

Key Points
Heme, a novel endogenous ligand shared by CLEC-2 and GPVI/FcRγ, activates human and murine platelets.
Platelet CLEC-2 and GPVI/FcRγ exacerbate RAKI in mice.
Abstract
There is increasing evidence that platelets participate in multiple pathophysiological processes other than thrombosis and hemostasis, such as immunity, inflammation, embryonic development, and cancer progression. A recent study revealed that heme (hemin)-activated platelets induce macrophage extracellular traps (METs) and exacerbate rhabdomyolysis-induced acute kidney injury (RAKI); however, how hemin activates platelets remains unclear. Here, we report that both C-type lectin-like receptor-2 (CLEC-2) and glycoprotein VI (GPVI) are platelet hemin receptors and are involved in the exacerbation of RAKI. We investigated hemin-induced platelet aggregation in humans and mice, binding of hemin to CLEC-2 and GPVI, the RAKI-associated phenotype in a mouse model, and in vitro MET formation. Using western blotting and surface plasmon resonance, we showed that hemin activates human platelets by stimulating the phosphorylation of SYK and PLCγ2 and directly binding to both CLEC-2 and GPVI. Furthermore, hemin-induced murine platelet aggregation was partially reduced in CLEC-2–depleted and FcRγ-deficient (equivalent to GPVI-deficient) platelets and almost completely inhibited in CLEC-2–depleted FcRγ-deficient (double-knockout) platelets. In addition, hemin-induced murine platelet aggregation was inhibited by the CLEC-2 inhibitor cobalt hematoporphyrin or GPVI antibody (JAQ-1). Renal dysfunction, tubular injury, and MET formation were attenuated in double-knockout RAKI mice. Furthermore, in vitro MET formation assay showed that the downstream signaling pathway of CLEC-2 and GPVI is involved in MET formation. We propose that both CLEC-2 and GPVI in platelets play an important role in RAKI development.
Visual Abstract
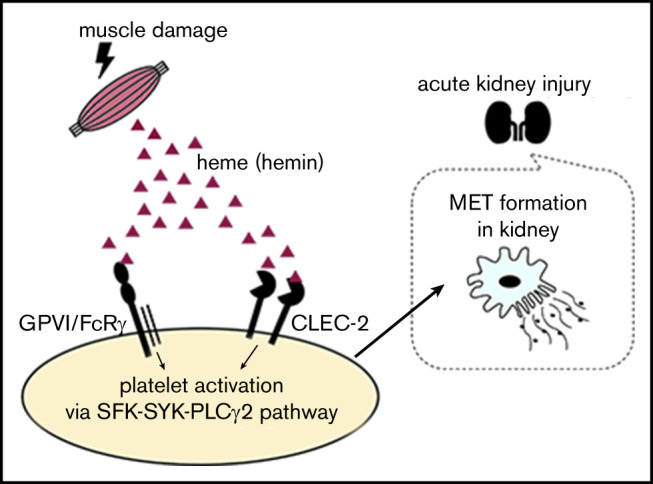
Introduction
Rhabdomyolysis may be caused by trauma, heat stroke, or medication and can lead to acute kidney injury (AKI).1 In rhabdomyolysis-induced AKI (RAKI), myoglobin-derived heme is immediately converted to hemin in the blood.2 Hemin induces multiple pathologic responses, such as inflammation, cellular damage, and endothelial activation, due to its ability to generate reactive oxidative species.3 Therefore, oxidative stress generated by hemin has been thought to directly exacerbate RAKI.4-6 A recent study proposed a novel mechanism of action for RAKI in which METs mediated by hemin-activated platelets promote renal tubule injury and lead to renal dysfunction; however, the mechanism of hemin-induced platelet activation remains unclear.7
Platelets are activated by multiple biological substances, which have corresponding receptors and downstream signaling cascades. C-type lectin-like receptor-2 (CLEC-2) and glycoprotein VI (GPVI) are platelet activation receptors with different endogenous ligands (podoplanin and collagen, respectively). Nevertheless, they share downstream signaling molecules, such as SYK, SLP-76, and PLCγ2. A number of recent studies have shown that CLEC-2 and GPVI are involved in various pathophysiological processes, including thrombosis, immunity, inflammation, embryonic development, and cancer progression.8-10 In the current study, we performed in vitro and in vivo analyses to elucidate the mechanism underlying hemin-induced platelet activation and its contribution to the exacerbation of RAKI. We propose that both CLEC-2 and GPVI are novel hemin receptors that activate platelets and are involved in RAKI development.
Methods
Preparation of hemin solution
Hemin was purchased from Nakalai Tesque (Kyoto, Japan). According to PubChem (https://pubchem.ncbi.nlm.nih.gov/), the molecular weight of hemin is 651.9 g/mol and the net charge is 0. Hemin was dissolved in 1% dimethyl sulfoxide/phosphate-buffered saline (PBS). For each experiment, we prepared the hemin solution from powder immediately before use and shaded the solution during experiments to maintain the quality.
Platelet aggregation
Washed human and murine platelets (2.0 × 108/mL) were prepared as previously described11 and stimulated with 0 to 10 μg/mL hemin, 0.125 or 0.25 µg/mL collagen-related peptide (CRP) (Peptide Institute, Osaka, Japan), 5 or 10 nM rhodocytin,12 0.25 U/mL thrombin (Haematologic Technologies, Essex, VT), or 250 nM phorbol 12-myristate 13-acetate (PMA) (Wako Pure Chemical, Osaka, Japan). Before stimulation, the washed platelets were incubated with 0.2 mM Arg-Gly-Asp-Ser (RGDS) (Peptide Institute, Osaka, Japan) for 5 minutes, 50 μM PP2 (Merck, Darmstadt, Germany) for 5 minutes, 10 µM SU6656 (Cayman Chemical, Ann Arbor, MI) for 30 minutes, 1 µM R406 (Cayman Chemical) for 1 minute, 0.8 µg/mL cobalt hematoporphyrin (Co-HP)13 for 2 minutes, or 10 µg/mL anti-mouse GPVI antibody (clone JAQ-1; Emfret Analytics, Eibelstadt, Germany) for 5 minutes. Platelet aggregation was evaluated by using MSM Hematracer 712 (LMS, Tokyo, Japan).
This study was approved by the Ethical Committee at the University of Yamanashi and the Animal Care and Use Committee at the University of Yamanashi. All participants provided written informed consent in accordance with the Declaration of Helsinki.
Western blotting
Western blotting was performed as previously described.14 Hemin-stimulated washed human platelets (5.0 × 108/mL) with or without PP2 were directly dissolved in sample buffer, separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and electrotransferred. The membranes were incubated with antibodies (1:1000) against phospho-human SYK (Y525/526) (Cell Signaling Technology, Beverly, MA) and phospho-human PLCγ2 (Cell Signaling Technology). The membranes were washed, followed by incubation with horseradish peroxidase–conjugated secondary antibody. The protein bands were visualized through chemiluminescence using enhanced chemiluminescence ECL prime western blotting detection reagents (GE Healthcare, Chicago, IL). The blots were subsequently stripped and reprobed with antibodies (1:1000) against human SYK (Santa Cruz Biotechnology, Dallas, TX) and human PLCγ2 (Santa Cruz Biotechnology). The bands were visualized in the same way. The protein bands were quantified by densitometry in ImageJ. The phosphorylation levels of SYK and PLCγ2 normalized by total SYK and PLCγ2, respectively, are presented relative to the mean phosphorylation levels of untreated controls.
