

Abstract
Despite improvements over the past 20 years, African Americans continue to have the highest incidence and mortality rates of colorectal cancer (CRC) in the US. While previous studies have found that copy number variations (CNVs) occur at similar frequency in African American and White CRCs, copy neutral loss of heterozygosity (cnLOH) has not been investigated. In the present study, we used publicly available data from The Cancer Genome Atlas (TCGA) as well as data from an African American CRC cohort, the Chicago Colorectal Cancer Consortium (CCCC), to compare frequencies of CNVs and cnLOH events in CRCs in the two racial groups. Using genotype microarray data, we analyzed large-scale CNV and cnLOH events from 166 microsatellite stable CRCs—31 and 39 African American CRCs from TCGA and the CCCC, respectively, and 96 White CRCs from TCGA. As reported previously, the frequencies of CNVs were similar between African American and White CRCs; however, there was a significantly lower frequency of cnLOH events in African American CRCs compared to White CRCs, even after adjusting for demographic and clinical covariates. Although larger differences for chromosome 18 were observed, a lower frequency of cnLOH events in African American CRCs was observed for nearly all chromosomes. These results suggest that mechanistic differences, including differences in the frequency of cnLOH, could contribute to clinicopathological disparities between African Americans and Whites. Additionally, we observed a previously uncharacterized phenomenon we refer to as small interstitial cnLOH, in which segments of chromosomes from 1–5 Mb long were affected by cnLOH.
Keywords: Copy number variation, copy-neutral loss of heterozygosity, African American colorectal cancer, The Cancer Genome Atlas, genotype and microarray data
Introduction
Despite the decrease in incidence and increase in survival of colorectal cancer (CRC) in the US over the past 20 years1, the health disparity affecting African American patients is still a major concern. African Americans continue to have the highest incidence and mortality rates of colorectal cancer in the US. Additionally, African American CRCs are more likely to present at a more advanced stage and to present with disease on the right side of the colorectum. The disparity remains even after correcting for socioeconomic factors, indicating that there is a biological component of CRC development in African Americans that contributes to these differences1.
Most CRCs are driven at least in part by a chromosomal instability that results in copy number variation (CNV). CNVs can be focal, affecting a small segment of the genome, but many studies focus on recurrent large-scale CNVs, which affect whole chromosome arms or entire chromosomes. These large-scale events are common in CRC and can result in dosage changes of oncogenes or tumor suppressors that drive tumorigenesis forward. Previous studies have investigated large-scale CNVs (i.e., chromosome gains and losses) in African American CRCs. These studies found that large-scale CNVs occur at similar frequencies in African American and White CRCs2–4. However, there is a source of chromosomal instability that has not yet been investigated, namely, copy-neutral loss of heterozygosity (cnLOH).
Tumor-specific cnLOH events are somatic mutations that cause reduction to homozygosity, that is, they generate regions of the chromosome that retain two copies of syntenic loci that are derived from either the maternal or paternal homolog in the tumor but that are bi-parental in the progenitor non-neoplastic tissue. Reduction to homozygosity can drive tumorigenesis if, for example, the genetic change results in the loss of a tumor suppressor gene function whereas two functional copies of the other genes are retained so that gene dosage is maintained. Large-scale cnLOH is generated by at least two different molecular mechanisms, including mechanisms based on homologous recombination and a chromosome non-disjunction (Figure 1). The first mechanism occurs via an inter-homolog recombination event that upon mitotic segregation results in chromosome-arm cnLOH (Figure 1A). Reduction to homozygosity via the homologous recombination mechanism frequently occurs as a tumor-initiating event at the APC gene5. The second mechanism occurs during mitosis due to syntelic attachments and centromere division failure, resulting in both chromatids segregating to one pole and neither chromatid to the other. A second chromosome non-disjunction event is required to eliminate the homolog or to reduplicate the chromosome to create a cell with a copy-neutral change (Figure 1B). These two genetic mechanisms result in cnLOH events that typically span a large segment of a chromosome arm (i.e., homologous recombination class) or the entire chromosome or chromosome arm (i.e., loss and reduplication). In contrast, chromosome-wide gains and losses arise predominantly from a simple chromosome non-disjunction mechanism.
Figure 1.
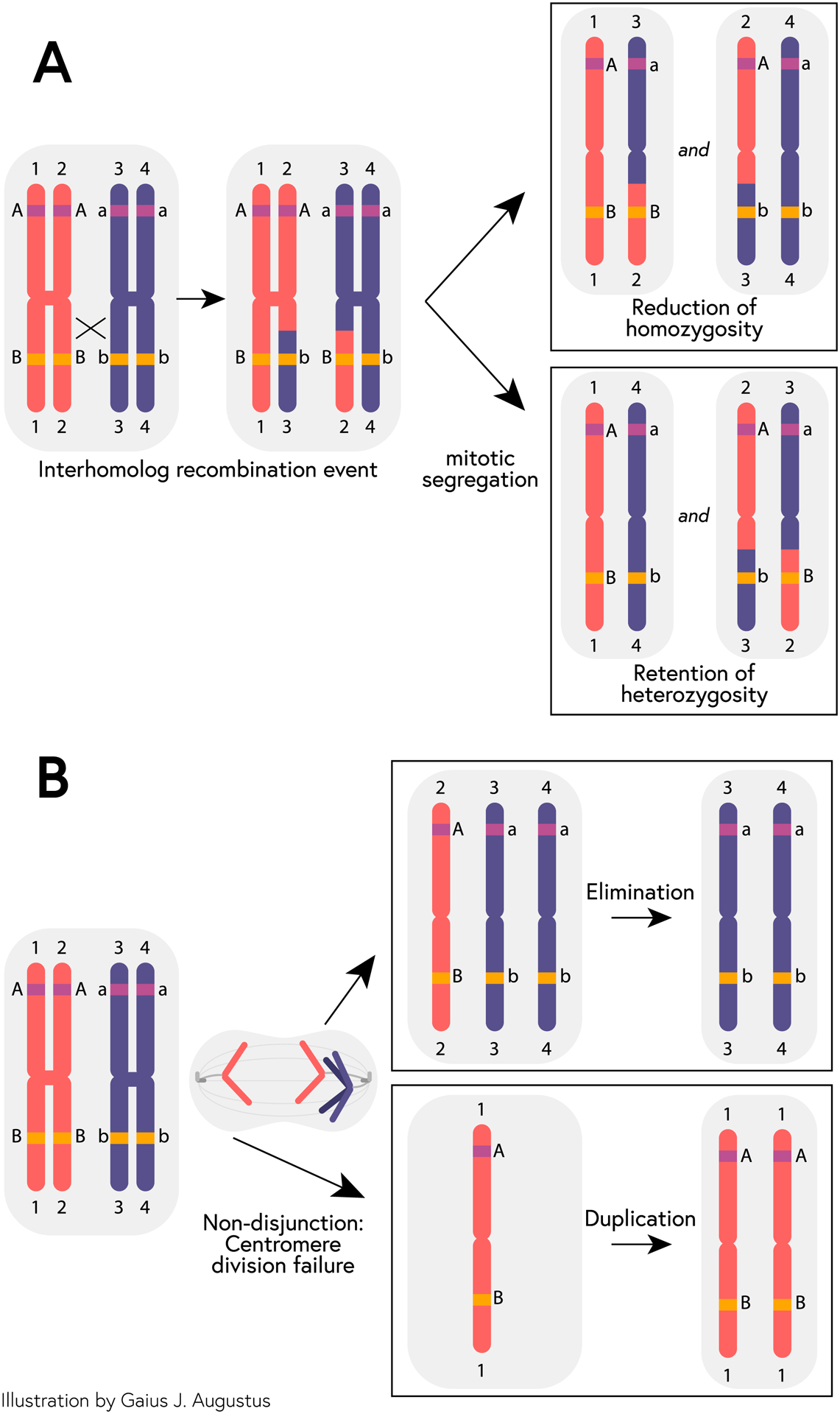
Model of mechanisms of copy-neutral loss of heterozygosity. (A) After an interhomolog recombination event affecting a given allele (i.e., the B allele, but not the A allele), mitotic segregation can result in either a reduction of homozygosity (from Bb in parental cell to BB or bb in daughter cell) or retention of heterozygosity (Bb in both parental and daughter cell). (B) A non-disjunction event during mitosis can lead to daughter cells with either an extra paternal chromatid or the loss of a paternal chromatid. Subsequent elimination of the single-copy chromosome in the former or duplication of the remaining chromosome in the latter results in cnLOH.
