

Summary
Ovarian cancer (OC) is the most lethal gynecological cancer. Faithful research models are indispensable to the progression of understanding OC etiology and therapy. Here, we provide a detailed protocol for establishing organoid cultures from patient OC biopsies. The organoids reproduce primary tumor- and patient-specific characteristics including phenotypic properties and genomic aberrations and exhibit patient-dependent responsiveness to drugs. OC-derived organoids provide powerful tools to gain deep insight into the cancer’s pathobiology and to screen patient-tumor drug sensitivity to progress toward personalized medicine.
For complete details on the use and execution of this protocol, please refer to Maenhoudt et al. (2020).
Subject areas: Cell culture, Cancer, Organoids
Graphical abstract
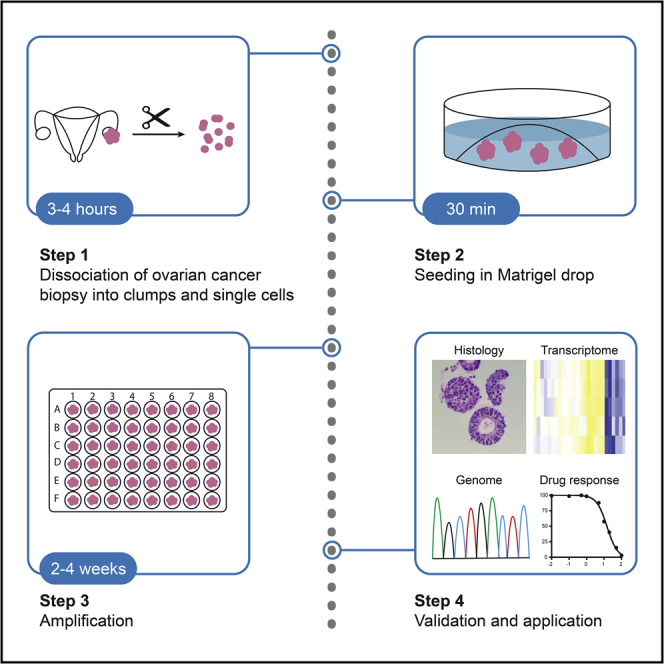
Highlights
-
•
Detailed protocol for the establishment and expansion of ovarian cancer organoids
-
•
Ovarian cancer organoids recapitulate key characteristics of the primary tumor
-
•
Personalized drug screening assays can be performed using ovarian cancer organoids
Ovarian cancer (OC) is the most lethal gynecological cancer. Faithful research models are indispensable to the progression of understanding OC etiology and therapy. Here, we provide a detailed protocol for establishing organoid cultures from patient OC biopsies. The organoids reproduce primary tumor- and patient-specific characteristics including phenotypic properties and genomic aberrations and exhibit patient-dependent responsiveness to drugs. OC-derived organoids provide powerful tools to gain deep insight into the cancer’s pathobiology and to screen patient-tumor drug sensitivity to progress toward personalized medicine.
Before you begin
-
1.
A complete list of reagents and labware can be found in the key resources table.
-
2.
Pre-warm a 48-well culture plate for 15–20 h in a 37°C incubator.
-
3.
All procedures, except for the paraffin-embedding of organoids, are performed under sterile conditions in a biological safety cabinet.
-
4.
Thaw Matrigel bottle for 15–20 h on ice and aliquot 1 mL in Eppendorf tubes.
-
5.
Thaw Matrigel aliquot on ice (4°C) for minimum 1 h before use.
-
6.
Cool centrifuges to 4°C.
-
7.
Before passaging of the organoids, cool 10 mL DMEM/F12 in a conical tube on ice.
