyn-LiG2) for at least 6 months even when administered at low titer (Figures 3C and 3E).
Figure 3.

Kinetics of luciferase expression in animals injected with the different amplicon vectors
(A–C) Time course of the BLI signal in mice injected with low titer (light blue) or high titer (dark blue) vAm-IE4/5-LiG2 (A); low titer (light green) or high titer (dark green) vAm-ECBA-LiG2 (B); low titer vAm-ESyn-LiG2 (red) or low titer vSAm-ESyn-LiG2 (purple) (C). (D and E) A comparison of the BLI signals produced by injection of the different amplicon vectors at the earliest (1 dpi, D) and latest (6 mpi, E) time points. 14 animals were injected with either low or high titer vAm-IE4/5-LiG2 vector, of which 8 were killed at 4 dpi, and 6 at 14 dpi. 28 animals per group were injected with other vectors, of which 16 were killed after at 4 dpi, 6 at 2 mpi, and 6 at 6 mpi. Therefore, data in (D) are the means ± SEM of 14 low or high titer vAm-IE4/5-LiG2 injected-mice and 28 animals of the other groups; data in (E) are the means ± SEM of 6 animals per group (but vAm-IE4/5-LiG2 injected-mice were not determined, n.d.). ∗∗p < 0.01; ∗∗∗p < 0.001; Mann-Whitney U test.
In sum, these data show that the kinetics of transgene expression depend on the dose of vector, the promoter, and the vector construction strategy.
Identification of transgene-expressing cells and spread of the amplicon vectors
We evaluated the GFP reporter gene expression to identify the cells expressing the transgenes and estimate the spread of the vectors from the site of injection. All four amplicon vectors (vAmIE4/5-LiG2, vAm-ECBA-LiG2, vAm-ESyn-LiG2, and vSAm-ESyn-LiG2) led to GFP expression in principal hippocampal cells, i.e., CA1, CA2, and CA3 pyramidal cells and, to a lesser extent, granular cells of the dentate gyrus (Figure 4). GFP-positive cells were not found in the contralateral hippocampi, suggesting the absence of retrograde transport (see Figure 4A for vAm-ESyn-LiG2).
Figure 4.

GFP expression and vector diffusion in the vAm-ESyn-LiG2 injected mouse hippocampus, at 4 dpi
(A) Representative coronal section of a mouse hippocampus of low titer vAm-ESyn-LiG2, near the level of amplicon vector injection. Note GFP-positive pyramidal cells in the injected hippocampus (asterisks), and absence of GFP signal in the contralateral hippocampus. (B) Representative images of serial coronal sections immunostained for GFP (green) and counterstained with DAPI (blue) covering about 900 μm (anterior to posterior) of an injected hippocampus, across the site of inoculation of high titer vAm-ECBA-LiG2. Horizontal bar in (A), 500 μm; horizontal bar in (B), 250 μm.
The spread of amplicon vectors in the rostral-caudal direction was estimated at 4 dpi in 4 animals injected with low and 4 injected with high titer vAm-IE4/5-LiG2 and in 6 animals of all other groups (see Figure 4B for vAm-ECBA-LiG2). One section every 300 μm across the hippocampus was analyzed by 2 researchers that were blind of the experimental group, to identify slices presenting GFP-positive cells. GFP-positive cells spanned 984 ± 82 μm (mean ± SEM of 10 animals) of the hippocampal thickness with the high titer amplicon vectors (vAm-IE4/5-LiG2 and vAm-ECBA-LiG2) and 548 ± 43 μm (mean ± SEM of 22 animals) of the hippocampal thickness with the low titer amplicon vectors.
Safety of amplicon vectors
We used multiple staining techniques and specific antibodies to assess the safety of amplicon vectors. In addition, we employed vehicle-injected mice as negative controls and mice injected with a toxic HSV vector carrying the ICP0 gene (JΔNI5 R0, employed at the titer of low titer amplicon vectors) as positive controls.28 First, Fluoro-Jade C (FJC) was employed to detect neuronal damage. Analysis revealed that no amplicon vectors evoked detectable toxicity for hippocampal neurons even when used at high titer (Figures 5C–5H). On the contrary, many FJC-positive cells were observed after JΔNI5 R0 injection (Figure 5B). Representative hippocampi at 4 dpi, when peak cytotoxicity is expected,28,29 are shown in Figures 5A–H. No FJC-positive cells were detected at later time points (2 mpi and 6 mpi; data not shown).
Figure 5.

Absence of detectable neurodegeneration and alterations in cytoarchitecture in the hippocampus of amplicon-injected animals
(A–H) Dorsal hippocampal, FJC-stained sections prepared from a negative control, vehicle-injected (A), a positive control, JΔNI R0 injected (B), and amplicon vector injected animals (C–H, as indicated), at 4 dpi. Note numerous FCJ-positive in the positive control, but not in other groups. (I–P) As above, hematoxylin and eosin stained. Obvious cell loss, cytoarchitecture damage, and cell infiltration in (J), but not in other groups. These images are representative of 5 section per animal, 6 animals per group except 4 animals per group in (C), (D) (K), and (L). Horizontal bar in (H) (for A–H panels), 500 μm; horizontal bar in (P) (for I–P panels), 320 μm.
Further experiments (hematoxylin and eosin staining, Figures 5I–5P) confirmed that only the JΔNI5 R0 vector caused damage to the hippocampal cytoarchitecture: extensive cell loss and degeneration of CA1 and CA3 pyramidal cells, together with damage in the dentate gyrus, were clearly visible 4 dpi (Figure 5J), whereas the hippocampal cytoarchitecture and morphology were preserved in vehicle and amplicon-vectors-injected mice (Figures 5I and 5K–5P). In addition, no hippocampal morphology changes or infiltrates of inflammatory cells were observed at 2 or 6 mpi in vehicle or amplicon-vector-injected mice (data not shown).
Finally, because old generation HSV vectors have been reported to be immunogenic and cause neuroinflammation, we assessed astrocytosis, microgliosis, and lymphocyte infiltration, respectively, using glial fibrillary acidic protein (GFAP), ionized calcium-binding adaptor molecule 1 (IBA-1), and CD45 immunofluorescence. Again, only the JΔNI5 R0 vector proved capable of increasing expression these three markers (Figures 6B, 6J, and 6R), whereas amplicon vectors yielded results identical to vehicle (Figure 6). Analysis at later time points (2 mpi and 6 mpi) did not reveal detectable changes in glial cell reactivity, microglia activation, or lymphocytes presence under any of the experimental conditions (data not shown).
Figure 6.