In the present study, we used publicly available data from The Cancer Genome Atlas (TCGA) as well as data from an African American CRC cohort, the Chicago Colorectal Cancer Consortium6 (CCCC), to compare frequencies and locations of cnLOH events in CRCs in the two racial groups. We hypothesized that differences in the frequency of cnLOH could contribute to the differing patterns of colorectal carcinogenesis between African Americans and Whites. We, therefore, investigated the frequency of cnLOH in African American CRCs and White CRCs. Surprisingly, we found that cnLOH occurs at a lower frequency in African American CRCs compared to White CRCs. Additionally, we identified a previously uncharacterized phenomenon we refer to as small interstitial cnLOH (si-cnLOH), in which small segments of chromosomes from 1–5 Mb long are affected by cnLOH.
Methods
Data acquisition
Fresh CRC tissue samples in the CCCC were collected from five major medical centers in Chicago between 2011 and 20126. Tumor samples and non-involved mucosal samples were obtained during surgery or endoscopy. Non-involved tissues were sampled from tissue >10 cm away from the tumor. These samples were cryopreserved in RNAlater. Microsatellite instability (MSI) status was determined in paired DNA samples from tumor and uninvolved tissue. The panel of mononucleotide markers included NR21, NR22, NR24, NR27, BAT25, and BAT26. Multiplex PCR amplified all markers, and PCR products were analyzed by capillary electrophoresis, as previously described7. The forward primers (Applied Biosystems, Foster City, CA) had a fluorescent tag (6FAM, HEX, NED) at the 5′ end to allow microsatellite detection by the ABI Prism 3100 Avant Genetic Analyzer (Applied Biosystems). The presence of peaks in the fluorescence profile of the amplified microsatellite DNA that were absent in a corresponding profile derived from normal mucosa of the same patient was interpreted as microsatellite instability. CRCs that exhibited MSI were excluded because it is known that the genomic instability that drives this subtype of CRC is a defect in mismatch repair, microsatellite unstable tumors tend to be chromosomally stable, and the frequencies of MSI in African Americans and Whites are similar8. Data on CNVs in tumor and normal samples from 39 microsatellite stable African American CRCs from the CCCC were acquired on the Affymetrix CytoScan HD microarray platform at the University of Illinois at Chicago Genomics Core.
Raw CNV data (CEL files) from microsatellite stable CRCs in TCGA (African American n = 31; non-Hispanic White n = 96, hereafter referred to as White) were obtained via the NCI’s Genomic Data Commons (GDC) website (June 8, 2017). These microarray data were generated on the Affymetrix GenomeWide SNP Array 6.0 platform. MSI status was determined by accessing clinical covariates available from the GDC.
The three cohorts were similar in sex, BMI, and tumor stage distributions (Table 1). The two African American cohorts were younger than the White cohort (mean in CCCC African Americans, 56.9 years; mean in TCGA African Americans, 61.4 years; mean in TCGA Whites, 67.2 years; p < 0.001, Table 1), a known clinical difference between the two ethnic groups1.
Table 1.
Clinical characteristics of patients in the three cohorts included in the study.
| CCCC African American N=39 |
TCGA African American N=31 |
TCGA White N=96 |
p-value | |
|---|---|---|---|---|
| Tumor stage: | 0.513 | |||
| Stage I | 5 (15.6%) | 5 (16.1%) | 15 (16.3%) | |
| Stage II | 11 (34.4%) | 6 (19.4%) | 30 (32.6%) | |
| Stage III | 10 (31.2%) | 10 (32.3%) | 33 (35.9%) | |
| Stage IV | 6 (18.8%) | 10 (32.3%) | 14 (15.2%) | |
| Age (years) | 56.9 (11.2) | 61.4 (12.2) | 67.2 (11.9) | <0.001 |
| Sex: | 0.555 | |||
| Female | 15 (39.5%) | 13 (41.9%) | 47 (49.0%) | |
| Male | 23 (60.5%) | 18 (58.1%) | 49 (51.0%) | |
| BMI: | 0.521 | |||
| Normal | 14 (36.8%) | 6 (23.1%) | 26 (32.9%) | |
| Underweight | 1 (2.63%) | 0 (0.00%) | 0 (0.00%) | |
| Overweight | 14 (36.8%) | 9 (34.6%) | 27 (34.2%) | |
| Obese | 9 (23.7%) | 11 (42.3%) | 26 (32.9%) |
BMI, body mass index
Data processing
Array data from TCGA and the CCCC were processed using Affymetrix Power Tools (APT, version 1.18.1). Population frequency of B allele (PFB) files were generated for each population (i.e., CCCC and TCGA African Americans and TCGA Whites) using compile_pfb.pl script provided by PennCNV (version 1.0.4). PennCNV was used to generate segments using default settings except for using our generated PFB files. Adjacent CNV segments were merged using PennCNV’s clean_cnv.pl script.
