

Key Points
MRD status by flow cytometry is associated with outcomes after decitabine plus venetoclax-based therapy in older/unfit patients with AML.
Negative MRD showed benefit across 1- to 4-month time points of assessment, irrespective of transplantation, as well as in adverse-risk AML.
Abstract
Assessment of measurable residual disease (MRD) provides prognostic information in acute myeloid leukemia (AML). However, the utility of MRD with venetoclax-based lower intensity regimens is unknown. We analyzed the prognostic value of achieving a negative MRD in older/“unfit” patients with AML receiving first-line therapy with 10-day decitabine and venetoclax. MRD was evaluated in bone marrow specimens using multicolor flow cytometry (sensitivity 0.1%). Ninety-seven patients achieving either a complete remission (CR) or CR with incomplete hematologic recovery (CRi) or morphologic leukemia-free state were included. Median age was 72 years (interquartile range, 68-78 years), and 64% had adverse-risk AML. Eighty-three patients achieved CR/CRi, and 52 (54%) became MRD negative. Median time to becoming MRD negative was 2.0 months (interquartile range, 0.9-3.1 months). Patients becoming MRD negative by 2 months had longer relapse-free survival (RFS) compared with those remaining MRD positive (median RFS, not reached vs 5.2 months; hazard ratio [HR], 0.31; 95% confidence interval [CI], 0.12-0.78; P = .004), longer event-free survival (EFS) (median EFS, not reached vs 5.8 months; HR, 0.25; 95% CI, 0.12-0.55; P < .001), as well as longer overall survival (OS) (median OS, 25.1 vs 7.1 months; HR, 0.23; 95% CI, 0.11-0.51; P < .001). Patients achieving an MRD-negative CR had longer OS compared with those with an inferior response (median OS, 25.1 vs 11.6 months; HR, 0.33; 95% CI, 0.19-0.58; P < .0005). Patients becoming MRD negative within 1 month had an improved OS compared with MRD-positive patients (median OS, 25.1 vs 3.4 months; HR, 0.15; 95% CI, 0.03-0.64; P < .0001). Differential impact of MRD status on survival outcomes persisted at a later 4-month time point of evaluation. In conclusion, MRD-negative status at 1, 2, and 4 months after starting therapy confers significantly better survival in older/unfit patients with AML receiving first-line therapy with 10-day decitabine and venetoclax. This trial was registered at www.clinicaltrials.gov as #NCT03404193.
Visual Abstract

Introduction
Measurable residual disease (MRD) assessment has been used for risk stratification and therapeutic decision-making in acute myeloid leukemia (AML).1-3 The prognostic value of MRD clearance after intensive chemotherapy and allogeneic hematopoietic stem cell transplantation (SCT) has been well described.4-6 Although the role of MRD testing in older or “unfit” patients treated with lower intensity regimens is less explored, previous studies have suggested potential prognostic and predictive utility of MRD for standard chemotherapy as well as epigenetic therapy in AML.7-9
Venetoclax-based lower intensity regimens have transformed the outcome of older/unfit patients with AML due to improved tolerability, higher response rates, and longer overall survival compared with hypomethylating agents and intensive chemotherapy.10-13 We now have the option of other lower intensity treatment approaches, both as standard and investigational treatment, and novel MRD-directed therapies are under investigation. Consequently, the utility of MRD assessment and its potential association with outcomes are of significant interest. To date, there are no published reports on the association of MRD response with outcomes using venetoclax-containing lower intensity regimens. Hence, we conducted this analysis to determine the prognostic value of MRD after frontline therapy with a 10-day decitabine plus venetoclax (DEC10-VEN)–based regimen in older/unfit patients with AML.
Methods
Bone marrow (BM) specimens were obtained at least after 1, 2, and 4 months of therapy for response assessment. MRD was assessed on the BM specimens using 8-color multiparametric flow cytometry (MFC) validated to a sensitivity level of 0.1% as previously described.6,14 Negative results were considered valid only if there had been acquisition of at least 200 000 events or a minimum of 200 CD34+ myeloid precursors. In cases in which a large number of events were obtained, or the AML had a distinct immunophenotype, the sensitivity might have reached below 0.1%.6
Patients were treated with decitabine 20 mg/m2 for 10 days for induction followed by decitabine for 5 days after achievement of complete remission (CR) or CR with incomplete hematologic recovery (CRi) with cycles repeated every 4 to 6 weeks. The venetoclax dose was 400 mg daily or equivalent, with appropriate dose reductions in the setting of concomitant azole antifungal therapy. Responses to therapy were defined per the European LeukemiaNet (ELN) 2017 criteria, and relapse was defined as re-emergence of >5% blasts in BM or peripheral blood in patients with a prior response.15 Relapse-free survival (RFS) was determined from date of achievement of CR/CRi until morphologic relapse, death, or censored at last follow-up. Event-free survival (EFS) was defined from start of therapy until relapse from CR/CRi, death, or censored at last follow-up, and compared with RFS, also included patients with a morphologic leukemia-free state (MLFS).5 Because this analysis focused on patients with a response, there were no patients with refractory disease as an event for EFS measurement.15 Overall survival (OS) was determined from the start of treatment until death or censored at last follow-up. Detailed results and the study protocol have been published previously.12 The data cutoff date for this analysis was 15 July 2020, and includes 5 additional patients and longer follow-up compared with our previously published report. These five additional patients included patients with newly diagnosed AML (n = 2) and patients with secondary AML with prior therapy for antecedent hematologic disorder (n = 3). The MD Anderson Institutional Review Board approved this study, and it was conducted in accordance with the Declaration of Helsinki.
