

Abstract
The Crk adaptor protein, a critical modifier of multiple signaling pathways, is overexpressed in many cancers where it contributes to tumor progression and metastasis. Recently, we have shown that Crk interacts with the peptidyl prolyl cis-trans isomerase, Cyclophilin A (CypA; PP1A) via a G219P220Y221 (GPY) motif in the carboxyl-terminal linker region of Crk, thereby delaying pY221 phosphorylation and preventing downregulation of Crk signaling. Here, we investigate the physiologic significance of the CypA/Crk interaction and query whether CypA inhibition affects Crk signaling in vitro and in vivo. We show that CypA, when induced under conditions of hypoxia, regulates Crk pY221 phosphorylation and signaling in cancer cell lines. Using nuclear magnetic resonance spectroscopy, we show that CypA binds to the Crk GPY motif via the catalytic PPII domain of CypA, and small-molecule nonimmunosuppressive inhibitors of CypA (Debio-025) disrupt the CypA–CrkII interaction and restores phosphorylation of Crk Y221. In cultured cell lines, Debio-025 suppresses cell migration, and when administered in vivo in an orthotopic model of triple-negative breast cancer, Debio-025 showed antitumor efficacy either alone or in combination with anti-PD-1 mAb, reducing both tumor volume and metastatic lung dispersion. Furthermore, when analyzed by NanoString immune profiling, treatment of Debio-025 with anti-PD-1 mAb increased both T-cell signaling and innate immune signaling in tumor microenvironment.
Implications:
These data suggest that pharmacologic inhibition of CypA may provide a promising and unanticipated consequence in cancer biology, in part by targeting the CypA/CrkII axis that regulates cell migration, tumor metastasis, and host antitumor immune evasion.
Introduction
CT10 regulator of kinase (v-Crk), originally identified as a transforming gene in the avian chicken tumor virus CT10 (1, 2), encodes a modular Src homology 2 (SH2) and Src homology 3 (SH3) domain-containing adaptor protein that mediates assemblages of protein–protein interactions downstream of tyrosine kinases (3, 4). By binding to tyrosine phosphorylated cytoskeletal proteins, such as paxillin and p130Cas/BCAR1 via the SH2 domain, and proline (PxxP)-containing proteins, including DOCK1 and C3G via the SH3N domain, Crk links signaling from integrins and growth factor receptors to regulate proliferation, motility, and survival (5–7). Pathophysiologically, when Crk is overexpressed in cancer cells, the adaptor protein function of Crk amplifies tyrosine phosphorylation–dependent signaling and can facilitate cell transformation, migration/invasion, and metastasis (8, 9). In addition to the SH2 and SH3N, the predominant cellular Crk isoform (c-Crk II) also encodes an approximately 50 amino acid proline-rich linker sequence between the SH3 domains and an atypical carboxyl-terminal SH3 domain (SH3C) that does not bind proline-rich sequences (5, 6, 10, 11). Both the SH3 linker sequence and SH3C functions as negative regulatory elements that curtail the adaptor protein function of c-Crk II, explaining why c-Crk II has less cell transformation and oncogenic activity compared with v-Crk (12). In the case for the negative regulatory activity of the SH3–SH3 linker sequence, this motif contains a consensus GPY221DHP224 that when (tyrosine) phosphorylated by either receptor tyrosine kinases (i.e., EGFR and PDGFR-B; refs. 13–15) or nonreceptor tyrosine kinases (i.e., Abl and Arg; refs. 16, 17), promote a conformation change via an intramolecular pTyr221-SH2 domain clamp that restricts the SH2 domain from binding other tyrosine phosphorylated proteins in trans, thereby inhibiting canonical Crk signaling (12). Reversible tyrosine phosphorylation of Tyr221 allows fine-tuning of CrkII adaptor function and permits dynamic regulation in signal transduction.
While Tyr221 phosphorylation/dephosphorylation represents a general on–off switch mechanism for the Crk adaptor function, we have recently described a separate level of regulation for CrkII, whereby CypA [a peptidyl prolyl cis-trans isomerase (PPIase) that catalyzes cis-trans isomerization of peptide bonds preceding proline residues] binds directly to the CrkII pTyr221 site via a G219P220 motif immediately adjacent to Tyr221 phosphorylation site (18). The GP duet of CrkII is a preferred substrate for CypA (19), and when bound by CypA, delays both EGFR- and Abl-mediated phosphorylation of Tyr221 phosphorylation in vitro and the subsequent negative regulation of the CrkII (18). On the basis of this arrangement, and the fact that CypA has been shown to be upregulated and overexpressed in a variety of human cancers, including breast cancer (20), non–small cell lung carcinoma (21), lung adenocarcinoma (22, 23), gastric cancer (24), pancreatic cancer (25), and melanoma (26), and linked with aggressive cancer phenotypes including increased cell proliferation, cell invasion, chemo-resistance (27), and hypoxia (28), we posit that CypA binds to Crk Y221 to delay negative regulation and augment Crk signaling. Indeed, previous findings that siRNA to CypA (29) suppress cancer cell growth and metastasis support a role for CypA in oncogenic processes, although it is not clear whether CypA inhibition indirectly impinges on Crk signaling pathways to drive oncogenesis.
While Crk has a well-established role in cancer, and has been intensely studied with respect to signaling downstream of integrins and growth factor receptors to control a variety of cellular functions important for the malignant phenotype, including proliferation, migration, and invasion (5, 6), recently we identified an unexpected role for Crk for regulating the antitumor immune response in mouse model for triple-negative breast cancer (30). We found that Crk knockout in the poorly immunogenic 4T1 cell line, generated by CRISPR/Cas9 gene editing, led to decreased epithelial-to-mesenchymal transition (EMT) and PD-L1 on the tumor cells, and broadly regulated the tumor microenvironment in an immune-competent syngeneic Balb/c model, including the enhanced infiltration of T lymphocytes, the elevation of cytotoxic effector cytokines, elevation in immune-surveillance cytokines and ILs, and decreased TH2 cells and tumor suppressive cytokines such as TGFβ (30).
Here, we explored the physiologic significance of the Crk/CypA interaction in cell lines and in an in vivo tumor model. Consistent with previous reports showing that many solid tumors can upregulate CypA expression (28, 31, 32), we show using unbiased mass spectrometry that CypA is upregulated in hypoxia, and in doing so, delays growth factor (EGF)-inducible tyrosine phosphorylation of CrkII. Using the low molecular weight CypA inhibitor, Debio-025 (33, 34), we show in vitro that Debio-025 dissociated CypA from Crk, and in cell lines decreased motility and invasion of cancer cells. Moreover, in vivo, oral gavage administration of Debio-025 shows antitumor and antimetastastic activity and can synergize with anti-PD-1 checkpoint therapy. Together, these data suggest that pharmacologic inhibition of CypA targets Crk indirectly by changing the kinetics of Crk Tyr221 phosphorylation to inhibit canonical Crk signaling. These data also show a potential therapeutic activity of Debio-025 in immune-oncology applications as a cancer repurposed drug that can target, in part, the Crk/CypA axis.
Materials and Methods
Protein isolation and immunoblotting
Whole-cell lysates of cells were prepared in HNTG buffer (HEPES 50 mmol/L, NaCl 150 mmol/L, Triton X-100 1%, and glycerol 10%) supplemented with Protease Inhibitor Cocktail (Sigma) and Phosphatase Inhibitor Cocktail (Cell Signaling Technology). Cells were scraped, incubated on ice for 10 minutes, and centrifuged at 12,000 rpm for 10 minutes. Cleared lysates were isolated, mixed with SDS containing Laemmli buffer boiled for 5 minutes, and resolved by SDS-PAGE. Immunoblotting was performed using monoclonal Crk (Cell Signaling Technology), CypA (Cell Signaling Technology), Crk pY221 (Cell Signaling Technology), HIF1α (NovusBio), and β-actin (Cell Signaling Technology) antibodies.
Cell lines and hypoxia conditions
MDA-MB-231, MDA-MB-468, HS683, and DU145 cells were purchased from ATCC. 4T1-luc2-GFP cells were purchased from Caliper Life Sciences. The cells were grown either in DMEM or RPMI supplemented with 10% FBS and 1% penicillin–streptomycin as suggested by ATCC. To induce hypoxia, cobalt chloride (CoCl2) was added at the indicated concentrations in the media or the cells were cultured in Hypoxia Incubator (Coy Laboratory Products). After thawing, cells were used for up to 8–10 passages and their authenticities were checked by short tandem repeat analysis according to the manufacturer’s protocol (GenePrint 10 System, Promega). Cells were routinely checked for Mycoplasma contamination using universal Mycoplasma Detection Kit (ATCC 30–1012K).
Mass spectrometry for hypoxia-induced protein estimation
MDA-MB-231 breast adenocarcinoma cells were cultured in normoxia or hypoxia conditions by culturing cells in common incubators with 5% CO2 or hypoxia incubators. The cells were cultured and passaged for 2–3 times, after which the lysates were prepared in HNTG buffer and after tryptic digestions and separation of peptides, samples were subjected to mass spectrometry at the Center for Advanced Proteomics Research at Rutgers University (Newark, NJ). For protein identification, a minimum of five peptides counts were used as threshold cutoff for each protein. Number of peptides enriched in hypoxic conditions is presented as heatmap.
Protein expression and nuclear magnetic resonance analysis
CrkII and CypA were expressed as described previously (18). Briefly, isotopically labeled samples were prepared by growing cells in M9 minimal medium supplemented with 1 g/L of 15NH4Cl and 2 g/L of glucose. CrkII and CypA constructs were grown at 37°C and protein synthesis was induced by addition of 0.25 mmol/L IPTG at OD600 ~0.4. Cells were lysed by sonication and the cytoplasmic fraction separated by centrifugation at 50,000 × g. The lysate was loaded onto Ni-NTA Agarose Resin (GE Healthcare) equilibrated with Tris buffer and 1 mol/L NaCl, pH 8. Protein was eluted with 400 mmol/L imidazole, and after TEV cleavage, the sample was concentrated and applied to a Superdex 75 size-exclude. All samples were monomeric in solution at concentrations used for the nuclear magnetic resonance (NMR) studies (0.3–0.5 mmol/L) as confirmed by multi-angle-light scattering. The NMR buffer used was 50 mmol/L potassium phosphate, 150 mmol/L NaCl, pH 6.5, 3 mmol/L 2-mercaptoethanol, and 0.5 mmol/L EDTA.