For CNV analysis, segments of gain and loss were aggregated to determine the proportion of each chromosome arm affected in each patient sample. Chromosome arms were then categorized as affected or unaffected by gain and loss, where having >27% of the arm affected was used as the threshold. This threshold was chosen because it was the mean percent affected amount across all chromosomes in all samples. Additionally, because gain and loss events can affect only part of a chromosome arm, a lower threshold was deemed appropriate to accurately capture chromosome arms affected by a copy number change. For the purposes of the present analysis, we did not differentiate between gain or loss of 1 or >1 copy (i.e., a copy number of 1 or 0, or a copy number of 3 or more). This analysis differs from the analysis reported in Xicola et al. (2018)4 due to the inclusion of African American TCGA CRC samples and the exclusion of MSI samples.
Similarly, to analyze large-scale cnLOH, segments of cnLOH were aggregated to determine the proportion of each chromosome arm affected. Chromosome arms were then categorized as affected or unaffected, where having >50% of the arm affected was used as the threshold. This threshold was chosen because cnLOH often affects most or all of a chromosome arm. It is also the threshold used by TCGA investigators9. For completeness, a 27% threshold was also assessed for cnLOH. This threshold did not change the overall results or interpretation (see below).
Small interstitial cnLOH analysis
Array data from CCCC tumor/normal pairs (i.e., tumor and normal samples from the same individual) were processed in APT using default settings. After identifying possible si-cnLOH events in the allelic difference data by visual inspection, we attempted to develop a method to automate the detection of si-cnLOH events. Several algorithms, including PennCNV and Paired PSCBS using Aroma (version 3.0.0), were assessed to determine an optimal program to complete segmentation of copy number data and identify small copy-neutral segments. Additionally, we developed a bioinformatic approach that used statistical and biological assumptions to extract si-cnLOH events from both non-segmented and segmented data (Supplementary Figure 1). For each algorithm, we produced a set of images that could be manually confirmed as potential si-cnLOH events. Our priority for this part of the study was to confirm the existence of these events. Therefore, the aim was to deploy a method of si-cnLOH detection that produced a reasonable number of images (<500 images) from the algorithm that we could evaluate allelic difference plots visually and subsequently test with SNP genotyping. Ultimately, the following method was determined to be optimal for this limited purpose.
Segments for cnLOH generated from APT were compared for each pair of samples. Non-tumor-specific cnLOH segments (i.e., cnLOH segments that appear in both tumor and normal samples) were removed using the tidygenomics R package (version 0.1.0). Upon inspection of remaining segments, we found a bimodal distribution where segments less than 5 Mb were most likely to be si-cnLOH events. We therefore selected regions that were tumor-specific and less than 5 Mb in length. Allelic difference plots of identified regions were generated for manual curation. A positive for a si-cnLOH was determined to be any region where heterozygosity exists in the normal sample but is lost in the tumor sample, and where the tumor sample retained two copies. Additionally, for a region to be classified as a si-cnLOH, there must not be cnLOH in the adjacent regions flanking it according to the segmentation data. Importantly, because the analysis is based on segmentation methods, start and end positions of si-cnLOH events are approximate.
Using this method, we generated 495 images. Of these 495, a rater classified each image (i.e., si-cnLOH) as “definitely”, “possibly”, or “not” a si-cnLOH. Images from the “definitely” and “possibly” class were then shown to an independent rater to reclassify. Images that both raters defined as “definitely” were included in the present study. Interrater reliability was >99%. From our 495 images from 39 samples, 465 images were classified as “not” si-cnLOH events, 7 were classified as “possibly” si-cnLOH events, and 24 were classified as “definitely” si-cnLOH events. The bioinformatics pipeline used to identify si-cnLOHs is provided in Supplementary Figure 1.
Validation of the microarray data was completed for 3 si-cnLOH events using PCR, restriction enzyme digestion, and agarose gel analysis to genotype SNPs inside the si-cnLOH and in the flanking regions in both tumor and non-involved samples. The SNPs that were genotyped and the associated primers designed for validation can be found in Supplementary Table 1.
To identify genes overlapping si-cnLOH locations, we used the GenomicRanges (version 1.34.0) and annotatr (version 1.8.0) under Bioconductor (version 3.7). To test whether specific genetic or functional pathways might be statistically over-represented among the genes affected, we used the Gene List Analysis protocol from PANTHER 15.0, which can be found at pantherdb.org (accessed February 25, 2020). To determine the mutational spectra in selected CRC tumor groups, we used the R package MutationalPatterns version 1.12.0 to create the 96-trinucleotide mutation count matrix based on single base substitutions from each somatic vcf file10. We then plotted the mutation spectrum to visualize the mean relative contribution of each of the six base substitution types over all samples. Error bars indicate standard deviation over all samples. Additional R (version 3.5.1) packages used for analysis of data include readr (version 1.3.1), ggplot2 (version 3.1.0), tidyr (version 0.8.2), dplyr (version 0.7.8), and stringr (version 1.3.1).
Statistical Analyses
Comparisons of events within categorical variables were determined by chi square or Fisher exact tests where appropriate. Comparison of number of events with categorical and continuous variables were determined by Wilcoxon Rank Sum tests or Spearman’s correlation, respectively. All p-values were adjusted for multiple testing using a Bonferroni correction. Linear regression models were used to determine associations between the number of chromosome arms affected by cnLOH in an individual and covariates. Covariates included age at diagnosis, sex, BMI, and tumor stage at diagnosis. Additional covariates for appropriate linear models included race (i.e., African American or White) and cohort (i.e., TCGA or CCCC).
Results
African Americans and Whites have similar frequencies of copy number gains and losses
The frequency of copy number gains and losses on each chromosome arm were compared in microsatellite stable CRCs from TCGA and the CCCC. African American and White CRCs had similar frequencies of chromosome-arm gains for all chromosomes as well as overall (Supplementary Table 2). Likewise, African American CRCs had similar frequencies of chromosome-arm losses as White CRCs for all chromosome arms (Supplementary Table 3). The overall frequency of copy number losses was significantly higher in TCGA African American CRCs compared to TCGA White CRCs (9.4% vs 6.3%, p = 0.01), but the frequency of chromosome losses in CCCC African American CRCs was slightly lower than in TCGA White CRCs (5.2% vs 6.3%, p = 1). No other comparison of chromosome losses was significantly different. Together, these results confirm findings of other studies2–4 that chromosome-arm copy number gains and losses occur at similar frequencies in African American and White CRCs.