Surface plasmon resonance spectrometry
The direct binding of hemin to CLEC-2 and GPVI was analyzed by using BIAcore X (BIAcore AB, Uppsala, Sweden). Human CLEC-2–human Fc, human GPVI–human Fc, and human Fc (control) recombinant proteins15 were covalently coupled to a CM chip (GE Healthcare). Various concentrations of hemin diluted in N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid–buffered saline with surfactant containing 1% dimethyl sulfoxide were perfused over the recombinant-coated surface at a rate of 10 µL/min at 25°C, and the changes in resonance unit (RU) were recorded. NaOH was used as a regeneration solution and injected to break the specific binding between the analyte and ligand. The RU showing hemin/CLEC-2 or hemin/GPVI interactions was calculated by subtracting RU of human Fc from that of human CLEC-2–human Fc or human GPVI–human Fc. The kinetics analysis was performed by using the Langmuir model.
Cell-based competitive binding assay
Human CLEC-2-T-REx 293 cells16 and GPVI-Jurkat cells17 were used to examine whether hemin competes with known ligands (podoplanin and CRP) in binding to the binding sites of CLEC-2 and GPVI. CLEC-2 expression was induced by adding 1 µg/mL doxycycline (BD Biosciences, San Jose, CA) or PBS to the medium. After 24 hours of treatment with doxycycline, T-REx 293 cells with or without CLEC-2 were suspended in PBS and incubated with 75 µg/mL of hemin for 20 minutes. Next, 2.5 µg/mL of human podoplanin–human Fc or human Fc was added to the cells and incubated for 1 hour. After washing, goat anti-human immunoglobulin G (IgG)-Alexa Fluor 488 (200:1) (Invitrogen, Carlsbad, CA) was added to detect the binding of human podoplanin–human Fc using the Accuri C6 flow cytometer (BD Biosciences). To detect CRP binding, we labeled CRP with Alexa Fluor 488 (CRP-AF488) using the Alexa Fluor 488 Antibody Labeling Kit (Invitrogen). Jurkat cells with or without GPVI were suspended in PBS and incubated with 75 µg/mL of hemin for 20 minutes. Thereafter, we added 0.1 µg/mL of CRP-AF488 and detected the binding of CRP using a flow cytometer.
Mice
FcRγ-deficient C57BL/6 mice were provided by the RIKEN Research Center for Allergy and Immunology (Yokohama, Japan).18 To obtain platelet-specific CLEC-2–depleted mice, we used an anti–CLEC-2 antibody (clone; 2A2B10).19 2A2B10 (8 mg/kg body weight) was intravenously or peritoneally injected into wild-type (WT) mice (C57BL/6; purchased from CLEA Japan, Tokyo, Japan). CLEC-2 levels were confirmed by using flow cytometry. Similarly, to prepare CLEC-2–depleted FcRγ-deficient (referred to as double-knockout [DKO] in this study) mice, anti–CLEC-2 antibody was administered to FcRγ-deficient mice.
RAKI model mice
Fifty percent glycerol (Sigma-Aldrich, St. Louis, MO) diluted in water was bilaterally injected (7.5 mL/kg body weight) into 8- to 10-week-old mouse quadriceps.20 To deplete platelet CLEC-2 expression, anti-mouse CLEC-2 antibody (8 mg/kg body weight)19 was injected intravenously 1 week before glycerol injection. Rat IgG (Cosmo Bio, Tokyo, Japan) was used as a control. Renal function was assessed by serum creatinine level 48 hours after glycerol injection. To evaluate renal tubular injury, mice were killed 48 hours after glycerol injection. Kidneys were fixed in 4% paraformaldehyde, embedded in paraffin, sliced at a thickness of 5 µm, and stained with periodic acid–Schiff. We defined tubular injury as intratubular cast formation, debris, loss of the brush border, and necrosis.21 Furthermore, we counted the number of injured tubules in 9 areas (668 × 524 µm) of the cortex.
For evaluation of MET formation, mice were killed 8 hours after glycerol injection. Kidneys were fixed in 4% paraformaldehyde, embedded in optimal cutting temperature compound (Sakura Finetek Japan, Tokyo, Japan), and cryosectioned at a thickness of 16 µm. Sections were blocked with 10% donkey serum for 30 minutes. We incubated specimens with 1:200 rabbit anti-CitH3 antibody (Ab5103, Abcam, Cambridge, MA) and 1:200 rat anti-F4/80 antibody (eBioscience, San Diego, CA) at 4°C overnight and then with 1:1000 Alexa Fluor 555–labeled anti-rabbit IgG antibody (Abcam) and 1:500 Alexa Fluor 488–labeled anti–rat IgG antibody (Life Technologies, Carlsbad, CA) for 90 minutes. After immunofluorescence staining, autofluorescence was suppressed by using TrueVIEW (Vector Laboratories, Burlingame, CA). Finally, nuclear staining was performed with 5 µg/mL 4′,6-diamidino-2-phenylindole (Sigma-Aldrich) for 5 minutes. We defined DNA-, F4/80-, and citrullinated histone H3 (CitH3)-positive MET fibers exceeding 20 µm in length as MET-like structures according to a previous report.7 We captured 5 representative fields (236 µm × 236 µm) per kidney and counted the MET-like structures (Olympus FV-1000).
Cell culture and induction of METs
THP-1 (human monocytic leukemia cell line, ATCC, Manassas, VA) was cultured in RPMI 1640 (Gibco Laboratories, Grand Island, NY) supplemented with 10% fetal bovine serum (Gibco) and penicillin/streptomycin (Lonza, Basel, Switzerland) at 37°C under 5% carbon dioxide. We incubated 1 × 105 THP-1 cells in 500 µL RPMI 1640 with 50 nM PMA for 3 days at 37°C under 5% carbon dioxide to differentiate these cells into macrophage-like cells. THP-1–derived macrophages were cultured without fetal bovine serum overnight and primed with 10 ng/mL tumor necrosis factor-α (Invitrogen) for 15 minutes. A total of 1 × 106/µL platelets were stimulated with 75 µg/mL hemin before adding 50 µL of hemin-activated platelet solution to THP-1–derived macrophages in the presence of 1 µM SYTOX Green (Life Technologies) and 1 µg/mL Hoechst33342 (Dojindo Laboratories, Kumamoto, Japan). To confirm the involvement of the SFK signaling pathway in platelet activation, PP2 was added to the washed platelets before hemin stimulation. As a positive control for MET formation, 500 nM PMA was added to THP-1–derived macrophages. We considered extracellular SYTOX Green–positive fibers as MET-like structures. Five fields (704 µm × 528 µm) were captured per well using a fluorescence microscope (Olympus IX71S1F-2), and the numbers of MET-like structures and total cells were counted. The percentage of MET-like structures in total cells was calculated.
Statistical analysis
Data are expressed as mean ± standard deviation unless otherwise noted. Statistical analyses were performed by using the Student t test, Tukey’s multiple comparison test, or the Holm-Šidák multiple-comparison test in GraphPad Prism 8 (GraphPad Software, La Jolla, CA). Differences with a P value <.05 were considered significant.