Absence of detectable neuroinflammatory reaction in the hippocampus of amplicon-injected animals
(A–H) Dorsal hippocampal, GFAP-stained sections prepared from a negative control, vehicle-injected (A), a positive control, JΔNI R0 injected (B), and various amplicon vector injected animals (C–H, as indicated), at 4 dpi. Note numerous GFAP-positive cells in the positive control, but not in other groups. (I–P) As above, IBA-1-stained. Note numerous IBA-1-positive cells in the positive control, but not in other groups. (Q–X) As above, CD45-stained. Again, note numerous CD45-positive cells in the positive control, but not in other groups. These images are representative of 5 section per animal, 6 animals per group except 4 animals per group in (C), (D), (K), (L), (S), and (T). Horizontal bar in (X) (for all panels), 70 μm.
In sum, histologic analysis suggests that none of the four amplicon vectors, when directly injected into the brain, induce any apparent hippocampal damage, even if applied at relatively high titers.
Amplicon vectors do not alter the electrophysiological properties of infected cells
To establish whether amplicon-infected cells are not only morphologically intact, but also physiologically healthy, we performed ex vivo and in vitro electrophysiology experiments on GFP-positive cells. Ex vivo experiments compared basal electrophysiological parameters of control, non-infected, and transduced (GFP-positive) hippocampal pyramidal neurons in slices prepared from sham-operated mice and mice injected with vSAm-ESyn-LiG2 (Figure 7A). None of the following physiological parameters was altered: resting membrane potential (RMP: −63.2 ± 1.5 mV in controls versus −59.2 ± 0.9 in GFP-positive cells; mean ± SEM, n = 4), input resistance (Rin: 108.5 ± 13.1 MΩ versus 99.7 ± 9.8), action potential threshold (APT: 38.8 ± 0.5 mV versus 37.0 ± 0.5), action potential amplitude (APA: 98.4 ± 1.2 versus 95.6 ± 0.4), or cumulative spike number (Figures 7B and 7C).
Figure 7.

Whole-cell patch clamp recordings in pyramidal neurons from hippocampal slices
(A) Representative image of GFP-expressing neurons from a hippocampal slice prepared 4 days after inoculation of the vSAm-ESyn-LiG2 amplicon vector (top, brightfield image; bottom, GFP fluorescence overlaid on the brightfield image). The recording pipette can be seen in the bottom image. (B) Representative traces from a naive, not transduced (NT) pyramidal neuron and a pyramidal neuron infected by vSAm-ESyn-LiG2, showing the AP firing pattern as response to square-wave depolarizing current injections (100 to 300 nA). (C) Number of APs fired in response to current injected steps. Data are the means ± SEM of 4 animals per group (value for each animal was the average of 2–3 cells).
We confirmed and extended these findings in vitro, in primary hippocampal cultures, focusing on the vSAm-ESyn-LiG2 vector. Rat primary hippocampal neurons at 8 days in vitro (DIV8) were mock-infected or infected with vSAm-ESyn-LiG2 at a multiplicity of infection (MOI) of one (Figure 8A), and whole-cell patch-clamp experiments were performed at DIV10/11. Current-clamp experiments displayed a similar voltage response in both infected and mock neurons (Figure 8B). Moreover, no significant differences were detected on the following basic electrophysiological parameters: resting membrane potential, action potential amplitude, after-hyperpolarization amplitude, action potential half-width, action potential threshold, and input resistance (Figures 8C–8H).
Figure 8.

Whole-cell patch clamp recordings from primary hippocampal neuronal cultures
(A) Representative image of native GFP signal in a vSAm-ESyn-LiG2 infected hippocampal neuron. Scale bar, 10 μm. (B) Current-clamp recordings of a hippocampal neuron showing voltage response to step current injections at various amplitudes in mock (NT) and vSAm-ESyn-LiG2 infected cells. (C–H) Basic electrophysiological parameters of mock (white circles; n = 16) and vSAm-ESyn-LiG2 infected neurons (purple circles; n = 7). AP, action potential; AHP, afterhyperpolarization.
Taken together, these experiments demonstrate that amplicon vectors, vSAm-ESyn-LiG2 in particular, do not alter the electrophysiological properties of infected hippocampal neurons.
Discussion
A high number of reports have dealt with different HSV-1-based vectors aiming to find an appropriate gene therapy delivery tool for CNS, but only a few have investigated amplicon vectors.30, 31, 32, 33, 34, 35 In the present study, we compared different promoters, titers, and amplicon vector backbones for their ability to drive transgene expression in CNS cells and for their safety. Our main findings are (1) that level and duration of transgene expression in vivo can be modulated by using different promoters, titers, and amplicon vector backbones; in particular, that the SAm technology can prevent silencing of amplicon-induced transgene expression, leading to very long-lasting expression; and (2) that all amplicon vectors are safe and do not alter the physiological properties of infected neurons. These findings will be discussed in detail below.
Promoters
A number of investigators have shown that transfer of genes into the CNS of mice and rats can be optimized using ubiquitous and/or cell-specific promoters.36,37 In the majority of these studies, however, viral promoters were employed and the duration or strength of transgene expression was assessed only in vitro, in cell cultures.37 We employed both viral and cellular promoters with expected different cell specificity and expression kinetics, and performed a systematic comparison in vivo, after direct injection in the hippocampus. Specifically, we tested the viral IE4/5 promoter to aim at a transient expression and two cellular promoters, ECBA and ESyn, to aim at a long-lasting expression. In addition, whereas the ECBA promoter is not cell-specific, the ESyn promoter is neuron-specific.21,38, 39, 40
Several reasons led us to choose the IE4/5 HSV-1 promoter for transient transgene expression.41 In general, the HSV immediate early genes (IE or α) are expressed very early in the viral life cycle. They are activated rapidly and transiently before any viral protein is synthesized, i.e., immediately following virus entry into the host cell.42 The results we obtained with the vAm-IE4/5-LiG2 vector are consistent with the natural activity of the IE4/5 promoter: a peak of transgene expression at 24 h post-infection (hpi) and then a very rapid drop. These results are similar to those described by other groups. When the IE4/5 promoter was employed to drive LacZ expression in cultured neurons using a HSV-1 vector, β-galactosidase peaked at 24 hpi, while it was absent at 2 weeks post-infection (wpi).43 An IE4/5 driven HSV-1 based amplicon vector was also used to transduce bovine monocytes, the transgene expression peaking at 20 hpi, but being still detectable at 90 hpi.18 Such results are also in line with our data, because the vAm-IE4/5-LiG2 vector-induced luciferase expression was still detectable at 3 dpi (i.e., 72 hpi), even if not at 5 dpi (120 hpi). As for cell specificity, apparently this was mostly neuronal in our hands, consistent with previous results in the cerebellum.44
For a long-term transgene expression, we employed an enhanced CBA promoter. In fact, we observed a long-term (6 months) expression of transgenes in neurons when using high titer vAm-ECBA-LiG2 vector, whereas at low titer transgene expression became almost undetectable in less than 1 month. These findings are in line with previously published studies. In particular, the CBA promoter in AAV-9 vectors has been reported to induce gene expression in vivo, in the adult mouse hippocampus, up to 4 wpi.45 In an AAV-2 vector, the CBA promoter induced transgene expression for 18 months in hippocampal and neocortical neurons, but not in astrocytes.20 Other studies, however, support the ability of the CBA promoter to express transgenes also in astrocytes, when engineered in a different AAV serotype.21 Therefore, the AAV serotype may be responsible for the cell specificity in these studies, even if capsid-promoter interactions may also contribute to determine it.46
To ensure a long-lasting transgene expression exclusively in neurons, we employed the human ESyn promoter, that has been reported to do so in multiple neuronal types not only when engineered in AAV,21,38 but also in lentivectors.39,40 In keeping with our findings, these experiments also demonstrate that ESyn promoter-endowed vectors are capable of expressing transgenes for a relatively long time. However, none of them was reported to achieve a steady and prolonged (months) level of transgene expression.