White CRCs have a higher frequency of cnLOH than African American CRCs for most chromosome arms
In order to determine if there was a difference in overall burden of cnLOH between African American and White CRCs, we compared the frequency of cnLOH across the genome in all samples. Chromosome arms in CCCC African American CRCs, TCGA African American CRCs, and combined African American CRCs were affected at a frequency of 2.6%, 6.5%, and 4.3%, respectively, whereas 11.8% of chromosome arms of White CRCs were affected by cnLOH (Figure 2, Supplementary Table 4, p < 0.001 for all comparisons).
Figure 2.
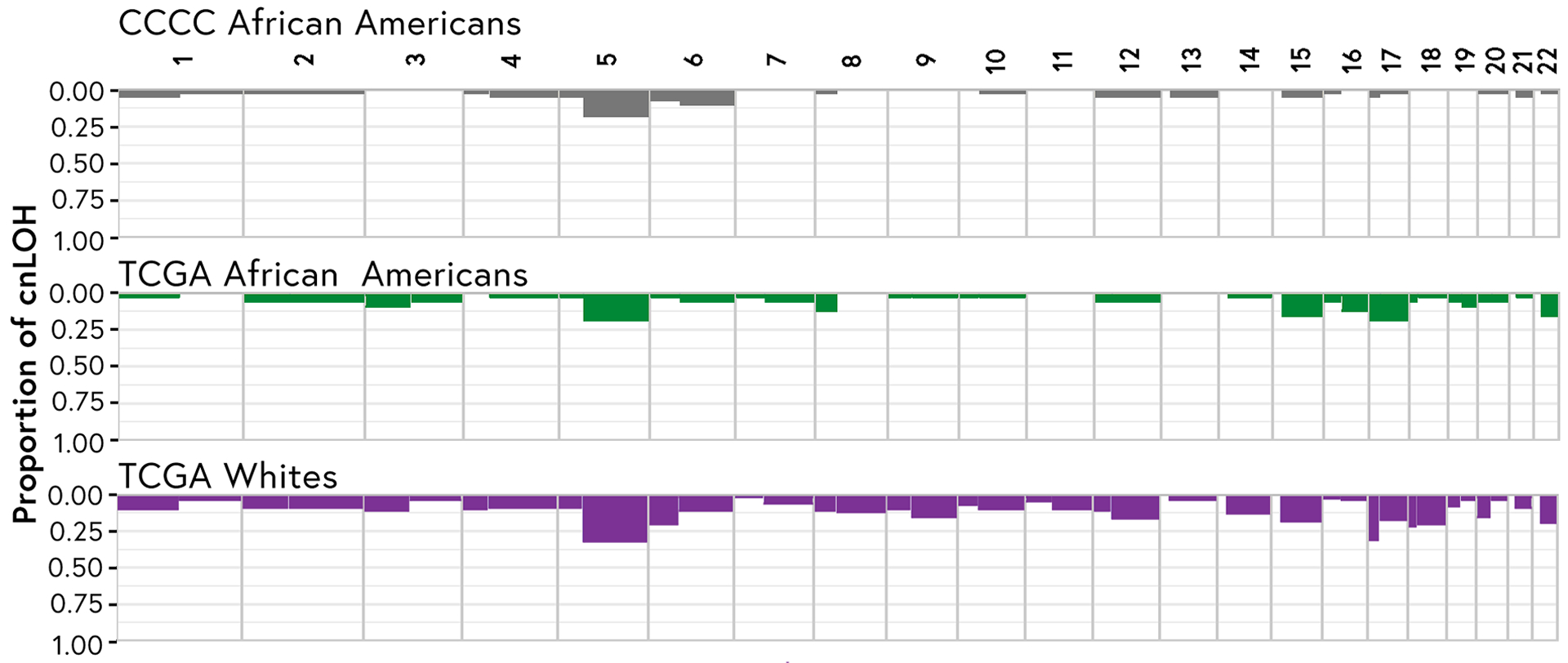
African American CRCs have a decreased frequency of cnLOH across the genome compared to White CRCs. Frequency of cnLOH by chromosome arm (expressed as the proportion of CRCs affected by cnLOH) is given for each chromosome arm in CCCC African Americans (top, grey), TCGA African Americans (middle, green), and TCGA Whites (bottom, purple).
To determine if the difference in cnLOH frequency was a global phenomenon or driven by a subset of chromosome arms, we compared the frequency of cnLOH on each chromosome arm between African Americans and Whites. Overall, White CRCs exhibited a 2.7-fold higher frequency of cnLOH compared to African American CRCs (Supplementary Table 4). The higher frequency of cnLOH on chromosome arm 18q in White CRCs was statistically significant, specifically compared to CRCs from the CCCC (White CRCs vs CCCC African American CRCs, >20.8 fold [0% vs 20.8%], p = 0.034; vs TCGA African American CRCs, 6.5-fold, p = 0.95; vs combined African American CRCs, 14.6-fold, p = 0.003; Supplementary Table 4). Similarly, there was a higher frequency of cnLOH on chromosome arm 18p. The higher frequency of cnLOH on chromosome arm 17p in White CRCs was statistically significant when compared to the frequency of CCCC African American CRCs but not of African American TCGA CRCs (6.1-fold, p = 0.03 vs 1.6-fold, p = 1.00; Supplementary Table 4). For no other comparison was the difference in frequency statistically significant after correction for multiple testing. Yet, large fold-change differences in cnLOH frequency were seen for many chromosomes.
Several chromosome arms showed greater than 2-fold differences between White and African American CRCs in all three African American vs. White comparisons, including 1p, 6p, 10p, 12q, and 20p (Supplementary Table 4). Chromosome arms 9p, 10q, and 18p showed a greater than 3-fold increased frequency of cnLOH events in White CRCs in comparison to African American CRCs in all three comparisons and chromosome arms 4p, 9q, 14q, and 18q showed a greater than 4-fold increased frequency in all three comparisons. Several chromosome arms had no cnLOH events in either African American group whereas events were found in White CRCs, specifically chromosome arms 8q, 11p, and 11q. With the exception of chromosome arms 16p, 16q, 19q, and 20q, all chromosome arms showed a higher frequency of cnLOH in White CRCs when compared to African American CRCs. For the most part, the greater fold difference in African American CRCs vs White CRCs was evident in both the CCCC and TCGA datasets. These data indicate that, although the fold difference between Whites CRCs and African American CRCs is demonstrably driven by oncogenesis for one specific chromosome, namely 18, where SMAD4 is localized, the higher frequency of cnLOH in Whites CRCs is a global phenomenon.
