

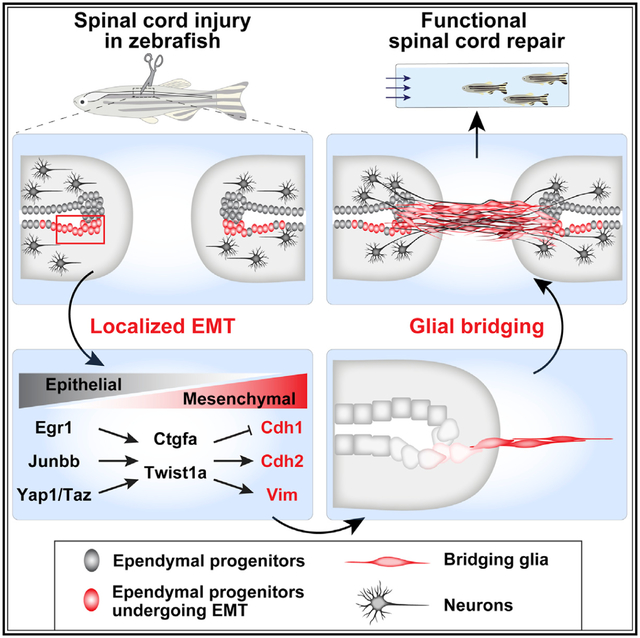
SUMMARY
Anti-regenerative scarring obstructs spinal cord repair in mammals and presents a major hurdle for regenerative medicine. In contrast, adult zebrafish possess specialized glial cells that spontaneously repair spinal cord injuries by forming a pro-regenerative bridge across the severed tissue. To identify the mechanisms that regulate differential regenerative capacity between mammals and zebrafish, we first defined the molecular identity of zebrafish bridging glia and then performed cross-species comparisons with mammalian glia. Our transcriptomics show that pro-regenerative zebrafish glia activate an epithelial-to-mesenchymal transition (EMT) gene program and that EMT gene expression is a major factor distinguishing mammalian and zebrafish glia. Functionally, we found that localized niches of glial progenitors undergo EMT after spinal cord injury in zebrafish and, using large-scale CRISPR-Cas9 mutagenesis, we identified the gene regulatory network that activates EMT and drives functional regeneration. Thus, non-regenerative mammalian glia lack an essential EMT-driving gene regulatory network that reprograms pro-regenerative zebrafish glia after injury.
Graphical Abstract
In Brief
Shaw et al. defined the molecular identity of regenerative zebrafish glia and performed cross-species comparisons with mammalian glia. They found that EMT localizes to zebrafish glial progenitors and distinguishes mammalian and zebrafish glia. They uncovered evidence that an EMT transcriptional module reprograms glial progenitors and promotes spinal cord repair in zebrafish.
INTRODUCTION
Traumatic spinal cord injuries (SCI) are incurable conditions that require long-term therapeutic, rehabilitative, and psychological interventions. Scarring and inherent resistance to neural regeneration present major hurdles to regenerative medicine (He and Jin, 2016; Silver, 2016; Sofroniew, 2018). In contrast to mammals, adult zebrafish possess an elevated regenerative capacity and reverse paralysis within 6–8 weeks of complete spinal cord (SC) transection (Becker et al., 1997; Goldshmit et al., 2012; Mokalled et al., 2016; Reimer et al., 2008). Zebrafish elicit efficient pro-regenerative glial cell responses in the absence of scarring (Goldshmit et al., 2012; Mokalled et al., 2016). Here, we explore regenerative processes that distinguish the zebrafish SC from mammalian SCs and enable its natural repair post injury.
Glial cell responses are thought to dictate SCI outcomes across species (Mokalled etal., 2016; O’Shea etal., 2017; Silver, 2016; Silver and Miller, 2004; Sofroniew, 2018). Following SC transection in zebrafish, specialized glial cells connect the severed SC ends and facilitate regeneration (Goldshmit et al., 2012; Mokalled et al., 2016). Conversely, mammalian astrocytes display heterogeneous and compartmentalized injury responses that are overshadowed by anti-regenerative scar-forming cells and inhibitory extracellular molecules (Dias and Göritz, 2018; Grimpe and Silver, 2004; Sofroniew, 2018). Reactive, scar-bordering astrocytes separate spared neurons from the non-neuronal lesion core in mammalian SCs. Unlike reactive astrocytes that localize to the neuronal compartment of the lesion, scar-bordering astrocytes possess properties of immature astroglia and their ablation impairs axon regeneration in mice (Anderson et al., 2016; Wanner et al., 2013). A small fraction of scar-bordering astrocytes is thought to form astroglial bridges that correlate with increased axon regrowth under genetic manipulations such as PTEN deletion in mice (Zukor et al., 2013). We envision that shifting mammalian glia toward a bridging phenotype could abate scarring and support axon regrowth. Practically, this outcome requires comprehensive cross-species comparisons and a detailed understanding of the cell fates and molecular networks that induce glial bridge formation.
Unlike CNS injuries, peripheral nerve injuries trigger pro-regenerative responses and functional repair in mammals. Glial cells are central to this regeneration process with repair Schwann cells migrating into the lesion, bridging severed nerve ends, and supporting distal innervation (Gomez-Sanchez et al., 2017; McDonald et al., 2006; Parrinello et al., 2010). Schwann cells undergo partial reprogramming from an epithelial fate to a more plastic mesenchymal fate to execute these regenerative functions (Arthur-Farraj et al., 2017; Clements et al., 2017). Glial bridging in zebrafish shares morphological and functional similarities with Schwann cell-mediated bridging after mammalian nerve injury. Yet, the extent of molecular similarities between these pro-regenerative glia remains to be determined.
Glial bridging mechanisms have remained underexplored, primarily due to the scarcity of genetic tools to label bridging glia and to manipulate bridging pathways in zebrafish. We previously showed that connective tissue growth factor a (ctgfa) is a central glial bridging factor and described the emergence of ctgfa+gfap+ bridging glia after SCI (Mokalled et al., 2016). Following injury, ctgfa expression is first broadly induced in ependymal radial glial progenitors, lining the central canal proximal tobothendsofthe lesion. During subsequent steps of regeneration, ctgfa transcripts localize to bridging glial cells and ventral ependymal progenitors. Genetic mutants in zebrafish ctgfa highlight its requirement during bridging and regeneration. Importantly, genetic and pharmacologic ctgfa overexpression are sufficient to promote bridging and functional SC repair. These findings poised us to provide a comprehensive understanding of glial bridging cell fate in zebrafish, and to perform cross-species comparisons between regenerative zebrafish glia and mammalian glia.
Our understanding and ability to manipulate cell fates boosted over the last decade. At the center of somatic cell reprogramming, epithelial-to-mesenchymal transition (EMT) emerged as a principal molecular mechanism that confers stem-like properties and drives cell fate transitions after injury (Jessen and Arthur-Farraj, 2019; Wilson et al., 2020; Ye and Weinberg, 2015), in addition to its established roles during embryogenesis and tumorigenesis. Twist, Zeb, and Snai transcription factors control EMT by downregulating cell-cell adhesion molecules, instigating loss of cell polarity while elevating cellular plasticity (Dongre and Weinberg, 2019). The transcriptome of zebrafish bridging glia suggested that ctgfa+ ependymal progenitors undergo EMT-dependent reprogramming into a proliferative, migratory, and mesenchymal cell fate (Mendez et al., 2010; Scarpa et al., 2015). Using CRISPR-Cas9 mutagenesis, here, we identify a gene regulatory network that activates twist-mediated EMT to promote glial bridging and show that an inducible twist expression enhances glial bridging and functional SC repair.
RESULTS
Molecular profiling of ctgfa+gfap+ cells after spinal cord injury
We devised a fluorescence-activated cell sorting and RNA deep sequencing (FACS-seq) strategy to profile the transcriptome of bridging glial cells using ctgfa and gfap (Figure 1). ctgfa expression demarcates bridging glia at the lesion core and their predicted progenitors in the ventral ependyma proximal to the lesion (Mokalled et al., 2016). To label bridging glia, we generated transgenic ctgfa:mCherry reporter lines in combination with the previously established gfap:EGFP transgene (Bernardos and Raymond, 2006) (Figure 1A). ctgfa:mCherry was broadly expressed in developing zebrafish animals, co-localizing with gfap:EGFP in the ventral floor plate at 5 days post fertilization (dpf) (Figures S1A–S1C). Consistent with its injury-induced expression, ctgfa:mCherry was not detectable in unlesioned adult SCs but was upregulated within the SC tissue after injury (Figure S1D). Longitudinal SC sections from adult ctgfa:mCherry;gfap:EGFP animals confirmed mCherry and EGFP expression in bridging glia as early as 5 days post injury (dpi) (Figure 1B). mCherry was also broadly induced in gfap+ ependymal progenitors at 1 week post injury (wpi) (Figure S1D) and localized to ventral ependymal cells at 10 dpi (Figure 1B). These expression patterns recapitulated the expression of endogenous ctgfa transcripts and of previously established ctgfa:EGFP reporters after SCI (Mokalled et al., 2016). We thus used the ctgfa/gfap dual reporter line to isolate ctgfa+gfap+ glial cells.
Figure 1. Molecular profiling of bridging glial cells in zebrafish.
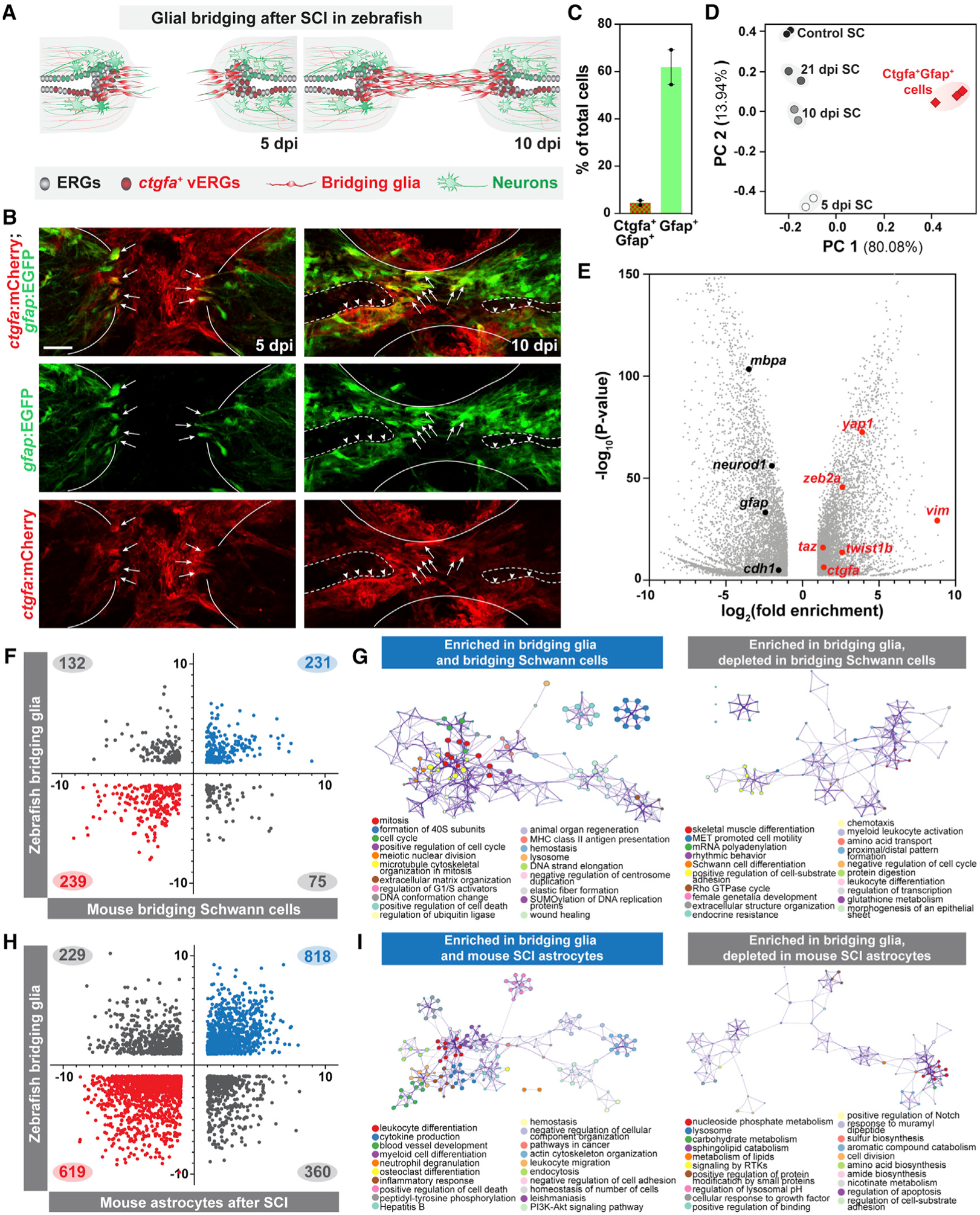
(A) Schematic representation of zebrafish SCs at 5 and 10 dpi. Ependymal radial glial (ERG) progenitors proliferate rostral and caudal to the lesion and the central canal expands proximal to the lesion. ctgfa is expressed in a subset of ERGs at 5 dpi (red) and concentrates to ventral ERGs (vERGs) at 10 dpi. Early bridging glia emerge at 5 dpi and a glial bridge is formed by 10 dpi.