In sum, these data support the notion that different promoters engineered in amplicon vectors can modulate duration of transgene expression only within a relatively short time frame (days to just a few weeks). By increasing the vector titer, as we did with those containing the ECBA promoter, it is possible to reach a long-term, apparently stable transgene expression, but only after a huge decay occurring in the first few days and then at levels slightly above background, i.e., entailing an initial risk of dose-dependent side effects and a later risk of insufficient effects.
Vector backbone
One possible explanation of the above results is that amplicon vector-induced transgene expression is silenced by the host cell.41 Other hypotheses cannot be excluded but seem very unlikely. The vector genome is not diluted in non-dividing cells like neurons, and death of infected neurons does not seem to occur with these amplicon backbones. If instead the issue is silencing, one possible approach to prolong it could be protecting the expression cassette against heterochromatin formation, for example using insulators or ubiquitous chromatin opening elements (UCOEs). Indeed, the A2UCUE, inserted in replication defective HSV-1 vectors to protect a luciferase cassette, ensured stronger but not more prolonged expression.25 In the vSAm-ESyn-LiG2 vector, we incorporated AT-rich and insulator-like sequences and also removed the bacterial DNA sequences that can trigger chromatin condensation,22,23 shielding transgenes from the negative heterochromatin effects of flanking viral sequences. We observed that, in comparison with the conventional vAm-ESyn-LiG2 amplicon, transgene expression displayed a much smaller initial decay and then remained high up to 6 months, even at low amplicon titer. These findings pose the basis for a new generation of amplicon vectors that ensure stable and long-lasting transgene expression, i.e., that overcome a critical limitation of previous backbones.
Safety
In terms of safety, all amplicon vectors tested in the present study were found to be safe at the used titers, independent of the promoter and backbone. To explore safety, we employed as a positive control the HSV-JDNIΔ5-R0 vector, which was previously reported to induce cell damage (specifically, morphological abnormalities, lymphocyte infiltration, neurodegeneration, and activation of astrocytes and microglia) in the inoculated brain tissue.28 The toxicity of the JΔNI5-R0 vector is due to expression of the ICP0 gene47 and, in fact, the JΔNI5 backbone (in which ICP0 is deleted) is not toxic to the brain tissue.28,48 In contrast with those injected with JΔNI5-R0, the hippocampi injected with the different amplicon vectors developed and used in the present study seemed intact, comparable with tissue injected with vehicle, even when high titers were employed. For further verifying safety, we also studied the functional impact of amplicon infection (in particular of the most innovative backbone, i.e., vSAm-ESyn-LiG2) by analyzing the electrophysiological properties of infected cells in vitro and ex vivo. Again, no detectable alteration could be observed.
Conclusions
Amplicon vectors have some distinct advantages for the gene therapy of neurological diseases, as compared with AAV- and LV-based vectors. Their most attractive feature is the ability to host huge amounts of foreign DNA, offering the opportunity not only to deliver very large genes or multiple genes, but also gene-expression control elements like cell-specific and inducible promoters.10 However, some important downsides, in particular the need of helper vectors for production and the short-term transgene expression, have so far precluded their development as a doable strategy for CNS gene therapy. One alternative to reduce helper virus contamination (employed in the present study) is the use of the LaL helper virus, in which the packaging sequence is deleted by Cre-lox specific-site recombination.14 However, no means were identified so far to prolong transgene expression, which may be due to the presence of bacterial sequences in the amplicon genome that cause transgene silencing by forming inactive chromatin.49 By removing these sequences and incorporating insulator-like sequences in the SAm vector, we managed to overcome this important limitation.
In conclusion, the amplicon vectors described here represent a flexible, efficient, and safe gene delivery system. The use of different promoters, with the addition of the backbones based on the SAm technology, offer opportunities for efficient short or prolonged gene expression in neuronal cells. Combination of ESyn or ECBA promoters with SAm amplicon vectors may find application for chronic gene therapy of neurological diseases, while amplicon vectors with the IE4/5 promoter may be useful if a strong and acute therapeutic gene expression is required.
Materials and methods
Viral vector construction
Amplicon plasmids
All plasmids were derived from pAm-ECBA-LiG2 (Figure 1B), containing an open reading frame (ORF) encoding the two reporter products, firefly luciferase (Luc) and GFP linked with an internal ribosomal entry site (IRES) element. This ORF is driven by a promoter (ECBA) that is similar to CBh, a previously described novel version of the CBA promoter,45 but harbors a synthetic chicken-β-actin/rabbit-β-globin intron instead of a minute virus of mice (MVM) intron, and terminates with a bovine growth hormone (BGH) polyadenylation signal. In addition to these elements, this plasmid contains a bacterial backbone with an ampicillin selection gene, bacterial origin of replication (Ori) and the HSV-1 elements essential for the production of amplicon vectors.12 In the pAm-IE4/5-LiG2 plasmid and pAm-ESyn-LiG2 plasmids, the ECBA promoter was removed and substituted by the HSV-1 IE 4/5 and ESyn promoters,19 respectively. The pSAm-ESyn-LiG2 plasmid was generated from the pAm-ESyn-LiG2 plasmid by the addition of two insulator sequences of about 0.5 Kb and a 2 Kb 70% AT-rich nucleotide sequence. The insulator sequence was from a genetically modified mouse tRNA gene (2tRNA-AT-IVS).50 The two tRNA insulators were engineered at each end of the HSV-1 CpG-rich amplicon sequences,51 in order to isolate the spread of epigenetic silencing from the viral sequence. The AT-rich sequence was placed between the insulator and the poly(A) sequence (Figure 1D). Before transfection for amplicon vector production, the bacterial DNA backbone was removed using rare-cutter restriction enzymes (PAC1, SFGI) on corresponding restriction sites flanking the amplicon gene expression cassettes, followed by a step of recircularization with ligase enzymes. This led to the creation of the different DNA minicircles, Am-IE4/5-LiG2, Am-ECBA-LiG2, Am-ESyn-LiG2, and SAm-ESyn-LiG2.
