

Abstract
Methods of error filtration and correction post-gene assembly are a major bottleneck in the synthetic biology pipeline. Current oligonucleotide purification strategies, including polyacrylamide gel electrophoresis and high-performance liquid chromatography, are often expensive and labor-intensive, give low mass recovery, and contain hazardous chemicals. To circumvent these limitations, we explored an enzymatic means of oligonucleotide purification using RecJ, which is the only known exonuclease to digest single-stranded DNA (ssDNA) in the 5′ to 3′ direction. As a potential application to remove failure strands generated in oligonucleotide synthesis, we found RecJ does not recognize the 5′ dimethoxytrityl blocking group and could therefore be used to specifically target and digest unblocked failure strands. In combination with ssDNA binding protein (SSBP), which acts to recruit RecJ via C-terminal recognition, secondary structure formation is precluded, allowing for enhanced RecJ processivity. Using this method to purify crude trityl-on oligonucleotides, we also found on average 30 units of RecJ with 0.5 μg of SSBP digests 53 pmol of 5′ hydroxylated ssDNA (60 min at 37 °C). With these parameters, the average purity is increased by 8%. As such, this novel method can be adapted to most laboratory practices, particularly those with DNA synthesis automation as a simple, inexpensive (<$4), and eco-friendly means of oligonucleotide trityl-on purification.
Graphical Abstract
Failure strands generated from the phosphoramidite method of synthesizing oligonucleotides present a problem in many fields of biology.1 These failures can have deleterious effects in downstream applications. As a result, time-consuming and expensive methods of error correction and filtration must be employed, particularly for gene synthesis and assembly.2 Methods used to filter out failures postsynthesis include polyacrylamide gel electrophoresis (PAGE),3 high-performance liquid chromatography (HPLC),4 and cartridge trityl-on purification (TOP).5 While these methods are very powerful, increasing oligonucleotide purity to >90%, they often incur high costs ($35−200), are labor-intensive, have long turnaround times and low mass recoveries (30−70%), and use hazardous chemicals (e.g., tetramethylethylenediamine, ammonium persulfate, acetonitrile, and trifluoroacetic acid).
Efforts have been made to minimize hazardous waste generated during purification. For example, Pokharel and Fang demonstrated a novel process of filtering out failure strands using methacrylation phosphoramidites.6 With their method of “catching by polymerization”, the full-length product (FLP) with methacrylation is copolymerized into a polyacrylamide gel, and failure strands are washed free. Acetic acid is then used to cleave the FLP into solution. Though they achieve high purity and recovery yield, methacrylation phosphoramidites are not yet commercially available, and the process requires four additional auxiliary positions for synthesis automation, which are added instead of the last base in sequence.
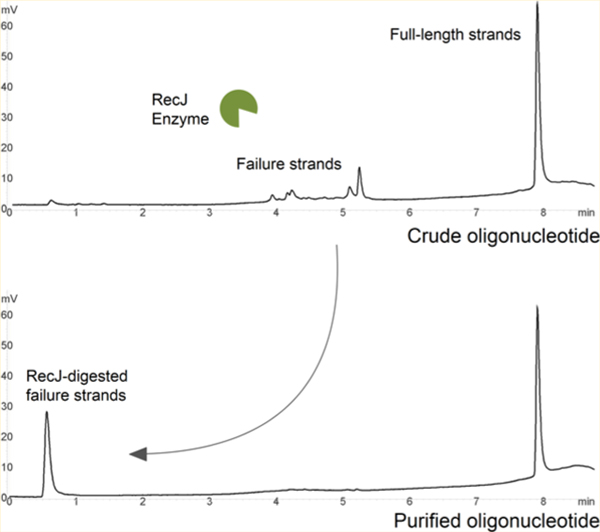
To circumvent limitations of current gel and column-based methods of purification, we explored an enzymatic approach to removing failure strands. The most logical avenue was to choose an enzyme with single-stranded DNA (ssDNA) exonuclease activity. One option we considered was Exo I, which digests 3′ hydroxylated ssDNA. However, this would pose a problem because once oligonucleotides are released from the solid support postsynthesis, there is no way to differentiate between full-length product and truncated failure strands in a crude, heterogeneous sample. We then considered using the dimethoxytrityl (trityl) group retained at the 5′ end of the FLP as a barrier to exonuclease activity. As it turns out, RecJ is the ideal candidate because it is the only known enzyme capable of digesting ssDNA from the 5′ direction.7 Also, on the basis of our research, RecJ does not recognize the trityl group of the FLP and will instead target trityl-off failure strands.