(B) mCherry and EGFP in ctgfa:mCherry;gfap:EGFP dual reporter line. Shown are longitudinal SC sections from adult animals at 5 and 10 dpi. For each time point, 10–12 SC sections were analyzed. Lines delineate the outer edges of the SC. Dashed lines outline the central canal. Arrows and arrowheads point to mCherry+EGFP+ bridging glia and vERGs, respectively.
(C and D) Capture and deep RNA-seq of bridging glia. ctgfa+gfap+ cells were sorted at 5 dpi. Bulk SC tissues from 5, 10, and 21 dpi, as well as uninjured control SCs were deep sequenced. mCherry+EGFP+ and EGFP+ cells comprised 3% and 60% of total dissociated cells, respectively (C). PCA scatter plot of gene expression shows the variances between biological replicates (D). x and y axes represent the percentages of variation explained by the principal components.
(E) Volcano plot representation of genes that are significantly enriched or depleted in sorted ctgfa+gfap+ cells at 5 dpi relative to non-sorted uninjured SC controls.
(F–I) Glial cell comparisons between zebrafish ctgfa+gfap+ cells and mammalian glial cells. The transcriptomes of ctgfa+gfap+ cells from zebrafish were cross-referenced with gene expression changes in bridging Schwann cells after nerve injury in mice (F), and in mouse astrocytes after SCI (H). Log2(fold enrichment) for zebrafish and mouse glia are shown on the y and x axes, respectively. Genes that are significantly changed in zebrafish and mice are shown. Genes that are enriched or depleted in both zebrafish and mice are shown in blue and red, respectively. Genes that are differentially changed between zebrafish and mice are shown in gray. Gene ontology analysis was performed on bridging glia enriched genes, subdivided based on their regulation in mouse bridging glia (G) or mouse SCI astrocytes (I). Scale bars, 50 μm.
To define the molecular signature of pro-regenerative bridging glia, we performed FACS-seq on ctgfa+gfap+ cells at 5 dpi and RNA-seq on bulk SC tissue at 5, 10, and 21 dpi (Figures 1C–1E). Complete SC transection was performed on ctgfa/gfap dual reporter animals. At 5 dpi, 2-mm SC tissue samples including the lesion site were harvested for sorting. Single reporter and wild-type animals were used as controls. We were able to collect mCherry+EGFP+ cells from animals with transected SCs but not from sham-injured animals, confirming the injury-induced nature of ctgfa and the emergence of ctgfa+gfap+ cells after injury. mCherry+EGFP+ cells comprised 3% of the cells dissociated at 5 dpi, while 60% of the cells were EGFP+ (Figure 1C). RNA samples from isolated ctgfa+gfap+ cells and bulk SC tissue at 5 dpi were deep sequenced. SC tissue samples from 10 and 21 dpi as well as uninjured control samples were sequenced (Figures 1D and S1E). Principal component and heatmap analyses revealed clustering of biological replicates, and highlighted the extent of molecular regeneration between 5, 10, and 21 dpi relative to control samples.
We performed differential gene expression analysis to identify genes that were enriched or depleted in bridging glia. Our FACS-seq approach enriched for ctgfa expression (Figure 1E). ctgfa is expressed in multiple cell types around the lesioned SC; yet, its expression is primarily confined to bridging glia and ventral ependymal progenitors within the dissected SC tissue. We thus observed an elevated ctgfa expression in ctgfa+gfap+ cells relative to the bulk SC tissue. Conversely, gfap is expressed in 60% of the total cells dissociated from SC tissue, but ctgfa+gfap+ cells comprise only a minor subset of gfap-expressing cells (Figure 1C). Consequently, gfap expression was attenuated in isolated ctgfa+gfap+ cells relative to that in the bulk SC tissue. Neuronal and oligodendrocytic markers, such as neurod1 and mbpa were depleted by FACS-seq, suggesting that isolated ctgfa+gfap+ cells were devoid of neurons and oligodendrocytes. We noted that EMT genes such as vimentin (vim), twist1, and zeb2 were enriched in ctgfa+gfap+ cells. These studies documented the first transcriptome for pro-regenerative bridging glia in zebrafish.
Molecular comparisons between zebrafish bridging glia and mammalian glia
We first investigated whether zebrafish bridging glia harbor molecular similarities with bridging Schwann cells in mammals. To test this hypothesis, we compared the transcriptomes of zebrafish bridging glia and murine repair Schwann cells (Clements et al., 2017) (Figures 1F and 1G; Table S1). Among genes that were enriched in bridging glia, 231 genes were also enriched in bridging Schwann cells, while 132 genes were depleted (Figure 1F). Similarities between zebrafish bridging glia and murine bridging Schwann cells included genes related to cell proliferation, wound healing, and regeneration (Figure 1G). These genes included the glial bridging factor ctgf (ccn2) and EMT-related genes, including snai2, tgfb1, and cdh1 (Table S1). Notably, Schwann cell identity genes (mbpa and sox10) were depleted in zebrafish, while bridging glia enriched transcription factors (egr1 and nr1d1) were depleted in murine bridging Schwann cells. These studies indicated that zebrafish bridging glia and bridging Schwann cells are distinct cell fates despite their morphological and functional similarities and suggested that mesenchymal gene expression correlates with regenerative bridging in both cell types.
We next compared the transcriptional profiles of zebrafish bridging glia to murine scar-bordering astrocytes after SCI (Anderson et al., 2016) (Figures 1H and 1I; Table S2). Based on the number of genes regulated in either zebrafish bridging glia or mouse astrocyte datasets alone, we expected about 3%–3.25% transcriptional similarities by chance. We found that 1,047 bridging glia enriched genes were significantly changed in mouse astrocytes. 818 of these genes followed similar trends in mice, accounting for 12% of the transcriptional similarities between bridging glia and astrocytes (Figure 1H). On the other hand, 979 bridging glia depleted genes were significantly changed in mouse astrocytes. 619 of these genes were also depleted in mouse astrocytes, accounting for 9% of the transcriptional similarities between bridging glia and astrocytes. Immune-related genes and transcription factors, including stat3, were enriched in both cell types (Herrmann et al., 2008; Okada et al., 2006), whereas genes related to extracellular structure organization, supramolecular fiber organization, and N-Glycan elongation were missing in zebrafish bridging glia (Figure 1I; Table S2). We also noted that bridging glia enriched for s100a10 expression, a marker of ischemic A2 astrocytes that are thought to promote neuronal survival and repair (Liddelow et al., 2017). Thus, despite their seemingly different injury responses, zebrafish bridging glia and mouse astrocytes possess vast transcriptional similarities that include astrocytic markers and transcription factors. Together, these genome-wide, cross-species comparisons highlighted molecular similarities and differences between zebrafish bridging glia and mammalian glial cells and provided molecular insights into the pro-regenerative cell fates that enable zebrafish SC repair.
Ventral ependymal progenitors undergo EMT during glial bridging
The transcriptome of zebrafish bridging glia revealed features of a mesenchymal transition. Hallmarks of EMT include concomitant downregulation of epithelial E-Cadherin (cdh1) and upregulation of N-cadherin (cdh2), driven by twist, zeb, and snai transcription factors. We comprehensively assessed the regulation of 120 epithelial and mesenchymal markers during bridging by FACS-seq and quantitative RT-PCR (qRT-PCR) (Figures 2A–2C, S2A, and S2B) (Chang et al., 2016). By averaging fold enrichment for each set of markers, we found that epithelial genes were downregulated (log2 enrichment of −0.2) and mesenchymal genes were enriched (log2 enrichment of 0.8) in isolated ctgfa+gfap+ cells (Figure 2B). We also grouped EMT genes based on their enrichment or depletion in isolated ctgfa+gfap+ cells (Figure 2C). 57% of epithelial markers were downregulated by FACS-seq, with 32% showing log2 enrichment <−1. On the other hand, 68% and 46% of mesenchymal markers were significantly enriched and had log2 enrichment >1, respectively. These findings suggested that a mesenchymal program is activated during glial bridging.
Figure 2. Ventral ependymal progenitors undergo EMT after SCI.

(A) Expression of EMT-related genes by FACS-seq. Heatmap representation of epithelial and mesenchymal gene expression in ctgfa+gfap+ cells. Color scale in the dendrogram represents log2(fold enrichment) in ctgfa+gfap+ cells relative to control SCs.
(B) Enrichment of EMT markers by FACS-seq. Log2(fold enrichment) was averaged for epithelial and mesenchymal genes.
(C) Distribution of EMT markers by FACS-seq. Upregulated genes were subdivided into log2(FE) >1 (Green) and log2(FE) = 0 to 1 (Light green). Downregulated genes were subdivided into log2(FE) = −1 to 0 (light red) and log2(FE) < −1 (red). Percent genes within each category are shown.
(D) RNAscope for twist1a in wild-type SCs at 1 and 2 wpi, and in uninjured controls. Hematoxylin staining (blue) was used as a counterstain. At 1 wpi, SC cross sections at the lesion site and distal to the lesion from the rostral side are shown. Arrowheads point to twist1a expression at the lesion core at 1 wpi. Arrows point to twist1a expression in ventral ependymal progenitors at 1 and 2 wpi.
(E–H) Cdh2 (E) and Cdh1 (G) immunostaining in ctgfa:EGFP zebrafish at 1 and 2 wpi, and in uninjured controls. SC cross sections are shown, and dotted lines delineate central canal edges. Arrowheads point to domains of co-expression of Cdh2 and EGFP (E), and domains of diminished Cdh1 expression and increased EGFP expression (G). Quantification of Cdh2 (F) and Cdh1 (H) expression in ctgfa- and ctgfa+ cells at 2 wpi are shown. ****p < 0.0001. Scale bars, 50 μm.
To visualize the spatial and temporal activation of EMT during SC regeneration, we characterized the expression of core EMT regulators at defined time points after SCI. twist1a transcripts were not detectable in the uninjured SC tissue (Figure 2D). At 1 wpi, we detected twist1a transcripts in the lesion core. twist1a expression localized to ventral ependymal progenitors at 1 and 2 wpi (Figures 2D, S2C, and S2D). In contrast with twist1a, twist1b expression did not show specific enrichment in ependymal cells after SCI. These results indicated that twist1a and ctgfa have overlapping gene expression patterns after SCI and suggested that twist1a directs localized mesenchymal transition in ctgfa+ ependymal progenitors during glial bridging. To test this hypothesis, we co-labeled mesenchymal Cdh2 or epithelial Cdh1 with ctgfa:EGFP. By immunohistochemistry, Cdh1 and Cdh2 showed dynamic and localized gene expression changes after SCI (Figures 2E–2H). Cdh2 was broadly upregulated at 1 and 2 wpi relative to uninjured controls and was more elevated in ctgfa+ relative to ctgfa− cells at 2 wpi (Figures 2E and 2F). Similarly, the mesenchymal marker Vimentin was globally upregulated at 1 wpi, showed preferential expression in ventral ependymal progenitors at 2 wpi, and localized to the progenitor motor domain by 3 wpi (Figure S2E). On the other hand, Cdh1 expression was increased in lateral ependymal cells after SCI but was preferentially downregulated in ctgfa+ cells at 1 and 2 wpi (Figures 2G and 2H). These results demonstrated that EMT is activated in ventral ependymal progenitors after SCI and suggested that twist1 reprograms ctgfa+ ependymal progenitors into a more mesenchymal cell fate during glial bridging.