The Cox proportional hazards regression model was used to evaluate the association between patient prognostic variables and OS. The Cox model for time-dependent variable was conducted to assess the effect of SCT on survival. Cumulative incidence of relapse was estimated by using the Fine-Gray competing risk approach, with death as a competing risk factor. The Fine-Gray competing risk regression model was used to assess the impact of clinical factors on relapse. Variables with P ≤ .1 under univariate analyses were included into the initial multivariable model. Backward model selection was used to remove variables from the model until all remaining variables were statistically significant with P < .05 and the MRD status was included in all models to assess its impact (eg, regression for RFS).
All analyses were conducted using Prism version 8.4 (GraphPad Software Inc., San Diego, CA), SAS version 9.4 (SAS Institute, Inc., Cary, NC), and R version 3.5.3 (R Foundation for Statistical Computing, Vienna, Austria).
Results
Between 20 January 2018 and 15 April 2020, we treated 118 older patients with newly diagnosed AML on this prospective phase 2 trial of DEC10-VEN. Ninety-seven (82%) patients achieved a response and were included in this analysis. The median age was 72 years (interquartile range, 69-78 years), 22 patients (23%) had an Eastern Cooperative Oncology Group performance score ≥2, and 55 patients (57%) had ELN adverse-risk disease (Table 1). Of 17 patients with FLT3-ITD/TKD (18%), 13 patients received FLT3 inhibitors in addition to DEC10-VEN, including gilteritinib (n = 5), sorafenib (n = 5), and midostaurin (n = 3). Of 21 patients with IDH1/2mut (22%), 2 patients received enasidenib in addition to DEC10-VEN. Of 97 patients, CR/CRi was achieved in 70% of patients (n = 83), and negative MRD status was achieved in 54% of patients (n = 52). The median time to achieve negative MRD was 2.0 months (interquartile range, 0.9-3.1 months). Sixteen patients underwent SCT after achievement of a response; 10 were MRD negative pre-SCT. The median OS of these responding patients was 14.1 months (95% confidence interval [CI], 11.6-25.1), and median RFS was 9.0 months (95% CI, 6.9-15.0). After a median follow-up of 20.2 months, 39 patients (40%) have relapsed, and 49 patients have died (Figure 1).
Table 1.
Baseline characteristics and outcomes of 97 responding patients with AML treated with DEC10-VEN
| Characteristic | N (%) or median [IQR] |
|---|---|
| Age, y | 72 [69-78] |
| ≥70 | 67 (69) |
| Male sex | 53 (55) |
| ECOG performance status ≥2 | 22 (23) |
| Peripheral blood blasts, % | 5 [0-24] |
| BM blasts, % | 38 [22-57] |
| Diagnosis | |
| De novo | 50 (52) |
| sAML with AHD | 31 (32) |
| Treatment naive | 12 (12) |
| Previously treated for AHD | 19 (20) |
| Therapy-related | 16 (16) |
| ELN 2017 risk group | |
| Favorable | 26 (27) |
| Intermediate | 16 (16) |
| Adverse | 55 (57) |
| ELN 2017 cytogenetic risk* | |
| Favorable | 0 (0) |
| Intermediate | 57 (59) |
| Adverse | 39 (40) |
| Mutations | |
| NPM1 | 28 (29) |
| FLT3-ITD/TKD | 17 (18) |
| IDH1/2 | 21 (22) |
| TP53 | 23 (24) |
| RUNX1 | 18 (19) |
| ASXL1 | 13 (13) |
| K/NRAS | 20 (21) |
| Prior therapies for AHD | 1 [1-2] |
| Hypomethylating agents | 17 (18) |
| Intensive chemotherapy (IC) | 3 (3) |
| Hypomethylating agents and IC | 2 (2) |
| SCT | 3 (3) |
| Outcomes | |
| CR | 59 (61) |
| CRi | 24 (25) |
| MLFS | 14 (14) |
| MRD negative by flow cytometry | 52 (54) |
| Median time to morphologic response, mo | 1.4 [1.1-2.6] |
| Median no. of cycles to response | 1 [1-2] |
| Median time to negative MRD, mo | 2.0 [0.9-3.1] |
AHD, antecedent hematologic disorder; ECOG, Eastern Cooperative Oncology Group; IQR, interquartile range; sAML, secondary acute myeloid leukemia.