White CRCs have more chromosome arms affected by cnLOH than African American CRCs after adjustment for covariates
We found that the mean number of cnLOH events per tumor was higher in TCGA White CRCs (mean 11.8%) than in both TCGA African American CRCs (mean 6.5%) or CCCC African American CRCs (2.9%; Figure 3). Univariate analysis revealed that the number of chromosome arms affected by cnLOH was higher in White CRCs than in African American CRCs (mean of White CRCs, 4.6; mean of African American CRCs, 1.7; p < 0.0001), but did not differ by sex (mean of female CRCs, 3.6; mean of male CRCs, 3.2; p = 1). In addition, age was not correlated with the number of chromosomes arms affected by cnLOH (rs(165) = 0.19, which is the Spearman correlation with n = 165).
Figure 3.

African American CRCs have fewer chromosome arms affected by cnLOH than White CRCs. Number of chromosome arms affected by cnLOH in each CRC (expressed as percentage of total chromosome arms) is represented as a dot on the graph, stratified into CCCC African Americans (left, grey), TCGA African Americans (middle, green), and TCGA Whites (right, purple). Boxes represent 25th, 50th, and 75th quartiles, while whiskers represent 1.5 interquartile range for that data series. Note that many samples had no arms affected and that the maximum percentage of arms affected for any individual was < 40%.
Because of possible differences between cohorts, we compared the African American CRCs from the CCCC and TCGA. The number of cnLOH-affected chromosome arms was not different in African Americans based on sex (mean of female CRCs, 2.3; mean of male CRCs, 1.4; p = 1). Additionally, we found no significant difference between CCCC and TCGA African American CRCs by cohort, though there was a trend toward fewer chromosome arms affected by cnLOH in CCCC African American CRCs than in African American TCGA CRCs (mean in CCCC, 1.1; mean in TCGA, 2.5; p = 0.10). The main difference between these cohorts was age (Table 1), therefore we tested for a correlation between age and number of chromosome arms affected by cnLOH in African American CRCs. Again, age was not correlated with the number of chromosome arms affected by cnLOH (rs(69) = 0.12, which is the Spearman correlation with n = 69).
We used multivariate linear regression models to determine if differences in the numbers of cnLOH events per CRC were associated with race after adjusting for covariates (Table 2). White race remained significantly associated with a higher frequency of cnLOH even after adjusting for age, BMI, tumor stage, sex, and cohort. After stratification by race, stage II CRC was associated with a higher frequency of cnLOH in African American CRCs after adjusting for other covariates. However, neither stage III nor stage IV CRC were associated with cnLOH. In the model of White CRCs, no variable was significantly associated with number of chromosome arms affected by cnLOH. The above associations remained whether we looked at single chromosome arm events or whole chromosome events. Overall, the above results indicate that African American CRCs have a reduced frequency of cnLOH, even after adjusting for age, sex, tumor stage, cohort, and BMI.
Table 2.
Linear model shows reduced cnLOH affected chromosomes in African Americans after adjustment for covariates. (1) All African American and White complete cases from TCGA and CCCC, (2) African American complete cases from TCGA and CCCC, (3) White complete cases from TCGA
| Dependent variable: | |||
|---|---|---|---|
| Number of arms affected by cnLOH | |||
| (1) | (2) | (3) | |
| Age (years) | 0.005 (−0.049, 0.060) | −0.012 (−0.061, 0.037) | 0.019 (−0.067, 0.105) |
| African American Race | −3.022*** (−4.668, −1.375) | ||
| Underweight BMI | 1.322 (−5.781, 8.425) | 0.539 (−3.648, 4.726) | |
| Overweight BMI | 0.086 (−1.399, 1.572) | 1.192 (−0.254, 2.638) | −1.157 (−3.467, 1.154) |
| Obese BMI | 0.171 (−1.356, 1.698) | 0.816 (−0.538, 2.171) | −0.131 (−2.522, 2.260) |
| Tumor Stage II | 0.679 (−1.247, 2.605) | 2.007** (0.287, 3.727) | −0.443 (−3.536, 2.650) |
| Tumor Stage III | 1.093 (−0.787, 2.974) | 0.125 (−1.515, 1.766) | 1.574 (−1.436, 4.585) |
| Tumor Stage IV | 1.508 (−0.652, 3.669) | 0.439 (−1.383, 2.260) | 2.299 (−1.362, 5.960) |
| TCGA Cohort | 0.828 (−1.042, 2.698) | 0.968* (−0.140, 2.076) | |
| Male Gender | −0.184 (−1.449, 1.081) | −0.195 (−1.390, 0.999) | 0.420 (−1.588, 2.429) |
| Intercept | 2.881 (−1.428, 7.189) | 0.416 (−3.027, 3.859) | 3.123 (−3.058, 9.305) |
| Observations | 135 | 58 | 77 |
p<0.1;
p<0.05;
p<0.001
To test whether specific somatic mutations might be driving the higher rate of cnLOH that was observed in White CRCs, we compared the most cancer-associated mutations in the 18 CRCs that exhibited the highest rate of cnLOH (9–15 events per tumor) with the bottom 27 that exhibited the lowest rate (0–1 events per tumor). No significant differences were detected amongst these gene lists. We also tested whether there was a difference in the mutational spectra of the top 18 and bottom 27 CRCs (Supplementary Figure 2). The two profiles were similar.
Small interstitial copy-neutral loss of heterozygosity (si-cnLOH)
Unexpectedly, during our investigation of large-scale cnLOH events, we also identified small regions of cnLOH that occur interstitially within the chromosome. In order to identify these si-cnLOH events, we compared microarray data from paired tumor and non-involved CRC samples from the CCCC. We identified 24 si-cnLOH events in 5 of 39 patients in the series (Table 3). These 5 patients had varying numbers of si-cnLOH events, ranging from 1 to 13 with a median of 3 and an average of 4.8 si-cnLOH events per sample. Out of the 24 si-cnLOH events, 6 occurred on chromosome 7 (across 3 samples), 4 occurred on chromosome 2 (across 3 samples), 3 on chromosome 8 (in one sample), and 2 occurred on chromosomes 1, 3, and 5 (with two events in 2 samples). The remaining events occurred on single chromosomes in single samples (chromosomes 9, 11, 12, 13, and 17). The average segment of chromosome subject to si-cnLOH was 1.6 Mb long (median 1.4 Mb) with the shortest being approximately 200 kb and longest 3.5 Mb.
Table 3.