Single-cell profiling of bridging glial cells after SCI
Our FACS-seq approach provided bulk transcriptional profiles for ctgfa+gfap+ ependymal progenitors and bridging glia. To uncover the extent of heterogeneity in these signatures, we performed single nuclear RNA-sequencing (snRNA-seq) on SC tissues from gfap:EGFP fish at 1 wpi using the 10x Genomics platform. This time point spans the initiation of glial bridges after SCI. 2-mm SC tissue sections spanning the lesion site were collected, and nuclei were isolated from 45 pooled SC tissues to account for individual variation (Matson et al., 2018). 33,185 SC nuclei were profiled and filtered by quality control analysis (Figure S3A). Unsupervised clustering of SC nuclei revealed major clusters of glial cells, neurons, oligodendrocytes, oligodendrocyte progenitor cells, and microglia/macrophages (Figure 3A) (Becht et al., 2018; Butler et al., 2018; Stuart et al., 2019; Zhang et al., 2014). Clusters representative of glial cells were further grouped into 10 glial clusters (Glial-0 to Glial-9) (Figure 3B). Initial differential gene expression highlighted Glial-7 and Glial-8 as putative clusters corresponding to bridging glia. ctgfa and twist1a were co-expressed in cluster Glial-8, while ctgfa and twist1b were co-expressed in cluster Glial-7 (Figure 3C). We noted that Glial-7 and Glial-9 co-clustered at lower and medium resolutions, suggesting that they share similar gene expression signatures. In fact, Glial-7 and Glial-9 differed mainly by their proliferative states, with Glial-9 expressing mki67 in addition to other proliferation markers (Figures 3C and S3C). To identify clusters of bridging glial cells independent of ctgf and twist expression, we performed genome-wide comparisons between glial cell clusters identified by snRNA-seq and bulk transcriptional profiles generated by FACS-seq. Markers of clusters Glial-7 and Glial-9 were enriched by FACS-seq with an average log2 enrichmentof 0.47 and 1.26, respectively (Figure 3D). When grouped based on their enrichment or depletion in isolated ctgfa+gfap+ cells, 62.5% and 80.95% of the genes that marked Glial-7 and Glial-9 clusters were upregulated by FACS-seq (Figure 3E). These results suggested that glial clusters 7, 8, and 9 encompass bridging glial cells.
Figure 3. Single cell profiling of glial cells during SC regeneration.
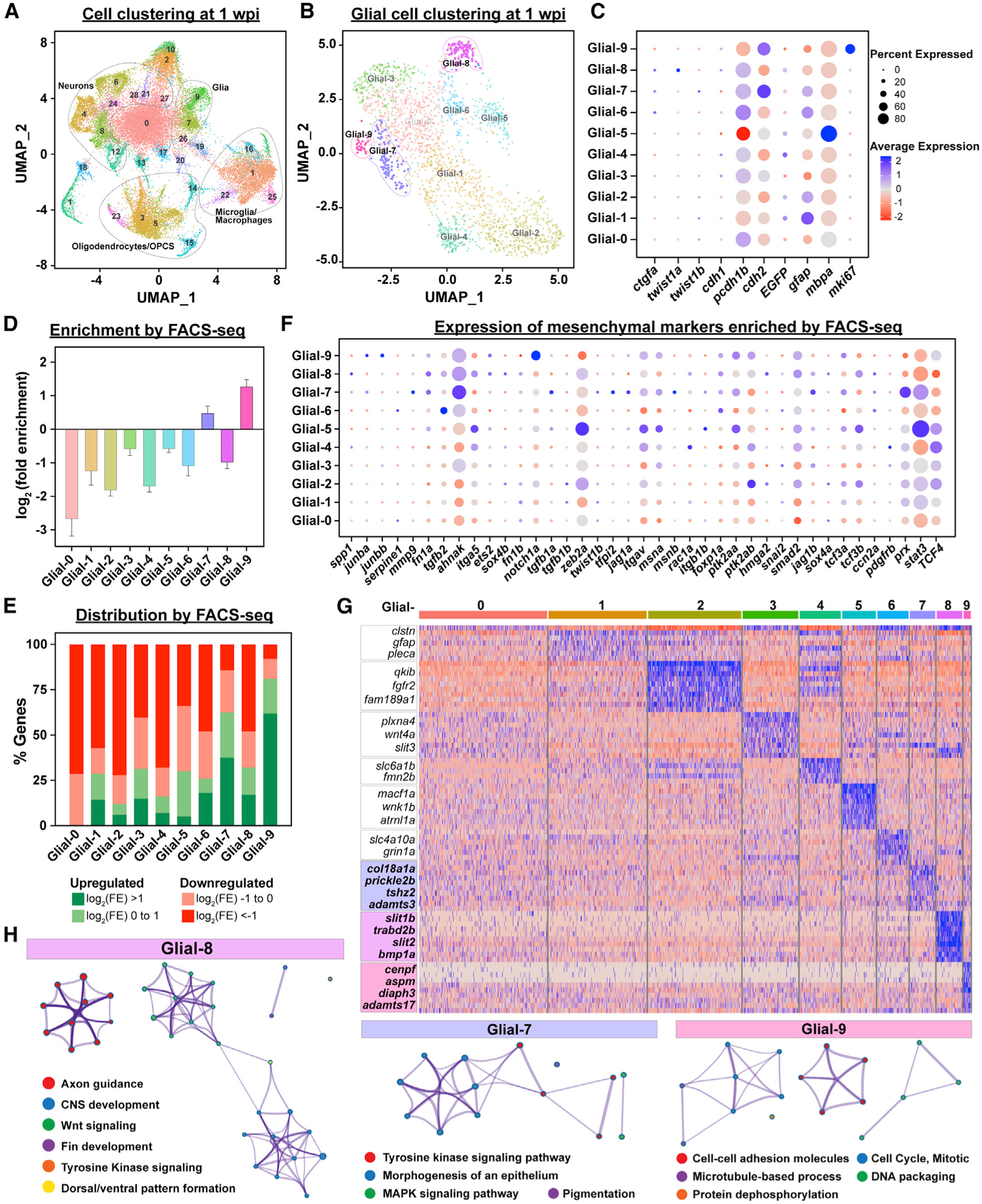
(A) UMAP of snRNA-seq data from SC tissues at 1 wpi.
(B) Sub-clustering and UMAP visualization of glial cell clusters at 1 wpi. Ten clusters of glial cells were identified and termed as Glial-0 to -9.
(C) Expression of canonical markers in each glial cluster. Dot colors represent average expression levels from low (red) to high (blue). Dot sizes represent the percent of positive cells for each gene.
(D) Enrichment of snRNA-seq-derived glial cell markers by FACS-seq. Glial cell markers were identified for each cluster by snRNA-seq analysis. Log2(fold enrichment) in sorted bridging glia relative to uninjured SCs were then then averaged for each cluster.
(E) Distribution of snRNA-seq-derived glial cell markers by FACS-seq. Markers of clusters Glial-0 to -9 were categorized based on their enrichment in ctgfa+gfap+ cells. Upregulated genes were subdivided into log2(FE) > 1 (Green) and log2(FE) = 0 to 1 (Light green). Downregulated genes were subdivided into log2(FE) = −1 to 0 (light red) and log2(FE) < −1 (red). Percent genes within each category are shown.
(F) Mesenchymal gene expression in glial clusters. A total of 39 mesenchymal markers that are enriched by FACS-seq are shown. Dot colors represent average expression levels from low (red) to high (blue). Dot sizes represent the percent of positive cells for each gene.
(G) Heatmap of genes enriched in clusters Glial-0 to -9. Blue indicates enriched genes and red indicates depleted genes.
(H) Gene ontology for markers of clusters Glial-7, -8, and -9.
FACS-seq analysis of isolated ctgfa+gfap+ cells revealed features of EMT during glial bridging. To further evaluate the role of EMT during bridging, we surveyed the glial cell clusters identified by snRNA-seq for mesenchymal marker expression. Focusing the analysis on markers that were enriched by FACS-seq (Figures 2A–2C), 53.8%, 46.2%, and 35.9% of mesenchymal genes were enriched in clusters Glial-7, Glial-8, and Glial-9 (Figures 3F and S3D). These results suggested clusters Glial-7, -8, and -9 have undergone a mesenchymal transition by 1 wpi. We next used differential gene expression to identify the top 10 markers for these glial clusters (Figure 3G). Concomitant with a pro-regenerative function for bridging glial cells, gene ontology classification suggested an axon guidance-related role for Glial-8 clustered cells (Figures 3H and S3B; Table S3). This analysis also indicated that cell adhesion and cell signaling pathways were active in Glial-7, -8, and -9. Our findings are consistent with the emergence of 3 subpopulations of bridging glia, suggesting that sub-clusters of ctgfa+twist1a+ ependymal progenitors, ctgfa+− twist1b+ glial cells, and a subset of proliferative glial cells adopt a mesenchymal fate during glial bridging.
An EMT-driving gene regulatory network during glial bridging
To identify transcriptional regulators necessary for glial bridging, we tested the roles of transcription factors that are enriched in bridging glia. A total of 9 transcription factors were chosen for mutagenesis (bach1a, bach1b, egr1, foxg1c, junba, junbb, nr1d1, spi1a, spi1b, taz, and yap1) (Figures S3E–S3G). cntf was selected as a positive control (Qin et al., 2013; Wang et al., 2014; Ye et al., 2004). For efficient and simultaneous gene targeting in adult zebrafish, we adapted a modified CRISPR-Cas9 protocol that achieved near-complete mutagenesis in F0 injected embryos (Hoshijima et al., 2019). CRISPR-Cas9-targeted animals were raised to adulthood, subjected to SCI, and swam against increasing water current inside an enclosed swim tunnel to screen for functional regeneration defects (Figure 4A) (Mokalled et al., 2016).
Figure 4. An EMT-driving gene regulatory network directs glial bridging.

(A) Pipeline for an in vivo CRISPR-Cas9 screen for glial bridging transcription factors.
(B) Functional recovery in CRISPR-Cas9-targeted animals 4 wpi. For each group of targeted animals, uninjected siblings were subjected to SCI and swim assays. Dots represent individual animals. Groups with significantly diminished swim function are shown in red.
(C and D) Glial bridging in CRISPR-Cas9-targeted animals. Gfap immunohistochemistry was performed at 4 wpi (C). Representative micrographs show Gfap+ bridges at the lesion site in bach1a;bach1b, egr1, junbb, and spi1a-targeted animals. Percent bridging was quantified for 10–14 animals per group (D).
(E and F) twist1a expression in CRISPR-Cas9-targeted animals. RNAscope was performed on SC tissues at 4 wpi (E). Dotted lines delineate central canal edges. Arrows point to ventral ependymal progenitors in distal SC sections. Normalized twist1a was quantified from 8–14 sections per group (F). ***p < 0.001; **p < 0.01; *p < 0.05. Scale bars, 50 μm.
We first assessed the validity of our CRISPR-Cas9 methods in the targeted adult animals. To maximize the effect of small indels on mutant gene function, CRISPR-Cas9 ribonucleoproteins (RNPs) were designed against genomic regions that encode their DNA-binding domains. Some genes were targeted using 2 sets of RNPs to maximize targeting efficiency. In addition to targeting individual genes, we combinatorially targeted transcription factor paralogs to account for putatively redundant effects on bridging. At 2 dpf, the rates of mutagenesis averaged to 83.6% per target site and 93.7% per gene (Figures S4A, S4C, and S4D). We then assessed the possibility and extent of negative selection that could impact mutagenesis rates in adult animals (Figure S4B). By capillary electrophoresis, the rates of mutagenesis averaged to 91.7% for bach1a, bach1b, foxg1c, junba, junbb, nr1d1, spi1a, taz, and yap1-targeted adults. However, 22% to 54% of cntf-, egr1-, and spi1b-injected adults showed lower mutagenesis efficiencies relative to their larval siblings. For these 3 genes, we selected 18 animals with the highest mutagenesis rates for SCI and regeneration assessment. The average mutagenesis rate for experimental animals hereafter was 92.5%. These results confirmed efficient mutagenesis in CRISPR-Cas9-injected animals and poised us to screen for adult regeneration phenotypes.
