

Key Points
T-cell reconstitution patterns in the bone marrow microenvironment correlate with patient outcomes.
Patients with worse outcomes have more T cells at the opposite ends of the differentiation spectrum (naive/terminally differentiated).
Abstract
The immediate postautologous stem cell transplant (ASCT) period in multiple myeloma represents a unique opportunity for long-term disease control because many patients have eradicated most of their disease but also a challenge because it is characterized by the increase of immune subsets detrimental to tumor immunosurveillance. The impact of the tumor immune microenvironment (iTME) in post-ASCT outcomes is not known. In this study, we included 58 patients undergoing upfront ASCT and evaluated their cellular and humoral iTME with cytometry by time of flight (CyTOF) and luminex, respectively, at day +60 to 100 post-ASCT. We identified 2 cellular iTME patterns. Group 1 was enriched in T-cell subsets at the opposite ends of the spectrum of T-cell differentiation compared with the rest of the patients, that is, cells already terminally differentiated (immune senescent or exhausted) and naive T cells. This group had worse hematologic responses post-ASCT, inferior survival, and shorter time to hematologic progression independent of established risk factors. No differences in the humoral iTME were noted between the 2 groups. In addition, no differences in the cellular/humoral iTME were noted according to high-risk fluorescence in situ hybridization status, early or late relapse. Finally, males had higher levels of natural killer cells negative for CD16, a key receptor mediating antibody-dependent cell cytotoxicity, a major mechanism of antitumor efficacy by therapeutic antibodies such as elotuzumab. Our findings suggest that T-cell iTME dysfunction post-ASCT, some of which could be reversible (exhaustion), correlates with worse outcomes. These results could be used to guide rational selection of post-ASCT maintenance/consolidation approaches in these patients.
Visual Abstract

Introduction
Autologous stem cell transplant (ASCT) after induction chemotherapy is considered standard of care for all eligible multiple myeloma (MM) patients. Despite the high levels of complete response (CR) with this approach, including minimal residual disease negative (MRD−) CR, the disease eventually relapses, and MM remains an incurable condition for most patients. The immediate post-ASCT period represents a unique opportunity for long-term disease control because many patients have eradicated most of their disease. It also represents a challenge because it is characterized by the increase of immunosuppressive subsets detrimental to tumor immunosurveillance.1 For this reason, standard maintenance therapies and ongoing clinical trials are utilizing agents such immunomodulatory drugs and CD38 monoclonal antibodies that target residual malignant plasma cells but also favorably reshape the tumor immune microenvironment (iTME). Understanding what drives early post-ASCT immunosuppression and how it relates to patient outcomes is key for developing novel therapies and utilizing existing therapies in a rational way in this setting.
Prior studies have identified peripheral blood immune subsets correlating with outcomes after ASCT.2,3 However, immune profiling was performed using oligoparametric techniques, such as flow cytometry, that cannot accurately capture the complexity of the immune system. Furthermore, the peripheral blood does not always reflect what happens in the bone marrow (BM), which represents the iTME for MM. Finally, many of these analyses were performed using a supervised or partially supervised approach, which could have biased the results.
In this study, we use mass cytometry and Luminex to comprehensively profile the cellular and humoral iTME, respectively, of one of the largest cohorts of patients with MM early post-ASCT. Unsupervised clustering identified 2 major patterns of cellular immune reconstitution, which correlated with patients’ outcomes independent of known risk factors.
