

Summary
mRNA degradation is connected to the translation process up to the degree that 5′-3′ mRNA degradation follows the last translating ribosome. To study 5′-3′co-translational mRNA decay and the associated ribosome dynamics, here we present an improved high-throughput 5′P degradome RNA sequencing protocol (HT-5Pseq). We exemplify its application in Saccharomyces cerevisiae, but in principle, it could be applied to any other eukaryotic organism. HT-5Pseq is easy, scalable, and uses affordable duplex-specific nuclease-based rRNA depletion.
For complete details on the use and execution of this protocol, please refer to Zhang and Pelechano (2021).
Subject areas: Sequencing, RNAseq, Gene Expression
Graphical abstract
Highlights
-
•
HT-5Pseq facilitates the investigation of 5′P mRNA degradome
-
•
HT-5Pseq is an easy, scalable, and affordable protocol
-
•
HT-5Pseq can be potentially applied in any eukaryotic organism
mRNA degradation is connected to the translation process up to the degree that 5′-3′ mRNA degradation follows the last translating ribosome. To study 5′-3′co-translational mRNA decay and the associated ribosome dynamics, here we present an improved high-throughput 5′P degradome RNA sequencing protocol (HT-5Pseq). We exemplify its application in Saccharomyces cerevisiae, but in principle, it could be applied to any other eukaryotic organism. HT-5Pseq is easy, scalable, and uses affordable duplex-specific nuclease-based rRNA depletion.
Before you begin
Overview of the protocol
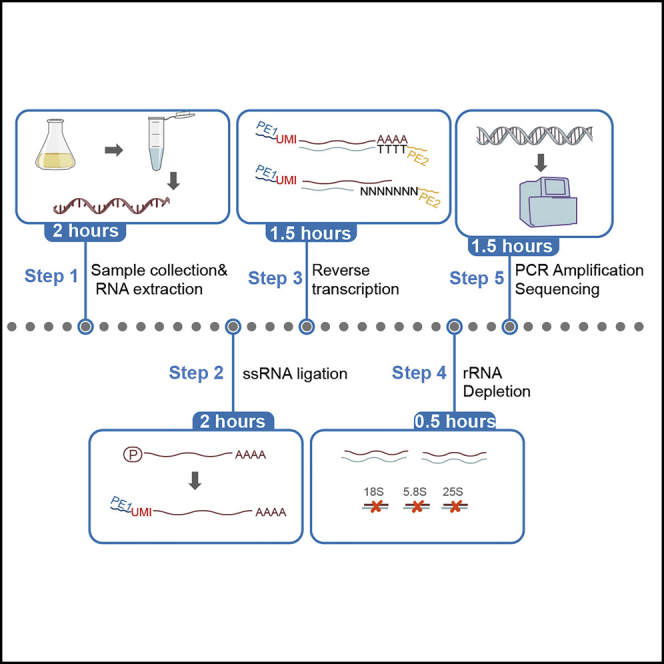
HT-5Pseq captures in vivo 5′P mRNA degradation intermediates by ligating an RNA oligo to the exposed 5′P. After ligation, the RNA molecules are reverse-transcribed to cDNA using oligo-dT and random hexamer as primers. After cDNA library generation, cDNA generated from abundant rRNA molecules is removed using duplex-specific nuclease (DSN) and custom designed DNA probes. The remaining cDNA molecules are used as template for Illumina compatible sequencing library preparation (Figure 1).
Figure 1.

Detailed HT-5PSeq library workflow
Prepare rRNA depletion probes mix
Timing: 1 h
-
1.Prepare the rRNA depletion probes mix:
-
a.Mix equal volumes of the equimolar probes.
-
b.Dilute probes mix from 200 μM to final 2 μM before use.
-
a.
Note: Here we use rRNA depletion probes (Table 1) designed for S. cerevisiae. However, it is possible to design rRNA depletion probes for the organism of interest.
Table 1.