Results
Hemin activates human platelet through the SFK-SYK-PLCγ2 pathway
To elucidate the role of platelets in RAKI, we first examined the molecular mechanism underlying platelet activation by heme (hemin). In aggregation assays, hemin increased light transmittance of washed human platelets in a dose-dependent manner (Figure 1A). RGDS, an inhibitory peptide of integrin-mediated platelet aggregation, almost completely blocked the hemin-induced increase in light transmittance. Similar results were obtained when platelets were stimulated with CRP or rhodocytin, ligands of GPVI, or CLEC-2, respectively (Figure 1B), indicating that hemin induces platelet aggregation but not agglutination. Hemin-induced platelet aggregation was also blocked by the SFK inhibitors PP2 and SU6656, as well as the R406 SYK inhibitor. In addition, western blot analysis showed that the phosphorylation of SYK and PLCγ2 was upregulated by hemin, and the upregulation was inhibited by PP2 to a basal level (Figure 1C-D). These results indicate that hemin activates human platelets through the SFK-SYK-PLCγ2 pathway.
Figure 1.
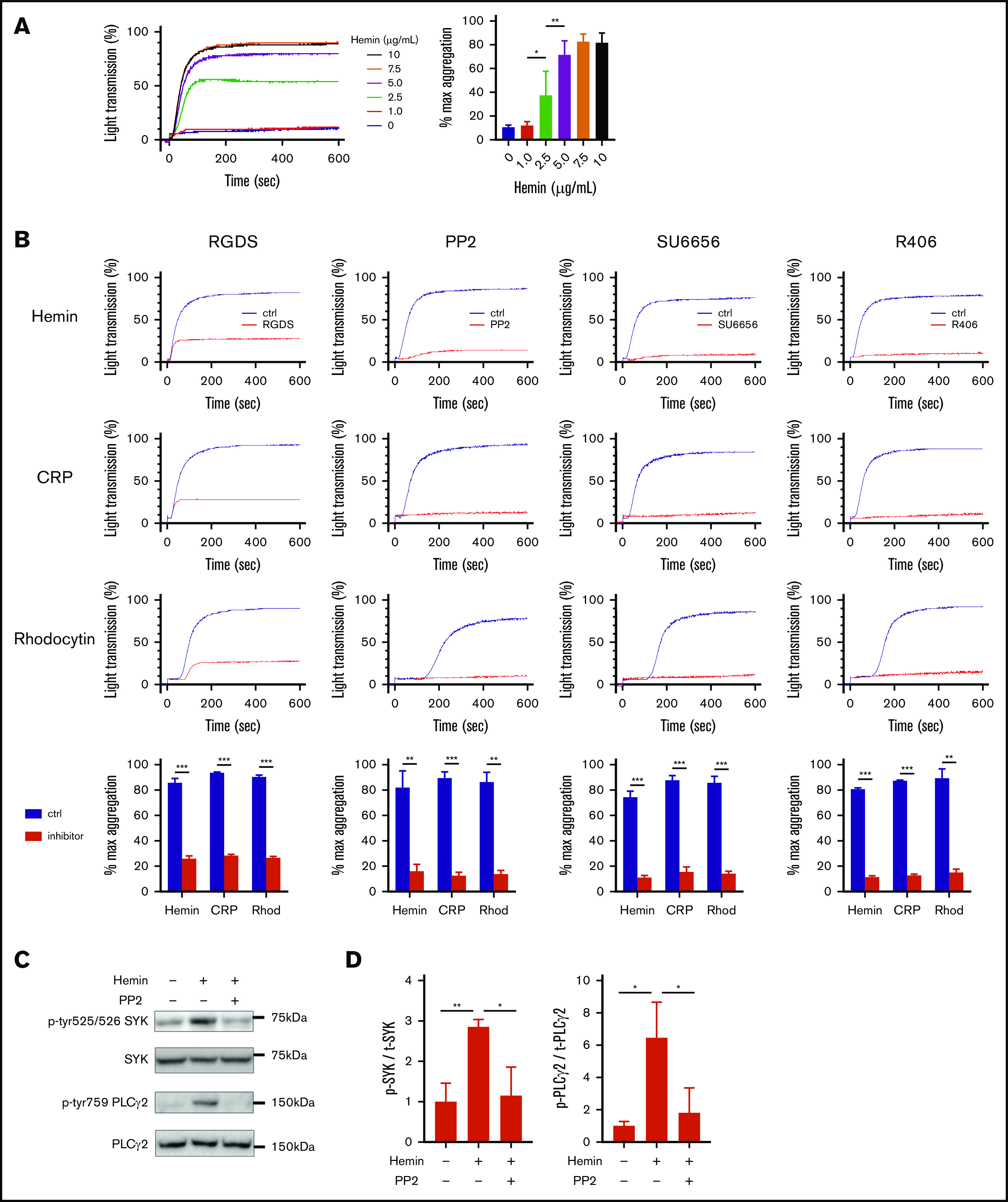
Hemin activates human platelet through the SFK-SYK-PLCγ2 pathway. (A) Hemin-induced platelet aggregation in humans. Washed human platelets were stimulated with 6 concentrations of hemin (0, 1.0, 2.5, 5.0, 7.5, and 10 μg/mL). The representative curves (left) and quantifications of maximum light transmission (right) are shown. (B) Blocking effect of 0.2 mM RGDS, 50 μM PP2, 10 µM SU6656, and 1 µM R406 on hemin (7.5 μg/mL)-, CRP (0.25 µg/mL)-, or rhodocytin (Rhod) (10 nM)-induced platelet aggregation. The representative curves and quantifications of maximum light transmission are shown. (C) Western blot analysis of phospho-human SYK (Y525/526), phospho-human PLCγ2 (Y759), human SYK, and human PLCγ2. Representative images of hemin-induced (7.5 μg/mL) phosphorylation of SYK and PLCγ2 and its inhibition by PP2 in human platelets are shown. (D) Quantitative analysis of SYK (left) and PLCγ2 (right) phosphorylation normalized by total SYK and PLCγ2, respectively. Data are the mean ± standard deviation (panels A, B, and D); n = 3 to 4. *P < .05, **P < .01, ***P < .001. Tukey’s multiple-comparison test for panels A and D, Student t test for panel B.
Among platelet activation receptors, CLEC-2 and GPVI require SFK-SYK-PLCγ2 signaling for platelet activation.22 Furthermore, Ca2+ flux and adenosine 5′-diphosphate release have an important role in CLEC-2–mediated or GPVI-mediated platelet aggregation, especially when stimulated with low concentrations of agonists. We therefore examined the dependence of hemin-induced platelet activation on Ca2+ flux and adenosine 5′-diphosphate release to confirm the similarity between hemin-induced and CLEC-2– and GPVI stimulation–induced platelet activation. BAPTA-AM, a membrane-permeable Ca2+ chelator, completely inhibited hemin-induced platelet aggregation. BAPTA-AM also completely inhibited CRP- or rhodocytin-induced platelet aggregation (supplemental Figure 1A). Although the inhibitory effect of apyrase was minimal when platelets were stimulated by a high concentration (7.5 µg/mL) of hemin, apyrase exhibited a noticeable inhibitory effect on platelet aggregation induced by a low concentration (3 µg/mL) of hemin (supplemental Figure 1B). A similar concentration-dependent effect of apyrase was observed in CRP- and rhodocytin-induced platelet aggregation.