The principle behind our method begins with the oligonucleotide synthesis cycle. During the coupling step, an incoming phosphoramidite, which is blocked at the 5′ end with a trityl group to prevent homopolymeric tract formation, is added to the growing strand. After the backbone is stabilized from a phosphite to phosphate bond during the oxidation step, the trityl group is cleaved with trichloroacetic acid. The cationic trityl group, which is orange in solution, is then used as a metric to determine the percent FLP generated at the end of each cycle. As a general rule, one of every 100 molecules will fail to couple, meaning that the average synthesis run is said to have a coupling efficiency (CE) of 99%.8 While there are several factors that may influence CE (synthesis parameters and quality of reagents), the longer the strand generated, the more failure strands will be produced. This is defined by the formula, (CE)n−1, where n is the number of bases added during synthesis minus 1 nucleoside preattached to the solid support [e.g., controlled-pore glass (CPG)]. For example, synthesis of a 40-base (40b) strand with a 99% CE will generate 68% FLP and 32% failure strands as opposed to synthesis of a 30b strand, which will yield 75% FLP with the same CE. Allowing these failure strands to continue through additional synthesis cycles will create internal deletions. To prevent this, acetic anhydride is used to “cap” the 5′-hydroxyl with an unreactive acetyl group. Only when samples are processed with ammonium hydroxide for cleavage off the solid support and for base deprotection are the capped failures deacylated to a 5′-hydroxyl. In a crude, unpurified sample, the final product is a mixed pool of FLP and various failure strands (n − 1, n − 2, n − 3, etc.). While the trityl group is typically cleaved from the FLP after the final step of the synthesis cycle, it can be retained. This is the underlying principle for cartridge-based trityl-on purification. For the TOP method,5 the hydrophobic trityl group binds strongly to the reverse-phase support (polymeric resin) packaged inside a cartridge. When the heterogeneous oligonucleotide pool is added, the trityl-on FLP (FLP-on) is retained by the hydrophobic resin, allowing unblocked failure strands to pass through. After the wash step, FLP-on is cleaved from the resin with trifluoroacetic acid and then eluted from the cartridge (trityl-off).
Our research offers a “green” method of trityl-on purification, one that is a reaction that occurs under aqueous conditions, rather than a mechanical means of truncated strand filtration (gel and column-based methods). Here we chose RecJ to target unblocked (trityl-off) failure strands because it is the only known exonuclease to digest ssDNA substrates with 5′ to 3′ polarity.7 Functions of this magnesium-dependent enzyme include (1) DNA mismatch repair (RecF recombinatorial pathway), where it excises the nicked strand at the break site,9,10 (2) 5′ exonuclease and hydrolytic deoxyribophospho-diesterase activity in the deoxyribose phosphate excision from abasic sites in double-stranded DNA (dsDNA),11 and (3) methyl-directed mismatch repair.12
We believe an enzymatic means of purification using RecJ has many potential benefits as an alternative to gel and column-based oligonucleotide purification. (1) It is eco-friendly (no hazardous chemicals). (2) It is simple to use (no special preparation or setup). (3) Samples can be pooled for gene synthesis and assembly. (4) ssDNA up to 1000 nucleotides in length can be digested to completion. (5) It is adaptable to any synthesis platform (single column, multiwell titer plate, or array). (6) It is compatible with most downstream biological assays and/or reactions.
In this article, we demonstrate a novel trityl-on purification method using RecJ as an efficient tool for digesting truncated failure strands (trityl-off) produced during oligonucleotide synthesis. We also examine how SSBP is used in combination with RecJ to enhance digestive processivity by recruiting RecJ and minimizing secondary structure formation.13−15 Finally, we address trityl stability and RecJ attack on popular 5′ oligonucleotide modifiers such as biotin, amine, and phosphate.
MATERIALS AND METHODS
In-House Oligonucleotide Synthesis.
Oligonucleotides (Table 1 of the Supporting Information, sections I and II) for RecJ−SSBP reactions and cartridge−TOP were synthesized in-house (Stanford Genome Technology Center) using a model 3900 DNA synthesizer (Applied Biosystems) with 1000 Å CPG columns (BioAutomation) for a 50 nanomole-scale synthesis. Cycle conditions were similar to the manufacturer’s recommended protocol, which included the following reagents: 3% trichloroacetic acid/dichloromethane (American International Chemicals), acetonitrile (BioAutomation), 0.02 M oxidizing solution (BioAutomation), cap A/B (Glen Research), 0.1 M solutions of dA, dC, dG, and dT (BioAutomation), and 0.25 M ethylthio-1H-tetrazole (Glen Research). Postsynthesis steps included cleavage of the strand from the support followed by base deprotection overnight at 55 °C with ammonium hydroxide (28−30%) (J. T. Baker). After lyophilization, oligonucleotides were resuspended, and the optical density for each was measured at 260 nm using the Nanodrop and Qubit reader for ssDNA. All samples were then normalized according to test parameters in 0.1× TE buffer and then analyzed in-house for purity using ion-pairing reverse-phase (RP) HPLC (Transgenomic WAVE system).
RecJ−SSBP Reactions.
RecJf (RecJ) used in this study is an exonuclease that catalyzes 5′ to 3′ degradation of ssDNA and is a recombinant of the native enzyme fused with maltose binding protein for increased water solubility. RecJ−SSBP test reactions used varying oligonucleotide concentrations, in units of RecJ and micrograms of SSBP, and contained the following reagents: RecJf [New England Biolabs (NEB)] (30 units/μL), ET SSB (Extreme Thermostable Single-Stranded DNA Binding Protein) (NEB) (0.5 μg/μL), and 1× NEBuffer 2 (NEB) [50 mM NaCl, 10 mM Tris-HCl, 10 mM MgCl2, and 1 mM DTT (pH 7.9) at 25 °C]; RNase/DNase-free water was added for a total volume of 20 μL. Samples were incubated in a thermal cycler (Verti-96, Applied Biosystems) for 30 and 60 min at 37 °C followed by heat inactivation (20 min at 70 °C).