Methods
We included patients with MM undergoing a preplanned, single ASCT within 12 months of diagnosis with available BM CD138− sorted or unsorted mononuclear cells and BM plasma at day +60 or day +100 after ASCT. All patients were conditioned with melphalan 200 mg/m2. All included patients had consented to have their BM samples and clinical data used for research purposes, and this study was approved by the Mayo Clinic Institutional Review Board and was conducted in accordance with the Declaration of Helsinki. The electronic medical record was reviewed to obtain clinical characteristics and treatment information for these patients. Hematologic responses were defined as per the international myeloma working group criteria4 and were classified into complete response (CR), very good partial response (VGPR), partial response (PR), no response, and progressive disease (PD). MRD status, when performed, was assessed by multicolor flow cytometry. Engraftment syndrome was defined using established criteria.5
Luminex analyses
Cytokine and chemokine protein levels in BM plasma were measured using Luminex xMAP technology. The multiplexing analysis was performed using the 65-plex immune monitoring kit (ProcartaPlex Thermo Fisher) on the Luminex 100 system. Raw data were analyzed per the manufacturer’s protocol. The following proteins were analyzed: cytokines: G-CSF, GM-CSF, IFN-α, IFN-γ, IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8 (CXCL8), IL-9, IL-10, IL-12p70, IL-13, IL-15, IL-16, IL-17A (CTLA-8), IL-18, IL-20, IL-21, IL-22, IL-23, IL-27, IL-31, LIF, M-CSF, MIF, TNF-α, TNF-β, TSLP; chemokines: BLC (CXCL13), ENA-78 (CXCL5), Eotaxin (CCL11), Eotaxin-2 (CCL24), Eotaxin-3 (CCL26), Fractalkine (CX3CL1), Gro-α (CXCL1), IP-10 (CXCL10), I-TAC (CXCL11), MCP-1 (CCL2), MCP-2 (CCL8), MCP-3 (CCL7), MDC (CCL22), MIG (CXCL9), MIP-1α (CCL3), MIP-1β (CCL4), MIP-3α (CCL20), SDF-1α (CXCL12); growth factors/regulators: FGF-2, HGF, MMP-1, NGFβ, SCF, VEGF-A; soluble receptors: APRIL, BAFF, CD30, CD40L (CD154), IL-2R (CD25), TNF-RII, TRAIL (CD253), TWEAK.
Mass cytometry
Our antibody panel included 36 antibodies directed against well-characterized surface markers of lymphoid cells (T cells, B cells, natural killer [NK] cells). Mass cytometry antibody–metal conjugate combinations are detailed in supplemental Table 1. Major lymphoid cell phenotypes are defined in supplemental Table 2. For primary conjugations, purified antibodies were obtained in carrier protein-free phosphate-buffered saline and labeled using the X8 polymer MaxPAR antibody conjugation kit (Fluidigm) according to the manufacturer’s protocol. The antibody panel was designed using the web-based Fluidigm panel designer to select channels with optimal signal and minimal background from oxidation, isotopic impurity, or abundance sensitivity. All antibodies were titrated to optimal staining concentrations using primary human BMs of patients with MM. Antibody master mixes were prepared fresh for each experiment. A reference sample was run with each experiment to evaluate for staining inconsistencies. All BM samples were processed identically. Mononuclear cells were obtained after ACK lysis of BM samples and were viably frozen in RPMI 1640, 20% fetal bovine serum, 10% dimethyl sulfoxide. Cryopreserved cells were resuscitated for mass cytometry analyses by rapid thawing and were rested in RPMI 1640 (20% fetal bovine serum) for 60 minutes prior to staining. Staining was performed using Fluidigm’s protocol. Briefly, 1 to 3 million cells were stained for viability with 5 mM cisplatin for 5 minutes at room temperature and quenched with cell staining medium (CSM; Fluidigm). Cells were then incubated for 10 minutes at room temperature with human FcR blocking reagent (Biolegend) and then stained with the surface antibody cocktail for 60 minutes at 4°C with gentle agitation. Finally, cells were washed twice with CSM, fixed with 1.6% paraformaldehyde, washed with CSM, and resuspended in 1:1000 solution of Iridium intercalator diluted in MaxPar Fix and Perm buffer (Fluidigm) for 20 minutes at room temperature. Prior to acquisition, cells were washed twice in CSM and twice in deionized water and were then diluted to a concentration of 0.5 million cells per milliliter in water containing 10% of EQ 4 Element Beads (Fluidigm). Cells were filtered through a 35-μm membrane prior to mass cytometry acquisition. Samples were then acquired on a Helios mass cytometer.