Hemin binds directly to both CLEC-2 and GPVI
Because the hemin-induced platelet activation seems mechanistically similar to the activation thorough CLEC-2 and/or GPVI, we hypothesized that hemin is an endogenous ligand for CLEC-2 and/or GPVI. Surface plasmon resonance was used to examine whether hemin directly binds to human CLEC-2 and/or human GPVI. Hemin was perfused on surfaces coated with human CLEC-2–human Fc or human GPVI–human Fc. The resonance unit, which indicates binding of hemin to the ligands (coated human CLEC-2–human Fc or human GPVI–human Fc), increased during perfusion of hemin solution and decreased after perfusion of hemin-free solution. The same experiment was performed by changing the ligand to human Fc alone. The RU curves were corrected for human Fc values. Interestingly, following the human Fc correction, the sensorgrams indicated direct and dose-dependent binding of hemin to both human CLEC-2 and human GPVI (Figure 2). Kinetics analysis showed that hemin binds to human CLEC-2 and human GPVI, with dissociation constant (KD) values of 0.915 µM and 29.4 µM, respectively.
Figure 2.
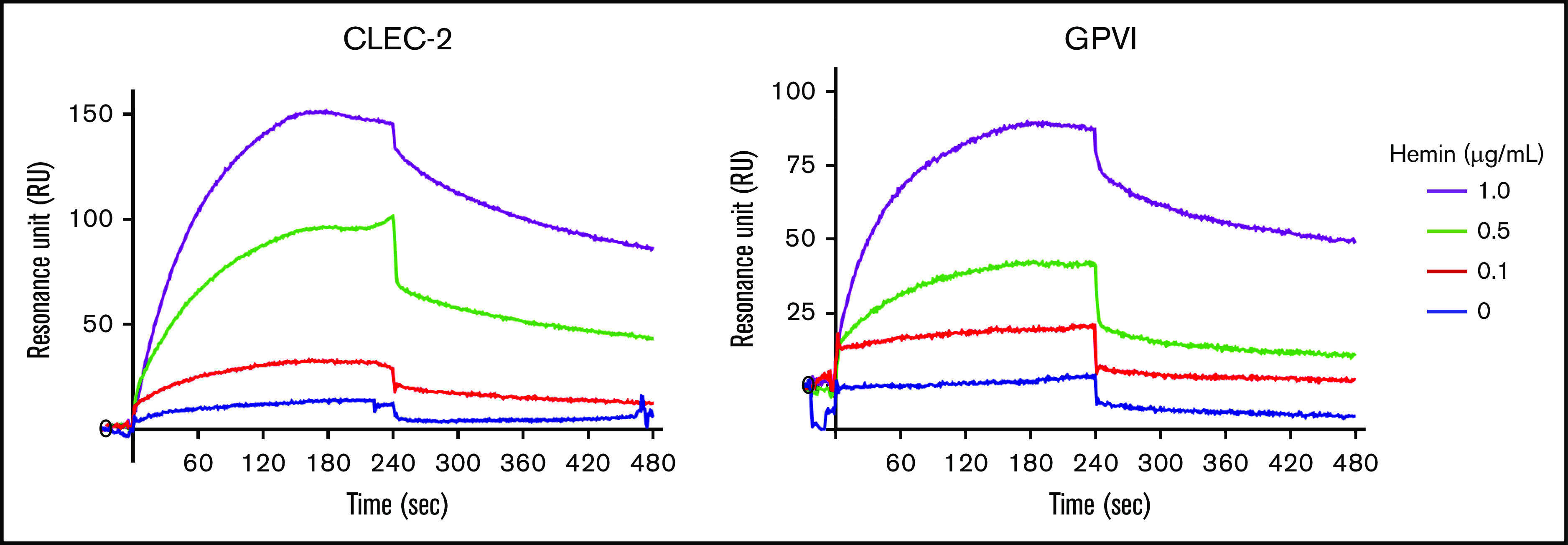
Hemin binds directly to both CLEC-2 and GPVI. Direct binding of hemin to immobilized human CLEC-2 (left) and human GPVI (right) evaluated by using the BIAcore X system. The changes in RU using different concentrations of hemin (0, 0.1, 0.5, and 1.0 μg/mL) are shown.
Both CLEC-2 and GPVI contribute to hemin-induced platelet activation
First, we confirmed that hemin induces platelet activation in mice and that this involves the SFK and SYK pathways. Hemin induced platelet aggregation in WT mice at similar concentrations to those observed in human subjects (Figure 3A). Furthermore, the inhibitory effects of PP2, SU6656, and R406 on hemin-induced platelet aggregation observed in mice were similar to the effects observed in humans (supplemental Figure 2). Next, we examined the dependency of hemin-induced platelet activation on mouse CLEC-2 and/or mouse GPVI. Platelet aggregation assay was further conducted by using CLEC-2–depleted platelets, FcRγ-deficient platelets (which lack GPVI expression), and CLEC-2–depleted FcRγ-deficient (referred to as DKO) platelets. Hemin-induced platelet aggregation was slightly but significantly decreased in CLEC-2–depleted platelets compared with that in WT platelets and further decreased in FcRγ-deficient platelets (Figure 3B). Importantly, in DKO platelets, hemin-induced (5.0 μg/mL) platelet aggregation was completely inhibited, and even the highest concentration of hemin (10 μg/mL) induced only marginal aggregation.
Figure 3.
