

Abstract
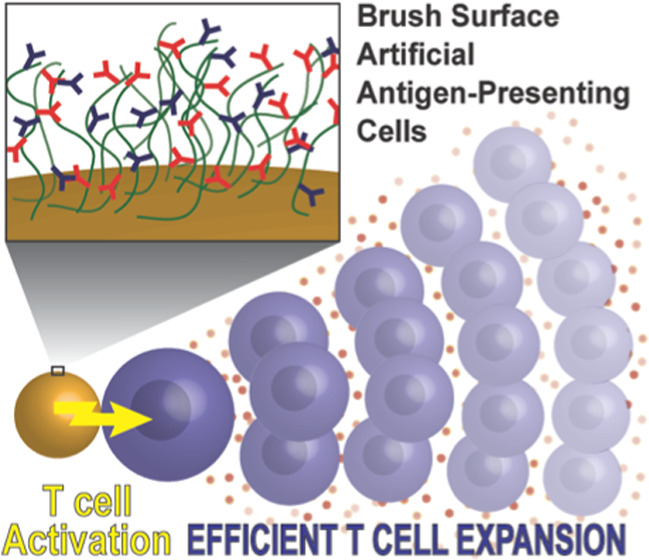
A variety of bioactive materials developed to expand T cells for adoptive transfer into cancer patients are currently evaluated in the clinic. In most cases, T cell activating biomolecules are attached to rigid surfaces or matrices and form a static interface between materials and the signaling receptors on the T cells. We hypothesized that a T cell activating polymer brush interface might better mimic the cell surface of a natural antigen-presenting cell, facilitating receptor movement and concomitant advantageous mechanical forces to provide enhanced T cell activating capacities. Here, as a proof of concept, we synthesized semiflexible polyisocyanopeptide (PIC) polymer-based immunobrushes equipped with T cell activating agonistic anti-CD3 (αCD3) and αCD28 antibodies placed on magnetic microbeads. We demonstrated enhanced efficiency of ex vivo expansion of activated primary human T cells even at very low numbers of stimulating antibodies compared to rigid beads. Importantly, the immunobrush architecture appeared crucial for this improved T cell activating capacity. Immunobrushes outperform current benchmarks by producing higher numbers of T cells exhibiting a combination of beneficial phenotypic characteristics, such as reduced exhaustion marker expression, high cytokine production, and robust expression of cytotoxic hallmarks. This study indicates that semiflexible immunobrushes have great potential in making T cell-based immunotherapies more effective.
Keywords: artificial antigen-presenting cell, polymer brush surface, adoptive cell therapy, polyisocyanopeptide, immunotherapy
1. Introduction
The human immune system has the capability to identify and clear infected or malignant cells,1 but in the case of cancer, the immune system often fails.2,3 Immunotherapies have therefore emerged to boost the immune system to better fight malignancies. As T cells play a central role in the clearance of cancer cells, many immunotherapies aim to increase tumor-specific T cell responses.4,5 For example, adoptive T cell therapy is aimed at strengthening the immune response by infusing autologous tumor-reactive T cells into cancer patients, thereby benefitting from their cytotoxic potential.6−10 To generate sufficient numbers of functional T cells, autologous T cells need to be activated and expanded ex vivo.
Current strategies for ex vivo T cell activation often make use of artificial antigen-presenting cells (aAPCs) that mimic the natural activation signals presented by antigen-presenting cells (APC) such as dendritic cells.11 For T cell activation, three main signals are important: (1) T cell receptor (TCR) stimulation;12 (2) co-stimulation, for example, by engagement of the co-stimulatory receptor CD28 on T cells;13,14 and (3) the secretion and binding of cytokines.15 A common design for aAPCs is the use of rigid synthetic scaffolds that present signals 1 and 2, often using agonistic anti-CD3 (αCD3) and αCD28 antibodies, respectively.11,16,17 Protocols using these aAPCs for T cell expansion are often complemented with soluble IL-2 as signal 3. Such rigid aAPCs have been developed in many shapes and sizes.11,18,19 Current standards for in vitro T cell activation are magnetic microbeads containing αCD3 and αCD28, most commonly Dynabeads with a diameter of 4.5 μm.20−22
Adoptive cell therapies (ACTs) exploiting Dynabeads or similar rigid aAPCs for T cell expansion have shown promising results in phase 1 and 2 clinical trials for hematological cancers and some solid cancers such as melanoma.6,7,23,24 However, the practical feasibility of ACT and treatment efficacy are still hampered by the extensive ex vivo expansion that is needed to obtain enough cells for reinfusion.25−28 This expansion is not only a time-consuming and expensive process that limits the widespread application of ACT, but can also yield T cells with poor persistence and limited functionality in vivo.26,28−31 These limitations could be a consequence of the fact that rigid spherical beads are poor mimics of natural APCs. APC membrane fluidity and receptor rearrangements play an important role during interaction with T cells and subsequent activation.32 A wide range of new strategies are being developed to improve ex vivo T cell expansion.33−36 To further boost adoptive cell therapies, it is crucial to develop better aAPCs that activate and expand T cells ex vivo at higher rates, while generating T cells with optimal phenotypes and high functionality. Therefore, it is crucial to increase our understanding of how aAPC design impacts T cell activation to improve the development of novel aAPC strategies.
Recently, we introduced a new type of aAPC based on soluble semiflexible polyisocyanopeptide (PIC) polymers with a length of around 400 nm. This scaffold is able to induce very potent and long-lasting T cell activation.37−40 The success of these aAPCs is attributed to the semiflexibility of the polymers, which is hypothesized to enable high motility of the signals that are attached. At the same time, the polymer backbone is stiff enough to present the signals in a stretched confirmation. The T cell activating capacity of PIC-based aAPCs is remarkable, especially given the fact that their size is far below the optimal 4–5 μm size range frequently used for aAPCs,18 which is advantageous, for example, for in vivo applications. In this work, we aim to combine the best of two worlds by grafting T cell activating PICs onto optimally sized magnetic microbeads, allowing easy separation from the expanded T cells. We hypothesize that due to their semiflexible nature, the grafted PICs will form a brush surface to more effectively present the activating signals, which will result in improved T cell expansion performance compared to rigid beads.
Here, we show the design and functionalization of PIC-grafted magnetic microbeads with αCD3 and αCD28 agonistic antibodies. We illustrate that the brush configuration of the polymer is crucial for efficient T cell activation and we optimized the T cell stimulating potential over a range of design parameters, such as antibody ratio and density. In addition, we demonstrate that these immunobrushes outperform rigid beads to which the same signals are directly attached to the surface. Finally, we show that the αCD3/αCD28 PIC immunobrush outperformed clinically approved expansion systems like CD3/CD28 Dynabeads or T Cell TransAct in expansion of primary T cells with beneficial phenotypic characteristics using standard operation procedures similar to those used for clinical application.
2. Experimental Section
2.1. Synthesis of Biotin End-Functional PIC
End-functional PIC were synthesized according to the previously reported protocol.41 Briefly, PICs with an allyl containing first block were prepared. For this, methoxy-, allyl-, and azide-terminated monomers were prepared as described according to the literature.37,41 For the polymerization of the first block, methoxy-terminated monomer and allyl-terminated monomer were dissolved in a 1:1 molar ratio in dry toluene obtained from an MBraun SPS 800 solvent system at a total monomer concentration of 27.7 mM. To this mixture, a 4 mM catalyst solution was added (Ni(ClO4)2·6H2O in 9:1 toluene/ethanol) with a monomer/catalyst ratio of 100:1 and was reacted for 10 min. Next, for the second block, a solution of azide monomer (0.66 mg, 0.0018 mmol) and methoxy monomer (19.34 mg, 0.054 mmol) in dry toluene (0.50 mL) was added and the polymerization was stirred overnight at room temperature (RT). Isocyanide consumption was confirmed by disappearance of the characteristic IR absorbance at 2140 cm–1. The polymers were precipitated three times in diisopropyl ether and air-dried overnight to give an off-white solid (15.35 mg, 77%). The helical backbone of the PIC was confirmed by circular dichroism spectroscopy of PIC solutions in phosphate-buffered saline (PBS) pH 7.4.43 The average polymer length was determined by atomic force spectroscopy (AFM, Figure S1) using a Nanoscope IV instrument (Bruker) and NSG-10 tapping mode tips (NT-MDT) and found to be 205 nm.37 The allyl functionalities in the first block were then converted to biotin via the nitrile imine-mediated tetrazole-ene cycloaddition (NITEC) reaction using a tetrazole-biotin compound as described before.41 Biotin conjugation was confirmed by measuring the fluorescence of the pyrazolines that are formed upon conjugation using a SPARK M10 plate reader from TECAN. The biotin end-functional PIC was then dissolved at 2 mg/mL in PBS and labeled overnight at 4 °C with dibenzocyclooctyne (DBCO)-conjugated Alexa Fluor 594 dye (Click Chemistry Tools) using a 1:500 ratio of dye/azide and used without purification.
