

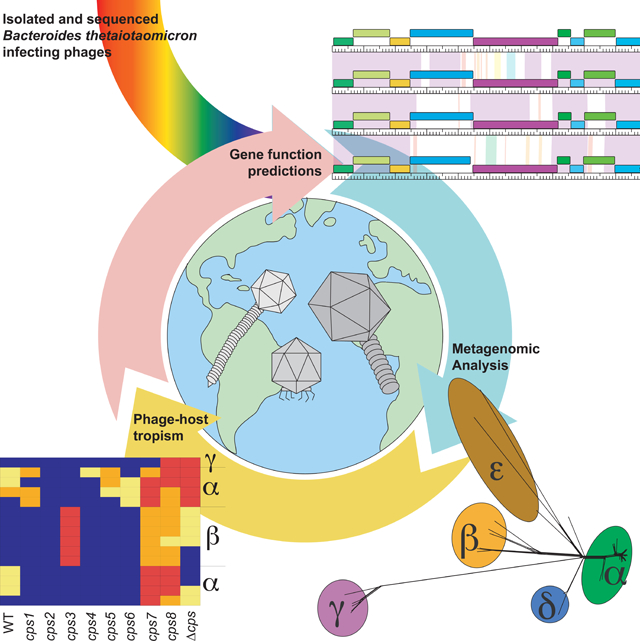
Summary
Our emerging view of the gut microbiome largely focuses on bacteria, while less is known about other microbial components such as bacteriophages (phages). Though phages are abundant in the gut, very few phages have been isolated from this ecosystem. Here, we report the genomes of 27 phages from the United States and Bangladesh that infect the prevalent human gut bacterium Bacteroides thetaiotaomicron. These phages are mostly distinct from previously sequenced phages with the exception of two, which are crAss-like phages. We compare these isolates to existing human gut metagenomes, revealing similarities to previously inferred phages and additional unexplored phage diversity. Finally, we use host tropisms of these phages to identify alleles of phage structural genes associated with infectivity. This work provides a detailed view of the gut’s “viral dark matter” and a framework for future efforts to further integrate isolation- and sequencing-focused efforts to understand gut-resident phages.
Graphical Abstract
eTOC blurb
Hryckowian and Merrill et al. provide genotypic and phenotypic data for 27 bacteriophage isolates that infect the prevalent human gut bacterium, Bacteroides thetaiotaomicron. They identify related bacteriophages in existing metagenomes and genes associated with infectivity. This work demonstrates the utility of integrating culture-based and computational efforts to understand gut-resident bacteriophages.
Introduction
Bacteriophages (phages) are abundant in free-living and host-associated microbial communities (microbiomes) (Brussow and Hendrix, 2002, Barr et al., 2013). Like other microbiome members (e.g. bacteria, fungi), the diversity and abundance of phages differ between healthy and diseased individuals. While some gut resident phages appear to be unique to individual humans and stable across long time scales (Shkoporov et al., 2019), others correlate with host disease status (Manrique et al., 2016, Duerkop et al., 2018). These observations highlight the possibility that phages play central roles in the structure and function of host-associated microbiomes and may therefore impact human health. Taken together with the burgeoning antibiotic resistance crisis, this possibility amplifies the importance of phage therapy as an alternative or supplement to existing paradigms of microbiome management (e.g. widespread antibiotic use).
Despite growing enthusiasm for phage-based therapeutics, gut-resident phages are poorly understood. Unlike bacteria, phages do not have conserved marker genes (e.g. the 16S rRNA marker gene) that enable phylogenetic classification and analysis. Instead, phage genomes must be inferred from metagenomic studies, either based on conservation of phage-like genes (e.g., terminase, DNA polymerase) (Grazziotin et al., 2017), sequence identity relative to known phage isolates (Roux et al., 2015), or by database-independent approaches (Ren et al., 2017). While powerful for general characterization of changes in the composition of phage communities, inter-study methodological variation (e.g. sample preparation, contig assembly, reference databases used) can impact a study’s conclusions to a greater extent than the treatment effects (e.g. health or disease status) (Gregory et al., 2019).
Furthermore, metagenomic approaches fail to provide definitive information on the bacterial hosts of these phages. To address this deficiency, many methods have been developed to predict the bacterial hosts of phages inferred from metagenomes. For example, homology searches, identification of CRISPR spacers, and co-occurrence analysis were used to make the prediction that the highly prevalent and abundant crAssphage infects bacteria in the phylum Bacteroidetes (Dutilh et al., 2014). This prediction was validated in part when a crAss-like phage (CrAss001) was isolated on Bacteroides intestinalis (Shkoporov et al., 2018). However, based on the divergence of CrAss001 from the prototypical crAssphage and the diversity of crAss-like phages (Yutin et al., 2018, Guerin et al., 2018), it is likely that other crAss-like phages infect other bacteria within the Bacteroidetes phylum. Furthermore, crAss-like phages can simultaneously be biomarkers of healthy and diseased states. For example, one crAss-like phage, IAS virus, is enriched in HIV+ individuals with low CD4 counts (Oude Munnink et al., 2014) while some crAss-like phages are stable over a 12-month period in healthy humans (Shkoporov et al., 2019).
CrAss001 is one of four sequenced phages confirmed to infect Bacteroides, the most abundant bacterial genus in the human gut microbiome. The other phages are B40–8 and B124–14 (which infect B. fragilis) and Hankyphage, which is a prophage in many Bacteroides strains (Benler et al., 2018, Ogilvie et al., 2012, Hawkins et al., 2008). Despite the prevalence of Hankyphage lysogens, Hankyphage was unable to form plaques on several Hankyphage-naïve Bacteroides species (Benler et al., 2018). Additionally, CrAss001 does not form robust plaques on B. intestinalis despite persisting at high levels in long-term co-culture with its host (Shkoporov et al., 2018). These observations suggest that unexplored factors influence Bacteroides-phage interactions.
Here, we report the genomes of 27 phages that infect B. thetaiotaomicron (18 previously described isolates (Porter et al., 2019) and 9 additional isolates; Table S1). By comparing these genomes with those of existing Bacteroides phage isolates and with phage genomes from publicly available metagenomic studies, we simultaneously reveal similarities among these phages and additional unexplored phage diversity. Finally, genomic analysis of the B. thetaiotaomicron-infecting phages in the context of their capsular polysaccharide (CPS)-specific host ranges reveals targets for future study of the structure-function relationships dictating phage host range. Together, this work demonstrates the utility and feasibility of efforts that integrate isolation- and computational-based methods. Future application of such an approach to other bacteria will enrich sequence databases by providing reference genomes and definitive host information, enable investigators to build phage-host experimental systems, and contribute to a growing collection of phages that may be used for therapeutic or biotechnological applications.
Results
Isolation and comparative analysis of 27 phages infecting B. thetaiotaomicron
Our study centers on phages isolated from four geographic locations, three within the US and one in Bangladesh (Figs. 1A, Table S1). Using a previously reported protocol for phage isolation (Porter et al., 2019), we isolated 9 bacteriophages from primary wastewater effluent from the Sand Island Wastewater Treatment Plant (Honolulu, Hawaii) or from sewer-adjacent pond water at two locations in Dhaka, Bangladesh. High titer stocks were prepared of these 9 phage isolates and of a subset of 18 phages from an existing collection of 71 B. thetaiotaomicron-infecting phages isolated from Ann Arbor, Michigan and San Jose, California (Porter et al., 2019). Phage genomes were sequenced and assembled (see STAR Methods).
Figure 1. Isolation and characterization of 27 Bacteroides thetaiotaomicron-infecting phages.