PAGE Setup and Analysis.
TBE (Tris-borate EDTA) buffer (1×) and 15% precast urea PAGE gels (Invitrogen) were used to analyze ssDNA samples processed with RecJ−SSBP. We used 5 μL of loading dye with urea (Invitrogen) mixed with a 5 μL sample and ran each gel for 60 min at 120 V. For visual analysis, the precast shell was cracked open and the gel was carefully added to 50 mL of 1× TBE buffer with 0.8 μL of ethidium bromide (Bio-Rad) for 10 min, mixing at 20 rpm. Samples were imaged using FluorChem HD2 (Alpha Innotech).
Cartridge Trityl-On Purification.
As a control, we processed in-house 40b strands using the Glen-Pak trityl-on purification cartridge (Glen Research),5 which was prepared as follows: to 50 μL post-synthesis oligonucleotides left in ammonium hydroxide solution, we added 450 μL of RNase/DNase-free water and 500 μL sodium chloride (100 mg/mL). The cartridge was first conditioned with 0.5 mL of acetonitrile (BioAutomation) followed by 1 mL of 2 M triethylammonium acetate (TEAA, Transgenomic) (1−2 drops per sec under vacuum). The oligonucleotide/salt solution was then added to the cartridge. One milliliter of the salt solution (5% acetonitrile in 100 mg/mL sodium chloride) was added twice to wash out failure strands. A 2% trifluoroacetic acid (TFA)/water mixture was then added (1 mL twice). Next, 2 mL of deionized water was added to rinse off the acid and salts from the cartridge. To elute the detritylated FLP, 1 mL of a 50% acetonitrile/water mixture containing 0.5% ammonium hydroxide was then added. Finally, the purified sample was added to a 1.5 mL Eppendorf tube and lyophilized (∼2 h at 60 °C using a vacuum centrifuge).
Sephadex Desalting Procedure.
Sephadex slurry was prepared by adding 5 g G-50 fine powder (GE Healthcare) to 75 mL RNase / DNase-free water, then allowed to set for 3 h. To an empty column (with a porous filter barrier at the bottom), 800 μL were added, then allowed to settle at room temperature (RT) for 5 min under gravity filtration (∼200 μL discarded as waste). The column was then centrifuged at 2.8k rpm for 2 min (Eppendorf 5415 D), and the flow-through discarded. The sample (20 μL) was then added to the middle of the resin bed and centrifuged again at 2.8k rpm for 2 min using a new 1.5 mL Eppendorf tube. The flow-through was saved for downstream processing and analysis.
RP HPLC Setup and Operation.
All samples were analyzed in-house using a Transgenomic Wave system (Transgenomic). Instrument setup consisted of a DNASep column (C-18) using reverse-phase ion-pairing buffers A and B [0.1 M TEAA (pH 7.0) and 0.1 M TEAA in a 75:25 (v/v) water/acetonitrile mixture, respectively] (ADS Biotec). Samples were processed at 80 °C with UV detection at 260 nm. Method and gradient parameters were optimized for best single-base resolution in addition to ideal separation of trityl-on and trityl-off samples (Figure 1 of the Supporting Information). Gradient parameter included the following: loading at 0 min, 82% A and 18% B; start gradient at 0.1 min, 82% A and 18% B; stop gradient at 5.1 min, 35% A and 65% B; start clean at 5.2 min, 10% A and 90% B; stop clean at 6.2 min, 10% A and 90% B; start equilibration at 6.3 min, 82% A and 18% B; stop equilibration at 6.8 min, 82% A and 18% B.
Standards for RecJ−SSBP Purification Analysis.
We compared the purity of samples treated with RecJ and SSBP to 40b samples subjected to PAGE (IDT) and trityl-on cartridge purification. Also, to determine the chromatographic region of failure strands (relative to the 40b target), we synthesized 39b, 38b, 35b, 31b, and 20b truncated variants (Figure 1 of the Supporting Information).
Calculations for Oligonucleotide Purity and Coupling Efficiency.
On the basis of the method used for sample processing with RP HPLC, we found a majority of failure peaks present between 1 and ∼4.5 min. To determine sample purity, we used the equation {[% area (A) of FLP-off + FLP-on]/[% A of FLP-off + FLP-on + total failures]} × 100, where the % A for each target peak was taken directly from the HPLC chromatogram analysis report. We did not include prematurely detritylated FLP in the total failure strands but instead added it to the total FLP-on to give a more accurate measure of purity. The total percent A of failure product was measured by adding all the peak values after 1 min up to the target FLP-off (∼4.5 min). The total percent hydrolysis product for each test sample (after treatment with RecJ−SSBP) was also adjusted by subtracting the peak percent area (before treatment) at the same time point (∼0.5 min). For samples where we calculated the relative abundance (picomoles of ssDNA), estimates were based on chromatographic evaluation of peaks that could be detected using the WAVE system. Coupling efficiencies were also back-calculated using the percent area of target full-length product derived from chromatographic analysis reports: ln(% A FLP/100) = ln(coupling efficiency)n−1. Our lab routinely reports coupling efficiencies of ≥99.5%.