Mass cytometry data analysis
Flow cytometry standard files were normalized and concatenated using the Fluidigm acquisition software. Flow cytometry standard files were uploaded to the Astrolabe Cytometry Platform (Astrolabe Diagnostics, Inc) where transformation, cleaning (doublets, debris), labeling, and unsupervised clustering (using FlowSOM)6 were done as previously described.7,8 Data were transformed using arcsinh with a cofactor of 5, and the marker intensities presented in the paper are all after transformation. A minimum of 86147 CD45+ events per file were used for clustering. Because the astrolabe platform is designed to overcluster data, astrolabe-identified cell clusters were sometimes merged so that the minimum median cluster size was not smaller than the minimum median doublet rate. As an additional method of robust cluster identification, we only considered clusters that formed discrete islands after viSNE visualization,9 a method that is sensitive to outliers and batch effects. Identified clusters were then exported for downstream statistical analyses. All immune subset frequencies are reported as a percent of CD45+ cells. Batch effects were handled as previously described.10 The Astrolabe cytometry platform applies the Ek'Balam cell labeling algorithm8 to each sample separately. Therefore, even if there is variance in the mean mass intensity of particular markers, the platform labels cell populations correctly. To avoid batch effects within the data analysis, the Astrolabe Cytometry Platform did not compare numerical intensities between samples. Each sample was analyzed separately, and then comparisons were made using cell frequencies. The underlying assumption was that a given subset was the same regardless of whether the underlying marker intensity had shifted; in other words, a T cell was defined as a T cell whether the CD3+ peak was centered on a transformed intensity of 4 or a transformed intensity of 6. This mirrors the approach used in manual gating analysis.
Statistical methods
Fisher's exact test was used to compare categorical variables and Wilcoxon rank sum (2 groups) or Kruskal-Wallis test (>2 groups) for continuous variables. Kaplan-Meier survival analysis was used to estimate the overall survival (OS) and time to progression (TTP), and we used the log-rank test to compare groups. A Cox proportional hazards model was used to evaluate the impact of variables of interest on OS/TTP and results presented as risk ratios with their respective 95% confidence interval. Hierarchical clustering of patients according to the abundance (% of CD45+ cells) of immune subsets was performed using the Ward’s minimum variance method. A 2-sided false discovery rate (FDR) adjusted P value of <.05 was considered significant when multiple comparisons were performed; otherwise, a P value of <.05 was considered significant. Statistical analysis was performed using JMP Pro statistical software version 14.1 (SAS Institute, Cary, NC).
Results
The clinical characteristics of the 58 patients included in the study are shown in Table 1. No patients were lost to follow-up. Because several patients were transplanted before MRD testing became standard at our institution, we only had MRD status in 12 patients with CR, of which 6 were MRD−.
Table 1.
Characteristics of patients
| Characteristic, N = 58 | Median (range) or n (%) |
|---|---|
| Female sex | 16 (28) |
| Age, y | 62 (41-75) |
| Diagnosis period | 6/2/2014 (1/29/2010-5/23/2018) |
| Follow-up of surviving patients for survival from ASCT, mo | 59 (24-110) |
| High-risk FISH* | 13 (22) |
| Type of induction therapy | |
| IMID-based | 43 (73) |
| At least triplet therapy | 49 (84) |
| Interval, diagnosis to ASCT, mo | 6 (1-11.6) |
| Hematologic response prior to ASCT | |
| CR | 19 (33) |
| VGPR | 19 (33) |
| PR | 14 (24) |
| < PR | 6 (10) |
| Engraftment syndrome | 6 (10) |
| Hematologic response post-ASCT | |
| CR | 24 (41) |
| VGPR | 17 (29) |
| PR | 11 (19) |
| <PR | 6 (10) |
| Maintenance therapy | |
| IMID based | 26 (45) |
| Single agent | 27 (47) |
| Combination therapy | 8 (13) |
| No maintenance† | 23 (40) |
| Outcomes post maintenance | |
| Time to relapse, mo | 17 (2-83) |
| Relapse within 12 mo | 14 (24) |
| Remission lasting >36 mo with maintenance | 10 (17) |
| Remission lasting >18 mo without maintenance | 11 (9) |
Defined as the presence of t(4;14), del(17/17p), t(14;16), or t(14;20).
Of which 3 were primary refractory and as a result switched therapy.
IMID, immunomodulatory drug (thalidomide, lenalidomide, pomalidomide); NR, no response/disease progression.
Mass cytometry identifies 2 major cellular immune reconstitution patterns after ASCT
We identified 26 lymphoid subsets early after ASCT. Their phenotypic characteristics and structure on a viSNE map are shown in Figure 1 and supplemental Figure 1, respectively. ViSNE maps for individual patients are also shown in supplemental Figure 2. To better understand patterns of immune reconstitution early post-ASCT, we clustered patients based on their cellular iTME (cytometry by time of flight [CyTOF]–identified immune subsets). Three groups were identified (Figure 2). Immune subset differences (using an FDR-corrected P value to account for multiple comparisons) across the 3 groups are shown in Figure 3. Group 2 was characterized by a robust early B-cell recovery compared with groups 1 and 3 with no other differences identified between groups 2 and 3.