2.2. Functionalization of Antibodies
Anti-human CD3 antibodies (clone OKT3, BioXcell) and anti-human CD28 antibodies (clone 9.3, BioXcell) were functionalized with dibenzocyclooctyne (DBCO) or biotin and fluorescent dyes as described previously.39,43 To ensure that the DBCO-functionalized antibodies contain the same amount of dyes as their biotin-functionalized counterparts, the antibodies were first conjugated with the dyes. The antibody storage buffer was converted to 50 mM borate buffer pH 8.5 using Amicon centrifugal filters (30 kDa cutoff, Merck), and the antibodies were reacted with 2.5 equiv of Atto 488-NHS ester (A488, Atto-TEC GmbH, for αCD3) and Alexa Fluor 647-NHS ester (AF647, Thermo Fischer Scientific, for αCD28) for 2 h at 4 °C. Dye-functionalized proteins were purified using a 5 mL 40K Zeba spin desalting column (Thermo Fischer Scientific). The batches of dye-functionalized antibodies were then redissolved in 50 mM borate buffer pH 8.5; half were reacted with 5 equiv DBCO-PEG4-NHS (Jena Bioscience) and the other half with 5 equiv NHS-PEG4-biotin (Jena Bioscience) for 2 h at 4 °C. The functionalized proteins were again purified using a 5 mL 40K Zeba spin desalting column. The degree of labeling (DOL) of DBCO and fluorescent dyes was determined using a NanoDrop 2000c spectrophotometer. The concentrations of protein, DBCO, and dye were calculated based on the absorbance at 280, 309, 501, and 650 nm and corrected for overlap in the absorbance spectra of the individual molecules. Typically, the DOL of DBCO was 2–4 and the DOL of the dyes was 1–3.
2.3. Conjugation of DBCO-Conjugated Antibodies to PIC
A 1 mg/mL solution of the biotin end-functional PIC in PBS (50 μL, 0.05 mg, 4.4 nmol azide monomer) was added to a solution of DBCO- and AF647-labeled αCD28 and DBCO- and A488-labeled αCD3 in PBS (450 μL) in the desired antibody ratios. The mixture was reacted overnight at RT. To prevent PIC cross-linking in the following steps, excess unreacted DBCO conjugated to the antibodies was quenched by addition of 1000 equiv azido propylamine from a 100 μM stock solution in dimethyl sulfoxide (DMSO) and was reacted for 2 h at room temperature. The conjugated polymer, when used for grafting, was then used without further purification as unreacted antibodies are removed by washing the beads after grafting. When used as soluble antibody-functionalized PIC for activation or for determination of antibody per PIC, they were purified using affinity purification according to the literature.43 The amount of antibody per PIC of purified polymer was determined by measuring the fluorescence of the respective labels on PIC, αCD3, and αCD28 and comparing it to trendlines of the pure labeled compounds.
2.4. Synthesis of PIC Beads and Ab Beads
Streptavidin-coated magnetic beads were prepared from 4.5 μm beads with a tosyl-activated surface (Dynabeads M450 Tosyl from Thermo Fischer Scientific). Tosyl bead suspension (1 mL) was washed three times with 50 mM borate buffer pH 8.5 using a magnetic separation rack from Westburg BV. The beads were resuspended in 1 mL of 20 mM streptavidin in borate buffer pH 8.5 and reacted overnight at room temperature. Next, the beads were washed 2× with 0.05% PBS Tween and 3× with PBS and resuspended in 1 mL of PBS. For the preparation of PIC beads or Ab beads, the supernatant was first removed using the magnet and then replaced with a solution of the desired biotin end-functional PIC or biotin-functionalized antibodies. The beads were incubated with the desired biotin compounds on a rotator overnight at 4 °C and washed with 2× 0.05% PBS Tween and 3× with PBS. The beads were analyzed by flow cytometry on a BD FACS Verse cytometer and via the previously reported stripping assay,41 with the exception that as a stripping solution, 3 mM biotin in 2% sodium dodecyl sulfate (SDS) in Milli-Q water was used. For the beads made for the antibody density series, only flow cytometry analysis was performed to determine antibody densities and PIC densities from the Ab/PIC ratio determined after polymer purification
For the preparation of flat PIC beads, a similar protocol was followed, but a PIC with random bound biotins instead of the end-functional PIC was used. To attach the biotin in a random manner, half of the azides in the PIC were converted to biotin, using DBCO-PEG4-biotin. This flexible poly(ethylene glycol) (PEG) spacer, combined with the semiflexibility of the PIC backbone and the flexible linkers connecting the antibodies to the PIC, should provide ample flexibility and accessibility of the antibodies to the T cells. The remaining azides were used for the conjugation of the antibodies. The cross-linked PIC beads were prepared by omitting the quenching of excess DBCO on the antibodies, after which the exact same protocol for PIC bead preparation was followed as above. For the flow cytometry analysis, single beads were gated using a plot of forward scatter versus side scatter, and mean fluorescence intensities (MFIs) of the A488 and AF647 labels were recorded using fluorescein isothiocyanate (FITC) and APC channel, respectively. The beads were stored as a suspension in PBS (1 mg/mL) and used within 2 weeks of preparation. Before using them to activate T cells, they were freshly washed three times with sterile PBS and then resuspended in sterile X-Vivo medium with 2% human serum at a concentration of 4 × 107 beads/mL and used directly in T cell activation assays.
2.5. Isolation of Primary Human Pan T Cells and Activation
Fresh peripheral blood mononuclear cells (PBMCs) were obtained by Ficoll density centrifugation of buffy coats obtained from healthy donors in accordance with recommendations of institutional guidelines. All subjects gave written informed consent in accordance with the Declaration of Helsinki. Pan T cells were isolated from peripheral blood leukocytes using the Pan T cell isolation kit (Miltenyi Biotec). T cell purity was determined by flow cytometry staining with CD3 (eBioscience) and was typically >98%. For the conditions where the proliferation after 3 days was measured, isolated T cells were stained with CellTrace Violet before activation. This staining was performed by incubating the cell suspension in 2.5 μM CellTrace Violet solution in PBS at room temperature for 10 min in the dark. The reaction was then quenched by adding an equal volume of fetal bovine serum, and cells were washed three times with X-Vivo medium containing 2% human serum.
The isolated T cells were cultured in 96-well u-bottom plates (Corning) using 105 cells per well in a final volume of 200 μL X-Vivo medium with 2% human serum. For the initial validation of the PIC beads, no IL-2 was added to the medium. For the expansion protocol, the medium was supplemented with 30 IU/mL IL-2 (Cell Genix). To stimulate cells, PIC beads were added (1 bead per cell, washed 3× in PBS): CD3/CD28 Dynabeads (Thermo Fischer Scientific) (1 bead per cell, washed 1× in PBS) or T Cell TransAct (Miltenyi Biotec) (1:100). The cells were incubated at 37 °C for a desired time. After 3, 6, 9, and 11 days of culture, the medium was taken off. T cells were split in half, and 200 μL of fresh medium with 30 IU/mL IL-2 was added.
2.6. IL-2 and IFNγ Analysis by Enzyme-Linked Immunosorbent Assay (ELISA)
After 20–24 h of cell culture, 125 μL of supernatant was taken from each condition and stored at −20 °C until used. Cytokine production was quantified using standard sandwich ELISA kits for human IFN γ (Invitrogen) and IL-2 (eBioscience) according to manufacturer’s protocol.
2.7. Determining T Cell Activation and Proliferation
On day 1, T cells were transferred to v-bottom 96-well plates (Corning) and stained with the Zombie Violet Fixable Viability Kit (Biolegend). To determine T cell activation, we performed cell surface staining with CD8-BV510, CD4-APC-Cy7, CD69-PE, and CD25-PE-Cy7 (all BD Biosciences). On day 3, we determined T cell proliferation by transferring T cells to v-bottom 96-well plates and staining with Fixable Viability Dye eFluor 780 (BD) followed by cell surface staining with CD4-PE (BD Biosciences) and CD8-BV510. All samples were acquired on the FACS Verse (BD Biosciences). The mean cell cycle of all T cells was determined as a measure for the average number of cell proliferation cycles. The mean cycle was calculated with the formula log2(f), where f is the CellTrace Violet mean fluorescence intensity (MFI) of all nonproliferated T cells divided by the CellTrace Violet MFI of all T cells.