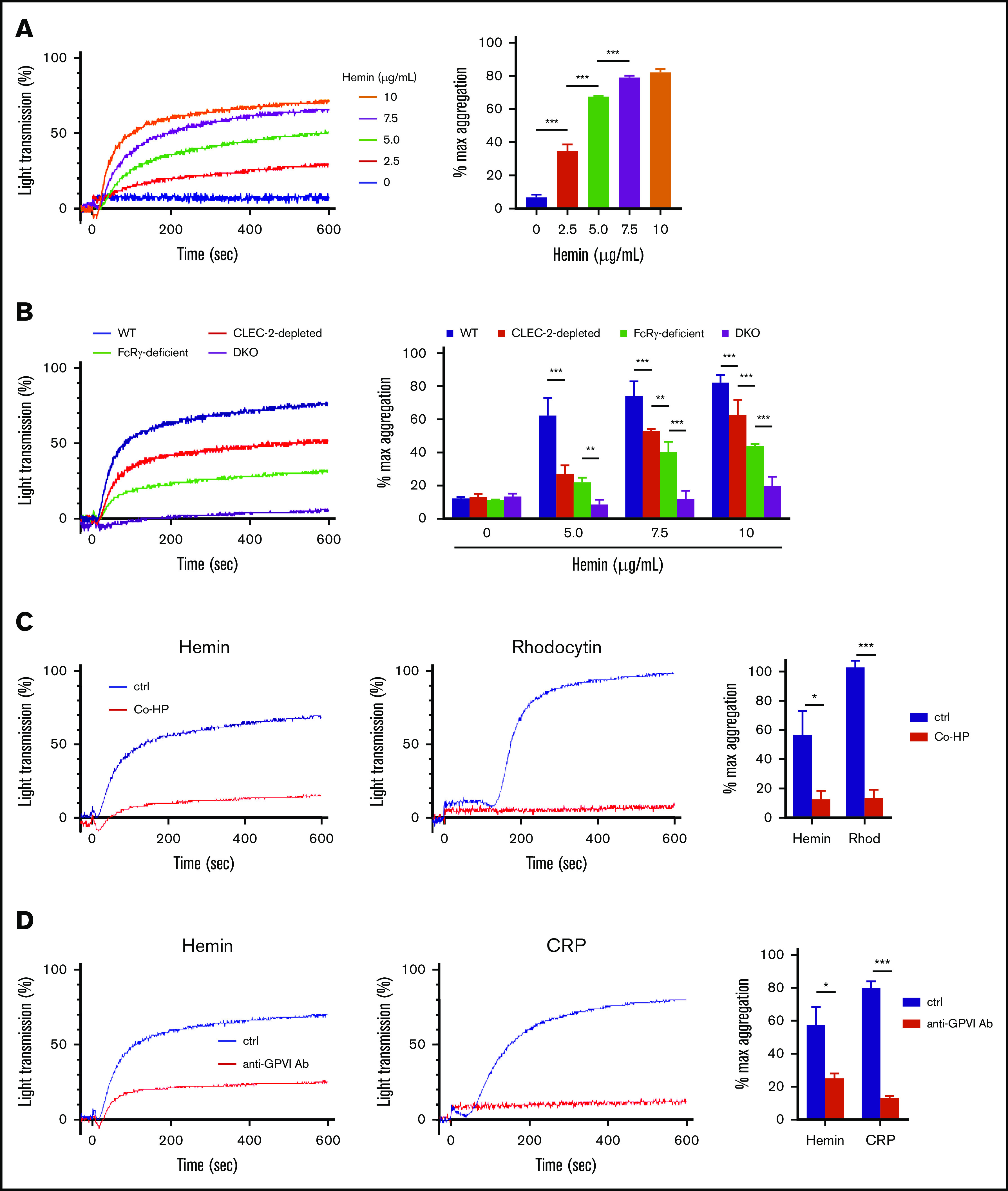
Contribution of CLEC-2 and GPVI to platelet activation by hemin. (A) Hemin-induced platelet aggregation in WT mice. Washed murine platelets were stimulated with 5 concentrations of hemin (0, 2.5, 5.0, 7.5, and 10 μg/mL). (B) Hemin-induced murine platelet aggregation using 4 concentrations of hemin (0, 5.0, 7.5, and 10 μg/mL) in WT (blue), CLEC-2–depleted (red), FcRγ-deficient (green), and CLEC-2–depleted FcRγ-deficient (purple) mice. The representative aggregation curves of platelets treated with 7.5 µg/mL hemin are shown. (C) Blocking effect of 0.8 µg/mL Co-HP or 1% dimethyl sulfoxide/PBS (control) on hemin (10 μg/mL)- or rhodocytin (Rhod) (5 nM)-induced platelet aggregation in FcRγ-deficient mice. The representative curves (left and middle) and quantifications of maximum light transmission (right) are shown. (D) Blocking effect of 10 µg/mL anti-GPVI antibody (ab) or control rat IgG (control) on hemin (10 μg/mL)- and CRP (0.125 µg/mL)-induced platelet aggregation in CLEC-2–depleted mice. The representative curves (left and middle) and quantifications of maximum light transmission (right) are shown. Data in the bar graphs (panels A-D) are mean ± standard deviation; n = 3. *P < .05, **P < .01, ***P < .001. Tukey’s multiple-comparison test was used for panels A and B, Student t test was used for panels C and D.
We confirmed platelet aggregation induced by 0.5 µg/mL CRP, 5 nM rhodocytin, or 0.05 U/mL thrombin as controls for our 4 platelet conditions. As expected, thrombin induced platelet aggregation in CLEC-2–depleted, FcRγ-deficient, and DKO platelets at equivalent levels as in WT platelets. On the other hand, responses of CLEC-2–depleted and GPVI-deficient platelets to CRP and rhodocytin, respectively, were preserved (data not shown). These findings indicate that hemin-induced platelet activation is highly dependent on both CLEC-2 and GPVI.
To elucidate whether hemin shares the CLEC-2 and GPVI binding sites with the known ligands, we used 2 competitive inhibitors against CLEC-2 and GPVI. One of them, Co-HP, was reported to competitively inhibit CLEC-2–podoplanin interaction.13 The second one is an anti-mouse GPVI monoclonal antibody, JAQ1, which reportedly recognizes an epitope identical with or similar to the CRP-binding site on GPVI and inhibits CRP-induced aggregation of murine platelets.23-25 To clearly detect the inhibitory effect of Co-HP and JAQ1, FcRγ-deficient and CLEC-2–depleted platelets were used in a platelet aggregation assay, respectively. Co-HP significantly inhibited hemin- or rhodocytin-induced platelet aggregation (Figure 3C). Similarly, JAQ-1 inhibited hemin- or CRP-induced platelet aggregation (Figure 3D). In contrast, low or no inhibitory effects of Co-HP or JAQ-1 were observed on thrombin- or PMA-induced platelet aggregation (supplemental Figure 3). In addition, cell-based competitive binding assay by flow cytometry showed that hemin significantly inhibits the binding of podoplanin or CRP to CLEC-2 or GPVI, respectively (supplemental Figure 4A-B). These results suggest that hemin shares the binding sites with known ligands for CLEC-2 and GPVI.
Renal damage and MET formation are reduced in DKO mice
A previous report showed that hemin-activated platelets exacerbate RAKI by inducing MET formation in renal tubules.7 Based on our findings regarding the capacity of hemin to activate human platelets by binding to both CLEC-2 and GPVI, and because hemin-induced platelet activation in mice was dependent on CLEC-2 and GPVI expression, we hypothesized that both CLEC-2 and GPVI are involved in RAKI. An RAKI mouse model was generated by injecting glycerol into the quadriceps of WT and DKO mice. We performed macroscopic observations for kidney ischemia, assessed serum creatinine levels, and used periodic acid–Schiff staining to identify tubular injuries. Severely ischemic, pale-appearing bilateral kidneys were observed in glycerol-injected WT mice, whereas nonischemic, deep-red kidneys were observed in glycerol-injected DKO mice, similar to the observation in water-injected non-rhabdomyolytic mice (Figure 4A). Serum creatinine levels of DKO mice were significantly lower than those of WT mice (Figure 4B). Furthermore, the percentage of injured tubules in WT mice was significantly higher than that in DKO mice (Figure 4C).
Figure 4.
