

Abstract
Glycosaminoglycans (GAGs) participate in a broad range of physiological processes, and their structures are of interest to researchers in structural biology and medicine. Although they are abundant in tissues and extracellular matrices, their structural heterogeneity makes them challenging analytes. Mass spectrometry, and more specifically, tandem mass spectrometry, is particularly well suited for their analysis. Many tandem mass spectrometry techniques have been examined for their suitability towards the structural characterization of GAGs. Threshold activation methods such as collision induced dissociation (CID) produce mainly glycosidic cleavages and do not yield a broad range of structurally informative cross-ring fragments. Considerable research effort has been directed at finding other means of dissociating gas-phase GAG ions to produce more comprehensive structural information. Here, we compare the structural information of GAGs obtained by charge transfer dissociation (CTD) and electron detachment dissociation (EDD). EDD has previously been applied to GAGs and is known to produce both glycosidic and cross-ring cleavages in similar abundance. CTD has not previously been used to analyze GAGs but has been shown to produce abundant cross-ring cleavages and no sulfate loss when applied to another class of sulfated carbohydrates like algal polysaccharides. In contrast to EDD, which is restricted to FTICR mass spectrometers, CTD can be implemented on other platforms, such as ion trap mass spectrometers (ITMS). Here, we show the capability of CTD-ITMS to produce structurally significant details of the sites of modification in both heparan sulfate (HS) and chondroitin sulfate (CS) standards ranging in length from degree of polymerization (dp) 4 to dp6. EDD and CTD both yield more structural information than CID, and yield similar fractional abundances to one another for glycosidic fragments, cross-ring fragments and neutral losses.
Keywords: Glycosaminoglycan, carbohydrate, tandem mass spectrometry, MS/MS, ion activation
INTRODUCTION
Glycosaminoglycans (GAGs) are linear poly-sulfated carbohydrates that exhibit a number of important biological functions, principally through their interactions with proteins.1–3 Their structural characterization presents a challenge to the analytical community, due to their high variability in the sites of sulfo modification, degree of polymerization, and hexuronic acid stereochemistry, a result of their enzymatic, non-template driven biosynthesis.4–6 Identifying sites of sulfation is essential to structure-function studies, as it influences the GAG-protein specific binding relationship.7 Structural changes in GAGs also directly effects the function of the GAG chain. For example, the physiochemical properties of certain GAGs classes are responsible for the viscoelastic properties of cartilage.8 Of the four established GAG classes, heparin (Hp) and heparan sulfate (HS) are the most diverse.4 Hp/HS have a high variability in sites of O-sulfation, N-sulfation, N-acetylation and hexuronic acid stereochemistry. Another class of sulfated GAGs, chondroitin sulfate (CS) and dermatan sulfate (DS), also vary in the sites of O-sulfation and hexuronic acid stereochemistry. Due to the heterogeneity of GAGs in naturally occurring material, they are difficult to analyze by methods such as nuclear magnetic resonance (NMR) and X-Ray diffraction.9–11 Disaccharide analysis using LC-MS can be used for analysis from small tissue volumes; however, the extent of enzymolysis in disaccharide analysis limits the structural information available for linkage positions, higher-order patterns and sulfation patterns.12–13 Advances in mass spectrometry, specifically soft ionization by electrospray (ESI) in combination with advanced methods of ion activation, have led to a more detailed understanding of GAG structure and function.10–11, 14
Fourier transform ion cyclotron resonance mass spectrometry (FTICR-MS) is a powerful tool for GAG structural characterization due to its high resolution and mass accuracy.9, 15 FTICR-MS can perform collisionally-activated, photo-induced, and electron-based tandem mass spectrometry (MS/MS) methods, which have all been shown to productively dissociate GAGs.15–30 Due to the acidic nature of the carboxyl groups and sulfo modifications on GAG chains, they are most efficiently analyzed by negative mode ionization using ESI.25 Mass spectrometry measurement of molecular weight provides details regarding the chain length, number of sulfo modifications, and the number of N-acetyl groups present in a GAG chain, however more detailed analysis is needed to assign the location of sulfo- modifications and N-acetylated hexosamine residues. MS/MS methods are useful analytical tools for obtaining this information. There are multiple electron-based activation methods for GAG analysis, and some of the more useful ones include electron induced dissociation (EID), electron detachment dissociation (EDD), and negative electron transfer dissociation (NETD).21, 24–29, 31–36 More recently ultraviolet photodissociation (UVPD) has been shown as a useful MS/MS tool for the analysis of GAGs.37
EDD has been extensively studied as a tool for the structural analysis of GAGs.17–18, 21, 24–29 EDD is accomplished by precursor selection and trapping of multiply-charged negative ions in the analyzer cell of an FTICR mass spectrometer, and bombarding these precursor ions with electrons of a moderate kinetic energy (19 eV). Fragmentation occurs via radical species produced by electron detachment, or from electronically excited even-electron precursor ions. The combination of these two pathways produces rich and complex mass spectra whose analysis provides considerable structural detail.25–26 A schematic representation of the EDD process is shown in Figure S1. EDD has been shown to produce both glycosidic and cross-ring fragmentation. Glycosidic cleavage products are used to assign the degree of sulfo modification and acetylation that is present in each monosaccharide, whereas cross-ring fragmentation is used to identify the sites of modification within a sugar residue.25–26, 29 EDD has also been shown to minimize sulfate decomposition compared to low-energy or threshold activation methods such as infrared multiphoton dissociation (IRMPD) and collision induced dissociation (CID).23, 25–27, 31
In the mid 2000s, Schlathölter’s group studied the gas-phase interactions between small biological ions and fast (1–200 keV) reagent ions, including H+, He+, He2+, O5+ and Xen+.38–40 Zubarev’s group enabled products of cation-cation reactions to be monitored in a 2D ion trap, 41 and the Jackson group extended the approach to other biological oligomers and negatively charged ions.42–47 The technique, now termed charge transfer dissociation (CTD), typically uses He+ cations and has some similarities to helium metastable atom-activated dissociation (MAD).42, 48–51 CTD exposes gas-phase precursor cations or anions to a beam of helium cations with kinetic energies in the range of 3–10 keV. Helium has a well-defined ionization energy (24.6 eV), which greatly exceeds the electron affinity of any organic molecule. The high kinetic energy provides sufficient energy to overcome any Coulombic barriers between the precursors, such as with cation-cation reactions, and the high electron affinity of helium cations provides excess energy for fragmentation.38–40, 47 The helium cations abstract electrons (charge transfer) from the precursor ions and cause the oxidized product ions to decompose via high-energy, radical-driven pathways. Recently, CTD was used to analyze sulfated oligosaccharides in both positive and negative ion modes.44–45 CTD is able to generate a- and x-ions from singly charged peptide cations, and cross-ring cleavages of oligosaccharides, which are typically not produced by CID.43–46 A schematic representation of the CTD process is shown in Figure S2. Here, we report the first application of CTD to the analysis of glycosaminoglycans. This work is motivated by the unique capabilities of CTD for fragmentation via radical pathways, and the potential to yield useful analytical information for the characterization of this challenging class of biomolecules. Moreover, the capability for deployment in ion trap mass spectrometers raises the potential to analyze GAGs using more widely available instrumentation compared to FTICR mass spectrometers that have been frequently used for GAG analysis to date.
EXPERIMENTAL
Preparation of Synthetic Heparan Sulfate Oligosaccharides
Heparan sulfate (HS) tetrasaccharide standards were prepared by chemical synthesis using a modular approach and further purified by silica gel column chromatography.18 Structures were confirmed by 1H NMR and accurate mass measurement by FTICR-MS.18
Preparation of Chondroitin Sulfate Oligosaccharides
Chondroitin sulfate A (CS-A) and dermatan sulfate (DS) oligosaccharides were prepared by partial enzymatic depolymerization from bovine trachea chondroitin sulfate A (Celsus laboratories, Cincinnati, OH, USA) and porcine intestinal mucosa dermatan sulfate (Celsus Laboratories) respectively. Full explanations of enzymatic depolymerization and desulfation have been reported previously.32, 36
Mass Spectrometry
0.1 mg/mL of each GAG standard was ionized by nanospray ESI with a spray voltage of 1.2 kV (pulled glass tip model Econo12-N; New Objective, Woburn, MA). All standards were analyzed in negative ion mode.
EDD spectra for all samples were collected on a 9.4T Bruker solariX XR Fourier transform ion cyclotron resonance mass spectrometer (Bremen, Germany) with a hollow cathode (HeatWave, Wasonville, CA, USA), which serves as the source of electrons for EDD. Multiply charged precursor ions were isolated in the quadrupole and accumulated for 2–3 s before injection into the dynamically harmonized FTICR cell. Ions were then irradiated for 1 s with 19 eV electrons. The cathode heater was set to 1.5 A and the extraction lens was set to 18.6 ± 0.4 V. 1 M points were acquired for each spectrum and 48 acquisitions were averaged for each stored spectrum. Internal calibration was performed using confidently assigned glycosidic cleavage product ions as internal calibrants.
CTD and CID experiments were performed on a modified Bruker AmaZon 3D ion trap (Bremen, Germany), with a custom vacuum chamber cover. A saddle field fast ion gun (VSW/ Atomtech, Macclesfield, UK) was installed on top of the ring electrode. The saddle field fast ion source was used as the helium cation source, which is described in detail elsewhere.42 Multiply charged precursor ions were activated with 7.5–8 keV helium cations for 100 ms for CTD. Helium gas flow in the ion gun was controlled via a variable leak valve and measured by the ion trap gauge; the readout was approximately 9.5 ×106 mbar. Precursor ions were accumulated for 20–60 ms in the ion trap before isolation. Each stored CTD spectrum was the average of 5 replicate scans, and the presented spectra are typically the result of 1.5–2 mins of spectral averaging. Internal calibration was performed using confidently assigned glycosidic cleavage product ions as internal calibrants.
For the experiments involving CTD with resonance ejection, precursor ions were first isolated and irradiated with helium cations to achieve CTD at the MS2 level. Any unreacted precursor ions were then resonantly ejected at the MS3 level using on-resonance excitation with an amplitude >3 V. In the absence of CTD at the MS2 level and the presence of the resonance ejection at the MS3 level, no fragment ions were observed, thereby verifying the absence of unwanted CID peaks during resonance ejection.
Product ions for CID, CTD and EDD were assigned using Glycoworkbench version 2.0, and in-house GAG analysis software.52–53 All product ions are reported using the annotation scheme proposed by Wolff and Amster, which was derived from the Domon and Costello nomenclature.26, 54 Fragmentation is illustrated in the molecular structure drawings using dashed lines through the structure and hash marks at the end to indicate the fragment ion. Open circles at the end of hash marks represent -SO3 loss, whereas a closed circle represents multiple -SO3 losses. Donut plots display percentages of the summed ion intensity of fragment ion types (glycosidic fragment, cross-ring fragment, glycosidic fragment with -SO3 loss, cross-ring fragment with -SO3 loss).
