

Abstract
AprD4 is a radical S-adenosyl-l-methionine (SAM) enzyme catalyzing C3′-deoxygenation of paromamine to form 4′-oxo-lividamine. It is the only 1,2-diol dehydratase in the radical SAM enzyme superfamily that has been identified and characterized in vitro. The AprD4 catalyzed 1,2-diol dehydration is a key step in the biosynthesis of several C3′-deoxy-aminoglycosides. While the regiochemistry of the hydrogen atom abstraction catalyzed by AprD4 has been established, the mechanism of the subsequent chemical transformation remains not fully understood. To investigate the mechanism, several substrate analogues were synthesized and their fates upon incubation with AprD4 analyzed. The results support a mechanism involving formation of a ketyl radical intermediate followed by direct elimination of the C3′-hydroxyl group rather than a gem-diol intermediate generated via 1,2-migration of the C3′-hydroxyl group to C4′. The stereochemistry of hydrogen atom incorporation after radical-mediated dehydration was also established.
Graphical Abstract
INTRODUCTION
The reductive deoxygenation of biological metabolites is a process found in countless biosynthetic pathways and accomplished by a mechanistically diverse array of enzyme catalysts and pathways.1–14 Arguably one of the best known motifs involves a two-step process where the C–O bond is first cleaved via Lewis acid-base chemistry to form a desaturated compound that is subsequently reduced in the second step, which typically involves hydride transfer from a biological reductant such as NADPH.8–10 The lyase activity in this process requires the presence of an acidic carbon suitably located relative to the oxy-leaving group so as to effect its elimination upon deprotonation. Consequently, such an acidic carbon must be introduced or otherwise activated, often by a neighboring electron withdrawing group or coupling with an organic cofactor.8–14 However, a number of dehydrations are also known to proceed despite the absence of an activated carbon. These reactions instead rely on oxidation of the substrate to form a radical intermediate that can only be generated via a limited number of specialized active site features such as a B12 coenzyme in glycerol dehydratase,15–19 or an amino acid radical species in ribonucleotide reductase20–21 among others.15–19,22–25 Following dehydration, the resulting product radical is subsequently reduced back to an even-electron state and if necessary reduced further in a subsequent step to complete the net deoxygenation.
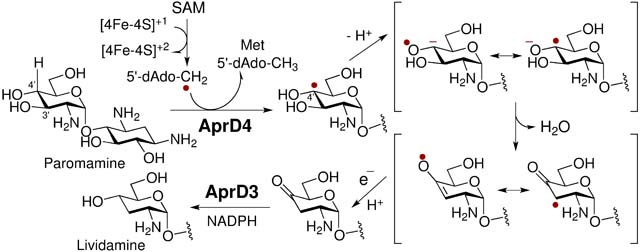
The C3′ deoxygenation of paromamine (1) also involves the formation of radical intermediates, because it too lacks an activating functional group on the glucosamine subunit. However, in contrast to previous examples, the initial, radical-mediated dehydration reaction involves a radical S-adenosyl-l-methionine (SAM) enzyme, AprD4.26–28 The resulting product, 4′-oxo-lividamine (3), retains the same oxidation state as the paromamine substrate and must therefore be reduced by its NADPH-dependent reductase partner AprD3 in order to generate the deoxygenated 3′-deoxy-pseudo-disaccharide lividamine (4, see Scheme 1).26–28 Although radical SAM enzymes have been shown to function as amino-lyases (e.g., TDP-4-amino-4,6-dideoxy-d-glucose deaminase, DesII),29–31 AprD4 is the only radical SAM hydroxy-lyase that has been characterized to date. Recent studies have also demonstrated that AprD4 along with AprD3 can catalyze C3′ deoxygenation of kanamycin C, kanamycin B and neamine (see Figure S1) via related pathways.28 The resulting absence of the C3′-hydroxyl group in each of these antibiotics leaves them inert to modification by 3′-phosphotransferases, which is a common mechanism of aminoglycoside-resistance in some bacterial strains.32–33 Moreover, AprD4 may also be involved in the biosynthesis of tobramycin,34 nebramycin 5′35 and lividomycin B36 given the structural resemblance of their pseudodisaccharide cores to that of paromamine. Therefore, a more complete understanding of the mechanism of AprD4 and its substrate specificity is anticipated to serve as a useful foundation for the development of more effective aminoglycoside antibiotic regimens.
Scheme 1.