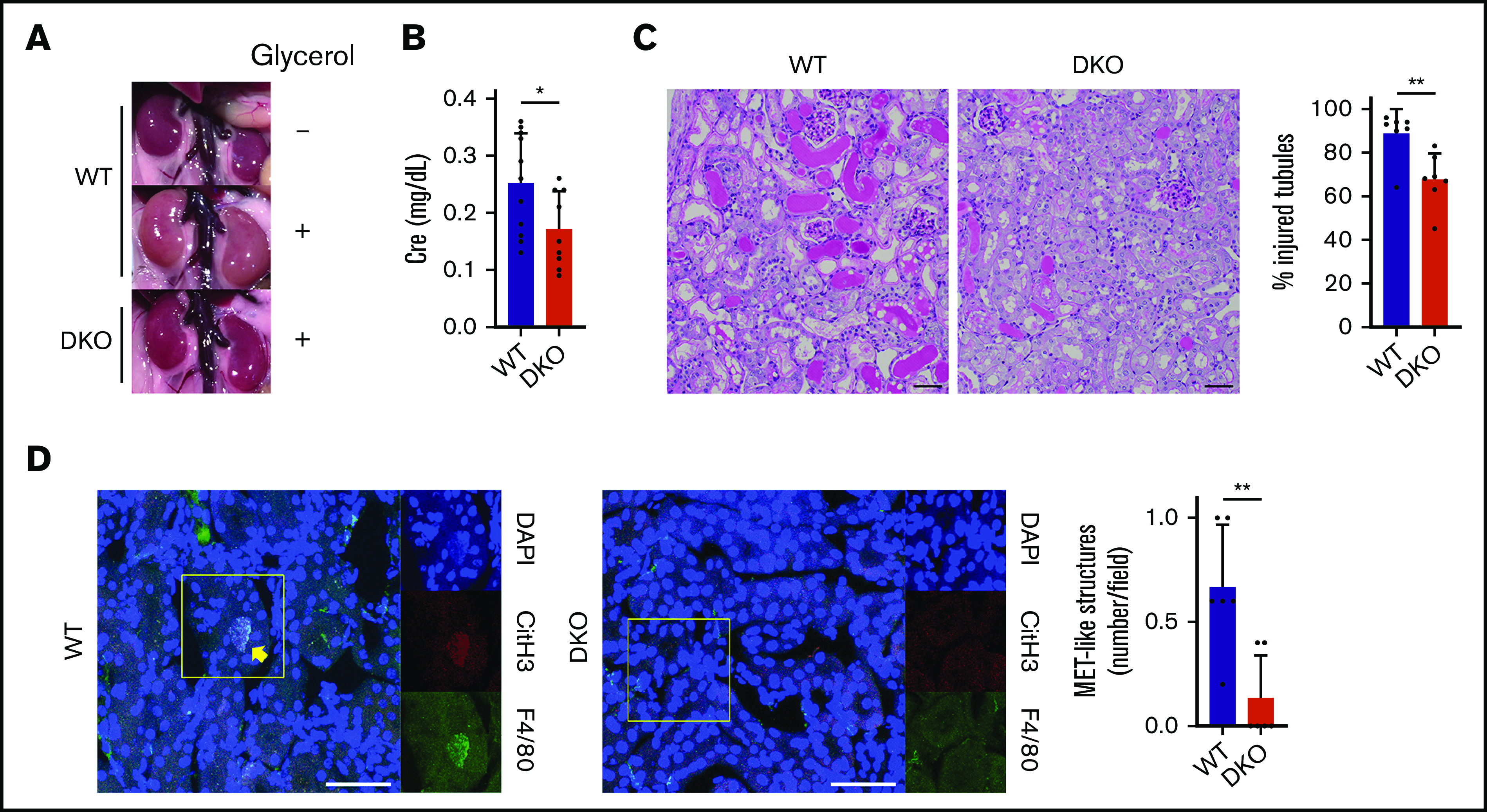
Renal damage and MET formation are reduced in DKO mice. Phenotypic analyses of RAKI WT and CLEC-2–depleted FcRγ-deficient (DKO) mice. (A) Representative macroscopic images of kidneys showing RAKI-induced ischemia. The top panel indicates water-injected (non-rhabdomyolytic) WT kidneys. (B) Serum creatinine (Cre) level at 48 hours after glycerol injection into WT and DKO mice. Data are expressed as mean ± standard deviation; n = 11 and 9, respectively. (C) Renal tubular injury evaluated by periodic acid–Schiff staining at 48 hours after glycerol injection into WT and DKO mice. Scale bars: 50 μm. Data are expressed as mean ± standard deviation; n = 7. (D) Immunofluorescence imaging of citrullinated histone H3 (CitH3, red), F4/80 (green), and 4′,6-diamidino-2-phenylindole (DAPI) (blue) for the detection of MET formation in renal tubules at 8 hours after glycerol injection into WT and DKO mice. The arrow indicates macrophages undergoing MET formation. Magnified single-color views of a framed area in each merged image are shown on the right. Scale bars: 50 μm. Data are expressed as mean ± standard deviation; n = 6. *P < .05, **P < .01.
We next performed immunofluorescence imaging of MET-like structure in RAKI-induced kidneys by detecting CitH3, F4/80, and DNA. The number of MET-like structures in RAKI-induced kidney was significantly reduced in DKO mice compared with that in WT mice (Figure 4D). These results strongly suggest that platelet CLEC-2 and GPVI/FcRγ have an important role in the exacerbation of RAKI.
MET formation by hemin-activated platelets requires activation of the SFK signaling pathway in platelets
To examine whether hemin-activated platelets promote MET formation via platelet SFK pathway, we used an in vitro MET formation assay using THP-1–derived macrophages. In the presence of hemin-activated platelets, 8.4% of MET-like structures were observed in macrophages differentiated from THP-1, which is comparable to the percentage of MET-like structures observed under stimulation with PMA (positive control). In contrast, hemin alone or resting platelets induced significantly fewer METs, comparable to the basal level. Importantly, METs induced by hemin-treated platelets were significantly inhibited by PP2 pretreatment of platelets, whereas PP2 did not inhibit PMA-induced MET formation (Figure 5). These results indicate that PP2 indirectly blocks MET formation by inhibiting platelet activation but does not directly affect THP-1-derived macrophages. Taken together, these results strongly suggest that generation of METs by hemin-activated platelets requires activation through the SFK-signaling pathway in platelets.
Figure 5.