To evaluate the outcomes of these mutations on SC regeneration, we assessed functional recovery in CRISPR-Cas9-injected animals. At 4 wpi, swim function was diminished in bach1a;bach1b, cntf, egr1, junbb, junba;junbb, spi1a, and taz/yap1-targeted animals relative to their respective uninjected siblings (Figure 4B). While bach1 paralogs and taz/yap were functionally redundant during SC repair, analysis of single mutants showed that mutations in junbb impaired functional regeneration. To confirm the regenerative phenotypes obtained in CRISPR-Cas9-injected animals, we generated stable mutant lines for egr1 (egr1stl667; Figure S4E) and junbb (junbbstl671 and junbbstl672; Figure S4F). Recapitulating egr1 and junbb crispant phenotypes, swim function was markedly compromised in stable egr1 and junbb mutants relative to their respective wild-type siblings at 4 wpi (Figure S4G). In the absence of injury, swim function was comparable between targeted bach1a/b, spi1a, taz/yap1, stable junbb mutants, and their respective controls under uninjured conditions (Figures S4G and S4H), suggesting that our CRISPR approach and phenotyping assays identified regeneration-specific phenotypes. In these assays, egr1 mutants showed a mild but significant decrease in swim function, swimming for ~40 min under increasing current velocity compared with a 55-min swim time in wild-type siblings. egr1 mutants completely failed to swim after SCI (Figure S4G). The cross-sectional area of glial bridges was significantly reduced in bach1a;bach1b, egr1, junbb, and spi1a-targeted animals at 4 wpi (Figures 4C and 4D), consistent with a glial bridging role for these transcription factors. These studies identified transcription factors that are required for glial bridging and functional SC repair.
bach1, egr1, and junb have been shown to regulate EMT and Ctgf signaling during development and/or tumorigenesis (Gervasi et al., 2012; Han et al., 2019; Sato et al., 2020; Wu et al., 2017). We thus tested whether similar regulatory mechanisms are deployed to promote tissue regeneration. By RNAscope and qRT-PCR, twist1a expression was significantly attenuated in egr1 and junbb-targeted animals (Figures 4E, 4F, and S4I). We noted that targeting spi1 and the bach1 transcriptional repressor resulted in respective decrease and increase in twist1 expression that approached significance (Figures 4F and S4I). These studies revealed a functionally validated gene regulatory network that directs EMT and glial bridging during SC regeneration.
Localized Yap activation in ependymal progenitors during glial bridging
Hippo signaling plays important roles in tissue regeneration in multiple tissue contexts. Yes-associated protein (Yap) and transcriptional co-activator with PDZ-binding motif (Taz) are downstream co-activators of Hippo signaling, regulating cell plasticity and organ growth during development and regeneration. Yap and Taz lack DNA-binding domains and control transcription by associating with the Tead DNA-binding transcription factors to promote cell proliferation and stem cell maintenance (Vassilev et al., 2001; Zhao et al., 2008). yap and taz were upregulated in injured SC tissue (Figure S2A). However, while taz was 2-fold enriched in isolated ctgfa+gfap+ cells, yap and the tead transcription factors were only mildly enriched by FACS-seq (Figure S2B), reflecting their ubiquitous expression across multiple SC cell types. For comprehensive analysis of Yap signaling, we examined the expression of previously described canonical and proliferative Yap target genes during glial bridging (Figures 5A–5C) (Zanconato et al., 2015). On an average, Yap targets were enriched in sorted ctgfa+gfap+ cells, with log2 enrichment of 0.4 for canonical targets and 1.3 for proliferative targets (Figure 5B). When grouped based on their enrichment or depletion by FACS-seq, 54% of canonical targets and 83% of proliferative targets were significantly enriched in isolated ctgfa+gfap+ cells (Figure 5C), suggesting that Yap signaling is activated after SCI.
Figure 5. Localized Yap activation promotes glial bridging and SC regeneration.

(A) Expression of Yap target genes in ctgfa+gfap+ cells. Genes were divided into canonical and proliferative genes. Color scale in the dendrogram represents log2(fold enrichment) by FACS-seq.
(B) Enrichment of Yap target genes by FACS-seq. Log2(fold enrichment) was averaged for canonical and proliferative targets.
(C) Distribution of Yap target genes by FACS-seq. Canonical and proliferative genes were categorized based on their enrichment in Ctgfa+Gfap+ cells. Upregulated genes were subdivided into log2(FE) > 1 (green) and log2(FE) = 0 to 1 (light green). Downregulated genes were subdivided into log2(FE) = −1 to 0 (light red) and log2(FE) < −1 (red). Percent genes within each category are shown.
(D) Yap expression in wild-type zebrafish at 2 wpi and uninjured controls. Lateral and ventral ependymal progenitors are shown in separate panels at 2 wpi. Central canals are constricted in uninjured SCs and expanded proximal to the lesion site after SCI. Arrowheads point to nuclei with attenuated Yap expression in lateral ERGs. Arrows point to nuclei with elevated Yap expression in ventral ERGs.
(E) Quantification of Yap expression and localization after SCI. Nuclear and cytoplasmic Yap were quantified in ventral and lateral ependymal domains at 2 wpi. Nuclear-to-cytoplasmic ratios were averaged for 2 sections per animal and 5 animals were used.
(F) Swim assays determined motor function recovery of hsp70:dsRed-dnYap (Tg+, SCI, red) and wild-type (Tg−, SCI, black) siblings at 2, 4, and 6 wpi. For controls, dnYap overexpressing (Tg+, uninjured, dashed red) and wild-type (Tg−, uninjured, dashed black) animals were analyzed. All groups were subjected to daily heat shocks. Statistical analyses of swim times are shown for injured dnYap relative to injured wild-type siblings.
(G) Anterograde axon tracing in dnYap-expressing zebrafish at 4 wpi. Biocytin axon tracer was applied rostrally and analyzed at 100 μm (proximal) and 500 μm (distal) caudal to the lesion. Representative traces of biocytin are shown for Tg+ and Tg− animals at the proximal level.
(H and I) Glial bridging in dnYap-expressing zebrafish. Gfap immunohistochemistry was performed at 10 dpi (H). Representative micrographs show Gfap+ bridges at the lesion site relative to the intact SC in dnYap-expressing (Tg+) and control siblings (Tg−). Percent bridging was quantified for 13 Tg− and 10 Tg+ animals (I) **p < 0.01; *p < 0.05. Scale bars, 10 μm (D) and 50 μm (G and H).
Yap and Taz require nuclear localization to exert their transcriptional regulatory functions. To identify domains of Yap/Taz activation after SCI, we performed immunohistochemistry for Yap and Taz on wild-type SC sections at 1, 2, and 3 wpi, as well as on uninjured controls. Consistent with transcriptomic data, Yap was broadly expressed in control and lesioned SCs between 1 and 3 wpi (Figure S5A). Yap sub-cellular localization revealed dynamic, domain-specific changes during regeneration. At 1 wpi, nuclear Yap was broadly expressed around the ependyma, suggesting global activation early after SCI (Figure S5A). At 2 wpi, Yap showed preferential nuclear localization in ventral ependymal progenitors relative to the lateral ependyma (Figures 5D and S5A). At 3 wpi, Yap was broadly downregulated but maintained its localization to ventral ependymal nuclei (Figure S5A). Quantification of nuclear and cytoplasmic Yap in ventral and lateral ependymal cells confirmed its nuclear enrichment (1.4-fold nuclear-to-cytoplasmic expression) in ventral progenitors at 2 wpi (Figure 5E). Yap was uniformly distributed in the nuclear and cytoplasmic compartments of lateral progenitors at the same time point (1-fold nuclear-to-cytoplasmic expression) (Figure 5E). On the other hand, we observed a uniformly elevated Taz expression at 1 and 2 wpi (Figure S5B). These experiments indicated that Yap and Taz were upregulated after SCI, and that Yap was preferentially activated in ventral ependymal progenitors during glial bridging.
Yap signaling is required for glial bridging and functional SC repair
Yap and Taz are functionally redundant in zebrafish, and dual Taz/Yap mutations impair embryonic body elongation during the mid-late stages of somitogenesis (Kimelman et al., 2017; Miesfeld et al., 2015). Using CRISPR-Cas9 mutagenesis, the taz/yap1-targeted animals that survived to adulthood showed SC regeneration defects (Figure 4B). To further understand the role of Yap signaling during SC regeneration, we expressed dominant negative Yap under the control of a heat-inducible promoter (hsp70:dsRed-dnYap) and assessed the outcomes of Yap inactivation on functional and anatomical SC repair (Mateus et al., 2015). hsp70:dsRed-dnYap zebrafish and wild-type siblings were subjected to SC transections followed by daily heat shocks to induce transgene expression. Swim assays revealed impaired functional recovery in dnYap-expressing fish at 4 and 6 wpi (Figure 5F). Swim capacity was comparable between uninjured dnYap-expressing and wild-type siblings after 6 weeks of daily heat shocks, indicating that dnYap expression was specifically detrimental in injury (Figure 5F). At 4 wpi, dnYap-expressing animals showed reduced axon regeneration by anterograde axon tracing (Figure 5G) and less glial bridging relative to wild-type siblings (Figure S5C). To assess the role of Yap signaling during glial bridge initiation, we examined glial bridging upon dnYap expression at 10 dpi (Figures 5H and 5I). At this time point, Gfap immunostaining revealed the formation of organized, circular bridges at the lesion core in control SC sections. Glial bridges were reduced by 50% and were irregularly shaped upon dnYap expression. These results revealed that Yap signaling promotes glial bridging and functional SC repair.
Yap regulates twist1-mediated EMT and ctgf-dependent bridging
The dynamics of Yap activation paralleled patterns of ctgfa and twist1a expression during glial bridging (Figures 2D and 5D) (Mokalled et al., 2016). To examine the correlation between Yap activation and Ctgfa expression, we injured ctgfa:EGFP transgenic zebrafish and performed time course analysis for Yap and EGFP (Figure S5D). At 1 wpi, nuclear Yap localized with ctgfa:EGFP proximal to the lesion. By 2 wpi, EGFP and nuclear Yap localized to ventral ependymal progenitors. At this time point, 38% of ctgfa:EGFP cells expressed nuclear Yap, while only 6% of EGFP− cells showed nuclear Yap expression (Figure S5E). To examine ctgfa expression upon Yap inactivation, we combined dnYap-expressing and ctgfa:EGFP transgenes. hsp70:dsRed-dnYap;ctgfa:EGFP (Tg+) fish and ctgfa:EGFP (Tg−) controls were subjected to SC transections and daily heat shocks to induce dnYap expression (Figure 6A). EGFP was expressed in ventral ependymal progenitors of ctgfa:EGFP controls at 10 dpi but was attenuated in dnYap-expressing animals (Figures 6B and 6C). We next assessed twist1a expression in dnYap-expressing SCs by RNAscope (Figures 6D and 6E). At 10 dpi, twist1a transcripts were detectable in the lesion cores and ventral ependymal progenitors of control SCs. Upon dnYap expression, twist1a was diminished in the lesion core and mislocalized to the outer edges of the lesion. Distal to the lesion, twist1a was reduced by 60% in ventral ependymal cells of dnYap-expressing animals (Figure 6E). These findings demonstrated that Yap promotes ctgfa and twist1a expression in ventral ependymal progenitors. ctgfa and twist1a are essential drivers of glial bridging and EMT (Dongre and Weinberg, 2019; Mokalled et al., 2016) (Figures S5F and S5G), indicating that Yap is required for ctgfa-dependent bridging and twist1a-driven EMT after SCI.
Figure 6. Yap signaling directs twist1a-driven EMT and ctgfa-dependent glial bridging.

(A–C) ctgfa reporter expression in ctgfa:EGFP;hsp70:dsRed-dnYap (Tg+) SCs. ctgfa:EGFP (Tg−) siblings were used as controls. All animals were subjected to SCI and daily heat shocks (+HS) (A). EGFP expression was assessed at 10 dpi (B). Dotted lines delineate central canal edges. For quantification, the area of EGFP fluorescence was calculated for 2 sections per animal (C).