-
8.Prepare the following media and sterile-filter using a 0.22 μm filter (see “materials and equipment” for detailed recipes):
-
a.Ovarian cancer organoid medium (OCOM; typically 20 mL)
-
b.Collection medium (10 mL)
-
c.Dissociation medium (8 mL)
-
d.Cryopreservation medium (20 mL)
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| DMEM/F12 | Gibco | 31330-038 |
| L-Glutamine | Thermo Scientific | 25030-024 |
| Penicillin-streptomycin (Pen/Strep) | Gibco | 15140-122 |
| A83-01 (TGFβ pathway inhibitor) | Sigma-Aldrich | SML0788 |
| Nicotinamide | Sigma-Aldrich | N0636 |
| N2 | Gibco | 17502-048 |
| B27 minus vitamin A | Gibco | 12587-010 |
| N-Acetylcysteine | Sigma-Aldrich | A7250 |
| 17-β Estradiol | Sigma-Aldrich | E2758 |
| SB203580 (p38 MAP kinase inhibitor) | Biotechne (Tocris) | 1264 |
| Human epidermal growth factor (EGF) | R&D Systems | 236-EG |
| Human noggin | PeproTech | 120-10C |
| Human R-spondin 1 (RSPO1) | PeproTech | 120-38 |
| Human insulin-like growth factor-I (IGF1) | PeproTech | 100-11 |
| Human hepatocyte growth factor (HGF) | PeproTech | 100-39 |
| Human Neuregulin-1 (NRG1) | PeproTech | 100-03 |
| InSolution Y-27632 (ROCK Inhibitor, RI) | Sigma-Aldrich | 688001 |
| Fetal bovine serum (FBS) | Sigma-Aldrich | F7524 |
| Dimethylsulfoxide (DMSO) | Sigma-Aldrich | D2650 |
| Phosphate buffered saline (PBS) | Gibco | 10010-015 |
| Collagenase IV | Gibco | 17104-019 |
| Matrigel (growth factor-reduced; phenol red-free) | Corning | 356231 |
| TrypLE express | Gibco | 12605-010 |
| Paraformaldehyde (PFA), 16% | Merck | 8.18715 |
| DNase | Sigma-Aldrich | D5025 |
| Ethanol absolute, ≥99.8% (EtOH) | Fisher Chemical | E/0650DF/15 |
| N-Butanol | Honeywell | 33065 |
| XTT (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide) | Invitrogen | X6493 |
| PMS (N-methyl dibenzopyrazine methyl sulfate) | J&K Scientific | 276473 |
| Biological samples | ||
| Biopsies derived from patients with ovarian cancer | N/A | N/A |
| Software and algorithms | ||
| GraphPad Prism V.8.0.1 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| 48-well flat bottom plates | Corning | 3548 |
| 96-well flat bottom plates | Thermo Scientific | 167008 |
| Steriflip-GP Sterile Centrifuge Tube Top Filter Unit, 0.22 μm | Millipore | SCGP00525 |
| Cryovials | Thermo Fisher Scientific | 375353 |
| Flat-bottom tubes of glass | Karl Hecht | 42779065 |
| 1.5 mL Eppendorf tube | Eppendorf | 0030120.086 |
| 15 mL Centrifuge tube | Corning | 430052 |
| 50 mL Conical tube | Corning | 430290 |
| 30 mL Conical tube | Fisher Scientific | 11339133 |
| Sterile 10 μL pipette tips with filter | Greiner | 771288 |
| Sterile 20 μL pipette tips with filter | Greiner | 774288 |
| Sterile 200 μL pipette tips with and without filter | Greiner | 739288 |
| Sterile 1000 μL pipette tips with filter | Greiner | 740288 |
| Pipette (P10, P20, P200, P1000) | Eppendorf or others | 2231300006 |
| Scalpel (surgical blade) | Swann-Morton | 0207 |
| Petri dish | Corning | 353002 |
| Plastic transfer pipette (3.5 mL) | Sarstedt | 86.1171.001 |
| Block heater | VWR | 460-2116 |
| 70 μm Cell strainer | Corning (Falcon) | 352350 |
| Stainless steel base molds | Simport | M474-2 |
| Millex-GV Syringe Filter Unit, 0.22 μm | Millipore | SLGV033R |
| Cryobox (Mr. Frosty Freezing Container) | Thermo Scientific | 5100-0001 |
| 37°C Water bath | Thermo Fisher | 15187015 |
| Shaking plate | VWR Collection | 444-2900 |
| 37°C and 5% CO2 tissue culture incubator | Thermo Fisher | 51026282 |
| Inverted microscope (such as Axiovert 40 CFL Trinocular) | Zeiss | N/A |
| Bunsen burner | N/A | N/A |
| Biological safety cabinets | Esco | AC2-6S8 |
| Chemical hood | N/A | N/A |
| HistoStar Embedding Workstation | Thermo Scientific | A81000001 |
| Cartridge burner | Usbeck | 1420 |
| Microplate reader VICTOR | PerkinElmer | 2030-0050 |
Materials and equipment
Ovarian cancer organoid medium
Prepare OCOM as follows:
-
•
Thaw frozen aliquots of growth factors on ice.
-
•
First bring DMEM/F12 into a conical tube, then add the growth factors.
-
•
Filter through a 0.22 μm filter.
-
•
Store the medium at 4°C (up to 2 weeks).