RESULTS AND DISCUSSION
Well-characterized standards were examined to test the capability of CTD to analyze GAGs. CTD results are compared to that of CID and EDD for a series of heparan sulfate tetrasaccharides, dermatan sulfate tetrasaccharide and hexasaccharide, and chondroitin sulfate A tetrasaccharide. Prior work in our group has demonstrated the capability of EDD on an FTICR to provide a complete set of fragment ions that can be used to assign the structural features of GAG samples. EDD fragmentation was enhanced using highly ionized precursor ions to retain labile sulfate half-ester modifications.14, 26, 55 For the data presented here, we have selected the optimal precursor charge state (as determined by EDD) to ensure complete deprotonation of sulfate groups, and selected precursors that lie outside the CTD chemical background range (i.e. >m/z 350). CTD has previously been shown to produce no sulfate decomposition for sulfo-peptides and sulfo-lipids, and minimal neutral loss for sulfo-oligosaccharides.43, 45 The previous data therefore suggested that CTD should be an effective ion activation method for GAG samples. The symbol nomenclature used for glycans is displayed in Figure S3.
Compound 1- HS Tetrasaccharide IdoA-GlcNAc-IdoA-GlcNAc6S-(CH2)5NH2
The MS/MS spectrum in Figure 1 shows the CTD results for the [M-2H]2- precursor (m/z 470.39) of the synthetic HS monosulfated tetrasaccharide IdoA-GlcNAc-IdoA-GlcNAc6S-(CH2)5NH2. For comparison, the EDD spectrum of the same precursor is presented in Figure S4. Both CTD and EDD produce a series of glycosidic fragments ions; however, EDD yields both reducing end and non-reducing end fragments for all the glycosidic cleavages. I.e. all the B, C, Y and Z ions are observed (Figures S4 and S5). Both CTD and EDD also produce copious cross-ring cleavages. EDD produced a 0,2A4 ion, which confirms the location of N-acetylation on the reducing-end glucosamine. Once the N-acetylation is confirmed on the reducing terminus, the presence of C3/Z1 fragment ions infer that the sulfate is located at the 6-O position.56–57 Though these fragment ions alone do not rule out the possibility of 3-O sulfation, the observation of N-acetylation rules out 3-O sulfation, which occurs only on GlcNS residues.57 CTD produced 2,4A4 and C3 fragments, which taken together confirm the presence of both N-Acetylation and 6-O sulfation. EDD produced more fragments than CTD on the uronic acid closest to the reducing end. Both EDD and CTD produced 1,5X2 and 3,5X2 ions, which together confirm the presence of N-acetylation on the glucosamine closest to the non-reducing end. Table S1 contains a list of identified fragment ions from CTD.
Figure 1.
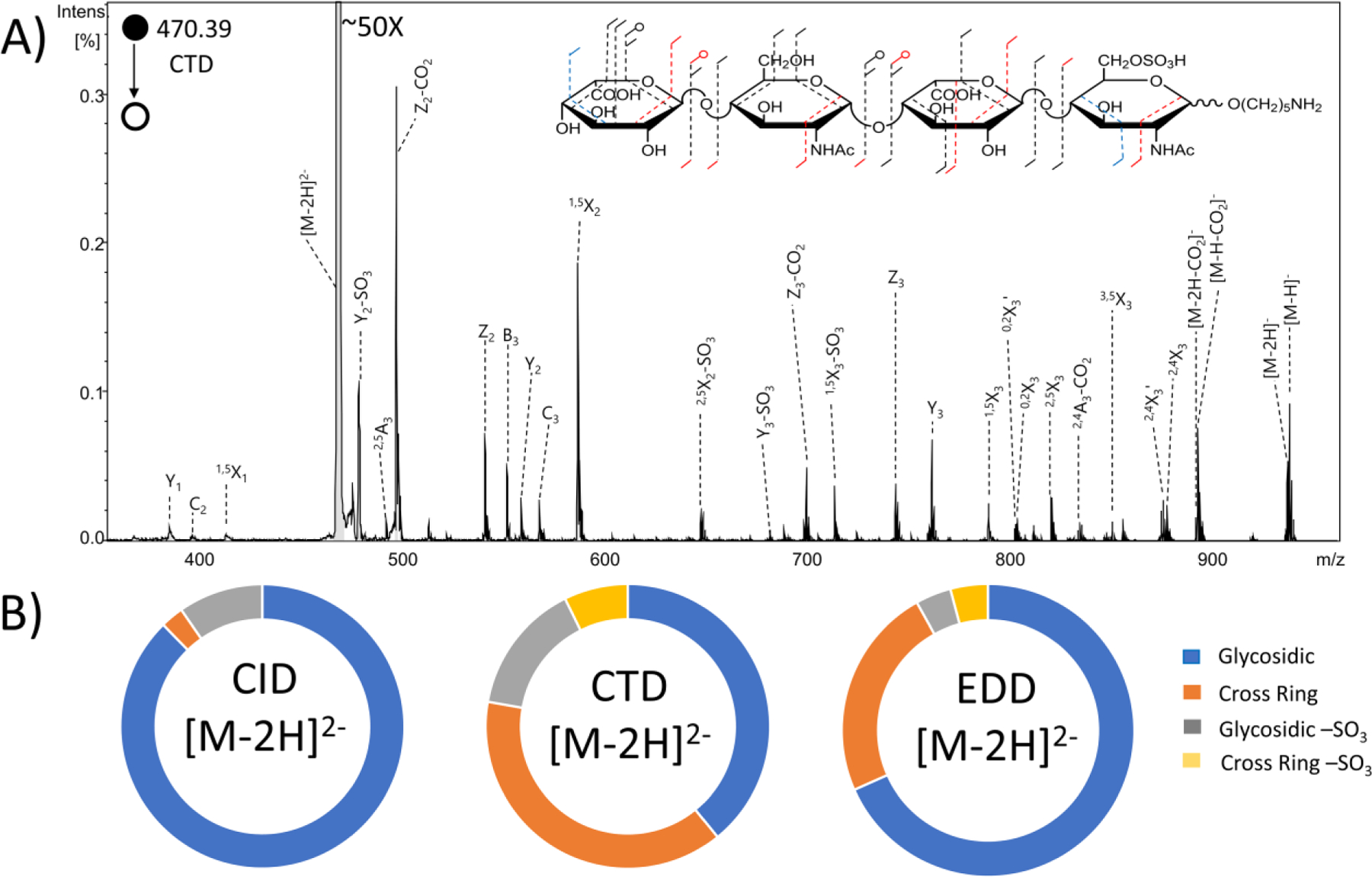
(A) CTD spectrum of the [M-2H]2− precursor of synthetic heparan sulfate tetramer standard IdoA-GlcNAc-IdoA-GlcNAc6S-O(CH2)5NH2. Molecular structure inset represents fragmentation seen from both CTD and EDD (black), CTD only (blue) and EDD only (red). (B) Fragment type intensity distribution for glycosidic, cross-ring, glycosidic sulfate loss and cross-ring sulfate loss fragments are shown.
The intensities of glycosidic, cross-ring and sulfate-loss fragment ions are compared for CID, CTD, and EDD in the donut plots shown in Figure 1B. EDD produced a lower abundance of cross-ring fragments (23.6%) and lower abundance of neutral sulfate losses (8.1%) than CTD (38.7% and 22.2%, respectively). MS/MS spectra of the same precursor using CID is also shown in Figure S4. The spectrum is dominated by glycosidic cleavages and neutral losses that provide ambiguous information about the locations of the different functional groups.
Compound 2- HS Tetrasaccharide GlcA-GlcNAc-GlcA2S-GlcNAc-(CH2)5NH2
The MS/MS spectrum in Figure 2 shows the CTD results for the [M-2H]2- precursor (m/z 469.14) of the synthetically produced HS monosulfated tetrasaccharide GlcA-GlcNAc-GlcA2S-GlcNAc-(CH2)5NH2. CID, CTD and EDD each produce fragments at every glycosidic bond (Figures S6 and S7). CTD and EDD produced more cross-ring cleavages than CID, as expected for a precursor with a protonated site. Past work investigating the effect of ionization state on CID fragmentation of GAGs showed that few informative fragments are seen with precursors that are not fully ionized.22 For this sample, CTD produced a greater number of cross-ring cleavages than EDD, specifically on the N-acetylglucosamine nearest to the non-reducing end. Neither EDD nor CTD produced cross-ring cleavages on the reducing end N-acetylglucosamine. However, both EDD and CTD produced B3 and C3 fragments, which confirm the presence of the N-acetylation.
Figure 2.
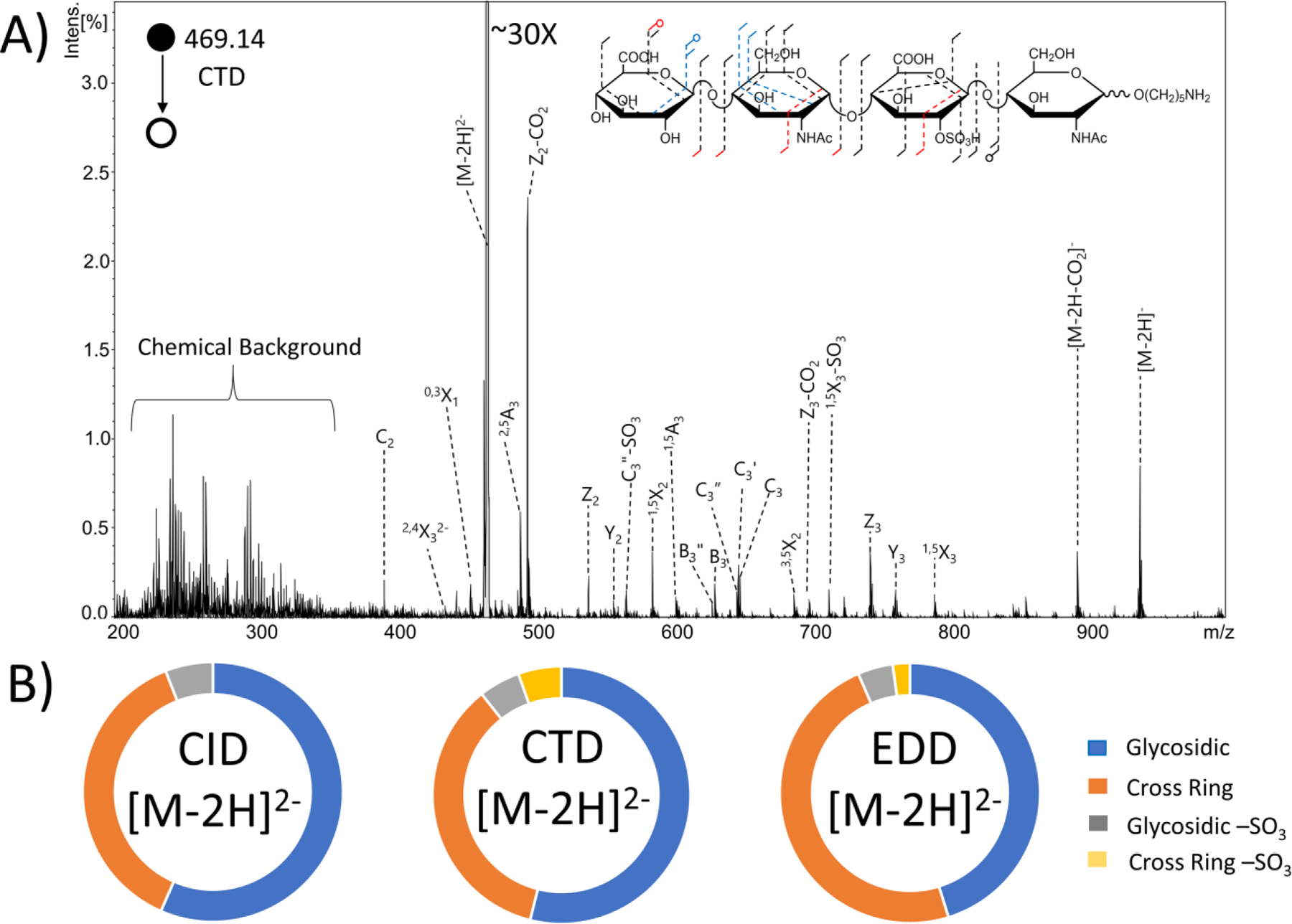
(A) CTD spectrum of the [M-2H]2− precursor of synthetic heparan sulfate tetramer standard GlcA-GlcNAc-GlcA2S-GlcNAc-O(CH2)5NH2. Molecular structure inset represents fragmentation seen from both CTD and EDD (black), CTD only (blue) and EDD only (red). (B) Fragment type distribution for glycosidic, cross-ring, glycosidic sulfate loss and cross-ring sulfate loss fragments are shown.