Energy Dot Plot Analysis.
Sequences used for secondary structure formation analysis were uploaded to http://unafold.rna.albany.edu/?q=mfold/DNA-Folding.
Oligonucleotide Yield Analysis.
Oligonucleotide yields were measured with the Qubit 2.0 fluorometer using the Qubit ssDNA Assay kit (Life Technologies), and the Nano-drop (ND-1000 spectrophotometer).
RESULTS AND DISCUSSION
Overview of the RecJ Trityl-On Purification Method.
Our enzymatic method of removing trityl-off failure oligonucleotides from crude synthesis material is composed of two basic steps: (1) RecJ digestion of failure strands and (2) detritylation of purified target FLP-on (Figure 1). This method is based on the principle that RecJ has specific 5′ exonuclease activity toward ssDNA containing a 5′-hydroxyl group. FLP protected by the dimethoxytrityl group retained postsynthesis is impervious to RecJ attack and therefore remains unaffected. Once the reaction is complete, target FLP-on is then detritylated using mild acid conditions.
Figure 1.

Method of RecJ digestion of oligonucleotide truncated failure strands in aqueous solution. Here, oligonucleotides are first synthesized trityl-on, removed from the instrument, cleaved, and deprotected according to the manufacturer’s protocol. In step 1 of the enzymatic purification method, the crude sample is treated with RecJ (green Pac-Man character) at 37 °C; while the trityl-on FLP is untouched (A), only those ssDNA molecules with a free 5′-hydroxyl group are digested (hydrolyzed) by RecJ (B). In step 2, a weak acid [0.1 M NaOAc (pH 3.5)] is added to the reaction mixture to detritylate FLP, yielding a 5′-hydroxyl group (C). The sample is then passed through a Sephadex (G-50 fine) column to remove acetate, trityl, and hydrolysis product. This yields a purified, desalted sample ready for downstream application. Each step is accompanied by a RP HPLC chromatogram depicting various oligonucleotide species present in the reaction.
RecJ has been shown to digest samples up to 1000 nucleotides long, with a minimum length requirement of seven nucleotides for adequate binding and digestion.15 In our method, 5′ unblocked oligonucleotides are digested down to the last 3′ mononucleotide during enzymatic hydrolysis. Using RP HPLC, hydrolysis product generation is measured by the appearance of the first major peak shown on the chromatogram (∼0.5 min); the percent area is proportional to the total percent areas of oligonucleotides digested by RecJ.
RecJ Processivity Optimization.
First, we determined optimal reaction conditions for RecJ digestion (processivity) of 5′ unblocked oligonucleotides in trityl-on samples. The objective here was to measure the average processivity of 30 units of RecJ, which is the stock solution amount of enzyme per microliter (NEB). To test this, we synthesized 10 unique 40b oligonucleotide samples trityl-on [S1−S10 (Table 1 of the Supporting Information, section I). We processed 500 pmol each with 30 units of RecJ (30 and 60 min incubation periods) at 37 °C. In our preliminary experiments, 500 pmol of crude trityl-on product yielded >90 pmol of unblocked oligonucleotides, providing an upper picomole limit that 30 units of RecJ alone could not fully digest. This allowed us room to adjust reaction parameters for optimal performance (Table 1). To improve RecJ processivity, we added 0.5 μg of SSBP (0.5 μg/μL stock solution, NEB) to a separate set of 10 samples using the same conditions at 30 and 60 min (each with 30 units of RecJ).
Table 1.
Summary of RecJ Processivity of Trityl-On Crude Samples over Time (30 and 60 min)a
| total digested (pmol) | failures digested (pmol) | percent failure decrease | percent purity increase | |||||
|---|---|---|---|---|---|---|---|---|
| 30 units of RecJ | 30 units of RecJ, 0.5 μg of SSBP | 30 units of RecJ | 30 units of RecJ, 0.5 μg of SSBP | 30 units of RecJ | 30 units of RecJ, 0.5 μg of SSBP | 30 units of RecJ | 30 units of RecJ, 0.5 μg of SSBP | |
| 30 min | ||||||||
| average | 14 | 36 | 11 | 25 | 22 | 49 | 2 | 6 |
| 60 min | ||||||||
| average | 22 | 53 | 17 | 35 | 31 | 53 | 4 | 8 |
Each condition tested the ability of RecJ to digest 5′ hydroxylated ssDNA generated from trityl-on samples starting with an average crude purity of 81% (based on 99.5% CE for 10 samples, each with ∼500 pmol of total material). Each time point (30 and 60 min incubation) shows an average processivity for 30 units of RecJ with or without 0.5 μg of SSBP. See also Tables 2 and 3 of the Supporting Information.
Table 1 shows that after 30 min, 30 units of RecJ alone digested an average of 14 pmol of total ssDNA (including failure strands and prematurely detritylated FLP). By adding 0.5 μg SSBP, processivity was increased 157% to an average of 36 pmol. After one hour of incubation, 22 pmol were digested per 30 units RecJ; here, with the addition of 0.5 μg of SSBP, RecJ processivity was increased 141% to 53 pmol. In the latter case, the sample purity was increased on average by 8%. Adding SSBP to 1 μg, however, had no marked impact on processivity after 60 min.