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM/F12 | - | 18.365 mL |
| L-glutamine (100×) | 1× | 200 μL |
| Pen/Strep (100×) | 1× | 200 μL |
| A83-01 (500 μM) | 0.25 μM | 10 μL |
| Nicotinamide (1 M) | 5 mM | 100 μL |
| N2 (100×) | 1× | 200 μL |
| B27 minus vitamin A (50×) | 1× | 400 μL |
| N-acetylcysteine (500 mM) | 1.25 mM | 50 μL |
| 17-β Estradiol (740 nM) | 10 nM | 270.27 μL |
| p38i (SB203580) (30 mM) | 1 μM | 0.67 μL |
| EGF (100 μg/mL) | 50 ng/mL | 10 μL |
| Noggin (250 μg/mL) | 100 ng/mL | 8 μL |
| RSPO1 (100 μg/mL) | 50 ng/mL | 10 μL |
| IGF1 (10 μg/mL) | 20 ng/mL | 40 μL |
| HGF (10 μg/mL) | 10 ng/mL | 20 μL |
| NRG1 (100 μg/mL) | 50 ng/mL | 10 μL |
| Y27632 (RI)a (10 mM) | 10 μM | 20 μL |
Y27632 (RI) is added to OCOM only for initiation of the organoid culture (i.e., at initial seeding of the dissociated tissue) and after each passaging (both until first medium refreshment), thus not for culture maintenance.
Collection medium
| Reagent | Fraction | Amount |
|---|---|---|
| DMEM/F12 | 88% | 8.8 mL |
| Pen/Strep | 2% | 0.2 mL |
| FBS | 10% | 1 mL |
Store at 4°C (up to 1 month).
Note: The added serum supports the viability of the collected tissue and has no negative impact on organoid establishment.
Dissociation medium
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM/F12 | - | 8 mL |
| Collagenase IV | 1 mg/mL | 8 mg |
This solution should be freshly prepared and used immediately.
Prepare dissociation medium as follows:
-
•
Bring 8 mg Collagenase IV into a 30 mL conical tube.
-
•
Add 8 mL of DMEM/F12 and mix by inverting the tube.
-
•
Place the dissociation medium in a 37°C water bath for 20 min before use to completely dissolve the Collagenase IV.
-
•
Filter through a 0.22 μm filter.
Cryopreservation medium
| Reagent | Fraction | Amount |
|---|---|---|
| DMEM/F12 | 60% | 6 mL |
| FBS | 30% | 3 mL |
| DMSO | 10% | 1 mL |
Store at 4°C protected from light (up to 1 month).
Step-by-step method details
Establishing organoids from patient OC biopsies
Timing: 3–4 h
With this protocol, we establish organoids from OC biopsies (primary or metastatic). Patient-derived biopsies (obtained under informed consent) are enzymatically and mechanically dissociated into small clumps and single cells. The resultant suspension is mixed with Matrigel, and droplets are deposited in a 48-well culture plate. After adding OCOM, organoids develop, grow and expand.
-
1.
The OC tissue biopsy is collected in a conical tube with 10 mL collection medium on ice and processed immediately.
-
2.
The tissue is transferred to a Petri dish in the biological safety cabinet and rinsed extensively with 5 mL PBS using a plastic transfer pipet to wash away blood and FBS.
-
3.
Repeat the rinsing until the PBS becomes clear.
-
4.The tumor biopsy is split into three parts, i.e., one for cryopreservation, one for fixation/histology and one for organoid formation (Figure 1). If the biopsy is small (≤ 5 mm3), it is recommended to use the complete tissue for organoid formation.Note: For PFA fixation of primary tumor (to perform immunohistological analysis; Figure 1)
-
a.Cut a piece (∼4 mm3) of the tumor tissue using a scalpel.
-
b.With a plastic transfer pipet, transfer the tissue piece to a 15 mL conical tube containing 3 mL of 4% PFA.
-
c.Incubate 15–20 h on ice for tissue fixation.
-
d.Next day, remove the PFA using a plastic transfer pipet.
-
e.Add 5 mL PBS to rinse the sample.
-
f.Place the tube on a shaking plate with mild shaking for 15 min.
-
g.Remove the PBS.
-
h.Repeat step e to g twice.
-
i.Add 2 mL Ethanol 70%.
 Pause point: The tissue can be stored in EtOH 70% at 4°C until further processing (not longer than 6 months).
Pause point: The tissue can be stored in EtOH 70% at 4°C until further processing (not longer than 6 months).
-
a.
-
5.
Mince the rest of the biopsy into small pieces (pieces of 1–2 mm3) using a scalpel.
-
6.Rinse again repeatedly with 3 mL PBS until the fluid becomes clear.Note: For cryopreservation of primary tissue (after mincing)
-
a.Add 1 mL of cryopreservation medium into one cryovial.
-
b.Add ± 10 pieces of tissue per vial and keep on ice.
-
c.Place the cryovials in a cryobox as soon as possible and freeze 15–20 h at −80°C.
-
d.Transfer the cryovials the following day to liquid nitrogen for long-term storage.
-
a.
-
7.
Collect all remaining tissue pieces into a 30 mL conical tube with 8 mL dissociation medium and incubate in a 37°C water bath for 1–2 h.
-
8.