rRNA depletion oligos for S. cerevisiae
| Oligo Name | Sequence 5′ to 3′ |
|---|---|
| 5.8S-1 | TATTCCAGGGGGCATGCCTGTTTGAGCGTCATT |
| 5S-1 | ACCATACGCGAAACTCAGGTGCTGCAATCT |
| 15S-1 | GTAAAAAATTTATAAGAATATGATGTTGGTTCA |
| 15S-2 | TCAGATTAAGCGCTAAATAAGGACATGACACAT |
| 18S-1 | GTTT GACCTCAAATCAGGTAGGAGTACCCGCTGAACT |
| 18S-2 | AAACTTTCAACAACGGATCTCTTGGTTCTCG |
| 18S-3 | TATCTGGTTGATCCTGCCAGTAGTCATATGCTTGTC |
| 18S-4 | CAAAGATTAAGCCATGCATGTCTAAGTATAAGC |
| 18S-5 | CAGTGAAACTGCGAATGGCTCATTAAATCAG |
| 18S-6 | CATGGTATAACTGTGGTAATTCTAGAGCTAATACATGC |
| 18S-7 | ACCCTTTGGAAGAGATGTATTTATTAGAT |
| 18S-8 | TGATGATTCATAATAACTTTTCGAATCGCATGGC |
| 18S-9 | TGGCGATGGTTCATTCAAATTTCTGCCCTATCAACTT |
| 18S-10 | TCCTAATTCAGGGAGGTAGTGACAATAAATAACGA |
| 18S-11 | TACCTTAACGAGGAACAATTGGAGGGCAAGTC |
| 18S-12 | AGCAGCCGCGGTAATTCCAGCTCCAATAGC |
| 18S-13 | CGTGTACTGGATTTCCAACGGGGCCTTT |
| 18S-14 | TTGAAAAAATTAGAGTGTTCAAAGCAGGCGT |
| 18S-15 | TTGCTCGAATATATTAGCATGGAATAATAGAAT |
| 18S-16 | GTAATGATTAATAGGGACGGTCGGGGGCAT |
| 18S-17 | TGCCAAGGACGTTTTCATTAATCAAGAACGA |
| 18S-18 | ACTATGCCGACTAGGGATCGGGTGGTGTT |
| 18S-19 | CTCGGCACCTTACGAGAAATCAAAGTC |
| 18S-20 | AGGTCCAGACACAATAAGGATTGACAGATTGA |
| 18S-21 | AGTTGGTGGAGTGATTTGTCTGCTTAATTGC |
| 18S-22 | CGCTACACTGACGGAGCCAGCGAGTCTAACC |
| 25S-1 | GACCTCAAATCAGGTAGGAGTACCCGCTGAACT |
| 25S-2 | GGAAAAGAAACCAACCGGGATTGCCTTAG |
| 25S-3 | CTTGGAACAGGACGTCATAGAGGGTGAGAATC |
| 25S-4 | AGAGTCGAGTTGTTTGGGAATGCAGCTCTAAGT |
| 25S-5 | CAGTGATGGAAAGATGAAAAGAACTTTGAAAAG |
| 25S-6 | TCTAACGTCTATGCGAGTGTTTGGGTGTAAAAC |
| 25S-7 | CAAGAGGTGCACAATCGACCGATCCTGATGT |
| 25S-8 | ATCGAACCATCTAGTAGCTGGTTCCTGCCGAAGT |
| 25S-9 | ACGTAGAGTTAAGGTGCCGGAATACACGCTC |
| 25S-10 | ATATGGATTCTTCACGGTAACGTAACTGAATG |
| 25S-12 | TCCACAGGAAGGAATAGTTTTCATGCCAGGTCGTAC |
| 25S-13 | TCAAAGTGAAGAAATTCAACCAAGCGCGGGTAA |
| 25S-14 | GAGGGTGTAGAATAAGTGGGAGCTTCGGCGC |
| 25S-15 | ATAGGGAACGTGAGCTGGGTTTAGACCGTCG |
| 25S-16 | CTCTTCCTATCATACCGAAGCAGAATTCGG |
| 25S-17 | ACACAATATAGATGGATACGAATAAGGCGTC |
| 25S-18 | TACTGATGAATGTTACCGCAATAGTAATTG |
| 25S-19 | GAGGAACAGTTCATTCGGATAA |
| 25S-20 | AATCATTTGTATACGACTTAGATGTACAACGGG |
| 21S-1 | GTAAAAAGTAGAATAATAGATTTGAAATA |
| 21S-2 | TAGATTTAAAGAGATAATCATGGAGTATAA |
| 21S-3 | TATAAACTAATAAAGATCAGG |
| 21S-4 | CCGTAATGTAGACCGACTCAGGTATGTAAGTA |
The mixed rRNA depletion oligos used in Step 27. The stock concentration is 200 μM. Working concentration is 2 μM.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Ethanol absolute ≥99.8% | VWR | 20821.330 |
| Glass beads, acid-washed | Sigma-Aldrich | G8772 |
| Acid-Phenol:Chloroform, pH 4.5 (with IAA, 125:24:1) | Thermo Fisher Scientific | AM9722 |
| dNTP set, 100 mM solution | Thermo Fisher Scientific | R0181 |
| Phenol solution. Saturated with 0.1M citrate buffer, pH 4.3 ± 0.2 | Sigma-Aldrich | P4682 |
| Chloroform:isoamyl alcohol 24:1 | Sigma-Aldrich | C0549 |
| Sodium acetate buffer solution, pH 5.3 | Sigma-Aldrich | S7899 |
| Glycoblue coprecipitant (15 mg/mL) | Thermo Fisher Scientific | AM9515 |
| Nuclease-free water, not DEPC treated | Thermo Fisher Scientific | AM9937 |
| RiboLock RNase inhibitor 40 000U/mL | Thermo Fisher Scientific | EO0382 |
| Turbo DNase kit | Thermo Fisher Scientific | AM1907 |