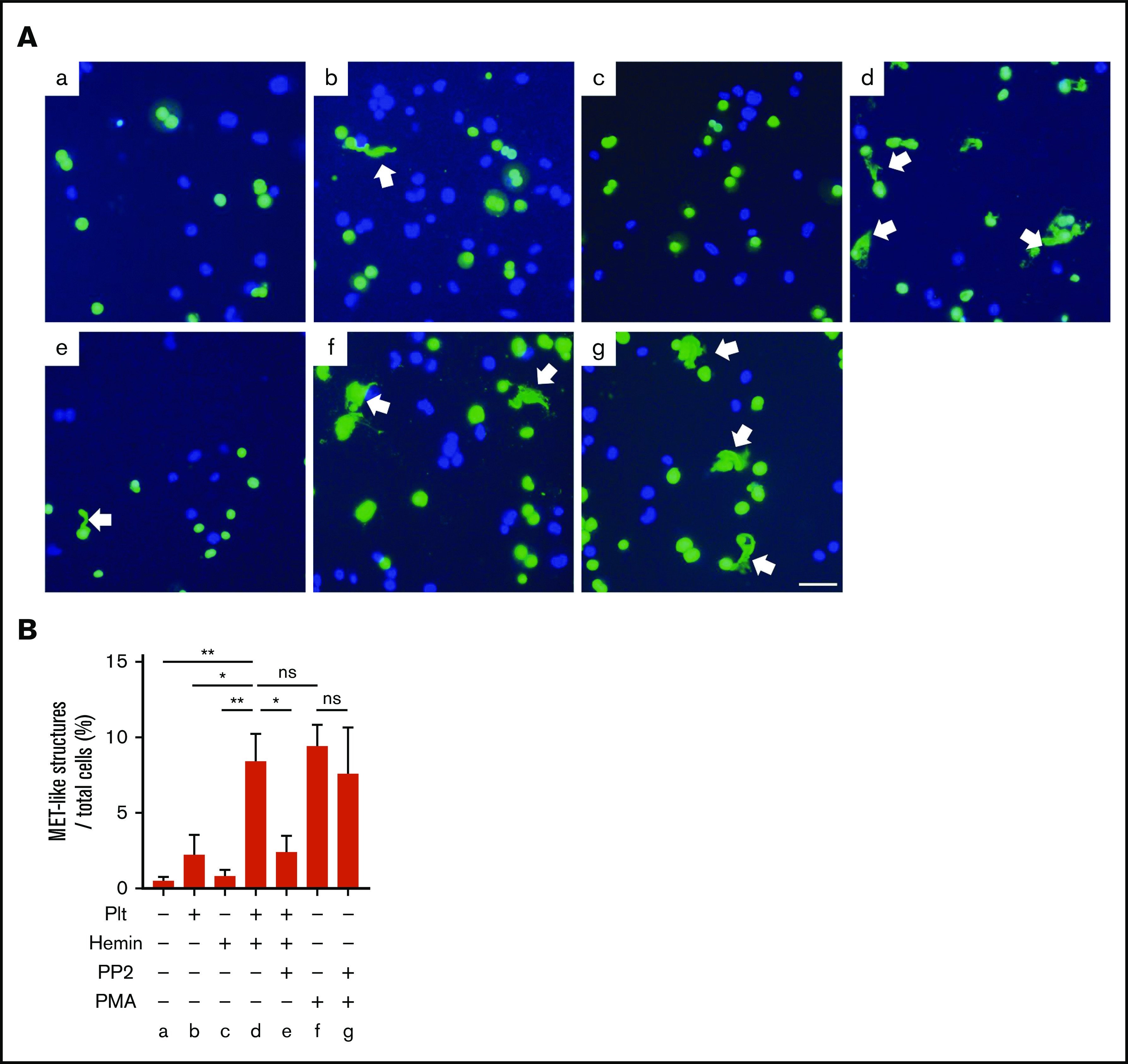
MET formation by hemin-activated platelets requires activation of the SFK pathway in platelets. (A) Macrophages differentiated from THP-1 were stimulated by resting platelets (b), hemin (c), hemin-activated platelets (d), hemin-activated platelets with PP2 (e), PMA (f), and PMA with PP2 (g). Representative images of SYTOX Green (green) and Hoechst 33342 (blue) staining are shown. Arrows indicate MET-like structures. Scale bars: 50 μm. (B) Quantification of MET-like structures. Data in the bar graph are expressed as mean ± standard error of the mean; n = 4. *P < .05, **P < .01. Holm-Šidák multiple-comparison test was used. ns, not significant.
Discussion
Rhabdomyolysis is a potentially fatal condition, and RAKI is a particularly serious complication. Myoglobin released from injured skeletal muscles has been thought to be the trigger for RAKI. In addition, macrophages have been shown to be involved in the pathogenesis of rhabdomyolysis.20 Although the precise molecular mechanism remains unclear, platelets are reportedly involved in this mechanism. Okubo et al7 presented a new mechanism underlying rhabdomyolysis-induced AKI, in which activated platelets by hemin released from necrotic muscle cells promote MET formation in kidneys. However, the mechanism by which hemin activates platelets has not been fully elucidated. In this study, we showed that hemin is a novel ligand of both CLEC-2 and GPVI and activates platelets through the SFK-SYK-PLCγ2 pathway. Furthermore, we showed that inhibition of platelet activation reduced MET formation in vitro, and that DKO mice exhibited reduced MET formation in RAKI-induced kidneys and attenuated renal dysfunction. This suggests that CLEC-2 and GPVI/FcRγ play an important role in the development of RAKI.
Surface plasmon resonance spectrometry, platelet aggregation assay, western blotting, and cell-based competitive binding assay indicated that hemin activates platelets through the SFK-SYK-PLCγ2 signaling pathway by binding to known ligand binding sites of CLEC-2 and GPVI. However, there are some unsolved issues. First, the precise hemin binding site on CLEC-2 and GPVI remains unknown. Further research investigating the crystal structure and analyzing the binding sites of a CLEC-2/GPVI mutant is required to identify the hemin binding sites. Second, other receptors might slightly contribute to hemin-induced platelet activation. There was still a small residual aggregation when platelet aggregation was inhibited by even a high concentration of PP2 (Figure 1B). In addition, murine platelet aggregation assay revealed that a high concentration of hemin slightly increased the light transmittance of DKO platelets (Figure 3B). Lastly, Co-HP and anti-GPVI antibodies failed to completely inhibit hemin-induced platelet aggregation (Figure 3C-D). These results suggest that other mechanisms, apart from those presented here, might be involved in platelet activation by hemin, although hemin mainly activates platelets via CLEC-2 and GPVI/FcRγ.
We confirmed that hemin-activated platelets induce MET formation in vitro and that hemin or resting platelets alone do not. Furthermore, we found that the induction of METs by hemin-activated platelets is attenuated by pretreatment of platelets with the SFK inhibitor PP2, whereas PP2 alone did not directly block MET induction by PMA (Figure 5B). Based on these results, we concluded that platelet CLEC-2 and GPVI downstream signals are involved in MET induction. However, it remains unclear how activated platelets induce METs. Mac-1, which is expressed on macrophages, is known to be important for neutrophil extracellular trap formation26 and binds to platelets via GPIbα.27 Thus, it is hypothesized that platelet GPIbα binds to Mac-1 on macrophages to induce METs. However, this hypothesis was not supported by previous findings7 and our results because resting platelets expressing GPIbα did not induce METs (Figure 5). Specific platelet surface membrane proteins that are upregulated upon platelet activation, such as activated integrin αIIbβ3 and P-selectin, may be involved in the induction of METs.
To the best of our knowledge, no guidelines for the management of rhabdomyolysis are available, nor have any randomized controlled trials of treatment been conducted. The main treatment is restricted to fluid replacement, although the type of fluid, its volume, and the timing of its initiation are being discussed.28 The current study provided a completely new treatment strategy for RAKI. Because we have shown that AKI is mild in DKO mice, anti–CLEC-2 and/or anti-GPVI therapy may reduce the exacerbation of AKI in patients with rhabdomyolysis.
In conclusion, we propose that hemin activates platelets by binding to both CLEC-2 and GPVI and that this pathway is involved in the exacerbation of RAKI. Although further research is needed to elucidate the precise mechanism of hemin-induced platelet activation and the mechanisms through which activated platelets induce METs, anti–CLEC-2 and/or anti-GPVI therapy might represent a new strategy for avoiding severe AKI in patients with rhabdomyolysis.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Takako Otsuka, Wakaba Iha, and Tokuhide Kimura from the University of Yamanashi for their technical assistance.
This study was supported in part by research grants from the Japanese Society of Hematology and the Japanese Association of Dialysis Physicians.
Footnotes
Requests for original data may be submitted to the corresponding author (Katsue Suzuki-Inoue; e-mail: katsuei@yamanashi.ac.jp).