On the non-reducing end N-acetylglucosamine, EDD produced a 0,2A2 fragment ion, which confirms the presence of the N-acetyl group. CTD produced 1,5X2 and 2,4X2 fragment ions, which confirm the presence of the N-acetyl group. For the glucuronic acid closest to the reducing end, EDD produced a 0,2A3 fragment, which confirms 2-O sulfation, and CTD produced 1,5A3 and 2,5A3 fragments, which also confirm 2-O sulfation for this residue. Incidentally, the biosynthesis of GAGs restricts sulfo-modification in uronic acid to the 2-O position, so only glycosidic cleavage is necessary to make this assignment.57 When comparing the overall intensity of fragment ions (Figure 2B), EDD produced a greater abundance of cross-ring fragments than CTD (48.3% and 35.5%); EDD also produced less-abundant sulfate decomposition in both glycosidic and cross-ring fragment ions (4.3% and 2.1% for EDD compared to 5.2% and 5.4% for CTD). Table S2 contains a list of identified fragment ions from CTD.
Compound 3- HS Tetrasaccharide GlcA-GlcNS6S-GlcA-GlcNS6S-(CH2)5NH2
The CID, CTD and EDD results for the [M-4H+Na]3- precursor (m/z 372.45) of the synthetic HS tetrasulfated tetrasaccharide GlcA-GlcNS6S-GlcA-GlcNS6S-(CH2)5NH2 are shown in Figure S8. EDD was able to break every glycosidic bond, however CTD was not able to break the central glycosidic bond without sulfate decomposition. Figure 3 shows fragment maps with the cleavages produced by CID, CTD and EDD and donut plots showing the intensity distributions of the principal fragment types. CTD produced a 0,2A2 cleavage, which confirms the location of the N-sulfation on the non-reducing end proximal glucosamine, however CTD did not yield any more cross-ring cleavages in this sugar residue to confirm 6-O sulfation. The Y3 fragment, in combination with the 0,2A2 fragment, can be used to determine the presence of a sulfate in this residue on either the 6-O or the 3-O position; 6-O sulfation is more likely, but 3-O sulfation cannot be disregarded.56 EDD also produced a 0,2A2 cleavage on the non-reducing end glucosamine, which confirms the presence of the N-sulfation, and a 1,5X2 fragment, which confirms an additional sulfate modification. However, like for CTD, the location of sulfation cannot be determined. Neither EDD nor CTD produced any informative cross-ring cleavages on the reducing end glucosamine. CTD and EDD yielded glycosidic fragment ions, which confirm the presence of two sulfates on the reducing end amino sugar. EDD produced a greater abundance of cross-ring fragments (26.7%) compared to either CID (2.1%) or CTD (5.7%) (Figure 3B). Table S3 contains a list of identified fragment ions from CTD. Previous work investigating the effect of the ionization state of GAGs on EDD fragmentation efficiency showed superior EDD efficiencies when at least one of the sulfate groups retains a proton or cation and is uncharged.26 When the GAG is not fully deprotonated, sulfate decomposition is minimized while maintaining informative fragment ions. The precursor ion chosen for these experiments results in four of the six ionizable sites ionized. A precursor ion with five ionized sites would be ideal for this sample, however the m/z value of the [M-5H]5– precursor would have fallen in the region of the CTD spectrum with chemical background (e.g. < m/z 350), and so this precursor was not selected. Na-H exchange is a valuable alternative to achieve higher levels of deprotonation. For this work, only Na+ adducts that were present within the original sample were used, and no additional Na+ was added. For future work, additional Na-H exchange precursors will be tested. Additionally, the chemical background could be minimized with an oil-free vacuum system and altering the beam conditions. Although the chemical background was minimized in Figure S2, the low mass cut-off during this experiment was approximately m/z 250, so the trapping efficiency for ions near or below this value was negligible. The high degree of sulfo loss and the lower abundance of informative cross-ring fragment ions could be attributable to the dissociation behavior of the less-than optimal precursor ion.
Figure 3.
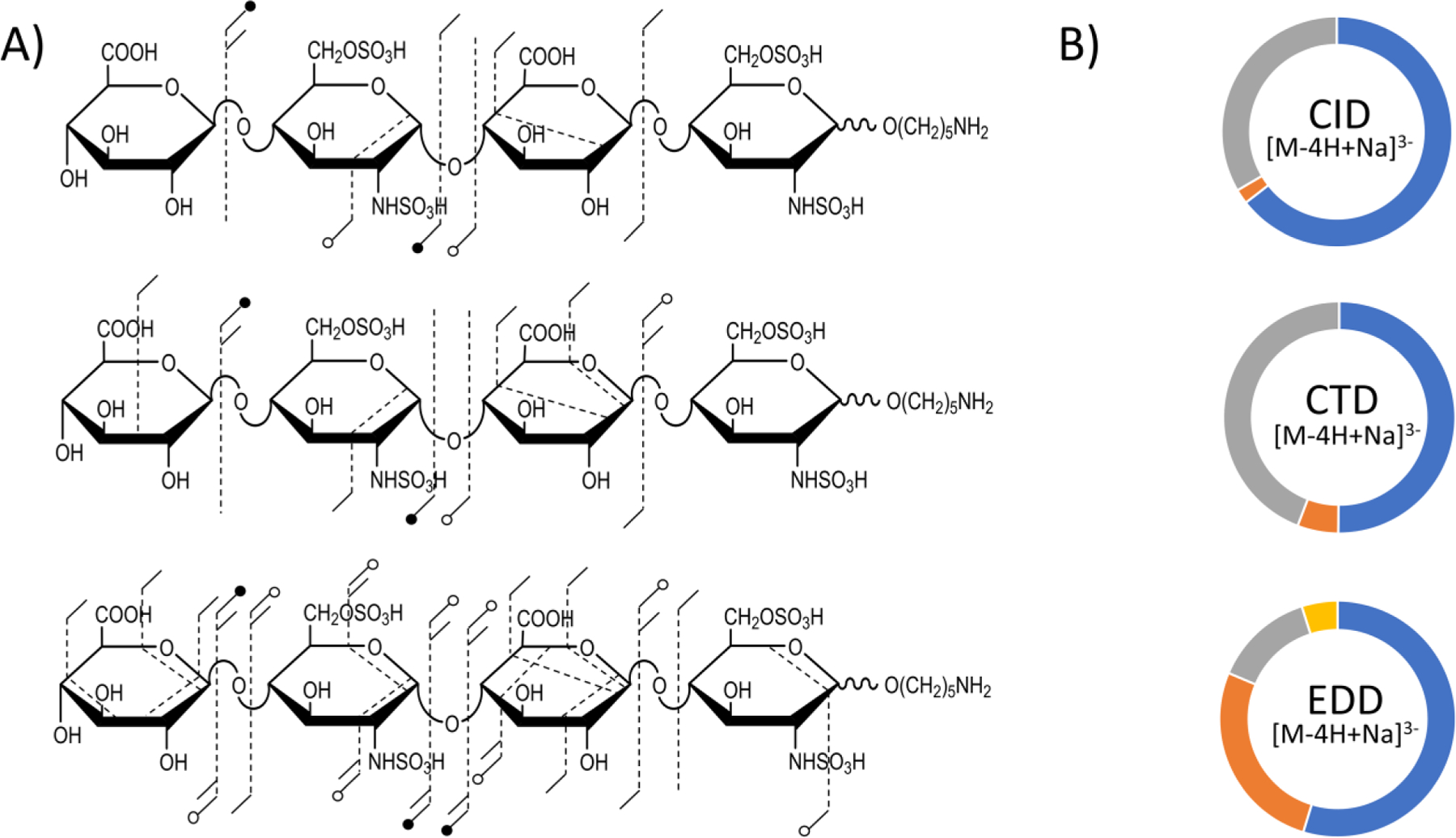
(A) Annotated molecular structures of the [M-4H+Na]3− of heparan sulfate tetramer standard GlcA-GlcNS6S-GlcA-GlcNS6S-O(CH2)5NH2. Fragment maps with results from CID, CTD and EDD are shown, respectively. (B) Fragment type distribution for glycosidic (blue), cross-ring (orange), glycosidic sulfate loss (gray) and cross-ring sulfate loss (yellow) fragments are shown.
Compound 4- HS Tetrasaccharide IdoA2S-GlcNS6S-IdoA-GlcNAc6S-(CH2)5NH2
The MS/MS spectra in Figure 4 show the CTD (top) and EDD (bottom) results for the [M-3H]3- precursor (m/z 378.47) of the synthetic HS tetrasulfated tetrasaccharide IdoA2S-GlcNS6S-IdoA-GlcNAc6S-(CH2)5NH2. Structural annotations for EDD and CTD are presented as well. CTD produced a 1,4A4 fragment ion, which confirms 6-O sulfation on the reducing end amino sugar. EDD produced a 3,5A4 cleavage, which also confirms this sulfation position. Both CTD and EDD produced Z2/Y2 and Z3/Y3 glycosidic cleavages, which confirm the presence of two sulfate modifications on the glucosamine residue closest to the non-reducing end. However, EDD produced no informative cross-ring cleavages on this glucosamine to confirm sulfate positions. In contrast, CTD produced a 1,4X2 cleavage on the non-reducing end glucosamine, which confirms sulfation at the 6-O position. CTD produced no additional cross-ring cleavages to confirm the location of the second sulfate on the glucosamine. However, since CTD confirmed the presence of 6-O sulfation on the non-reducing end amino sugar, one can infer that the other sulfate is on the N position, especially because 3-O sulfation requires N-sulfation to be present.57
Figure 4.