(D and E) twist1a expression in hsp70:dsRed-dnYap and control wild-type siblings. All animals were subjected to daily heat shocks (+HS). RNAscope was performed at 10 dpi (D). Arrowheads point to the lesion core; arrows point to ventral ependymal progenitors in distal SC sections. For quantification, normalized twist1a area was calculated for 2 sections per animal (E).
(F and G) Swim assays determined motor function recovery of hsp70:Twist1a-2A-EGFP (Tg+, red) and wild-type (Tg−, black) siblings at 2, 4, and 6 wpi. Both groups were subjected to SCI and daily heat shocks (F). Average swim activities are shown for Twist1a-expressing (Tg+, red) and control animals (Tg−, teal) at 6 wpi in the absence of water current and at a water current velocity of 20 cm/s (G).
(H and I) Glial bridging in Twist1a-expressing zebrafish at 10 dpi. Representative immunohistochemistry shows the Gfap+ bridge at the lesion site relative to the intact SC in Twist1a-expressing (Tg+) and control siblings (Tg−) (H). Percent bridging was calculated for 10 Tg− and 9 Tg+ animals (I).
(J and K) Cell proliferation in Twist1a-expressing and control siblings. PCNA staining was performed on Tg− and Tg+ animals at 10 dpi (J). Percent PCNA+ cells was quantified for 10 Tg− and 9 Tg+ animals at the lesion, in whole SC tissues, and around the ependyma (K). Percent
(L) Schematic Model shows injury-induced EMT regulates ependymal cell reprogramming into bridging glia. Transcriptional modulators Egr1, Junbb, Yap, and Taz direct Ctgfa and Twist1a expression, which then induce the expression of mesenchymal genes, including Cdh2 and Vim, while epithelial markers such as Cdh1 are reduced. ***p < 0.001; **p < 0.01; *p < 0.05; ns, not significant. Scale bars, 50 μm.
Twist1a expression promotes glial bridging and functional SC repair
To examine the regenerative effects of EMT activation, we expressed the EMT-driving Twist1a transcription factor under the control of a heat-inducible promoter (hsp70:Twist1a-2A-EGFP) and assessed the outcomes of Twist1a expression on functional and anatomical SC repair. hsp70:Twist1a-2A-EGFP zebrafish and wild-type siblings were subjected to SC transections followed by daily heat shocks to induce transgene expression (Figure 6F). Swim assays revealed improved functional recovery in Twist1a-expressing fish at 4 and 6 wpi (Figure 6G). Functional recovery was even more pronounced in Twist1-expressing animals by tracking the swim activity under a steady current of 20 cm/s (Figure 6G). By immunostaining, glial bridging and ependymal cell proliferation were increased by 2-fold in Twist1a-expressing animals relative to their wild-type siblings (Figures 6H–6K). These results indicated that activation of twist1a-driven EMT promotes glial bridging and functional regeneration after SCI.
DISCUSSION
This study identified an essential EMT-driving gene regulatory network that regulates differential regenerative capacity between mammals and zebrafish after SC injury (Figures 6L and S6B). The combination of bulk, single cell, and comparative transcriptomics provided comprehensive characterization of pro-regenerative glial cell fates in zebrafish and revealed EMT as a hallmark of glial bridging. At the center of glial bridging is a multi-nodal gene regulatory network that is necessary and sufficient to promote twist-mediated EMT, ctgf-dependent glial bridging, and functional SC repair.
Our findings support the emergence of lineage-restricted ependymal progenitors during SC regeneration in zebrafish. Consistent with the lineage restriction within ependymal cells, domains of ventro-lateral ependymal progenitors reminiscent of the developing progenitor motor neuron (PMN) domain were shown to give rise to regenerating motor neurons in injured adult fish (Reimer et al., 2008). Our study highlights a niche of ventral ependymal cells that express activated Yap, Ctgf, and Twist1 after SCI. Ependymal radial glial cells are organized into a monolayer of polarized, epithelial-like cells in uninjured SCs. Shortly after injury, these cells undergo cell shape changes and become highly proliferative. Using genetic loss and gain of function, our study indicates that ventral ependymal progenitors undergo EMT-mediated reprogramming after injury and that this cellular reprogramming is required and sufficient to promote glial bridging. Decreased expression of Cdh1 marks the loss of epithelial cell fate in a first and essential step toward mesenchymal transition (Huang et al., 2012). Cdh1 downregulation in ctgfa+ ependymal cells is concomitant with increased expression of mesenchymal markers, including Vim and Cdh2 (Wheelock et al., 2008). EMT is often linked to increased plasticity and stem cell activation during tissue regeneration (Jessen and Arthur-Farraj, 2019; Wilson et al., 2020). We propose that EMT plays a dual regenerative role during reprogramming of ependymal progenitors, by suppressing their epitheliality in response to injury and enhancing their plasticity during regeneration.
Complete SC transection in zebrafish results in pronounced mechanical stress, manifested by prolonged expansion of the central canal proximal to the lesion from the rostral and caudal ends. Our results support a model in which Yap senses these mechanical stresses, resulting in ependymal cell activation and glial bridging. Yap is regulated by various upstream regulators, including cell polarity and adhesion proteins. Of high relevance to tissue regeneration, Yap relays cellular stress into transcriptional responses by surveying the levels of filamentous actin (Aragona et al., 2013; Dupont et al., 2011). Active Yap signaling maintains stemness in various stem cell types, including neural and glial progenitors, while Yap activation is sufficient to revert differentiated cells back to a tissue-specific stem/progenitor cell state (Cao et al., 2008; Panciera et al., 2016). Our study is consistent with Yap orchestrating regenerative responses and enhancing cell plasticity in response to the mechanical stresses associated with SCI. Why and how Yap activation confines to specific niches of progenitor cells warrant further investigation.
Our findings shed light on glial bridging as an effective, natural mechanism of SC repair. Transcriptomes of zebrafish bridging glia were more similar to scar-bordering astrocytes than to bridging Schwann cells in mammals. The transcription factors required for glial bridging were enriched in mouse astrocytes, while the EMT-related genes identified in zebrafish were similarly regulated in mouse Schwann cells (Figure S6). We thus propose that zebrafish bridging glia possess a hybrid molecular identity that combines increased EMT-mediated plasticity with an astrocyte-like cell identity. We suggest that further investigation into bridging glial cell fate will springboard translational applications to improve bridging and regeneration in the mammalian CNS.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Mayssa Mokalled (mmokalled@wustl.edu).
Materials Availability
All plasmids and zebrafish mutant and transgenic lines in this study are available upon request.
Data and Code Availability
The accession number for the FAC-seq and snRNA-seq data reported in this paper is GEO: GSE164945.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Zebrafish
Adult zebrafish of the Ekkwill, Tubingen, and AB strains were maintained at the Washington University Zebrafish Core Facility. All animal experiments were performed in compliance with IACUC institutional animal protocols. Male and female animals between 3 and 9 months of ~2 cm in length were used. Experimental fish and control siblings of similar size and equal sex distribution were used for all experiments. SC transection surgeries and regeneration analyses were performed in a blinded manner, and 2 to 4 independent experiments were repeated using different clutches of animals. Transected animals from control and experimental groups were housed in equal numbers (4–7 fish) in 1 liter tanks. The following previously published zebrafish strains were used: Tg(−5.5Kb-ctgfa:EGFPpd96) (Mokalled et al., 2016), Tg(gfap:EGFPmi2002) (Bernardos and Raymond, 2006), and Tg(hsp:dsRed-dnYap) (Mateus et al., 2015). Newly constructed strains are described below.
Generation of Tg(ctgfa:mCherry) zebrafish
The following primers were used to amplify a 5.5 Kb genomic region upstream of the ctgfa translational start site: ClaI forward primer 5’-atcgattttggctactttcagctaagactgg-3’ and ClaI reverse primer 5’-atcgattctttaaagtttgtagcaaaaagaaa-3’. The genomic fragment was cloned into PCR2.1-TOPO vector, then subcloned into ClaI digested PCS2-mCherry-Nitroreductase plasmid to generate ctgfa:mCherry-Nitroreductase clone. The clone was co-injected into one-cell stage wild-type embryos with I-SceI. Three founders were isolated and propagated. This line is referred to hereafter as ctgfa:mCherry as the inducible Nitroreductase cassette was not used in this study. The full name of this line is Tg(ctgfa:mCherry-NTR)stl650. ctgfa:mCherry animals were analyzed as hemizygotes, or crossed into gfap:EGFP transgene (mi2002) (Bernardos and Raymond, 2006) to generate ctgfa:mCherry; gfap:EGFP animals.
Generation of Tg(hsp70:Twist1a-2A-EGFP) zebrafish
The following forward primers were used to amplify twist1a cDNA cassette: ClaI forward primer 5’-ccATCGATaggcagcaatagcgtcagat-3’ and ClaI reverse primer 5’-ccatcgatttctttaaagtttgtagcaaaaaaga-3’. The genomic fragments were cloned into PCR2.1-TOPO vector, then subcloned into ClaI digested PCS2-hsp70–2A-EGFP plasmid. Clones were co-injected into one-cell stage wild-type embryos with I-SceI. A minimum of 3 founders were isolated and propagated for each transgene. The full name of these lines are Tg(hsp70:Twist1a-2A-EGFP)stl654. Animals were analyzed as hemizygotes.
Spinal cord transection
Zebrafish were anaesthetized using MS-222. Fine scissors were used to make a small incision that transects the SC 4 mm caudal to the brainstem region. Complete transection was visually confirmed at the time of surgery. Injured animals were also assessed at 2 or 3 dpi to confirm loss of swim capacity post-surgery. For sham injuries, animals were anaesthetized and fine scissors were used to transect skin and muscle tissues without inducing SCI.
METHOD DETAILS
FACS sorting and RNA sequencing
Two mm SC sections, including the lesion site plus additional rostral and caudal tissue proximal to the lesion, were collected from adult injured zebrafish at 5 dpi. Control tissue sections were collected from uninjured siblings. Three biological replicates were used. Each replicate represents cells that were sorted from 40–50 pooled SCs. Wild-type, ctgfa:mCherry, and gfap:EGFP animals were lesioned and dissociated to set up FACS gates. Tissues were dissociated using 0.05% Trypsin for 5 min at room temperature, and subjected to 3 washes with HBSS solution. Cell supernatants were triturated in DMEM media with 20% fetal bovine serum using a 1000 μL pipette and applied to a 100 μm cell strainer (MidSci, cat# 100ICS). Dissociated cells were pelleted at 500 G for 5 min, resuspended in HBSS with 2% serum, and sorted using a MoFlo Cell sorter machine. Sorted cells were collected into serum-supplemented DMEM prior to RNA extraction. Total RNA was prepared using NucleoSpin RNA Plus XS (Clontech, cat# 740990). Total RNA was also prepared from two mm SC sections at 5, 10, 21 dpi, and from uninjured controls for bulk RNA sequencing. For bulk sequencing, 10 SCs from wild-type animals were pooled for each replicate and time point. Two biological replicates were used for control, 10 dpi, and 21 dpi SCs. TruSeq libraries were prepared and sequenced on Illumina HiSeq 3000 using 50 bp single-end reading strategy. Quality QC and trimming of adapters and short sequences were performed using Fastx. Sequencing reads were mapped to the zebrafish genome (Zv11) using Bowtie2, then assembled and quantified using the Cufflinks and Cuffdiff algorithms. Genes with log2(fold enrichment) between −1 and 1, or with −log10(P-value) less than 1 were considered insignificant. FACS experiments were performed at the Siteman Cancer Center Flow Cytometry shared Resource. RNA sequencing was performed at the Genome Technology Access Center at Washington University. Analysis was performed in collaboration with the Bioinformatics Core at the Center for Regenerative Medicine at Washington University. Bulk sequencing data are accessible through GEO Series accession number GSE164945.
Single nuclear RNA sequencing
gfap:EGFP transgenic animals were subjected to SCI and 2-mm SC tissue sections spanning the lesion site were collected at 1 wpi. A total of 33185 nuclei were isolated from 45 pooled SC tissues as previously described (Matson et al., 2018). For single nuclear library preparation on the 10x Genomics platform, the Chromium Single Cell 3′ GEM Library and Gel Bead Kit v3 (cat# 1000092) and the Chromium Chip B Single Cell Kit (cat# 1000074) were used according to the manufacturer’s instructions in the Chromium Single Cell 3′ Reagents Kits V3 User Guide. The resulting cDNA libraries were quantified on an Agilent Tapestation and sequenced on Illumina HiSeq 3000.