Figure 1.

Heatmap demonstrating the phenotypic characteristics of all identified immune subsets. The scale represents normalized median marker intensity.
Figure 2.

Immune reconstitution patterns after ASCT. Grouping of patients after hierarchical clustering based on their cellular immune microenvironment (CyTOF identified immune subsets expressed as % of CD45+ cells) according to subset abundance (blue = low; red = high; a detailed legend of the corresponding numerical values for each group is presented in supplemental Figure 3).
Figure 3.
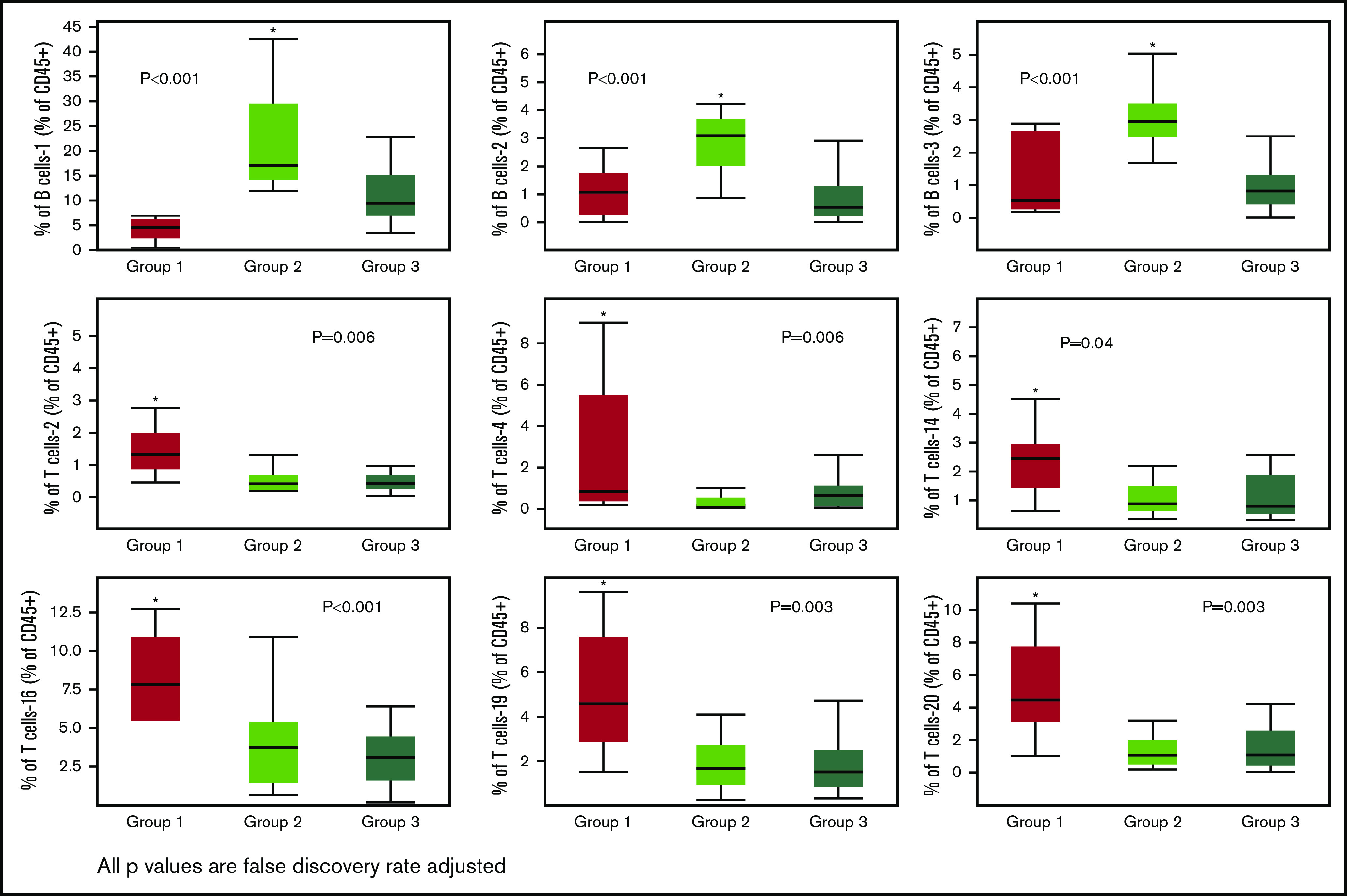
Immune subsets significantly (FDR adjusted P value < .05) different across the 3 groups. Only 1 group per figure was different from the other two (indicated by the asterisks).