One patient had insufficient metaphases.
Figure 1.

Schema showing overall population and patients included in this analysis.
In this study, DEC10-VEN offered high rates of negative MRD in responding patients with de novo AML (62%) and intermediate-risk cytogenetics (67%), but rates were modest in patients with secondary AML, therapy-related AML, and adverse-risk cytogenetics (33%-53%) (Figure 2A). High rates of negative MRD ranging from 50% to 79% were achieved in several mutational subgroups, including NPM1mut, FLT3mut, IDH1/2mut, and RUNX1mut while patients with adverse-risk mutations, including TP53mut, ASXL1mut, and K/NRASmut, had lower rates of negative MRD of 26% to 46% (Figure 2B). A competing risk analysis for cumulative incidence of relapse with death as a competing event showed a numerically lower rate of relapse in patients achieving CR/CRi with negative MRD at 2 months compared with those who were MRD positive (37% vs 53%; P = .086) (Figure 2C). Patients achieving CR/CRi who were MRD negative by 2 months after starting therapy had a significantly longer RFS compared with patients who remained MRD positive (median RFS, not reached vs 5.2 months; hazard ratio [HR], 0.31; 95% CI, 0.12-0.78; P = .004) (Figure 2D). Patients with any response and negative MRD by 2 months had significantly longer EFS, as well as OS, compared with those who remained MRD positive. Median EFS was not reached vs 5.8 months (HR, 0.25; 95% CI, 0.12-0.55; P < .001) (Figure 2E), and median OS was 25.1 vs 7.1 months (HR, 0.23; 95% CI, 0.11-0.51; P < .001) (Figure 2F).
Figure 2.
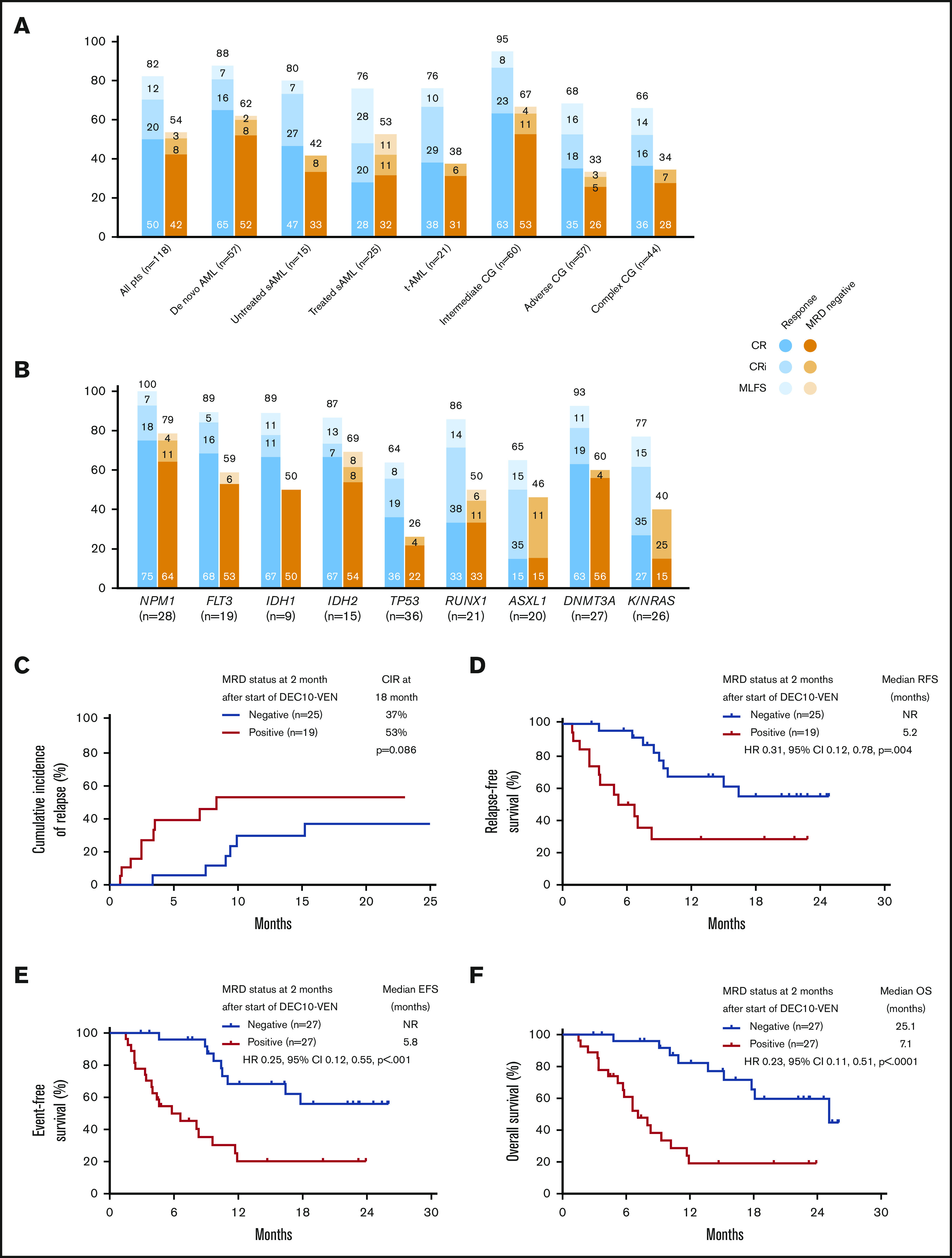
Rates of negative MRD and survival outcomes. Assessment of patients in clinical subgroups (A) and mutational subgroups (B) of AML treated with DEC10-VEN; outcomes according to MRD status at 2 months, including competing risk analysis for cumulative incidence of relapse (CIR) with death as a competing event (C), RFS (D), EFS (E), and OS (F). CG, cytogenetics; NR, not reached; pts, patients; sAML, secondary AML from antecedent hematologic disorder; t-AML, therapy-related AML.