| T4 RNA ligase 1 | NEB | M0204L |
| SuperScript™ II Reverse Transcriptase | Thermo Fisher Scientific | 18064071 |
| Phusion®High-Fidelity PCR Master Mix | NEB | M0531S |
| AMPure XP | Beckman Coulter | A63881 |
| RNAClean XP | Beckman Coulter | A63987 |
| Duplex-specific nuclease | Evrogen | EA002 |
| Critical commercial assays | ||
| High Sensitivity DNA kit | Agilent | 5067-4626 |
| Qubit™ dsDNA HS Assay Kit | Thermo Fisher Scientific | Q32854 |
| Qubit™ RNA HS assay kit | Thermo Fisher Scientific | Q32852 |
| Deposited data | ||
| The raw and processed sequencing data | This paper | GEO: GSE152375 |
| Experimental models: organisms/strains | ||
| Saccharomyces cerevisiae strains: BY4741:(MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) | NA | NA |
| Oligonucleotides | ||
| See Tables 1 and 2 | This paper | N/A |
| Software and algorithms | ||
| Fivepseq package | GitHub | Nersisyan et al., 2020 |
| bcl2fastq v2.20.0 | Illumina | https://emea.support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html |
| Cutadapt | GitHub | https://github.com/marcelm/cutadapt/ |
| UMI-tools | GitHub | Smith et al., 2017 |
| STAR 2.7.0 | GitHub | Dobin et al., 2013 |
| DESeq2 | Bioconductor | Love et al., 2014 |
| Subread package | GitHub | Liao et al., 2014 |
| IGV | https://igv.org | Thorvaldsdóttir et al., 2013 |
| RStudio version 3.5.0 | RStudio, Inc., Boston, MA | N/A |
| Equipment | ||
| Refrigerated benchtop centrifuge | N/A | N/A |
| Thermo-block | N/A | N/A |
| Agilent Bioanalyzer 2100 | Agilent | Bioanalyzer 2100 |
| Chemical hood | N/A | N/A |
| Qubit Fluorometer | Invitrogen | QuBit 2.0 |
| Magnet stand for PCR tubes | N/A | N/A |
| Vortex mixer | Scientific Industries | Vortex-Genie 2 |
Materials and equipment
LET lysis buffer
| Reagent | Final Concentration |
|---|---|
| Tris, pH 8.0 | 25 mM |
| EDTA, pH 8.0 | 20 mM |
| LiCl | 100 mM |
Filter sterilize the LET lysis buffer. Use RNAse-free water. Possible for long term storage at 20°C–25°C.
Step-by-step method details
RNA extraction
-
1.Obtain total RNA from sample of interest
-
a.Harvest 1.5 mL cells from mid-log grown yeast (OD600 density ~ 0.6–0.8) using a standard bench microcentrifuge at 30 s, 8000 rpm at room temperature.
-
b.Extract total RNA using phenol-chloroform extraction method.
-
i.Set up the following tubes:
- Tube 1: 500 μL phenol:chloroform:IAA (125:24:1)
- Tube 2: 500 μL chloroform:IAA (24:1)
- Tube 3: 40 μL 3M NaOAc (made with nuclease-free H2O)
-
ii.Add (approx. 200 μL) of glass beads and 150 μL LET in yeast cell pellet.
-
iii.Add 150 μL phenol. Vortex at 20°C–25°C in vortex mixer for 2 min at top speed.
-
iv.Add 250 μL nuclease-free H2O and 250 μL phenol:chloroform:IAA.
-
v.Vortex in vortex mixer for additional 2 min followed by centrifugation at 4°C for 2 min at 14,000 × g.
-
vi.Remove aqueous phase (approx. 450 μL) and add to Tube #1. Vortex 30 sec and spin 1 min at 14,000 × g.
-
vii.Remove aqueous phase and add to Tube #2. Vortex 30 sec and spin 1 min at 14,000 rpm.
-
viii.Remove aqueous phase (approx. 400 μL) and add to Tube #3 mix well. Add 1 mL 95% (vol/vol) ethanol, mix and place at −20°C/−80°C, 30 min.
-
ix.Collect RNA by centrifugation at 4°C for 20 min at 14,000 × g.