Every 20 min, mechanically assist the enzymatic dissociation by pipetting the sample up and down with a glass Pasteur pipet to break up bulky clumps. When large pieces are no longer present, further dissociate the clumps every 20 min using a Pasteur pipet narrowed by fire-polishing (Figure 2).
-
9.
Add 50 μL DNase (0.2 mg/mL) to the suspension and incubate for 1 min at 20°C–22°C.
-
10.
Stop the enzymatic activity by adding ice-cold 8 mL DMEM/F12 + 10% FBS.
-
11.
In case of remaining fragments, filter the solution through a 70 μm cell strainer to collect the dissociated cells (together with some small cell clumps left). The persisting large tissue fragments on the cell strainer should be discarded.
CRITICAL: Remaining large tissue pieces should be discarded, as they do not give rise to organoids and will lead to more cell death after plating in the Matrigel droplet.
-
12.
Centrifuge the filtered suspension at 220 g for 5 min at 4°C.
-
13.
Remove the supernatant and resuspend the pellet in 1 mL DMEM/F12 (20°C–22°C) and transfer the suspension to an Eppendorf tube.
-
14.
Centrifuge at 220 g for 5 min at 4°C.
-
15.
Resuspend the pellet in 500 μL DMEM/F12 (20°C–22°C).
-
16.
Count the cells using an automated cell counter or hemocytometer. The (few) remaining small cell clumps can be ignored for counting.
Note: After this step, primary tissue cells can be collected for RNA and DNA extraction.
-
17.
Centrifuge at 220 g for 5 min at 4°C.
-
18.
Calculate the number of wells that can be plated with 30,000 cells per 20 μL droplet at one droplet per well.
-
19.Take off the correct amount of supernatant and resuspend in ice-cold Matrigel to obtain a ratio of 70% Matrigel/30% cell suspension (in DMEM/F12) for plating; keep on ice.
-
a.For instance, if 600,000 cells are present in the suspension, calculate for 20 droplets each containing 30,000 cells. 20 droplets of 20 μL make 400 μL. Hence, leave 120 μL of the cell suspension in the Eppendorf tube, resuspend the pellet and add 280 μL of Matrigel.
-
a.
-
20.
Dispense 20 μL droplets per well in a pre-warmed 48-well culture plate.
-
21.
Put the plate upside down in the 37°C/5% CO2 incubator for 20 min to allow the Matrigel to solidify and ensure in-gel distribution of the cells (i.e., avoid sedimentation at the bottom).
-
22.
Prewarm OCOM in a 37°C water bath.
-
23.
Add RI (10 μM) to the medium.
-
24.
Add 250 μL warm OCOM to each well and return the plate to the 37°C/5% CO2 incubator.
-
25.
Refresh the culture medium every 2–3 days (without RI).
Note: The organoids develop and grow steadily over time (Figure 3), in a timespan depending on patient tumor (typically, within 2–4 weeks).
Figure 1.

Processing of the OC biopsy
Tissue in collection medium is transferred to a Petri dish. The biopsy is rinsed multiple times with PBS and then cut with a scalpel into small pieces, which are subjected to further dissociation (for organoid development), fixation (for immunohistological analysis) and cryopreservation.
Figure 2.

Fire-polishing of the glass Pasteur pipet
The pipet is exposed to a Bunsen flame while constantly rotated (left panel) until its edges are polished and the opening narrowed down to about half its original diameter (right panel; opening before (left) and after (right)).
Figure 3.

Organoid development from OC
Representative brightfield images are shown at different days (d) after seeding. Scale bar, 200 μm.
Passaging OC organoids
Organoids are passaged when they are fully formed, i.e., reaching a diameter of around 200–300 μm and achieving appropriate confluency (Figure 4). The split ratio is 1:3 to 1:5 for a 48-well culture plate, according to organoid confluency.
-
26.
Place the required number of Matrigel aliquots on ice to thaw.
-
27.Remove the medium from the wells with organoids that need to be split.
-
a.Within the same culture condition, you can pool the organoids from up to 4 confluent wells.
-
a.
-
28.Add 400 μL of ice-cold DMEM/F12 per well and pipette with a p1000 tip up and down multiple times to release the organoids from the Matrigel.
-
a.Gently scratch the bottom of the well with the pipette tip, to help detach the Matrigel.
-
b.Avoid making bubbles, although do pipet vigorously enough.
-
c.If multiple wells are pooled, transfer the 400 μL from the first well to the next (and so on) to release the organoids from the Matrigel of all wells to be pooled.
-
a.
-
29.
Collect the resuspended organoids in a 1.5 mL Eppendorf tube and put on ice.
-
30.Rinse all wells from which organoids were collected once with 400 μL DMEM/F12.
-
a.Double-check the wells of the culture plate under the light microscope to ensure that all organoids are collected. If not, rinse again with 400 μL DMEM/F12.
-
a.
-
31.