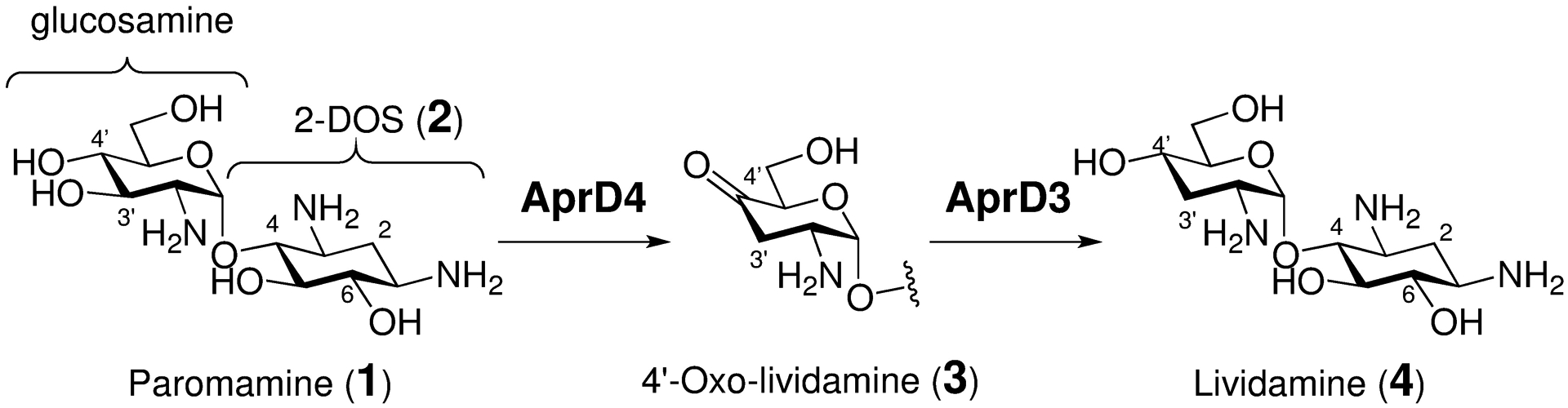
C3′-deoxygenation of paromamine (1) to lividamine (4) catalyzed by AprD4 and AprD3.
RESULTS AND DISCUSSION
Like other members of the radical SAM enzyme superfamily, AprD4 harbors a characteristic [4Fe-4S] cluster in the active site. However, unlike most members of this superfamily, AprD4 is characterized by a unique CX3CX3C [4Fe-4S] binding motif rather than the canonical CX3CX2C motif.37–38 The reaction of a typical radical SAM enzyme is initiated by electron transfer from the reduced [4Fe-4S]1+ cluster to SAM leading to reductive homolysis of the latter (5) concomitant with the generation of methionine (6) and a 5′-deoxyadenosyl radical (5′-dAdoCH2•) equivalent (7). The latter serves to abstract a hydrogen atom from the substrate thereby activating the substrate for its subsequent radical-mediated transformation to 4′-oxo-lividamine (3) (Scheme 2).39–42 The well characterized DesII is a closely related enzyme, which catalyzes the deamination of TDP-4-amino-4,6-dideoxy-d-glucose (49) to the corresponding 4,6-dideoxy-3-ketoglucose (53) (see Scheme 4).29–31
Scheme 2.
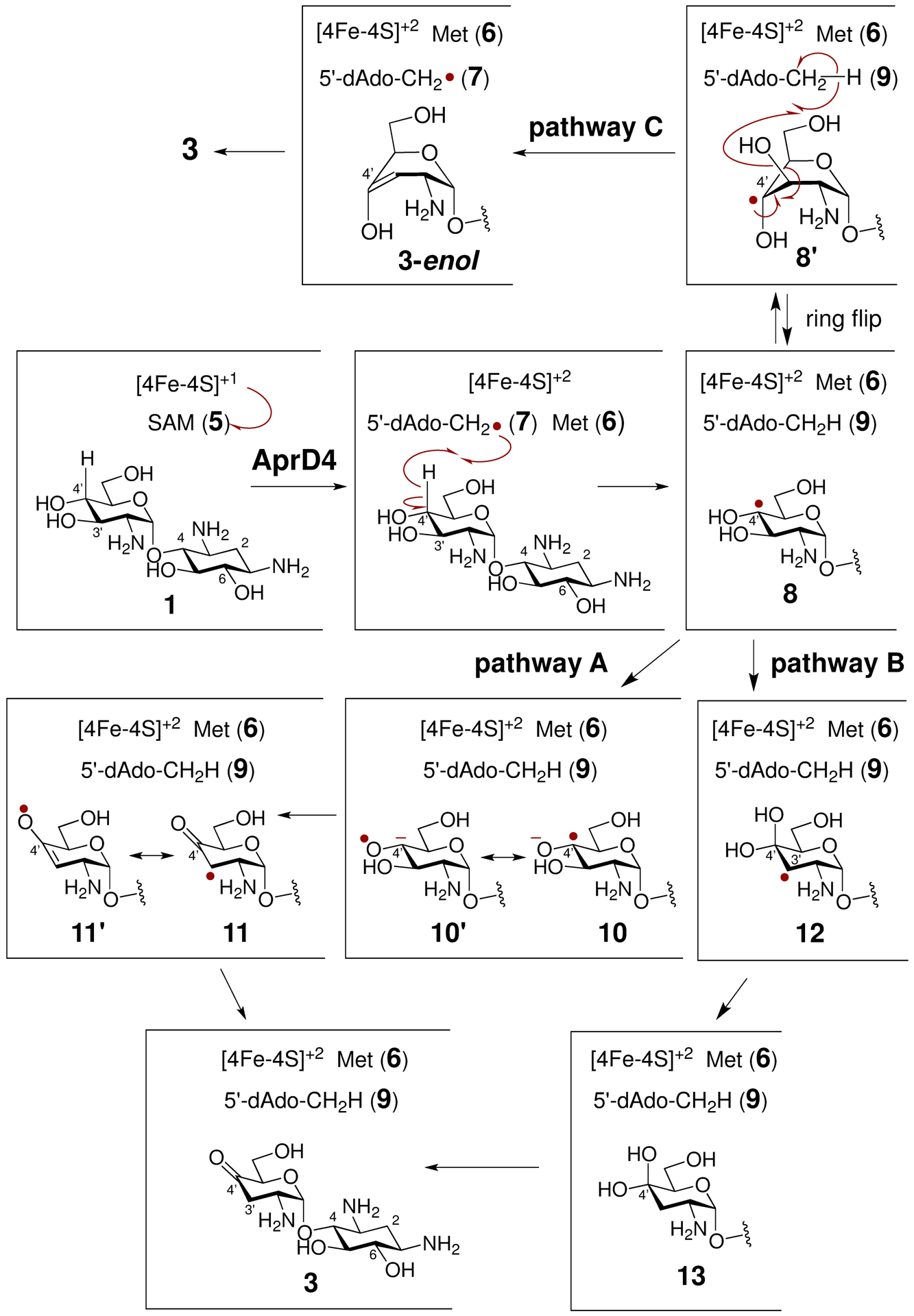
Proposed mechanism for AprD4-catalyzed 1,2-diol dehydration of paromamine (1). For the sake of brevity, the aminocyclitol 2-deoxystreptamine (2-DOS) (2) unit is not shown in some compounds in Scheme 2.