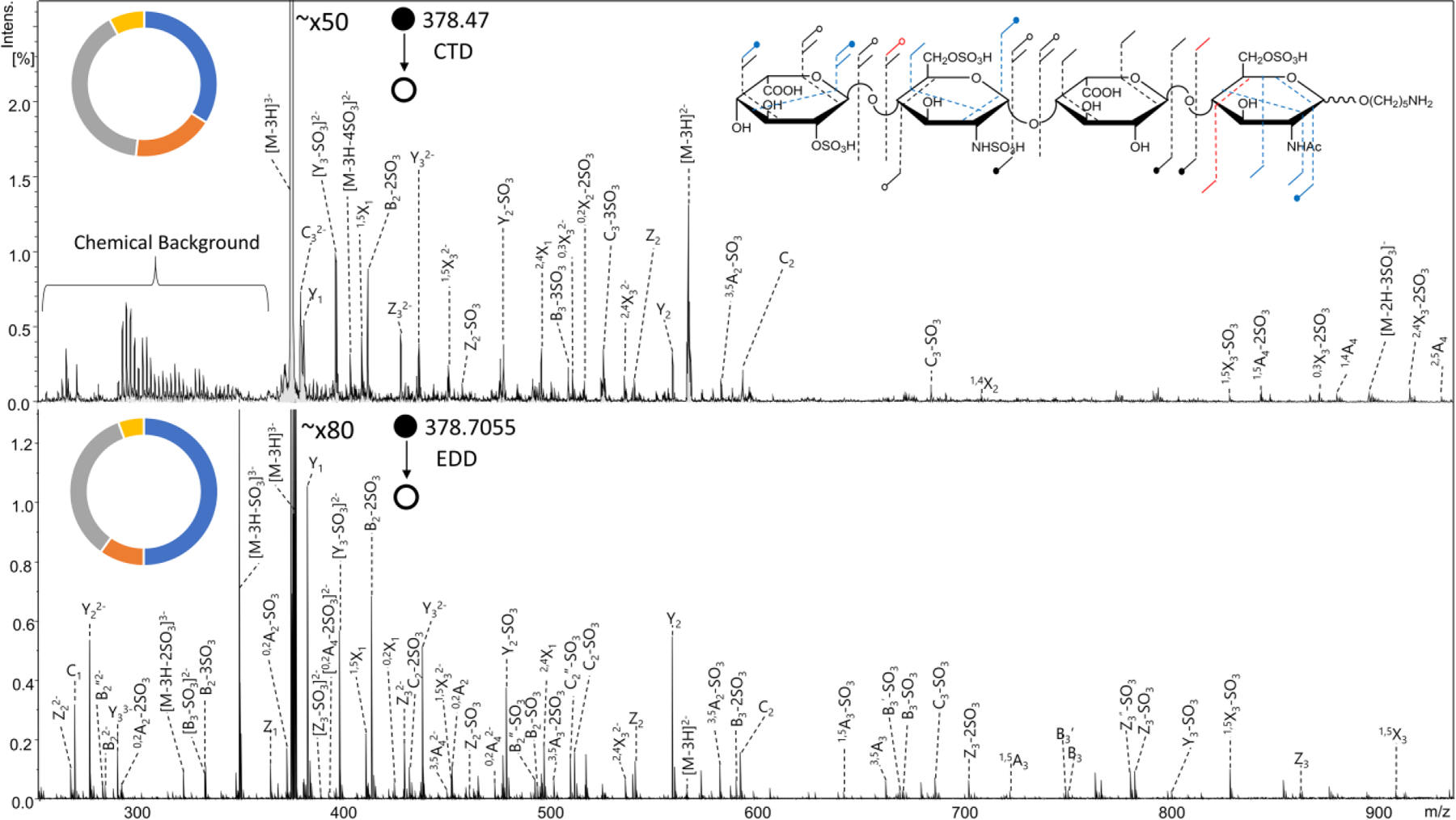
MS/MS spectra of the [M-3H]3- precursor of synthetic heparan sulfate tetramer standard IdoA2S-GlcNS6S-IdoA-GlcNAc6S-O(CH2)5NH2. (top) CTD MS/MS spectrum (bottom) EDD MS/MS spectrum. Molecular structure inset represents fragmentation seen from both CTD and EDD (black), CTD only (blue) and EDD only (red). Fragment type distribution for glycosidic (blue), cross-ring (orange), glycosidic sulfate loss (gray) and cross-ring sulfate loss (yellow) fragment ions are shown.
The final sulfate modification is located on the non-reducing end uronic acid. The known biosynthetic pathway requires that sulfation on uronic acid is at the 2-O position, and only CTD confirmed this location of the sulfate modification by producing a 0,3X3 ion. EDD and CTD both produced 1,5X3 and 2,4X3 fragment ions, which, when combined, confirm the 2-O location for the sulfate modification on the uronic acid. CTD produced slightly more abundant cross-ring fragments than EDD (17.9% and 9.8%, respectively); however, both CTD and EDD produced a notable abundance of ions containing sulfate decomposition, specifically from glycosidic cleavages. Sulfate decomposition accounted for 48.1% of assigned fragments for CTD and 40.2% for EDD. Table S4 contains a list of identified fragment ions from CTD. The abundance of sulfate loss can possibly be attributed to the precursor selection, which has one less ionized site than the number of sulfates on the GAG standard. As stated previously, the ideal precursor for minimizing sulfate decomposition has one more ionized site than the number of sulfate modifications on a GAG.26 Precursor selection was limited by the chemical background of the CTD experiments, which, when the lower m/z limit is set to m/z 250, tends to maximize between m/z 250–300 and tail off towards m/z 350. As mentioned previously, precursors with Na-H exchange can substitute for higher charge states without entering the m/z range of the chemical background. For this preliminary comparison between EDD and CTD, no additional Na+ was added to the GAG samples, so the presented results might not necessarily reflect the best-case scenarios for structural elucidation using EDD or CTD.
Compound 5- DS Tetrasaccharide ΔHexA-GalNAc4S-IdoA-GalNAc4S-OH
Dermatan sulfate (DS) tetrasaccharide is disulfated and is known to have 4-O sulfation on both N-acetyl galactosamines. MS/MS spectra from CID, CTD and EDD, as well as annotated molecular structures and donut charts, are shown in Figures S9 and S10. Unfortunately, neither EDD nor CTD produce cross-ring fragments that can distinguish 4-O from 6-O sulfation. However, both CTD and EDD produced B3 and C3 fragment ions to confirm the presence of a sulfate modification on the reducing end amino sugar. EDD yielded B2 and Y3 fragment ions, which also confirms the presence of a sulfate modification on the non-reducing end amino sugar. CTD produced glycosidic fragment ions breaking the central glycosidic bond (Z2, Y2, C2), but no glycosidic fragments to distinguish the location of the sulfate modification from the non-reducing end amino sugar and uronic acid. Table S5 contains a list of identified fragment ions from CTD.
The lack of cross-ring fragmentation on the amino sugars has been reported previously when using EDD and other radical-based ion activation techniques.28, 36–37 The proposed mechanism by Wolff et al. suggests that a radical site first forms on the uronic acid carboxylate, either at the initiation of electron detachment or from hydrogen rearrangement. The change in linkage from β−1,4 in HS standards to β−1,3 in CS/DS standards appears to be a major factor in the differences in fragmentation between the two GAG subclasses. Whereas the neutral losses of SO3 from CTD fragments of highly sulfated algal carrageenans appear to be favored by certain positions,39 the sulfate losses in HS, CS and DS seem to be less predictable and multiple sulfate losses are observed in both CTD and EDD.
An added benefit to using an ion trap instrument for CTD experiments is the ability to perform resonance ejection to remove unwanted ions from the trap. This can lead to an extension of the dynamic range of the ion trap instrument.58 In this case, any residual precursor ions remaining after the CTD reaction were resonantly ejected before the mass acquisition scan to prevent space charge effects—such as peak broadening—on fragment ions with lower m/z values than the precursor. By ejecting the unreacted precursor ions, the previously suppressed fragment ions became well resolved peaks that could be assigned to specific fragments. A comparison of CTD of the [M-2H]2- precursor (m/z 458.46) of DS tetramer with and without resonance ejection is shown in Figure 5. There was an overall increase in the abundance and number of fragment ions produced; however, no new cross-ring cleavages on either amino sugar were observed. Resonance ejection CTD produced detectable fragments corresponding to Y3 and Z3 fragment ions, possibly because of a trace amount of resonance excitation (CID) during resonance ejection.59 When combined with the Y2, Z2 and C2 ions from CTD activation, these data confirm a sulfate modification on the non-reducing end amino sugar and not the non-reducing end uronic acid. CTD with resonance ejection yielded a handful of more informative fragmentation, but at a cost of a slightly higher degree of sulfate decomposition (17.8% compared to 16.2% for CTD without resonance ejection) (Figure 5C). As mentioned previously, the chemical background of CTD is an ongoing issue requiring improvement in future work.
Figure 5.