2.8. T Cell Expansion and CD4/CD8 Ratio
To quantify expansion of the T cells over time, we transferred T cells on days 1, 7, and 14 to 96-well v-bottom plates and performed a cell surface staining with CD8-BV510 and CD4-APC Cy7. The cells were acquired and the cell number was quantified on an MACS Quant flow cytometer (Miltenyi Biotec) together with a propidium iodide staining (Miltenyi Biotec) to assess cell viability. The cell number was corrected for the number of times the cells were split during cell culture, and the fold expansion was calculated relative to the number of T cells put in the wells on day 0 (50.000).
2.9. T Cell Phenotype
On day 14, we transferred the cells to a 96-well v-bottom plate and performed flow cytometry stainings to assess the phenotype of the T cells. All of the cells were stained with Fixable Viability Dye eFluor 780, followed by staining according to the following panels:
-
(1)
Memory phenotype—Cell surface: CD8-PE-Cy7 (Biolegend), CD95-BV421 (Biolegend), CD45RA-BV510 (Biolegend), CCR7-PE (BD Biosciences).
-
(2)
Exhaustion—Cell surface: CD4-BV421 (Biolegend), CD8-PECy7, PD-1-BV510 (BD Biosciences), TIM-3-PerCP-eFluor 710 (eBioscience).
To measure cytokine production and cytotoxicity, T cells were first restimulated for 5.5 h with 20 ng/mL phorbol myristate acetate (PMA) (Calbiochem), 1 μg/mL ionomycin (Sigma-Aldrich), 10 μg/mL Brefeldin A (BFA) (Sigma-Aldrich), and 2 μM monensin (eBioscience). For the cytotoxicity panel, at the same time, cells were incubated with CD107a-PeCy5 (BD Biosciences) for 5.5 h. Next, T cells were transferred to a 96-well v-bottom plate and stained with Fixable Viability Dye eFluor 780, followed by staining according to the following panels:
-
(1)
Cytokine production—Cell surface: CD8-BV510, intracellular: IFNy-BV421 (BD Biosciences), IL-2-PE (eBioscience) TNFα-PerCP/Cy5.5 (eBioscience).
-
(2)
Cytotoxicity—Cell surface: CD4-BV421, intracellular: Granzyme B-PE (Biolegend).
For intracellular stainings, cells were fixated and permeabilized using a Fixation/Permeabilization Solution Kit (BD Biosciences). All samples were acquired on the FACS Verse, and the gates were set using fluorescence minus one (FMO) controls.
2.10. Statistics
Data are expressed as mean ± standard error of the mean (SEM).
Statistical analyses were performed in GraphPad Prism 8.0.2 software using the appropriate testing methods, as indicated in the figure legends. Normality was tested using the D’Agostino–Pearson omnibus normality test. Statistical significance was defined as a two-sided significance level of <0.05 indicated as *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001. Flow cytometry data were analyzed using FlowJo software version X 10.0.7r2 (Tree Star).
3. Results
3.1. PIC Immunobrushes Effectively Stimulate T Cells
To synthesize T cell activating PIC immunobrushes, we applied a methodology we recently developed to graft PIC polymers to various surfaces.41 Here, we aimed to synthesize PIC block copolymers containing orthogonal functional groups to introduce T cell activating signals on one side of the polymer and use the other side to attach the polymers to 4.5 μm magnetic beads (Figure 1a). The first block contained allyl functional groups, which were subsequently modified with tetrazole-biotin via photoinduced nitrile imine-mediated tetrazole-ene cycloaddition (NITEC) reaction. The second, larger block contains azide functional groups, which can be used to “click” dibenzocyclooctyne (DBCO)-modified antibodies via a strain-promoted azide alkyne cycloaddition reaction (SPAAC) (Figure 1a).42 The ratio between the average length of the biotin and azide functional blocks was 1:20, with an average total length of 205 nm as determined with atomic force microscopy (Figure S1). Next, DBCO-modified αCD3 and αCD28 antibodies were “clicked” to the PIC block copolymers in different ratios (Table S1 for all formulations) to establish the optimal ratio for T cell activation and expansion. Subsequently, these antibody-functionalized polymers were grafted onto streptavidin-coated 4.5-μm-sized magnetic beads, to obtain the αCD3/αCD28 PIC brush beads to be characterized for their T cell activating capacities (Figure 1a).
Figure 1.

Synthesis and characterization of PIC immunobrush beads. (a) Schematic overview of the preparation of PIC brush beads (top), Ab beads (bottom left), flat PIC beads (bottom center), and cross-linked PIC beads (bottom right). (b, c) Production of IFNγ (b, n = 3 in two independent experiments) and IL-2 (c, n = 4 in two independent experiments) after 20–24 h of human T cells stimulated with directly coupled antibody beads or PIC beads. (d) Mean cell cycle of proliferated human T cells on day 3, stimulated directly coupled antibody beads or PIC beads n = 4 in two independent experiments. (b–d) A paired t test was performed on log2-transformed data. (e, f) IFNγ and IL-2 production of human T cells after 20–24 h stimulated with PIC beads with reduced mobility, n = 2 in one independent experiment. An RM one-way analysis of variance (ANOVA) was performed on log2-transformed data with Geisser–Greenhouse correction, followed by Dunnett’s multiple comparison test.
Having synthesized the αCD3/αCD28 PIC immunobrush beads, we set out to analyze their ability to stimulate primary human T cells (CD4+ and CD8+ Pan T cells). The performance of the PIC immunobrush beads was compared to that of beads with similar quantities and ratios of αCD3 and αCD28 antibodies directly attached to the particle surface (Ab beads, Figure 1a) (PIC beads: 4.2 ratio αCD28/αCD3, 12.2 ng of total antibody/106 beads; Ab beads: 3.6 ratio αCD28/αCD3, 14.5 ng of total antibody/106 beads). No soluble IL-2 was added to the culture, to establish the pure effect of the PIC beads on T cell activation. We observed that the IFNγ production of T cells stimulated with PIC beads was remarkedly upregulated (3.1-fold) after 1 day of stimulation compared to Ab beads (Figure 1b). Even more pronounced, IL-2 production of T cells stimulated with the PIC beads was increased 17-fold compared to exposure to Ab beads (Figure 1c). Three days of T cell stimulation with PIC beads led to a high proliferative character, similar to that of T cells cultured together with Ab beads (Figure 1d).
Next, to investigate the impact of immobilization of PIC on their performance, we compared the T cell activation potential of αCD3/αCD28 PIC immunobrush beads with soluble αCD3/αCD28 PICs.37,38,40 Human T cells were stimulated with similar amounts of αCD3 and αCD28 immobilized on PIC beads (1 bead per cell) or soluble PIC43 having similar antibody concentration and ratios (Table S1). Compared to stimulation with soluble PIC, 1 day stimulation with PIC beads led to 1.7- and 1.9-fold increases in IFNγ and IL-2 production, respectively (Figure S2a,b). The proliferation of PIC bead-stimulated T cells was increased 2.5-fold (Figure S2c). Together, these results indicate that clustering the αCD3/αCD28 PIC together on microbeads greatly enhances their ability to activate T cells.
We argued that the brush design of our PIC immunobrush beads (i.e., the polymers connected to the surface on one side at a high enough density that PIC polymers are oriented perpendicular to the bead surface) is a key factor in the observed increased activation of T cells. We hypothesize that, compared to antibodies directly coupled to beads, antibodies coupled to PICs have much more mobility due to the semiflexible nature of the PIC. Thus, they are better capable to follow receptor rearrangements, similar to ligands in a membrane of a natural APC, a characteristic that appeared to be very important in T cell activation.44−47 To substantiate this hypothesis, instead of installing biotins at one end of the PIC, we functionalized PICs with biotins randomly throughout the entire polymer (Figure 1a, bottom center). When bound to 4.5 μm streptavidin beads, the complete polymer chain is expected to be attached to the bead surface (flat PIC beads, Table S1), which reduces the ligand mobility compared to PICs attached to the surface with a single end. In addition, we aimed to rigidify the PIC brush structure by allowing the cross-linking of the single-end surface-bound αCD3/αCD28 PICs. To this end, we grafted beads with αCD3/αCD28 PICs for which the step to quench the excess DBCO on the antibodies was omitted. This allows reaction with the excess azides on adjacent PICs on the bead to induce cross-linking without the need for additional antibody modification (cross-linked PIC, Figure 1a, bottom right, and Table S1). Cross-linking of the PICs on the beads will reduce the PIC mobility and could have influenced accessibility of antibodies, which are present within the brush. We observed that primary human T cells stimulated with flat or cross-linked PIC beads did not produce substantial amounts of IFNγ and IL-2 after 1 day of culture, whereas the PIC immunobrush beads produced large amounts of these cytokines (Figure 1e,f). Together, these results indeed indicate that the brush design and ligand mobility are important factors in the observed increase in T cell activation after PIC bead stimulation.