Real-time cell migration assay
Real-time cell migration assay was performed using XCELLi-gence RTCA DP. Briefly, cells were serum starved overnight with 0.5% FBS-containing media. A total of 40,000 cells/well were added in top well of XCELLigence RTCA DP plate in 100 μL volume in starvation media, while 180 μL of complete media with 10% FBS was added in bottom chamber as chemo-attractant. For Debio-025 experiments, the drug was added in the starvation media while seeding cells in the XCELLigence plate. For the hypoxia experiment, cells were pretreated with CoCl2 for 24 hours and then subjected to XCELLigence assay.
CypA knockdown
Transient transfection of plasmid encoding short hairpin RNA (shRNA) targeting CypA was performed using specific shRNA obtained from Santa Cruz Biotechnology (catalog no.: sc-142741-SH) according to the manufacturer’s protocol. shRNA plasmids that encodes scrambled shRNA sequence were used as control (sc-108060). Seventy-two hours after transient transfection, cells were harvested for confirmation of knockdown using Western blotting and migration assays.
In vivo experiments
For the in vivo studies, 6-week-old, female Balb/C mice from The Jackson Laboratory were used. A total of 1 × 105 4T1 wild-type cells were injected in the mammary fat pad of each mice. The tumors were palped every 3 days, and body weight and tumor volumes were measured. At the end of the 6 weeks or when tumor size reached 2 cm, the mice were sacrificed, and tumors and lungs were harvested. Debio-025 was administered at indicated concentrations every 3 days via oral gavage. No significant differences in the mice body weight were observed because of drug treatment. Anti-PD-1 mAb (BioXCell, clone: 29F.1A12) or isotype IgG antibodies were administered (intraperitoneally) every 3 days at 200 mg/kg/day dosage in the combination experiments with a total of four administrations per group/study starting at day 10. For estimation of metastasis, lungs were washed with PBS thrice and added to Bouin solution. Pulmonary metastases were counted using stereomicroscope by two investigators separately. All the procedures involving animal care use were approved by IACUC of Rutgers University (Newark, NJ).
NanoString immune-profiling analysis
Total RNA was isolated from three primary tumors/group from each group: placebo + isotype, isotype + Debio-025, placebo + anti-PD-1, and Debio-025 + anti-PD-1 using RNeasy Plus Total RNA Isolation Kit (Qiagen). All the RNA samples passed quality control (assessed by OD 260/280). Samples were subjected to analysis by nCounter murine PanCancer Immune Profiling Panel according to the manufacturer’s protocol at NYU Genomic Center (NanoString Technologies). Normalization of raw data was performed using the nSolver 3.0 Analysis Software (NanoString Technologies). RNA samples were subjected to direct gene expression analysis by measure counts of mRNA/per sample using murine nCounter PanCancer Immune Profiling Panel. Multiplex assay consisting of 770 murine inflammatory response genes were analyzed using nSOLVER Analysis software 3.0 by the methods described previously. Successfully counted Fields of view (FOV) counts for each gene were normalized using average values of FOV counts from 15 housekeeping genes. The gene expression (represented in FOV counts) for each gene for all groups were calculated, and resulting data were presented using GraphPad Prism software for statistical analysis.
The Cancer Genome Atlas and tumor immune dysfunction and exclusion analyses
RNA-sequencing (RNA-seq) datasets from multiple cancer types from The Cancer Genome Atlas (TCGA) and paired normal tissues from GTEx datasets were analyzed for PPIA (CypA) expression using Gene Expression Profiling Interactive Analysis tool (reference). Log2 (TPM+1) scale was used to represent expression of CypA across cancer and normal tissues. Survival analyses were performed using Kaplan–Meier plotter tool (35). To define high and low expression of CypA in tumor RNA-seq samples, the cohorts were divided into two groups according to the median (or top and bottom quartile) expression of the PPIA gene. Follow-up threshold were set to prevent exclusion of all patients from survival analysis. Tumor immune estimation resource (TIMER) analysis was performed to estimate the exclusion of immune cell populations with expression of CypA as described previously (36). Tumor immune dysfunction and exclusion (TIDE) analyses were performed to estimate survival benefit in context of cytotoxic T-lymphocyte infiltration in CypA high and low tumors as described previously (37).
Statistical analyses
Differences between groups in all in vivo experiments were examined for statistical significance using a two-tailed Student t test. Difference in tumor growth was assessed by two-way ANOVA using with repeated measures. Bartlett test was used to determine whether the datasets followed Gaussian distribution. Unpaired t test with Welch correction was used for datasets with Gaussian distribution, while Mann–Whitney test was performed for non-Gaussian datasets. GraphPad PRISM was used to perform statistical analyses and chart the graphs. P < 0.05 was considered significant.
Results
Hypoxia-inducible CypA expression suppresses EGF-induced Crk Y221 phosphorylation
CrkII, an SH2 and SH3 domain–containing adaptor protein, has important functions for cytoskeletal remodeling following extracellular activation of integrins and growth factor receptors such as β1 integrin or EGFR (13, 38, 39). Previously we showed that Crk Y221 phosphorylation and signaling is regulated by CypA (PP1A), a PPIase that is overexpressed in a wide range of cancers, binds GP motif, and delays Crk Y221 phosphorylation (18). Here, by analyzing pan-cancer RNA-seq expression data from 9,736 tumors and 8,587 normal samples (obtained through TCGA and GTex datasets) the expression of CypA mRNA is significantly upregulated in multiple human cancers including breast, colon, prostate, and lung cancer (Fig. 1A). In addition, distant metastasis–free survival analysis using 1,746 breast cancer patient’s microarray datasets showed that higher level of CypA is associated with poor patient survival outcomes (Fig. 1B). Because CypA expression has been linked to hypoxia and hypoxia-inducible factor-1α (HIF1α), via the arrangement of tandem HIF1α responsive elements in the CypA promoter region and can be a way to acutely alter CypA levels (28, 40), we first examined CypA and other members of the PPIase family by unbiased LC/MS-MS techniques in MDA-MB-231 tumor cells under normoxia and hypoxia (Fig. 1C). Interestingly, under these conditions, CypA/PP1A was most robustly upregulated at the protein level compared with the PPI/FKBP family members, although several members (PPIF, PPIG, and others) showed finite upregulation under hypoxia, indicating this phenomenon is not exclusive to PPIA.
Figure 1.
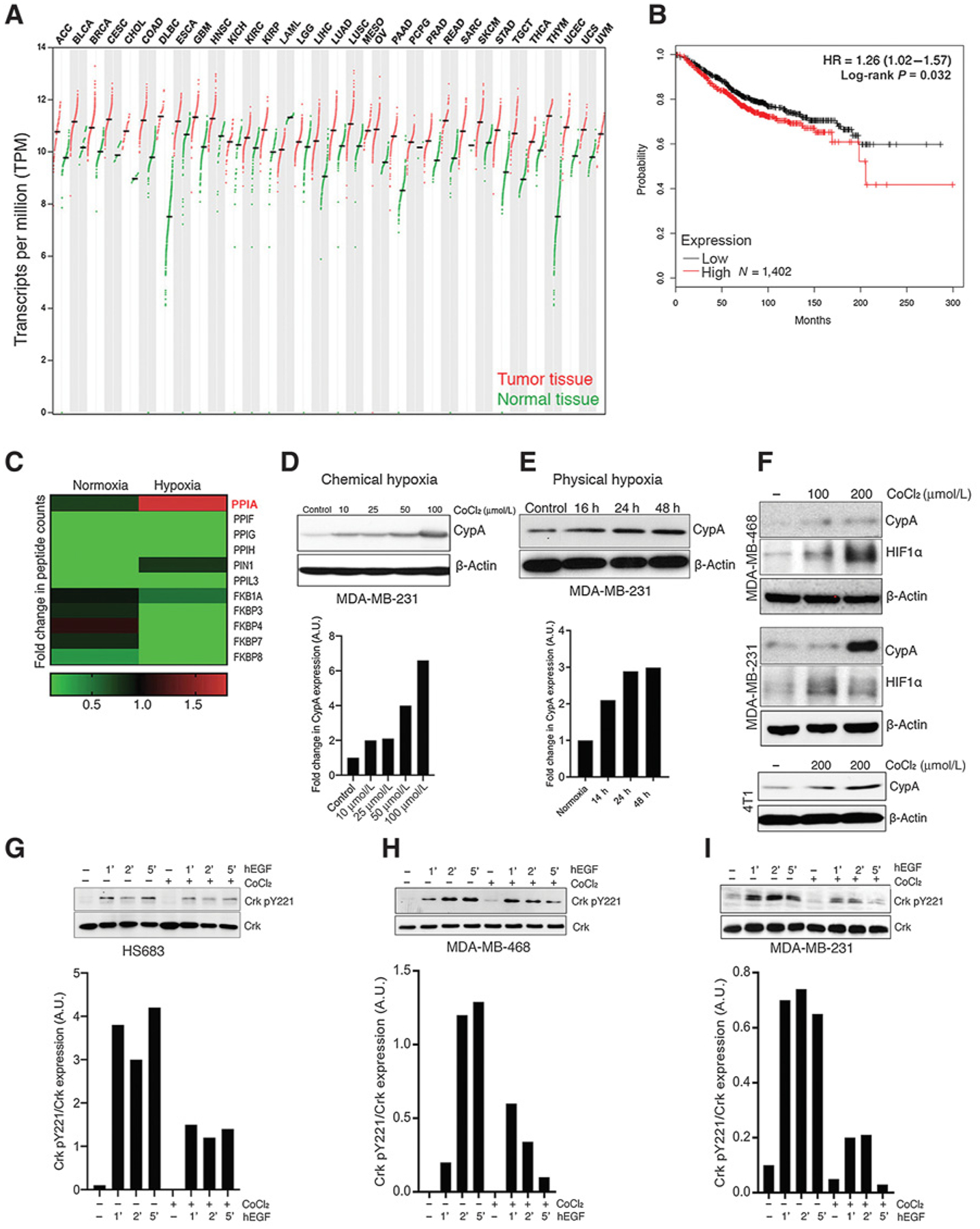
Hypoxia induces CypA expression and suppresses EGF-induced Crk Y221 phosphorylation. A, Expression of CypA in human cancers by TCGA RNA-seq data analysis. Log2 (TPM+1) scale of transcript per million of CypA in indicated number of normal and cancerous tissues are shown. B, Distant metastasis–free survival of breast cancer tumor RNA-seq data analyzed by CypA expression [high (red; N = 1035) and low (black; N = 711) expression] and presented in Kaplan–Meier curve. HR and P value calculated using Cox regression analysis are indicated. C, Protein expression of multiple proline-prolyl isomerase upon induction of hypoxia by mass spectrometric analysis. Relative change in peptide counts of each proline-prolyl isomerase in normoxic and hypoxic conditions presented in heatmap. D and E, Western blot analysis of CypA gene expression in MDA-MB-231 by chemical hypoxia (CoCl2 treatment; a) and physical hypoxia (hypoxia chamber; b). F, Western blotting analysis of hypoxia-induced CypA and HIF1α expression in MDA-MB-231, MDA-MB-468, and 4T1 cells. Representative images from Western blot analysis of Crk Y221 phosphorylation upon induction of hypoxia in HS683 (G), MDA-MB-468 (H), and MDA-MB-231 cells (I). Densitometric analysis using ImageJ is shown below each panel. The bar graphs represent mean expression of Y221 phosphorylated versus total Crk in each group from three independent experiments. Cells were pretreated with CoCl2 overnight in serum-starved medium followed by EGF stimulation for indicated time points, lysates were made and probed for Crk Y221 phosphorylation. ACC, adrenocortical carcinoma; BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, cholangiocarcinoma; COAD, colon adenocarcinoma; DLBC, lymphoid neoplasm diffuse large B-cell lymphoma; ESCA, esophageal carcinoma; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LAML, acute myeloid leukemia; LGG, brain lower grade glioma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; MESO, mesothelioma; OV, ovarian serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; PCPG, pheochromocytoma and paraganglioma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; SARC, sarcoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; TGCT, testicular germ cell tumors; THCA, thyroid carcinoma; THYM, thymoma; UCEC, uterine corpus endometrial carcinoma; UCS, uterine carcinosarcoma; and UVM, uveal melanoma.