Numbers of si-cnLOH events and their chromosome locations in the patients in whom they were identified.
| Patient F95 | Patient 20018 | Patient C1 | Patient R27 | Patient U83 | Total Events | |
|---|---|---|---|---|---|---|
| Chr 7 | 4 | - | 1a | 1 | - | 6 |
| Chr 2 | 1 | 1 | 2a | - | - | 4 |
| Chr 8 | 3 | - | - | - | - | 3 |
| Chr 1 | 1a | - | - | - | 1 | 2 |
| Chr 3 | 1 | 1 | - | - | - | 2 |
| Chr 5 | 2 | - | - | - | - | 2 |
| Chr 9 | - | 1 | - | - | - | 1 |
| Chr 11 | 1 | - | - | - | - | 1 |
| Chr 12 | - | - | 1 | - | - | 1 |
| Chr 13 | - | - | - | 1 | - | 1 |
| Chr 17 | - | - | 1 | - | - | 1 |
| Total | 13 | 3 | 5 | 2 | 1 |
si-cnLOH was validated by genotyping SNPs inside and outside of region
In order to validate the microarray data, we interrogated 3 si-cnLOH events (Table 4) using PCR, restriction enzyme digestion, and agarose gel analysis to genotype SNPs inside the segment affected by si-cnLOH and in the flanking regions in both tumor and non-involved samples. In each case, tumor heterozygosity was retained outside of the si-cnLOH but lost inside the segment affected by si-cnLOH (Table 4). Two of the five CRCs that exhibited si-cnLOH events overlapped with CRCs from which we had mutation data from our previous exome sequencing study4. Although the 24 regions encompassed by si-cnLOH events overlapped with some gene and promoter regions (Supplementary Tables 5 & 6), there were no mutations in the two CRCs from which we had exome sequence data that overlapped with regions affected by si-cnLOH events. Moreover, gene list enrichment analysis using the Panther algorithm (pantherdb.org) failed to uncover associations with functional pathways. We therefore conclude that these events predominantly occur randomly with respect to somatic SNVs associated with the clonal evolution of the tumor.
Table 4.
Allelic difference plots, patient IDs, chromosomal location, and rsIDs of SNPs validated within si-cnLOHs

|
Discussion
The present study is the first to report that cnLOH is less frequent in African American CRCs than in White CRCs. The lower frequency of cnLOH was detected in two independent cohorts of African American CRCs in comparison to a larger series of White CRCs from TCGA. A lower frequency was observed with TCGA African American CRCs compared to TCGA White CRCs, arguing against the idea that a detection difference between the array platforms used in the CCCC and TCGA explains the effect. However, we acknowledge that including a series of CRCs from Chicago whites for analysis on the CytoScan array would have strengthened the results. In our multivariate models, race was the dominant predictor of lower cnLOH, whereas age, sex, BMI, and tumor stage did not predict lower cnLOH. These results indicate that African American CRCs are less likely to exhibit this particular form of genomic instability. We found no compensatory increase in copy number gains or losses, consistent with previous studies’ assessment of CNVs between White and African American CRCs2–4. Thus, the results suggest that the overall frequency of chromosome instability is lower in African American CRCs than in White CRCs.
We found that the fold difference in cnLOH frequency was greatly elevated over the average fold difference for specific chromosomes that carry genes important in tumor development, such as chromosome 18. However, a fold difference >1 was seen for almost every chromosome comparing White CRCs to African American CRCs, indicating that the difference is likely the result of a genome-wide effect. In our univariate analysis, CCCC African American CRCs were associated with less frequent cnLOH events than TCGA African American CRCs (Supplementary Table 4, 2.8% in CCCC CRCs vs 6.5% in TCGA CRCs, p < 0.0001). Differences between the two African American cohorts include older age in TCGA than in the CCCC as well as more stage IV and less stage II disease in the TCGA cohort. After adjusting for age, tumor stage, and other covariates, the association was no longer significant (Table 3). Overall, our findings lend credence to a hypothesis in which clonal evolution in African American CRCs develops via different genetic mechanisms than the evolution in White CRCs. The lower frequency of cnLOH in African American CRCs could reflect an innately higher chromosome stability in African American cells, for example because there is a lower rate of mitotic recombination. Alternatively, African American cells could be less tolerant of aneuploidy, although the similar frequencies of CNVs in White and African American CRCs argues against this idea.
We are not the first to suggest a mechanistic difference in the development of CRC in African Americans. Work by our and the Cleveland group identified SNVs in genes in African American CRCs that are rarely observed in White CRCs4,11,12. These independent studies present evidence that the tumorigenic processes may differ by ethnic group, but interpretation of results is limited because the sample size of these studies are relatively small and therefore possibly subject to selection bias.
We are not aware of any known carcinogenic exposures that induce cnLOH that could explain the higher cnLOH in Whites compared to African Americans. Another possibility is that the carcinogenic mechanisms that operate in African Americans favor epigenetic over chromosome mutational mechanisms. We recently reported that early-onset CRCs in African Americans more frequently develop by an epigenetic mechanism4, however, we found no association between age and cnLOH, which may argue against this idea.
The present study is also the first to describe and characterize a novel type of chromosomal mutation in CRC, the si-cnLOH event. There is, as far as we could find, no previous evidence of si-cnLOH events in CRC. Previous studies in lymphoma have mentioned the existence of si-cnLOH events in leukemias13,14, noting that tumor-normal pairs are necessary for their validation. Tumor-normal pairs are necessary because regions of homozygosity (ROHs) exist in the germline15. An ROH is a relatively small segment of the genome in which there is an excess of contiguous SNVs that are homozygous. ROHs are thought to result from gene-conversion-like recombination events in evolutionary history16,17. We obtained paired samples for every patient included in our CCCC cohort in order to identify tumor-specific si-cnLOH events by removing germline ROHs that were present in the normal patient’s DNA. We also found that the Affymetrix GenomeWide 6.0 Array, used in most genomic CNV studies, including TCGA’s CRC study, did not provide adequate resolution to identify si-cnLOH events. Here, we used the Affymetrix CytoScan HD array, which provided higher resolution and less variability, allowing us to detect these previously elusive events.
The 5 of 39 CRCs which exhibited si-cnLOH events each had a differing frequency of events. No si-cnLOH was recurrent, suggesting that it is an event that affects random segments of the genome. A mechanism to generate a tumor-specific si-cnLOH is currently unknown, but the mechanism is very likely the same as that which creates ROHs in the germline. We speculate that si-cnLOH events are generated by a break-induced replication (BIR) mechanism in which sequences are copied conservatively from the homologous chromosome to repair a double strand break (Figure 4). BIR has been demonstrated in mammalian cells18–20. BIR that copies from the sister chromatid results in no change in genetic constitution, but when the homologous chromosome is used for repair, an ROH results. Repair using the homologous chromosome should be less likely to occur than using the sister chromatid, which agrees with the general rarity of these events. BIR has been documented in mitosis-associated DNA synthesis—a repair process that is stimulated by conditions of replication stress20.