3.2. PIC Immunobrush Beads Effectively Stimulate T Cells
Having established the importance of the brush design of our PIC beads and the benefits for T cell activation, we next optimized the ratio of the αCD3 and αCD28 antibodies conjugated to the PIC immunobrush. This parameter is known to affect T cell activation.17,48 A series of Ab beads and PIC immunobrush beads with αCD28/αCD3 ratios between 0.3 and 5 were prepared (Table S1 for all formulations). Human T cells were stimulated with one bead per cell ratio for both Ab beads and PIC beads. No significant differences in the proliferation of T cells after 3 days of culture was observed between the different αCD28/αCD3 ratios, independent of the platform used, with a maximum proliferation rate already reached at the lowest αCD28/αCD3 ratio for both Ab beads and PIC beads (Figure S3a). By contrast, PIC beads induced much higher amounts of cytokines (IFNγ and IL-2) over the full range of ratios, compared to the Ab beads (Figure 2a,b). Although a maximum cytokine secretion was reached at 0.75 (IFNγ) and 1.4 (IL-2) αCD28/αCD3 ratios for both Ab and PIC beads, the plateau secretion levels for IFNγ and IL-2 were 1.9- and 4.5-fold higher for PIC beads, respectively. Based on these results we selected αCD28/αCD3 ratios higher than 2.5 for further experiments to guarantee robust and reproducible T cell stimulation and cytokine production.
Figure 2.

Influence of αCD28/αCD3 ratio and density. (a, b) IFNγ and IL-2 concentrations in the supernatant of T cells stimulated with Ab or PIC beads with different αCD28/αCD3 ratios after 24 h of culture n = 3 in two independent experiments. (c, d) IFNγ and IL-2 concentrations in the supernatant of T cells stimulated with Ab or PIC beads with different total antibody densities after 24 h of culture n = 4 and 3, respectively, in two independent experiments. (e) Percentage of IFNγ producing T cells after 7 days of culture with Ab or PIC beads over a full range of antibody densities. (f) Mean fluorescence intensities (MFI) of IFNγ produced by T cells after 7 days of culture with Ab or PIC beads over a full range of antibody densities. (g) Percentage of IL-2 producing T cells after 7 days of culture with Ab or PIC beads over a full range of antibody densities. (h) Mean fluorescent intensities (MFI) of IL-2 produced by T cells after 7 days of culture with Ab or PIC beads over a full range of antibody densities n = 2 in one independent experiment for (e)–(h).
Next, the effect of antibody density was investigated, which is another main parameter that affects T cell activation.38,49 To this end, we synthesized Ab beads and PIC immunobrush beads with a fixed αCD28/αCD3 ratio of around 3, but varied the density of the antibodies on the beads. The density was controlled by addition of variable amounts of either biotinylated αCD28/αCD3 to obtain Ab beads, or DBCO-functionalized αCD28/αCD3 coupled to biotinylated brush PIC to generate PIC immunobrush beads (Table S1). Stimulation of T cells with both types of beads reached the same level of maximum proliferation (Figure S3b). The PIC beads, however, reached this maximum at lower antibody densities (3.5 instead of 22 ng antibody per million beads). For the IFNγ and IL-2 production (Figure 2c,d), a higher maximum was observed for the PIC immunobrush beads, which was reached at lower antibody densities compared to Ab beads. Altogether, these data demonstrate that αCD3/αCD28 PIC immunobrush beads outperform Ab beads, requiring considerably less antibodies to achieve robust T cell activation with higher levels of cytokine production.
To investigate the origin of the increased cytokine levels induced by PIC immunobrush beads, we performed intracellular cytokine staining on T cells to determine cytokine production at the single-cell level by flow cytometry. We observed that PIC beads induced a higher percentage of IFNγ-positive T cells in comparison to Ab bead stimulated T cells (Figure 2e). In contrast, the amount of IFNγ as measured by the mean fluorescence intensity (MFI) produced by individual T cells is similar for both Ab and PIC beads (Figure 2f), indicating that the higher IFNγ levels observed in the culture supernatant are a result of the induction of a larger population of IFNγ-producing cells. Interestingly, for IL-2, the situation is inverse. Here, no clear differences in the percentage of IL-2 producing T cells are found between stimulation with Ab or PIC beads for all tested densities (Figure 2g), but an approximately 2-fold higher IL-2 MFI was measured in T cells stimulated with PIC beads (Figure 2h). These findings indicate that activation of T cells via PIC immunobrushes affects the production of the different cytokines via different mechanisms.
3.3. Ex Vivo Polyclonal Expansion of Primary Human T Cells
Having determined the optimal parameters for our PIC immunobrush bead design (αCD28/αCD3 ratios higher than 2.5, and a minimal antibody density of around 12 ng/million beads), we next evaluated their capability to expand T cells according to a clinically used protocol. CD3/CD28 Dynabeads and T Cell TransAct are two systems that are frequently used for ex vivo expansion of T cells for adoptive T cell therapies in the clinic. The CD3/CD28 Dynabeads are 4.5 μm magnetic beads equipped with αCD3 and αCD28 antibodies, whereas T Cell TransAct is a colloidal polymeric nanomatrix conjugated to humanized αCD3 and αCD28.50 We compared these two systems to our PIC beads with optimized αCD28/αCD3 ratio and density (Table S1) for their T cell expansion performance. For the commercial systems, manufacturer protocols were followed to ensure optimal conditions. In addition, T cell cultures were supplemented with IL-2 (30 IU/mL). After 1 day of culture, PIC beads and Dynabeads induced a large (>80%) population of CD69+CD25+ T cells, indicating that T cells are activated (Figure S4). By contrast, stimulation with T Cell TransAct only led to 50% of CD69+CD25+ T cells (Figure S4). In line with our previous findings, PIC beads induced high amounts of IFNγ in the supernatant after 1 day of culture, whereas T cells stimulated with CD3/CD28 Dynabeads or T Cell TransAct secreted much less cytokine (Figure 3a). PIC beads and Dynabeads, but not T Cell TransAct, induced high production of IL-2 by the T cells (Figure 3b). All systems induced robust proliferation and high viability of T cells compared to unstimulated cells after 3 days, although stimulation with PIC beads and Dynabeads induced the highest proliferation in terms of the proportion of T cells that divided and their average numbers of cycles (Figures 3c, S5, and S6). After 14 days of culture, all three systems induced robust expansion, with a 75-fold expansion of T cells for the PIC beads, on par with T Cell TransAct and greater than Dynabeads in our hands (Figure 3d). Considering that the amount of stimulating antibodies per bead is far greater on the Dynabeads versus the PIC beads (∼45031vs 13 ng/bead), this difference is remarkable and shows the importance of the method of presentation of T cell activating signals.
Figure 3.

Polyclonal ex vivo expansion of human T cells. (a, b) IFNγ and IL-2 concentrations in the supernatant of human T cells stimulated with different expansion systems after 1 day, respectively; n = 8 in four independent experiments. Data were analyzed with an RM one-way ANOVA with Geisser–Greenhouse correction, followed by Dunnett’s multiple comparison test. (c) Mean cell cycle of proliferated T cells on day 3, stimulated with PIC beads, Dynabeads, or T Cell TransAct; n = 6 (T cell transact) in three independent experiments or n = 8 (all others) in four independent experiments. Data were analyzed using a mixed-effects analysis, with the Geisser–Greenhouse correction, followed by Dunnett’s multiple-comparisons test (d) fold expansion of Pan T cells after 7 and 14 days of culture. Fold expansion was calculated relative to the number of T cells put into the wells on day 0 (50.000); n = 4 (T cell Transact) or n = 5 (all others) in two independent experiments. Significance was analyzed with an RM two-way ANOVA followed by Sidak’s multiple-comparisons test on log2-transformed data.
3.4. Phenotype and Function of Ex Vivo Expanded Human T Cells
Next, we evaluated the phenotype and function of the T cells expanded with PIC immunobrush beads, Dynabeads, or T Cell TransAct. First, the ratio of CD4+ and CD8+ cells was determined on different days during the 14 days of culture. In the first week of culture, no differences in the CD4/CD8+ ratio were observed irrespective of the type of stimulation (ratio of 2), but after 10 days, the population of T cells treated with PIC beads or Dynabeads was skewed towards CD8+ cells (ratio, 0.3–0.4) (Figure 4a). Treatment with T Cell TransAct yielded similar numbers of CD4+ and CD8+ T cells (ratio 0.9) on day 14.