To test hypoxia induction of CypA in cancer cells more formally, we subjected MDA-MB-231 cells to chemical hypoxia (CoCl2 induction; ref. 41) and physical hypoxia (2% atmospheric oxygen; Fig. 1D and E) and tested effects of hypoxia-induced CypA on kinetics of Crk Y221 phosphorylation (Fig. 1G–I). Notably, we observed that in both chemical and physical conditions of hypoxia, CypA protein expression in MDA-MB-231, MDA-MB-468, and mouse 4T1 cells, was induced in a dose- and time-dependent manner (Fig. 1D and E) that also correlated with HIF1α expression (Fig. 1F). Previously, we showed that CypA and Crk interaction could suppress Crk Y221 phosphorylation in vitro. We next assessed whether CypA, pathophysiologically induced under conditions of acute experimental hypoxia, could also suppress the kinetics of Crk phosphorylation in cells lines, when Hs683, MDA-MB-468, and MDA-MB-231 cells were subjected to chemical hypoxia and serum starvation as above, followed by EGF stimulation to induce Crk Y221 phosphorylation. As shown in Fig. 1G–I, while EGF stimulation enhances Crk Y221 phosphorylation in a time-dependent manner, Crk Y221 phosphorylation is suppressed by hypoxia induction. Together, these data indicate that hypoxia induction enhances CypA expression and suppresses EGF-induced canonical Crk signaling mediated by Crk Y221 phosphorylation in cancer cell lines.
Debio-025 disrupts the Crk–CypA complex formation by a CypA-specific interaction
We hypothesized that because hypoxia provides a pathophysiologic setting for upregulation of CypA, therefore subsequent alteration in the Crk signaling axis that regulates cell migration, invasion, and metastasis would affect these phenotypes. To address this experimentally, we employed Debio-025 (alisporivir), a previously described CypA inhibitor and nonimmunosuppressive cyclosporin A analog originally developed as a selective HCV inhibitor because HCV replication depends on the PPIase activity of CypA (33, 34). We tested the ability of Debio-025 to block Crk–CypA complex formation using NMR spectroscopy on purified proteins. As such, we first determined the effect of CypA on CrkII (Fig. 2A). As previously shown, CypA uses its catalytic site to bind to CrkII at the GP motif. This binding can be observed by a titration experiment of 15N-labeled CrkII (1–304) with unlabeled CypA. Resonance around Pro220 which include Tyr221, (the primary phosphorylation site of CrkII) and Ala222 are completely broadened out indicating direct binding (Fig. 2D–F). To test whether Debio-025 can displace CypA from the CrkII–CypA complex, Debio-025 was titrated against the 15N-labeled CrkII and unlabeled CypA preformed complex. As shown in Fig. 2C, CypA is completely displaced by the inhibitor, whereby resonances for Y221 and Ala222 reappear (Fig. 2D–F). As a control, to test whether Debio-025 interacts with CrkII, Debio-025 was titrated to 15N-labeled CrkII (1–304). As shown in Fig. 2B, no perturbations were observed indicating that CrkII does not interact with the inhibitor. Rather, Debio-025 binds with high affinity to the CypA catalytic site abolishing the binding of CypA and CrkII (Fig. 2G–I). Together, these data indeed show that Debio-025 blocks the Crk–CypA complex formation by specifically targeting CypA.
Figure 2.
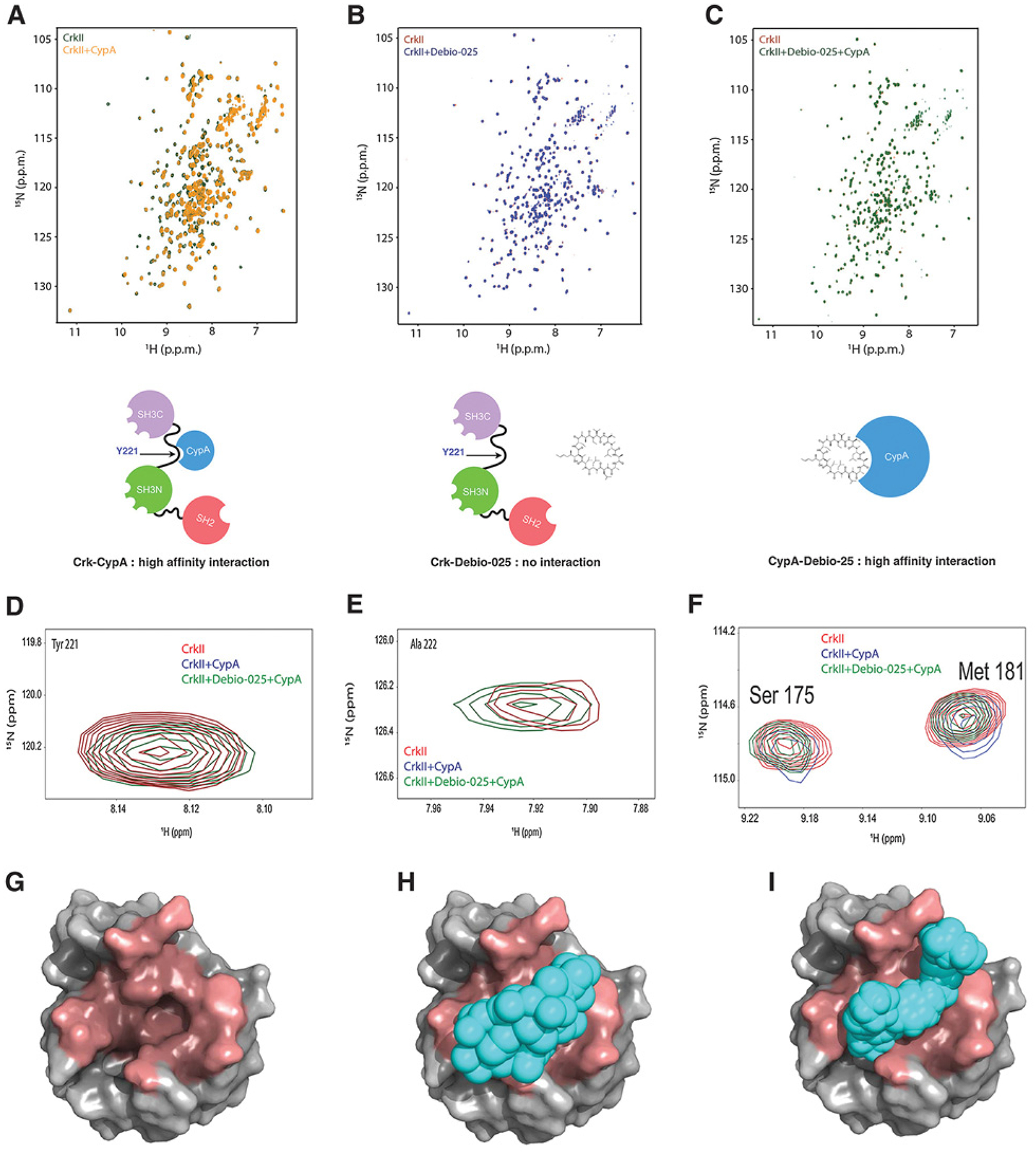
Debio-025 disrupts the Crk–CypA complex formation by a CypA-specific interaction. A, Overlaid 1H-15N Heteronuclear Single Quantum Coherence (HSQC) NMR spectra of labeled CrkII (green) and labeled CrkII titrated with equimolar unlabeled CypA (yellow). B, Overlaid 1H-15N HSQC NMR spectra of labeled CrkII (red) and labeled CrkII titrated with equimolar Debio-025. C, Overlaid 1H-15N NMR spectra of labeled CrkII (red) and labeled CrkII titrated with equimolar unlabeled CypA–Debio-025 complex (green). Schematic models are shown for each condition below. D–F, Zoomed view of 1H-15N HSQC NMR spectra of residues of CrkII showing interaction with CypA. G–I, Molecular models showing CrkII and Debio-025 occupy the same binding site on CypA. CypA (gray, PDB:ID 5HSV) shown as a surface representation and binding pocket for Debio-025 and CrkII (dark red; G), Debio-025 (cyan) bound to CypA (H), and CrkII peptide (216 - PEPGPYAQP - 224; cyan) bound to CypA (PDB ID: 2ms4; I). CrkII and Debio-025 occupies the same binding site on CypA.