Patients who underwent SCT had longer OS compared with patients who did not or could not undergo SCT, with a median OS not reached vs 12.1 months (HR, 0.30; 95% CI, 0.15-0.58; P = .013) (Figure 3A). Among patients who underwent SCT, relapse occurred in 2 out of 5 patients who had positive MRD before SCT vs in 1 out of 10 patients who were MRD negative (P = .170; 1 patient was not evaluable for MRD). Even after censoring for SCT, RFS, EFS, and OS were significantly longer in patients who were MRD negative by 2 months (Figure 3B-D). In addition, patients achieving any response who became MRD negative by 1 month after starting therapy had an 85% reduction in risk of death, with a median OS of 25.1 months compared with patients who achieved a response but remained MRD positive who had a median OS of 3.4 months (HR, 0.15; 95% CI, 0.03-0.64; P < .0001) (Figure 3E). Patients meeting the ELN 2017–defined response of CR MRD-negative at any time point had a longer median OS of 25.1 months compared with those with an inferior response (ie, MRD-positive CR or MRD-positive or negative CRi/MLFS) who had a median OS of 11.6 months (HR, 0.33; 95% CI, 0.19-0.58; P < .0005) (Figure 3F). Among patients with ELN adverse-risk disease, achievement of negative MRD at any time conferred a longer median EFS of 13.5 months compared with patients who never achieved negative MRD who had a median EFS of 5.8 months (HR, 0.34; 95% CI, 0.18-0.63; P = .001). There was no significant difference in EFS between ELN favorable risk patients who became MRD negative at any time vs those who never became MRD negative (median EFS, 29.6 months vs not reached; HR, 0.88; 95% CI, 0.09-8.29; P = .906).
Figure 3.