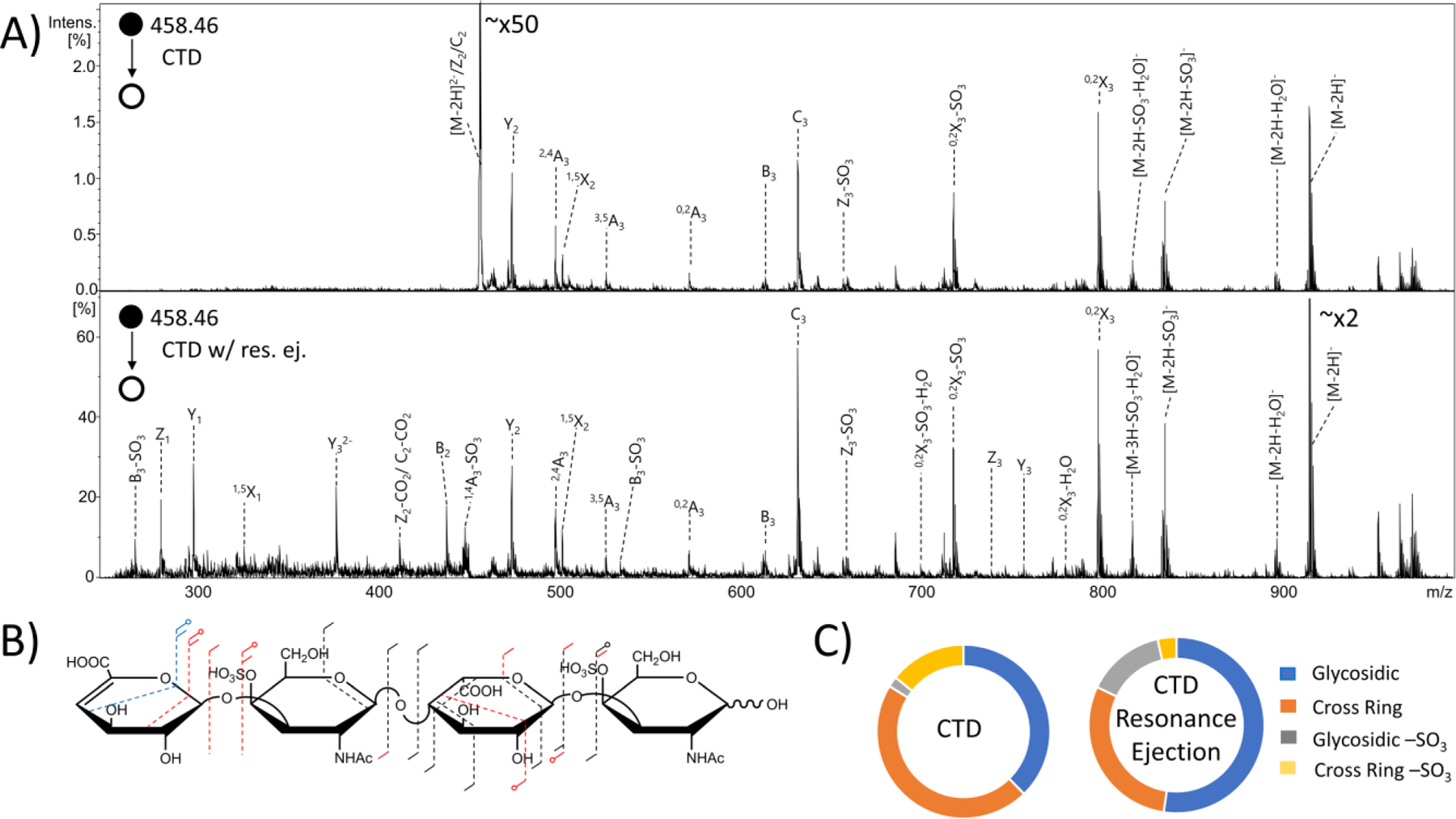
MS/MS spectra of the [M-2H]2- precursor of dermatan sulfate tetramer standard ΔHexA-GalNAc4S-IdoA-GalNAc4S-OH. (A) (top) CTD MS/MS spectrum (bottom) CTD MS/MS spectrum with resonance ejection. (B) Molecular structure inset represents fragmentation seen from both CTD and CTD with resonance ejection (black), CTD only (blue) and CTD with resonance ejection only (red). (C) Fragment type distribution for glycosidic, cross-ring, glycosidic sulfate loss and cross-ring sulfate loss fragment ions are shown.
Compound 6- DS Hexasaccharide ΔHexA-GalNAc4S-IdoA-GalNAc4S-IdoA-GalNAc4S-OH
DS hexasaccharide contains three sulfate modifications, all at the 4-O position on the three amino sugars. MS/MS results from CID, CTD and EDD experiments of the [M-3H]3- precursor (m/z 458.65), as well as annotated molecular structures and donut charts, are shown in Figures S11 and S12. Similar to the DS tetramer results, neither EDD nor CTD produced the necessary cross-ring cleavages to confirm the location of the sulfate modifications at the 4-O position on the amino sugars. EDD produced all necessary glycosidic cleavages to confirm that the sulfate modifications are on the amino sugars. CTD produced the necessary glycosidic cleavages to confirm that two of the sulfates are on amino sugars; however, like the DS tetramer, CTD did not produce the necessary glycosidic cleavages to determine if the final sulfate is on the non-reducing end amino sugar or the non-reducing end uronic acid. Table S6 contains a list of identified fragment ions from CTD. For hexameric oligosaccharides, in comparison to the tetramers examined above, the increase in the number of possible fragmentation peaks leads to spectral congestion in the region above m/z 800. As a result of the lower resolving power of the ion trap mass spectrometer, a number of lower abundance ions were unable to be assigned in this region of the mass spectrum. By comparison, the EDD-FTICR measurements on the same sample provide well-resolved peaks that are easily assigned.
CTD with resonance ejection was also performed on this compound. Results comparing CTD with resonance ejection and CTD without resonance ejection are shown in Figure 6. Resonance ejection CTD produced a Z5 fragment ion, which confirms the final sulfate modification is located on the amino sugar. Although resonance ejection hardly changes the relative abundance of fragments with m/z values above the precursor m/z value, CTD with resonance ejection provides additional peaks below the m/z of the precursor and changes the overall distribution of fragment types. For example, CTD with resonance ejection produced a greater percentage of glycosidic cleavage intensity, but less cross-ring cleavage intensity than CTD without resonance ejection, as shown in Figure 6C. The overall intensity of sulfo loss peaks increased slightly with resonance ejection CTD (13.4% with resonance ejection versus 11.2% without).
Figure 6.
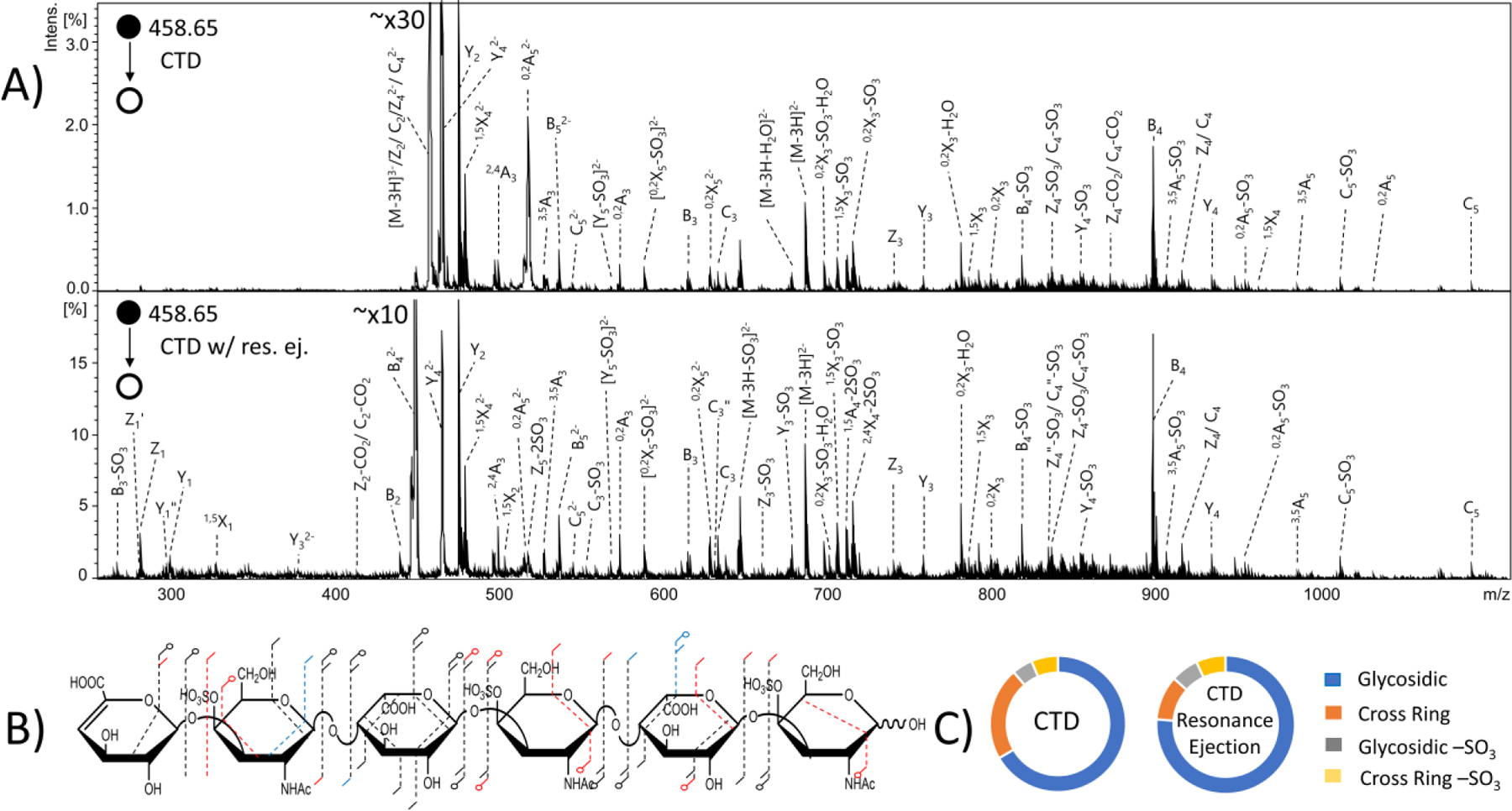
MS/MS spectra of the [M-3H]3- precursor of dermatan sulfate hexamer standard ΔHexA-GalNAc4S-IdoA-GalNAc4S-IdoA-GalNAc4S-OH. (A) (top) CTD MS/MS spectrum (bottom) CTD MS/MS spectrum with resonance ejection. (B) Molecular structure inset represents fragmentation seen from both CTD and CTD with resonance ejection (black), CTD only (blue) and CTD with resonance ejection only (red). (C) Fragment type distribution for glycosidic, cross-ring, glycosidic sulfate loss and cross-ring sulfate loss fragment ions are shown.
Compound 7- CS-A Hexasaccharide ΔHexA-GalNAc4S-GlcA-GalNAc4S-GlcA-GalNAc4S-OH
Chondroitin sulfate A (CS-A) hexasaccharide has three sulfate modifications at the 4-O position of the amino sugar residues. MS/MS data from CID, CTD and EDD experiments of the [M-3H]3- precursor (m/z 458.42) and annotated molecular structures are shown in Figures S13 and S14. Similar to the DS samples, the majority of the cross-ring fragmentation of CS-A was on the uronic acid residues, and no cross-ring fragments could distinguish 6-O and 4-O sulfation by either EDD or CTD. EDD produced all the necessary glycosidic cleavages to confirm sulfate modifications on the amino sugar residues and eliminated the possibility of sulfation on the uronic acid residues. CTD produced the necessary glycosidic cleavages to confirm sulfate modifications on the reducing end amino sugar and central amino sugar. CTD did not produce any glycosidic fragments to distinguish the location of the remaining sulfate modification which could be on either the non-reducing end uronic acid or the adjacent amino sugar. Table S7 contains a list of identified fragment ions from CTD.
CTD with resonance ejection was performed on this compound (Figure S15). CTD with resonance ejection yielded the glycosidic cleavages necessary (Y5 and Z5) to determine that the final sulfate is located on the non-reducing end amino sugar. When comparing the overall fragment ion intensity between CTD, CID, EDD and CTD with resonance ejection for this compound (Figure 7), CTD and EDD produced a similar intensity of cross-ring fragment ions, whereas CID and resonance ejection CTD both produced a significantly lower intensity of the cross-ring fragment ions but a much higher intensity of glycosidic cleavages. CTD with resonance ejection produced a much lower intensity of sulfate decomposition than CTD without resonance ejection.