Scheme 4.

Comparison of proposed mechanisms for AprD4 and DesII.
Two mechanisms have been proposed to account for the AprD4-catalyzed dehydration of 1 to 4′-oxo-lividamine (3) as shown in Scheme 2.26 Both mechanisms involve initial hydrogen-atom abstraction from the C4′ position of paromamine (1) by the 5′-deoxyadenosyl radical equivalent 7.26–28 The resulting substrate radical (8) may be deprotonated to form a ketyl radical (10/10′) that subsequently undergoes β-elimination of the 3′-OH group to yield the keto/enol radical (11/11′) (Scheme 2, pathway A). One-electron reduction of 11/11′ would then complete the dehydration. This mechanism is similar to those of (R)-2-hydroxyacyl-CoA dehydratase24–25 as well as glycyl radical-dependent diol-dehydratase19,22–23 and has been argued for on the basis of the AprD4 crystal structure with bound substrate.43 Alternatively, the initially formed substrate radical (8) may undergo a radical-induced 1,2-hydroxyl migration to form the gem-diol radical 12 (Scheme 2 pathway B), a process analogous to that of the B12-dependent diol-dehydratases and ethanolamine ammonia lyases.15–19 After reduction, the gem-diol intermediate 13 could then eliminate water to form 4′-oxo-lividamine (3). Although initial hydrogen-atom abstraction from the C4′ position has been established based on deuterium transfer from [4′−2H]-paromamine to 7 to yield [5′−2H]-5′-dAdo (9),26 a complete picture of the AprD4 catalytic cycle remains to be described. A third mechanism suggested by a reviewer can also be envisioned that involves ring flipping of 8 followed by abstraction of a hydrogen atom from 5′-dAdo (9) by a 3′-hydroxy radical leaving group from 8′ to yield an enol product (3-enol) and regenerate 7 (Scheme 2 pathway C). However, because multiple incorporation of deuterium into 5′-dAdo is not observed with [4′−2H]-paromamine in vitro,26 SAM is not regenerated from the 5′-dAdo radical (7), which would need to be reduced to 5′-dAdo.
To investigate the mechanism of AprD4, several substrate analogues (14–21, Figure 1A) were synthesized. Compound 14 was designed to distinguish pathways A and B, because the C4′-methoxy group will prevent ionization of the initial substrate radical thereby impeding formation of a ketyl radical intermediate (vis-a-vis 8→10/10′). In contrast, the C4′-methoxy group should have much less of an impact on radical-induced 1,2-migration of the C3′-hydroxyl group to give 12. As shown in Figure 1C (trace b), when 1 mM 14 was incubated with 0.05 mM AprD4, 0.05 mM AprD3, 2 mM SAM, 1 mM NADPH, 10 mM DTT and 2 mM sodium dithionite in 50 mM NH4HCO3 (pH 7.8) for 16 h, neither the 4′-hydroxy-4′-methoxy hemiacetal nor lividamine product was observed. Both enzymes were active as formation of 4 was observed when incubated with paromamine (1) (Figure 1B, traces a and b).
Figure 1.