Figure 4.
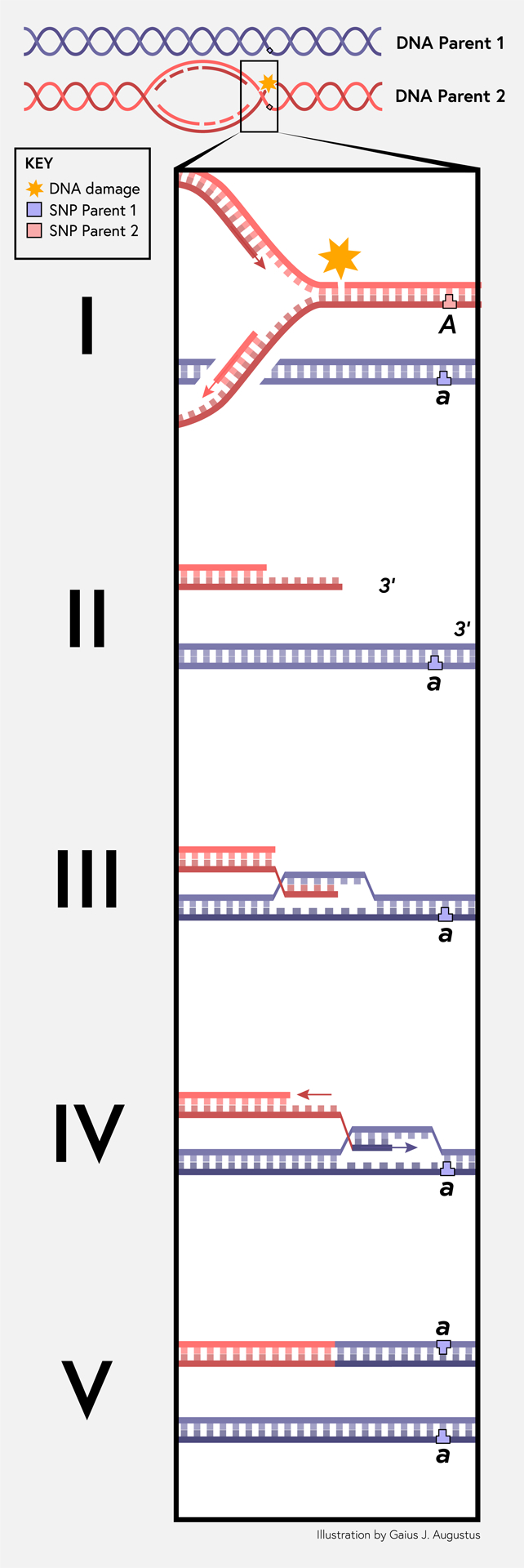
Break-induced DNA replication model for the generation of small-interstitial, copy-neutral loss of heterozygosity (si-cnLOH). (I) DNA replication encounters DNA damage that leads to a replication-associated, single-ended DNA break. The letters variants A and a represent a DNA polymorphism at a locus that is different between the homologous chromosomes. (II) Normally, repair of the replication-associated break is carried out by homologous recombination using the closely apposite sister chromatid; however, on rare occasions, repair is carried out using the homologous chromosome instead (represented by the blue lines). (III) The homologous recombination protein RAD51 catalyzes invasion of the single-stranded DNA with a 3’ end, pairing with the homologous sequence and forming a displacement loop. (IV) The 3’ hydroxyl from the invading DNA strand is available for incorporation by DNA polymerase delta. At the same time as polymerase is extending the invading strand, a DNA helicase displaces the nascent DNA behind polymerase. This generates a moving bubble. As the nascent DNA is displaced, the nascent DNA is replicated by lagging strand synthesis. (V) The break-induced replication mechanism copies variants from the homologous chromosome onto the chromosome that suffered the replication-associated DNA break by conservative DNA synthesis. This copy mechanism maintains copy number and reduces variants to homozygosity. The copy mechanism ends at some point when the invading strand is fully displaced, is recaptured by the sister chromatid, and meets a replication fork that converges upon it from the opposite direction.
We recognize that the method of identification for si-cnLOH events is in need of further development to improve sensitivity and specificity of results. Because of this, we do not claim that the current study is fully representative of the actual frequency of si-cnLOH events in CRC. The actual frequency could be higher than what is presented in this study, both in the number of events and in the number of patients affected. Improved methods are needed to meet the challenge of detecting and verifying these small copy-neutral events.
The rate of si-cnLOH mutation events in normal cell division is unknown; however, given the apparently high frequency of events in patient F95 (n = 13) and the range of frequencies seen in the 5 patients exhibiting the si-cnLOH phenotype, it seems plausible that elevated frequencies of si-cnLOH events could be a novel although rare form of chromosome instability and that cancer-specific events may be identified in future studies. Alternatively, persons with increased si-cnLOH events were inadvertently exposed to agents that cause increased replication stress. While si-cnLOH events appear to be random and there is no association with functional genetic pathways, we cannot rule out the possibility that some si-cnLOH events drive tumor progression. Further studies are necessary to determine the role si-cnLOH events play, if any, in colorectal tumorigenesis, and to determine whether the frequency of si-cnLOH events differs by race.
In summary, we found that cnLOH is less frequent in African American CRCs than in White CRCs, suggesting a biological difference associated with ancestry in the frequency of cnLOH events in tumorigenesis. The results add to mounting evidence that genetic mechanisms in CRC initiation and progression are different in African Americans compared to Whites. In addition, we are the first to describe a novel genetic mechanism in CRC based on si-cnLOH, which may comprise a new form of genomic instability.
Supplementary Material
Supplementary Figure 1. Bioinformatics pipeline used to identify regions of the genome subject to small, interstitial copy-neutral loss of heterozygosity (si-cnLOH). DNA samples from patient CRCs were processed on the Affymetrix CytoScan HD platform and the raw copy number files in CEL format were processed using Affymetrix Power Tools to identify genomic segments with regions of homozygosity (1). Regions of homozygosity that were present in both the tumor and normal specimen were filtered out using the tidygenomics application (2). Segments of homozygosity larger than 5 million base pair in length were then filtered out to eliminate regions of homozygosity that were generated by mitotic recombination or mitotic mechanisms (3). The results of the analysis were tabulated, visualized in allelic difference plots, and manually inspected for accuracy.
Supplementary Figure 2. Mutational spectrum of the 18 CRCs with 9 or more cnLOH events (High) compared to the 27 tumors with 0–1 cnLOH events (Low).