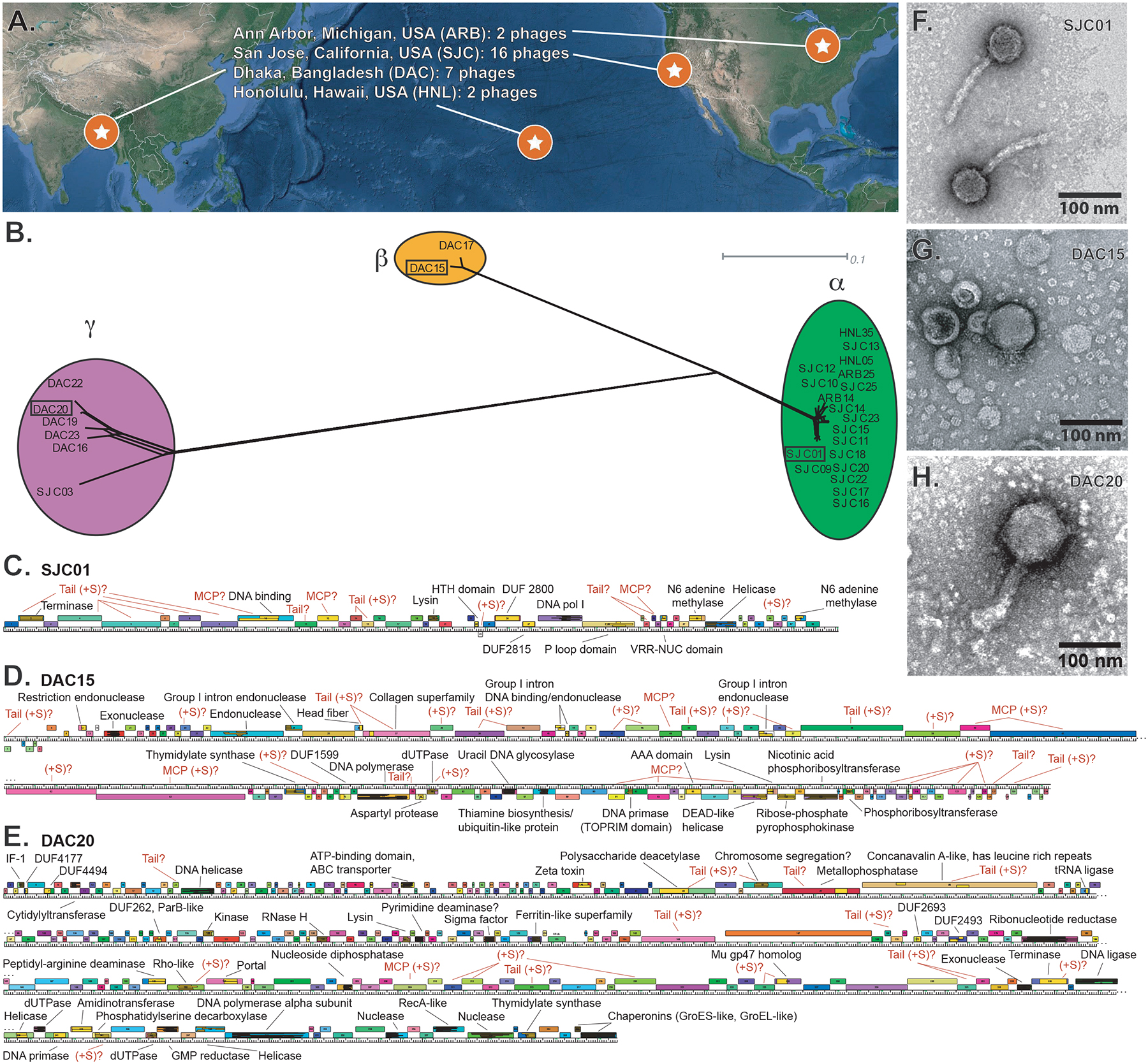
(A) Phages were isolated from wastewater from 3 locations in the United States and from 2 locations in Dhaka, Bangladesh. (B) Network phylogeny analysis of phage genomes, compared according to shared gene content using Phamerator and Splitstree (see STAR Methods). Colored ellipses indicate groups of phages according to cluster assignment, assigned by vConTACT2. The scale bar indicates 0.1 substitutions per site. (C-E) Annotated genome maps of representative members of each cluster (SJC01, DAC15, and DAC20). Genes are represented as colored boxes and conserved domains are inlaid yellow boxes within genes. If a gene has a conserved domain, it is annotated in black text. iVireons was used to predict structural genes as described in STAR Methods and are annotated in red as predicted tail, major capsid, or general structural (tail, MCP, +S, respectively). (F-H) Transmission electron micrographs of SJC01, DAC15, and DAC20 show morphological differences between these representatives of the phage clusters. See also Tables S1–S6 and Figures S1–S3.
Phages were grouped into three distinct genome clusters (α, β, γ) with vConTACT2 (Bin Jang et al., 2019). Phage genomes were then annotated and compared on the basis of shared gene phamily (pham) membership (Cresawn et al., 2011). Phams are groups of related protein-encoding genes where pham membership is built and expanded when a candidate protein shares ≥32.5% identity or blastp e-value ≤1e-50 with one or more existing members of the pham. A dendrogram was built based on the presence or absence of each pham in each phage to visualize and validate the genome cluster assignments (Figs. 1B). Clusters α, β, γ have genomes that are on average 38kb +/− 0.4kb, 99kb +/− 0.3kb, and 177kb +/− 4.5kb, respectively, and exhibit extensive genomic mosaicism (Figs. S1–S3; Table S1). Genome maps of representatives of each of these clusters are shown in Figs. 1C–E. tRNAs were detected in cluster β and γ phages (n=12–13 and n=2–3, respectively) but not in cluster α phages (Tables S1, S2).
While there is a high degree of intra-cluster sequence identity, there are only two phams shared between cluster β and γ representatives. Consistent with observations from previously isolated phages (Hatfull and Hendrix, 2011), the majority (roughly 80%) of phams in these B. thetaiotaomicron-infecting phages have no detectable conserved domains or known functions (Tables S3–S5).
Transmission electron microscopy of one representative from each cluster reveals distinct virion morphologies. Based on these representatives, cluster α phages are siphoviruses, cluster β phages are podoviruses, cluster γ phages are myoviruses, and the capsid sizes of these phages scale with genome size (Figure 1F–H).
Comparative analysis of B. thetaiotaomicron phages with existing Bacteroides phage isolates.
We compared these 27 B. thetaiotaomicron-infecting phages to 4 other previously sequenced Bacteroides-infecting phages (Benler et al., 2018, Ogilvie et al., 2012, Hawkins et al., 2008, Shkoporov et al., 2018) (Fig. 2). We noted extensive shared phams (n=53) and genome organization between the cluster β phages (DAC15 and DAC17) and CrAss001 (Fig. 2, Fig. S4, Table S6), reinforcing predictions that at least a subset of crAss-like phages prey on Bacteroides. Few phams are shared between the other isolated B. thetaiotaomicron-infecting phages and the previously isolated Bacteroides-infecting phages (Table S6). Furthermore, B40–8 and B124–14 are members of a separate cluster (cluster δ) and Hankyphage is a singleton with no isolated relatives (Fig. 2). These cluster assignments are validated by vConTACT2 (Bin Jang et al., 2019) (see STAR Methods). No RefSeq phage genomes from the ProkaryoticViralRefSeq94-Merged database were grouped into clusters with these 31 isolated phages.
Figure 2. Network phylogeny of 31 Bacteroides-infecting phages based on gene content.
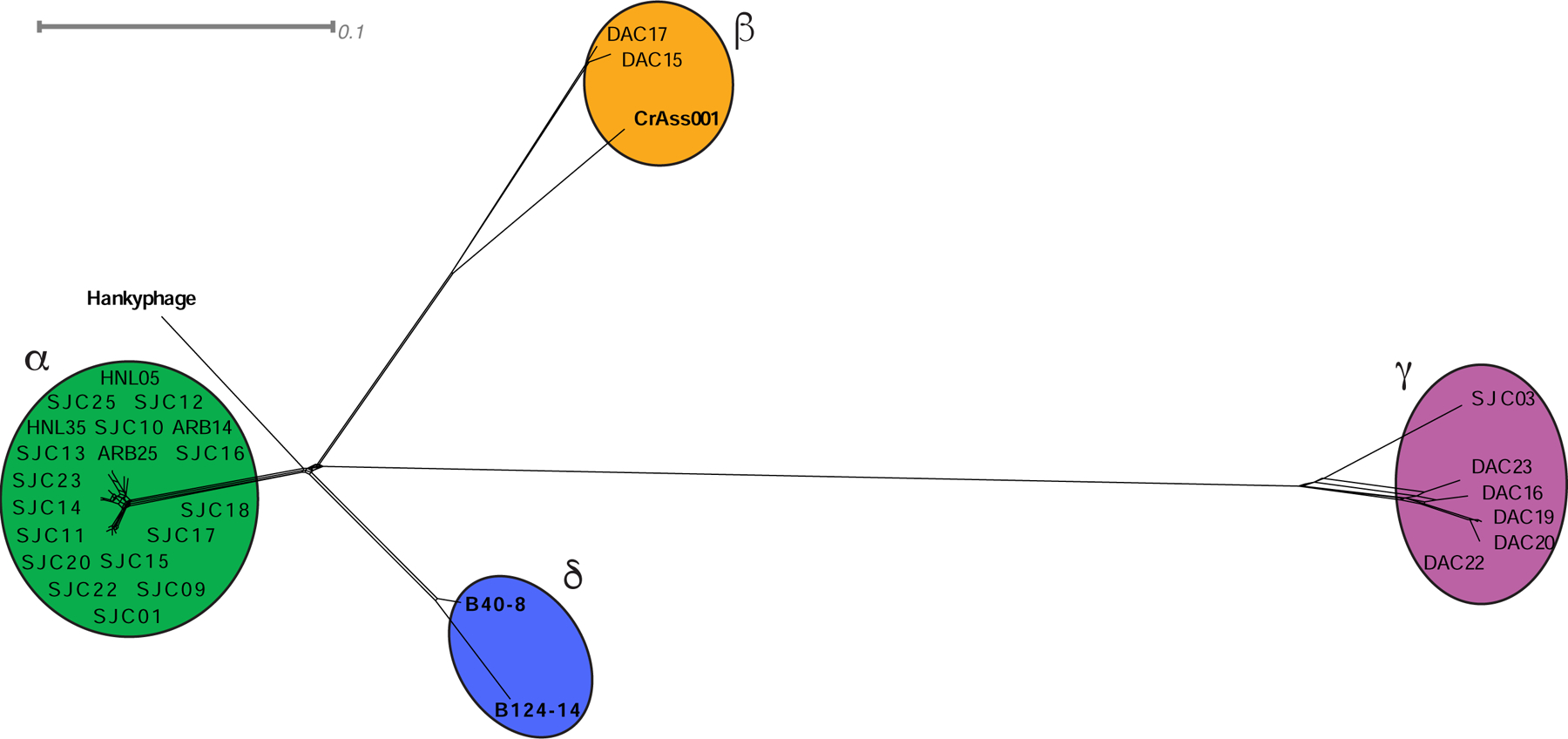
The genomes of 31 Bacteroides infecting phages were compared according to shared gene content using Phamerator and Splitstree, (see STAR Methods). Colored ellipses indicate groups of phages according to cluster assignment, assigned by vConTACT2. The scale bar indicates 0.1 substitutions per site. See also Table S6 and Figure S4.