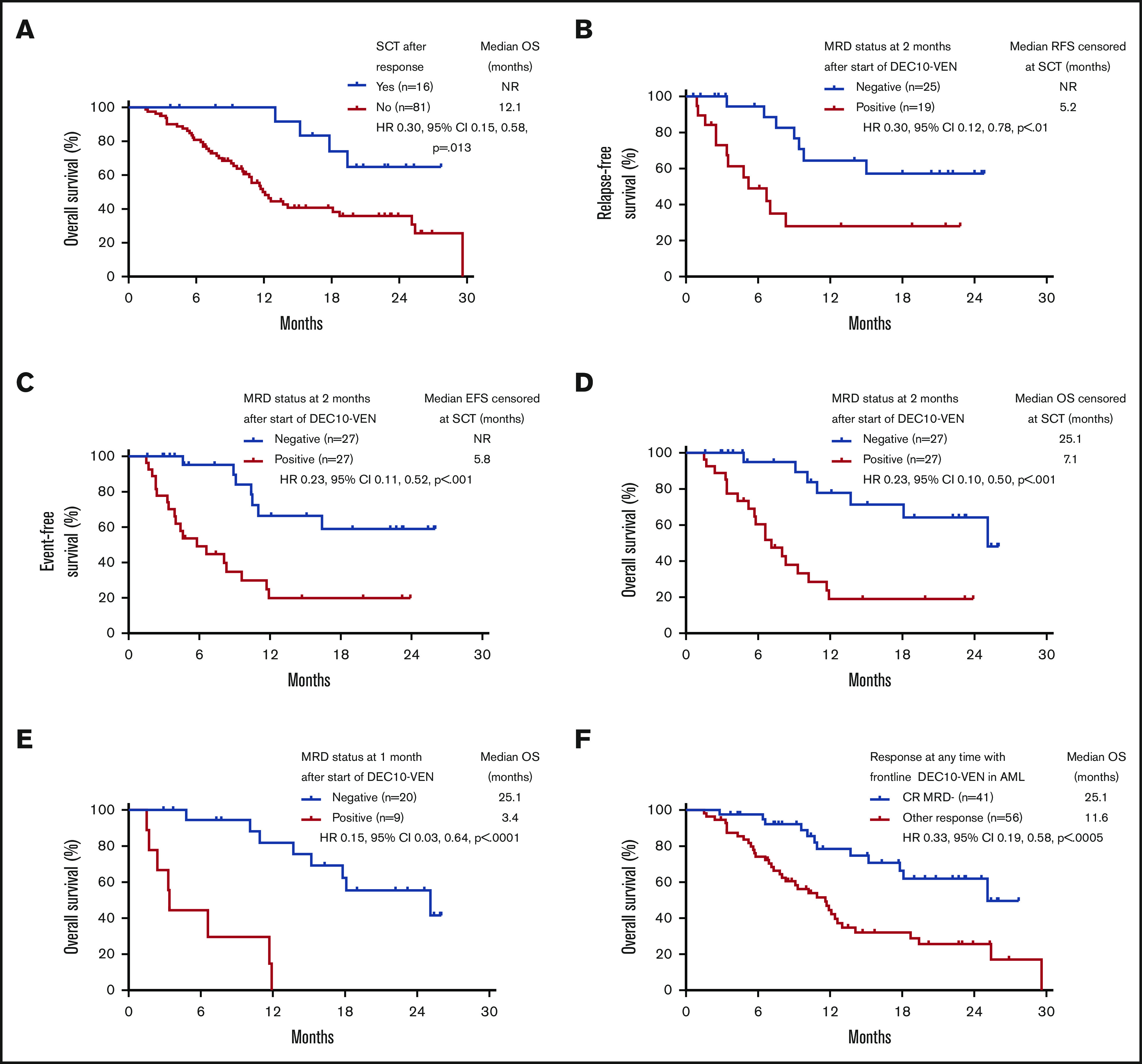
Survival according to SCT. (A) OS of responding patients according to allogeneic SCT. Outcomes with censoring at SCT, including RFS (B), EFS (C), OS (D), OS according to MRD status in patients achieving response at 1 month (E), and OS in patients achieving CRwith negative MRD (CR MRD–) at any time compared with any other response (F). NR, not reached.
Comparison of outcomes according to MRD status at 4 months yielded results similar to those at the 2-month time point. Patients who achieved negative MRD by 4 months had longer RFS (median RFS, not reached vs 6.4 months; HR, 0.34; 95% CI, 0.17-0.67; P < .001) (supplemental Figure 1A), longer EFS (median EFS, not reached vs 8.3 months; HR, 0.31; 95% CI, 0.17-0.55; P < .001) (supplemental Figure 1B), and longer OS (median OS, 25.1 vs 10.2 months; HR, 0.39; 95% CI, 0.21-0.71; P < .0010 (supplemental Figure 1C). Additional analysis showed that among patients with negative MRD at any time, those achieving CR had OS comparable to patients with CRi or MLFS (25.1 vs 18.7 months; HR, 0.70; 95% CI, 0.24-2.01; P = .63) (supplemental Figure 2). Among patients with persistent MRD, patients with CR had a trend toward longer OS compared with CRi/MLFS (median OS, 11.6 vs 8.0 months; HR, 0.47; 95% CI, 0.23-0.99; P = .061) (supplemental Figure 3). Factors associated with a higher risk of death on multivariate analysis for OS included MRD-positive status at 2 months (HR, 3.81; 95% CI, 1.48-9.81; P = .006) and TP53mut (HR, 3.90; 95% CI, 1.62-9.42; P = .003) (supplemental Table 1). Factors associated with a lower risk of death included IDH1/2mut (HR, 0.09; 95% CI, 0.02-0.40; P = .002) and attainment of CR/CRi (HR, 0.26; 95% CI, 0.09-0.74; P = .011). Multivariate analysis for RFS showed that TP53mut was associated with a very high risk of relapse (HR, 79.98; 95% CI, 13.04-478.53; P < .001) (supplemental Table 2).