Figure 4.

Phenotype and function of human T cells expanded with PIC immunobrush beads on day 14. (a) Ratio of CD4+–CD8+ human T cells after expansion n = 2–5 in one or two independent experiments. Data were analyzed using a mixed-effects model, followed by a Dunnett’s multiple-comparisons test on log2-transformed data. (b, c) CD4+ and CD8+ differentiation of human T cells in to different memory subsets after expansion with PIC beads, Dynabeads, or T Cell TransAct, respectively; n = 2 in one independent experiment. (d) CD4+ and (e) CD8+ cytotoxic character of human T cells after expansion; n = 5 in two independent experiments. Statistics: CD4+ T cells: CD107–GZMB+ PIC beads vs Dynabeads p < 0.05. CD8+ T cells: CD107–GZMB– PIC beads vs Dynabeads p < 0.05, CD107–GZMB+ PIC beads vs T cell Transact p < 0.01, CD107+GZMB+ PIC beads vs T cell Transact p < 0.05. (f, g) CD4+ and CD8+ exhaustion character of human T cells after expansion, respectively; n = 5 in two independent experiments. Statistics: CD4+ T cells: PD-1–TIM3– PIC beads vs T cell Transact p < 0.01, PD-1–TIM3+ PIC beads vs T cell Transact p < 0.05, PD-1+TIM-3+ PIC vs T cell Transact p < 0.01. CD8+ T cells: PD-1–TIM3+ PIC beads vs T cell Transact p < 0.01, PD-1+TIM3+ PIC vs T cell Transact p < 0.01. (h, i) Intracellular expression of 0, 1, 2, or 3 cytokines (IL-2, IFNγ and/or TNFα) in CD4+ (h) and CD8+ (i) T cell; n = 5 in two independent experiments. Statistics: CD4+ T cells: three markers PIC vs Dynabeads p < 0.05. CD8+ T cells: 1 marker PIC vs T cell Transact p < 0.05, 0 markers PIC vs T cell Transact p < 0.05. (b–i) Significance was analyzed with an RM two-way ANOVA with Geisser–Greenhouse correction, followed by Dunnett’s multiple-comparisons test.
After 14 days of culture T cells, the memory phenotype of the T cells was determined according to the linear T cell differentiation model, which assumes that T cells linearly progress from naïve T cells (Tn) into T stem cell memory (Tscm), T central memory (Tcm), T effector memory (Tem), and finally T terminal effector (Tte) cells (Figure S7).51 During this progression, T cells lose self-renewal potential but gain effector functions. CD4+ T cells stimulated with PIC beads induced a Tte phenotype in about 55% of the cells, but also significant Tscm and Tem populations (both ∼20%) were observed. Dynabead stimulation led to a comparable differentiation of CD4+ T cells, with a bit less (∼45%) Tte cells. On the other hand, T Cell TransAct led to the predominant formation of a Tte population of around 75% for the CD4+ T cells (Figure 4b). PIC bead stimulation of CD8+ T cells led to a population of ∼75% Tem and ∼20% Tte cells. For Dynabeads, a slightly smaller Tem population of ∼65% and similar amounts of Tte T cells (∼25%) were observed compared to stimulation with PIC beads. As for the CD4+ T cells, stimulation with T Cell TransAct led to mostly CD8+ T cells with a Tte phenotype (∼65%) and less Tem cells (∼10%) (Figure 4c). These data indicate that T cells stimulated with either PIC beads or Dynabeads progress into less differentiated states compared to T cells stimulated with T Cell TransAct. This behavior potentially has important consequences, as less differentiated cells tend to have a higher proliferative capacity and improved persistence after adoptive cell transfer in vivo.52
To determine the potential cytotoxic capacity of the generated T cells, we analyzed the upregulation of CD107a and Granzyme B, well-known markers for cytotoxic potential (Figure S8). We observed after 14 days of culture that a large proportion of both CD4+ T cells (Figure 4d) and CD8+ T cells (Figure 4e) co-expressed CD107a and granzyme B after stimulation with PIC beads, CD3/CD28 Dynabeads, or T Cell TransAct. This finding indicates that all three systems are capable of inducing T cells with a cytotoxic phenotype, although T cell TransAct induced a slightly higher cytotoxic population, in line with their more pronounced Tte phenotype.
Next, we evaluated the degree of T cell exhaustion, a loss of effector functions due to T cell overstimulation, by analyzing the upregulation of immune checkpoint proteins programmed cell death protein 1 (PD-1) and T cell immunoglobulin mucin-3 (TIM3) after 14 days (Figure S9). Although PD-1 and TIM3 can be viewed as activation markers because they are upregulated rapidly on activated T cells following stimulation, they are considered as markers for exhausted T cells when they remain high on stimulated T cells for a prolonged period of time. Co-expression of multiple exhaustion markers can indicate dysfunctional T cell behavior.53 CD4+ T cells co-cultured with T Cell TransAct showed the highest proportion of PD-1+TIM3+ T cells (12.5%), suggesting that a more exhausted phenotype is induced in CD4+ T cells than PIC beads or Dynabeads (Figure 4f). Similarly, for CD8+ T cells, T Cell TransAct also induced the highest proportion of cells with an exhaustion phenotype, as we observed that only ∼60% of CD8+ T cells stimulated with T Cell TransAct were PD-1–TIM3–, in contrast to around 90% for T cells stimulated with either PIC beads or Dynabeads (Figure 4g).
Finally, we examined the intracellular production of cytokines IL-2, IFNγ, and TNFα in restimulated T cells on day 14 as a measure of their effector function (Figure S10). Even though T cells stimulated with T Cell TransAct displayed a higher proportion of PD-1+TIM3+ cells, a significant proportion of both CD4+ (Figures 4h and S11a) and CD8+ (Figures 4i and S11b) T cells co-produced all three of these cytokines following stimulation with PIC beads, Dynabeads, and T Cell TransAct. These data suggest that all three of these systems are capable of generating functional T cells with cytokine-producing capabilities.
4. Discussion
The synthesis and characterization of PIC immunobrush beads functionalized with T cell stimulating antibodies (αCD3/αCD28) are reported. This novel semiflexible polymer brush surface aAPC platform results in more efficient activation of T cells compared to aAPCs with a similar core but with the antibodies directly attached to the bead surface instead. We have shown that T cell expansion is highly dependent on the characteristics of the material that delivers the activation cues to the T cells and that the brush orientation of the CD3/CD28 PIC attached to the bead is crucial for the increased T cell activation. Hypothetically, the brush design results in a better mimic of the natural dendritic cell–T cell interaction. In contrast to many other aAPCs used54−58 to induce T cell activation and proliferation, the semiflexible brush beads support movement and lateral rearrangement of receptors over the membrane of T cells, thereby facilitating receptor clustering. This reorganization of receptors and subsequent downstream signaling are of utmost importance to obtain optimal T cell activation.45,59−61 Additionally, the antibodies bound to the PIC immunobrush will deliver a different mechanical signal to the T cell compared to antibodies directly attached to the rigid core of a static bead. The semiflexible nature of the polymer might play a big role here as it is known that (local) mechanical cues can greatly influence T cell activation.32,62 Very soft substrates (≤0.5 Pa)62 do result in lower T cell activation as well as very stiff substrates (>2 MPa),63 which indicates that there is an optimal range of stiffnesses where T cell activation is highest. With the semiflexible character of the polymer, we most likely are in this optimal range. This notion is supported by the finding that expansion of T cells stimulated with αCD3/αCD28 functionalized “soft” beads was higher compared to treatment with more rigid Dynabeads.64,65 This result is in line with our findings where T cells stimulated with PIC immunobrush beads proliferated and expanded much better compared to T cells stimulated with rigid beads. These characteristics could also be the basis for the observation that coupling αCD3 and αCD28 antibodies to PIC immunobrushes led to lower antibody density thresholds to achieve full T cell activation compared to rigid scaffolds. In line with this, our PIC immunobrush beads required 34-fold less antibody compared to Dynabeads to obtain higher numbers of expanded T cells. The density and ratio of the αCD3/αCD28 antibodies on aAPCs have been shown to be important design parameters influencing T cell activation.66,67 The semiflexible nature of the brush could result in an increased effective multivalency with a higher effective number of antibodies bound to the T cells.38
Furthermore, we observed a dramatic increase in cytokine secretion by T cells stimulated with PIC immunobrush beads. While the detected amounts of both secreted IFNγ and IL-2 were higher, the origins of the increased concentrations are different. Compared to stimulation with Ab beads, the number of IFNγ producing T cells is increased, while for IL-2, the number of producing T cells is similar, but rather the amount of cytokine produced per cell is enhanced. This contradicts previous work that describes the digital secretion of IL-2 by CD4+ T cells triggered by a single peptide major histocompatibility complex (pMHC) ligand. Increasing the number of pMHC did not lead to increased IL-2 secretion per cell but instead enhanced the proportion of responding T cells.68 This might suggest that an alternative mechanism is engaged with respect to IL-2 production following polyclonal stimulation of T cells with PIC beads compared to antigen-specific stimulation using soluble pMHC. As such, a more in-depth analysis, for example, by looking at the (mechanical) signaling pathways that lead to cytokine production, is needed to determine the mechanism behind this finding.