Authorship
Contribution: S. Oishi and N.T. designed the study; S. Oishi, N.T., S. Otake, N.O., T. Sasaki, T. Shirai, and Y.Y. performed the experiments; H.S. and T.I. provided important materials; K.T. and T.K. provided important insights and advice; and S. Oishi, N.T., and K.S.-I. were responsible for assembling the figures and writing the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Katsue Suzuki-Inoue, Department of Clinical and Laboratory Medicine, Faculty of Medicine, University of Yamanashi, 1110 Shimokato, Chuo, Yamanashi 409-3898, Japan; e-mail: katsuei@yamanashi.ac.jp.
References
- 1.Bosch X, Poch E, Grau JM. Rhabdomyolysis and acute kidney injury. N Engl J Med. 2009;361(1):62-72. [DOI] [PubMed] [Google Scholar]
- 2.Cannon JB, Yunker MH, Luoma N. The effect of aggregation inhibitors and antioxidants on the stability of hemin solutions. PDA J Pharm Sci Technol. 1995;49(2):77-82. [PubMed] [Google Scholar]
- 3.Chiabrando D, Vinchi F, Fiorito V, Mercurio S, Tolosano E. Heme in pathophysiology: a matter of scavenging, metabolism and trafficking across cell membranes. Front Pharmacol. 2014;5:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolisetty S, Zarjou A, Agarwal A. Heme oxygenase 1 as a therapeutic target in acute kidney injury. Am J Kidney Dis. 2017;69(4):531-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zager RA. Combined mannitol and deferoxamine therapy for myohemoglobinuric renal injury and oxidant tubular stress. Mechanistic and therapeutic implications. J Clin Invest. 1992;90(3):711-719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boutaud O, Moore KP, Reeder BJ, et al. Acetaminophen inhibits hemoprotein-catalyzed lipid peroxidation and attenuates rhabdomyolysis-induced renal failure. Proc Natl Acad Sci USA. 2010;107(6):2699-2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okubo K, Kurosawa M, Kamiya M, et al. Macrophage extracellular trap formation promoted by platelet activation is a key mediator of rhabdomyolysis-induced acute kidney injury. Nat Med. 2018;24(2):232-238. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki-Inoue K, Tsukiji N, Shirai T, Osada M, Inoue O, Ozaki Y. Platelet CLEC-2: roles beyond hemostasis. Semin Thromb Hemost. 2018;44(2):126-134. [DOI] [PubMed] [Google Scholar]
- 9.Rayes J, Watson SP, Nieswandt B. Functional significance of the platelet immune receptors GPVI and CLEC-2. J Clin Invest. 2019;129(1):12-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suzuki-Inoue K. Platelets and cancer-associated thrombosis: focusing on the platelet activation receptor CLEC-2 and podoplanin. Blood. 2019;134(22):1912-1918. [DOI] [PubMed] [Google Scholar]
- 11.Inoue O, Hokamura K, Shirai T, et al. Vascular smooth muscle cells stimulate platelets and facilitate thrombus formation through platelet CLEC-2: implications in atherothrombosis. PLoS One. 2015;10(9):e0139357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shin Y, Morita T. Rhodocytin, a functional novel platelet agonist belonging to the heterodimeric C-type lectin family, induces platelet aggregation independently of glycoprotein Ib. Biochem Biophys Res Commun. 1998;245(3):741-745. [DOI] [PubMed] [Google Scholar]
- 13.Tsukiji N, Osada M, Sasaki T, et al. Cobalt hematoporphyrin inhibits CLEC-2-podoplanin interaction, tumor metastasis, and arterial/venous thrombosis in mice. Blood Adv. 2018;2(17):2214-2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suzuki-Inoue K, Inoue O, Frampton J, Watson SP. Murine GPVI stimulates weak integrin activation in PLCgamma2-/- platelets: involvement of PLCgamma1 and PI3-kinase. Blood. 2003;102(4):1367-1373. [DOI] [PubMed] [Google Scholar]
- 15.Suzuki-Inoue K, Kato Y, Inoue O, et al. Involvement of the snake toxin receptor CLEC-2, in podoplanin-mediated platelet activation, by cancer cells. J Biol Chem. 2007;282(36):25993-26001. [DOI] [PubMed] [Google Scholar]
- 16.Chaipan C, Soilleux EJ, Simpson P, et al. DC-SIGN and CLEC-2 mediate human immunodeficiency virus type 1 capture by platelets. J Virol. 2006;80(18):8951-8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suzuki-Inoue K, Tulasne D, Shen Y, et al. Association of Fyn and Lyn with the proline-rich domain of glycoprotein VI regulates intracellular signaling. J Biol Chem. 2002;277(24):21561-21566. [DOI] [PubMed] [Google Scholar]
- 18.Park SY, Ueda S, Ohno H, et al. Resistance of Fc receptor- deficient mice to fatal glomerulonephritis. J Clin Invest. 1998;102(6):1229-1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shirai T, Inoue O, Tamura S, et al. C-type lectin-like receptor 2 promotes hematogenous tumor metastasis and prothrombotic state in tumor-bearing mice. J Thromb Haemost. 2017;15(3):513-525. [DOI] [PubMed] [Google Scholar]
- 20.Belliere J, Casemayou A, Ducasse L, et al. Specific macrophage subtypes influence the progression of rhabdomyolysis-induced kidney injury. J Am Soc Nephrol. 2015;26(6):1363-1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang X, Zhao W, Zhang L, et al. The role of complement activation in rhabdomyolysis-induced acute kidney injury. PLoS One. 2018;13(2):e0192361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ozaki Y, Suzuki-Inoue K, Inoue O. Novel interactions in platelet biology: CLEC-2/podoplanin and laminin/GPVI. J Thromb Haemost. 2009;7(suppl 1):191-194. [DOI] [PubMed] [Google Scholar]
- 23.Nieswandt B, Bergmeier W, Schulte V, Rackebrandt K, Gessner JE, Zirngibl H. Expression and function of the mouse collagen receptor glycoprotein VI is strictly dependent on its association with the FcRgamma chain. J Biol Chem. 2000;275(31):23998-24002. [DOI] [PubMed] [Google Scholar]
- 24.Nieswandt B, Schulte V, Bergmeier W, et al. Long-term antithrombotic protection by in vivo depletion of platelet glycoprotein VI in mice. J Exp Med. 2001;193(4):459-469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schulte V, Snell D, Bergmeier W, Zirngibl H, Watson SP, Nieswandt B. Evidence for two distinct epitopes within collagen for activation of murine platelets. J Biol Chem. 2001;276(1):364-368. [DOI] [PubMed] [Google Scholar]
- 26.Silva JC, Rodrigues NC, Thompson-Souza GA, Muniz VS, Neves JS, Figueiredo RT. Mac-1 triggers neutrophil DNA extracellular trap formation to Aspergillus fumigatus independently of PAD4 histone citrullination. J Leukoc Biol. 2020;107(1):69-83. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, Gao H, Shi C, et al. Leukocyte integrin Mac-1 regulates thrombosis via interaction with platelet GPIbα [published correction appears in Nat Commun. 2017;8:16124]. Nat Commun. 2017;8(1):15559.28555620 [Google Scholar]
- 28.Chavez LO, Leon M, Einav S, Varon J. Beyond muscle destruction: a systematic review of rhabdomyolysis for clinical practice. Crit Care. 2016;20(1):135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.