Acknowledgements
This work was supported by grants from the National Cancer Institute (U01 CA153060 and P30 CA023074, NAE; the Cancer Biology Training Grant T32 CA009213, GJA). The results shown here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. The content of this article is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health. We thank Mary Yagle and Johnathan Blohm for their assistance in SNP genotyping. Zarema Arbieva and the University of Illinois at Chicago Genomics Core in the Research Resources Core performed the hybridization and initial analysis of CytoScan HD arrays.
Footnotes
Data availability
The data that support the findings of this study are available from TCGA. Restrictions apply to the availability of these data, which were used under license for this study. Data are available at https://gdc.cancer.gov/ with approved access through the NIH database of Genotypes and Phenotypes (dbGAP). The data from the Chicago Colorectal Cancer Consortium part of this study are available from the corresponding author upon reasonable request.
References
- 1.Augustus GJ, Ellis NA. Colorectal Cancer Disparity in African Americans: Risk Factors and Carcinogenic Mechanisms. Am J Pathol. 2018;188:291–303. doi: 10.1016/J.AJPATH.2017.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Varadan V, Singh S, Nosrati A, et al. ENVE: a novel computational framework characterizes copy-number mutational landscapes in African American colon cancers. Genome Med. 2015;7(1):69. doi: 10.1186/s13073-015-0192-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ashktorab H, Schaffer AA, Daremipouran M, et al. Distinct genetic alterations in colorectal cancer. PLoS One. 2010;5(1). doi: 10.1371/journal.pone.0008879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xicola RM, Manojlovic Z, Augustus GJ, et al. Lack of APC somatic mutation is associated with early-onset colorectal cancer in African Americans. Carcinogenesis. 2018;39(11):1331–1341. doi: 10.1093/carcin/bgy122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Melcher R, Hartmann E, Zopf W, et al. LOH and copy neutral LOH (cnLOH) act as alternative mechanism in sporadic colorectal cancers with chromosomal and microsatellite instability. Carcinogenesis. 2011;32(4):636–642. doi: 10.1093/carcin/bgr011 [DOI] [PubMed] [Google Scholar]
- 6.Xicola RM, Gagnon M, Clark JR, et al. Excess of proximal microsatellite-stable colorectal cancer in african americans from a multiethnic study. Clin Cancer Res. 2014;20(18):4962–4970. doi: 10.1158/1078-0432.CCR-14-0353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xicola RM, Llor X, Pons E, et al. Performance of different microsatellite marker panels for detection of mismatch repair-deficient colorectal tumors. J Natl Cancer Inst. 2007; 99(3):244–252. doi: 10.1093/jnci/djk033 [DOI] [PubMed] [Google Scholar]
- 8.Ashktorab H, Ahuja S, Kannan L, et al. A meta-analysis of MSI frequency and race in colorectal cancer. Oncotarget. 2016;7(23):34546–34557. doi: 10.18632/oncotarget.8945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muzny DM, Bainbridge MN, Chang K, et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blokzijl F, Janssen R, van Boxtel R, Cuppen E. MutationalPatterns: comprehensive genome-wide analysis of mutational processes. Genome Med. 2018; 10(1):33. doi: 10.1186/s13073-018-0539-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guda K, Veigl ML, Varadan V, et al. Novel recurrently mutated genes in African American colon cancers. Proc Natl Acad Sci U S A. 2015;112(4):1149–1154. doi: 10.1073/pnas.1417064112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ashktorab H, Daremipouran M, Devaney J, et al. Identification of novel mutations by exome sequencing in African American colorectal cancer patients. Cancer. 2015;121(1):34–42. doi: 10.1002/cncr.28922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheung K-JJ, Delaney A, Ben-Neriah S, et al. High Resolution Analysis of Follicular Lymphoma Genomes Reveals Somatic Recurrent Sites of Copy-Neutral Loss of Heterozygosity and Copy Number Alterations that Target Single Genes. Genes Chromosomes Cancer. 2010;49(March):669–681. doi: 10.1002/gcc.20780 [DOI] [PubMed] [Google Scholar]
- 14.O’Keefe C, McDevitt MA, Maciejewski JP. Copy neutral loss of heterozygosity: a novel chromosomal lesion in myeloid malignancies. Blood. 2010;115(14):2731–2739. doi: 10.1182/blood-2009-10-201848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pemberton TJ, Absher D, Feldman MW, Myers RM, Rosenberg NA, Li JZ. Genomic patterns of homozygosity in worldwide human populations. Am J Hum Genet. 2012;91(2):275–292. doi: 10.1016/j.ajhg.2012.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J-C, Ross L, Mahon LW, et al. Regions of homozygosity identified by oligonucleotide SNP arrays: evaluating the incidence and clinical utility. Eur J Hum Genet. 2015;23(5):663–671. doi: 10.1038/ejhg.2014.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kearney HM, Kearney JB, Conlin LK. Diagnostic Implications of Excessive Homozygosity Detected by SNP-Based Microarrays: Consanguinity, Uniparental Disomy, and Recessive Single-Gene Mutations. Clin Lab Med. 2011;31(4):595–613. doi: 10.1016/j.cll.2011.08.003 [DOI] [PubMed] [Google Scholar]
- 18.Costantino L, Sotiriou SK, Rantala JK, et al. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science. 2014; 343(6166):88–91. doi: 10.1126/science.1243211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhowmick R, Minocherhomji S, Hickson ID. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol Cell. 2016;64(6):1117–1126. 10.1016/j.molcel.2016.10.037. [DOI] [PubMed] [Google Scholar]
- 20.Sotiriou SK, Kamileri I, Lugli N, et al. Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks. 2016;64(6):1127–1134. doi: 10.1016/j.molcel.2016.10.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Bioinformatics pipeline used to identify regions of the genome subject to small, interstitial copy-neutral loss of heterozygosity (si-cnLOH). DNA samples from patient CRCs were processed on the Affymetrix CytoScan HD platform and the raw copy number files in CEL format were processed using Affymetrix Power Tools to identify genomic segments with regions of homozygosity (1). Regions of homozygosity that were present in both the tumor and normal specimen were filtered out using the tidygenomics application (2). Segments of homozygosity larger than 5 million base pair in length were then filtered out to eliminate regions of homozygosity that were generated by mitotic recombination or mitotic mechanisms (3). The results of the analysis were tabulated, visualized in allelic difference plots, and manually inspected for accuracy.
Supplementary Figure 2. Mutational spectrum of the 18 CRCs with 9 or more cnLOH events (High) compared to the 27 tumors with 0–1 cnLOH events (Low).