Identification of phages related to isolated B. thetaiotaomicron phages in existing metagenomes.
Because the majority of phage-focused work in the gut microbiome field is based on metagenomic sequencing, we wondered if relatives of the sequenced B. thetaiotaomicron-infecting phage isolates could be found in existing metagenomes. To identify relatives of these phages, we used the protein search feature of SearchSRA (Torres et al., 2017, Levi et al., 2018, Towns et al., 2014, Stewart et al., 2015, Buchfink et al., 2015b, Langmead and Salzberg, 2012) to map 100,000 subsampled reads from each of the ~100,000 metagenomes in the Sequence Read Archive (SRA) onto representatives of clusters α, β, and γ (SJC01, DAC15, and DAC20, respectively). We identified 812 candidate metagenomes in the SRA where at least one of the representative phage genomes was covered by reads at an estimated read depth of >15% (given the true sequencing depth of the sample) and the percent of the genome detected was >20% (Fig. 3A–C). We subsequently focused on human gut-derived metagenomes possessing sequences that are SJC01-like (>50% detected, >30x estimated coverage), DAC15-like (>40% detected, >15x estimated coverage), or DAC20-like (>20% detected) genomes for further analysis (Table S7). These metagenomes were downloaded from NCBI and assembled. Contigs containing significant hits (blastp e-value <1e-3) for >25% of the genes in SJC01, DAC15, or DAC20 were compared to the genomes of the isolated Bacteroides-infecting phages described above. See STAR Methods for a more detailed description of this method of identifying Phage in SearchSRA (PhiSh). Several PhiSh genomes were identified which are related to SJC01, including previously uncharacterized genomes (PhiSh01 – PhiSh03, PhiSh05 – PhiSh07)(Monaco et al., 2016, He et al., 2017, Liu et al., 2016, Zheng et al., 2017, Guthrie et al., 2017) and a genome previously identified in a study examining the rapid evolution of the human gut virome (PhiSh04) (Minot et al., 2013). We also noticed that HSC01, a genome of a phage predicted to infect Bacteroides caccae (Reyes et al., 2013) is related to SJC01 (Figs. 3DE; Table S8). All of these SJC01-like PhiSh genomes are grouped into cluster α by vConTACT2.
Figure 3. Identification of Phage in SearchSRA (PhiSh) related to isolated B. thetaiotaomicron-infecting phages.
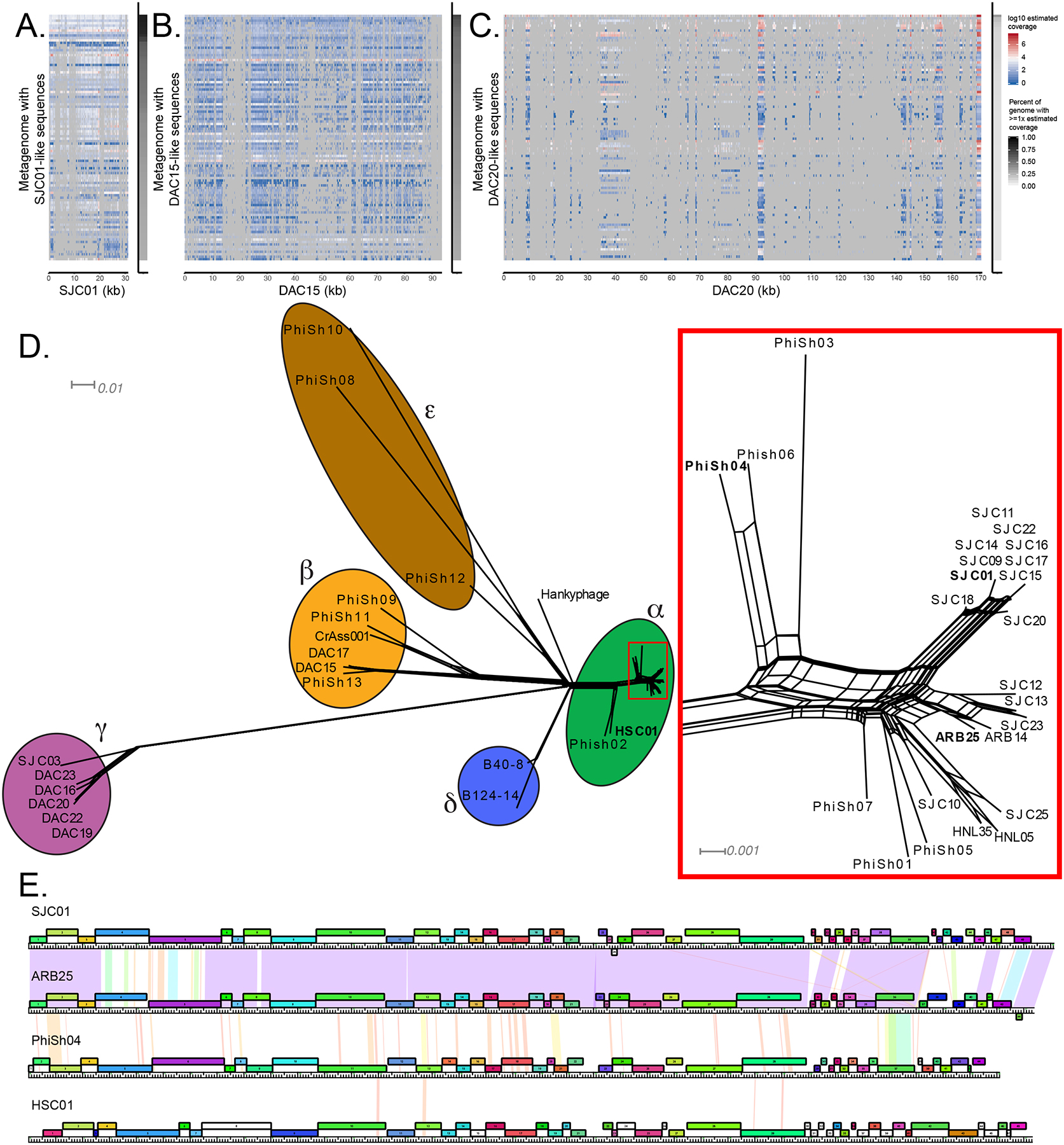
Representatives of each genome cluster (SJC01, DAC15, DAC20) were used to query the entire NCBI SRA using SearchSRA (see STAR Methods). (A-C) Log10-transformed coverage depth of the 100 best hits identified via SearchSRA (tDNA mode) to SJC01, DAC15, and DAC20, respectively. Hits are ranked along the y-axis based on percent coverage by reads. The x-axis represents genome positions of SJC01, DAC15, and DAC20, respectively. The percentage of SJC01, DAC15, and DAC20 genomes detected (≥ 1 read) in each metagenome is indicated by the gray shaded column on the right of each panel. (D) Network phylogeny of Bacteroides-infecting phage genomes described in Figure 2 and related genomes identified in publicly available metagenomes. Genomes were compared according to shared gene content using Phamerator and Splitstree (see STAR Methods). Colored ellipses indicate groups of phages according to cluster assignment, assigned by vConTACT2. The subset of cluster alpha phages enclosed in a rectangle is shown in greater detail at the right-hand side of the panel. Phages highlighted in panel E are in bold. The scale bars indicate 0.01 and 0.001 substitutions per site for the main tree and the cluster alpha subset, respectively. (E) Genome maps of 4 cluster α phages (SJC01, ARB25, PhiSh04, and HSC01). The genes are color-coded according to pham membership and are numbered. Pairwise nucleotide identity is represented as shading between genomes. The color of this shading represents the degree of sequence similarity with violet being the most similar (BLASTN score = 0), progressing through the color spectrum from indigo, blue, green, yellow, orange, to red, which is the least similar (BLASTN score = 10−4). Regions with no shading indicate no similarity with a BLASTN score greater than 10−4. See also Tables S6–S8.
Six DAC15-like genomes were also identified (PhiSh08 – PhiSh13) (Table S8). Five of these genomes (PhiSh08 – PhiSh12) were previously identified in a study aimed at identifying crAss-like phages in human fecal metagenomes (Guerin et al., 2018) while PhiSh13 represents a previously unidentified crAss-like phage genome (He et al., 2017). Importantly, these DAC15-like PhiSh genomes are diverse (they can be classified into the previously described candidate crAss-like genera 6, 7, and 10; Table S8) and are differentially clustered by vConTACT2 (clusters β and ε), demonstrating that the PhiSh identification approach can detect genomes closely and distantly related to the PhiSh bait genome used (Fig. 3D).
We then sought to predict the bacterial hosts of these PhiSh using CRISPR spacer analysis. CRISPR protospacers were identified with the JGI IMG/VR Spacer Database (Paez-Espino et al., 2019). PhiSh02, PhiSh04, and Phish06 are predicted to infect within the Bacteroides genus while hosts for the other PhiSh (Table S8) and B. thetaiotaomicron-infecting phage isolates (Table S1) were not predicted, highlighting potential shortcomings of this approach given current databases.