RecJ−SSBP Trityl-On Purification Method Step 1: RecJ Digestion of Failure Strands.
At the first step of the method outlined in Figure 1, all samples were processed trityl-on using the reaction parameters just described. To more closely examine how RecJ and SSBP behave toward 5′ unblocked ssDNA, we added each to a 40b sample (trityl-on) to show the disappearance of the trityl-off product following incubation. Analysis also included the relative abundance of failure strands, prematurely detritylated FLP (FLP-off), trityl-on peak (FLP-on), and the hydrolyzed product of RecJ digestion (free nucleotides) (Figure 2). To study this reaction, we chose ion-pairing RP HPLC, which is ideal for separating the hydrophobic dimethyoxytrityl-on target oligonucleotides from both the hydrolysis and trityl-off products. While anion-exchange chromatography is the gold standard for oligonucleotide single-base resolution, we successfully optimized the ion-pair RP HPLC system to yield adequate peak separation between all relevant species. We synthesized a series of 5′ truncated strands (39b, 38b, 35b, 31b, and 20b) to confirm where along the x-axis of the chromatogram most of the failure strands are present (Figure 1 of the Supporting Information). For all samples, we targeted only detectable peaks between 1 and ∼4.5 min (target FLP-off). We also purchased PAGE-purified 40b controls (IDT) for target FLP-off size confirmation.
Figure 2.

RP HPLC chromatograms of a trityl-on oligonucleotide before and after treatment with RecJ and SSBP. With a 10 μM reaction mixture, each peak was assigned a picomole (pm) value based on its relative abundance. Chromatogram B shows crude [40b (Table 1 of the Supporting Information, section II)] product before treatment with 19.5% failure product (II), 17.7% prematurely detritylated FLP (III), and 62.8% trityl-on FLP (IV). Following treatment with 60 units of RecJ and 0.5 μg of SSBP (37 °C for 60 min), chromatogram A shows 32.6% hydrolysis product, 1.9% remaining failure product (II), 2.9% prematurely detritylated FLP (III), and 62.6% trityl-on FLP (IV). Numbers on each chromatogram indicate where sample peaks were detected.
The crude sample (40b) before purification (Figure 2B) shows 200 pmol total in a volume of 20 μL (10 μM), which yielded 38.9 pmol of failure product (II), 35.4 pmol of prematurely detritylated FLP (III), and 125.7 pmol trityl-on FLP (IV). For chromatogram A (after purification), the hydrolysis product (I) was measured at 32.6% (65.2 pmol). The amount consumed by RecJ in the reaction closely matches this value at 64.7 pmol [74.3 pmol minus 9.6 pmol (failure + FLP-off before and after, respectively)]. Failure product has also been reduced by 90% (from 38.9 to 3.8 pmol). To obtain a more accurate estimate of the purity, we added total picomoles of FLP-off to total picomioles of FLP-on (see Materials and Methods), which we calculated for this sample at 97.1%.
RecJ−SSBP Trityl-On Purification Method Step 2: Detritylation of Purified Target FLP-On.
After RecJ digestion of 5′ unblocked oligonucleotides, the trityl group of the FLP is removed by a weak acid. For this, we used 0.1 M NaOAc (80 μL, pH 3.5), incubated for 60 min at RT (total volume of 100 μL). It has been reported that standard treatment with acetic acid (80% in water, pH 2.4) causes depurination (release of adenosine and guanosine by hydrolysis at the β-N-glycosidic bond). Krotz and colleagues found that using NaOAc as a milder trityl cleaving agent significantly reduces the level of possible damage to DNA during the detritylation process.16 Therefore, we substituted 80% acetic acid with 0.1 M NaOAc for all experiments.
Following detritylation, the sample mixed with NaOAc was lyophilized and then desalted to remove acetate, trityl, and hydrolysis product (Figure 3). For this step, we used Sephadex G-50 (fine) packaged inside a column, which is routinely used for polymerase chain reaction cleanup where unincorporated triphosphate nucleotides are captured within the bead matrix. The final purity of this 40b sample was measured to be 95.3%.
Figure 3.

Purity analysis of the fully deblocked 40b strand before and after treatment with RecJ and SSBP. (A) Two superimposed RP HPLC chromatograms of the 40b strand (200 pmol total) before (I) and after (II) purification with 60 units of RecJ and 0.5 μg of SSBP. Both samples were treated with 0.1 M NaOAc (pH 3.5) postreaction to demonstrate the purity of final detritylated product. (B) PAGE image of the same two samples before (I) and after (II) purification with the disappearance of truncated failure product. Gel analysis was performed on 15% PAGE with urea (120 V for 60 min). Both chromatograms were offset for the sake of clarity and have been moved along the x-axis; the control target FLP peak matched up with the target FLP peak in Figure 2. The larger peak for the untreated FLP-off sample is due to the addition of prematurely detritylated FLP [127.5 pmol + 35.4 pmol (Figure 2B)].
RecJ can also be heat-inactivated postpurification (70 °C for 20 min). While this step is optional, treatment with NaOAc at pH 3.5 and lyophilization (60 min, 60 °C), should be enough to completely stop RecJ processivity before downstream application.