Debio-025 treatment inhibits cell migration in a CypA-dependent manner
To investigate whether Debio-025 alters cell migration, a characteristic phenotypic outcome mediated by canonical Crk signaling, DU145 prostate cancer cell line, MDA-MB-231 breast cancer cell line, and Hs683 GBM cell line were serum starved (to reduce Crk Y221 phosphorylation) and subsequently treated with EGF and either 10 μmol/L Debio-025 or vehicle as control and subjected to realtime cell migration analysis using XCELLigence. Under these conditions, Debio-025 treatment suppresses cell migration in all three cancer cell lines (5-fold decrease in DU145 and Hs683 and more than 2-fold decrease in MDA-MB-231 cells; Fig. 3A). To further test the requirement of CypA for Debio-025–mediated suppression of cell migration, CypA was targeted by shRNA knockdown in MDA-MB-231 cells (Fig. 3B) and subjected the CypA-knockdown cells or scramble-knockdown (control) cells to cell migration analysis post-Debio-025 treatment. Interestingly, while Debio-025 suppresses cell migration of control cells, it loses the ability to suppress migration of CypA-knockdown cells (Fig. 3C). We also tested whether Debio-025 affects the cell proliferation of MDA-MB-231 cells, DU145, and Hs683 cell lines using ATP-based cell proliferation analysis (Fig. 3D). Debio-025 does not affect the cell proliferation of the tumor cell lines further reaffirming that the change in cell index in the XCELLigence-based experiments were primarily due to cell migration capacity of cells and not because of increased cell proliferation. These results indicate a functional inhibition of Crk signaling by disrupting the CrkII–CypA complex with Debio-025.
Figure 3.
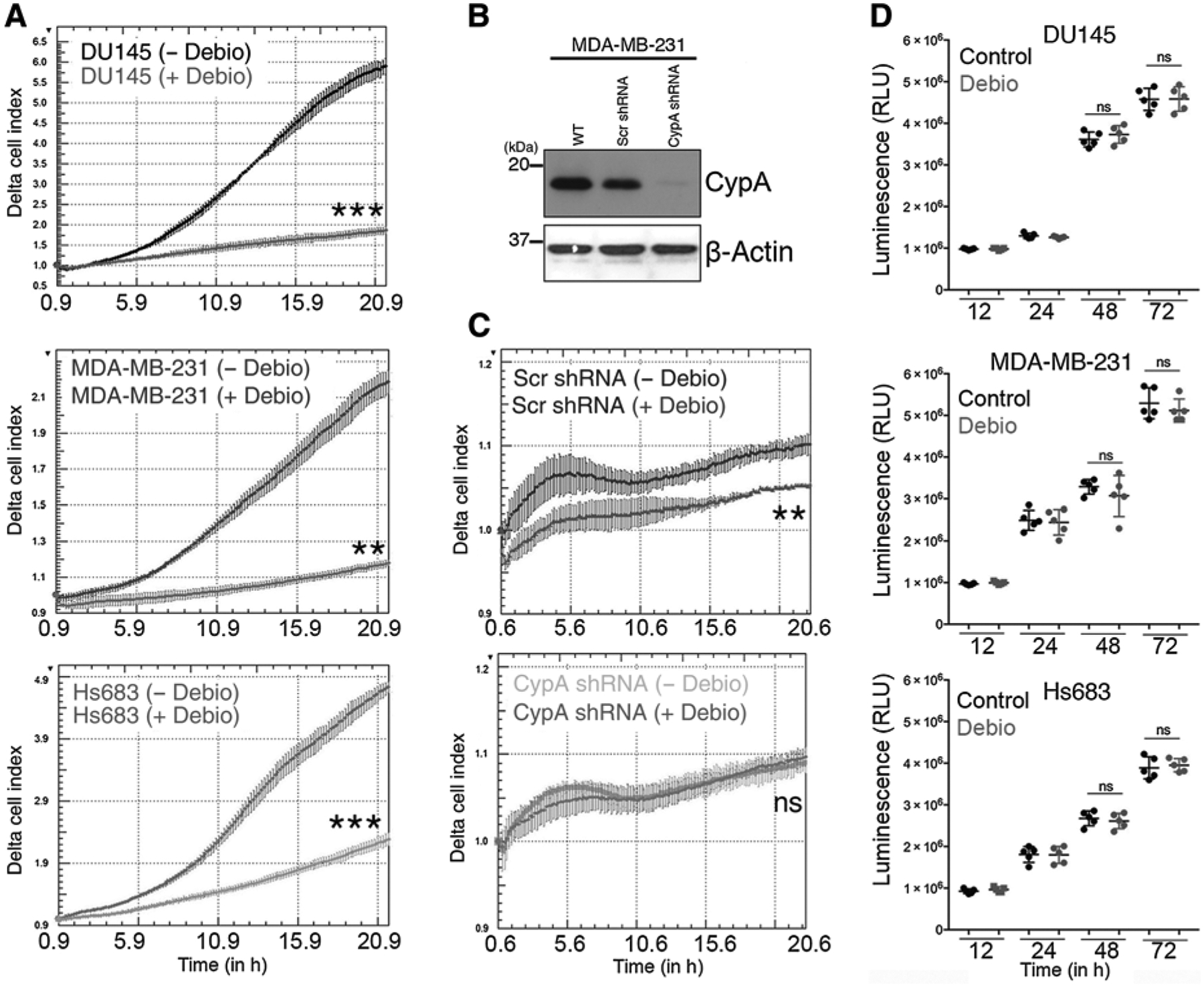
Debio-025 treatment inhibits cell migration in a CypA-dependent manner. A, Tumor cell migration in response to Debio-025 treatment. DU145, MDA-MB-231, and Hs683 cells were assessed for tumor cell migration using XCELLigence assay toward serum gradient. B, Western blotting analysis of CypA knockdown using shRNA in MDA-MB-231 cells. C, Determination of effect of scramble (Scr) and CypA knockdown on migration of MDA-MB-231 cells. D, Cell proliferation assays using CellTiter-Glo luminescent cell viability assay on DU145, MDA-MB-231, and Hs683 cells to test the effect of the Debio-025 on cell proliferation of cancer cells. Error bars, SD; all P values are based on one-sided Student t tests. **, P < 0.001; ***, P < 0.001; ns, nonsignificant; WT, wild-type.
Debio-025 suppresses 4T1 tumor growth and metastasis and enhances efficacy of anti-PD-1 immune checkpoint blockade therapy
As noted above, Crk is overexpressed in many cancers including breast cancer and drives tumor growth and metastasis. We have recently shown that genetic ablation of Crk suppresses tumor immune evasion and enhance responsiveness to anti-PD-1 therapy (30). While CypA augments Crk signaling in biochemical and cell biological outcomes, it is not clear whether increased CypA in a pathologic in vivo setting would enhance tumor growth and metastasis. To assess a potential benefit of targeting CypA with a small-molecule inhibitor in a tumor model, we used oral formulation of Debio-025 in a 4T1 triple-negative breast cancer model. In a therapeutic experiment, 7 days after tumor cell injection, mice were treated every 3 days for five times with different dosage of Debio-025 or placebo control by oral gavage (Fig. 4A). We found that Debio-025 treatment significantly decreases 4T1 tumor growth at 50, 80, and 100 mg/kg/day doses and enhances survival of tumor-bearing mice (Fig. 4A and B). While higher doses of Debio-025 modestly decrease body weight, the mice recover completely on withdrawal of the drug (Fig. 4C). Importantly, tumor weights and pulmonary metastases per mice at the end of the study were significantly suppressed by the Debio-025 treatment (Fig. 4D and E). Representative images of tumor and lung metastasis are shown (Fig. 4D and E, right). To further test whether the differences in the tumor growth in the Debio-025–treated group were due to a tumor intrinsic survival and proliferation defect, the 4T1 tumor cell line was subjected to in vitro cell proliferation assay in presence of different doses of Debio-025. Debio-025 treatment does not inhibit the cell proliferation capacity of 4T1 cells (Fig. 4F), consistent with the results with other cancer cell lines (Fig. 3D), indicating a role for the tumor cell extrinsic mechanism in suppression of tumor growth and metastasis. Concomitant with suppressed tumor growth, splenomegaly, a phenotype linked with tumor growth, was also suppressed by Debio-025 treatment (Fig. 4G). Together, these results show that a novel nonimmunosuppressive CypA inhibitor, Debio-025, can suppress triple-negative breast tumor growth and metastasis by tumor extrinsic mechanism(s).
Figure 4.