We observed that the B. thetaiotaomicron-infecting phage isolates described in this study do not encode integrases (Figures S1–S3). Similarly, the PhiSh genomes we identified do not encode integrases (Supplementary Data 1) and were not identified as parts of larger contigs containing recognizable sequences of bacterial chromosomes (Table S8). Further, a subset of PhiSh genomes (i.e., with “flag=1” or “flag=3” in the contig name; Table S8) were assembled as stand-alone contigs (no connectivity with other contigs, or as circularized contigs (indicating a complete phage genome), respectively. Other connectivity levels are observed for another subset of PhiSh genomes (i.e., with “flag=0” in the contig name; Table S8) which likely a results from genomes that are either incomplete or heterogenous (e.g., a mixture of related genomes causing the assembly graph to diverge). Taken together, these data suggest that the phage isolates and related PhiSh described here are lytic phages.
Only partial γ-like PhiSh genomes were identified (Fig. 3C). The lack of full-length γ-like PhiSh genomes may be due to insufficient sequencing depth of the original studies or the presence of highly divergent phages which share subsets of genes with cluster γ phages.
Identification of infection-associated phams
Previously, we demonstrated that multiple phase-variable chromosomal loci, including those encoding capsular polysaccharides (CPS), modify bacteriophage susceptibility in B. thetaiotaomicron (Porter et al., 2019). However, phage-encoded determinants of host tropism in these phages were previously unexplored. When the CPS specificities of these phages are compared with genome cluster membership (Fig. 1B, Table S1), relationships between host range and genome cluster membership become evident (Fig. 4A). For example, cluster γ phages tend to be most restrictive in their host range, primarily infecting cps7, cps8, and acapsular strains. Cluster β phages are similarly restricted in their host range but are unique in their ability to efficiently infect B. thetaiotaomicron cps3. Some cluster α phages have promiscuous host ranges while other cluster α phages have restrictive host ranges (more similar to those of the cluster β and cluster γ phages). This variation in host range among cluster α phages prompted us to search for phams that are associated with different infection patterns.
Figure 4. Prediction of infection-associated phams (IAPs) in Bacteroides-infecting phages.
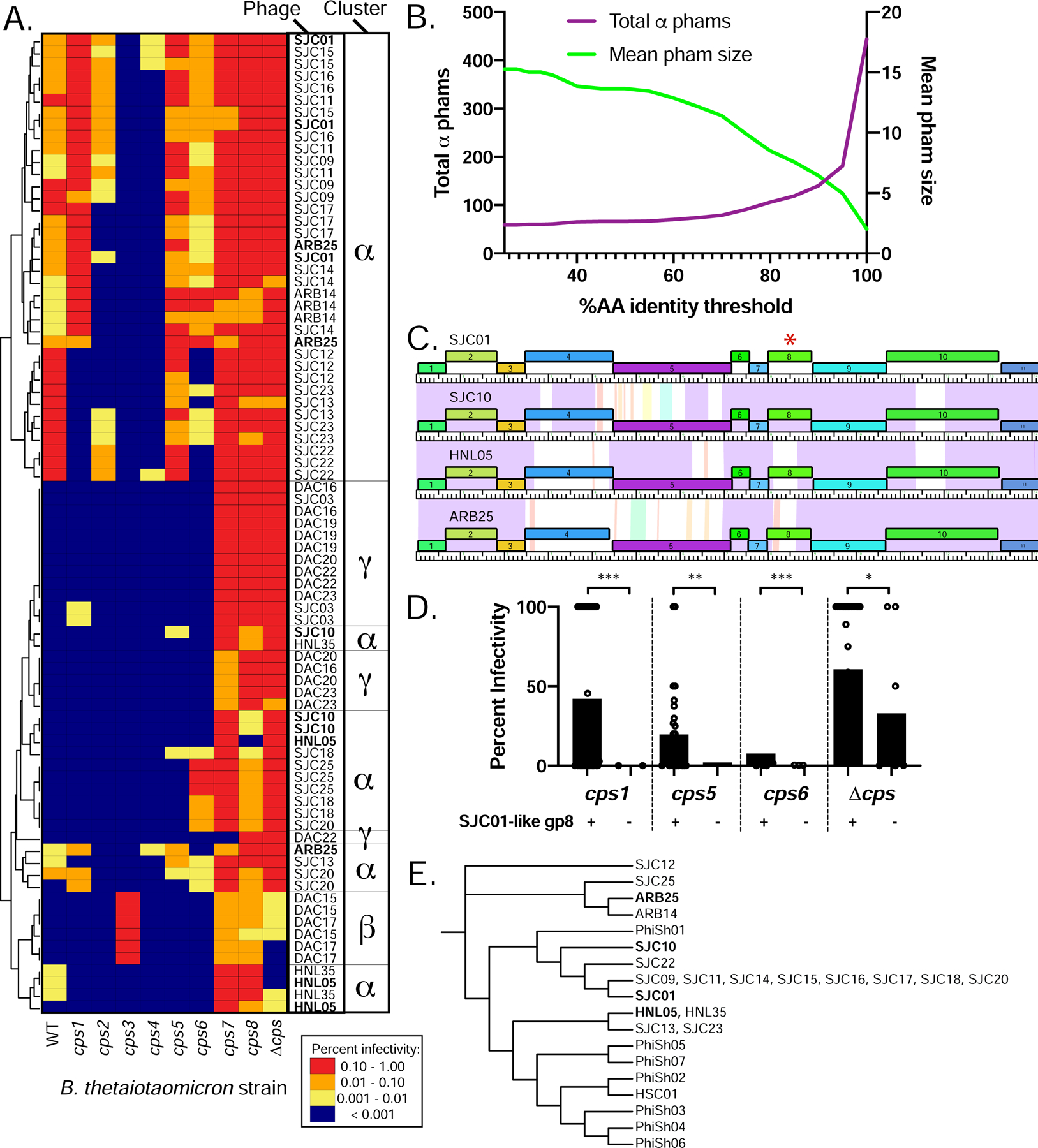
(A) Host range of phages on strains of Bacteroides thetaiotaomicron VPI-5482 expressing a variety of CPS (WT, wild type), a single CPS (cps1-cps8 strains) or no CPS (Δcps, acapsular). Tenfold serial dilutions of phage lysates ranging from approximately 106 to 103 plaque-forming units (PFU)/mL were spotted onto top agar plates containing each of the 10 bacterial strains. Plates were then incubated overnight, and plaques on each host were counted. Phage titers (PFU/ml) were calculated for each host and normalized to the titer on the “preferred host strain” for each replicate (individual replicates are shown to highlight variation between replicates, n=3 per phage). The phages were then clustered based on their plaquing efficiencies on the different strains (see STAR Methods). Each row in the heat map corresponds to one of three individual experimental replicates with a phage, whereas each column corresponds to one of the 10 host strains. (B) Changes in the total number of phams and average pham size as a function of percent amino acid identity. (C) Partial genome maps of 4 cluster α phages (SJC01, SJC10, HNL05, and ARB25) highlighting variation in gp4, gp5, and gp8. The genes are color coded according to pham membership at standard cutoffs and are numbered. Pairwise nucleotide identity is represented as shading between genomes. The color of this shading represents the degree of sequence similarity with violet being the most similar (BLASTN score = 0), progressing through the color spectrum from indigo, blue, green, yellow, orange, to red, which is the least similar (BLASTN score = 10−4). Regions with no shading indicate no similarity with a BLASTN score greater than 10−4. The red asterisk highlights gp8 from these phages. Data corresponding to these 4 phages in panels A and E are in bold. (D) Phages containing SJC01-like gp8 were compared against phages containing the alternative allele of gp8 (85% AA identity threshold) in terms of infectivity on bacterial strains highlighted in panel A. SJC01 gp8 is associated with higher infectivity of B. thetaiotaomicron cps1, cps5, cps6, and Δcps as assessed by Mann-Whitney U Test (p<0.05 = *, p<0.01 = **, p<0.001 = ***). (E) gp8 from cluster α isolates and the gene in the same position in cluster α genomes identified from metagenomes (PhiSh01–07, and HSC01) were aligned using ClustalW and a dendrogram of these alleles was created using The Interactive Tree of Life (see STAR Methods).
We noted two major themes driving genomic variation among the cluster α phages: variation between shared predicted structural components in these phages, such as gene products (gps) 4, 5, and 8; and mosaicism in genes at the 3’ end of the genomes, representing genes encoding small hypothetical proteins and genes encoding predicted DNA methylases (Fig. S1). Therefore, we considered the possibility that allelic variation and presence/absence of phams could contribute to differences in host range among the phages. To account for each of these possibilities, we used an algorithm to identify infection-associated phams (IAPs). Specifically, we computed phams at alternative cutoffs such that membership was dictated by varying levels of amino acid (AA) identity between 25 and 100% (see STAR Methods). As the threshold value increases, the total number of phams increases, with a concomitant decrease in mean pham membership (Fig. 4B and (Cresawn et al., 2011)). With the possibility that different thresholds may reveal allelic variants that correspond to infectivity, we compared these alternative pham tables with infection thresholds. This approach identified 662 total phams across all 64 infectivity/pham threshold comparisons in the 19 cluster α phages. Of these, 135 were identified as IAPs.