Although venetoclax dose reductions with concomitant azole antifungal use were planned per protocol, 30 patients did not receive the intended dose reduction of venetoclax during cycle 1 and were exposed to a relatively higher-than-recommended dose. The rate of negative MRD at any time point in patients who received a higher-than-recommended dose of venetoclax was 60% compared with 52% in patients who received the recommended dose of venetoclax (18 of 30 vs 34 of 66; P = .439).
Discussion
Given the prognostic value of MRD in AML, the role of SCT for MRD-positive patients, novel therapies leading to high response rates, and emerging immunotherapies with potential to eradicate MRD, the evaluation and monitoring of MRD have become important aspects of standard clinical care. Our results showed high rates of MRD-negative remissions with DEC10-VEN across multiple clinical and mutational subgroups and strong prognostic value of MRD on RFS, EFS, and OS. The magnitude of benefit conferred by negative MRD status was similar across 1-, 2-, and 4-month time points. Although MRD-negative rates were lower in ELN adverse-risk mutations, achievement of MRD-negative status still conferred OS benefit in these patients.
With necessary caution for cross-trial comparisons, our results showed a higher rate of negative MRD with a DEC10-VEN in a more adverse-risk AML population compared with venetoclax with azacitidine or 5-day decitabine (54% vs 30%).12,16 In addition, improved survival with a median OS of more than 2 years can provide the opportunity to evaluate treatment de-escalation for patients achieving negative MRD after 1 or 2 cycles of therapy. This is relevant for regimens with venetoclax plus hypomethylating agents, which have substantial myelotoxicity and 30% to 40% grade 3/4 infectious complications.10,12,17 However, such de-escalation approaches would need further evaluation in controlled settings, with close monitoring, and follow-up BM evaluations to detect MRD relapse, before we can recommend broader adoption of such approaches. Conversely, poor RFS, EFS, and OS in MRD-positive patients, and high mortality due to relapse (67%, n = 33/49), highlight the urgent need for novel agents to eradicate MRD in this population.
Among FLT3mut patients, 76% received FLT3 inhibitors with venetoclax and decitabine, 89% of patients achieved CR/CRi/MLFS, and 56% of responders achieved negative MRD by MFC. Early follow-up in the treatment-naive subgroup of these FLT3mut patients revealed an 1-year OS of 80%, suggesting a potential favorable impact with the addition of FLT3 inhibitors compared with historical outcomes.18 However, longer follow-up is needed, and with only 4 patients not receiving FLT3 inhibitors, the numbers were limited to evaluate the incremental benefit of such “triplet” approach.
Limitations of the current study include the retrospective nature of this analysis and the small sample size which restricted statistical power. In addition, the ability to measure MRD varies greatly according to the expertise and the technique used by the individual centers and the lack of standardization of MFC assays to measure MRD in AML. Hence, prospective studies are needed to confirm these findings. Although we defined relapse morphologically on BM evaluation instead of using MRD, retrospective data showing comparable OS with morphologic versus MRD relapse support this choice.19 Of 97 patients reported in this study, 13 patients received FLT3 inhibitors, and 2 patients received enasidenib. This should be taken into consideration when interpreting these results because these targeted therapies can be potential confounders for the survival outcomes reported. We have reported MRD using next-generation sequencing from this trial previously, and additional analyses are ongoing.12,20
In conclusion, attainment of MRD-negative status at 1 and 2 months after starting therapy is associated with significantly better OS in older patients with AML with intermediate- and adverse-risk cytogenetics receiving frontline therapy with the DEC10-VEN regimen. These data warrant prospective investigation of the association of MRD clearance with outcomes in AML when using lower intensity strategies.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the patients, their caregivers, and members of the study team involved in this trial.
This study was supported in part by the MD Anderson Cancer Center Support Grant CA016672 and the Research Project Grant Program (R01CA235622) from the National Cancer Institute, National Institutes of Health.
Footnotes
At this time, individual patient-level data cannot be shared outside of the authors’ institution.
Authorship
Contribution: A.M., C.D.D., M.Y.K., H.M.K., and F.R. contributed to conception and design;
C.D.D., M.Y.K., H.M.K., and F.R. provided administrative support; C.D.D., S.A.W., J.J., T.M.K., N.G.D., N.J.S., M.Y., N.P., G.B., P.B., G.C.I., A.F., E.J.J., N.J., G.G.-M., M.O., K.T., G.M.-B., L.M., J.A.B., P.A.T., S.V., K.S., M.A., C.R.R., K.S.M., S.P., H.M.K., M.Y.K., and F.R. provided study materials or patients; K.S.M., S.P., and A.M. were responsible for collection and assembly of data; A.M., C.D.D., S.A.W., J.J., W.Q., J.N., M.Y.K., and F.R. were responsible for data analysis and interpretation; A.M., C.D.D., M.Y.K., and F.R. wrote the manuscript; and all authors contributed to critical revision of the manuscript for important intellectual content, and reviewed and approved the final version of the manuscript.