Figure 7.
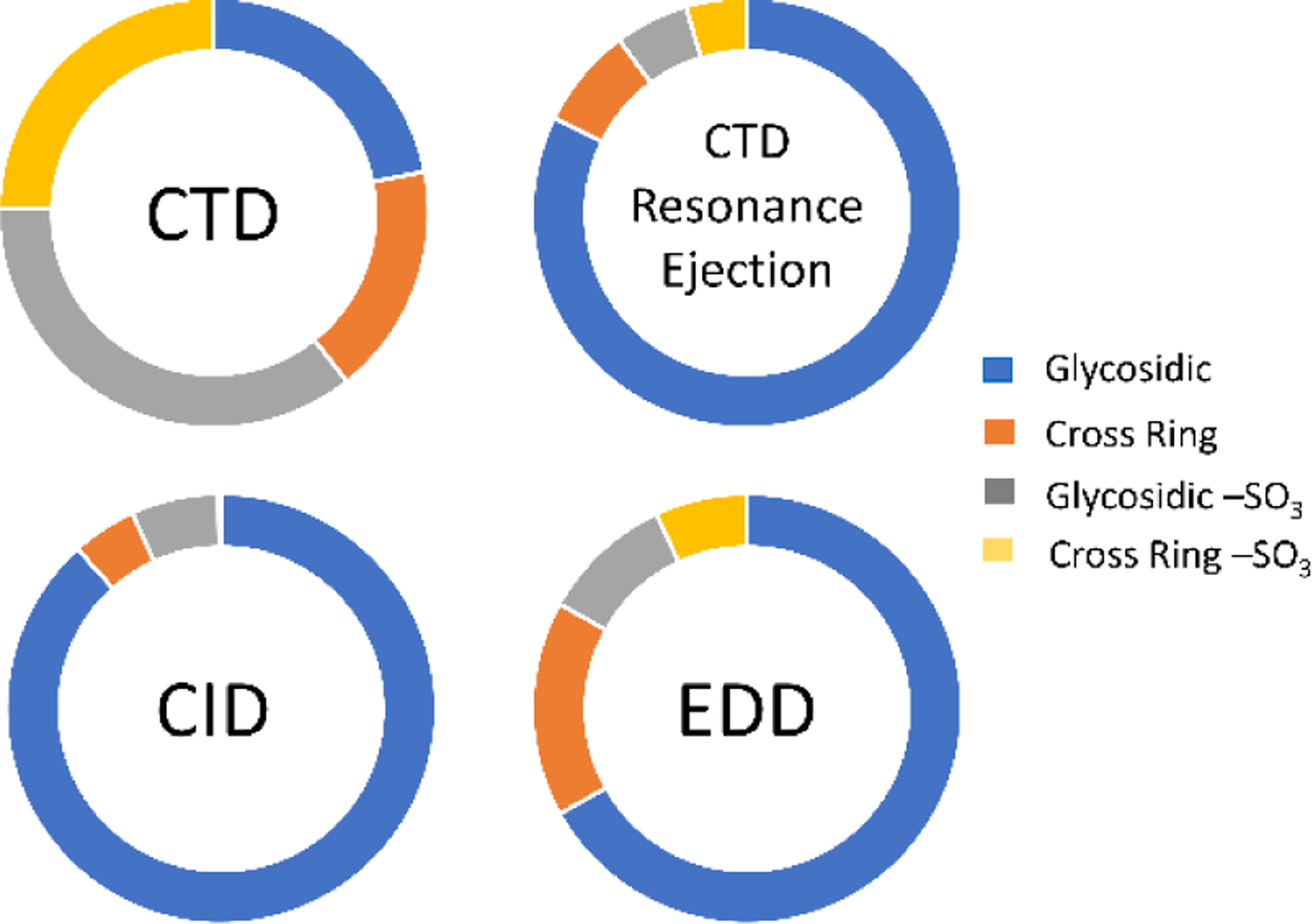
MS/MS results of the [M-3H]3- precursor of chondroitin sulfate hexamer standard ΔHexA-GalNAc4S-GlcA-GalNAc4S-GlcA-GalNAc4S-OH. Fragment type distribution for glycosidic, cross-ring, glycosidic sulfate loss and cross-ring sulfate loss fragment ions are shown.
CS-A and DS are isomers, differing in stereochemistry at the C5 position of the uronic acid. CS-A contains glucuronic acid whereas DS contains iduronic acid. Extensive work has been done to investigate the differences produced from MS/MS of these isomers.32, 60–61 In general, several selected fragment ions exhibit intensity differences that correlate with glucuronic acid versus iduronic acid. The selected precursor ion was significant in yielding these diagnostic differences. Although CTD did not provide differences in intensity of the fragment ions previously shown to be diagnostic in EDD,32, 60–61 CTD of CS-A had a significantly lower intensity for B4 fragment ions than DS, as shown in Figure 8 for the [M-3H]3- precursor (m/z 458). Therefore, the stereochemical differences between CS-A and DS provide readily distinguishable spectral differences. Significantly more work would be necessary to elucidate whether or not CTD provides any general trends between spectral and stereochemical differences.
Figure 8.
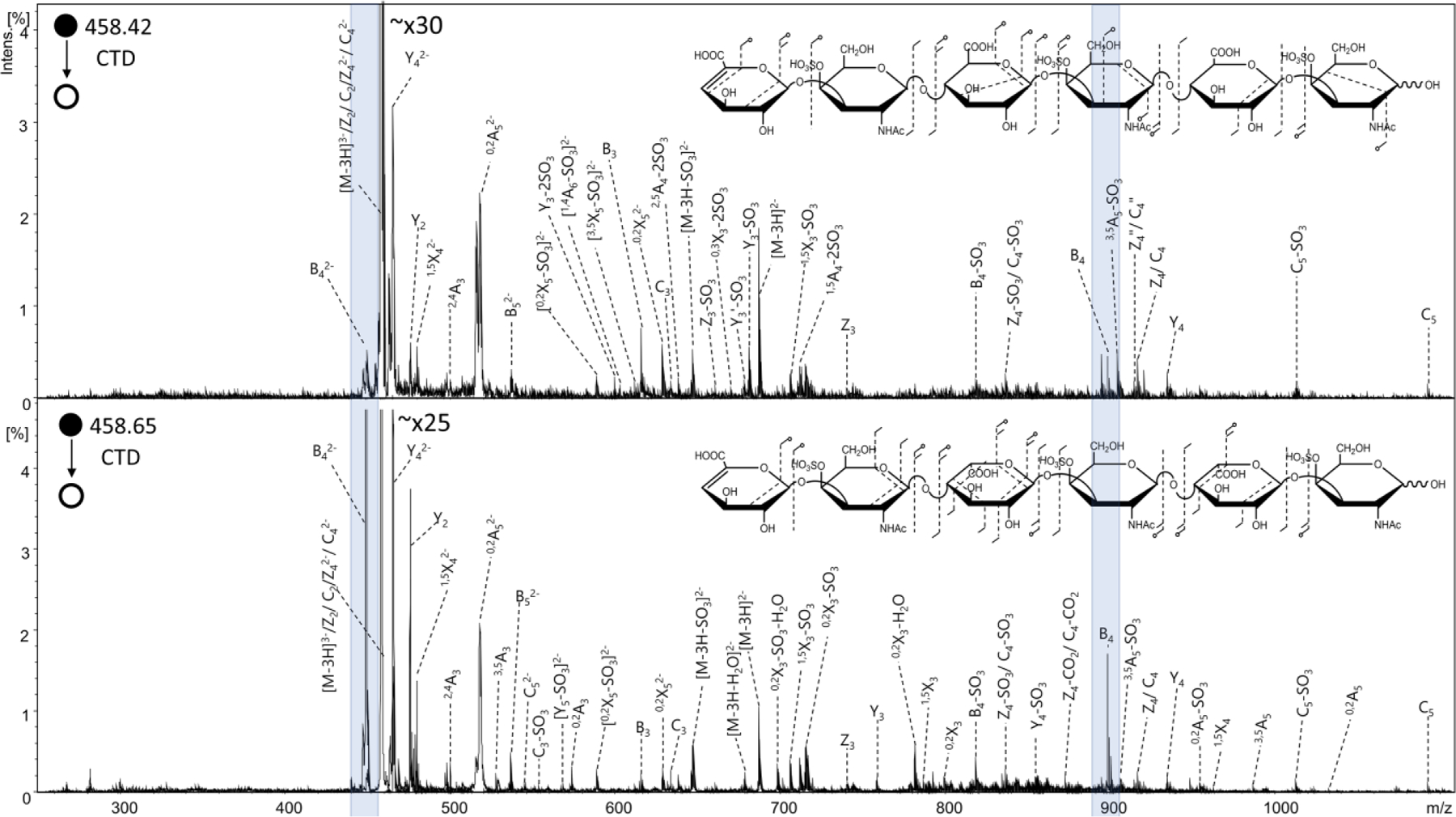
CTD spectra of the [M-3H]3- precursor of (top) chondroitin sulfate hexamer ΔHexA-GalNAc4S-GlcAA-GalNAc4S-GlcA-GalNAc4S-OH and (bottom) dermatan sulfate hexamer standard ΔHexA-GalNAc4S-IdoA-GalNAc4S-IdoA-GalNAc4S-OH. Molecular structure inset represents fragmentation seen from CTD MS/MS results of chondroitin sulfate (top) and dermatan sulfate (bottom). Blue highlights indicate the B4 fragment ions shown to have a higher intensity for dermatan sulfate than chondroitin sulfate.
CONCLUSIONS
The data shows that charge transfer dissociation (CTD) is a powerful tool for sequencing GAG polysaccharides with minimal sulfate decomposition. CTD uses a 100 ms activation time with 1.5–2 min of signal averaging, making it more efficient to collect a data set for CTD than EDD. For CS, DS and HS standards investigated here, CTD performance was comparable to that of EDD, and both methods were able to establish the location of sulfate modifications from cross-ring cleavages. For CS/DS standards, CTD and EDD both fell short of being able to assign the site of sulfation as 4-O versus 6-O in the amino sugar residues.
In addition to a reduction in cross-ring fragmentation, CTD produces fewer product ions than EDD, however the products formed by CTD in general yield the same structural information. For additional information, CTD can be paired with resonance ejection to observe lower-abundance product ions. CTD as implemented here produces a chemical background which interferes with the observation of fragments in the low m/z region (i.e. m/z 200–350). This results in loss of information in this mass range; however, this was overcome in the resonance ejection experiments, which did yield fragment ions in the low m/z region. The necessity of a properly ionized precursor ion was shown for highly sulfated GAG standards, where both EDD and CTD are susceptible to sulfate decomposition from the activation of less-than-optimal precursor species. The extent of deprotonation of the precursors can be overcome by using solution-phase Na-H exchange to ionize acidic sites. For this preliminary test of CTD for GAG analysis, no additional Na+ was added to the GAG standards. Further studies to investigate the ability for sodiated precursors to produce a higher abundance of informative cross-ring fragments will be performed.