The remaining differences across the 3 groups were exclusively driven by T-cell subsets in group 1. Group 1 was characterized by higher levels of the T-cell-2 subset, a double-negative, terminally differentiated subset and the T-cell-4 subset, resembling NKT cells (CD56+) but also with markers of immune senescence/terminal differentiation (CD27/CD28−, KLRG1+, CD57+) and low levels of TIGIT expression. Other T-cell subsets differentially increased in group 1 were T-cell-14, -16, -19, and -20 subsets. Of these, the T-cell-16 and -19 subsets were CCR5 expressing subsets with features of terminal differentiation (CD27/CD28−, KLRG1+) and, in the case of T-cell-16, exhaustion (TIGIT+). T-cell subset -20 had a phenotype characteristic of naive T cells (CCR7/CD45RA+, CD45RO−). No differences in NK-cell subset reconstitution patterns were identified. Finally, no cytokine differences were noted across the 3 groups (data not shown).
These data suggest that early immune reconstitution in a subset of patients post-ASCT is enriched in T-cell subsets at the opposite ends of the spectrum of T-cell differentiation, that is, cells already terminally differentiated (immune senescent) or naive T cells.
Immune reconstitution patterns early after ASCT correlate with patient outcomes
We then evaluated if there were differences in immune subsets or cytokines according to patient characteristics and outcomes. We considered the following characteristics as dichotomous variables (using an FDR-corrected P value to account for multiple comparisons): sex, high-risk fluorescence in situ hybridization (FISH), CR status after ASCT, relapse within 12 months from ASCT, and relapse within 18 months (unmaintained) or 36 months (maintained) post-ASCT. We found no differences with the exception of sex and CR status. Males had higher levels of the CD16− NK-cell-3 subset compared with females (median 1.9% vs 0.7%, FDR-corrected P value of .02). Patients in a hematologic CR had lower levels of the T-cell-13 subset (median 3.2% vs 5.2%, FDR-corrected P value of .03). When considering all the clinical characteristics included in Table 1, patients in group 1 were less likely to achieve a hematologic CR (10% vs 48%, P = .04) and more likely to have a hematologic PR or PD (80% vs 19%, P < .001) post-ASCT compared with the rest of the cohort.
Subsequently, we evaluated whether immune reconstitution patterns post-ASCT correlate with OS or TTP. Group 1 had worse OS from diagnosis and from ASCT and worse TTP from ASCT (Figure 4). Univariable and multivariable analyses revealed that immune group 1 and high-risk FISH were independent predictors of OS (Table 2).
Figure 4.
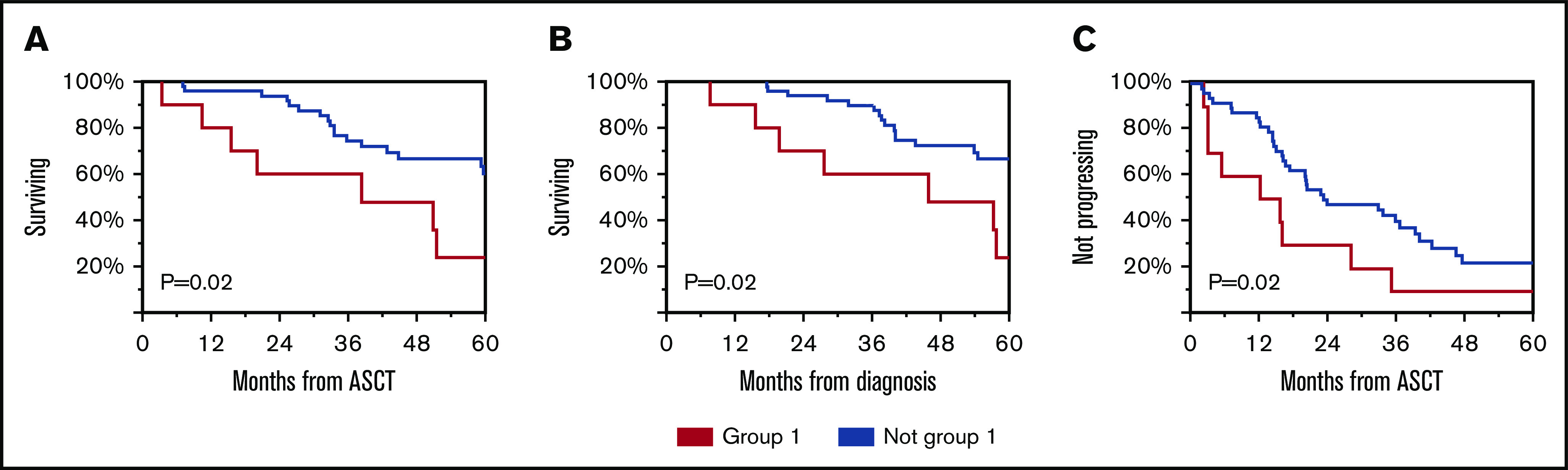
Clinical outcomes of patients according to immune reconstitution pattern. OS from ASCT (A), diagnosis (B), and TTP from ASCT (C) for group 1 compared with others.