Cell lines and virus
The cell lines employed in this study were Gli36 cells (a human glioblastoma cell line), Vero cells (African green monkey kidney epithelial cell line), and trans-complementing Vero cells for amplification and purification of HSV-1 based amplicon vectors (ICP4 Vero cells; ICP4/Cre Vero cells).13,51 All cell lines were propagated in Dulbecco’s minimum essential medium (DMEM, Lonza, Switzerland) supplemented with 10% fetal bovine serum (FBS, Invitrogen Gibco, USA), 100 U/mL penicillin, and 100 mg/mL streptomycin (Invitrogen). Cells were maintained at 37°C in a humidified incubator containing 5% CO2.
Amplicon vectors
Amplicon vectors were produced by transfecting 5 μg of each minicircle amplicon into trans-complementing Vero cells using the jetPRIME reagent (Polyplus-transfection, France). Cells were superinfected the following day with the LaLΔJ helper virus at a MOI of 0.5 plaque-forming units (PFU)/cell in medium M199 (Gibco) supplemented with 1% FBS and 1% penicillin/streptomycin. 3 days later, cells were harvested and amplicon viral particles were extracted by several rounds of freeze/thaw and sonication.14 To calculate the purity of production, we titrated amplicon and helper particles to obtain transduction units (TU)/mL by using the cell number counting assay on Gli36 cells, and PFU/mL by using trans-complementing Vero cells. Several rounds of infections and productions were performed to obtain high quantity amplicon particles and a final infection-production step was performed on trans-complementing purifying Vero cells to obtain a final high purity working stock of amplicon vectors. The degree of purity was greater than 99% for all amplicon vectors. All virus stocks were checked for absence of revertant helper viruses on Vero cells. All amplicon vectors were also tested on Gli36 cells to ensure they were able to induce GFP expression and capable of forming plaques.
In vivo experiments
Animals
Male ICR (CD-1) mice (30–40 g; Envigo, Italy) were used for in vivo experiments. Mice were housed 5 per cage with environmental enrichment objects, under standard conditions: constant temperature (22°C–24°C) and relative humidity (55%–65%), 12 h light/dark cycle, and free access to food and water. Experiments involving animals were conducted in accordance with European Community (EU Directive 2010/63/EU), national and local laws and policies. The IACUC of the University of Ferrara approved this study and the Italian Ministry of Health authorized it (D.M., 715/2016-PR). We adhered to the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines.52 All efforts were made to minimize animal suffering and to improve animal wellbeing.
Vector injections
Mice were stereotaxically injected with 1 μL of the different amplicon vectors into the dorsal hippocampus under general anesthesia induced with ketamine/xylazine (43 and 7 mg/kg in saline pH 7.4, intraperitoneally, i.p.) and maintained with isoflurane 1.4% in oxygen (flow 1.2 mL/min) using the following coordinates: A –2.0, L –1.5, D +1.9 mm from bregma, nose bar at 0.0 mm. Infusion was performed via a glass needle (80 μm internal diameter at the tip) connected to a 25 μL Hamilton syringe, using a gradient flow 0.3–1.0 μL/min in 2 min 12 s. After the injection, the glass needle was left in place for an additional minute to allow the diffusion of vector particles, before being slowly withdrawn from the brain. All animals were given a post-operatory analgesic and antibiotic treatment (tramadol 5 mg/kg in saline, i.p. and enrofloxacin 10 mg/kg in saline, i.p.) for 2 days.
14 mice were injected with low and 14 with high titer vAm-IE4/5-LiG2, whereas 28 animals were injected with each of the other vectors. Randomly identified subgroups of animals were killed the day after undergoing BLI imaging at selected time points (4 dpi, 2 mpi, 6 mpi) for electrophysiological and histological analyses, as detailed below. Another group of 15 animals was infused with 1 μL of vehicle (UltraMEM, Gibco) and killed at the same time points (5 per time point) as a negative control for histological analyses. Using the same procedure, 5 animals were injected with 1 μL of HSV-JΔNI5-R0 4.0 × 108 PFU/mL, a HSV vector capable to induce neurotoxicity;28 these mice were killed at 4 dpi and used as a positive control for cell damage in immunohistochemistry assays.
In vivo BLI imaging
Mice were scanned in an IVIS100 in vivo imaging system (Perkin Elmer) at multiple time points after vector injection (Figure 2). The experimenter who IVIS scanned the animals was blinded to the vector used. Anesthesia was performed in an induction chamber with 2% isoflurane (Farmavet, Italy) in 100% oxygen at a flow rate of 1 mL/min and maintained in the IVIS with a 1.5% isoflurane mixture at 0.5 mL/min. 15 min before each imaging session, mice were i.p. injected with D-luciferin (250 μL, 150 mg/kg; Promega) dissolved in phosphate-buffered saline (PBS, pH 7.4, Gibco), placed in the anesthesia induction chamber and then placed in prone position into the imaging chamber. Two consecutive 5 min frames were acquired around the maximum signal, i.e., 15–20 min after D-luciferin injection. Photon emission was quantified using the Living Image software and reported as photon flux (p/s) from a 1.5 cm2 circular region of interest (ROI) on the head after subtraction of the BLI signal emitted by back fur from a 1.5 cm2 circular ROI. The limit of detection was 104 p/s. Statistical analyses were performed using the Prism (GraphPad Software, La Jolla, CA, USA) software.
Histology
As stated above, animals were killed at different time points for histological analyses. Of the 14 animals injected with either low or high titer vAm-IE4/5-LiG2, 8 were killed at 4 dpi and 6 at 14 dpi. Of the 28 animals in each of the other groups, 16 were killed at 4 dpi, 6 at 2 mpi, and 6 at 6 mpi. Of the 16 killed at 4 dpi in these groups, 4 per group were employed for electrophysiology (see below). The brains of all others were processed for either freezing or paraffin-embedding (50% for each preparation), as described below. Frozen sections were then employed for FJC, hematoxylin and eosin, and NeuroTrace staining, paraffin sections for GFP, GFAP, IBA-1, and CD45 immunofluorescence.
Fluoro Jade-C, hematoxylin and eosin, and NeuroTrace
Mice were deeply anesthetized by i.p. injection of a ketamine/xylazine mixture (100 and 15 mg/kg in pH 7.4 saline, respectively) and perfused transcardially with ice-cold PBS (pH = 7.4) followed by ice-cold 4% paraformaldehyde (PFA) in PBS. Brains were extracted, fixed for 1 h in PFA 4% at 4°C and then immersed into a 30% sucrose solution in PBS (4°C) overnight, for cryopreservation. Brains were then frozen in −50°C isopentane and stored at −80°C until being cut in 20 μm thick slices using a cryostat (Leica Biosystems, Germany) and mounted onto polarized slides (Superfrost slides, Bio-Optica).