Centrifuge at 220 g for 5 min at 4°C.
-
32.
Prewarm an aliquot of TrypLE Express (supplemented with RI at 10 μM) in a 37°C water bath. 500 μL TrypLE is needed per Eppendorf tube of organoids.
-
33.
Carefully remove the supernatant from the pelleted organoids.
-
34.
Add 500 μL of prewarmed TrypLE to each Eppendorf tube containing the organoid pellet. Swirl tubes gently so that organoids are mixed with the TrypLE.
-
35.
Incubate in a 37°C water bath for 10 min (dense organoids; see below) or 5 min (low-cohesive and cystic organoids; see below).
-
36.
Add 500 μL of ice-cold DMEM/F12 to inactivate TrypLE activity.
-
37.
Centrifuge at 220 g for 5 min at 4°C.
-
38.
Remove the supernatant.
-
39.
Resuspend the organoid pellet in 700 μL ice-cold DMEM/F12.
-
40.
Place a P200 tip (no filter) over a P1000 tip (with filter) and pipet up and down for 25 times to mechanically disrupt the organoids.
Note: Adjust the volume of the pipette so that most of the medium volume (with organoids) is aspirated without air to avoid making bubbles.
Note: Check under the light microscope whether single cells are obtained. If some undispersed structures remain (Figure 5), further apply mechanical dissociation to obtain individual cells.
Figure 4.

Confluent organoid cultures in a well of a 48-well culture plate, ready to be passaged
Representative brightfield images are shown. Scale bar, 200 μm. Regarding the different morphologies, see “expected outcomes”.
Figure 5.

Dissociation of OC organoids
When undispersed organoid structures are still present (as shown), further mechanical disruption is needed to eventually obtain single cells. Scale bar, 200 μm.
-
41.
Centrifuge at 220 g for 5 min at 4°C.
-
42.
Resuspend the pellet in 500 μL DMEM/F12 (20°C–22°C).
-
43.
Count the cells using an automated cell counter or hemocytometer.
-
44.
Calculate the number of wells that can be plated and the amount of cell suspension and Matrigel needed as described above (step 18 of organoid establishment).
-
45.
Resuspend the pellet in 70% Matrigel and keep on ice.
-
46.
Dispense 20 μL droplets, containing 30,000 cells per well, in a pre-warmed 48-well culture plate.
-
47.
Incubate the plate upside down in the 37°C/5% CO2 incubator for 20 min.
-
48.
Meanwhile, pre-warm OCOM in a 37°C water bath.
-
49.
Add RI (10 μM) to the medium.
-
50.
Add 250 μL warm OCOM to each well and return the plate to the 37°C/5% CO2 incubator.
-
51.
Refresh the culture medium every 2–3 days and passage the organoids again at confluence (i.e., every 14–28 days).
Note: Some organoid lines are passageable for a long period (up to one year and more), while other lines show short-term passeagability (up to 4 passages, i.e., 8–16 weeks), which depends on the patient tumor. Organoids keep characteristics during passaging.
Cryopreserving OC organoids
Follow the passaging protocol until step 37.
-
52.After centrifugation, resuspend the pellet in 1 mL cryopreservation medium, transfer into a cryovial and put on ice.
-
a.The cryovial may contain organoids from 1 to 4 confluent wells.
-
a.
-
53.
Place the cryovials in a cryobox and transfer to −80°C.
-
54.
The next day, transfer the samples to liquid nitrogen for long-term storage.
Thawing OC organoids
-
55.
Remove the cryovial form the liquid nitrogen tank and put on ice. Immediately proceed with the thawing protocol.
-
56.
Thaw the solution with the cryopreserved OC organoids in a 37°C water bath for maximum 2 min.
-
57.
Transfer the content to a 15 mL conical tube with ice-cold 10 mL DMEM/F12/10% FBS.
-
58.
Rinse the cryovial with 1 mL DMEM F12/10% FBS to ensure that all pieces are recovered and transferred.
-
59.
Centrifuge at 220 g for 5 min at 4°C.
-
60.
Resuspend the pellet with 1 mL ice-cold DMEM/F12.
-
61.
Transfer the suspension to a 1.5 mL Eppendorf tube.
-
62.
Centrifuge at 220 g for 5 min at 4°C.
-
63.
Resuspend the pellet in 500 μL ice-cold DMEM/F12.
-
64.
Count the cells using an automated cell counter or hemocytometer.
-
65.
Calculate the number of wells that can be plated and the amount of cell suspension and Matrigel needed as described above (step 18 of organoid establishment).
-
66.
Resuspend the pellet in 70% Matrigel and keep on ice.
-
67.
Dispense 20 μL droplets, containing 30,000 cells per well, in a pre-warmed 48-well culture plate.
-
68.
Incubate the plate upside down in the 37°C/5% CO2 incubator for 20 min.