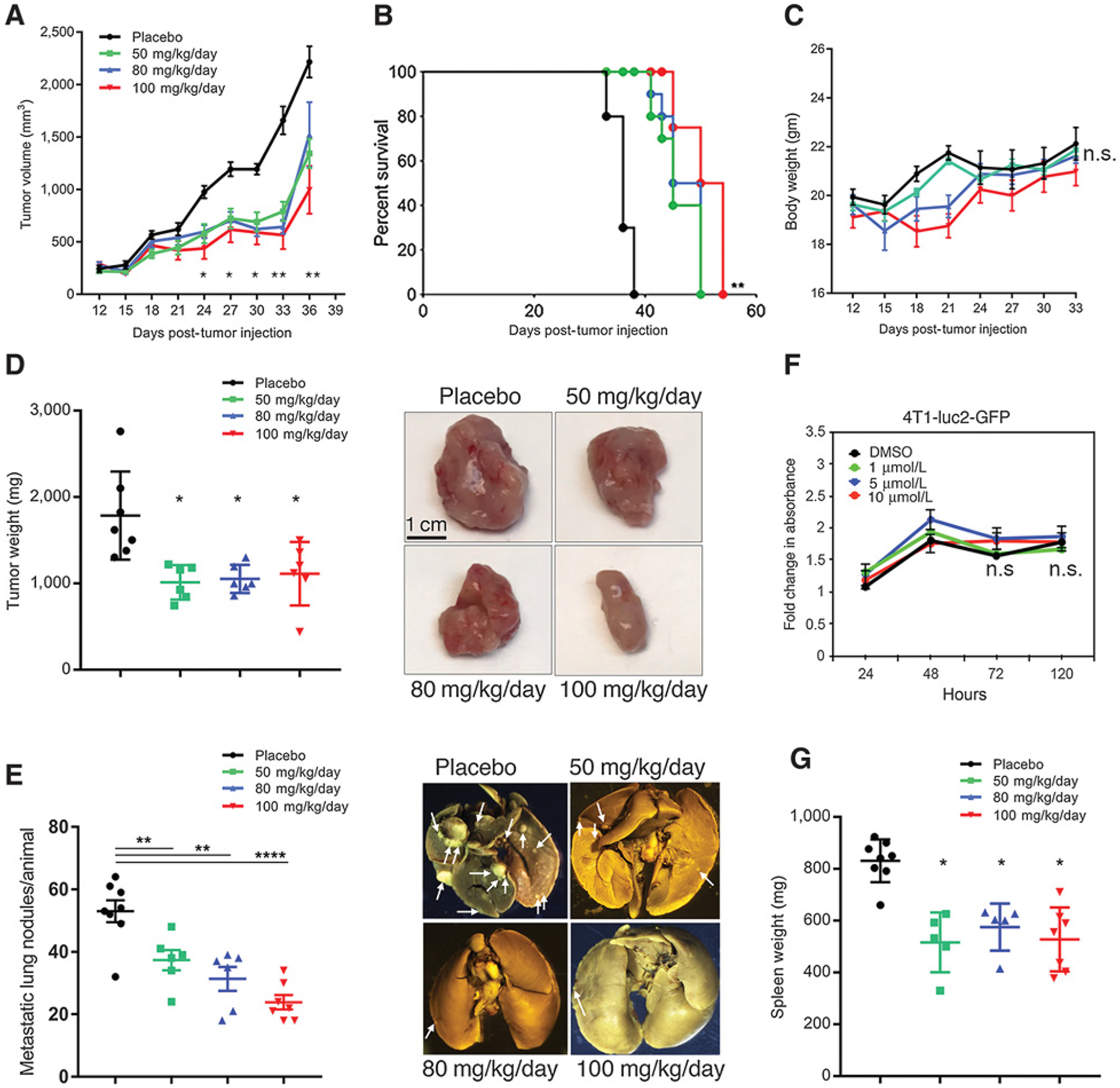
Debio-025 suppresses primary tumor growth and metastasis in murine breast cancer model. A, Tumor growth of wild-type 4T1 cells in mice administered with 50, 80, or 100 mg/kg/day of Debio-025 or vehicle control. B, Kaplan–Meier curve showing percentage survival of tumor-bearing mice upon Debio-025 treatment. C, Body weight analysis of 4T1 tumor–bearing mice upon administration of vehicle or indicated dosage of Debio-025. D, Tumor weight of 4T1 tumor–bearing mice at the end of 36 days upon indicated Debio-025 treatment. Representative tumors sizes from Debio-025–administered groups or vehicle treatment (n = 8/group) are shown. E, Metastasis of 4T1 tumor–bearing mice at the end of 36 days upon indicated Debio-025 treatment with representative metastatic lung nodules from the four treatment groups. F, Cell growth assay using MTT to estimate change in rate of cell proliferation upon treatment with indicated concentrations of Debio-025 for 4 days. G, Change in spleen weight upon Debio-025 treatment in tumor-bearing mice. *, P < 0.05; **, P < 0.001; ****, P < 0.0001; ns, nonsignificant.
Clinical and therapeutic relevance of CypA expression in tumor immunity
Hence, to investigate CypA-dependent tumor cell extrinsic mechanisms that may lead to suppression of tumor growth, we performed a systematic analysis of immune infiltrates in human breast tumor microenvironment using TIMER analysis and human breast cancer TCGA datasets (42). We found that in all breast cancer TCGA datasets (breast invasive carcinoma, luminal, basal, and Her2) mRNA levels of CypA negatively corelated with intratumoral infiltration of CD8 T cell, macrophages, and dendritic cells, indicating a direct role for higher CypA expression in suppression of tumor immunity human breast cancer (Fig. 5A). To examine the clinical benefit in survival of patients based on CypA expression levels and calculated levels of CD8 T-cell infiltration, TIDE analysis was utilized to provide data-driven gene signatures of T-cell dysfunction computed from large amount of cancer clinical datasets including TCGA and METABRIC (37). All patients’ datasets were divided according to expression level of CypA (higher or lower than mean expression values of all patients). The impact of CypA expression and calculated levels of CTL were used to compute overall and disease-free survival of patients. As indicated, the infiltration of CTL in patients with lower expression of CypA significantly improved overall and disease-free survival (Fig. 5B). Higher expression of CypA negatively correlated with CTL (Fig. 5A) and showed no benefit in improving patient survival (Fig. 5B).
Figure 5.
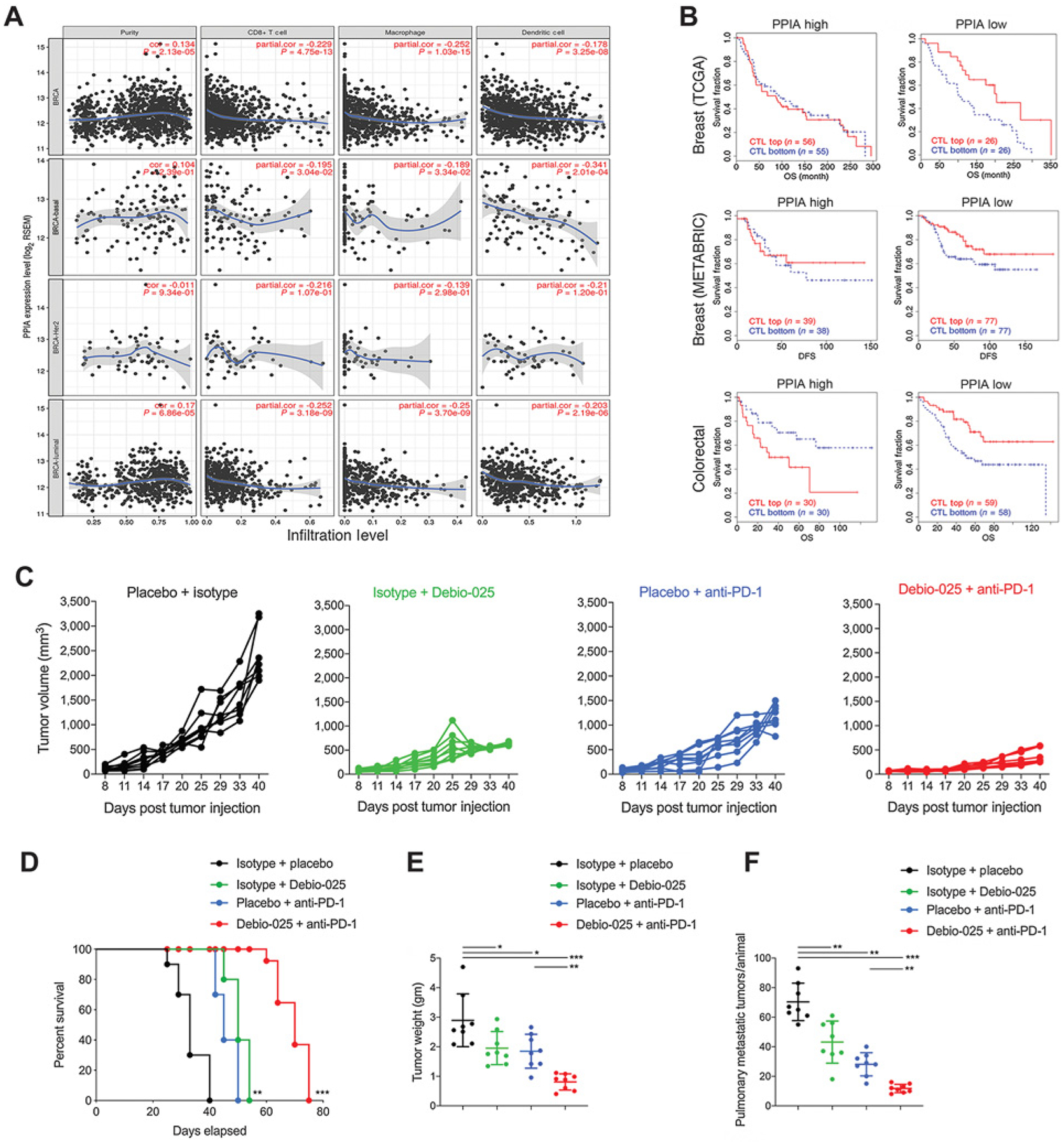
CypA expression negatively correlates with cytotoxic immune cell populations and clinical response to checkpoint blockade and targeting CypA provides enhanced therapeutic response with immunotherapy. A, TIMER analysis plots for breast cancer–sequencing data from TCGA plotted and classified for overall breast cancer specimens (BRCA), basal subtype (BRCA-basal), Her2-negative subtype (BRCA-Her2), and luminal subtype (BRCA-luminal). Adjusted for purity of tumor samples sequencing data, correlation plots show CypA expression and extent of infiltration levels of CD8+ T cells, macrophage, and dendritic cells inferred from sequencing data. B, TIDE analysis plots for estimation of CypA as a gene expression biomarker to predict the clinical response to immune checkpoint blockade in CypA-high and -low expressing breast tumors (top, TCGA cohort), (middle, METABRIC cohort), and colorectal tumors (bottom, TCGA cohort). Estimation of efficacy of Debio-025 anti-PD-1 combination therapy in suppressing primary and metastatic 4T1 tumors: Debio-025 and anti-PD-1 or isotypes and vehicle control on 4T1 tumor growth were administered (see Materials and Methods for details) to estimate changes in primary tumor growth (C), survival (D), tumor weight (E), and pulmonary metastasis (F; n = 6–8/group). Error bars, SD; all P values are based on one-sided Student t tests or two-way repeated measure ANOVA. *, P < 0.05; **, P < 0.001; ***, P < 0.001; DFS, disease-free survival; OS, overall survival.