As with any trityl-on purification method, failures may carry over into the fully processed sample. These include depurinated strands, base additions, and 3′ and internal deletions, all of which could still retain their trityl groups. Internal deletions, the most common of these failures, are derived from incomplete detritylation and capping inefficiency.17 Though sample purity in Figure 2A post-treatment was calculated to be 97.1%, the 1.8% drop to 95.3% following detritylation may be due to these trityl-on failures. These failures are often imbedded within and around the target trityl-on peak itself (Figure 2A,B IV); because of the hydrophobic−charge interaction between the trityl group and HPLC column absorbent, longer trityl-on strands are actually less hydrophobic than shorter ones, which elute later from the column.18 Furthermore, as shown in chromatograms I and II of Figure 3A, peaks 20 and 12 appear to be single-base additions. While this may be the case, we were unable to define clear n + 1 bands in Figure 3B even with overexposure of the gel image. However, if we do include peak value 12 (Figure 3A II) with the total failure product of the treated sample, the final purity becomes 94.6%. This still translates into a 16.6% increase in purity (8.3%/30 units RecJ, 0.5 μg SSBP).
To minimize trityl-on synthesis failures, we suggest (1) optimizing the capping step, (2) applying a milder deblocking agent (3% dichloroacetic acid), (3) using a less acidic, more nucleophilic activator [4,5-dicyanoimidazole (DCI)], and (4) choosing the appropriate solid support for the length of DNA synthesized. If the strands are too long, narrow-channel pores will cause steric hindrance, thus shearing DNA at the 3′ end from the surface of the support.
Finally, we compared the purity of sample II of Figure 3A with that of cartridge (TOP)- and PAGE-purified samples (95.3, 95.0, and 96.6%, respectively). The PAGE-purified sample was also used as a control to match the target strand size on RP HPLC with test samples processed using RecJ and SSBP.
To purify crude trityl-on product, <500 pmol total (40b) on a per reaction basis, our method can be very cost-effective. This is compared with current column and gel-based methodologies, which require much greater starting yields to offset mass recovery loss. For example, we estimate it costs <$4 to digest ∼50 pmol of total 5′ hydroxylated ssDNA (failure product and FLP-off in the crude trityl-on sample; the cost includes 30 units of RecJ and 0.5 μg of SSBP, which was determined by stock reagents purchased from NEB). For purifying larger quantities of crude trityl-on ssDNA (≥500 pmol), adding more units of RecJ may be necessary to achieve a final purity of ≥95%. This is also the case for purifying longer strands at the same concentration, which have a greater percentage of failure product (assuming a 99.5% CE). Because the RecJ stock solution contains 50% glycerol, this could interfere with enzyme processivity. Therefore, we suggest increasing the reaction volume (e.g., 50−100 μL) and/or performing dialysis on RecJ to remove excess glycerol before application.
Benefits of Adding SSBP to RecJ in the Reaction Mixture for Increased Processivity.
During our tests, we found that RecJ alone failed to digest certain oligonucleotides [S11 (Table 1 of the Supporting Information, section III)] after incubation for 30 min at 37 °C (Figure 4A, lane 2). Upon closer inspection using the mfold software package,19 which predicts nucleic acid folding and hybridization, we discovered that S11 was strongly predisposed to forming secondary structures where the 5′ end is locked in a double-stranded complex, rendering it inaccessible to RecJ attack (Figure 4B, structure I). Compared to more relaxed sequences with 5′ overhangs [S12 (Table 1 of the Supporting Information, section III)], secondary structure formation is much less stable, forming only poorly ordered structures easily disrupted by elevated temperatures (Figure 4B, structures II−IV). While the energy plot for S12 (Figure 4B, structure III) shows a 5′ end duplex, it is far less stable than that formed by S11 (structure I) (−3.04 and −11.49 kcal/mol, respectively).
Figure 4.

Effects of oligonucleotide secondary structure formation on RecJ processivity. The PAGE image (A) demonstrates how S11, because of its increased level of secondary structure formation, is resistant to digestion by RecJ alone (lane 2) compared to treatment with RecJ and SSBP in combination (lane 3); lanes 4−6 show S12 before and after RecJ and RecJ−SSBP treatment (30 min, 37 °C). (B) Energy dot plots for S11 (I) and S12 (II−IV). The ΔG Gibbs free energy for S11 (37 °C) was measured at −11.49 kcal/mol (I)17 and for S12 at −3.74 kcal/mol (II), −3.04 kcal/mol (III), and −2.83 kcal/mol (IV). For tests run (A), 1 μM reactions were used accordingly: 30 units of RecJ (lanes 2 and 5), 30 units of RecJ and 0.5 μg of SSBP (lanes 3 and 6), and controls for S11 and S12 in which neither RecJ nor SSBP was applied (lanes 1 and 4). Gel analysis was performed on 15% PAGE with urea (120 V for 60 min). (Off) labels in panel A denote trityl-off oligonucleotide strands.
When we added 0.5 μg of SSBP to the reaction mixture with 30 units of RecJ, S11 was fully digested (Figure 4A, lane 3). This suggests a beneficial interaction between RecJ and SSBP. The recognition of ssDNA by SSBP is largely electrostatic, where the negatively charged phosphates of the DNA backbone interact with the positively charged amino acids of SSBP (from Escherichia coli), in particular, the lysine residues.14 Furthermore, DNA unwinding by SSBP disrupts secondary structure formation, where the melting temperature of duplex DNA is depressed; this is most prominent for AT-rich regions.