The developed PIC immunobrush beads were benchmarked against two frequently used products for ex vivo expansion of T cells for clinical adoptive therapies, namely, CD3/CD28 Dynabeads and T Cell TransAct. Compared to Dynabeads, our platform resulted in 50% more expanded T cells, remarkedly with only 3% of the amount of antibody present on Dynabeads.31 This larger population of expanded T cells activated with our PIC immunobrush beads showed a phenotype and potential functionality—as judged by cytotoxic markers, cytokine production, and exhaustion markers—similar to T cells expanded with Dynabeads, showing the potential of our system. T Cell TransAct induced a more differentiated, terminal effector phenotype, which is considered to be less favorable, as it is shown before that less differentiated cells persist and proliferate more after adoptive cell transfer in vivo.52
Our platform is highly modular and not limited to microbeads, PICs, or αCD3/αCD28. Previously we successfully coupled different cytokines and biomolecules to the PICs,38,39 and a next step would be the incorporation of peptide major histocompatibility complex (MHC) complexes for the antigen-specific expansion of rare T cell populations. Furthermore, we have shown before that PICs can be easily coupled to different types of scaffolds such as nanoparticles or flat surfaces.41 Finally, other types of flexible and semiflexible polymers could be used as brushes to investigate the effect of polymer stiffness on T cell activation.
5. Conclusions
In summary, here, we developed T cell activating PIC polymer immunobrush magnetic beads as a potent addition to the aAPC arsenal. This approach synergizes the best of two worlds, being the enhanced T cell activating capacity of agonistic antibodies conjugated to semiflexible PIC polymers, with the reported optimal size (4.5 μm) and ease of purification of the magnetic beads. We demonstrate that the brush architecture is crucial for the T cell activating capacity. The PIC immunobrush beads induced high ex vivo expansion of functional T cells with low numbers of stimulating antibodies and outperformed current benchmarks by producing higher numbers of cells exhibiting a combination of beneficial phenotypic characteristics, such as reduced expression of exhaustion markers, high cytokine production, and robust expression of cytotoxic hallmarks. The ex vivo expansion of T cells is an important process in many immunotherapies, including adoptive T cell transfer. Several types of materials functionalized with biomolecules to activate and expand T cells have been developed and are being evaluated in clinical trials. Therefore, our PIC immunobrush strategy could contribute in making T cell-based immunotherapies more effective. Furthermore, our platform is highly modular, not restricted to beads, antibodies or even PICs and consequently of interest to the broader materials science community.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.0c21994.
AFM analysis of PIC length; overview of all formulations used in this study; comparison with soluble PIC; influence of αCD28/αCD3 ratio and density on proliferation; CD69 and CD25 expression of stimulated T cells; viability of expanded T cells; and detailed overview of intracellular cytokine production (PDF)
Author Contributions
R.H., J.W., and D.V. contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by a PhD grant from the NWO Gravity Program Institute for Chemical Immunology, ERC Adv. Grant PATHFINDER (269019) and the Oncode Institute. C.F. is recipient of the NWO Spinoza award and ERC Adv. Grant ARTimmune (834618). M.V. is recipient of ERC Starting grant CHEMCHECK (679921) and a Gravity Program Institute for Chemical Immunology tenure track grant by NWO.
The authors declare no competing financial interest.
Supplementary Material
References
- Dunn G. P.; Bruce A. T.; Ikeda H.; Old L. J.; Schreiber R. D. Cancer Immunoediting: From Immunosurveillance to Tumor Escape. Nat. Immunol. 2002, 3, 991–998. 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- Pauken K. E.; Wherry E. J. Overcoming T Cell Exhaustion in Infection and Cancer. Trends Immunol. 2015, 36, 265–276. 10.1016/j.it.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake C. G.; Jaffee E.; Pardoll D. M. Mechanisms of Immune Evasion by Tumors. Adv. Immunol. 2006, 90, 51–81. 10.1016/s0065-2776(06)90002-9. [DOI] [PubMed] [Google Scholar]
- Anusha A.; Kumar S.; Kaushik S.; Jyoti A. Cancer Immunotherapy. J. Pharm. Sci. Res. 2017, 9, 662–666. [Google Scholar]
- Restifo N. P.; Dudley M. E.; Rosenberg S. A. Adoptive Immunotherapy for Cancer: Harnessing the T Cell Response. Nat. Rev. Immunol. 2012, 12, 269–281. 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley M. E.; Gross C. A.; Somerville R. P. T.; Hong Y.; Schaub N. P.; Rosati S. F.; White D. E.; Nathan D.; Restifo N. P.; Steinberg S. M.; Wunderlich J. R.; Kammula U. S.; Sherry R. M.; Yang J. C.; Phan G. Q.; Hughes M. S.; Laurencot C. M.; Rosenberg S. A. Randomized Selection Design Trial Evaluating Cd8+-Enriched Versus Unselected Tumor-Infiltrating Lymphocytes for Adoptive Cell Therapy for Patients with Melanoma. J. Clin. Oncol. 2013, 31, 2152–2159. 10.1200/JCO.2012.46.6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg S. A.; Yang J. C.; Sherry R. M.; Kammula U. S.; Hughes M. S.; Phan G. Q.; Citrin D. E.; Restifo N. P.; Robbins P. F.; Wunderlich J. R.; Morton K. E.; Laurencot C. M.; Steinberg S. M.; White D. E.; Dudley M. E. Durable Complete Responses in Heavily Pretreated Patients with Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svane I. M.; Verdegaal E. M. Achievements and Challenges of Adoptive T Cell Therapy with Tumor-Infiltrating or Blood-Derived Lymphocytes for Metastatic Melanoma: What Is Needed to Achieve Standard of Care?. Cancer Immunol. Immunother. 2014, 63, 1081–1091. 10.1007/s00262-014-1580-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- June C. H. Principles of Adoptive T Cell Cancer Therapy. J. Clin. Invest. 2007, 117, 1204–1212. 10.1172/JCI31446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- June C. H.; Riddell S. R.; Schumacher T. N. Adoptive Cellular Therapy: A Race to the Finish Line. Sci. Transl. Med. 2015, 7, 280ps7 10.1126/scitranslmed.aaa3643. [DOI] [PubMed] [Google Scholar]
- Eggermont L. J.; Paulis L. E.; Tel J.; Figdor C. G. Towards Efficient Cancer Immunotherapy: Advances in Developing Artificial Antigen-Presenting Cells. Trends Biotechnol. 2014, 32, 456–465. 10.1016/j.tibtech.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Garvin J. E.; Koretzky G. A.; Jordan M. S. T Cell Activation. Annu Rev Immunol. 2009, 27, 591–619. 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boesteanu A. C.; Katsikis P. D. Memory T Cells Need Cd28 Costimulation to Remember. Semin. Immunol. 2009, 21, 69–77. 10.1016/j.smim.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe A. H. Mechanisms of Costimulation. Immunol. Rev. 2009, 229, 5–11. 10.1111/j.1600-065X.2009.00784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantrell D. Signaling in Lymphocyte Activation. Cold Spring Harb. Perspect. Biol. 2015, 7, a018788 10.1101/cshperspect.a018788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal L. R.; Bailey S. R.; Wyatt M. M.; Bowers J. S.; Majchrzak K.; Nelson M. H.; Haupt C.; Paulos C. M.; Varela J. C. The Basics of Artificial Antigen Presenting Cells in T Cell-Based Cancer Immunotherapies. J. Immunol. Res. Ther. 2017, 2, 68–79. [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Kurlander R. J. Comparison of Anti-Cd3 and Anti-Cd28-Coated Beads with Soluble Anti-Cd3 for Expanding Human T Cells: Differing Impact on Cd8 T Cell Phenotype and Responsiveness to Restimulation. J. Transl. Med. 2010, 8, 104 10.1186/1479-5876-8-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mescher M. F. Surface Contact Requirements for Activation of Cytotoxic T Lymphocytes. J. Immunol. 1992, 149, 2402–2405. [PubMed] [Google Scholar]