For evaluation of neuronal damage, 5 representative frozen sections per animal spanning 1 mm from the site of vector inoculation (one section every 0.5 mm) were stained with FJC as previously described.53 Briefly, sections were rehydrated in PBS for 5 min and incubated in a solution containing 1% NaOH in 80% ethanol (5 min), in 70% ethanol (2 min), and then in distilled water (2 min). Thereafter, they were incubated for 10 min in 0.06% potassium permanganate, washed for 2 min in distilled water, and transferred into a 0.001% FJC staining solution in 0.1% acetic acid. After staining, sections were washed three times in distilled water and dried for 30 min at 50°C. Coverslips were mounted using the (distyrene, plasticizer and xylene (DPX) mountant for histology (Sigma).
To assess hippocampal cytoarchitecture and the presence of immune cells infiltrates, we incubated 5 other frozen sections per animal (as above) in Mayer’s hematoxylin solution 0.1% (Fluka, 5 min), washed in water (5 min), incubated in alcohol eosin solution 0.5% (Diapath, 2 min) and dehydrated. Coverslips were mounted using the DPX mountant for histology (Sigma).
To reveal the neurons, we stained 5 other frozen sections per animal (as above) with red NeuroTrace. Briefly, sections were washed for 10 min in PBS plus 0.1% Triton X-100, washed twice in PBS for 5 min, and then incubated with the NeuroTrace solution (1:150 in PBS, N21482, Invitrogen) for 20 min at room temperature, washed again for 10 min in PBS plus 0.1% Triton X–100 and then twice in PBS for 5 min. These slices were stained after collecting the pictures of the native GFP signal under the fluorescence microscope. Finally, the coverslips were mounted using the Shur/Mount (Bio-Optica).
Immunofluorescence
Animals were killed by decapitation under isoflurane anesthesia. Brains were removed, immersed for 48 h in 10% neutralized formalin, and then paraffin embedded in an automated tissue processor (VTP-300, Bio-Optica, Italy). Serial sections of 6 μm were cut with a microtome (Leica RM21225RT, Germany) and mounted onto polarized slides (Superfrost slides, Bio-Optica). Immunohistochemistry was performed on every 50th section across the dorsal hippocampus. After dewaxing (two washes in xylol for 10 min, 5 min in ethanol 100%, 5 min in ethanol 95%, 5 min in ethanol 80%) and rehydration in PBS, antigens were unmasked by immersing in a 0.1 M sodium citrate and 0.1 M citric acid solution (18 ml/L and 82 mL/L in distilled water) and microwaving at 750 Watt for 5 min and twice at 350 Watt for 5 min. After washing in PBS, sections were incubated with Triton X-100 (Sigma; 0.3% in PBS at room temperature for 10 min), washed twice in PBS, and incubated with a mixture of 5% bovine serum albumin (BSA) and 5% goat serum for 30 min.
Sections were then incubated overnight in a humid chamber at 4°C with the primary antibodies: GFP 1:50 (rabbit polyclonal, SC-8334, Santa Cruz), GFAP 1:100 (rabbit polyclonal, G9269, Sigma), IBA-1 1:200 (rabbit polyclonal, 234003, Synaptic System). The GFP antibody was revealed using a goat anti-rabbit, Alexa Fluor 488-conjugated, secondary antibody (1:500; A11001, Invitrogen), while GFAP and IBA-1 were detected using a goat anti-rabbit Alexa Fluor 594-conjugated monoclonal antibody (1:500; A11012, Invitrogen). CD45 was revealed using the biotin-streptavidin system composed of biotin anti-mouse/human CD45R/B220 antibody (1:100; 103204, BioLegend) associated with Alexa Fluor 594 Streptavidin (1:250; 405240, BioLegend). All secondary antibodies were incubated at room temperature for 2.5 h. Finally, sections were washed in PBS, counterstained with 0.0001% 4’,6-diamidino-2-phenylindole (DAPI, Sigma) for 10 min, and washed again. Coverslips were mounted using Shur/Mount (Bio-Optica).
Patch-clamp recordings
Hippocampal slices
Ex vivo patch clamp recordings were performed on CA1 pyramidal neurons in transversal slices of the hippocampus of naive and pSAm-ESyn-LiG2 injected mice, at 4 dpi. Animals were deeply anesthetized with isoflurane and decapitated. Brains were rapidly removed and hippocampi dissected in ice-cold gassed (95% O2 and 5% CO2) artificial cerebrospinal fluid (aCSF) composed of the following: 124 mM NaCl, 2.75 mM KCl, 1.25 mM NaH2PO4, 1.3 mM MgCl2, 2 mM CaCl2, 26 mM NaHCO3, 11 mM D-glucose. Hippocampi were sliced coronally around the vector injection area into 200 μm thick slices using a vibratome (DSK, T1000, Dosaka, Japan), equipped with a sapphire blade (Tedpella, USA). After recovery for at least 90 min at room temperature, slices were individually transferred into a Petri dish and the CA1 region was disconnected from the CA3 region by surgical cut. Slices were then transferred to a recording chamber positioned on an upright microscope (Axioskop; Zeiss, Göttingen, Germany) and superfused continuously through a peristaltic pump with warmed (Warner Instruments in-line heater TC324-C) aCSF, containing (in mM) 124 NaCl, 2.5 KCl, 1.2 NaH2PO4, 24 NaHCO3, 5 HEPES, 2 MgSO4, 2 CaCl2, and 12.5 D-glucose and saturated with 95% O2, 5% CO2 (pH 7.3). The rate of superperfusion was 2 mL/min, such that a complete exchange of the recording chamber volume occurred in approximately 1 min. Recordings were carried out at 30°C–31°C. Slices were allowed to equilibrate for at least 15 min before beginning the recordings. Neurons were visualized by infrared Dodt gradient contrast (DGC) video microscopy with a NIR-IR camera mounted coaxially with the light path. GFP-expressing neurons were visually identified by luminescence, emitted by illuminating the slice with an HBO 100 W lamp coaxial with the microscope objective. Light was filtered with an optical filter set for the detection of GFP green fluorescence (Zeiss).
Patch pipettes were prepared from thick-walled borosilicate glass (Hilgemberg GmbH, Malsfeld, Germany) on a P-2000 Flaming-Brown puller (Sutter Instruments, Novato, CA, USA). Whole-cell patch clamp recordings were performed using pipettes filled with a solution containing the following (in mM): 124 KH2PO4, 2 MgCl2, 10 NaCl, 10 HEPES, 0.5 EGTA, 2 Na2-ATP, 0.02 Na-GTP, pH 7.2, adjusted with KOH (~8); osmolarity, 290 mOsM; tip resistance, 2–3 MΩ. Gigaseals (>3 GΩ) were formed in a voltage-clamp configuration. Whole-cell access was achieved by rupturing the membrane with negative pressure. After a waiting period of 4–5 min, to allow for neuron dialysis with the pipette solution, we switched to a current-clamp configuration to confirm the identity of pyramidal neurons by recording responses to negative and positive current pulses evoking repetitive AP firing, to calculate the cumulative spikes number and to identify AP threshold. Cumulative spikes were calculated when, following a depolarizing step, the number of evoked APs was at least 6. Threshold was defined as the voltage when the rate of rise of the AP upstrokes first exceeded 15 mV/ms. Cell input resistance (Rin) stability was monitored throughout the experiments by periodic application of short (20 ms) hyperpolarizing pulses (−5 mV). This value was in the range of 90–130 MΩ and the access resistance, estimated from the amplitude of the uncompensated capacitive transient (6–10 MΩ) was not adjusted. When access resistance increased above 10%, recordings were halted. Membrane potential was not adjusted for the pipette liquid junction potential (~8 mV).