-
69.
Meanwhile, pre-warm OCOM in a 37°C water bath.
-
70.
Add RI (10 μM) to the medium.
-
71.
Add 250 μL warm OCOM to each well and return the plate to the 37°C/5% CO2 incubator.
-
72.
Refresh the culture medium every 2–3 days and passage the organoids again at confluence (i.e., every 14–28 days).
Note: The growing efficiency of cryopreserved OC organoid lines following thawing is 100%.
Fixing OC organoids in PFA
-
73.
Remove the medium from the wells with organoids that need to be analyzed.
-
74.
Add 400 μL of ice-cold DMEM/F12 to the well, and per well pipette with a p1000 tip up and down multiple times to release the organoids from the Matrigel.
Note: Organoids from multiple wells can be pooled.
-
75.
Collect the organoids in a 1.5 mL Eppendorf tube and put on ice.
-
76.
Centrifuge at 220 g for 5 min at 4°C.
-
77.
Remove the supernatant (containing the Matrigel as dissolved in the ice-cold DMEM/F12) and resuspend the organoid pellet in 1 mL of 4% PFA in the chemical hood.
-
78.
Incubate for 1 h at 20°C–22°C.
-
79.
Centrifuge at 220 g for 5 min at 4°C.
-
80.
Wash 3 times by resuspending the pellet in 1 mL PBS and centrifuging at 200 g for 5 min at 4°C to remove the rinsing solution.
-
81.
Resuspend the pellet in 1 mL of EtOH 70%.
-
82.
Organoids can be stored in EtOH 70% at 4°C for up to 1 month.
Paraffin-embedding OC organoids
-
83.
Work in a chemical hood.
-
84.
Pellet the PFA-fixed organoids in 70% EtOH at 220 g for 5 min at 4°C.
-
85.
Remove the supernatant and resuspend in 1 mL of 100% EtOH with a plastic transfer pipet.
-
86.
Mark glass flat-bottom tubes using tape, to avoid wiping out with EtOH (Figure 6A).
-
87.
Fill the flat-bottom tubes with ∼4 mL 100% EtOH.
-
88.
Transfer the organoids into the flat-bottom tube with a plastic transfer pipet and pipet up and down a few times.
Tip: Wash the Eppendorf tube with 100% EtOH to ensure that all organoids are transferred.
-
89.
Let the organoids settle down by gravity during 30 min at 20°C–22°C.
-
90.
Discard most of the 100% EtOH and rinse again with 100% EtOH. Pipet up and down with the plastic transfer pipet when adding the EtOH.
-
91.
Let the organoids settle down again for 30 min at 20°C–22°C.
-
92.
Repeat washing steps 90 and 91 once.
-
93.
Rinse the organoids with N-Butanol and let them settle down for 30 min at 20°C–22°C.
-
94.
Perform 2 more washing steps of 30 min with N-Butanol.
-
95.
Remove the N-Butanol as much as possible.
-
96.
The following steps are carried out with the HistoStar Embedding Workstation.
-
97.Pre-warm the heating block in the paraffin of the HistoStar (Figure 6B).
-
a.The glass tubes, containing the organoids, will stay in the heating block during the paraffin incubation.
-
a.
-
98.
Resuspend the organoids in warm paraffin solution of the HistoStar Embedding Workstation and incubate for 30 min at 60°C.
-
99.
Perform 3 consecutive incubation steps with warm paraffin.
-
100.
Discard ¾ of the paraffin and let the organoids settle at the bottom of the tube.
-
101.
Light up a cartridge burner, warm a metal mold and keep it warm in the HistoStar (Figures 6C and 6D).
-
102.
Warm the glass Pasteur pipet with the cartridge burner and transfer rapidly all organoids in melted paraffin from the glass tube to the warmed metal mold.
-
103.
Move all the organoids to the center of the metal mold with blunt forceps.
-
104.
Put the metal mold on a cold plate of the HistoStar for 10 sec.
-
105.
Put the cassette on the metal mold and cover the cassette with warm paraffin of the Histostar.
-
106.
Let it cool down for 20 min on the cold plate.
-
107.
Remove the metal mold and store the paraffin blocks at 4°C until further processing.
Figure 6.

Technical aspects of paraffin embedding of OC organoids
(A) Glass flat-bottom tubes are labeled with tape.
(B) The heating block, containing the glass tubes, is put in the paraffin of the HistoStar to heat up.
(C) A cartridge burner is used to warm a metal mold.
(D) The cassette is put on the metal mold, covered with paraffin and placed on the cold plate to cool down.
Drug screening using OC organoids
Follow the passaging protocol until step 43.
-
108.
Calculate the number of wells that can be plated with 2,000 cells per 3 μL droplet and calculate the amount of cell suspension and Matrigel needed.
-
109.