Unbiased cell clustering
Sequenced nuclei reads were aligned to a custom zebrafish reference (GRCz11) using Cellranger v3.0.1 software (10X Genomics). This reference was constructed from the Ensemble genome build (http://www.ensembl.org/Danio_rerio/Info/Index) and modified to recognize reads containing introns (https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/advanced/references). Further, the GFP transgene was appended to the final genome reference build. Aligned reads were analyzed using the Seurat package v3.1.4 (Butler et al., 2018) within R v3.6.3. Cell quality was assessed using commonly used QC matrices (Ilicic et al., 2016). Cells that have unique number of genes between 200 to 4000 and a mitochondrial gene percentage less than five were filtered. The “SCTransform” function was used for normalization, scaling and finding variable features. SCTransform returned 3000 highly variable features for downstream analysis (Hafemeister and Satija, 2019). During normalization, confounding sources of variation from mitochondrial mapping percentage were removed. PCA analysis was performed on scaled data using highly variable genes. Significant principal components were determined using “ElbowPlot” function. Forty principal components were selected to create a Shared Nearest Neighbour (SNN) graph using the “FindNeighbours” function. Next, clustering was performed by the Louvain algorithm using the function “FindClusters” and the resolution parameter was set to one. Uniform Manifold Approximation and Reduction (UMAP) was used for non-linear dimensional reduction of the first 40 principle components and to visualize the data in two-dimensional space (“RunUMAP” function) (Becht et al., 2018). Data was graphed using different plot function (such as “Dimplot”, “VlnPlot”, “FeaturePlot”, “DoHeatmap”) to view the cell cluster identity and marker gene expression. Differential gene expression for individual cluster was identified using Wilcoxon rank sum tests in the “FindAllMarkers” function. Marker genes detected in >20% of clustered cells and with a logFC threshold of at least 0.25 were selected. Only positive markers were reported. Cluster identity was determined by comparing top markers of each clusters with the markers of different cell types identified in the mouse cerebral cortex (Zhang et al., 2014).
Subsetting glial clusters
Glial cell clusters (Clusters- 7, 9 & 26, Figure 3A) were identified in the complete dataset. The function “subset” was used to create a Seurat object Glial cells. The subset was again normalized and scaled using “SCTransform” function. Twenty principle components were used for clustering and the resolution parameter was set to 0.9. Further analysis was done as described above for unbiased clustering. The top 50 markers for cluster Glial-7, -8, ad -9 were obtained using the function “FindAllMarkers”. snRNA-seq data are accessible through GEO Series accession number GSE164945.
Gene ontology
Gene Ontology analysis was performed using Metascape (Zhou et al., 2019) (Tables S1–S3). Input and analysis species were set as D. rerio. Express analysis was performed for the gene ontology. Metascape identified all statistically enriched terms (includes GO biological processes, Reactome gene set and KEGG pathway), and calculated accumulative hypergeometric p-values and enrichment factors. Significant terms were hierarchically clustered into a tree based on Kappa-statistical similarities among their gene memberships. A kappa score of 0.3 was applied to cast the tree into term clusters. Selected subset of representative terms from this cluster were used to generate a network layout. Each term is represented by a circle node, where its size is proportional to the number of input genes fall into that term, and its color represent its cluster identity. Terms with a similarity score > 0.3 are linked by an edge (the thickness of the edge represents the similarity score). The network is visualized with Cytoscape (v3.1.2) with “force-directed” layout and with edge bundled for clarity. One term from each cluster is selected to have its term description shown as label.
Histology
Sixteen μm cross or 20 μm longitudinal cryosections of paraformaldehyde-fixed SC tissues were used. Tissue sections were imaged using a Zeiss AxioVision compound microscope for in situ hybridization and RNAscope, or a Zeiss LSM 800 confocal microscope for immunofluorescence. in situ hybridization probes for twist1a were subcloned after amplification from 2 dpf zebrafish cDNA into PCR2.1-TOPO vectors (twist1a forward primer 5’-tgtgattgctctgctgttcc-3’, twist1a reverse primer 5’-ggtgaggcgattagcttctg-3’). Linearized vectors were used to generate the digoxygenin labeled cRNA probes. in situ hybridizations were performed as previously described. RNAscope was performed according to the manufacturer’ protocol (Advanced Cell Diagnostics, 322300).
For immunohistochemistry, tissue sections were circumscribed with a hydrophobic barrier pen and rehydrated in PBT (0.1% Tween-20 in PBS). After 2 × 5 min washes in PBT, sections were treated with blocking agent (5% goat serum in PBT) for 1 hour at room temperature. For nuclear antigens, sections were treated with 0.2% TritonX-100 in PBT for 5 minutes and washed thoroughly in PBT prior to the blocking step. Sections were incubated overnight with indicated primary antibodies diluted in blocking agent, washed in PBT, and treated for 1 hour in secondary antibodies diluted in blocking agent. Following washes, sections were incubated in 1 μg/mL of Hoechst, washed in PBS, and mounted in Fluoromount-G mounting media. Primary antibodies used in this study were chicken anti-GFP (AVES, 1020, 1:1000), mouse anti-GFAP (ZIRC, Zrf1, 1:1000), rabbit anti-dsRed (Clontech, 632496, 1:250), rabbit anti-Cdh1 (Genetex, GTX125890; 1:250), rabbit anti-Cdh2 (Genetex, GTX125885, 1:500), mouse anti-Vim (Millipore, MAB3400, 1:500), rabbit anti-Yap (Cell Signaling, 4912S, 1:100), rabbit anti-Taz (Cell Signaling, 8418S, 1:100), and rabbit anti-PCNA (Genetex, GTX124496, 1:500). Secondary antibodies (Invitrogen, 1:200) used in this study were Alexa Fluor 488, Alexa Fluor 594 goat anti-rabbit, anti-mouse, or anti-chicken antibodies. For PCNA staining, sections were treated in citrate buffer (10 mM citric acid, 0.05% Tween-20, pH 6.0) at 98 °C for 20 minutes prior to staining.
Swim capacity assays
Zebrafish were exercised in groups of 8–12 in a 5L swim tunnel respirometer device (Loligo, cat# SW100605L, 120V/60Hz). After 10 minutes of acclimation inside the enclosed tunnel, water current velocity was increased every two minutes and fish swam against the current until they reached exhaustion. Exhausted animals were removed from the chamber without disturbing the remaining fish. Swim time and current velocity at exhaustion were recorded. Results were expressed as means ± SEM.
Axon tracing
Anterograde axon tracing was performed on adult fish at 4 wpi. Fish were anaesthetized using MS-222 and fine scissors were used to transect the cord 4 mm rostral to the lesion site. Biocytin-soaked Gelfoam Gelatin Sponge was applied at the new injury site (Gelfoam, Pfizer, cat# 09–0315-08; Biocytin, saturated solution, Sigma, cat# B4261). Fish were euthanized 6 hours post-treatment and Biocytin was histologically detected using Alexa Fluor 594-conjugated Streptavidin (Molecular Probes, cat# S-11227).
Protein-Protein network generation
To identify functional interactions, the human orthologs of the glial bridging factor Ctgf, the transcription factor hits identified by CRISPR/Cas9 screening, and EMT components were used as a source list. The web-based LENS tool (https://hagrid.dbmi.pitt.edu/LENS/) was used to map out the interactions between these genes based on previously confirmed and published interactions.
CRISPR/Cas9 Mutagenesis
CRISPR/Cas9 design and mutagenesis was performed as previously described (Hoshijima et al., 2019). To avoid phenotypes that might cause lethality prior to adulthood, genes were prescreened for maternal supply using the Zebrafish Gene Expression Atlas (https://www.ebi.ac.uk/gxa/experiments/E-ERAD-475). Genes that are expressed zygotically, but not supplied maternally were not selected for analysis. ZFIN was used to identify additional genes with morphant or mutant phenotypes that would cause embryonic lethality. To mutant candidate genes, crRNA guide RNA sequences were selected using CHOPCHOP (https://chopchop.cbu.uib.no/) (Table S4). Only sequences with no off-target sites with 3 or fewer mismatches elsewhere in the genome were selected. To maximize the effect of small indels, target sites were chosen against the DNA binding sites of targeted transcription factors. For most genes, an additional second target site was selected within an early exon. For genes with only one or two exons, only one site was selected (foxg1c, spi1a, and spi1b). Lyophilized Alt-R tracrRNA and crRNA gRNAs (IDT, cat# 1072534) were reconstituted according to the manufacturer’s specifications (100 μM stocks at −20°C). Prior to the day of injection, crRNA and tracrRNA were mixed at a final concentration of 50 μM and annealed by heating to 95°C and then gradual cooling to 25°C (−0.1°C/second). The resulting dgRNA duplexes were stored at −20°C until use. Alt-R S.p. Cas9 nuclease V3 (IDT, cat# 1081059, supplied at 61.7 μM in 50% glycerol) was diluted with Cas9 dilution buffer (1 M HEPES (pH 7.5), 2 M KCl) to a working concentration of 25 μM and stored in single use aliquots at −80°C. On the day of injection, annealed dgRNA duplexes were diluted 1:1 in duplex buffer (IDT, cat# 11–05-01–03) to a working concentration of 25 μM. Equal volumes of dgRNA and Cas9 were incubated at 37°C for 5 minutes. For samples where 2 sites were being targeted at once, Cas9 protein was added in equal molar amounts to the total concentration of gRNA. CRISPR/Cas9 solutions were maintained at room temperature during injections. Tubingen wild-type embryos were injected with 1 nL of CRISPR/Cas9 solution at the one-cell stage and grown to adulthood for SC surgeries and functional analysis.
Capillary electrophoresis and NGS
Capillary electrophoresis was used to calculate the % indel for each CRISPR/Cas9 target site. For DNA extraction, whole 2 dpf larvae or ~3 mm of excised adult tail fins were added to 50 mM NaOH in 50 μL (larvae) or 100 μL (adult fin). DNA samples were incubated at 95°C for 20 minutes and then rapidly cooled to 4°C. DNA extractions were neutralized by adding 5 μL (larvae) or 10 μL (adult fins) of 1 M Tris-HCl (pH 8.0). Small 100–250 bp PCR products were generated using NEB Taq Polymerase (cat# M0273) with gene-specific primers in a volume of 10 μL in Pryme PCR semi-skirted PCR plates (MidSci, cat# AVRT1). Samples were diluted to 24 μL with TE dilution buffer (Agilent) and loaded into the 5200 Fragment Analyzer System (Agilent, cat# M5310AA). Capillary electrophoresis was carried out using the Agilent Fragment Analyzer Qualitative DNA Kit (cat# DNF-905-K1000) according to the manufacturer’s specifications. For controls, 3 wild-type uninjected siblings were amplified. Peaks within 1 bp of wild-type peaks were considered wild types. Percent indel was calculated by dividing total mutated peak signal by total wild-type peak signal. Because of significant noise due to primers (<70 bp) and non-specific products (>200 bp) in the wild-type samples, only signals between 70 and 200 bp were used. To confirm mutagenesis rates, capillary electrophoresis and next generation sequencing were performed for the same cohort of targeted animals at 2 dpf. DNA extracts from larvae or adult fins were directly submitted to the Washington University Genome Engineering & iPSC Center (GEiC) for next generation sequencing (NGS) using the same primer sets as for capillary electrophoresis. The average mutagenesis rates were comparable across experiments. The rate of mutagenesis derived by capillary electrophoresis is underestimated for indels that result in <2 bp deletion.