Recordings were made using an AXON Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA) interfaced with a PC through Digidata 1440A (Molecular Devices, Sunnyvale, CA, USA). Data were acquired and analyzed using the Clampfit 9.2 software (Molecular Devices, Sunnyvale, CA, USA).
Primary cultures
Primary cultures of hippocampal neurons were prepared according to Ryan & Smith54 from 1- to 2-day-old Sprague–Dawley rats (n = 3). After cutting the hippocampi into small sections, the tissue was incubated into Hank’s solution containing 3.5 mg/mL trypsin type IX (Merck-Sigma) and 0.5 mg/mL DNase type IV (Calbiochem, La Jolla, CA, USA) for 5 min. The pieces were then mechanically dissociated in a Hank’s solution supplemented with 12 mM MgSO4 and 0.5 mg/mL DNase IV. After centrifugation, cells were plated onto poly-ornithine coated coverslips and maintained in minimal essential medium supplemented with 20 mM glucose, B27 (Life Technologies, Carlsbad, CA, USA), 2 mM glutamax and 5% fetal clone III (FCIII; Hyclone, South Logan, UT, USA) for 3 h. After adhesion, cells were maintained in Neurobasal Medium (ThermoFisher, Carlsbad, CA, USA) supplemented with 2 mM glutamax, B27 and 5 μM 1-β-D-cytosine-arabinofuranoside (Merck-Sigma). Cultures were maintained at 37°C in a 5% CO2 humidified incubator and used between 7 and 12 days after plating. Neurons were infected with the vSAm-ESyn-LiG2 vector at 8 days in vitro (DIV), with a MOI of 1, by adding the corresponding viral volume in the culture medium for 1 h at 37 °C in a 5% CO2 incubator. Patch-clamp experiments were performed 48–72 h after transduction.
Electrophysiological recordings in vitro were then performed as follows: individual slides with plated neurons were submerged in a recording chamber mounted on the stage of an upright BX51WI microscope (Olympus) equipped with differential interference contrast optics (DIC) and an optical filter set for the detection of GFP (Semrock, Rochester, NY, USA). Slides were continuously perfused (3–5 mL/min) with aCSF containing (in mM): 124 NaCl, 2.5 KCl, 1.2 NaH2PO4, 24 NaHCO3, 5 HEPES, 2 MgSO4, 2 CaCl2, and 12,5 D-glucose, saturated with 95% O2, 5% CO2 (pH 7.3) at room temperature. GFP-expressing neurons were visually identified by emitted fluorescence. Whole-cell patch clamp recordings were performed using pipettes filled with a solution containing the following (in mM): 124 KH2PO4, 2 MgCl2, 10 NaCl, 10 HEPES, 0.5 EGTA, 2 Na2-ATP, 0.02 Na-GTP (pH 7.2, adjusted with KOH; tip resistance: 5–7 MΩ). All recordings were performed using a MultiClamp 700B amplifier interfaced with a PC through Digidata 1440A (Molecular Devices, Sunnyvale, CA, USA). Current-clamp traces were sampled at a frequency of 10 kHz and low-pass filtered at 2 kHz. Data were acquired using the pClamp10 software (Molecular Devices) and analyzed with Prism 8 (GraphPad Software, La Jolla, CA, USA).
Acknowledgments
We wish to thank to Anna Binaschi, Giovanna Paolone, and Sara Carli for help with experiments in animals. This work was supported by funding from the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement 602102 (EPITARGET; to M. Simonato and H.B.).
Author contributions
M. Soukupová, S.Z., P.T., S.I., M.B., S.C., B.B., and S.F. carried out the experiments in animals; P.T., C.P.-B., and H.B. designed and prepared the vectors; M. Soukupová wrote the manuscript with support from P.T., H.B., and M. Simonato; M. Simonato supervised the project.
Declaration of interests
The authors declare no competing interests.
References
- 1.Simonato M., Manservigi R., Marconi P., Glorioso J. Gene transfer into neurones for the molecular analysis of behaviour: focus on herpes simplex vectors. Trends Neurosci. 2000;23:183–190. doi: 10.1016/s0166-2236(99)01539-8. [DOI] [PubMed] [Google Scholar]
- 2.Simonato M., Bennett J., Boulis N.M., Castro M.G., Fink D.J., Goins W.F., Gray S.J., Lowenstein P.R., Vandenberghe L.H., Wilson T.J. Progress in gene therapy for neurological disorders. Nat. Rev. Neurol. 2013;9:277–291. doi: 10.1038/nrneurol.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen S.H., Haam J., Walker M., Scappini E., Naughton J., Martin N.P. Recombinant Viral Vectors as Neuroscience Tools. Curr. Protoc. Neurosci. 2019;87:e67. doi: 10.1002/cpns.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choudhury S.R., Hudry E., Maguire C.A., Sena-Esteves M., Breakefield X.O., Grandi P. Viral vectors for therapy of neurologic diseases. Neuropharmacology. 2017;120:63–80. doi: 10.1016/j.neuropharm.2016.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duan D. Systemic delivery of adeno-associated viral vectors. Curr. Opin. Virol. 2016;21:16–25. doi: 10.1016/j.coviro.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kafri T. Lentivirus vectors: difficulties and hopes before clinical trials. Curr. Opin. Mol. Ther. 2001;3:316–326. [PubMed] [Google Scholar]
- 7.Martin-Rendon E., Azzouz M., Mazarakis N.D. Lentiviral vectors for the treatment of neurodegenerative diseases. Curr. Opin. Mol. Ther. 2001;3:476–481. [PubMed] [Google Scholar]
- 8.Nanou A., Azzouz M. Gene therapy for neurodegenerative diseases based on lentiviral vectors. Prog. Brain Res. 2009;175:187–200. doi: 10.1016/S0079-6123(09)17513-1. [DOI] [PubMed] [Google Scholar]
- 9.Ingusci S., Cattaneo S., Verlengia G., Zucchini S., Simonato M. A Matter of Genes: The Hurdles of Gene Therapy for Epilepsy. Epilepsy Curr. 2019;19:38–43. doi: 10.1177/1535759718822846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ingusci S., Verlengia G., Soukupova M., Zucchini S., Simonato M. Gene Therapy Tools for Brain Diseases. Front. Pharmacol. 2019;10:724. doi: 10.3389/fphar.2019.00724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017;377:1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 12.Epstein A.L. Progress and prospects: biological properties and technological advances of herpes simplex virus type 1-based amplicon vectors. Gene Ther. 2009;16:709–715. doi: 10.1038/gt.2009.42. [DOI] [PubMed] [Google Scholar]