-
x.Wash pellet with 500 μL 70% (vol/vol) cold ethanol and re-centrifuged for 10 min.
-
xi.Drain supernatant and air-dry pellet for ~ 3 min.
-
xii.Resuspend pellet in 10 μL nuclease-free water.
-
i.
-
a.
CRITICAL: Phenol and Chloroform are acute toxic. Be careful when handling phenol and chloroform, always wear gloves, lab coat and perform manipulations following local safety regulations in fume hood.
-
2.
Check RNA quality by running a Bioanalyzer RNA gel or an agarose gel.
Note: Any alternative approach producing high-quality RNA may be used instead.
Removal of DNA from RNA samples
Any contaminant DNA is removed from the sample.
-
3.
Starting with 6 μg of total RNA, prepare the following mix and incubate the samples for 20 min at 37°C.
| Reagent | Final Concentration | Amount |
|---|---|---|
| TURBO DNAse buffer (10×) | 1× | 1 μL |
| TURBO DNAse (2 U/μL) | 0.06 U/μL | 0.3 μL |
| Ribolock (40 U/μL) | 1.2 U/μL | 0.3 μL |
| Sample RNA | 0.7 μg/μL | 8.4 μL |
| Total | 10 μL |
Note: It is possible to lower starting material to 500 ng in total. However, low input material usually decreases library complexity and increase the PCR duplicates.
-
4.
Add 2 μL of TURBO DNAse inactivation reagent and incubate 5 min at 20°C–25°C (tapping once in a while).
-
5.
Centrifugate at 14,000 × g for 2 min at 20°C–25°C and transfer the supernatant to a clean precooled tube.
Note: This pellet is normally quite loose, repeat the centrifugation if it is resuspended and avoid the carryover of any DNase inactivation reagent.
-
6.
Ethanol precipitate the DNA-free RNA by adding 2.5 volumes (with respect to the sample volume) of 95% (vol/vol) ethanol, a 1/10 volume of 3 M sodium acetate, 1 μL of glycoblue. Mix sample by gently inverting and incubate it for minimum 30 min at −20°C/−80°C.
Pause point: The ethanol precipitation can be left 16–18 h at −20°C/−80°C.
-
7.
Centrifugate at 14,000 × g for 30 min at 4°C to precipitate the RNA.
-
8.
Wash the pellet with 500 μL of cold 70% (vol/vol).
-
9.
Centrifugate 14,000 × g for 10 min at 4°C.
-
10.
Remove the remaining ethanol, air-dry pellet for 3 min and resuspend it in 1.8 μL of RNAse-free water.
Note: If the next step is the single-strand RNA ligation, RNA can be directly resuspended in ligation mix (step 11) and top up RNAse-free water to 10 μL .
Single-strand RNA ligation
5′ phosphate molecules are ligated with RNA oligos including unique molecular identifiers (UMI).
-
11.
Prepare a 10 μL reaction mix with the components listed below:
| Reagent | Final Concentration | Amount |
|---|---|---|
| T4 RNA ligase buffer (10×) | 1× | 1 μL |
| rP5 _RND oligo (100 μM) | 10 μM | 1 μL |
| ATP (10 mM) | 1 mM | 1 μL |
| Ribolock (40 U/μL) | 0.8 U/μL | 0.2 μL |
| T4 RNA ligase 1 (10 U/μL) | 1 U/μL | 1 μL |
| PEG8000 (50%) | 20% | 4 μL |
| DNA-free RNA sample | 1.8 μL | |
| Total | 10 μL |
-
12.
Incubate sample at 25°C for 2 h.
-
13.
Increase the sample volume with RNase-free water to 40 μL.
-
14.Purify the sample using 1.8× volumes of RNAClean XP beads, as described by the manufacturer’s instruction.
-
a.Add 72 μL of RNAClean XP beads and mix sample by several times pipetting up and down.
-
b.Incubate at 20°C–25°C for 5 mins until RNA bind to beads.
-
a.
-
15.Place the PCR tubes at the magnets stand and wait till the solution is clear (~2 mins)
-
a.Remove the supernatant.
-
b.Wash beads twice with 200 μL of freshly made 70% (vol/vol) ethanol.
-
a.
-
16.Remove the ethanol and let the beads slightly dry for 1 min.
-
a.Elute samples in 12 μL RNase-free water.
-
a.
Note: avoid over drying the beads as that might result in sample loss.
Reverse transcription
cDNA library is transcribed by the defined ratio of oligo-dT and random hexamer.
-
17.