- Hickey J. W.; Vicente F. P.; Howard G. P.; Mao H. Q.; Schneck J. P. Biologically Inspired Design of Nanoparticle Artificial Antigen-Presenting Cells for Immunomodulation. Nano Lett. 2017, 17, 7045–7054. 10.1021/acs.nanolett.7b03734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C.; Økern G.; Rehan S.; Beagley L.; Lee S. K.; Aarvak T.; Schjetne K. W.; Khanna R. Ex Vivo Expansion of Human T Cells for Adoptive Immunotherapy Using the Novel Xeno-Free Cts Immune Cell Serum Replacement. Clin. Transl. Immunol. 2015, 4, e31 10.1038/cti.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trickett A.; Kwan Y. L. T Cell Stimulation and Expansion Using Anti-Cd3/Cd28 Beads. J. Immunol. Methods 2003, 275, 251–255. 10.1016/S0022-1759(03)00010-3. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Wu W.; Wan T.; Liu Y.; Peng G.; Chen Z.; Zhu H. Impact of Polyclonal Anti-Cd3/Cd28-Coated Magnetic Bead Expansion Methods on T Cell Proliferation, Differentiation and Function. Int. Immunopharmacol. 2013, 15, 129–137. 10.1016/j.intimp.2012.10.023. [DOI] [PubMed] [Google Scholar]
- Junker N.; Andersen M. H.; Wenandy L.; Dombernowsky S. L.; Kiss K.; Sørensen C. H.; Therkildsen M. H.; Von Buchwald C.; Andersen E.; Straten P. T.; Svane I. M. Bimodal Ex Vivo Expansion of T Cells from Patients with Head and Neck Squamous Cell Carcinoma: A Prerequisite for Adoptive Cell Transfer. Cytotherapy 2011, 13, 822–834. 10.3109/14653249.2011.563291. [DOI] [PubMed] [Google Scholar]
- Stevanović S.; Draper L. M.; Langhan M. M.; Campbell T. E.; Kwong M. L.; Wunderlich J. R.; Dudley M. E.; Yang J. C.; Sherry R. M.; Kammula U. S.; Restifo N. P.; Rosenberg S. A.; Hinrichs C. S. Complete Regression of Metastatic Cervical Cancer after Treatment with Human Papillomavirus-Targeted Tumor-Infiltrating T Cells. J. Clin. Oncol. 2015, 33, 1543–1550. 10.1200/JCO.2014.58.9093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong C. S. M.; Dardalhon V.; Devaud C.; Taylor N.; Darcy P. K.; Kershaw M. H. Car T-Cell Therapy of Solid Tumors. Immunol. Cell Biol. 2017, 95, 356–363. 10.1038/icb.2016.128. [DOI] [PubMed] [Google Scholar]
- Redeker A.; Arens R. Improving Adoptive T Cell Therapy: The Particular Role of T Cell Costimulation, Cytokines, and Post-Transfer Vaccination. Front. Immunol. 2016, 7, 345 10.3389/fimmu.2016.00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee C. Adoptive T Cell Therapy: Addressing Challenges in Cancer Immunotherapy. J. Transl. Med. 2005, 3, 17 10.1186/1479-5876-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs C. S.; Rosenberg S. A. Exploiting the Curative Potential of Adoptive T-Cell Therapy for Cancer. Immunol. Rev. 2014, 257, 56–71. 10.1111/imr.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher B.; Packard B. S.; Read E. J.; Carrasquillo J. A.; Carter C. S.; Topalian S. L.; Yang J. C.; Yolles P.; Larson S. M.; Rosenberg S. A. Tumor Localization of Adoptively Transferred Indium-111 Labeled Tumor Infiltrating Lymphocytes in Patients with Metastatic Melanoma. J. Clin. Oncol. 1989, 7, 250–261. 10.1200/JCO.1989.7.2.250. [DOI] [PubMed] [Google Scholar]
- Griffith K. D.; Read E. J.; Carrasquillo J. A.; Carter C. S.; Yang J. C.; Fisher B.; Aebersold P.; Packard B. S.; Yu M. Y.; Rosenberg S. A. In Vivo Distribution of Adoptively Transferred Indium-111-Labeled Tumor Infiltrating Lymphocytes and Peripheral Blood Lymphocytes in Patients with Metastatic Melanoma. J. Natl. Cancer Inst. 1989, 81, 1709–1717. 10.1093/jnci/81.22.1709. [DOI] [PubMed] [Google Scholar]
- Cheung A. S.; Zhang D. K. Y.; Koshy S. T.; Mooney D. J. Scaffolds That Mimic Antigen-Presenting Cells Enable Ex Vivo Expansion of Primary T Cells. Nat. Biotechnol. 2018, 36, 160–169. 10.1038/nbt.4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W.; Tamzalit F.; Chaudhuri P. K.; Black C. T.; Huse M.; Kam L. C. T Cell Activation and Immune Synapse Organization Respond to the Microscale Mechanics of Structured Surfaces. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 19835–19840. 10.1073/pnas.1906986116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster A. E.; Rooney C. M. Improving T Cell Therapy for Cancer. Expert Opin. Biol. Ther. 2006, 6, 215–229. 10.1517/14712598.6.3.215. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Zhang M.; Ramos C. A.; Durett A.; Liu E.; Dakhova O.; Liu H.; Creighton C. J.; Gee A. P.; Heslop H. E.; Rooney C. M.; Savoldo B.; Dotti G. Closely Related T-Memory Stem Cells Correlate with in Vivo Expansion of Car.Cd19-T Cells and Are Preserved by Il-7 and Il-15. Blood 2014, 123, 3750–3759. 10.1182/blood-2014-01-552174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvec A. R.; Ecker C.; Kong H.; Winters E. A.; Glover J.; Varela-Rohena A.; Riley J. L. Improved Expansion and in Vivo Function of Patient T Cells by a Serum-Free Medium. Mol. Ther.--Methods Clin. Dev. 2018, 8, 65–74. 10.1016/j.omtm.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen A.-M.; Borelli G.; Hoel H. J.; Lislerud K.; Gaudernack G.; Kvalheim G.; Aarvak T. Ex Vivo Expansion Protocol for Human Tumor Specific T Cells for Adoptive T Cell Therapy. J. Immunol. Methods 2010, 355, 52–60. 10.1016/j.jim.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Mandal S.; Eksteen-Akeroyd Z. H.; Jacobs M. J.; Hammink R.; Koepf M.; Lambeck A. J. a.; van Hest J. C. M.; Wilson C. J.; Blank K.; Figdor C. G.; Rowan A. E. Therapeutic Nanoworms: Towards Novel Synthetic Dendritic Cells for Immunotherapy. Chem. Sci. 2013, 4, 4168–4174. 10.1039/c3sc51399h. [DOI] [Google Scholar]
- Hammink R.; Mandal S.; Eggermont L. J.; Nooteboom M.; Willems P. H. G. M.; Tel J.; Rowan A. E.; Figdor C. G.; Blank K. G. Controlling T-Cell Activation with Synthetic Dendritic Cells Using the Multivalency Effect. ACS Omega 2017, 2, 937–945. 10.1021/acsomega.6b00436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermont L. J.; Hammink R.; Blank K. G.; Rowan A. E.; Tel J.; Figdor C. G. Cytokine-Functionalized Synthetic Dendritic Cells for T cell Targeted Immunotherapies. Adv. Ther. 2018, 1, 1800021 10.1002/adtp.201800021. [DOI] [Google Scholar]
- Mandal S.; Hammink R.; Tel J.; Eksteen-Akeroyd Z. H.; Rowan A. E.; Blank K.; Figdor C. G. Polymer-Based Synthetic Dendritic Cells for Tailoring Robust and Multifunctional T Cell Responses. ACS Chem. Biol. 2015, 10, 485–492. 10.1021/cb500455g. [DOI] [PubMed] [Google Scholar]