Resuspend the pellet in 70% Matrigel and keep on ice.
-
110.
Dispense one 3 μL droplet (containing ∼2,000 cells) per well in a pre-warmed 96-well flat-bottom culture plate.
-
111.
Incubate the plate upside down in the 37°C/5% CO2 incubator for 10 min.
-
112.
Meanwhile, pre-warm OCOM in a 37°C water bath.
-
113.
Add RI (10 μM) to the medium.
-
114.
Add 100 μL warm OCOM to each well and return the plate to the 37°C/5% CO2 incubator.
-
115.
Refresh the culture medium every 2–3 days and allow the organoids to form for 2–4 weeks.
-
116.
When organoids are fully formed and the culture is confluent, apply a concentration dilution series of tested drugs or control (vehicle) to the organoid cultures (each dose in triplicate wells).
-
117.
Refresh the drug at the tested dilutions after 48 h.
-
118.Assess cell viability after 72 h of drug treatment using the XTT assay
-
a.Prepare 1 mg/mL XTT labeling mixture by dissolving XTT powder in DMEM/F12 and adding 1/1000 electron-coupling reagent PMS
-
b.Pre-warm the mixture for 30 min in a 37°C water bath, vortex every 10 min
-
c.Add 100 μL XTT labeling mixture per organoid well and incubate for 4 h at 37°C/5% CO2
-
d.Measure the absorbance of the samples using a microplate reader at 450 nm.
-
a.
-
119.
Data analysis and calculation of IC50 values can be done with GraphPad Prism (V8.0.1) (see quantification and statistical analysis for detailed calculations).
Expected outcomes
Organoid development
We provide a detailed protocol to establish organoid cultures starting from human OC biopsies. The organoids, typically forming within 2–4 weeks, show inter-patient morphological heterogeneity encompassing dense, cystic and low-cohesive phenotypes (Figure 7). The organoids recapitulate the parent tumor’s marker expression and mutational landscape, and display tumor-specific sensitivity to chemotherapeutic drugs clinically used.
Figure 7.

Different morphological phenotypes of OC organoids, i.e., dense (left), cystic (middle) and low-cohesive (right)
Representative images of individual organoids (brightfield) are shown. Scale bar, 200 μm.
Details of organoid characterization can be found in our recent publication (Maenhoudt et al., 2020). It is recommended to validate each new organoid line (against the original tumor) minimally by hematoxylin-eosin staining to identify high-grade nuclear atypia, and by immunofluorescence staining to examine the expression of OC-associated markers (such as PAX8, cytokeratin-7 and p53 for HGSOC). The organoids typically contain more than 90% tumor (epithelial) cells. The patient-derived OC organoids provide powerful tools to study the cancer’s pathobiology as well as a platform for drug discovery and (personalized) drug screening.
Quantification and statistical analysis
IC50 in drug screening assay
After measuring the absorbance in the drug screening assay using XTT, data are analyzed using Graphpad Prism software (V8.0.1). See Table 1 for an example dataset. An XY table is then created (Table 2), with the logarithm of the concentration of the drug into X and the response (i.e., percentage viability based on absorbance) into Y. To analyze the data, apply nonlinear regression analysis with dose-response inhibition. To determine the IC50, the equation ‘log(drug/inhibitor) vs response – variable slope (4 parameters)’ is used.
Table 1.
Example dataset
| Drug (μM) | Absorbance | ||
|---|---|---|---|
| Control | 1.191 | 1.211 | 0.933 |
| 0.01 μM | 0.917 | 1.286 | 1.102 |
| 0.1 μM | 1.090 | 1.144 | 1.051 |
| 0.5 μM | 0.973 | 1.201 | 1.154 |
| 1 μM | 1.153 | 1.136 | 0.988 |
| 5 μM | 0.952 | 1.126 | 0.844 |
| 10 μM | 0.651 | 0.574 | 0.698 |
| 20 μM | 0.493 | 0.398 | 0.452 |
| 50 μM | 0.233 | 0.119 | 0.159 |
| 100 μM | 0.033 | 0.041 | 0.022 |
Table 2.
Calculation of the percentage viability based on absorbance
| Log(Drug μM) | Average absorbance | Viability (%) |
|---|---|---|
| Control | 1.112 | 100.000 |
| -2.000 | 1.102 | 99.100 |
| -1.000 | 1.095 | 98.501 |
| -0.301 | 1.109 | 99.790 |
| 0.000 | 1.092 | 98.261 |
| 0.699 | 0.974 | 87.616 |
| 1.000 | 0.641 | 57.661 |
| 1.301 | 0.448 | 40.270 |
| 1.699 | 0.170 | 15.322 |
| 2.000 | 0.032 | 2.879 |
Measured absorbance using a microplate reader at 450 nm (step 118d).
-
1.