Among the IAPs is cluster α gp8, which is present in all cluster α phages, exhibits sequence variation among these phages, and is predicted to encode a tail protein (Figs. 1C, 4C, S1). At the 85% AA identity cutoff, gp8 is grouped into two distinct phams. Phages that have the SJC01-like variant infect the cps1, cps5, cps6, and Δcps strains more efficiently than those that do not (Fig. 4D). Analysis of this IAP in the context of metagenome-derived cluster α-like phages reveals additional variation not represented in our isolates (Fig. 4E). Interestingly, the variants of this IAP in PhiSh02 and HSC01 contain Bacteroides-Associated Carbohydrate Binding Often N-terminal (BACON) domains (See (Reyes et al., 2013) and Supplementary Data 1). These combined observations suggest a role for this IAP in differential recognition of complex polysaccharides (e.g. capsular polysaccharides) in Bacteroides-infecting phages.
We were unable to identify IAPs in the cluster beta and cluster gamma phages. We expect that this is due to the small number of cluster beta and gamma representatives isolated (n=2 and n=6, respectively) and intra-cluster similarities in host range (Figure 4A).
Discussion
In this work, we integrate phenotypic and genomic characterization of isolated phages with metagenomic analysis to highlight several opportunities for future study of gut-resident phages. In particular, though metagenome-focused studies of phages continue to generate tremendous insights into the composition and dynamics of viromes in the gut and other ecosystems, they are limited in scope due to a lack of definitive connections between predicted phages and their bacterial hosts. Several approaches have been developed to predict phage host range (Edwards et al., 2016). These approaches have been validated in part, notably for CrAss001, which was isolated on B. intestinalis after predictions that crAss-like phages infect members of the phylum Bacteroidetes (Dutilh et al., 2014, Yutin et al., 2018). We further validate these predictions with two more crAss-like phages, DAC15 and DAC17, which infect B. thetaiotaomicron. This brings the total number of published crAss-like phage isolates to three (Figs. 2, S4). As more crAss-like phages are isolated, we anticipate that existing discrepancies relating to the roles of these phages in the gut (e.g. some crAss-like phages are associated with disease (Oude Munnink et al., 2014) while others are stably maintained in healthy individuals (Shkoporov et al., 2019)) will be disentangled with controlled experimental approaches.
Similarly, based on our genomic and metagenomic analysis of cluster α phages, we show that two previously reported phage genomes (PhiSh04 and HSC01) are related despite differences in temporal dynamics and predicted host range (Fig. 3D) (Reyes et al., 2013, Minot et al., 2013). This raises questions about which phage or bacterially encoded genes are responsible for differences in host range and phage population dynamics. Some insights come from experiments using the cluster α phage ARB25. ARB25 is stably maintained in bi-colonization with its host in gnotobiotic mice for months and the mechanisms used by B. thetaiotaomicron to evade ARB25 include differential expression of CPS and other cell surface features (Porter et al., 2019). HSC01, unlike PhiSh04 or ARB25, does not stably co-exist with its predicted host (B. caccae) in gnotobiotic mice (Reyes et al., 2013), suggesting that although HSC01 is closely related to phages that are stably maintained, it may have distinct ecological impacts in the gut. Alternatively, it is possible that other members of the gut microbiome affect the relationship between these phages and their hosts.
By combining work that involves phage isolation, sequencing, and phenotypic characterization, with metagenomic analyses, we hope to reciprocally inform these studies (e.g., by adding phages and information on IAPs to publically available databases) and to provide the reagents necessary to experimentally test hypotheses using the broad toolkit available in the gut microbiome field (e.g., by probing phage-host interactions using gnotobiotics and molecular genetics). Future isolation efforts can be further optimized with high throughput approaches (e.g. robotics and automated liquid handling) or as part of educational efforts like those pioneered by the SEA-PHAGES program (Hanauer et al., 2017), which would simultaneously crowd source the effort while providing training opportunities for the next generation of microbiome scientists. Together, this integration will allow for a more comprehensive consideration of the interactions that occur between phages and their hosts at the population, individual, and molecular scales.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Justin Sonnenburg (jsonnenburg@stanford.edu).
Materials Availability
Phages described in this study are available upon request from the Lead Contact.
Data and Code Availability
The Genomes of the phage isolates used in this study (also described in Table S1) are uploaded to NCBI, BioProject ID PRJNA606391. Supplementary Data 1–3, containing Genbank and fasta files of the PhiSh genomes; code and data to allow an exact reproduction of the IAP identification method; and a tutorial for identifying PhiSh, respectively, are accessible at https://purl.stanford.edu/vz665fs9726
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains and culture conditions.
The bacterial strains used in this study (B. thetaiotaomicron VPI-5482 and isogenic cps mutants) are listed in the Key Resources Table. Frozen stocks of these strains were maintained in 25% glycerol at −80°C and were routinely cultured in an anaerobic chamber (Coy) under 5% H2, 10% CO2, 85% N2 at 37°C in Bacteroides Phage Recovery Medium (BPRM), as described previously (Porter et al., 2019): per 1 liter of broth, 10 g meat peptone, 10 g casein peptone, 2 g yeast extract, 5 g NaCl, 0.5 g L-cysteine monohydrate, 1.8 g glucose, and 0.12 g MgSO4 heptahydrate were added; after autoclaving and cooling to approximately 55 °C, 10 ml of 0.22 μm-filtered hemin solution (0.1% w/v in 0.02% NaOH), 1 ml of 0.22 μm-filtered 0.05 g/ml CaCl2 solution, and 25 ml of 0.22μm-filtered 1 M Na2CO3 solution were added. For BPRM agar plates, 15 g/L agar was added prior to autoclaving and hemin and Na2CO3 were added as above prior to pouring the plates. For BPRM top agar used in soft agar overlays, 3.5 g/L agar was added prior to autoclaving. Hemin, CaCl2, and Na2CO3 were added to the top agar as above immediately before conducting experiments. Bacterial strains were routinely struck from the freezer stocks onto BPRM agar and grown anaerobically for up to 2 days. A single colony was picked for each bacterial strain, inoculated into 5 mL BPRM, and grown anaerobically overnight to provide the starting culture for experiments.
KEY RESOURCES TABLE
METHOD DETAILS
Bacteriophage isolation from primary wastewater effluent and sewer-adjacent pond water
The bacteriophages described in this study were isolated from primary wastewater effluent from the Ann Arbor, Michigan Wastewater Treatment Plant and from the San Jose-Santa Clara Regional Wastewater Treatment Facility, as described previously (Porter et al., 2019). For the current study, phages were isolated from primary wastewater effluent from the Sand Island Wastewater Treatment Plant (Honolulu, Hawaii) or from sewer-adjacent pond water in Dhaka, Bangladesh (Table S1). Water samples were centrifuged at 5,500 rcf for 10 minutes at room temperature to remove any remaining solids. The supernatant was then sequentially filtered through 0.45 μm and 0.22 μm polyvinylidene fluoride (PVDF) filters. This processed primary effluent was concentrated up to 500-fold via 100 kDa PVDF size exclusion columns.
Initial screening for plaques was done using a soft agar overlay method where 50 μL of the concentrated primary effluent was combined with 0.5 mL overnight culture and 4.5 mL BPRM top agar and poured onto a standard circular petri dish [100 mm × 15 mm]. Soft agar overlays were incubated anaerobically at 37 °C overnight. To promote a diverse collection of phages, no more than 5 plaques from the same plate were plaque purified and a diversity of plaque morphologies were selected as applicable.
Single, isolated plaques were picked into 100 μL phage buffer (prepared as an autoclaved solution of 5 ml of 1 M Tris pH 7.5, 5 ml of 1 M MgSO4, 2 g NaCl in 500 ml with ddH2O). Phages were plaque purified using a 96-well plate-based method, where serial dilutions were prepared in 96-well plates and 1 μL of each dilution was spotted onto a solidified top agar overlay. This procedure was repeated at least 3 times to plaque purify each phage.
High titer phage stocks were generated by flooding a soft agar overlay on a plate that yielded a “lacey” pattern of bacterial growth (near confluent lysis). Following overnight incubation of each plate, 5 ml of sterile phage buffer was added to the plate to re-suspend the phage. After at least 2 hours of incubation at room temperature, the lysate was spun at 5,500 rcf for 10 minutes to clear debris and then filter sterilized through a 0.22 μm PVDF filter. For more details on phages used in this work, see Table S1.
Phage genome sequencing and assembly
DNA was extracted from high-titer phage lysates and sequencing libraries were prepared using the Ultra II FS Kit (New England Biolabs) or for ARB14 and ARB25, the TruSeq Nano DNA LT Kit (Illumina). Libraries were quantified using a BioAnalyzer (Agilent) and subsequently sequenced using 150-base single-end reads (Illumina MiSeq), or for ARB14 and ARB25, 250-base paired-end reads (Illumina MiSeq). Phage genomes were assembled using Geneious version 9.1.5 with default options after trimming reads with an error probability limit of 0.05. All genomes published here circularized during assembly. Phage genomes belonging to the same cluster were rearranged to have identical 5’ ends. Coverage for each assembly was calculated by mapping reads onto each assembled genome using bowtie2 (Langmead and Salzberg, 2012) (--very-sensitive) and then using jgi_summarize_ban_contig_depths from the MetaBAT2 tool (Kang et al., 2019) to calculate mean coverage depth.