Quantitative real time PCR
For qRT-PCR, RNA was collected from whole spinal cord or FACS-sorted cells as described in FACS sorting and RNA sequencing. For cDNA synthesis, 500 ng (whole spinal cord) or 25 ng (FACS-sorted cells) was converted into cDNA with the Maxima First Strand cDNA Synthesis Kit (ThermoFisher, cat# K1672) according to manufacturer’s specifications. Quantitative PCR was completed using the Luna polymerase master mix (NEB, cat# M3003) using gene-specific primers (Table S5). Primers were designed to flank introns and were confirmed to not amplify project from genomic DNA (except for junbb, which does not have introns). To determine primer efficiency, a standard curve was generated for each primer set using cDNA pooled from 1, 3, and 5 dpf wild-type embryos. qRT-PCR was performed on a Bio-Rad CFX Connect Real-Time System. Expression fold change for each gene of interest was calculated using the ΔCq method and normalized to the expression fold change of eF1α expression compared to controls. For the wild-type bulk spinal cord injury time course (Figures S2B and S3G) and ctgfa+gfap+ FACS-sorted cells (Figures S2A, S2B, and S3G), 6 biological replicates were used. For F0 injected crispants versus uninjected control siblings (Figure S4I), two technical replicates of 3 biological replicates were used. For ctgfa mutant versus wild-type control siblings, (Figure S5G), 2 technical replicates of 2 biological replicates were used.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification
All sample sizes (n) are indicated for the number of animals used in each experiment. Surgeries, swims, and analysis were completed blind to condition. Quantification was performed on single-plane images. Quantification was performed on single-plane images. To quantify colocalization of the two ctgfa reporter transgenes (Figure S1B), the “Colocalization Threshold” plug in in Fiji was ran using Costes automatic thresholding to calculate Pearson’s Correlation Coefficient. This plug-in generates a plot of all pixel intensities within an image. A linear regression is performed on this plot, which provides a ratio of the intensities of the 2 channels across all pixels in the image or region of interest. This plug-in automatically runs Costes method of unbiased, reproducible auto-threshold determination, which is based on an iterative procedure to determine what threshold for each channel would give an R-value of 0 in the scatterplot. Any pixel that is above that threshold in both channels is considered ‘colocalized’. Information on this plug-in can be found at https://imagej.net/Colocalization_Analysis#Colocalization_Threshold. To quantify Cdh fluorescence intensity (Figures 2F, 2H, S2E, and S2F), Costes automatic thresholding was performed in the ctgfa:EGFP channel to define ctgfa+ cells. The absolute fluorescence value in the Cdh channel was measured in ctgfa+ and ctgfa− ROI’s. To calculate glial bridging (Figures 4D, 5I, 6I, and S5C), GFAP immunohistochemistry was performed on serial transverse sections. The cross-sectional area of the glial bridge and the area of the intact SC rostral to the lesion were measured using ImageJ software. Bridging was calculated as a ratio of these measurements. Mann Whitney tests were performed using Prism software to determine statistical significance between groups. To quantify twist1a+ area (Figures 4F and 6E), the Measure function in Fiji was used after Costes automatic thresholding. Values are normalized to the average twist1a+ area in controls. To calculate Nuclear/Cytoplasmic Yap (Figure 5E), the nuclear Hoechst channel was thresholded and used to define nuclear particles. The fluorescence intensity of the green (Yap+) channel for each particle was calculated using the Analyze Particles function in Fiji. Nuclear particles were then subtracted to generate cytoplasmic ROIs, which were also measured in the green channel with the Analyze Particles function. For calculation of axon growth index (Figure 5G), biocytin-labeled axons were quantified using the “threshold” and “particle analysis” tools in the ImageJ software (26). Four sections per fish at 0.5 (proximal) and 2 (distal) mm caudal to the lesion core, and 2 sections 1 mm rostral to the lesion, were analyzed. Axon growth was normalized to the efficiency of Biocytin labeling rostral to the lesion for each fish. The axon growth index was then normalized to the control group for each experiment. To calculate the % of Yap+ctgfa+ cells (Figure S5E), Yap+ nuclei were counted manually blind to condition. To calculate ctgfa:GFP+ area (Figure 6C), the GFP channel was thresholded blind to condition using a standard threshold % for all control and experimental samples. The thresholded area was calculated using the Measure function in Fiji. The GFP+ area was divided by the cross-sectional area of the spinal cord.
To represent average swim activity (Figure 6G), experimental and control animals were tracked inside a swim tunnel using the Loli-track software (from Loligo). Tracking was performed in the absence of water current (0 cm/sec) for 5 min, and at 20 cm/sec water current for 5 min. Swim activity was averaged for each genotype. To calculate PCNA+ nuclei (Figure 6K), the nuclear Hoechst channel was thresholded to define nuclear particles. Any nuclear particle with an average fluorescence value >40 was defined as PCNA+.
Statistical tests
For violin plots, solid lines indicate the group median; dotted lines indicate the 25th and 75th quartiles. All statistical tests were performed using GraphPad Prism. Where two groups are compared, student’s t-test (with Welch’s correction, where appropriate) was used (Figures 2F, 2H, 5E, 5G, 5I, 6C, 6E, 6I, 6K, S2B, S3G, S4G, S5C, S5E, and S5G). Where three or more groups were compared, one-way ANOVA with appropriate corrections for multiple comparisons were used (Figures 4B, 4D, 4F, S2A, S3F, S4H, and S4I). For swim analyses in Figures 5F and 6G Mann-Whitney tests were performed.
Molecular comparisons between zebrafish bridging glia and mammalian glia
In the tables below, the “Number of genes” represents genes that are either upregulated (log2 FC >1) or downregulated (log2 FC <1) in each dataset with P-value <0.01. “Probability of gene occurence” represents the proportion of upregulated (or downregulated) genes relative to the whole genome. For each comparison, the “Probability of co-occurrence” represents the probability for a gene to be upregulated (or downregulated) in 2 datasets. “Actual gene similarities” represents the proportion of genes are that are actually upregulated (or downregulated) in 2 datasets relative to the total number of aligned genes.
| zebrafish bridging glia | mouse SCI asctrocytes | Combined datasets | ||||
|---|---|---|---|---|---|---|
| Number of genes | Probability of gene occurrence | Number of genes | Probability of gene occurrence | Probability of co-occurrence | Actual co-occurrence frequency | |
| For upregulated genes | 4917 | 0.25 | 2476 | 0.13 | 0.0325 | 0.12 |
| For downregulated genes | 5934 | 0.3 | 1934 | 0.1 | 0.03 | 0.09 |
| Total | 19419 | 18814 | 6875 aligned genes | |||
| zebrafish bridging glia | mouse bridging Schwann cells | Combined datasets | ||||
| Number of genes | Probability of gene occurrence | Number of genes | Probability of gene occurrence | Probability of co-occurrence | Actual co-occurrence frequency | |
| For upregulated genes | 4917 | 0.25 | 859 | 0.038 | 0.0095 | 0.06 |
| For downregulated genes | 5934 | 0.3 | 1134 | 0.05 | 0.015 | 0.06 |
| Total | 19419 | 22370 | 3811 aligned genes | |||
ADDITIONAL RESOURCES
FACS-seq and snRNA-seq data
The data presented and analyzed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE164945 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164945).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Chicken anti-GFP | AVES | Cat# AS10 1012; RRID: AB_10754353 |
| Mouse anti-GFAP | ZIRC | Cat# zrf-1; RRID: AB_10013806 |
| Rabbit anti-dsRed | Clontech | Cat# 632496; RRID: |
| Rabbit anti-Cdh1 | Genetex | Cat# GTX125890; RRID: AB_11167551 |
| Rabbit anti-Cdh2 | Genetex | Cat# GTX125885; RRID: AB_2885609 |
| Mouse anti-Vim | Millipore | Cat# MAB3400; RRID: AB_10551174 |
| Rabbit anti-Yap | Cell Signaling | Cat# 4912S; RRID: AB_2218911 |
| Rabbit anti-Taz | Cell Signaling | Cat# 8418S; RRID: AB_10950494 |
| Rabbit anti-PCNA | Genetex | GTX124496; RRID: AB_11161916 |
| Alexa Fluor 488 goat anti-rabbit | Invitrogen | Cat# A-11008; RRID: AB_143165 |
| Alexa Fluor 594 goat anti-rabbit | Invitrogen | Cat# A-11072; RRID: AB_142057 |
| Alexa Fluor 488 goat anti-chicken | Invitrogen | Cat# A-11039; RRID: AB_142924 |
| Alexa Fluor 488 goat anti-mouse | Invitrogen | Cat# A-21121; RRID: AB_2535764 |
| Alexa Fluor 594 goat anti-mouse | Invitrogen | Cat# A-21125; RRID: AB_141593 |
| Alexa Fluor 594-conjugated Streptavidin | Molecular Probes | Cat# S-11227; RRID: AB_2313574 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Gelfoam Gelatin Sponge | Pfizer | Cat# 09–0315-08 |
| Biocytin, saturated solution | Sigma | Cat# B4261 |
| Alt-R tracrRNA | IDT | Cat# 1072534 |
| Alt-R crRNA (see Table S4 for sequences) | IDT | N/A |
| Duplex buffer | IDT | Cat# 11–05-01–03 |
| Alt-R S.p. Cas9 nuclease v3 | IDT | Cat# 1081059 |
| Taq Polymerase | NEB | Cat# M0273 |
| Critical Commercial Assays | ||
| NucleoSpin RNA Plus XS Kit | Clontech | Cat# 740990 |
| Chromium Single Cell 3’ GEM Library and Gel Bead Kit v3 | 10x Genomics | Cat# 1000092 |
| Chromium Chip B Single Cell Kit | 10x Genomics | Cat# 1000074 |
| 5200 Fragment Analyzer System | Agilent | Cat# M5310AA |
| Fragment Analyzer Qualitative DNA Kit | Agilent | Cat# DNF-905-K1000 |
| Maxima First Strand cDNA Synthesis Kit | ThermoFisher | Cat# K1672 |
| Luna qPCR master mix | NEB | Cat# M3003 |
| Experimental Models: Organisms/Strains | ||
| Zebrafish, Tg(-5.5Kb-ctgfa:EGFP)pd96 | Mokalled et al., 2016 | N/A |
| Zebrafish, Tg(gfap:EGFP)mi2002 | Bernardos and Raymond, 2006 | N/A |
| Zebrafish, Tg(hsp:dsRed-dnYap) | Mateus et al., 2015 | N/A |
| Zebrafish, Tg(ctgfa:mCherry-NTR)stl650 | This study | N/A |
| Zebrafish, Tg(hsp70:twist1a-2A-EGFP)stl654 | This study | N/A |
| Oligonucleotides | ||
| ctgfa_ClaI_F: atcgattttggctactttcagctaagactgg |
This study | N/A |
| ctgfa_ClaI_R: atcgattctttaaagtttgtagcaaaaagaaa |
This study | N/A |
| twist1a_ClaI_F: ccatcgataggcagcaatagcgtcagat |
This study | N/A |
| twist1a_ClaI_R: ccatcgatttctttaaagtttgtagcaaaaaaga |
This study | N/A |
| twist1a_ISH_F: tgtgattgctctgctgttcc |
This study | N/A |
| twist1a_ISH_R: ggtgaggcgattagcttctg |
This study | N/A |
| qPCR primers (see Table S4 for sequences) | This study | N/A |
| Recombinant DNA | ||
| PCS2-ctgfa:mCherry-Nitroreductase | This study | N/A |
| PCS2-hsp70:twist1a-2A-EGFP | This study | N/A |
| PCR2.1-twist1a | This study | N/A |
| Software and Algorithms | ||
| Seurat package v3.1.4 | Butler et al., 2018 | https://satijalab.org/seurat/ |
| R v3.6.3 | https://cran.r-project.org/ | |
| Other | ||
| 100 mm cell strainer | MidSci | Cat# 100ICS |
| Vannas scissors | World Precision Instruments | Cat# 14003-G |
| 5L swim tunnel respirometer device | Loligo | Cat# SW100605L, 120/60Hz |
| Vitrectomy Scissors, Vertical Opening, 60 deg, 20G | World Precision Instruments | Cat# WP504240 |
| Pryme PCR Semi-Skirted PCR plates | MidSci | Cat# AVRT1 |
| Metascape Gene Ontology Analysis | Zhou et al., 2019 | https://metascape.org/ |
| LENS protein-protein interaction network tool | Handen and Ganapthiraju, 2015 | https://hagrid.dbmi.pitt.edu/LENS |
| CHOPCHOP | Labun et al., 2019 | https://chopchop.cbu.uib.no/ |
| Sequence data | This study | GEO Series accession number GSE164945 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164945) |
Highlights.