To experimentally examine a causal effect of intratumoral CypA expression in suppressing tumor-infiltrating immune cell populations, we used anti-PD-1 immunotherapy model. Previously, we have shown that Crk signaling regulates PD-L1 expression on 4T1 cells and that genetic ablation of Crk may increase response to anti-PD-1 checkpoint therapy (30). As Debio-025 targets Crk signaling via CypA, we hypothesized that suppression of CypA–Crk axis by Debio-025 treatment might amplify response to anti-PD-1 treatment by enhancing antitumor immune cell populations in tumor. To test this, mice with palpable tumors were administered every 3 days for four times with 50 mg/kg/day Debio-025 (oral) and 200 mg/kg/day of anti-PD1 (i.p.). While Debio-025 and anti-PD-1 showed suppression in tumor growth and metastasis, both of these phenotypes showed synergistic therapeutic benefit and enhanced survival of tumor-bearing mice upon combination of Debio-025 with anti-PD-1 treatment (Fig. 5C–F). These results indicated a role for CypA targeting by nonimmunosuppressive Debio-025 inhibitor for control of tumor growth, metastasis, and enhancing response to anti-PD-1 checkpoint treatment in breast cancer model.
Combination of Debio-025 with anti-PD-1 immune checkpoint blockade therapy amplifies CD8 T-cell and innate immune cells infiltration and effector response
T-cell exhaustion in breast cancer provides a tumor immune evasion mechanism and provides resistance mechanism to anti-PD-1 immunotherapy. To experimentally examine our observations from the TIMER analysis, indicating a negatively correlation between CypA expression and breast tumor intrinsic CD8 T-cell and innate immune cell populations (macrophages and dendritic cells), we subjected the three tumors/group from Debio-025 and anti-PD-1 combination therapy experiment to unbiased RNA-based tumor immune profiling using NanoString analysis. Analysis of FOV (gene counts) for 700 immune-related genes revealed that intratumoral CD8 T-cells population was synergistically improved by combination of Debio-025 with anti-PD-1. In addition, expression of multiple bonafide genes characteristic of effector T-cell response (Cd2, Glycam1, Cd33, Cd44, Cd69, and Cd27) were upregulated by combination of Debio-025 and anti-PD-1 group as compared with anti-PD-1 treatment alone (Fig. 6A). Similarly, expression of cytotoxic serine protease Granzyme B and mitogenic cytokine IL2 secreted by effector CD8 T cells were enhanced significantly in Debio-025 and anti-PD-1 combination group as compared with anti-PD-1 alone, indicating enhanced cytotoxic and activation status of CD8 T cells in Debio-025 and anti-PD-1 combination therapy group.
Figure 6.
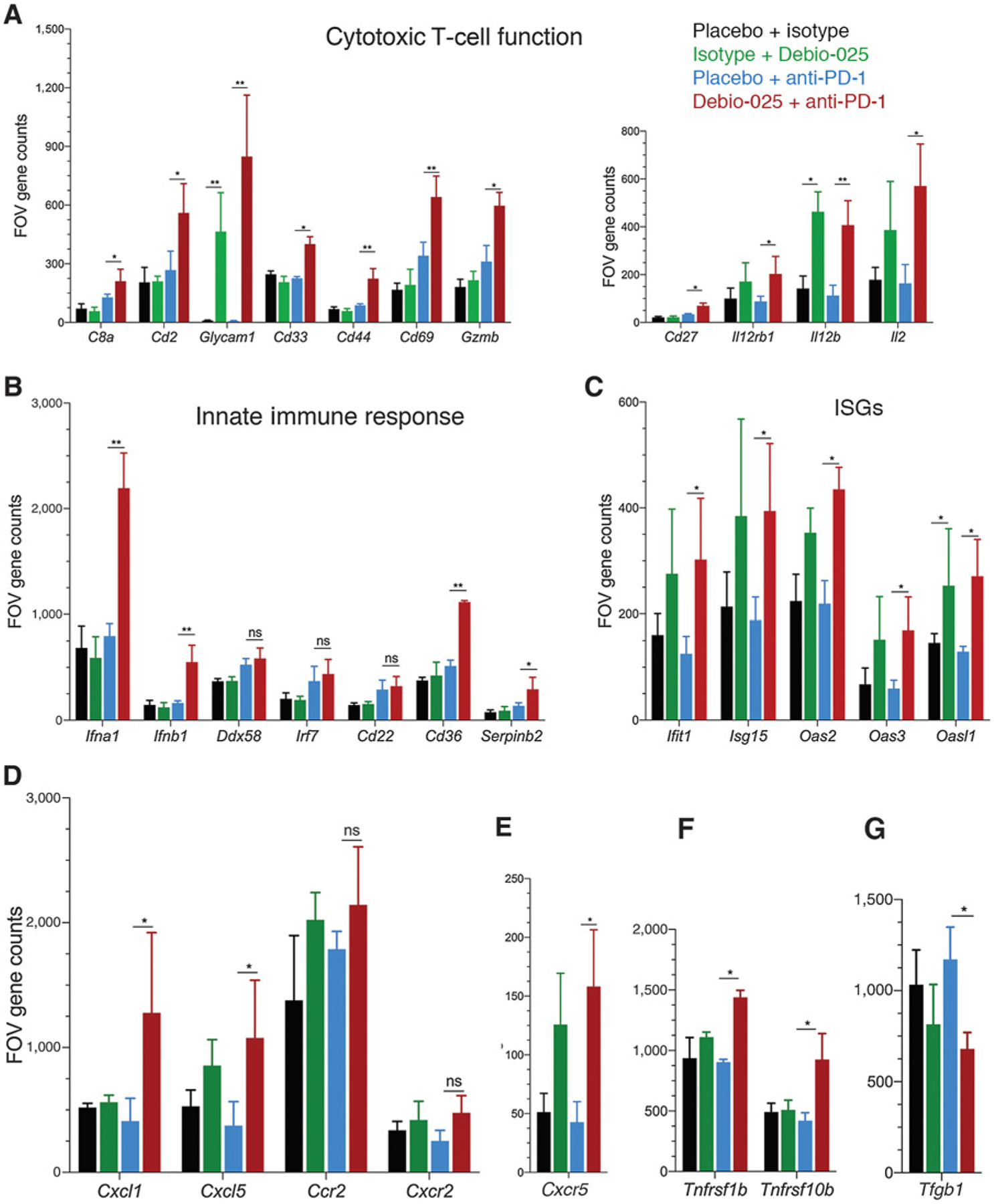
Effect of Debio-025 and anti-PD-1 combinatorial therapy on enrichment of antitumoral immune cell response. Gene expression of immune-related genes using NanoString pan-cancer immune profiling panel was performed from tumors harvested at the end of the study from placebo + isotype, placebo + anti-PD-1, isotype + Debio-025, and Debio-025 + anti-PD-1 treatment groups and subjected to NanoString analysis. FOV counts per gene from each sample were calculated from normalized expression presented using nSolver software. A, FOV counts for T-cell markers and cytokines related to T-cell function are shown (CD8a, Cd2, Glycam1, Cd33, Cd44, Cd69, Gzmb, Cd27, Il12rb1, Il12b, and Il2). B, FOV counts from RNA-based NanoString analysis for innate immune response markers are shown for each treatment group (Cd22, Cd36, Ddx58, Ifna1, Ifna4, Ifnb1, and Irf7). C, IFN-stimulated gene (ISG) expressions between different groups are shown (Ift1, Isg15, Oas2, Oas3, and Oasl1). Immuno-attractant chemokines (Cxcl1 and Cxcl5; D) and receptors (Ccr2 and Cxcr2) and Cxcr5 (E), antitumor chemokine receptors (Tnfrsf10b and Tnfrsf1b; F), and TGFβ (G) gene expressions between each group are shown. RNA expression values are presented in FOV counts and graphically represented by GraphPad Prism. Error bars indicate SD. Statistically significant differences are indicated in each case. *, P < 0.05; **, P < 0.01; versus vehicle group (n = 3 per group; Student two-tailed t test; ns, nonsignificant).