Conflict-of-interest disclosure: A.M. reports research funding from Celgene. C.D.D. reports personal fees from AbbVie, Agios, Novartis, Immune-Onc, Daiichi Sankyo, Celgene, Jazz Pharmaceuticals, and Notable Labs, outside the submitted work. N.G.D. reports grants from AbbVie, Genentech, Astellas, Daiichi Sankyo, Pfizer, BMS, ImmunoGen, Novimmune, and Forty Seven; and personal fees from AbbVie, Genentech, Astellas, Daiichi Sankyo, Pfizer, BMS, ImmunoGen, Jazz Pharmaceuticals, Trillium, Forty Seven, Gilead, Kite, and Novartis. N.J.S. reports grants from Takeda Oncology and Astellas; and personal fees from Takeda Oncology, AstraZeneca, and Amgen. N.P. reports personal fees from Pacylex Pharmaceuticals, Incyte, LFB Biotechnologies, Roche Diagnostics, and Blueprint Medicines; grants and other from Affymetrix; grants from SagerStrong Foundation; personal fees and other from Novartis; personal fees, nonfinancial support, and other from Stemline Therapeutics and AbbVie; personal fees and nonfinancial support from Celgene, Mustang Bio, and DAVA Oncology; and other from Samus Therapeutics, Cellectis, Daiichi Sankyo, and Plexxikon, outside the submitted work. G.B. has received research funding from AbbVie, Incyte, Janssen, GSK, Cyclacel, and Oncoceutics, Inc.; consultancy and research funding from BioLineRx; and consultancy for NKarta and PTC Therapeutics. P.B. reports research grant and personal fees from Incyte, Celgene, CTI Biopharma, Kartos Therapeutics, and Blueprint Medicines; and research grants from Constellation Pharmaceuticals, NS Pharma, Promedior, Pfizer, and Astellas. G.C.I. received research funding from Celgene; and served on an advisory board for Novartis. E.J.J. reports consultancy research funding from Takeda, BMS, Adaptive, Amgen, AbbVie, Pfizer, and Cyclacel Ltd; and research grants with Amgen, AbbVie, Spectrum, BMS, Takeda, Pfizer and Adaptive. N.J. reports the following: consultancy, honoraria, or membership on an entity’s Board of Directors or advisory committees, and research funding from AbbVie, Pharmacyclics, an AbbVie company, Genentech, Pfizer, ADC Therapeutics, AstraZeneca, Servier, Verastem, Precision Biosciences, and Adaptive Biotechnologies; consultancy, honoraria, or membership on an entity’s Board of Directors or advisory committees from Janssen Pharmaceuticals, Inc.; and research funding from BMS, Incyte, and Cellectis. K.T. has received personal fees for service on advisory boards of Symbio Pharmaceuticals, GSK, and Celgene. P.A.T. reports research funding to institution and consultancy/honorarium paid to institution from Pharmacyclics, AbbVie, and Amgen; consultancy/honorarium paid to institution from Genentech, Gilead Sciences, and Janssen-Cilag; and research funding to institution from Pfizer. K.S. reports honoraria from Otsuka; and consultancy for Pfizer. M.A. reports consultancy, patents, and royalties from Daiichi Sankyo, Inc.; patents licensed, royalty bearing, and research funding from Jazz Pharmaceuticals; consultancy for Celgene, Amgen, AstraZeneca, and 6 Dimensions Capital; equity ownership in Reata, Aptose, Eutropics, Oncoceutics, and Oncolyze; equity ownership and membership on an entity’s Board of Directors or advisory committees for Senti Biosciences; research funding from the Breast Cancer Research Foundation, CPRIT, and the NCI/National Institutes of Health; and membership on an entity’s Board of Directors or advisory committees for the Center for Drug Research & Development, Cancer UK, NCI-CTEP, German Research Council, Leukemia Lymphoma Society, NCI-RDCRN (Rare Diseases Clinical Research Network), CLL Foundation, and BioLineRx. H.M.K. reports grants and other from AbbVie, Agios, Amgen, ImmunoGen, and Pfizer; grants from Ariad, Astex, BMS, Cyclacel, Daiichi Sankyo, Jazz Pharmaceuticals, and Novartis; and other from Actinium and Takeda, outside the submitted work. M.Y.K. has received grants from the National Institutes of Health, the NCI, AbbVie, Genentech, Stemline Therapeutics, Forty Seven, Eli Lilly, Cellectis, Calithera, Ablynx, and AstraZeneca; consulting/honorarium from AbbVie, Genentech, F. Hoffmann–La Roche, Stemline Therapeutics, Amgen, Forty Seven, and Kisoji; clinical trial support from Ascentage; and stocks/royalties in Reata Pharmaceutical. F.R. reports personal fees and research grants from AbbVie. The remaining authors declare no competing financial interests.