Annotation and comparative analyses of B. thetaiotaomicron infecting phages
Protein-coding genes and tRNAs were predicted and annotated using DNA-Master default parameters (http://cobamide2.pitt.edu/), which incorporates Genemark (Besemer and Borodovsky, 2005), Glimmer (Delcher et al., 1999), and tRNAscan-SE (Lowe and Eddy, 1997). Phage genomes were clustered together using vConTACT2 and the ProkaryoticViralRefSeq94-Merged database with default parameters (Bin Jang et al., 2019). Phage genomes were annotated and compared on the basis of shared gene phamily (pham) membership with Phamerator using default parameters (Cresawn et al., 2011). Phams are groups of related protein-encoding genes where pham membership is built and expanded when a candidate protein shares ≥32.5% identity or blastp e-value ≤1e-50 with one or more existing members of the pham. Phage genome ends and packaging strategies for cluster β phages were inferred using PhageTerm (Garneau et al., 2017) which identified clear direct terminal repeats (DTRs). PhageTerm was unable to identify DTRs or cohesive ends in the cluster α or γ phages, possibly indicating a headful packaging strategy. The large terminase proteins share significant similarity (BLASTP e-value <1e-3) with the PBSX-family of large terminases, which also use a headful packaging strategy (Table S1) (Anderson and Bott, 1985). To predict virion structural genes, iVireons was used with default parameters (Seguritan et al., 2012). Protein-coding genes were classified as “predicted structural genes” (e.g. general structural, tail, or capsid, annotated in Fig. 1) for genes with score 0.7 and above. To visualize genome-level relationships among phages, pham tables were processed with Janus (http://cobamide2.pitt.edu/) and Splitstree (Hudson and Bryant, 2006) using default parameters. CRISPR protospacers were identified and used as the basis for host prediction of the isolated B. thetaiotaomicron phages and PhiSh genomes with the JGI IMG/VR Spacer Database (Paez-Espino et al., 2019). A spacer-protospacer match was considered to be relevant if at least 95% identity is shared over the entire length of the spacer. Matches were not identified for the B. thetaiotaomicron-infecting phage isolates (Table S1). Matches to PhiSh genomes are shown in Table S8. Genomes of the B. thetaiotaomicron-infecting phage isolates described in Table S1 are uploaded to NCBI (BioProject ID PRJNA606391).
Quantitative host range analysis
Host range analysis was carried out as previously described (Porter et al., 2019). Briefly, high titer phage stocks were prepared on their “preferred host strain,” which is the strain yielding the highest titer of phages in a pre-screen of phage host range (Table S1). Lysates were then diluted to approximately 106 PFU/mL, were added to the wells of a 96-well plate, then further diluted to 105, 104, and 103 PFU/mL. One microliter of each dilution was plated onto solidified top agar overlays containing wildtype B. thetaiotaomicron, acapsular B. thetaiotaomicron, or B. thetaiotaomicron expressing a single capsule (see Key Resources Table). After spots dried, plates were incubated anaerobically for 15–24 hours prior to counting plaques. Phage titers were normalized to the “preferred host strain.” Three independent replicates were performed for each phage/host pair and are represented individually in Figure 4A. The heatmaps and dendrogram were generated using the “heatmap” function in the “stats” package of R (version 3.4.0), which employs unsupervised hierarchical clustering (complete linkage method) to group similar phage infection profiles, with branch length in the dendrogram at the left of Figure 4A indicating degree of similarity between infection profiles.
Infection associated pham identification
We defined an infection-associated pham (IAP) as a pham that (1) was found in every phage of a given cluster (α, β, and γ; see Fig. 1) that infected the B. thetaiotaomicron isolate in question, but (2) was not found in every phage of the same cluster. Criterion (1) is a stringent threshold. For example, if 10 different phages infected a given bacterial strain, but only 9 shared a particular pham, it would fail criterion (1). Criterion (2) was included to eliminate core genes.
We employed two important thresholds when identifying IAPs. The first of these is an infection threshold - the normalized percentage of infectivity a given phage on a given isolate as described in STAR METHODS section ‘Quantitative host range analysis’. Here, a stringent threshold is 100%, which considers “infection” to be a case where the phage generates as many plaques on a given B. thetaiotaomicron strain as it does on its preferred host strain. A permissive threshold is 1% - here a phage would have to cause 1/100th as many plaques as it did on its preferred host. The second of these is the pham identity threshold - the percentage sequence identity that two genes must share to be counted as in the same pham. This clustering is described in STAR Methods section ‘Annotation and comparative analyses of B. thetaiotaomicron infecting phages.’ Here, a stringent clustering threshold is 100%, where genes sharing 100% sequence identity are grouped in the same pham. A permissive threshold would be 1%. The lower this threshold, the more disparate the sequences that are grouped together.
We computed our IAP identification algorithm using as thresholds each member of the product set of [1%, 5%, 10%, 50%] X [25%, 27.5%, 30%, 35%, 40%, 45%, 50%, 60%, 70%, 75%, 80%, 85%, 90%, 95%, 100%] (infection threshold and pham identity threshold, respectively). Code and data are available as Supplementary Data 2, which provides a simple python script and the accompanying data allowing exact reproduction of the method.
Comparisons of cluster α gp8 and homologs from metagenome-derived cluster α genomes (PhiSh01-PhiSh07, HSC01) were conducted using Clustal Omega (Sievers et al., 2011) and visualized using The Interactive Tree of Life (Letunic and Bork, 2019) with default parameters.
Transmission Electron Microscopy
High titer phage lysates of representatives from each genome cluster (SJC01, DAC15, DAC20) were precipitated overnight at 4°C with gentle rocking in a solution of 1M NaCl and 10% w/v PEG8000. Phages were then precipitated via centrifugation (5500×g for 10 minutes at 4°C). Six milliliters of phage buffer was added to the pellet and broken with gentle agitation and swirling and the mixture was incubated overnight at 4°C with gentle rocking. The following day, the sample was centrifuged at 5500×g for 10 minutes at 4°C. CsCl was slowly added to the supernatant and gently dissolved via gentle swirling (final concentration 75% w/v solution). Samples were centrifuged at 26,000 RPM for 24 hours at 5°C. Phage bands were extracted and stored at 4°C.
CsCl-banded lysates were applied directly to glow discharged Carbon Type-B 200 mesh copper grids. Samples were allowed to adsorb to the grids for 3 minutes and were subsequently washed with 2 drops of ultrapure water. Three drops of uranyl acetate (1% w/v in water) were applied to the grid and the third drop was maintained on the grid for 1 minute. Filter paper was used to remove the majority of the uranyl acetate and allowed to dry at room temperature. Samples were then viewed at 120 kV on a JEOL JEM-1400 transmission electron microscope and images were collected using a Gatan Orius digital camera.
Comparative genomic analyses between isolated B. thetaiotaomicron infecting phages, other isolated Bacteroides-infecting phages, and PhiSh genomes.
Genomes of representatives of each genome cluster (SJC01, DAC15, DAC20) were queried against the entire SRA using SearchSRA (Torres et al., 2017, Levi et al., 2018, Towns et al., 2014, Stewart et al., 2015, Buchfink et al., 2015b, Langmead and Salzberg, 2012). To determine whether these genome clusters are found in human gut metagenomes, one representative from each cluster (SJC01, DAC15, DAC20) was queried using SearchSRA using the “protein search” option. SearchSRA uses DIAMOND blastx to query 100,000 reads from each of ~100,000 metagenomes publicly available in NCBI SRA against a single query amino acid sequence. The input data for each representative phage genome consisted of a single amino acid sequence consisting of every translated gene in order of appearance in the genome, separated by “XXX”. This input format was required when the analysis was conducted (July 24, 2019).
Data were retrieved from SearchSRA in the typical BLAST M8 format (one file per NCBI metagenome aligned to the reference phage) and parsed into BED format. BEDTools (Quinlan, 2014) coverage was used to calculate the coverage depth of each base pair along the genome. These tables were read into R 3.6.2. For each sequence run (SRR) that had ≥1 read aligning to a query amino acid sequence, SRAdb (Zhu et al., 2013) was used to get the associated sample accession number (SRS) and other related sample metadata. Coverage data from sequencing runs belonging to the same sample were combined, and then average coverage depth and detection (% of bases with ≥ 1x coverage) was calculated for each metagenome sample mapped.
For each metagenome sample mapped where the number of reads sequenced was >10000, the estimated true coverage depth of the reference phage in that metagenome sample was calculated as # spots sequenced*SearchSRA average coverage / 100000. To determine whether to assemble a given metagenome and search for a relative of a given representative phage, we filtered the list of metagenome samples based on whether the estimated real coverage was >15% and the percent of the genome detected was >20%. This list was filtered further by selecting only human gut metagenomes and by selecting samples where coverage and detection were the highest (Table S7).
Metagenomes were downloaded from NCBI SRA using parallel-fastq-dump 0.6.5 (https://github.com/rvalieris/parallel-fastq-dump). For each metagenome assembled, reads were trimmed using BBDuk (https://sourceforge.net/projects/bbmap/) 38.69 (parameters ref=adapters,phix threads=$(($coreNum - 2)) ktrim=r k=23 mink=11 hdist=2 tpe tbo qtrim=rl trimq=20 minlen=55) and assembled using MEGAHIT v1.2.9 (--mem-flag 2 -k-list 21,29,39,49,59,69,79,89,99) for all samples, or -k-list 21,29,39,49,59,69,79,89,99,109,119,129,139,149 if read length was >=2×250bp.