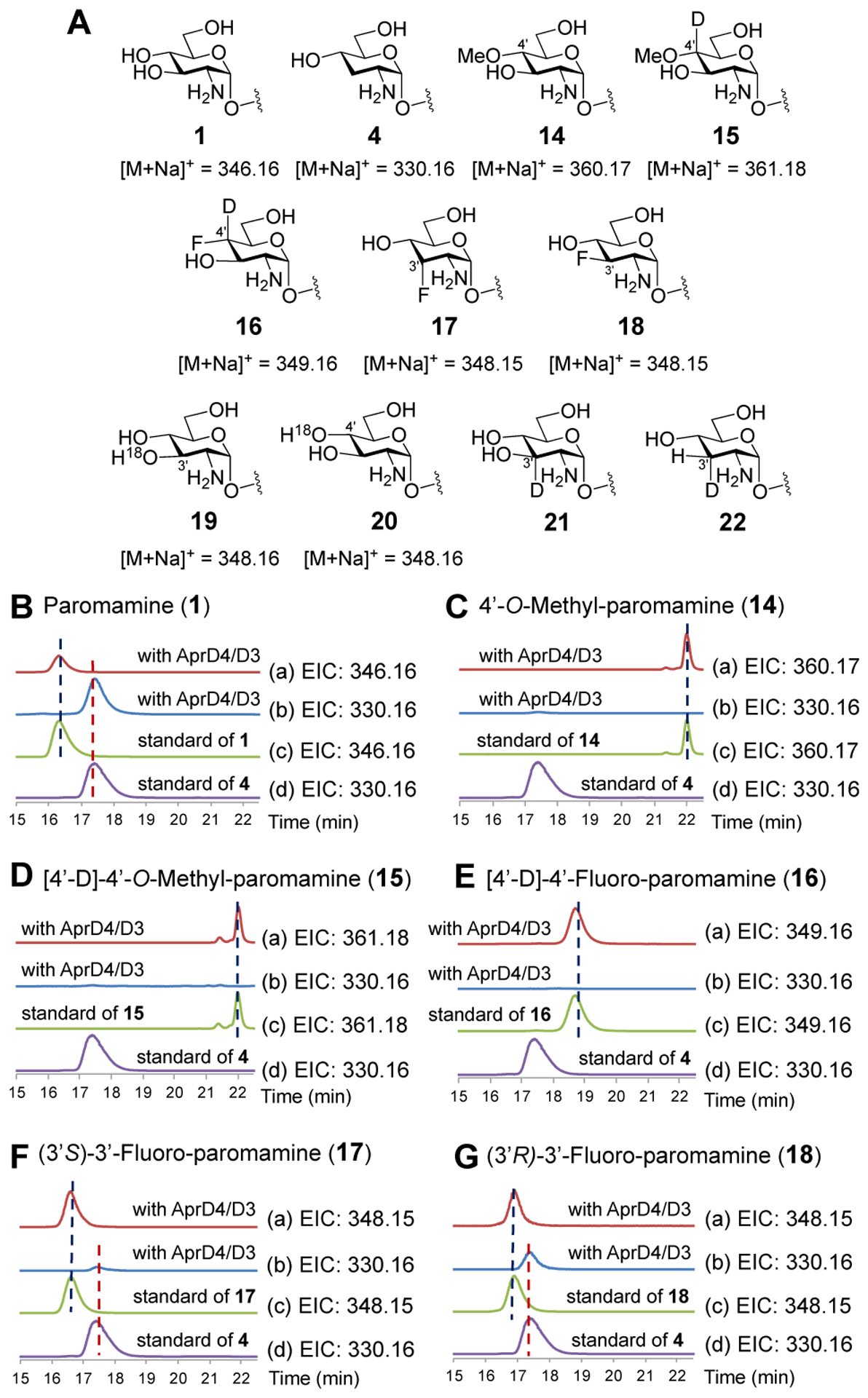
Mechanistic study of 1,2-diol dehydration catalyzed by AprD4. (A) Structures and exact masses of paromamine (1), substrate analogues 14–21 and lividamine (4). (B)-(G) LC-MS analysis of AprD4 and AprD3 assay shown with (a) extracted ion chromatogram (EIC) of [M+Na]+ corresponding to the respective substrate in each assay; (b) EIC of [M+Na]+ = 330.16 (corresponding to 4) in the assay; (c) EIC of [M+Na]+ corresponding to substrate standard; (d) EIC of [M+Na]+ = 330.16 for lividamine (4) standard. AprD4 and AprD3 were incubated with (B) paromamine (1), (C) 14, (D) 15, (E) 16, (F) 17, or (G) 18.
However, this result could also be a consequence of interference of the C4′ methoxy group with the H atom abstraction step. Indeed, little deuterium was transferred to 5′-dAdo (9) when the C4′ deuterated isotopolog 15 was used in place of 14 (Figure 1D, trace b, Table 1, entry 3, and Figure S2D). In this experiment, any deuterium transfer is assumed to be from the C4′ of 15 to the 5′-methyl group of 5′-dAdo (9). Moreover, increasing the concentration of 15 to 5 mM had only a small effect on the deuterium enrichment of 5′-dAdo (Table 1, entry 4, and Figure S2E), and only the low-level formation of unlabeled 14 was observed instead of lividamine (see Figures S3A/S3B). These results imply that the majority of 5′-dAdo produced in the presence of 14 and 15 is due to uncoupled reduction of SAM, which is commonly observed among the radical SAM enzymes.39–42,44 According to the AprD4 structure recently reported by Zhang and Nicolet,43 the main chain amide N-atom of the active site residue Ala27 is positioned near the 4′-OH group of paromamine (1). This residue may therefore engage in a steric clash with the bulky 4′-OMe group of 14/15 thereby hampering hydrogen atom abstraction from C4′.
Table 1.
Deuterium incorporation into 5′-dAdo (9) during the incubation of substrate analogues with AprD4/AprD3.
| Entry | Substrate | obs. A253/A252a | % Fraction of labeled 5′-dAdob |
|---|---|---|---|
| 1 | 1 | 0.105 ± 0.001 | NA |
| 2 | 14 | 0.101 ± 0.001 | NA |
| 3 | 15 | 0.113 ± 0.000 | 0.8 ± 0.1 |
| 4 | 15 (5 mM) | 0.179 ± 0.001 | 6.9 ± 0.1 |
| 5 | 16 | 0.276 ± 0.003 | 14.6 ± 0.3 |
| 6 | 16 (5 mM) | 0.474 ± 0.005 | 27.0 ± 0.4 |
The MS signal for protonated 5′-dAdo at 253.1 m/z (m + 1) was integrated and divided by that at 252.1 m/z (m) to obtain the observed signal intensity ratio A253/A252. This ratio largely reflects natural abundance 13C in the absence of labeled substrate (entry 1). Therefore, the ratio R of C5′-deuterated versus unlabeled 5′-dAdo observed in the presence of labeled substrate can be obtained by subtracting from the observed A253/A252 ratio that of entry 1.45
The fraction of labeled 5′-dAdo is then given by R/(R+1) and is listed as a percentage (NA indicates no significant labeling). All ratios were measured from three separate assays each.