Bulk and single cell transcriptomes of regenerative zebrafish glia were generated
Pathways that distinguish regenerative zebrafish glia from mammalian glia were defined
An injury-induced EMT transcriptional module localizes to zebrafish glial progenitors
EMT transcription factors direct mesenchymal fate and regenerative bridging in zebrafish
ACKNOWLEDGMENTS
We thank V. Cavalli, A. Johnson, H. McNeill, K. Monk, and L. Solnica-Krezel for discussion, Y. Hou, D. Grunwald, and A. Stratman for sharing protocols and reagents, T. Li and B. Zhang for Bioinformatics analysis, and the Washington University zebrafish Shared Resource for animal care. This research was supported by the W.M. Keck Post-doctoral Fellowship (to D.K.S), the Washington University Center of Regenerative Medicine T32 (T32 EB028092 to D.K.S), grants from NIH (R01 NS113915 to M.H.M.), the Curators of the University of Missouri (Spinal Cord Injury and Disease Training Program to M.H.M.), the McDonnell Center for Cellular Neuroscience (to M.H.M.), and the Institute for Clinical and Translational Sciences at Washington University (to M.H.M.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.devcel.2021.01.017.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPORTING CITATION
The following reference appears in the supplemental information: Cao et al., 2016.
REFERENCES
- Anderson MA, Burda JE, Ren Y, Ao Y, O’Shea TM, Kawaguchi R, Coppola G, Khakh BS, Deming TJ, and Sofroniew MV (2016). Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, and Piccolo S (2013). A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 154, 1047–1059. [DOI] [PubMed] [Google Scholar]
- Arthur-Farraj PJ, Morgan CC, Adamowicz M, Gomez-Sanchez JA, Fazal SV, Beucher A, Razzaghi B, Mirsky R, Jessen KR, and Aitman TJ (2017). Changes in the coding and non-coding transcriptome and DNA methylome that define the Schwann cell repair phenotype after nerve injury. Cell Rep. 20, 2719–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2018). Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- Becker T, Wullimann MF, Becker CG, Bernhardt RR, and Schachner M (1997). Axonal regrowth after spinal cord transection in adult zebrafish. J. Comp. Neurol 377, 577–595. [DOI] [PubMed] [Google Scholar]
- Bernardos RL, and Raymond PA (2006). GFAP transgenic zebrafish. Gene Expr. Patterns 6, 1007–1013. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Navis A, Cox BD, Dickson AL, Gemberling M, Karra R, Bagnat M, and Poss KD (2016). Single epicardial cell transcriptome sequencing identifies caveolin 1 as an essential factor in zebrafish heart regeneration. Development 143, 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Pfaff SL, and Gage FH (2008). YAP regulates neural progenitor cell number via the TEA domain transcription factor. Genes Dev 22, 3320–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H, Liu Y, Xue M, Liu H, Du S, Zhang L, and Wang P (2016). Synergistic action of master transcription factors controls epithelial-to-mesenchymal transition. Nucleic Acids Res 44, 2514–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements MP, Byrne E, Camarillo Guerrero LF, Cattin AL, Zakka L, Ashraf A, Burden JJ, Khadayate S, Lloyd AC, Marguerat S, and Parrinello S (2017). The wound microenvironment reprograms Schwann cells to invasive mesenchymal-like cells to drive peripheral nerve regeneration. Neuron 96, 98–114.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias DO, and Göritz C (2018). Fibrotic scarring following lesions to the central nervous system. Matrix Biol 68–69, 561–570. [DOI] [PubMed] [Google Scholar]
- Dongre A, and Weinberg RA (2019). New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol 20, 69–84. [DOI] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. (2011). Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183. [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, and Lash AE (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucl Acids Res 30, 207–210, 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervasi M, Bianchi-Smiraglia A, Cummings M, Zheng Q, Wang D, Liu S, and Bakin AV (2012). JunB contributes to Id2 repression and the epithelial-mesenchymal transition in response to transforming growth factor-β. J. Cell Biol 196, 589–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldshmit Y, Sztal TE, Jusuf PR, Hall TE, Nguyen-Chi M, and Currie PD (2012). Fgf-dependent glial cell bridges facilitate spinal cord regeneration in zebrafish. J. Neurosci 32, 7477–7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Sanchez JA, Pilch KS, van der Lans M, Fazal SV, Benito C, Wagstaff LJ, Mirsky R, and Jessen KR (2017). After nerve injury, lineage tracing shows that myelin and Remak Schwann cells elongate extensively and branch to form repair Schwann cells, which shorten radically on remyelination. J. Neurosci 37, 9086–9099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimpe B, and Silver J (2004). A novel DNA enzyme reduces glycosaminoglycan chains in the glial scar and allows microtransplanted dorsal root ganglia axons to regenerate beyond lesions in the spinal cord. J. Neurosci 24, 1393–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafemeister C, and Satija R (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Zhang Y, Niu C, Guo J, Li J, Wei X, Jia M, Zhi X, Yao L, and Meng D (2019). BTB and CNC homology 1 (Bach1) promotes human ovarian cancer cell metastasis by HMGA2-mediated epithelial-mesenchymal transition. Cancer Lett 445, 45–56. [DOI] [PubMed] [Google Scholar]
- Handen A, and Ganapthiraju MK (2015). LENS: web-based lens for enrichment and network studies of human proteins. BMC Med Genomics 8, 10.1186/1755-8794-8-S4-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, and Jin Y (2016). Intrinsic control of axon regeneration. Neuron 90, 437–451. [DOI] [PubMed] [Google Scholar]
- Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, and Sofroniew MV (2008). STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J. Neurosci 28, 7231–7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshijima K, Jurynec MJ, Klatt Shaw D, Jacobi AM, Behlke MA, and Grunwald DJ (2019). Highly efficient CRISPR-Cas9-based methods for generating deletion mutations and F0 embryos that lack gene function in zebrafish. Dev. Cell 51, 645–657.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang RY, Guilford P, and Thiery JP (2012). Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J. Cell Sci 125, 4417–4422. [DOI] [PubMed] [Google Scholar]
- Ilicic T, Kim JK, Kolodziejczyk AA, Bagger FO, McCarthy DJ, Marioni JC, and Teichmann SA (2016). Classification of low quality cells from single-cell RNA-seq data. Genome Biol 17, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KR, and Arthur-Farraj P (2019). Repair Schwann cell update: adaptive reprogramming, EMT, and stemness in regenerating nerves. Glia 67, 421–437. [DOI] [PubMed] [Google Scholar]
- Kimelman D, Smith NL, Lai JKH, and Stainier DY (2017). Regulation of posterior body and epidermal morphogenesis in zebrafish by localized Yap1 and Wwtr1. eLife 6, e31065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labun K, Montague TG, Krause M, Torres Cleuren YN, Tjeldnes H, and Valen E (2019). CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucl Acids Res 47, 171–174, 10.1093/nar/gkz365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung W-S, Peterson TC, et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateus R, Lourenço R, Fang Y, Brito G, Farinho A, Valério F, and Jacinto A (2015). Control of tissue growth by Yap relies on cell density and F-actin in zebrafish fin regeneration. Development 142, 2752–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson KJE, Sathyamurthy A, Johnson KR, Kelly MC, Kelley MW, and Levine AJ (2018). Isolation of adult spinal cord nuclei for massively parallel single-nucleus RNA sequencing. J. Vis. Exp 140, 58413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald D, Cheng C, Chen Y, and Zochodne D (2006). Early events of peripheral nerve regeneration. Neuron Glia Biol 2, 139–147. [DOI] [PubMed] [Google Scholar]
- Mendez MG, Kojima S, and Goldman RD (2010). Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J 24, 1838–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesfeld JB, Gestri G, Clark BS, Flinn MA, Poole RJ, Bader JR, Besharse JC, Wilson SW, and Link BA (2015). Yap and Taz regulate retinal pigment epithelial cell fate. Development 142, 3021–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokalled MH, Patra C, Dickson AL, Endo T, Stainier DY, and Poss KD (2016). Injury-induced ctgfa directs glial bridging and spinal cord regeneration in zebrafish. Science 354, 630–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, and Okano H (2006). Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat. Med 12, 829–834. [DOI] [PubMed] [Google Scholar]
- O’Shea TM, Burda JE, and Sofroniew MV (2017). Cell biology of spinal cord injury and repair. J. Clin. Invest 127, 3259–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panciera T, Azzolin L, Fujimura A, Di Biagio D, Frasson C, Bresolin S, Soligo S, Basso G, Bicciato S, Rosato A, et al. (2016). Induction of expandable tissue-specific stem/progenitor cells through transient expression of YAP/TAZ. Cell Stem Cell 19, 725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello S, Napoli I, Ribeiro S, Wingfield Digby P, Fedorova M, Parkinson DB, Doddrell RD, Nakayama M, Adams RH, and Lloyd AC (2010). EphB signaling directs peripheral nerve regeneration through Sox2-dependent Schwann cell sorting. Cell 143, 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S, Zou Y, and Zhang CL (2013). Cross-talk between KLF4 and STAT3 regulates axon regeneration. Nat. Commun 4, 2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimer MM, Sörensen I, Kuscha V, Frank RE, Liu C, Becker CG, and Becker T (2008). Motor neuron regeneration in adult zebrafish. J. Neurosci 28, 8510–8516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Matsumoto M, Saiki Y, Alam M, Nishizawa H, Rokugo M, Brydun A, Yamada S, Kaneko MK, Funayama R, et al. (2020). BACH1 promotes pancreatic cancer metastasis by repressing epithelial genes and enhancing epithelial-mesenchymal transition. Cancer Res 80, 1279–1292. [DOI] [PubMed] [Google Scholar]
- Scarpa E, Szabó A, Bibonne A, Theveneau E, Parsons M, and Mayor R (2015). Cadherin switch during EMT in neural crest cells leads to contact inhibition of locomotion via repolarization of forces. Dev. Cell 34, 421–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver J (2016). The glial scar is more than just astrocytes. Exp. Neurol 286, 147–149. [DOI] [PubMed] [Google Scholar]
- Silver J, and Miller JH (2004). Regeneration beyond the glial scar. Nat. Rev. Neurosci 5, 146–156. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV (2018). Dissecting spinal cord regeneration. Nature 557, 343–350. [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev A, Kaneko KJ, Shu H, Zhao Y, and DePamphilis ML (2001). TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev 15, 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Zhang S, Zhang AF, Yang ZY, and Li XG (2014). Sodium hyaluronate-CNTF gelatinous particles promote axonal growth, neurogenesis and functional recovery after spinal cord injury. Spinal Cord 52, 517–523. [DOI] [PubMed] [Google Scholar]
- Wanner IB, Anderson MA, Song B, Levine J, Fernandez A, Gray-Thompson Z, Ao Y, and Sofroniew MV (2013). Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J. Neurosci 33, 12870–12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheelock MJ, Shintani Y, Maeda M, Fukumoto Y, and Johnson KR (2008). Cadherin switching. J. Cell Sci 121, 727–735. [DOI] [PubMed] [Google Scholar]
- Wilson MM, Weinberg RA, Lees JA, and Guen VJ (2020). Emerging mechanisms by which EMT programs control stemness. Trends Cancer 6, 775–780. [DOI] [PubMed] [Google Scholar]
- Wu WS, You RI, Cheng CC, Lee MC, Lin TY, and Hu CT (2017). Snail collaborates with EGR-1 and SP-1 to directly activate transcription of MMP 9 and ZEB1. Sci. Rep 7, 17753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Cao L, Cui R, Huang A, Yan Z, Lu C, and He C (2004). The effects of ciliary neurotrophic factor on neurological function and glial activity following contusive spinal cord injury in the rats. Brain Res 997, 30–39. [DOI] [PubMed] [Google Scholar]
- Ye X, and Weinberg RA (2015). Epithelial-mesenchymal plasticity: a central regulator of cancer progression. Trends Cell Biol 25, 675–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, and Piccolo S (2015). Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol 17, 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of Glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, et al. (2008). TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 22, 1962–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zukor K, Belin S, Wang C, Keelan N, Wang X, and He Z (2013). Short hairpin RNA against PTEN enhances regenerative growth of corticospinal tract axons after spinal cord injury. J. Neurosci 33, 15350–15361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the FAC-seq and snRNA-seq data reported in this paper is GEO: GSE164945.