Using an ion trap instrument presents an additional difficulty for GAG analysis. The low resolution of ion trap spectrometers increases the difficulty in confidently assigning fragment ions. In many cases multiple fragment ions can fall within a few Da of each other, thereby creating the need for high resolution MS in combination with CTD. CTD has been achieved with linear ion trap mass spectrometers,41–42 so coupling with an Orbitrap should be a relatively trivial exercise. CTD with high resolution MS would alleviate the resolution discrepancies between CTD and EDD in the present manuscript. Overall, the product ions produced by CTD of GAG standards does yield informative structure information for characterizing GAGs, and presents an additional approach for the analysis of this challenging compound class.
Supplementary Material
Supporting Information
Figure S1, Schematic of EDD; Figure S2, Schematic of CTD; Figure S3, Symbol nomenclature for glycans; Figure S4, CID, CTD and EDD spectra of compound IdoA-GlcNAc-IdoA-GlcNAc6S-(CH2)5NH2; Figure S5, donut plots and fragment maps of compound IdoA-GlcNAc-IdoA-GlcNAc6S-(CH2)5NH2; Figure S6, CID, CTD and EDD spectra of compound GlcA-GlcNAc-GlcA2S-GlcNAc-(CH2)5NH2; Figure S7, donut plots and fragment maps of compound GlcA-GlcNAc-GlcA2S-GlcNAc-(CH2)5NH2; Figure S8 CID, CTD and EDD spectra of compound GlcA-GlcNS6S-GlcA-GlcNS6S-(CH2)5NH2; Figure S9 CID, CTD and EDD spectra of compound ΔHexA-GalNAc4S-IdoA-GalNAc4S-OH; Figure S10, donut plots and fragment maps of compound ΔHexA-GalNAc4S-IdoA-GalNAc4S-OH; Figure S11 CID, CTD and EDD spectra of compound ΔHexA-GalNAc4S-IdoA-GalNAc4S-IdoA-GalNAc4s-OH; Figure S12, donut plots and fragment maps of compound ΔHexA-GalNAc4S-IdoA-GalNAc4S-IdoA-GalNAc4S-OH; Figure S13 CID, CTD and EDD spectra of compound ΔHexA-GalNAc4S-GlcA-GalNAc4S-GlcAA-GalNAc4S-OH; Figure S14, fragment maps of compound ΔHexA-GalNAc4S-GlcA-GalNAc4S-GlcA-GalNAc4S-OH; Figure S15 CTD and CTD with resonance ejection spectra of compound ΔHexA-GalNAc4S-GlcA-GalNAc4S-GlcA-GalNAc4S-OH; Tables S1- S7, lists of identified fragment ions from CTD for all GAG standards.
ACKNOWLEDGEMENTS
LEP and IJA gratefully acknowledge financial support from the National Institutes of Health, P41GM103390 and U01CA231074. GPJ acknowledges financial support from the National Science Foundation (NSF) under Grant No. CHE-1710376 and the National Institutes of Health (NIH) under Grant No. 1R01GM114494–01. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the NSF or NIH. The authors would like to acknowledge Geert- Jan Boons (Complex Carbohydrate Research Center, University of Georgia) for providing synthetic heparan sulfate samples that were utilized during this study, as well as Robert Linhardt and Fuming Zhang (Rensselaer Polytechnic Institute) for providing CS-A and DS samples that were utilized during this study.
Footnotes
The authors declare no competing financial interest.
Contributor Information
Lauren E. Pepi, Department of Chemistry, University of Georgia, Athens, Georgia, 30602, United States
Zachary J. Sasiene, C. Eugene Bennett Department of Chemistry, West Virginia University, Morgantown, West Virginia, 26506, United States.
Praneeth M. Mendis, C. Eugene Bennett Department of Chemistry, West Virginia University, Morgantown, West Virginia, 26506, United States
Glen P. Jackson, C. Eugene Bennett Department of Chemistry and Department of Forensic and Investigative Science, West Virginia University, Morgantown, West Virginia, 26506, United States.
I. Jonathan Amster, Department of Chemistry, University of Georgia, Athens, Georgia, 30602, United States.
REFERENCES
- (1).Clegg DO; Reda DJ; Harris CL; Klein MA; O’Dell JR; Hooper MM; Bradley JD; Bingham CO; Weisman MH; Jackson CG; Lane NE; Cush JJ; Moreland LW; Schumacher HR; Oddis CV; Wolfe F; Molitor JA; Yocum DE; Schnitzer TJ; Furst DE; Sawitzke AD; Shi H; Brandt KD; Moskowitz RW; Williams HJ, Glucosamine, Chondroitin Sulfate, and the Two in Combination for Painful Knee Osteoarthritis. New Engl J Med 2006, 354, 795–808. [DOI] [PubMed] [Google Scholar]
- (2).Sugahara K; Kitagawa H, Recent advances in the study of the biosynthesis and functions of sulfated glycosaminoglycans. Curr Opin Struct Biol 2000, 10, 518–527. [DOI] [PubMed] [Google Scholar]
- (3).Watanabe H; Yamada Y; Kimata K, Roles of Aggrecan, a Large Chondroitin Sulfate Proteoglycan, in Cartilage Structure and Function. J Biochem 1998, 124, 687–693. [DOI] [PubMed] [Google Scholar]
- (4).Esko JD; Lindahl U, Molecular diversity of heparan sulfate. J Clin Invest 2001, 108, 169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Varki A; Cummings RD; Esko JD; Freeze HH; Stanley P; Bertozzi CR; Hart GW; Etzler ME, Essentials of Glycobiology. NY, 2009; Vol. 2. [PubMed] [Google Scholar]
- (6).Ly M; Laremore TN; Linhardt RJ, Proteoglycomics: Recent Progress and Future Challenges. Omics 2010, 14, 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhao Y; Singh A; Li L; Linhardt RJ; Xu Y; Liu J; Woods RJ; Amster IJ, Investigating changes in the gas-phase conformation of Antithrombin III upon binding of Arixtra using traveling wave ion mobility spectrometry (TWIMS). Analyst 2015, 140, 6980–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sugahara K; Mikami T; Uyama T; Mizuguchi S; Nomura K; Kitagawa H, Recent advances in the structural biology of chondroitin sulfate and dermatan sulfate. Curr Opin Struct Biol 2003, 13, 612–620. [DOI] [PubMed] [Google Scholar]
- (9).Zhou W; Hakansson K, Structural Characterization of Carbohydrates by Fourier Transform Tandem Mass Spectrometry. Curr Proteomics 2011, 8, 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zaia J, Glycosaminoglycan glycomics using mass spectrometry. Mol Cell Proteomics 2013, 12, 885–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Staples GO; Zaia J, Analysis of Glycosaminoglycans Using Mass Spectrometry. Curr Proteomics 2011, 8, 325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Turiák L; Tóth G; Ozohanics O; Révész Á; Ács A; Vékey K; Zaia J; Drahos L, Sensitive method for glycosaminoglycan analysis of tissue sections. Journal of Chromatography A 2018, 1544, 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Sun X; Li L; Overdier KH; Ammons LA; Douglas IS; Burlew CC; Zhang F; Schmidt EP; Chi L; Linhardt RJ, Analysis of total human urinary glycosaminoglycan disaccharides by liquid chromatography–tandem mass spectrometry. Anal Chem 2015, 87, 6220–6227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zaia J, Principles of mass spectrometry of glycosaminoglycans. J Biomacromol Mass Spectrom 2005, 1, 3–36. [Google Scholar]
- (15).Amster IJ, Fourier Transform Mass Spectrometry. J Mass Spectrom 1996, 31, 1325–1337. [Google Scholar]
- (16).Agyekum I; Pepi L; Yu Y; Li J; Yan L; Linhardt RJ; Chen S; Amster IJ, Structural elucidation of fucosylated chondroitin sulfates from sea cucumber using FTICR-MS/MS. Eur J Mass Spectrom 2017, 24, 157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Agyekum I; Patel AB; Zong C; Boons G-J; Amster IJ, Assignment of hexuronic acid stereochemistry in synthetic heparan sulfate tetrasaccharides with 2-O-sulfo uronic acids using electron detachment dissociation. Int J Mass Spectrom 2015, 390, 163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Agyekum I; Zong C; Boons G-J; Amster IJ, Single Stage Tandem Mass Spectrometry Assignment of the C-5 Uronic Acid Stereochemistry in Heparan Sulfate Tetrasaccharides using Electron Detachment Dissociation. J. Am Soc Mass Spectrom 2017, 28, 1741–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Chi L; Wolff JJ; Laremore TN; Restaino OF; Xie J; Schiraldi C; Toida T; Amster IJ; Linhardt RJ, Structural Analysis of Bikunin Glycosaminoglycan. J Am Chem Soc 2008, 130, 2617–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Laremore TN; Leach FE; Solakyildirim K; Amster IJ; Linhardt RJ, Glycosaminoglycan Characterization by Electrospray Ionization Mass Spectrometry Including Fourier Transform Mass Spectrometry. Method Enzymol 2010, 478, 79–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Leach FE 3rd; Arungundram S; Al-Mafraji K; Venot A; Boons GJ; Amster IJ, Electron Detachment Dissociation of Synthetic Heparan Sulfate Glycosaminoglycan Tetrasaccharides Varying in Degree of Sulfation and Hexuronic Acid Stereochemistry. Int J Mass Spectrom 2012, 330, 152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kailemia MJ; Li L; Ly M; Linhardt RJ; Amster IJ, Complete Mass Spectral Characterization of a Synthetic Ultralow-Molecular-Weight Heparin Using Collision-Induced Dissociation. Anal Chem 2012, 84, 5475–5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kailemia MJ; Ruhaak LR; Lebrilla CB; Amster IJ, Oligosaccharide analysis by mass spectrometry: a review of recent developments. Anal Chem 2014, 86, 196–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Leach FE; Xiao Z; Laremore TN; Linhardt RJ; Amster IJ, Electron detachment dissociation and infrared multiphoton dissociation of heparin tetrasaccharides. Int J Mass Spectrom 2011, 308, 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wolff JJ; Amster IJ; Chi L; Linhardt RJ, Electron detachment dissociation of glycosaminoglycan tetrasaccharides. J Am Soc Mass Spectrom 2007, 18, 234–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wolff JJ; Laremore TN; Busch AM; Linhardt RJ; Amster IJ, Influence of charge state and sodium cationization on the electron detachment dissociation and infrared multiphoton dissociation of glycosaminoglycan oligosaccharides. J Am Soc Mass Spectrom 2008, 19, 790–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wolff