In response to these complications, the analogue 16 carrying both a deuterium and a less sterically hindered fluoro substituent at C4′ was also prepared (Scheme 3A). The key steps included fluorination with diethylaminosulfur trifluoride (DAST) and coupling with 26 to form the pseudo-disaccharide 27, which after alkaline hydrolysis and hydrogenation gave 16.46 It was expected that redistribution of electron density on the initially formed substrate radical (i.e., 8→10/10′) necessary to promote elimination of the C3′-hydroxyl group would be prohibited by the 4′-fluoro group, whereas 1,2-migration (i.e., 8→12) leading to 4′-oxo-lividamine via the 4′-fluoro-4′-hydroxyl intermediate should not be markedly affected.47 Once again, no discernable formation of lividamine was observed (Figure 1E, trace b); however, in contrast to the experiments with 14 and 15, enrichment of 5′-dAdo (9) with deuterium was much more robust (Table 1, entries 5 & 6, and Figures S2F &2G), though not as significant as in the case of [4′−2H]-paromamine, where in a previous work nearly all the observed 5′-dAdo had been found to become labeled.26 Furthermore, formation of unlabeled 16 was detected by LC-MS (see Figures S3D and S3E). These findings imply that a substrate radical is indeed generated during the reaction of AprD4 with 16 but does not undergo conversion to 4′-oxo-lividamine.
Scheme 3.

Synthetic schemes for the preparation of [4′−2H]-4′-deoxy-4′-fluoro-paromamine (16), (3′S)-3′-deoxy-3′-fluoro-paromamine (17) and (3′R)-3′-deoxy-3′-fluoro-paromamine (18).
To further evaluate the mechanistic alternatives, (3′S)-3′-deoxy-3′-fluoro-paromamine (17) and (3′R)-3′-deoxy-3′-fluoro-paromamine (18) were prepared according to Schemes 3B and 3C. The fluoro substituent at C3′ in both cases was introduced in a nucleophilic substitution reaction (29→30 or 35→36) using tetra-n-butylammonium fluoride (TBAF) as the fluorine donor.48 Due to the increased electronegativity of the fluoro substituent, the C3′-F moiety of 17 and 18 was anticipated to be less prone to 1,2-migration compared to the C3′-OH group of 1 (see Scheme 4, 46→48→12).49 However, the fluoro substituent was expected to be a good leaving group thereby facilitating the conversion of 17 and/or 18 to 4′-oxo-lividamine (3) via an E1cB-type elimination analogous to pathway A. Indeed, when either 17 or 18 was incubated with AprD4 and AprD3, lividamine (4) production was observed (Figures 1F–G, trace b) based on LC-MS analysis and derivatization with benzyl chloroformate (see Figure S4). More lividamine appeared to be produced in the assay of 18 versus 17, consistent with 18 more closely matching the stereochemistry of paromamine. These observations collectively appear to be more suggestive of a mechanism involving a C4′ ketyl radical intermediate as opposed to a 1,2-migration. Furthermore, the conversion of 17 to 4′-oxo-lividamine is also inconsistent with the mechanism in Pathway C of Scheme 2, because this analog lacks an oxygen or a fluorine as the H-atom acceptor with the requisite stereochemistry at C3′.
Finally, compounds 19 and 20 with 18O-labeling at C3′ and C4′, respectively, were also prepared. If the reaction proceeds via a ketyl radical intermediate (Scheme 2 pathway A), then no retention of the 18O labeling in lividamine would be anticipated using 19 as the substrate. However, if the reaction proceeds via a 1,2-migration (pathway B), then partial retention of the 18O label would be expected due to the 16O,18O-gem-diol intermediate. Excess AprD3 was used in the assays to minimize the extent of solvent exchange into the ketone moiety of 3. Incubation of 19 with various ratios of AprD3/AprD4 in H2O resulted in no retention of the 18O-label in lividamine (Table S1 entries 1–4). Furthermore, incubation of unlabeled paromamine (1) in buffered H218O (>90% enriched) showed minimal (<2%) wash-in of H218O (Table S1 entries 5–8). In contrast, incubation of 20 with AprD4 and AprD3 led to significant (>97%) retention of 18O under the same assay conditions (Table S1 entries 9–12) indicating that the C4′–O bond of paromamine remains intact during the catalytic cycle. While 1,2-migration and stereoselective elimination of the C3′ hydroxyl group from C4′ of the putative gem-diol 12 in the enzyme active site cannot be definitively ruled out, such a mechanism would not explain the lack of AprD4 activity with the C4′-fluoro analog 16. Furthermore, if the reaction were to proceed via a gem-diol intermediate, then at least one of the two diastereomers 17 or 18 would likely have failed to produce 4′-oxo-lividamine.