- 13.Epstein A.L. HSV-1-derived amplicon vectors: recent technological improvements and remaining difficulties--a review. Mem. Inst. Oswaldo Cruz. 2009;104:399–410. doi: 10.1590/s0074-02762009000300002. [DOI] [PubMed] [Google Scholar]
- 14.Zaupa C., Revol-Guyot V., Epstein A.L. Improved packaging system for generation of high-level noncytotoxic HSV-1 amplicon vectors using Cre-loxP site-specific recombination to delete the packaging signals of defective helper genomes. Hum. Gene Ther. 2003;14:1049–1063. doi: 10.1089/104303403322124774. [DOI] [PubMed] [Google Scholar]
- 15.de Silva S., Bowers W.J. Herpes Virus Amplicon Vectors. Viruses. 2009;1:594–629. doi: 10.3390/v1030594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oehmig A., Fraefel C., Breakefield X.O., Ackermann M. Herpes simplex virus type 1 amplicons and their hybrid virus partners, EBV, AAV, and retrovirus. Curr. Gene Ther. 2004;4:385–408. doi: 10.2174/1566523043346129. [DOI] [PubMed] [Google Scholar]
- 17.Jacobs A., Breakefield X.O., Fraefel C. HSV-1-based vectors for gene therapy of neurological diseases and brain tumors: part II. Vector systems and applications. Neoplasia. 1999;1:402–416. doi: 10.1038/sj.neo.7900056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwyzer M., Fischer-Bracher C., Fraefel C., Bächi T., Nuñez R., Engels M., Ackermann M. Transduction of Vero cells and bovine monocytes with a herpes simplex virus-1 based amplicon carrying the gene for the bovine herpesvirus-1 Circ protein. Vet. Microbiol. 2002;86:165–174. doi: 10.1016/s0378-1135(01)00500-4. [DOI] [PubMed] [Google Scholar]
- 19.Hioki H., Kameda H., Nakamura H., Okunomiya T., Ohira K., Nakamura K., Kuroda M., Furuta T., Kaneko T. Efficient gene transduction of neurons by lentivirus with enhanced neuron-specific promoters. Gene Ther. 2007;14:872–882. doi: 10.1038/sj.gt.3302924. [DOI] [PubMed] [Google Scholar]
- 20.Klein R.L., Hamby M.E., Gong Y., Hirko A.C., Wang S., Hughes J.A., King M.A., Meyer E.M. Dose and promoter effects of adeno-associated viral vector for green fluorescent protein expression in the rat brain. Exp. Neurol. 2002;176:66–74. doi: 10.1006/exnr.2002.7942. [DOI] [PubMed] [Google Scholar]
- 21.Rincon M.Y., de Vin F., Duqué S.I., Fripont S., Castaldo S.A., Bouhuijzen-Wenger J., Holt M.G. Widespread transduction of astrocytes and neurons in the mouse central nervous system after systemic delivery of a self-complementary AAV-PHP.B vector. Gene Ther. 2018;25:83–92. doi: 10.1038/s41434-018-0005-z. [DOI] [PubMed] [Google Scholar]
- 22.Hsieh C.L. In vivo activity of murine de novo methyltransferases, Dnmt3a and Dnmt3b. Mol. Cell. Biol. 1999;19:8211–8218. doi: 10.1128/mcb.19.12.8211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu J., Zhang F., Xu S., Fire A.Z., Kay M.A. he extragenic spacer length between the 5’ and 3’ ends of the transgene expression cassette affects transgene silencing from plasmid-based vectors. Mol. Ther. 2012;20:2111–2119. doi: 10.1038/mt.2012.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fitzsimons H.L., Mckenzie J.M., During M.J. Insulators coupled to a minimal bidirectional tet cassette for tight regulation of rAAV-mediated gene transfer in the mammalian brain. Gene Ther. 2001;8:1675–1681. doi: 10.1038/sj.gt.3301582. [DOI] [PubMed] [Google Scholar]
- 25.Han F., Miyagawa Y., Verlengia G., Ingusci S., Soukupova M., Simonato M., Glorioso J.C., Cohen J.B. Cellular Antisilencing Elements Support Transgene Expression from Herpes Simplex Virus Vectors in the Absence of Immediate Early Gene Expression. J. Virol. 2018;92:e00536-18. doi: 10.1128/JVI.00536-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johansen J., Tornøe J., Møller A., Johansen T.E. Increased in vitro and in vivo transgene expression levels mediated through cis-acting elements. J. Gene Med. 2003;5:1080–1089. doi: 10.1002/jgm.444. [DOI] [PubMed] [Google Scholar]
- 27.Steinwaerder D.S., Lieber A. Insulation from viral transcriptional regulatory elements improves inducible transgene expression from adenovirus vectors in vitro and in vivo. Gene Ther. 2000;7:556–567. doi: 10.1038/sj.gt.3301139. [DOI] [PubMed] [Google Scholar]
- 28.Verlengia G., Miyagawa Y., Ingusci S., Cohen J.B., Simonato M., Glorioso J.C. Engineered HSV vector achieves safe long-term transgene expression in the central nervous system. Sci. Rep. 2017;7:1507. doi: 10.1038/s41598-017-01635-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paradiso B., Marconi P., Zucchini S., Berto E., Binaschi A., Bozac A., Buzzi A., Mazzuferi M., Magri E., Navarro Mora G. Localized delivery of fibroblast growth factor-2 and brain-derived neurotrophic factor reduces spontaneous seizures in an epilepsy model. Proc. Natl. Acad. Sci. USA. 2009;106:7191–7196. doi: 10.1073/pnas.0810710106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adrover M.F., Guyot-Revol V., Cheli V.T., Blanco C., Vidal R., Alché L., Kornisiuk E., Epstein A.L., Jerusalinsky D. Hippocampal infection with HSV-1-derived vectors expressing an NMDAR1 antisense modifies behavior. Genes Brain Behav. 2003;2:103–113. doi: 10.1034/j.1601-183x.2003.00015.x. [DOI] [PubMed] [Google Scholar]