- Voerman D.; Schluck M.; Weiden J.; Joosten B.; Eggermont L. J.; van den Eijnde T.; Ignacio B.; Cambi A.; Figdor C. G.; Kouwer P. H. J.; Verdoes M.; Hammink R.; Rowan A. E. Synthetic Semiflexible and Bioactive Brushes. Biomacromolecules 2019, 20, 2587–2597. 10.1021/acs.biomac.9b00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dommerholt J.; Rutjes F. P. J. T.; van Delft F. L. Strain-Promoted 1,3-Dipolar Cycloaddition of Cycloalkynes and Organic Azides. Top. Curr. Chem. 2016, 374, 16 10.1007/s41061-016-0016-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammink R.; Eggermont L. J.; Zisis T.; Tel J.; Figdor C. G.; Rowan A. E.; Blank K. G. Affinity-Based Purification of Polyisocyanopeptide Bioconjugates. Bioconjugate Chem. 2017, 28, 2560–2568. 10.1021/acs.bioconjchem.7b00398. [DOI] [PubMed] [Google Scholar]
- Yokosuka T.; Kobayashi W.; Sakata-Sogawa K.; Takamatsu M.; Hashimoto-Tane A.; Dustin M. L.; Tokunaga M.; Saito T. Spatiotemporal Regulation of T Cell Costimulation by Tcr-Cd28 Microclusters and Protein Kinase C Theta Translocation. Immunity 2008, 29, 589–601. 10.1016/j.immuni.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokosuka T.; Saito T. The Immunological Synapse, Tcr Microclusters, and T Cell Activation. Curr. Top. Microbiol. Immunol. 2010, 340, 81–107. [DOI] [PubMed] [Google Scholar]
- Pageon S. V.; Tabarin T.; Yamamoto Y.; Ma Y.; Nicovich P. R.; Bridgeman J. S.; Cohnen A.; Benzing C.; Gao Y.; Crowther M. D.; Tungatt K.; Dolton G.; Sewell A. K.; Price D. A.; Acuto O.; Parton R. G.; Gooding J. J.; Rossy J.; Rossjohn J.; Gaus K. Functional Role of T-Cell Receptor Nanoclusters in Signal Initiation and Antigen Discrimination. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, E5454–E5463. 10.1073/pnas.1607436113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin M. L. The Immunological Synapse. Cancer Immunol. Res. 2014, 2, 1023–1033. 10.1158/2326-6066.CIR-14-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalamasz D.; Long S. A.; Taniguchi R.; Buckner J. H.; Berenson R. J.; Bonyhadi M. Optimization of Human T-Cell Expansion Ex Vivo Using Magnetic Beads Conjugated with Anti-Cd3 and Anti-Cd28 Antibodies. J. Immunother. 2004, 27, 405–418. 10.1097/00002371-200409000-00010. [DOI] [PubMed] [Google Scholar]
- Matic J.; Deeg J.; Scheffold A.; Goldstein I.; Spatz J. P. Fine Tuning and Efficient T Cell Activation with Stimulatory Acd3 Nanoarrays. Nano Lett. 2013, 13, 5090–5097. 10.1021/nl4022623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casati A.; Varghaei-Nahvi A.; Feldman S. A.; Assenmacher M.; Rosenberg S. A.; Dudley M. E.; Scheffold A. Clinical-Scale Selection and Viral Transduction of Human Naïve and Central Memory Cd8+ T Cells for Adoptive Cell Therapy of Cancer Patients. Cancer Immunol. Immunother. 2013, 62, 1563–1573. 10.1007/s00262-013-1459-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahnke Y. D.; Brodie T. M.; Sallusto F.; Roederer M.; Lugli E. The Who’s Who of T-Cell Differentiation: Human Memory T-Cell Subsets. Eur. J. Immunol. 2013, 43, 2797–2809. 10.1002/eji.201343751. [DOI] [PubMed] [Google Scholar]
- Busch D. H.; Fräßle S. P.; Sommermeyer D.; Buchholz V. R.; Riddell S. R. Role of Memory T Cell Subsets for Adoptive Immunotherapy. Semin. Immunol. 2016, 28, 28–34. 10.1016/j.smim.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry E. J.; Kurachi M. Molecular and Cellular Insights into T Cell Exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenblock E. R.; Fadel T.; Labowsky M.; Pober J. S.; Fahmy T. M. An Artificial Antigen-Presenting Cell with Paracrine Delivery of Il-2 Impacts the Magnitude and Direction of the T Cell Response. J. Biol. Chem. 2011, 286, 34883–34892. 10.1074/jbc.M111.276329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenblock E. R.; Fahmy T. M. A Comprehensive Platform for Ex Vivo T-Cell Expansion Based on Biodegradable Polymeric Artificial Antigen-Presenting Cells. Mol. Ther. 2008, 16, 765–772. 10.1038/mt.2008.11. [DOI] [PubMed] [Google Scholar]
- Sunshine J. C.; Perica K.; Schneck J. P.; Green J. J. Particle Shape Dependence of Cd8+ T Cell Activation by Artificial Antigen Presenting Cells. Biomaterials 2014, 35, 269–277. 10.1016/j.biomaterials.2013.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer R. A.; Sunshine J. C.; Perica K.; Kosmides A. K.; Aje K.; Schneck J. P.; Green J. J. Biodegradable Nanoellipsoidal Artificial Antigen Presenting Cells for Antigen Specific T-Cell Activation. Small 2015, 11, 1519–1525. 10.1002/smll.201402369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadel T. R.; Sharp F. A.; Vudattu N.; Ragheb R.; Garyu J.; Kim D.; Hong E.; Li N.; Haller G. L.; Pfefferle L. D.; Justesen S.; Herold K. C.; Fahmy T. M. A Carbon Nanotube-Polymer Composite for T-Cell Therapy. Nat. Nanotechnol. 2014, 9, 639–647. 10.1038/nnano.2014.154. [DOI] [PubMed] [Google Scholar]
- Alarcón B.; Mestre D.; Martínez-Martín N. The Immunological Synapse: A Cause or Consequence of T-Cell Receptor Triggering?. Immunology 2011, 133, 420–425. 10.1111/j.1365-2567.2011.03458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto-Tane A.; Saito T. Dynamic Regulation of Tcr-Microclusters and the Microsynapse for T Cell Activation. Front. Immunol. 2016, 7, 255 10.3389/fimmu.2016.00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T.; Yokosuka T. Immunological Synapse and Microclusters: The Site for Recognition and Activation of T Cells. Curr. Opin. Immunol. 2006, 18, 305–313. 10.1016/j.coi.2006.03.014. [DOI] [PubMed] [Google Scholar]
- Saitakis M.; Dogniaux S.; Goudot C.; Bufi N.; Asnacios S.; Maurin M.; Randriamampita C.; Asnacios A.; Hivroz C. Different Tcr-Induced T Lymphocyte Responses Are Potentiated by Stiffness with Variable Sensitivity. eLife 2017, 6, e23190 10.7554/eLife.23190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor R. S.; Hao X.; Shen K.; Bashour K.; Akimova T.; Hancock W. W.; Kam L. C.; Milone M. C. Substrate Rigidity Regulates Human T Cell Activation and Proliferation. J. Immunol. 2012, 189, 1330–1339. 10.4049/jimmunol.1102757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert L. H.; Goebrecht G. K. E.; De Leo S. E.; O’Connor R. S.; Nunez-Cruz S.; Li T.-D.; Yuan J.; Milone M. C.; Kam L. C. Improving T Cell Expansion with a Soft Touch. Nano Lett. 2017, 17, 821–826. 10.1021/acs.nanolett.6b04071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majedi F. S.; Hasani-Sadrabadi M. M.; Thauland T. J.; Li S.; Bouchard L.-S.; Butte M. J. Augmentation of T-Cell Activation by Oscillatory Forces and Engineered Antigen-Presenting Cells. Nano Lett. 2019, 19, 6945–6954. 10.1021/acs.nanolett.9b02252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcassian D.; Depoil D.; Rudnicka D.; Liu M.; Davis D. M.; Dustin M. L.; Dunlop I. E. Nanoscale Ligand Spacing Influences Receptor Triggering in T Cells and Nk Cells. Nano Lett. 2013, 13, 5608–5614. 10.1021/nl403252x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perica K.; Kosmides A. K.; Schneck J. P. Linking Form to Function: Biophysical Aspects of Artificial Antigen Presenting Cell Design. Biochim. Biophys. Acta, Mol. Cell Res. 2015, 1853, 781–790. 10.1016/j.bbamcr.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Brameshuber M.; Zeng X.; Xie J.; Li Q.-j.; Chien Y.-h.; Valitutti S.; Davis M. M. A Single Peptide-Major Histocompatibility Complex Ligand Triggers Digital Cytokine Secretion in Cd4(+) T Cells. Immunity 2013, 39, 846–857. 10.1016/j.immuni.2013.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.