The final step of the AprD4 catalyzed reaction involves one-electron reduction of the product radical (e.g., 11/11′) to afford 4′-oxo-lividamine (3) and may involve hydrogen atom transfer to C3′ from an active site residue such as tyrosine or cysteine.50 When [3′−2H]-paromamine (21, Figure 1A) was prepared and reacted with AprD4/AprD3, the C3′ of the product (22) was found to have the S-configuration such that the radical substitution proceeds with the hydrogen atom adding to the same face from which the C3′-OH leaves (Figure S5). This result is in agreement with a recent report that a deuterium is incorporated equatorially at C3′ of lividamine when the reaction is run in buffered D2O.28 This is also consistent with the proposal that Tyr216 of AprD4 is responsible for the transfer of the solvent-exchangeable hydrogen atom to C3′ of the radical intermediate based on the crystal structure of AprD4 and experiments demonstrating the reduced activity of the Y216F and Y216H mutants.43
CONCLUSION
In summary, AprD4 catalyzed dehydration of paramomine is more consistent with a ketyl radical intermediate (pathway A) rather than a radical-mediated 1,2-migration (pathway B). Elimination of 18O from C3′−18O paromamine and retention of 18O during dehydration of C4′−18O paromamine demonstrate that the C3′ hydroxyl group is selectively eliminated during this enzymatic reaction. Incubation of AprD4 with the substrate analogues 14–16 further show that the hydroxyl group at the C4′ position of paromamine (1) is necessary for elimination of the C3′ hydroxyl group despite not being necessary for substrate radical generation. These observations favor direct elimination the C3′ hydroxyl group from a ketyl radical intermediate, which is also consistent with the observation that AprD4 can function as an effective dehydrofluorinase when presented with the 3′-deoxy-3′-fluoro analogs of paromamine (17 and 18). This mechanistic model involving a ketyl radical intermediate is likewise consistent with the conclusions drawn from the AprD4 structural analysis43 as well as the mechanisms proposed for several other radical-mediated dehydratases including ribonucleotide reductase,20–21 1,2-propanediol dehydratases,19 and hydroxyacyl-CoA dehydratases.24–25
The proposed mechanism for AprD4 also bears some resemblance to that of DesII, which is a radical SAM lyase responsible for C4 deamination (Scheme 4, 49→53) during the biosynthesis of TDP-desosamine.29–31 In this case, a conformational adjustment of the substrate is proposed upon binding of 49 in the DesII active site so as to facilitate hyperconjugation between the C–N bond at C4 and the π-system at C3 in 51/52 and thus promote elimination.51–52 A similar model of catalysis is thus expected to hold for AprD4 as well (46→47/47’→11/11’).43
Supplementary Material
ACKNOWLEDGMENT
We thank an anonymous reviewer for suggesting an alternative mechanism (pathway C in Scheme 2). We also thank Dr. Mark Ruszczycky for his critical review and help of preparing the manuscript. This work was supported by grants from the National Institutes of Health (GM035906) and the Welch Foundation (F-1511).
ABBREVIATIONS
- DesII
TDP-4-amino-4,6-dideoxy-d-glucose deaminase or TDP-4-amino-4,6-dideoxy-d-glucose 4-amino-lyase
- AprD4
paromamine 3’-dehydratase or paromamine 3’-hydroxy-lyase
- AprD3
4’-oxo-lividamine 4’-reductase
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental details, including synthesis of compounds, activity assays, analytic methods, and NMR spectra (PDF).
REFERENCES
- (1).Anderson JA; Lin B-K; Williams HJ; Scott AI Deoxygenation of phenolic natural products. Enzymatic conversion of emodin to chrysophanol. J. Am. Chem. Soc 1988, 110, 1623–1624. [Google Scholar]
- (2).Viviani F; Gaudry M; Marquet A Melanin biosynthesis: A study of polyphenol deoxygenation. J. Chem. Soc. Perkin Trans 1 1990, 1255–1259. [Google Scholar]
- (3).Vidal‐Cros A; Viviani F; Labesse G; Boccara M; Gaudry M Polyhydroxynaphthalene reductase involved in melanin biosynthesis in Magnaporthe grisea. Purification, cDNA cloning and sequencing. Eur. J. Biochem 1994, 219, 985–992. [DOI] [PubMed] [Google Scholar]
- (4).Thompson JE; Fahnestock S; Farralli L; Liao D-I; Valenti B; Jordan DB The second naphthol reductase of fungal melanin biosynthesis in Magnaporthe grisea. J. Biol. Chem 2000, 275, 34867–34872. [DOI] [PubMed] [Google Scholar]
- (5).Simpson TJ; Weerasooriya MKB NMR studies of tautomerism in the fungal melanin biosynthesis intermediate 1,3,8-trihydroxynaphthalene. J. Chem. Soc. Perkin Trans 1 2000, 2771–2775. [Google Scholar]
- (6).Henry KM; Townsend CA Ordering the reductive and cytochrome P450 oxidative steps in demethylsterigmatocystin formation yields general insights into the biosynthesis of aflatoxin and related fungal metabolites. J. Am. Chem. Soc 2005, 127, 3724–3733. [DOI] [PubMed] [Google Scholar]