- 31.Cheli V.T., Adrover M.F., Blanco C., Rial Verde E., Guyot-Revol V., Vidal R., Martin E., Alché L., Sanchez G., Acerbo M. Gene transfer of NMDAR1 subunit sequences to the rat CNS using herpes simplex virus vectors interfered with habituation. Cell. Mol. Neurobiol. 2002;22:303–314. doi: 10.1023/A:1020720001865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Falcicchia C., Trempat P., Binaschi A., Perrier-Biollay C., Roncon P., Soukupova M., Berthommé H., Simonato M. Silencing Status Epilepticus-Induced BDNF Expression with Herpes Simplex Virus Type-1 Based Amplicon Vectors. PLoS ONE. 2016;11:e0150995. doi: 10.1371/journal.pone.0150995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frazer M.E., Hughes J.E., Mastrangelo M.A., Tibbens J.L., Federoff H.J., Bowers W.J. Reduced pathology and improved behavioral performance in Alzheimer’s disease mice vaccinated with HSV amplicons expressing amyloid-beta and interleukin-4. Mol. Ther. 2008;16:845–853. doi: 10.1038/mt.2008.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lim F., Palomo G.M., Mauritz C., Giménez-Cassina A., Illana B., Wandosell F., Díaz-Nido J. Functional recovery in a Friedreich’s ataxia mouse model by frataxin gene transfer using an HSV-1 amplicon vector. Mol. Ther. 2007;15:1072–1078. doi: 10.1038/sj.mt.6300143. [DOI] [PubMed] [Google Scholar]
- 35.Katsu-Jiménez Y., Loría F., Corona J.C., Díaz-Nido J. Gene Transfer of Brain-derived Neurotrophic Factor (BDNF) Prevents Neurodegeneration Triggered by FXN Deficiency. Mol. Ther. 2016;24:877–889. doi: 10.1038/mt.2016.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kügler S. Tissue-Specific Promoters in the CNS. Methods Mol. Biol. 2016;1382:81–91. doi: 10.1007/978-1-4939-3271-9_6. [DOI] [PubMed] [Google Scholar]
- 37.Powell S.K., Rivera-Soto R., Gray S.J. Viral expression cassette elements to enhance transgene target specificity and expression in gene therapy. Discov. Med. 2015;19:49–57. [PMC free article] [PubMed] [Google Scholar]
- 38.Kügler S., Lingor P., Schöll U., Zolotukhin S., Bähr M. Differential transgene expression in brain cells in vivo and in vitro from AAV-2 vectors with small transcriptional control units. Virology. 2003;311:89–95. doi: 10.1016/s0042-6822(03)00162-4. [DOI] [PubMed] [Google Scholar]
- 39.Kuroda H., Kutner R.H., Bazan N.G., Reiser J. A comparative analysis of constitutive and cell-specific promoters in the adult mouse hippocampus using lentivirus vector-mediated gene transfer. J. Gene Med. 2008;10:1163–1175. doi: 10.1002/jgm.1249. [DOI] [PubMed] [Google Scholar]
- 40.Dittgen T., Nimmerjahn A., Komai S., Licznerski P., Waters J., Margrie T.W., Helmchen F., Denk W., Brecht M., Osten P. Lentivirus-based genetic manipulations of cortical neurons and their optical and electrophysiological monitoring in vivo. Proc. Natl. Acad. Sci. USA. 2004;101:18206–18211. doi: 10.1073/pnas.0407976101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harkness J.M., Kader M., DeLuca N.A. Transcription of the herpes simplex virus 1 genome during productive and quiescent infection of neuronal and nonneuronal cells. J. Virol. 2014;88:6847–6861. doi: 10.1128/JVI.00516-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Glorioso J.C. Herpes simplex viral vectors: late bloomers with big potential. Hum. Gene Ther. 2014;25:83–91. doi: 10.1089/hum.2014.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Geller A.I. A system, using neural cell lines, to characterize HSV-1 vectors containing genes which affect neuronal physiology, or neuronal promoters. J. Neurosci. Methods. 1991;36:91–103. doi: 10.1016/0165-0270(91)90142-m. [DOI] [PubMed] [Google Scholar]
- 44.Gimenez-Cassina A., Wade-Martins R., Gomez-Sebastian S., Corona J.C., Lim F., Diaz-Nido J. Infectious delivery and long-term persistence of transgene expression in the brain by a 135-kb iBAC-FXN genomic DNA expression vector. Gene Ther. 2011;18:1015–1019. doi: 10.1038/gt.2011.45. [DOI] [PubMed] [Google Scholar]
- 45.Gray S.J., Foti S.B., Schwartz J.W., Bachaboina L., Taylor-Blake B., Coleman J., Ehlers M.D., Zylka M.J., McCown T.J., Samulski R.J. Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Hum. Gene Ther. 2011;22:1143–1153. doi: 10.1089/hum.2010.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Powell S.K., Samulski R.J., McCown T.J. AAV Capsid-Promoter Interactions Determine CNS Cell-Selective Gene Expression In Vivo. Mol. Ther. 2020;28:1373–1380. doi: 10.1016/j.ymthe.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cuchet D., Ferrera R., Lomonte P., Epstein A.L. Characterization of antiproliferative and cytotoxic properties of the HSV-1 immediate-early ICPo protein. J. Gene Med. 2005;7:1187–1199. doi: 10.1002/jgm.761. [DOI] [PubMed] [Google Scholar]
- 48.Miyagawa Y., Marino P., Verlengia G., Uchida H., Goins W.F., Yokota S., Geller D.A., Yoshida O., Mester J., Cohen J.B., Glorioso J.C. Herpes simplex viral-vector design for efficient transduction of nonneuronal cells without cytotoxicity. Proc. Natl. Acad. Sci. USA. 2015;112:E1632–E1641. doi: 10.1073/pnas.1423556112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suzuki M., Kasai K., Saeki Y. Plasmid DNA sequences present in conventional herpes simplex virus amplicon vectors cause rapid transgene silencing by forming inactive chromatin. J. Virol. 2006;80:3293–3300. doi: 10.1128/JVI.80.7.3293-3300.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ebersole T., Kim J.H., Samoshkin A., Kouprina N., Pavlicek A., White R.J., Larionov V. tRNA genes protect a reporter gene from epigenetic silencing in mouse cells. Cell Cycle. 2011;10:2779–2791. doi: 10.4161/cc.10.16.17092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Epstein A.L. HSV-1-based amplicon vectors: design and applications. Gene Ther. 2005;12(Suppl 1):S154–S158. doi: 10.1038/sj.gt.3302617. [DOI] [PubMed] [Google Scholar]
- 52.Kilkenny C., Browne W.J., Cuthill I.C., Emerson M., Altman D.G. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8:e1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmued L.C., Stowers C.C., Scallet A.C., Xu L. Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res. 2005;1035:24–31. doi: 10.1016/j.brainres.2004.11.054. [DOI] [PubMed] [Google Scholar]
- 54.Ryan T.A., Smith S.J. Vesicle pool mobilization during action potential firing at hippocampal synapses. Neuron. 1995;14:983–989. doi: 10.1016/0896-6273(95)90336-4. [DOI] [PubMed] [Google Scholar]