Prepare a reaction mix as the components listed below:
| Reagent | Final Concentration | Amount |
|---|---|---|
| Total ligated RNA | 10.6 μL | |
| 5PSeq RT oligo (20 μM) | 1 μM | 1 μL |
| 5PSeq dT oligo (0.05 μM) | 0.5 nM | 0.2 μL |
| dNTPs (10 mM) | 0.05 mM | 0.1 μL |
| Total | 12.8 μL |
-
18.
Denature the sample at 65°C for 5 min. Then place on ice directly.
-
19.
To each tube, add 6.2 μL of mixture containing the following components:
| Reagent | Final Concentration | Amount |
|---|---|---|
| First-strand buffer (5×) | 1× | 4 μL |
| DTT (100 mM) | 10 mM | 2 μL |
| Ribolock (40 U/μL) | 0.4 U/μL | 0.2 μL |
| Total | 6.2 μL |
-
20.
Add 1 μL of SuperScript II reverse transcriptase to each tube.
-
21.
Incubate the sample at 25°C for 10 min, 42°C for 50 min and inactivate reaction at 70°C for 15 min.
Note: Any commonly reverse transcriptase could be used instead of SuperScript II.
Remove template RNA
Removing any excess of RNA, avoids that cDNA-RNA duplexes are degraded during duplex-specific nuclease (DSN) treatment.
-
22.
Remove the template RNA by adding 100 mM NaOH (8 μL) to the sample (from step 21), and incubate at 65°C for 20 min.
-
23.
Neutralized sample by adding 100 mM Tris-HCl, pH = 7.0 (8 μL).
Note: It is also possible to remove excess RNA in cDNA-RNA duplex by using RNaseH.
-
24.Purify the sample using 1.8× volumes of Ampure XP beads, as described by the manufacturer’s instruction.
-
a.Add 64.8 μL of Ampure XP beads and mix sample by several times pipetting up and down.
-
b.Incubate at 20°C–25°C for 5 mins until samples bind to beads.
-
a.
-
25.Place the PCR tubes at the magnets stand and wait till the solution is clear (~2 mins).
-
a.Remove the supernatant.
-
b.Wash beads twice with 200 μL of freshly made 70% (vol/vol) ethanol.
-
a.
-
26.
Remove the residual ethanol and allow the beads to slightly dry for 1 min. Elute in 8 μL of RNase-free water.
Note: avoid over drying the beads as that might result in sample loss. If Ampure XP beads is not available, alternative beads such as MagSi magnetic beads (cat: MDKT00010075) can also be used.
Duplex-specific nuclease (DSN) for rRNA depletion
rRNA depletion is based on the designed DNA probes targeting ribosomal RNA (i.e., RDN18, RDN25, RDN5.8, RDN5).
-
27.
Set up the following 16 μL reaction:
| Reagent | Final Concentration | Amount |
|---|---|---|
| Yeast rRNA depletion Probes (2 μM) | 0.5 μM | 4 μL |
| DSN master buffer (4×) | 1× | 4 μL |
| Sample from 26 | 8 μL | |
| Total | 16 μL |
-
28.
Denature sample for 2 min at 98°C using thermocycler.
-
29.
Incubate the sample for 5 min at 68°C.
-
30.
Add pre-warmed (2 min at 68°C) mix containing the following components:
| Reagent | Final Concentration | Amount |
|---|---|---|
| DSN enzyme (1 U/μL) | 0.25 U/ μL | 1 μL |
| DSN master buffer(4×) | 1× | 1 μL |
| H2O | 2 μL | |
| Total | 4μL |
-
31.
Mix sample by pipetting several times and incubate for 20 min at 68°C in a thermocycler.
-
32.
To inactivate DSN enzyme, add 20 μL of DSN stop solution (2×), mix contents and spin the tube briefly in a micro-centrifuge.
-
33.
Incubate the sample for 10 min at 68°C temperature.
-
34.Purify the sample using 1.8× volumes of Ampure XP beads, as described by the manufacturer’s instruction.
-
a.Add 72 μL of Ampure XP beads and mix sample by several times pipetting up and down.
-
b.Incubate at 20°C–25°C for 5 mins until samples bind to beads.
-
a.
-
35.Place the PCR tubes at the magnets stand and wait till the solution is clear (~2 min).
-
a.Remove the supernatant.
-
b.Wash beads twice with 200 μL of freshly made 70% (vol/vol) ethanol.
-
a.
-
36.
Remove the residual ethanol and allow the beads to slightly dry for 1 min. Elute in 9.6 μL of RNase-free water.
Note: avoid over drying the beads as that might result in sample loss.
Library PCR amplification
PCR is used to generate an Illumina compatible sequencing library
-
37.