- (7).Schaä MA; Husain SM; Ferlaino S; Muä M Tautomers of anthrahydroquinones: Enzymatic reduction and implications for chrysophanol, monodictyphenone, and related xanthone biosyntheses. J. Am. Chem. Soc 2012, 134, 14742–14745. [DOI] [PubMed] [Google Scholar]
- (8).Draeger G; Park S-H; Floss HG Mechanism of the 2-deoxygenation step in the biosynthesis of the deoxyhexose moieties of the antibiotics granaticin and oleandomycin. J. Am. Chem. Soc 1999, 121, 2611–2612. [Google Scholar]
- (9).Chen H; Agnihotri G; Guo Z; Que NLS; Chen XH; Liu H -w. Biosynthesis of mycarose: Isolation and characterization of enzymes involved in the C-2 deoxygenation. J. Am. Chem. Soc 1999, 121, 8124–8125. [Google Scholar]
- (10).Chan DI; Vogel HJ Current understanding of fatty acid biosynthesis and the acyl carrier protein. Biochem. J 2010, 430, 1–19. [DOI] [PubMed] [Google Scholar]
- (11).He X; Agnihotri G; Liu H -w. Novel enzymatic mechanisms in carbohydrate metabolism. Chem. Rev 2000, 100, 4615–4661. [DOI] [PubMed] [Google Scholar]
- (12).He X; Liu H -w. Mechanisms of enzymatic C–O bond cleavages in deoxyhexose biosynthesis Curr. Opin. Chem. Biol 2002, 6, 590–597. [DOI] [PubMed] [Google Scholar]
- (13).Hong L; Zhao Z; Liu H.-w. Characterization of SpnQ from the spinosyn biosynthetic pathway of Saccharopolyspora spinosa: Mechanistic and evolutionary implications for C-3 deoxygenation in deoxysugar biosynthesis. J. Am. Chem. Soc 2006, 128, 14262–14263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lin C-I; McCarty RM; Liu H -w. The biosynthesis of nitrogen-, sulfur-, and high-carbon chain-containing sugars. Chem. Soc. Rev 2013, 42, 4377–4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Toraya T Cobalamin-dependent dehydratases and a deaminase: Radical catalysis and reactivating chaperones. Arch. Biochem. Biophys 2014, 544, 40–57. [DOI] [PubMed] [Google Scholar]
- (16).Semialjac M; Schwarz H Computational exploration of rearrangements related to the vitamin B12-dependent ethanolamine ammonia lyase catalyzed transformation. J. Am. Chem. Soc 2002, 124, 8974–8983. [DOI] [PubMed] [Google Scholar]
- (17).Wetmore SD; Smith DM; Bennett JT; Radom L Understanding the mechanism of action of B12-dependent ethanolamine ammonia-lyase: Synergistic interactions at play. J. Am. Chem. Soc 2002, 124, 14054–14065. [DOI] [PubMed] [Google Scholar]
- (18).Bridwell-Rabb J; Grell TAJ; Dreenan CL A rich man, poor man story of S-adenosylmethionine and cobalamin revisited. Annu. Rev. Biochem 2018, 87, 555–584. [DOI] [PubMed] [Google Scholar]
- (19).Levin BJ; Balskus EP Characterization of 1,2-propanediol dehydratases reveals distinct mechanisms for B12-dependent and glycyl radical enzymes. Biochemistry 2018, 57, 3222–3226. [DOI] [PubMed] [Google Scholar]
- (20).Stubbe J; van der Donk WA Ribonucleotide reductases: radical enzymes with suicidal tendencies. Chem. Biol 1995, 2, 793–801. [DOI] [PubMed] [Google Scholar]
- (21).Wei Y; Funk MA; Rosado LA; Baek J; Drennan CL; Stubbe J The class III ribonucleotide reductase from Neisseria bacilliformis can utilize thioredoxin as a reductant. Proc. Natl. Acad. Sci 2014, 111, E3756–E3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kovačević B; Barić D; Babić D; Bilić L; Hanževački M; Sandala GM; Radom L; Smith DM Computational tale of two enzymes: Glycerol dehydration with or without B12. J. Am. Chem. Soc 2018, 140, 8487–8496. [DOI] [PubMed] [Google Scholar]
- (23).Bridwell-Rabb J; Drennan CL; Vitamin B12 in the spotlight again. Curr. Opin. Chem. Biol 2017, 37, 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Buckel W; Martins BM; Messerschmidt A; Golding BT Radical-mediated dehydration reactions in anaerobic bacteria. Biol. Chem 2005, 386, 951–959. [DOI] [PubMed] [Google Scholar]
- (25).Buckel W Radical and electron recycling in catalysis. Angew. Chem. Int. Ed 2009, 48, 6779–6787. [DOI] [PubMed] [Google Scholar]
- (26).Kim HJ; LeVieux J; Yeh Y-C; Liu H -w. C3′-Deoxygenation of paromamine catalyzed by a radical S-adenosylmethionine enzyme: Characterization of the enzyme AprD4 and its reductase partner AprD3. Angew. Chem. Int. Ed 2016, 55, 3724–3728; Angew. Chem. 2016, 128, 3788–3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Lv M; Ji X; Zhao J; Li Y; Zhang C; Su L; Ding W; Deng Z; Yu Y; Zhang Q Characterization of a C3 deoxygenation pathway reveals a key branch point in aminoglycoside biosynthesis. J. Am. Chem. Soc 2016, 138, 6427–6435. [DOI] [PubMed] [Google Scholar]
- (28).Kudo F; Tokumitsu T; Eguchi T Substrate specificity of radical S-adenosyl-l-methionine dehydratase AprD4 and its partner reductase AprD3 in the C3’-deoxygenation of aminoglycoside antibiotics. J. Antibiot 2017, 70, 423–428. [DOI] [PubMed] [Google Scholar]