It has been reported that Deinococcus radiodurans RecJ and SSBP play an essential role as a co-complex in processing broken DNA ends for homologous recombination.20 On the basis of crystallography, Cheng and colleagues defined a mechanistic pathway describing how RecJ binds to double-strand breaks to initiate resection and exonuclease activity in the 5′ to 3′ direction. Here, the 5′ terminus of ssDNA is anchored to a phosphate binding pocket above the RecJ active site for hydrolysis. A helical gateway at the entrance to this active site precludes entry of dsDNA. This explains why when strands similar to S11 form a duplex at the 5′ terminus, they are not recognized by RecJ alone.
Moreover, it has been found that SSBP attaches to RecJ via the C-terminal region, which may help recruit RecJ for enhanced processivity.14 Therefore, SSBP provides a valuable asset for enhancing RecJ exonuclease activity for digesting crude oligonucleotide failures by allowing access to 5′ strands prone to secondary structure formation.
Effects of Temperature on Trityl Stability over Time.
While the main contributor to effective detritylation is the buffer pH, we also observed that trityl stability is affected by temperature. To demonstrate this, we exposed three trityl-on oligonucleotide samples (Table 1 of the Supporting Information, section III) to RT and 37 °C conditions over a 24 h period (5 μM ssDNA in water and 1× buffer 2 at pH 7; same as the RecJ−SSBP reaction conditions). During their incubation, aliquots were taken at defined time points for RP HPLC analysis to determine percent detritylation (Figure 2 of the Supporting Information). Here, even at RT, FLP products began a slow detritylation process around 10 min, and after 1 day, >6% had already lost their trityl group. At 37 °C, noticeable detritylation occurred after 3 h, and by 24 h, nearly 25% of the FLP was detritylated. Aliquots of these samples from the same study were kept frozen at −20 °C (RecJ−SSBP reaction conditions) and showed no significant detritylation over a period of 7 days. If the target FLP sample is completely blocked at the 5′ end prior to incubation, a negligible amount of FLP-on may begin to detritylate at 37 °C within 30 min to 1 h.
As we have learned, RecJ does not discriminate between prematurely detritylated FLP and trityl-off failure strands and will digest both. If the trityl group is cleaved before or during incubation, this product is now free to react with RecJ and SSBP. This is clearly shown in Figure 2, where digestion of prematurely detritylated FLP makes up 54% of the hydrolysis product. This suggests trityl stability plays a key role in overall RecJ−SSBP efficiency and final FLP yield. Therefore, proper storage and handling of trityl-on material are necessary to aid RecJ and SSBP in targeting and digesting only failure strands in a reaction.
Susceptibility of 5′ Modified Oligonucleotides to RecJ Attack.
We also investigated whether the more popular 5′ modifiers such as biotin, amine, and phosphate would be impervious to RecJ attack. For this test, we added 30 units RecJ and 0.5 μg SSBP to 20 pmol each of HPLC-purified T20b strands (IDT) modified at the 5′ end to either biotin, amine, or phosphate (60 min at 37 °C). We then subjected each sample to RP HPLC and found that all three labeled oligonucleotides were completely digested (Figure 3 of the Supporting Information).
The digestion of oligonucleotides possessing a 5′ phosphate corroborates previous reports.15 Our test results also confirm this exonuclease activity on biotin and amine-modified oligonucleotides. Synthesis reagent vendors such as Glen Research offer these specialty monomers trityl-on, which may be processed with the RecJ−SSBP method of purification. This method is also beneficial for removing unmodified species, which will allow for higher efficiency in downstream assays. Also, when commercially available, the methacrylation phosphoramidite-modified oligonucleotides mentioned earlier6 may prove to be adaptable to our RecJ−SSBP protocol.
CONCLUSION
Our method of enzymatic trityl-on oligonucleotide purification presents a novel approach to removing truncated failure product in an aqueous solution. On the basis of standard trityl-on synthesis conditions, we have shown that on average, 30 units of RecJ with 0.5 μg of SSBP digests 53 pmol of trityl-off material (60 min at 37 °C), increasing the sample purity by 8%. On the basis of these observations, one can use the coupling efficiency and target oligonucleotide concentration to estimate how many units RecJ may be necessary to increase the final purity to ≥95%. Because RecJ targets all species of 5′ hydroxylated ssDNA, the overall sample purity may vary depending on the ratio of failure strands and prematurely detritylated FLP present in the reaction mixture. As such, special care must be taken to adequately handle and store trityl-on postsynthesis material. Avoiding high temperatures and low pH buffers, may further improve RecJ processivity and final target FLP yield. Our results also suggest that SSBP enhances the performance of RecJ by first binding to ssDNA and then recruiting RecJ for oligonucleotide failure strand digestion. Furthermore, SSBP disrupts secondary structure formation, allowing RecJ greater access to the 5′ end. Additional benefits of the RecJ−SSBP method of trityl-on purification include (1) the simplicity of use and setup, (2) the ability to digest strands up to 1000 nucleotides long, (3) the adaptability to any synthesis platform (single column, multiwell titer plate, or array), and (4) the compatibility with most downstream biological assays. We believe the RecJ−SSBP method can be applied to any laboratory practice, particularly one with DNA synthesis automation. Finally, this method provides an inexpensive (<$4 to digest ∼53 pmol of total 5′ hydroxylated ssDNA) and eco-friendly addition to high-fidelity gene synthesis and assembly pipelines for both industry and academia.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Drs. Colin James Bell Harvey, Raeka Aiyar, and Curt Fischer (Stanford Genome Technology Center) for their invaluable feedback and edits regarding the manuscript. The authors also thank Dr. Agustin Sanchez at the Peptide and Nucleic Acid Facility (Stanford University) for his expert advice interpreting HPLC chromatogram results.