Calculate the logarithm of the concentration of the drug
-
2.
Calculate the average absorbance per drug concentration.
-
3.Calculate the percentage viability based on absorbance. The control condition is set as 100% viability. The equation used to calculate the viability percentage is:
-
4.
Using Graphpad Prism software, an XY table is created, with the logarithm of the concentration of the drug into X and the viability (%) into Y.
-
5.
An XY graph is created.
-
6.
To analyze the XY data, ‘Nonlinear Regression (curve fit)’ analysis is performed. The equation used is ‘log [inhibitor] vs response - Variable slope (four parameters)’ (Figure 8).
Figure 8.

Example of a drug dose-response curve
Mean data points (each dot represents the mean of 3 technical replicates) are displayed for each concentration analyzed. The IC50 value is calculated using GraphPad Prism software.
Limitations
The efficiency of deriving OC organoids still shows room for improvement (e.g., ∼40% for high grade serous OC; see Maenhoudt et al., 2020). Organoid development efficiency may depend on type and grade, cell composition and patient origin of the highly heterogenous OC. Importantly, successful organoid formation is strongly dependent on the viability of the cells after dissociation, which can be assessed using the trypan blue exclusion test. It is critical that the dissociation time does not exceed 2 h. Therefore, progress must regularly be checked under the light microscope. Once the organoids form, it is important to passage them at the appropriate time (confluency) to avoid cell death. In addition, nutrient exhaustion should be prevented by refreshing culture medium every 2–3 days.
Of note, the degree of tumor cell content of the original biopsy (including the presence of necrotic tissue), which may depend on the specific tumor and/or on the spatial region of the tumor where the biopsy was taken, may also influence organoid derivation efficiency.
Troubleshooting
Problem 1
No organoid formation after dissociation of the OC biopsy or after thawing cryopreserved OC tissue (steps 25 and 72).
Potential solution
Viability of the cryopreserved tissue could be low due to inadequate cryopreservation. Make sure the cryopreservation medium has the correct composition and the tissue pieces are constantly kept on ice while cryopreserving. The tissue should be cut in small pieces (1–2 mm3) for appropriate penetration of the DMSO.
Prolonged dissociation time decreases cell viability. We recommend enzymatic dissociation for not longer than 2 h. Once single cells and (few) cell clusters are visible under the microscope, immediately stop the enzymatic reaction.
Adding RI is essential for organoid (re-)formation. Failing to add RI in the medium will result in fewer organoids. Therefore, always add RI in the Matrigel/cell suspension at seeding of tissue cells and clumps, and at passaging.
The correct composition of the organoid medium is critical. Multiple freeze-thaw cycles of the growth factors should be avoided and medium can only be kept up to 2 weeks at 4°C.
Problem 2
Organoids do not regrow after passaging (step 51).
Potential solution
If not enough organoids are present in a well for passaging, several wells can be pooled together.
Limit TrypLE incubation to 10 min, as prolonged enzymatic reaction reduces cell viability.
While mechanically splitting the organoids, pre-wet the tips with FBS which helps to avoid adherence of the organoids inside the tip and (major) loss of structures.
It is important that large organoid clumps are dissociated into small cell clusters or single cells. Strong trituration by pipetting up and down (with a maximum of 300 times) may be needed, which does not negatively affect cell viability.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Prof. Dr. Hugo Vankelecom (hugo.vankelecom@kuleuven.be).
Material availability
This study generated high grade serous OC organoid lines. Availability is restricted by MTA.
Data and code availability
The published article includes all datasets or codes generated or analyzed during this study.
Acknowledgments
This work was supported by grants from the KU Leuven Research Fund and from the Fund for Scientific Research-Flanders (FWO). N.M. is supported by a PhD Fellowship from the FWO (11A1120N). Histological instruments and microscopes could be used from InfraMouse (VIB-KU Leuven). We thank Dr. Frédérickx (Vankelecom Lab) for critically reading the protocol.
Author contributions
N.M. designed the concepts and experiments, performed the experiments and the data analysis, interpreted the results, and co-wrote the manuscript. H.V. designed and supervised the project, co-developed the concepts and ideas, co-designed the experiments, co-analyzed and co-interpreted the data, and wrote the manuscript. Both co-authors critically read and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Nina Maenhoudt, Email: nina.maenhoudt@kuleuven.be.
Hugo Vankelecom, Email: hugo.vankelecom@kuleuven.be.
References
- Maenhoudt N., Defraye C., Boretto M., Jan Z., Heremans R., Boeckx B., Hermans F., Arijs I., Cox B., Van Nieuwenhuysen E. Developing organoids from ovarian cancer as experimental and preclinical models. Stem Cell Reports. 2020;14:717–729. doi: 10.1016/j.stemcr.2020.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The published article includes all datasets or codes generated or analyzed during this study.