To identify contigs in the metagenome assemblies that might be putative relatives of the representative phages, we used DIAMOND 0.9.24 (Buchfink et al., 2015a) to build a blastx database containing all individual amino acid sequences from all three representative genomes. DIAMOND blastx queries consisted all contigs from a single metagenome assembly. Individual contigs containing significant (e <= 0.001) hits for >25% of the genes from a given representative phage genome were reoriented to align the 5’ ends with isolated phage genomes and then included in subsequent Phamerator analysis (Table S8). See Supplementary Data 1 for Genbank and fasta files of the PhiSh genomes. A tutorial for performing this analysis can be found as Supplementary Data 3.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using Graphpad Prism 8.4. Details of specific analyses, including statistical tests used, are found in applicable figure legends.
Supplementary Material
Table S1. Isolation and genomic details of Bacteroides thetaiotaomicron phages used in this study. Related to Figure 1.
Table S3. Conserved domains identified in cluster alpha phages. Related to Figure 1.
Table S4. Conserved domains identified in cluster beta phages. Related to Figure 1.
Table S5. Conserved domains identified in cluster gamma phages. Related to Figure 1.
Table S6. Pham table including genomes of Bacteroides-infecting phage isolates and genomes identified in previous metagenomic studies. Related to Figures 1–3.
Table S7. SRA Samples containing PhiSh genomes. Related to Figure 3.
Table S8. PhiSh genome details. Related to Figure 3.
Highlights.
27 Bacteroides thetaiotaomicron-infecting phages from two continents were isolated
Genome sequencing of isolates reveals unexplored diversity of these phages
Relatives of these isolates were identified in existing metagenomic datasets
Phage genes associate with host capsular polysaccharide tropism
Acknowledgements
We thank Jackson Gardner for assistance with host range analyses; Dylan Maghini for assistance with lysate preparation for electron microscopy; Gayatri Vithanage, Lyle Shizumura, Greig Steward, and Ned Ruby for logistical assistance in phage isolation from Sand Island Wastewater Treatment Plant; and John Perrino for transmission electron microscopy expertise. This work was funded by NIH grants (GM099513 and DK096023 to ECM; DP5OD019893 to EJN, DK085025 and AT00989203 to JLS), an NIH postdoctoral NRSA (5T32AI007328 to AJH), a Stanford University School of Medicine Dean’s Postdoctoral Fellowship (AJH), the NIH Cellular Biotechnology Training Program (T32GM008353 to NTP), by a NCCR ARRA Award (1S10RR026780-01 to Stanford University Cell Sciences Imaging Facility), and by a National Science Foundation Graduate Research Fellowship (DGE-114747 to BDM). JLS is a Chan Zuckerberg Biohub Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- ANDERSON LM & BOTT KF 1985. DNA packaging by the Bacillus subtilis defective bacteriophage PBSX. J Virol, 54, 773–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARR JJ, AURO R, FURLAN M, WHITESON KL, ERB ML, POGLIANO J, STOTLAND A, WOLKOWICZ R, CUTTING AS, DORAN KS, SALAMON P, YOULE M & ROHWER F 2013. Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc Natl Acad Sci U S A, 110, 10771–6. DOI: 10.1073/pnas.1305923110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENLER S, COBIAN-GUEMES AG, MCNAIR K, HUNG SH, LEVI K, EDWARDS R & ROHWER F 2018. A diversity-generating retroelement encoded by a globally ubiquitous Bacteroides phage. Microbiome, 6, 191. 10.1186/s40168-018-0573-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- BESEMER J & BORODOVSKY M 2005. GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res, 33, W451–4. 10.1093/nar/gki487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIN JANG H, BOLDUC B, ZABLOCKI O, KUHN JH, ROUX S, ADRIAENSSENS EM, BRISTER JR, KROPINSKI AM, KRUPOVIC M, LAVIGNE R, TURNER D & SULLIVAN MB 2019. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat Biotechnol, 37, 632–639. DOI: 10.1038/s41587-019-0100-8 [DOI] [PubMed] [Google Scholar]
- BRUSSOW H & HENDRIX RW 2002. Phage genomics: small is beautiful. Cell, 108, 13–6. DOI: 10.1016/s0092-8674(01)00637-7 [DOI] [PubMed] [Google Scholar]
- BUCHFINK B, XIE C & HUSON DH 2015a. Fast and sensitive protein alignment using DIAMOND. Nat Methods, 12, 59–60. DOI: 10.1038/nmeth.3176 [DOI] [PubMed] [Google Scholar]
- CRESAWN SG, BOGEL M, DAY N, JACOBS-SERA D, HENDRIX RW & HATFULL GF 2011. Phamerator: a bioinformatic tool for comparative bacteriophage genomics. BMC Bioinformatics, 12, 395. DOI: 10.1186/1471-2105-12-395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DELCHER AL, HARMON D, KASIF S, WHITE O & SALZBERG SL 1999. Improved microbial gene identification with GLIMMER. Nucleic Acids Res, 27, 4636–41. DOI: 10.1093/nar/27.23.4636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUERKOP BA, KLEINER M, PAEZ-ESPINO D, ZHU W, BUSHNELL B, HASSELL B, WINTER SE, KYRPIDES NC & HOOPER LV 2018. Murine colitis reveals a disease-associated bacteriophage community. Nat Microbiol. DOI: 10.1038/s41564-018-0210-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUTILH BE, CASSMAN N, MCNAIR K, SANCHEZ SE, SILVA GG, BOLING L, BARR JJ, SPETH DR, SEGURITAN V, AZIZ RK, FELTS B, DINSDALE EA, MOKILI JL & EDWARDS RA 2014. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat Commun, 5, 4498. DOI: 10.1038/ncomms5498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS RA, MCNAIR K, FAUST K, RAES J & DUTILH BE 2016. Computational approaches to predict bacteriophage-host relationships. FEMS Microbiol Rev, 40, 258–72. DOI: 10.1093/femsre/fuv048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARNEAU JR, DEPARDIEU F, FORTIER LC, BIKARD D & MONOT M 2017. PhageTerm: a tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci Rep, 7, 8292. DOI: 10.1038/s41598-017-07910-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRAZZIOTIN AL, KOONIN EV & KRISTENSEN DM 2017. Prokaryotic Virus Orthologous Groups (pVOGs): a resource for comparative genomics and protein family annotation. Nucleic Acids Res, 45, D491–d498. DOI: 10.1093/nar/gkw975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- GREGORY AC, ZABLOCKI O, HOWELL A, BOLDUC B & SULLIVAN MB 2019. The human gut virome database. bioRxiv. doi: 10.1101/655910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUERIN E, SHKOPOROV A, STOCKDALE SR, CLOONEY AG, RYAN FJ, SUTTON TDS, DRAPER LA, GONZALEZ-TORTUERO E, ROSS RP & HILL C 2018. Biology and Taxonomy of crAss-like Bacteriophages, the Most Abundant Virus in the Human Gut. Cell Host Microbe, 24, 653–664.e6. DOI: 10.1016/j.chom.2018.10.002 [DOI] [PubMed] [Google Scholar]
- GUTHRIE L, GUPTA S, DAILY J & KELLY L 2017. Human microbiome signatures of differential colorectal cancer drug metabolism. NPJ Biofilms Microbiomes, 3, 27. DOI: 10.1038/s41522-017-0034-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- HANAUER DI, GRAHAM MJ, BETANCUR L, BOBROWNICKI A, CRESAWN SG, GARLENA RA, JACOBS-SERA D, KAUFMANN N, POPE WH, RUSSELL DA, JACOBS WR JR., SIVANATHAN V, ASAI DJ & HATFULL GF 2017. An inclusive Research Education Community (iREC): Impact of the SEA-PHAGES program on research outcomes and student learning. Proc Natl Acad Sci U S A, 114, 13531–13536. DOI: 10.1073/pnas.1718188115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- HATFULL GF & HENDRIX RW 2011. Bacteriophages and their genomes. Curr Opin Virol, 1, 298–303. DOI: 10.1016/j.coviro.2011.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAWKINS SA, LAYTON AC, RIPP S, WILLIAMS D & SAYLER GS 2008. Genome sequence of the Bacteroides fragilis phage ATCC 51477-B1. Virol J, 5, 97. DOI: 10.1186/1743-422X-5-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- HE Q, GAO Y, JIE Z, YU X, LAURSEN JM, XIAO L, LI Y, LI L, ZHANG F, FENG Q, LI X, YU J, LIU C, LAN P, YAN T, LIU X, XU X, YANG H, WANG J, MADSEN L, BRIX S, WANG J, KRISTIANSEN K & JIA H 2017. Two distinct metacommunities characterize the gut microbiota in Crohn’s disease patients. Gigascience, 6, 1–11. DOI: 10.1093/gigascience/gix050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- HICKEY CA, KUHN KA, DONERMEYER DL, PORTER NT, CHUNSHENG J, CAMERON EA, JUNG H, KAIKO GE, WEGORZEWSKA M, MALVIN NP, GLOWACKI RWP, HANSSON GC, ALLEN PM, MARTENS EC, STAPPENBECK TS 2015. Colitogenic Bacteroides thetaiotaomicron Antigens Access Host Immune Cells in a Sulfatase-Dependent Manner via Outer Membrane Vesicles. Cell Host & Microbe, 17, 672–80. DOI: 10.1016/j.chom.2015.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUDSON DH & BRYANT D 2006. Application of Phylogenetic Networks in Evolutionary Studies. Mol Biol Evol. 23, 254–67. DOI: 10.1093/molbev/msj030 [DOI] [PubMed] [Google Scholar]