- (29).Szu P-H; Ruszczycky MW; Choi S.-h.; Yan F; Liu H.-w. Characterization and mechanistic studies of DesII: A radical S-Adenosyl-l-methionine enzyme involved in the biosynthesis of TDP-d-desosamine. J. Am. Chem. Soc 2009, 131, 14030–14042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ruszczycky MW; Choi S.-h.; Liu H.-w. Stoichiometry of the redox neutral deamination and oxidative dehydrogenation reactions catalyzed by the radical SAM enzyme DesII. J. Am. Chem. Soc 2010, 132, 2359–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ruszczycky MW; Choi S.-h.; Liu H.-w. EPR-kinetic isotope effect study of the mechanism of radical-mediated dehydrogenation of an alcohol by the radical SAM enzyme DesII. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 2088–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Wright GD Aminoglycoside-modifying enzymes. Curr. Opin. Microbiol 1999, 2, 499–503. [DOI] [PubMed] [Google Scholar]
- (33).Houghton JL; Green KD; Chen W; Garneau-Tsodikova S The Future of aminoglycosides: the end or renaissance? ChemBioChem 2010, 11, 880–902. [DOI] [PubMed] [Google Scholar]
- (34).Kharel MK; Basnet DB; Lee HC; Liou K; Woo JS; Kim B-G; Sohng JK Isolation and characterization of the tobramycin biosynthetic gene cluster from Streptomyces tenebrarius. FEMS Microbiol. Lett 2004, 230, 185–190. [DOI] [PubMed] [Google Scholar]
- (35).Koch KF; Davis FA; Rhoades JA Nebramycin: separation of the and identification of factors 4, 5, and 5′. J. Antibiot 1973, 26, 745–751. [DOI] [PubMed] [Google Scholar]
- (36).Mori T; Kyotani Y; Watanabe I; Oda T Chemical conversion of lividomycin A into lividomycin B. J. Antibiot 1972, 25, 149–150. [DOI] [PubMed] [Google Scholar]
- (37).Sofia HJ; Chen G; Hetzler BG; Reyes-Spindola JF; Miller NE Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001, 29, 1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38). https://www.uniprot.org/uniprot/Q2MFI7.
- (39).Broderick JB; Duffus BR; Duschene KS; Shepard EM Radical S‑adenosylmethionine enzymes. Chem. Rev 2014, 114, 4229–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Vey JL; Drennan CL Structural insights into radical generation by the radical SAM superfamily. Chem. Rev 2011, 111, 2487–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Young AP; Bandarian V TYW1: A radical SAM enzyme involved in the biosynthesis of wybutosine bases. Meth. Enzymol 2018, 606, 119–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Ruszczycky MW; Zhong A; Liu H -w. Following the electrons: Peculiarities in the catalytic cycles of radical SAM enzymes. Nat. Prod. Rep 2018, 35, 615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Liu W-Q; Amara P; Mouesca J-M; Ji X; Renoux O; Martin L; Zhang C; Zhang Q; Nicolet Y 1,2-Diol dehydration by the radical SAM enzyme AprD4: A matter of proton circulation and substrate flexibility. J. Am. Chem. Soc 2018, 140, 1365–1371. [DOI] [PubMed] [Google Scholar]
- (44).Duschene KS; Veneziano SE; Silver SC; Broderick JB Control of radical chemistry in the AdoMet radical enzymes. Curr. Opin. Chem. Biol 2009, 13, 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Wang B; Sun G; Anderson DR; Jia M; Previs S; Anderson VE Isotopologue distributions of peptide product ions by tandem mass spectrometry: quantitation of low levels of deuterium incorporation. Anal. Biochem 2007, 367, 40–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Greenberg WA; Priestley ES; Sears PS; Alper PB; Rosenbohm C; Hendrix M; Hung S-C; Wong C-H Design and synthesis of new aminoglycoside antibiotics containing neamine as an optimal core structure: Correlation of antibiotic activity with in vitro inhibition of translation. J. Am. Chem. Soc 1999, 121, 6527–6541. [Google Scholar]
- (47).Lin G-M; Choi S-H; Ruszczycky MW; Liu H -w. Mechanistic investigation of the radical S-adenosyl-l-methionine enzyme DesII using fluorinated analogues. J. Am. Chem. Soc 2015, 137, 4964–4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Yoshitomi M; Toshiaki N; Arata Y; Keiichi U Stereoselective synthesis of 3′-deoxy-3′-fluoroadenosine. Bull. Chem. Soc. Jpn 1989, 62, 2119–2120. [Google Scholar]
- (49).O’Hagan D Understanding organofluorine chemistry. An introduction to the C–F bond. Chem. Soc. Rev 2008, 37, 308–319. [DOI] [PubMed] [Google Scholar]
- (50).Lilla EA; Yokoyama K Carbon extension in peptidylnucleoside biosynthesis by radical SAM enzymes. Nat. Chem. Biol 2016, 12, 905–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Ko Y; Ruszczycky MW; Choi S-H; Liu H -w. Mechanistic studies of the radical S-adenosylmethionine enzyme DesII with TDP-d-fucose. Angew. Chem. Int. Ed 2015, 54, 860–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Ruszczycky MW; Liu H -w. Mechanistic enzymology of the radical SAM enzyme DesII. Isr. J. Chem 2015, 55, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.