To amplify the library by PCR, prepare the following mix:
| Reagent | Final Concentration | Amount |
|---|---|---|
| Phusion high-fidelity PCR master-mix (2×) | 1× | 10 μL |
| Illumina compatible PE1.0 (10 μM) | 0.1 μM | 0.2 μL |
| Illumina compatible PE2_MPX (10 μM) | 0.1 μM | 0.2 μL |
| Sample (from step 36) | 9.6 μL | |
| Total | 20 μL |
Table2.
Oligonucleotides used for HT-5Pseq library preparation
| Primer name | Purpose | Sequence (5′-3′) |
|---|---|---|
| RNA_rP5_RND | To add UMI and common sequence to the RNAs (step 11) | rCrUrUrUrCrCrCrUrArCrArCrGrArCrGrCrUrCrUrUrCrCrGrArUrCrU rNrNrNrNrNrNrNrN |
| 5Pseq-dT | To do reverse transcription by using oligo-dT and add common sequence to the libraries (step 17) | GTGACTGGAGTTCAGACGTGTGCTC TTCCGATCT TTTTTTTTTT |
| 5Pseq-RT | To do reverse transcription by using random hexamer and add common sequence to the libraries (step 17) | GTGACTGGAGTTCAGACGTGTGCTC TTCCGATCT NNNNNN |
| Illumina compatible PE1.0 | To generate library by PCR (step 37) | ATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACG ACGCTCTTCCGATC∗T |
| PE2_MPX_01 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_02 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATACATCGGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_03 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATGCCTAAGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_04 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATTGGTCAGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_05 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATCACTGTGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_06 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATATTGGCGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_07 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATGATCTGGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_08 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATTCAAGTGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_09 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATCTGATCGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_10 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATAAGCTAGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_11 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATGTAGCCGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_12 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATTACAAGGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_13 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATTTGACTGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_14 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATGGAACTGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_15 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATTGACATGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_16 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATGGACGGGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_17 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATCTCTACGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_18 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATGCGGACGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_19 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATTTTCACGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_20 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATGGCCACGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_21 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATCGAAACGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_22 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATCGTACGGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_23 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATCCACTCGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_24 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATGCTACCGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_25 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATATCAGTGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_26 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATGCTCATGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_27 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATAGGAATGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_28 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATCTTTTGGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_29 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATTAGTTGGTGACTGGAGT TCAGACGTGTGCTCTTCCGATC∗T |
| PE2_MPX_30 | To add multiplex barcode by PCR (step 37) | CAAGCAGAAGACGGCATACGAGATCCGGTGGTGACTGGAG TTCAGACGTGTGCTCTTCCGATC∗T |
refers to S-linkage between the two bases r refers to a RNA base. Barcodes are identified in bold black. Sequences are represented in 5′ to 3′orientation. # Oligonucleotide sequences © 2006–2021 Illumina, Inc. All rights reserved.
Note: Any good quality used thermostable DNA polymerase used for sequencing library preparation could be used instead.
-
38.
Run the following PCR program:
| PCR Cycling Conditions | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Initial Denaturation | 98°C | 30 sec | 1 |
| Denaturation | 98°C | 20 sec | 14–18 cycles |
| Annealing | 65°C | 30 sec | |
| Extension | 72°C | 30 sec | |
| Final extension | 72°C | 7 min | 1 |
| Hold | 4°C | ||
-
39.
Bring the volume to 100 μL with nuclease-free water.
-
40.
Add 0.7× Ampure XP beads to the sample to bind the long DNA molecules. For 100 μL of sample, add 70 μL of beads and mix by pipetting. Incubate at 20°C–25°C for 5 mins.
-
41.
Place the PCR tube on the magnets stand and wait till the solution is clear. Transfer the supernatant to a new tube.
-
42.
Add with 0.2× Vol. (respect to the original sample volume) of Ampure XP beads (20 μL respect to 100 μL) to the recovered supernatant from step 41. Mix the sample by pipetting up and down and incubate at 20°C–25°C for 5 mins.
-
43.Place the PCR tubes at the magnets stand and wait till the solution is clear (~2 mins).
-
a.Remove the supernatant
-
b.Wash beads twice with 200 μL of freshly made 70% (vol/vol) ethanol.
-
a.
-
44.
Remove the residual ethanol and allow the beads to slightly dry for 1 min. Elute in 10 μL in water or EB buffer (10 mM Tris-HCl, pH 8.0).
-
45.
Measure the final library concentration by Qubit using the dsDNA HS assay kit and check the size distribution by Bioanalyzer.
-
46.
Sequence the libraries using pair-end 75 cycles Illumina NextSeq 500.