As the results from TIMER analysis (Fig. 5A) predicted an inverse correlation between CypA expression and innate immune cell (macrophages and dendritic cells) populations, we analyzed expression of genes characteristic of this pathway from the aforementioned NanoString data. We found that Ifna1, Ifnb1, and Cd36 genes that are involved in type I IFN response were upregulated in Debio-025 + anti-PD-1 combination group as compared with anti-PD-1 treatment alone (Fig. 6B). Serpinb2 (plasminogen activator inhibitor-2) that is commonly induced during inflammatory processes and upregulated in activated macrophages were amplified in combination group. Consistent with increase in type I IFN genes, IFN-stimulated genes Ifit1, Isg15, Oas2, Oas3, and Oasl1, which mediate antiviral immunity, were also significantly upregulated by both Debio-025 single agent and Debio-025 + anti-PD-1 combination groups (Fig. 6C). Although Ddx58, Irf7, and Cd22 innate immune genes showed a trend of additive increased expression in combination group, it did not reach significance. The analysis of tumor microenvironment expression of cytokines, chemokines, and chemokine receptors was extended; whereby neutrophil-recruiting cytokines Cxcl1 (expressed on activated macrophages) and Cxcl5 (produced after stimulation of cells with inflammatory cytokines such as TNFα and IL1) were upregulated in combination group as compared with anti-PD-1 alone (Fig. 6D). Consistently, Cxcr5 (promotes tumor infiltration and proinflammatory functions of CD8 T cells), Tnfrsf1b (TNF receptor 1b), and Tnfrsf10b (TRAIL receptor 2) that promotes TNFα-induced apoptosis were also upregulated by Debio-025 and anti-PD-1 combination (Fig. 6E and F). Conversely, expression of tumor-promoting anti-inflammatory cytokine TGFβ was significantly suppressed by combination therapy (Fig. 6G). Taken together, these results suggest a direct role for targeting CypA to enhance tumor immunogenicity and also identify a potential strategy for improving tumor response to anti-PD-1 therapy.
Discussion
Previously, we showed that CypA, a PPIase upregulated in cancers and a key determinant to malignant transformation and metastasis, binds to the GPY221 auto-inhibitory motif in CrkII to delay Crk Tyr221 phosphorylation and the subsequent downregulation in Crk signaling (18). These results, together with observations that both CypA and Crk are substantially upregulated in clinically high-grade cancers, suggest that targeting the CypA axis with nonimmunosuppressive cyclosporin A derivatives might be an effective approach for cancer therapeutics, in part by targeting the Crk/CypA axis. Here, we show that Debio-025 (alisporivir), a nonimmunosuppressive small-molecule CypA inhibitor, is effective to inhibit Crk/CypA interactions by binding to the catalytic site of CypA. Debio-025 is effective in blocking cell migration of tumor cells, and biochemically can target CrkII by delaying the kinetics of Crk Y221 phosphorylation, a read-out of Crk adaptor protein function. In vivo, Debio-025 has notable antitumorigenic and antimetastatic activity in a 4T1 mouse orthotopic model of triple-negative breast cancer. When combined with immune checkpoint therapeutic anti-PD-1 mAb, Debio-025 cooperates to enhance antitumor and antimetastatic activity and induces an immunogenic response including the activation of cytotoxic T-cell and innate immunity. These studies identify an unanticipated function for CypA inhibition in the immuno-oncology applications that can be explored in a broader range of tumor types.
While CypA is ubiquitously expressed, and has pleotropic activities that control protein dynamics and folding under normal physiologic conditions, it is also substantially upregulated in a variety of solid cancers (43) and is a direct target of HIF1α, indicating CypA’s role in the adaptation of the tumor microenvironment to hypoxia (28). Mechanistically, the oncogenic roles of CypA are likely to be complex and multifactorial. Elevated levels of CypA can act on general protein folding pathways (44, 45) acting as “foldases” to accommodate increased rates of protein translation (observed in cancers) or can act in a chaperone capacity to stabilize oncogenic client proteins from ubiquitin-mediated degradation (27, 46). However, emerging studies also indicate that CypA targets specific signaling and oncogenic pathways to facilitate cancer progression. For example, it has been observed that CypA regulates Jak2/Stat5 signaling in the mammary epithelium and that CypA knockout, or CypA inhibitors (Cyclosporin A or NIM811) blocked prolactin-dependent Jak2 phosphorylation and subsequent breast cancer proliferation, motility, invasion, and metastatic progression in estrogen receptor–positive tumors (47). Other studies have shown that CypA promotes non–small cell lung cancer via p38 MAPK (22), and in glioma, CypA can maintain glioma-initiating cell stemness by regulating Wnt/β-catenin signaling (48). Interestingly, in addition to its intracellular role, CypA can also be secreted from cells in response to inflammation and hypoxia (46, 49) and, in doing so, act on specific extracellular receptors and matrix metalloproteinases such as CD147, MMP-3, and MMP-9 (29, 43) to alter the tumor microenvironment. In this study, we show that CypA targets the canonical auto-inhibitory switch mechanism (Tyr221 phosphorylation) that is required for downregulating Crk adaptor function and suppressing migration and invasion. Evidence for a functional biological role for the Crk Y221/Crk is supported by our studies that hypoxia acutely increases CypA levels and concomitantly delays pTyr221 phosphorylation in response to EGF. This supports the idea that in addition to increased CypA in many solid cancers, hypoxia may additionally target the Crk/CypA axis to increase migration and invasion of cancer cells.
While Crk has been intensely studied with respect to its central role in cell motility and invasion, and in metastasis in vivo, we have recently shown using CRISPR-Cas9–mediated knockout of Crk in the poorly immunogenic breast adenocarcinoma 4T1 model, that Crk knockout suppresses EMT and PD-L1 expression on tumor cells, and acts additively with anti-PD-1 therapy (30). Interestingly, Crk-knockout tumors showed significant increase in intratumoral immune infiltration of CD8+ and CD3+ cells, elevations in immunogenic cytokines. Cell autonomous effects of Crk knockout are reminiscent of studies by Jiang and colleagues, showing that inhibitors of focal adhesion kinase (FAK; ref. 50) increases tumor surveillance and synergizes with anti-PD-1 checkpoint therapeutics (51). Consistent with this idea, studies employing genome-scale CRISPR-Cas9 screens to identify mechanisms of tumor cell resistance have identified Crk as a candidate marker that unsensitizes tumor cells to T-cell killing (52). Hence, signaling at focal adhesions, such as the activation of Crk and FAK may act as “signaling hubs” for cell intrinsic immune escape. While it has been experimentally difficult to target Crk, either using phosphopeptide mimetics to target the SH2 domain or proline-rich peptide mimetics to target the SH3 domain, as these consensus sequences target overlapping SH2/SH3 domains. Likewise, although miR-126 has been shown to be downregulated in several cancers and can target Crk, this miR-126 also has significant off-target effects and lacks ease of delivery of into tumor cells (53–55). The current findings that CypA inhibitors Debio-025 partially pheno-copy Crk knockout together with fact that Crk is regulated in trans by CypA might have translational significance, given the previous development and FDA approval of Debio-025 for other applications such as HCV replication.
In summary, our results indicated that Crk, via its tyrosine 221 auto-inhibitory motif, represents a functionally relevant target for CypA and may be targeted by small-molecule CypA inhibitors such as Debio-025. A correlation was observed between elevated CypA levels and cancer grade and furthermore, mouse models indicate that in vivo administration of Debio-025 alone or in combination with anti-PD-1 checkpoint promote significant tumor suppression in a triple-negative breast cancer model. The models here support further preclinical investigations using nonimmunosuppressive CypA inhibitors such as Debio-025 for repurposing into oncology applications.