Funding
This work was funded by National Institutes of Health Grant 2P01HG000205-24 to R.D.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b00010.
Three tables and three figures mentioned in the text (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Kosuri S, and Church GM (2014) Large-scale de novo DNA synthesis: technologies and applications. Nat. Methods 11, 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Mueller S, Coleman JR, and Wimmer E. (2009) Putting Synthesis into Biology: A Viral View of Genetic Engineering through De Novo Gene and Genome Synthesis. Chem. Biol 16, 337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Vorndam AV, and Kerschner J. (1986) Purification of Small Oligonucleotides by Polyacrylamide-Gel Electrophoresis and Transfer to Diethylaminoethyl Paper. Anal. Biochem 152, 221–225. [DOI] [PubMed] [Google Scholar]
- (4).Warren WJ, and Vella G. (1995) Principles and Methods for the Analysis and Purification of Synthetic Deoxyribonucleotides by High-Performance Liquid-Chromatography. Mol. Biotechnol 4, 179–199. [DOI] [PubMed] [Google Scholar]
- (5).Glen-PakTM Cartridges: DNA and RNA Purification. http://www.glenresearch.com/Technical/GlenPak_UserGuide.pdf.
- (6).Pokharel D, and Fang SY (2016) Polymerizable phosphoramidites with an acid-cleavable linker for eco-friendly synthetic oligodeoxynucleotide purification. Green Chem. 18, 1125–1136. [Google Scholar]
- (7).Lovett ST, and Kolodner RD (1989) Identification and Purification of a Single-Stranded-DNA-Specific Exonuclease Encoded by the Recj Gene of Escherichia-Coli. Proc. Natl. Acad. Sci. U. S. A 86, 2627–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Temsamani J, Kubert M, and Agrawal S. (1995) Sequence identity of the n-1 product of a synthetic oligonucleotide. Nucleic Acids Res. 23, 1841–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ivancic-Bace I, Salaj-Smic E, and Brcic-Kostic K. (2005) Effects of recJ, recQ, and recFOR mutations on recombination in nucleasedeficient recB recD double mutants of Escherichia coli. J. Bacteriol 187, 1350–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Li GM (2008) Mechanisms and functions of DNA mismatch repair. Cell Res. 18, 85–98. [DOI] [PubMed] [Google Scholar]
- (11).Dianov G, Sedgwick B, Daly G, Olsson M, Lovett S, and Lindahl T. (1994) Release of 5′-terminal deoxyribose-phosphate residues from incised abasic sites in DNA by the Escherichia coli RecJ. protein. Nucleic Acids Res. 22, 993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Viswanathan M, Burdett V, Baitinger C, Modrich P, and Lovett ST (2001) Redundant exonuclease involvement in Escherichia coli methyl-directed mismatch repair. J. Biol. Chem 276, 31053–31058. [DOI] [PubMed] [Google Scholar]
- (13).Kowalczykowski SC, Bear DG, and Von Hippel PH (1981) Single-Stranded DNA Binding Proteins. Enzymes 14, 373–444. [Google Scholar]
- (14).Sigal N, Delius H, Kornberg T, Gefter ML, and Alberts B. (1972) A DNA-unwinding protein isolated from Escherichia coli: its interaction with DNA and with DNA polymerases. Proc. Natl. Acad. Sci. U. S. A 69, 3537–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Han ES, Cooper DL, Persky NS, Sutera VA Jr., Whitaker RD, Montello ML, and Lovett ST (2006) RecJ exonuclease: substrates, products and interaction with SSB. Nucleic Acids Res. 34, 1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Krotz AH, Cole DL, and Ravikumar VT (2003) Synthesis of antisense oligonucleotides with minimum depurination. Nucleosides, Nucleotides Nucleic Acids 22, 129–134. [DOI] [PubMed] [Google Scholar]
- (17).Gilar M, and Bouvier ESP (2000) Purification of crude DNA oligonucleotides by solid-phase extraction and reversed-phase high-performance liquid chromatography. J. Chromatogr A 890, 167–177. [DOI] [PubMed] [Google Scholar]
- (18).Oliver RWA (1989) HPLC of macromolecules: A practical approach, IRL Press, Oxford, U.K. [Google Scholar]
- (19).Zuker M. (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Cheng KY, Xu H, Chen XY, Wang LY, Tian B, Zhao Y, and Hua YJ (2016) Structural basis for DNA 5′-end resection by RecJ. eLife 5, e14294. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.