Correspondence: Farhad Ravandi, Department of Leukemia, The University of Texas MD Anderson Cancer Center, 1400 Holcombe Blvd, Unit 428, Houston, TX 77030; e-mail: fravandi@mdanderson.org.
References
- 1.Schuurhuis GJ, Heuser M, Freeman S, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2018;131(12):1275-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Short NJ, Ravandi F. How close are we to incorporating measurable residual disease into clinical practice for acute myeloid leukemia? Haematologica. 2019;104(8):1532-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ravandi F, Walter RB, Freeman SD. Evaluating measurable residual disease in acute myeloid leukemia. Blood Adv. 2018;2(11):1356-1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jongen-Lavrencic M, Grob T, Hanekamp D, et al. Molecular minimal residual disease in acute myeloid leukemia. N Engl J Med. 2018;378(13):1189-1199. [DOI] [PubMed] [Google Scholar]
- 5.Ivey A, Hills RK, Simpson MA, et al. ; UK National Cancer Research Institute AML Working Group . Assessment of minimal residual disease in standard-risk AML. N Engl J Med. 2016;374(5):422-433. [DOI] [PubMed] [Google Scholar]
- 6.Shah MV, Jorgensen JL, Saliba RM, et al. Early post-transplant minimal residual disease assessment improves risk stratification in acute myeloid leukemia. Biol Blood Marrow Transplant. 2018;24(7):1514-1520. [DOI] [PubMed] [Google Scholar]
- 7.Freeman SD, Virgo P, Couzens S, et al. Prognostic relevance of treatment response measured by flow cytometric residual disease detection in older patients with acute myeloid leukemia. J Clin Oncol. 2013;31(32):4123-4131. [DOI] [PubMed] [Google Scholar]
- 8.Boddu P, Jorgensen J, Kantarjian H, et al. Achievement of a negative minimal residual disease state after hypomethylating agent therapy in older patients with AML reduces the risk of relapse. Leukemia. 2018;32(1):241-244. [DOI] [PubMed] [Google Scholar]
- 9.Ragon BK, Daver N, Garcia-Manero G, et al. Minimal residual disease eradication with epigenetic therapy in core binding factor acute myeloid leukemia. Am J Hematol. 2017;92(9):845-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617-629. [DOI] [PubMed] [Google Scholar]
- 11.Wei AH, Montesinos P, Ivanov V, et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: a phase 3 randomized placebo-controlled trial. Blood. 2020;135(24):2137-2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DiNardo CD, Maiti A, Rausch CR, et al. 10-Day decitabine with venetoclax for newly diagnosed intensive chemotherapy ineligible, and relapsed or refractory acute myeloid leukaemia: a single-centre, phase 2 trial. Lancet Haematol. 2020;7(10):e724-e736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maiti A, Qiao W, Sasaki K, et al. Venetoclax with decitabine vs intensive chemotherapy in acute myeloid leukemia: a propensity score matched analysis stratified by risk of treatment-related mortality. Am J Hematol. 2021;96(3):282-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu J, Jorgensen JL, Wang SA. How do we use multicolor flow cytometry to detect minimal residual disease in acute myeloid leukemia? Clin Lab Med. 2017;37(4):787-802. [DOI] [PubMed] [Google Scholar]
- 15.Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DiNardo CD, Pratz KW, Letai A, et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol. 2018;19(2):216-228. [DOI] [PubMed] [Google Scholar]
- 18.Maiti A, DiNardo CD, Ravandi F, et al. Venetoclax, FLT3 inhibitor and decitabine in FLT3mut acute myeloid leukemia: subgroup analysis of a Phase II trial. Blood. 2020;136(suppl 1):53-55. [Google Scholar]
- 19.Shih L, Othus M, Gardner K, et al. Relation between measurable residual disease and morphologic relapse in AML. J Clin Oncol. 2020;38(suppl 15):7535. [Google Scholar]
- 20.Maiti A, Rausch CR, Cortes JE, et al. Outcomes in molecular subgroups and resistance patterns with ten-day decitabine and venetoclax (DEC10-VEN) in acute myeloid leukemia. Blood. 2019;134(suppl_1):645.31262782 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.