Acknowledgments
We thank the members of the Birge laboratory for helpful discussions. We thank Anne Vaslin-Chessex and Gregoire Vuagniaux at Debiopharm for kindly providing the Debio-025 for in vitro and in vivo experiments. This work was supported in part by NIH R01 CA165077 to R.B. Birge. We acknowledge Rutgers Society of Research Scholars award to S. Kumar.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Matsuda M, Mayer BJ, Fukui Y, Hanafusa H. Binding of transforming protein, P47gag-crk, to a broad range of phosphotyrosine-containing proteins. Science 1990;248:1537–9. [DOI] [PubMed] [Google Scholar]
- 2.Mayer BJ, Hamaguchi M, Hanafusa H. A novel viral oncogene with structural similarity to phospholipase C. Nature 1988;332:272–5. [DOI] [PubMed] [Google Scholar]
- 3.Hanafusa H Protein phosphorylation and cell transformation. Zhonghua Min Guo Wei Sheng Wu Ji Mian Yi Xue Za Zhi 1991;24:1–9. [PubMed] [Google Scholar]
- 4.Mayer BJ, Hanafusa H. Mutagenic analysis of the v-crk oncogene: requirement for SH2 and SH3 domains and correlation between increased cellular phosphotyrosine and transformation. J Virol 1990;64:3581–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Birge RB, Kalodimos C, Inagaki F, Tanaka S. Crk and CrkL adaptor proteins: networks for physiological and pathological signaling. Cell Commun Signal 2009;7:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feller SM. Crk family adaptors-signalling complex formation and biological roles. Oncogene 2001;20:6348–71. [DOI] [PubMed] [Google Scholar]
- 7.Liu D. The adaptor protein Crk in immune response. Immunol Cell Biol 2014;92:80–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar S, Fajardo JE, Birge RB, Sriram G. Crk at the quarter century mark: perspectives in signaling and cancer. J Cell Biochem 2014;115:819–25. [DOI] [PubMed] [Google Scholar]
- 9.Sriram G, Birge RB. Emerging roles for crk in human cancer. Genes Cancer 2010; 1:1132–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsuda M, Tanaka S, Nagata S, Kojima A, Kurata T, Shibuya M. Two species of human CRK cDNA encode proteins with distinct biological activities. Mol Cell Biol 1992;12:3482–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reichman CT, Mayer BJ, Keshav S, Hanafusa H. The product of the cellular crk gene consists primarily of SH2 and SH3 regions. Cell Growth Differ 1992;3: 451–60. [PubMed] [Google Scholar]
- 12.Kobashigawa Y, Sakai M, Naito M, Yokochi M, Kumeta H, Makino Y, et al. Structural basis for the transforming activity of human cancer-related signaling adaptor protein CRK. Nat Struct Mol Biol 2007;14:503–10. [DOI] [PubMed] [Google Scholar]
- 13.Hashimoto Y, Katayama H, Kiyokawa E, Ota S, Kurata T, Gotoh N, et al. Phosphorylation of CrkII adaptor protein at tyrosine 221 by epidermal growth factor receptor. J Biol Chem 1998;273:17186–91. [DOI] [PubMed] [Google Scholar]
- 14.Kurokawa K, Mochizuki N, Ohba Y, Mizuno H, Miyawaki A, Matsuda M. A pair of fluorescent resonance energy transfer-based probes for tyrosine phosphorylation of the CrkII adaptor protein in vivo. J Biol Chem 2001;276:31305–10. [DOI] [PubMed] [Google Scholar]
- 15.Kumar S, Lu B, Davra V, Hornbeck P, Machida K, Birge RB. Crk tyrosine phosphorylation regulates PDGF-BB-inducible Src activation and breast tumorigenicity and metastasis. Mol Cancer Res 2018;16:173–83. [DOI] [PubMed] [Google Scholar]
- 16.Feller SM, Knudsen B, Hanafusa H. c-Abl kinase regulates the protein binding activity of c-Crk. EMBO J 1994;13:2341–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ren R, Ye ZS, Baltimore D. Abl protein-tyrosine kinase selects the Crk adapter as a substrate using SH3-binding sites. Genes Dev 1994;8:783–95. [DOI] [PubMed] [Google Scholar]
- 18.Saleh T, Jankowski W, Sriram G, Rossi P, Shah S, Lee KB, et al. Cyclophilin A promotes cell migration via the Abl-Crk signaling pathway. Nat Chem Biol 2016; 12:117–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nath PR, Dong G, Braiman A, Isakov N. Immunophilins control T lymphocyte adhesion and migration by regulating CrkII binding to C3G. J Immunol 2014; 193:3966–77. [DOI] [PubMed] [Google Scholar]
- 20.Zheng J, Koblinski JE, Dutson LV, Feeney YB, Clevenger CV. Prolyl isomerase cyclophilin A regulation of Janus-activated kinase 2 and the progression of human breast cancer. Cancer Res 2008;68:7769–78. [DOI] [PubMed] [Google Scholar]
- 21.Howard BA, Furumai R, Campa MJ, Rabbani ZN, Vujaskovic Z, Wang XF, et al. Stable RNA interference-mediated suppression of cyclophilin A diminishes non-small-cell lung tumor growth in vivo. Cancer Res 2005;65:8853–60. [DOI] [PubMed] [Google Scholar]
- 22.Guo Y, Jiang M, Zhao X, Gu M, Wang Z, Xu S, et al. Cyclophilin A promotes non-small cell lung cancer metastasis via p38 MAPK. Thorac Cancer 2018;9:120–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakano N, Sakashita S, Matsuoka R, Murata Y, Shiba-Ishii A, Kobayashi N, et al. Cyclophilin A expression and its prognostic significance in lung adenocarcinoma. Pathol Int 2017;67:555–63. [DOI] [PubMed] [Google Scholar]
- 24.Feng W, Xin Y, Xiao Y, Li W, Sun D. Cyclophilin A enhances cell proliferation and xenografted tumor growth of early gastric cancer. Dig Dis Sci 2015;60: 2700–11. [DOI] [PubMed] [Google Scholar]
- 25.Li M, Zhai Q, Bharadwaj U, Wang H, Li F, Fisher WE, et al. Cyclophilin A is overexpressed in human pancreatic cancer cells and stimulates cell proliferation through CD147. Cancer 2006;106:2284–94. [DOI] [PubMed] [Google Scholar]
- 26.Al-Ghoul M, Brück TB, Lauer-Fields JL, Asirvatham VS, Zapata C, Kerr RG, et al. Comparative proteomic analysis of matched primary and metastatic melanoma cell lines. J Proteome Res 2008;7:4107–18. [DOI] [PubMed] [Google Scholar]
- 27.Lee J. Role of cyclophilin a during oncogenesis. Arch Pharm Res 2010;33:181–7. [DOI] [PubMed] [Google Scholar]
- 28.Zhang H, Chen J, Liu F, Gao C, Wang X, Zhao T, et al. CypA, a gene downstream of HIF-1α, promotes the development of PDAC. PLoS One 2014;9:e92824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qian Z, Zhao X, Jiang M, Jia W, Zhang C, Wang Y, et al. Downregulation of cyclophilin A by siRNA diminishes non-small cell lung cancer cell growth and metastasis via the regulation of matrix metallopeptidase9. BMCCancer 2012;12:442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar S, Davra V, Obr AE, Geng K, Wood TL, De Lorenzo MS, et al. Crk adaptor protein promotes PD-L1 expression, EMT and immune evasion in a murine model of triple-negative breast cancer. Oncoimmunology 2017;7:e1376155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi KJ, Piao YJ, Lim MJ, Kim JH, Ha J, Choe W, et al. Overexpressed cyclophilin A in cancer cells renders resistance to hypoxia- and cisplatin-induced cell death. Cancer Res 2007;67:3654–62. [DOI] [PubMed] [Google Scholar]
- 32.Seko Y, Fujimura T, Taka H, Mineki R, Murayama K, Nagai R. Hypoxia followed by reoxygenation induces secretion of cyclophilin A from cultured rat cardiac myocytes. Biochem Biophys Res Commun 2004;317:162–8. [DOI] [PubMed] [Google Scholar]
- 33.Coelmont L, Hanoulle X, Chatterji U, Berger C, Snoeck J, Bobardt M, et al. DEB025 (alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A. PLoS One 2010;5:e13687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Landrieu I, Hanoulle X, Bonachera F, Hamel A, Sibille N, Yin Y, et al. Structural basis for the non-immunosuppressive character of the cyclosporin A analogue Debio 025. Biochemistry 2010;49:4679–86. [DOI] [PubMed] [Google Scholar]
- 35.Nagy A, Lánczky A, Menyhárt O, Győrffy B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci Rep 2018;15:9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS, et al. A web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res 2017; 77:E108–E110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 2018;10:1550–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katayama H, Hashimoto Y, Kiyokawa E, Nakaya M, Sakamoto A, Machinami R, et al. Epidermal growth factor-dependent dissociation of CrkII proto-oncogene product from the epidermal growth factor receptor in human glioma cells. Jpn J Cancer Res 1999;90:1096–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takino T, Tamura M, Miyamori H, Araki M, Matsumoto K, Sato H, et al. Tyrosine phosphorylation of the CrkII adaptor protein modulates cell migration. J Cell Sci 2003;116:3145–55. [DOI] [PubMed] [Google Scholar]
- 40.Mao M, Yu X, Ge X, Gu R, Li Q, Song S, et al. Acetylated cyclophilin A is a major mediator in hypoxia-induced autophagy and pulmonary vascular angiogenesis. J Hypertens 2017;35:798–809. [DOI] [PubMed] [Google Scholar]
- 41.Guo M, Song LP, Jiang Y, Liu W, Yu Y, Chen GQ. Hypoxia-mimetic agents desferrioxamine and cobalt chloride induce leukemic cell apoptosis through different hypoxia-inducible factor-1alpha independent mechanisms. Apoptosis 2006;11:67–77. [DOI] [PubMed] [Google Scholar]
- 42.Li B, Severson E, Pignon JC, Zhao H, Li T, Novak J, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol 2016; 17:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Obchoei S, Wongkhan S, Wongkham C, Li M, Yao Q, Chen C. Cyclophilin A: potential functions and therapeutic target for human cancer. Med Sci Monit 2009;15:RA221–32. [PubMed] [Google Scholar]
- 44.Lang K, Schmid FX. Protein-disulphide isomerase and prolyl isomerase act differently and independently as catalysts of protein folding. Nature 1988;331: 453–55. [DOI] [PubMed] [Google Scholar]
- 45.Wang P, Heitman J. The cyclophilins. Genome Biol 2005;6:226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nigro P, Pompilio G, Capogrossi MC. Cyclophilin A: a key player for human disease. Cell Death Dis 2013;4:e888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Volker SE, Hedrick SE, Feeney YB, Clevenger CV. Cyclophilin A function in mammary epithelium impacts Jak2/Stat5 signaling, morphogenesis, differentiation, and tumorigenesis in the mammary gland. Cancer Res 2018;78: 3877–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang G, Shen J, Sun J, Jiang Z, Fan J, Wang H, et al. Cyclophilin A maintains glioma-initiating cell stemness by regulating Wnt/β-catenin signaling. Clin Cancer Res 2017;23:6640–9. [DOI] [PubMed] [Google Scholar]
- 49.Jin ZG, Melaragno MG, Liao DF, Yan C, Haendeler J, Suh YA, et al. Cyclophilin A is a secreted growth factor induced by oxidative stress. Circ Res 2000;87:789–96. [DOI] [PubMed] [Google Scholar]
- 50.Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med 2016;22:851–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Symeonides SN, Anderton SM, Serrels A. FAK-inhibition opens the door to checkpoint immunotherapy in pancreatic cancer. J Immunother Cancer 2017;5:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 2018;359:770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frampton AE, Krell J, Jacob J, Stebbing J, Castellano L, Jiao LR. Loss of miR-126 is crucial to pancreatic cancer progression. Expert Rev Anticancer Ther 2012;12: 881–4. [DOI] [PubMed] [Google Scholar]
- 54.Lu W, Tao X, Fan Y, Tang Y, Xu X, Fan S, et al. LINC00888 promoted tumorigenicity of melanoma via miR-126/CRK signaling axis. Onco Targets Ther 2018;11:4431–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yue S, Shi H, Han J, Zhang T, Zhu W, Zhang D. Prognostic value of microRNA-126 and CRK expression in gastric cancer. Onco Targets Ther 2016;9:6127–35. [DOI] [PMC free article] [PubMed] [Google Scholar]