- KANG DD, LI F, KIRTON E, THOMAS A, EGAN R, AN H & WANG Z 2019. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ, 7, e7359. DOI: 10.7717/peerj.7359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANGMEAD B & SALZBERG SL 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods, 9(4):357–9 DOI: 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LETUNIC I & BORK P 2019. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res, 47, W256–w259. DOI: 10.1093/nar/gkz239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEVI K, RYNGE M, EROMA A & EDWARDS RA 2018. Searching the Sequence Read Archive using Jetstream and Wrangler. Proceedings of the Practice and Experience on Advanced Research Computing. DOI: 10.1145/3219104.3229278 [DOI] [Google Scholar]
- LIU W, ZHANG J, WU C, CAI S, HUANG W, CHEN J, XI X, LIANG Z, HOU Q, ZHOU B, QIN N & ZHANG H 2016. Unique Features of Ethnic Mongolian Gut Microbiome revealed by metagenomic analysis. Sci Rep, 6, 34826. DOI: 10.1038/srep34826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOWE TM & EDDY SR 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res, 25, 955–64. DOI: 10.1093/nar/25.5.955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MANRIQUE P, BOLDUC B, WALK ST, VAN DER OOST J, DE VOS WM & YOUNG MJ 2016. Healthy human gut phageome. Proc Natl Acad Sci U S A, 113, 10400–5. DOI: 10.1073/pnas.1601060113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTENS EC, CHIANG HC, GORDON JI 2008. Mucosal Glycan Foraging Enhances Fitness and Transmission of a Saccharolytic Human Gut Bacterial Symbiont. Cell Host & Microbe, 4, 447–57. DOI: DOI: 10.1016/j.chom.2008.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MINOT S, BRYSON A, CHEHOUD C, WU GD, LEWIS JD & BUSHMAN FD 2013. Rapid evolution of the human gut virome. Proc Natl Acad Sci U S A, 110, 12450–5. DOI: 10.1073/pnas.1300833110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MONACO CL, GOOTENBERG DB, ZHAO G, HANDLEY SA, GHEBREMICHAEL MS, LIM ES, LANKOWSKI A, BALDRIDGE MT, WILEN CB, FLAGG M, NORMAN JM, KELLER BC, LUEVANO JM, WANG D, BOUM Y, MARTIN JN, HUNT PW, BANGSBERG DR, SIEDNER MJ, KWON DS & VIRGIN HW 2016. Altered Virome and Bacterial Microbiome in Human Immunodeficiency Virus-Associated Acquired Immunodeficiency Syndrome. Cell Host Microbe, 19, 311–22. DOI: 10.1016/j.chom.2016.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- OGILVIE LA, CAPLIN J, DEDI C, DISTON D, CHEEK E, BOWLER L, TAYLOR H, EBDON J & JONES BV 2012. Comparative (meta)genomic analysis and ecological profiling of human gut-specific bacteriophage phiB124–14. PLoS One, 7, e35053. DOI: 10.1371/journal.pone.0035053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- OUDE MUNNINK BB, CANUTI M, DEIJS M, DE VRIES M, JEBBINK MF, REBERS S, MOLENKAMP R, VAN HEMERT FJ, CHUNG K, COTTEN M, SNIJDERS F, SOL CJ & VAN DER HOEK L 2014. Unexplained diarrhoea in HIV-1 infected individuals. BMC Infect Dis, 14, 22. DOI: 10.1186/1471-2334-14-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAEZ-ESPINO D, ROUX S, CHEN IA, PALANIAPPAN K, RATNER A, CHU K, HUNTEMANN M, REDDY TBK, PONS JC, LLABRES M, ELOE-FADROSH EA, IVANOVA NN & KYRPIDES NC 2019. IMG/VR v.2.0: an integrated data management and analysis system for cultivated and environmental viral genomes. Nucleic Acids Res, 47, D678–d686. DOI: 10.1093/nar/gky1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- PORTER NT, HRYCKOWIAN AJ, MERRILL BD, FUENTES JJ, GARDNER JO, GLOWACKI RWP, SINGH S, CRAWFORD RD, SNITKIN ES, SONNENBURG JL & MARTENS EC 2019. Multiple phase-variable mechanisms, including capsular polysaccharides, modify bacteriophage susceptibility in Bacteroides thetaiotaomicron. bioRxiv. doi: 10.1101/521070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUINLAN AR 2014. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr Protoc Bioinformatics, 47, 11.12.1–34. DOI: 10.1002/0471250953.bi1112s47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- REN J, AHLGREN NA, LU YY, FUHRMAN JA & SUN F 2017. VirFinder: a novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome, 5, 69. DOI: 10.1186/s40168-017-0283-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- REYES A, WU M, MCNULTY NP, ROHWER FL & GORDON JI 2013. Gnotobiotic mouse model of phage-bacterial host dynamics in the human gut. Proc Natl Acad Sci U S A, 110, 20236–41. DOI: 10.1073/pnas.1319470110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROUX S, ENAULT F, HURWITZ BL & SULLIVAN MB 2015. VirSorter: mining viral signal from microbial genomic data. PeerJ, 3, e985. DOI: 10.7717/peerj.985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEGURITAN V, ALVES N JR., ARNOULT M, RAYMOND A, LORIMER D, BURGIN AB JR., SALAMON P & SEGALL AM 2012. Artificial neural networks trained to detect viral and phage structural proteins. PLoS Comput Biol, 8, e1002657. DOI: 10.1371/journal.pcbi.1002657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHKOPOROV AN, CLOONEY AG, SUTTON TDS, RYAN FJ, DALY KM, NOLAN JA, MCDONNELL SA, KHOKHLOVA EV, DRAPER LA, FORDE A, GUERIN E, VELAYUDHAN V, ROSS RP & HILL C 2019. The Human Gut Virome Is Highly Diverse, Stable, and Individual Specific. Cell Host Microbe, 26, 527–541.e5. DOI: 10.1016/j.chom.2019.09.009 [DOI] [PubMed] [Google Scholar]
- SHKOPOROV AN, KHOKHLOVA EV, FITZGERALD CB, STOCKDALE SR, DRAPER LA, ROSS RP & HILL C 2018. PhiCrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat Commun, 9, 4781. DOI: 10.1038/s41467-018-07225-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIEVERS F, WILM A, DINEEN D, GIBSON TJ, KARPLUS K, LI W, LOPEZ R, MCWILLIAM H, REMMERT M, SODING J, THOMPSON JD & HIGGINS DG 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol, 7, 539. DOI: 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEWART CA, COCKERILL TM, FOSTER I, HANCOCK D, MERCHANT N, SKIDMORE E, STANZIONE D, TAYLOR J, TUECKE S, TURNER G, VAUGHN M & GAFFNEY NI 2015. Jetstream: a self-provisioned, scalable science and engineering cloud environment. Proceedings of the 2015 XSEDE Conference: Scientific Advancements Enabled by Enhanced Cyberinfrastructure. DOI: 10.1145/2792745.2792774 [DOI] [Google Scholar]
- TORRES PJ, EDWARDS RA & MCNAIR KA 2017. {PARTIE}: a partition engine to separate metagenomic and amplicon projects in the Sequence Read Archive. Bioinformatics. 33(15): 2389–91 DOI: 10.1093/bioinformatics/btx184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOWNS J, COCKERILL T, DAHAN M, FOSTER I, GAITHER K, GRIMSHAW A, HAZLEWOOD V, LATHROP S, LIFKA D, PETERSON GD, ROSKIES R, SCOTT JR & WILKINS-DIEHR N 2014. XSEDE: Accelerating Scientific Discovery. Computing in Science Engineering. DOI: 10.1109/MCSE.2014.80 [DOI] [Google Scholar]
- YUTIN N, MAKAROVA KS, GUSSOW AB, KRUPOVIC M, SEGALL A, EDWARDS RA & KOONIN EV 2018. Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat Microbiol, 3, 38–46. DOI: 10.1038/s41564-017-0053-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHENG S, SHAO S, QIAO Z, CHEN X, PIAO C, YU Y, GAO F, ZHANG J & DU J 2017. Clinical Parameters and Gut Microbiome Changes Before and After Surgery in Thoracic Aortic Dissection in Patients with Gastrointestinal Complications. Sci Rep, 7, 15228. DOI: 10.1038/s41598-017-15079-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHU Y, STEPHENS RM, MELTZER PS & DAVIS SR 2013. SRAdb: query and use public next-generation sequencing data from within R. BMC Bioinformatics, 14, 19. DOI: 10.1186/1471-2105-14-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Isolation and genomic details of Bacteroides thetaiotaomicron phages used in this study. Related to Figure 1.
Table S3. Conserved domains identified in cluster alpha phages. Related to Figure 1.
Table S4. Conserved domains identified in cluster beta phages. Related to Figure 1.
Table S5. Conserved domains identified in cluster gamma phages. Related to Figure 1.
Table S6. Pham table including genomes of Bacteroides-infecting phage isolates and genomes identified in previous metagenomic studies. Related to Figures 1–3.
Table S7. SRA Samples containing PhiSh genomes. Related to Figure 3.
Table S8. PhiSh genome details. Related to Figure 3.
Data Availability Statement
The Genomes of the phage isolates used in this study (also described in Table S1) are uploaded to NCBI, BioProject ID PRJNA606391. Supplementary Data 1–3, containing Genbank and fasta files of the PhiSh genomes; code and data to allow an exact reproduction of the IAP identification method; and a tutorial for identifying PhiSh, respectively, are accessible at https://purl.stanford.edu/vz665fs9726