JJ; Laremore TN; Leach FE; Linhardt RJ; Amster IJ, Electron Capture Dissociation, Electron Detachment Dissociation and Infrared Multiphoton Dissociation of Sucrose Octasulfate. Eur J Mass Spectrom 2009, 15, 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wolff JJ; Laremore TN; Busch AM; Linhardt RJ; Amster IJ, Electron detachment dissociation of dermatan sulfate oligosaccharides. J Am Soc Mass Spectrom 2008, 19, 294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wolff JJ; Chi L; Linhardt RJ; Amster IJ, Distinguishing glucuronic from iduronic acid in glycosaminoglycan tetrasaccharides by using electron detachment dissociation. Anal Chem 2007, 79, 2015–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Yu Y; Duan J; Leach FE; Toida T; Higashi K; Zhang H; Zhang F; Amster IJ; Linhardt RJ, Sequencing the Dermatan Sulfate Chain of Decorin. J Am Chem Soc 2017, 139, 16986–16995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wolff JJ; Laremore TN; Aslam H; Linhardt RJ; Amster IJ, Electron-induced dissociation of glycosaminoglycan tetrasaccharides. J Am Soc Mass Spectrom 2008, 19, 1449–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Leach FE; Ly M; Laremore TN; Wolff JJ; Perlow J; Linhardt RJ; Amster IJ, Hexuronic Acid Stereochemistry Determination in Chondroitin Sulfate Glycosaminoglycan Oligosaccharides by Electron Detachment Dissociation. J. Am Soc Mass Spectrom 2012, 23, 1488–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Leach FE; Wolff JJ; Laremore TN; Linhardt RJ; Amster IJ, Evaluation of the experimental parameters which control electron detachment dissociation, and their effect on the fragmentation efficiency of glycosaminoglycan carbohydrates. Int J Mass Spectrom 2008, 276, 110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wolff JJ; Leach FE; Laremore TN; Kaplan DA; Easterling ML; Linhardt RJ; Amster IJ, Negative electron transfer dissociation of glycosaminoglycans. Anal Chem 2010, 82, 3460–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Leach FE; Wolff JJ; Xiao Z; Ly M; Laremore TN; Arungundram S; Al-Mafraji K; Venot A; Boons G-J; Linhardt RJ; Amster IJ, Negative electron transfer dissociation Fourier transform mass spectrometry of glycosaminoglycan carbohydrates. Eur J Mass Spectrom 2011, 17, 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Leach FE; Riley NM; Westphall MS; Coon JJ; Amster IJ, Negative Electron Transfer Dissociation Sequencing of Increasingly Sulfated Glycosaminoglycan Oligosaccharides on an Orbitrap Mass Spectrometer. J. Am Soc Mass Spectrom 2017, 28, 1844–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Klein DR; Leach FE; Amster IJ; Brodbelt JS, Structural Characterization of Glycosaminoglycan Carbohydrates Using Ultraviolet Photodissociation. Anal Chem 2019, 91, 6019–6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Bari S; Sobocinski P; Postma J; Alvarado F; Hoekstra R; Bernigaud V; Manil B; Rangama J; Huber B; Schlathölter T, Fragmentation of α-and β-alanine molecules by ions at Bragg-peak energies. The Journal of Chemical Physics 2008, 128, 02B623. [DOI] [PubMed] [Google Scholar]
- (39).Bari S; Hoekstra R; Schlathölter T, Peptide fragmentation by keV ion-induced dissociation. Physical Chemistry Chemical Physics 2010, 12, 3376–3383. [DOI] [PubMed] [Google Scholar]
- (40).Maclot S; Capron M; Maisonny R; Ławicki A; Méry A; Rangama J; Chesnel JY; Bari S; Hoekstra R; Schlathölter T, Ion‐Induced Fragmentation of Amino Acids: Effect of the Environment. ChemPhysChem 2011, 12, 930–936. [DOI] [PubMed] [Google Scholar]
- (41).Chingin K; Makarov A; Denisov E; Rebrov O; Zubarev RA, Fragmentation of Positively-Charged Biological Ions Activated with a Beam of High-Energy Cations. Anal Chem 2014, 86, 372–379. [DOI] [PubMed] [Google Scholar]
- (42).Hoffmann WD; Jackson GP, Charge Transfer Dissociation (CTD) Mass Spectrometry of Peptide Cations Using Kiloelectronvolt Helium Cations. J. Am Soc Mass Spectrom 2014, 25, 1939–1943. [DOI] [PubMed] [Google Scholar]
- (43).Li P; Jackson GP, Charge Transfer Dissociation (CTD) Mass Spectrometry of Peptide Cations: Study of Charge State Effects and Side-Chain Losses. J. Am Soc Mass Spectrom 2017, 28, 1271–1281. [DOI] [PubMed] [Google Scholar]
- (44).Ropartz D; Li P; Fanuel M; Giuliani A; Rogniaux H; Jackson GP, Charge Transfer Dissociation of Complex Oligosaccharides: Comparison with Collision-Induced Dissociation and Extreme Ultraviolet Dissociative Photoionization. J. Am Soc Mass Spectrom 2016, 27, 1614–1619. [DOI] [PubMed] [Google Scholar]
- (45).Ropartz D; Li P; Jackson GP; Rogniaux H, Negative Polarity Helium Charge Transfer Dissociation Tandem Mass Spectrometry: Radical-Initiated Fragmentation of Complex Polysulfated Anions. Anal Chem 2017, 89, 3824–3828. [DOI] [PubMed] [Google Scholar]
- (46).Li P; Kreft I; Jackson GP, Top-Down Charge Transfer Dissociation (CTD) of Gas-Phase Insulin: Evidence of a One-Step, Two-Electron Oxidation Mechanism. J. Am Soc Mass Spectrom 2018, 29, 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Li P; Jackson GP, Charge transfer dissociation of phosphocholines: gas-phase ion/ion reactions between helium cations and phospholipid cations. J Mass Spectrom 2017, 52, 271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Li P; Hoffmann WD; Jackson GP, Multistage Mass Spectrometry of Phospholipids using Collision-Induced Dissociation (CID) and Metastable Atom-Activated Dissociation (MAD). Int J Mass Spectrom 2016, 403, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Berkout VD; Doroshenko VM, Fragmentation of phosphorylated and singly charged peptide ions via interaction with metastable atoms. Int J Mass Spectrom 2008, 278, 150–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Cook SL; Collin OL; Jackson GP, Metastable atom‐activated dissociation mass spectrometry: leucine/isoleucine differentiation and ring cleavage of proline residues. J Mass Spectrom 2009, 44, 1211–1223. [DOI] [PubMed] [Google Scholar]
- (51).Cook SL; Jackson GP, Metastable atom-activated dissociation mass spectrometry of phosphorylated and sulfonated peptides in negative ion mode. J. Am Soc Mass Spectrom 2011, 22, 1088–1099. [DOI] [PubMed] [Google Scholar]
- (52).Ceroni A; Maass K; Geyer H; Geyer R; Dell A; Haslam SM, GlycoWorkbench: a tool for the computer-assisted annotation of mass spectra of glycans. J Proteome Res 2008, 7, 1650–1659. [DOI] [PubMed] [Google Scholar]
- (53).Duan J; Jonathan Amster I, An Automated, High-Throughput Method for Interpreting the Tandem Mass Spectra of Glycosaminoglycans. J. Am Soc Mass Spectrom 2018, 29, 1802–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Domon B; Costello CE, A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconjugate J 1988, 5, 397–409. [Google Scholar]
- (55).Zaia J; Costello CE, Tandem Mass Spectrometry of Sulfated Heparin-Like Glycosaminoglycan Oligosaccharides. Anal Chem 2003, 75, 2445–2455. [DOI] [PubMed] [Google Scholar]
- (56).Thacker BE; Xu D; Lawrence R; Esko JD, Heparan sulfate 3-O-sulfation: a rare modification in search of a function. Matrix Biol 2014, 35, 60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Rabenstein DL, Heparin and heparan sulfate: structure and function. Nat Prod Rep 2002, 19, 312–331. [DOI] [PubMed] [Google Scholar]
- (58).Williams JD; Cox KA; Cooks RG; McLuckey SA; Hart KJ; Goeringer DE, Resonance Ejection Ion Trap Mass Spectrometry and Nonlinear Field Contributions: The Effect of Scan Direction on Mass Resolution. Anal Chem 1994, 66, 725–729. [Google Scholar]
- (59).Charles MJ; McLuckey SA; Glish GL, Competition between resonance ejection and ion dissociation during resonant excitation in a quadrupole ion trap. J. Am Soc Mass Spectrom 1994, 5, 1031–1041. [DOI] [PubMed] [Google Scholar]
- (60).Zaia J; Li X-Q; Chan S-Y; Costello CE, Tandem mass spectrometric strategies for determination of sulfation positions and uronic acid epimerization in chondroitin sulfate oligosaccharides. J. Am Soc Mass Spectrom 2003, 14, 1270–1281. [DOI] [PubMed] [Google Scholar]
- (61).Kailemia MJ; Patel AB; Johnson DT; Li LY; Linhardt RJ; Amster IJ, Differentiating chondroitin sulfate glycosaminoglycans using collision-induced dissociation; uronic acid cross-ring diagnostic fragments in a single stage of tandem mass spectrometry. Eur J Mass Spectrom 2015, 21, 275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Figure S1, Schematic of EDD; Figure S2, Schematic of CTD; Figure S3, Symbol nomenclature for glycans; Figure S4, CID, CTD and EDD spectra of compound IdoA-GlcNAc-IdoA-GlcNAc6S-(CH2)5NH2; Figure S5, donut plots and fragment maps of compound IdoA-GlcNAc-IdoA-GlcNAc6S-(CH2)5NH2; Figure S6, CID, CTD and EDD spectra of compound GlcA-GlcNAc-GlcA2S-GlcNAc-(CH2)5NH2; Figure S7, donut plots and fragment maps of compound GlcA-GlcNAc-GlcA2S-GlcNAc-(CH2)5NH2; Figure S8 CID, CTD and EDD spectra of compound GlcA-GlcNS6S-GlcA-GlcNS6S-(CH2)5NH2; Figure S9 CID, CTD and EDD spectra of compound ΔHexA-GalNAc4S-IdoA-GalNAc4S-OH; Figure S10, donut plots and fragment maps of compound ΔHexA-GalNAc4S-IdoA-GalNAc4S-OH; Figure S11 CID, CTD and EDD spectra of compound ΔHexA-GalNAc4S-IdoA-GalNAc4S-IdoA-GalNAc4s-OH; Figure S12, donut plots and fragment maps of compound ΔHexA-GalNAc4S-IdoA-GalNAc4S-IdoA-GalNAc4S-OH; Figure S13 CID, CTD and EDD spectra of compound ΔHexA-GalNAc4S-GlcA-GalNAc4S-GlcAA-GalNAc4S-OH; Figure S14, fragment maps of compound ΔHexA-GalNAc4S-GlcA-GalNAc4S-GlcA-GalNAc4S-OH; Figure S15 CTD and CTD with resonance ejection spectra of compound ΔHexA-GalNAc4S-GlcA-GalNAc4S-GlcA-GalNAc4S-OH; Tables S1- S7, lists of identified fragment ions from CTD for all GAG standards.