Note: For pair-end 75 cycles sequencing in this case, using 60 bp for read1 and 15 bp for read2. Read2 will identify the molecule primed by either oligo-dT or random hexamer. In general, we recommend at least 6 million raw reads per yeast sample. Any alternative Illumina platform could be used instead of a NextSeq 500. Read sequencing length can be altered depending on the complexity of the genome of interest and the ability to uniquely map reads to the genome.
Expected outcomes
This protocol will generate sequencing libraries of 5′P mRNA degradation intermediates, detailed workflow is shown in Figure 1. In the final HT-5Pseq library, the average size is expected to be around 450 bp, including 150 bp Illumina adapter sequences (Figure 2). The expected concentration of library can be 0.5–2 ng/μL. The sequencing depth required will depend on the library complexity and analysis requirement. By mapping HT-5Pseq reads to the reference genome, the expected results are as following: 1) reads coverage is distributed along the whole mRNA regions (Figure 3); 2) rRNA contamination of HT-5Pseq library is less than 12%–20% (Figure 4); 3) A clear 3-nt pattern can be observed with respect to specific codons, including start and stop codon at metagene level (Figure 5); 4) Codon-specific/amino acid specific pausing can be extracted with respect to specific codons.
Figure 2.

Example of a size-selected HT-5PSeq pool libraries (step 45)
Figure 3.

Example of HT-5PSeq reads visualized by IGV (Thorvaldsdóttir et al., 2013) for S. cerevisiae
Two Biological replicates are shown.
Figure 4.

Distribution of mRNA, rRNA reads in S. cerevisiae HT-5Pseq after rRNA depletion
NonDSN refers to control libraries omitting DSN rRNA depletion. NonProbe refers to libraries treated with DSN but omitting the depletion oligos (Data from (Zhang and Pelechano, 2021)). Two biological replicates are shown.
Figure 5.

HT-5Pseq reveals ribosome dynamics at codon resolution (Data from (Zhang and Pelechano, 2021))
(A) Metagene analysis for 5 ´P read coverage relative to arginine (CGA). Dotted lines at -17 corresponding to the expected 5′ end of protected ribosome located at A site.
(B) 3-nt periodicity shown in 5′P read by Fourier transform calculation.
Quantification and statistical analysis
Here we provide a potential bioinformatic pipeline for 5′Pseq data.
-
•
De-multiplex raw data using the indexing information: Using bcl2fastq (v2.20.0) for base-calling. We recommend allowing 1 mismatch in index 1 and 1 mismatch in index2.
-
•
Trim sequencing adaptor: Use cutadapt V1.16 to trim sequencing adapter (-a AGATCGGAAGAGCACACGTCTGAACTCCAGTC).
-
•
Extract UMI: Use UMI-tools (v0.5.4) to extract 8-nt random barcodes on the 5′ ends of reads. These UMI information will be used to remove PCR duplicates.
-
•
Align sequencing reads: Use star/2.7.0 (Dobin et al., 2013) to align 5′-end reads to reference genome (SGD R64-1-1 for S. cerevisiae genome). For mapping the 5′-ends reads to the genome, we recommend using the parameter --alignEndsType Extend5pOfRead1 to exclude soft-clipped bases on the 5′end.
-
•
Remove PCR duplicates: Use UMI-tools (v0.5.4) to remove duplicated 5′ ends of read introduced by PCR during library preparation.
-
•
Quantify transcripts: Use Subread package (featureCounts) (Liao et al., 2014) to count mRNA, tRNA, rRNA and snRNA and snoRNA transcripts. Use DESeq2 packages from R and Bioconductor (Love et al., 2014) to perform differential gene expression analysis.
-
•
Analysis 5′ ends positions: Use Fivepseq package to map 5′ ends with respect to start, stop codon and codons at metagene level (Nersisyan et al., 2020).
Limitations
Although HT-5PSeq offers high quality degradome information at a fraction of the costs and with significantly decreased hands-on time in comparison with standard 5PSeq (Pelechano et al., 2015), this approach has several limitations that need to be accounted for.
Firstly, the main limitation is that HT-5Pseq approach focus on the subpopulation of 5′ end of mRNA undergoing decay. Therefore, any exposed 5′P end of molecule can be captured independent of their relationship with co-translation degradation process.
Next, HT-5Pseq measures the kinetics competition of 5′-3′ degradation machinery and ribosome, therefore we do not recommend this approach to directly measure absolute translation rates. However, we have shown that the last translating ribosome during co-translational decay can infer the general ribosome dynamics (Pelechano et al., 2015).
Thirdly, as the abundance of 5′P end molecule depends on both translation and mRNA stability, any factor involved in those process can affect the observed 5′P seq profile. For example, when investigating mRNA degradation profiles in xrn1Δ the ribosome associated 3-nt pattern is greatly decreased (Pelechano et al., 2015). In xrn1Δ the observed 5′P profiles reflects a combination of transcription start site mapping (as expected from the exposed 5′P after decapping) complemented by other endonucleolitica cleavages events. In addition, HT-5Pseq libraries may vary in library complexity as a result of the variation of fractions on mRNA degradation intermediates present in a sample in respect to the total RNA. For example, HT-5PSeq libraries from xrn1Δ cells are in general more complex, as 5′P mRNA degradation intermediates are not efficiently removed and thus represent a higher proportion of the total RNA population. To control for this, we add UMI during the RNA ligation step. If a lower fraction of mRNA degradation intermediates is expected, we recommend increasing the amount of total RNA starting material.
Troubleshooting
Problem 1
RNA degradation
Potential solution
-
1)
When handling with RNA samples, always keep RNA on the ice.
-
2)
Check the RNA integrity of RNA extraction.
-
3)
Use RNase inhibitor during the protocol (steps 11 and 19) and aliquot RNase-free reagents.
-
4)
Perform RNA extraction with phenol-chloroform (step 1) as fast as possible.
Problem 2
Low yield DNA library (step 45)
Potential solution
-
1)
If starting RNA is less, increase the starting RNA amount.
-
2)
Inefficient removal of RNA template (step 22), this will loss the cDNA library after DSN treatment. Optimize RNA removal steps (step 22).
-
3)
Increase few PCR cycles in PCR amplification steps (step 38). Final PCR cycles should be less than 20 cycles.
-
4)
For bead cleanup, do not over dry the beads. That might lead to sample loss.
Problem 3
Large number of rRNA reads
Potential solution
-
1)
DNA leftover in RNA sample can potential saturate DSN enzymatic activity (step 27). This may decrease the rRNA depletion efficiency. Perform DNase treatment to RNA samples (step 3).
-
2)
Optimize DSN for rRNA depletion if using custom depletion probes (step 27–36). Optimize mix annealing temperature (steps 29–30) based on the melting temperature of newly designed probes. Mix samples with probes by pipetting and keep them on the thermocycle at the selected temperature.
Problem 4
Large number of PCR duplicates
Potential solution
-
3)
Increase the input RNA material to increase the complexity.
-
4)
Decrease the final PCR cycles (from step 38) that will increase the useful reads.
Problem 5
Biased 5′P reads to 3′ end
Potential solution
-
5)
If the 5′P reads biased towards to 3′, decrease the usage of oligo-dT in reverse-transcription (step 17) to get more homogenous distribution profile.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Vicent Pelechano (vicente.pelechano.garcia@ki.se) .
Materials availability
This study did not generate new unique materials nor reagents.
Data and code availability
The raw and processed sequencing data are deposited at GEO with accession number GSE152375.
Acknowledgments
We thank all members of the Pelechano, Kutter and Friedländer laboratories for useful discussions. We kindly thank Lilit Nersisyan and Jingwen Wang for bioinformatic support. Computational analysis was performed on resources provided by SNIC through Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX). This project was funded by the Swedish Foundation’s Starting Grant (Ragnar Söderberg Foundation), a Wallenberg Academy Fellowship (KAW 2016.0123), the Swedish Research Council (VR 2016-01842), Karolinska Institutet (SciLifeLab Fellowship, SFO and KI funds) and a Joint China-Sweden mobility grant (STINT, CH2018-7750) to V.P. Y.Z. is funded by a fellowship from the China Scholarship Council.
Author contributions
V.P. and Y.Z. conceived the project. Y.Z. developed the methods and performed the experiments and analysis. Y.Z. and V.P. wrote the protocol.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Yujie Zhang, Email: yujie.zhang@ki.se.
Vicent Pelechano, Email: vicente.pelechano.garcia@ki.se.
References
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:1–21. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nersisyan L., Ropat M., Pelechano V. Improved computational analysis of ribosome dynamics from 5′P degradome data using fivepseq. NAR Genom. Bioinform. 2020;2:1–12. doi: 10.1093/nargab/lqaa099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V., Wei W., Steinmetz L.M. Widespread co-translational RNA decay reveals ribosome dynamics. Cell. 2015;161:1400–1412. doi: 10.1016/j.cell.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T., Heger A., Sudbery I. UMI-tools: Modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Research. 2017;27:491–499. doi: 10.1101/gr.209601.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H., Robinson J.T., Mesirov J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Pelechano V. Cell Reports Methods; 2021. High-Throughput 5′P Sequencing Enables the Study of Degradation-Associated Ribosome Stalls. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw and processed sequencing data are deposited at GEO with accession number GSE152375.