Table 2.
Univariable and multivariable analyses on OS and TTP
| Univariable | Multivariable | ||||
|---|---|---|---|---|---|
| Characteristic | Risk ratio (range) | P | Characteristic | Risk ratio (range) | P |
| OS | |||||
| Group 1 vs not | 2.5 (1.1-5.6) | .02 | Group 1 vs not | 3.5 (1.5-8.3) | .009 |
| Age | 1.4 (0.22-10.7) | .7 | |||
| High-risk disease* | 5.51(2.3-11.2) | <.0001 | High-risk disease* | 6.4 (2.8-14.9) | <.0001 |
| CR post-ASCT | 0.83 (0.38-1.76) | .6 | |||
| PR/PD vs not | 1.9 (0.9-4) | .1 | |||
| Received maintenance therapy | 2.18 (0.96-4.94) | .06 | |||
| TTP | |||||
| Group 1 vs not | 2.1 (1.01-2.1) | .04 | Group 1 vs not | 2.2 (1.1.-4.6) | .03 |
| Age | 0.26 (0.1-1.5) | .12 | |||
| High-risk disease* | 2.5 (1.2-5.2) | .01 | High-risk disease* | 2.7 (1.3-5.6) | .008 |
| CR post-ASCT | 1.1 (0.6-2) | .8 | |||
| PR/PD vs not | 1.8 (0.99-3.4) | .06 | |||
| Received maintenance therapy | 0.9 (0.5-1.6) | .7 | |||
Defined as the presence of t(4;14), del(17/17p), t(14;16), or t(14;20).
Discussion
In this study, we identified 2 major reconstitution patterns of the cellular iTME early post-ASCT in patients with MM treated with an upfront ASCT. We find that a group of patients is characterized by higher levels of naive and terminally differentiated T cells, some of which are expressing markers of T-cell exhaustion. This group is characterized by worse hematologic responses after ASCT and inferior OS and TTP independent of high-risk FISH.
The presence of T cells at the 2 opposite ends of the T-cell differentiation spectrum, that is, naive and terminally differentiated, in the group with the worst outcomes suggests that the iTME of these patients is enriched in subsets unable to mediate effective clearance of malignant cells. Naive T cells have not been yet activated to engage antigens. On the opposite spectrum, terminally differentiated effector T cells have expanded in response to antigens, presumably against MM cells in this case, because these patients had higher levels of residual disease, but were unable to control the tumor. We were intrigued to note CCR5 expression in several of these T-cell subsets. Maximal T-cell recruitment and T-cell–mediated antitumor responses rely on CCR5 expression in both CD4+ and CD8+ T cells.11,12 CCR5 expression on CD4 T cells is essential for activation of antigen-presenting cells and the resulting enhanced CD8 T cell cross-priming and tumor rejection in mouse models of malignancies.11 This is thought to be mediated by the expression of CCR5 ligands CCL3 and CCL4, which are also produced by malignant plasma cells in the iTME of MM and are thought to be implicated in osteoclastogenesis.11,13 Therefore, these immune cells are likely attracted in the iTME by residual malignant plasma cells. Infiltration by CCR5+ lymphocytes is common in the microenvironment of other tumors14,15 and has been associated with favorable prognosis.15 We noted features of T-cell exhaustion (PD-1 and TIGIT) in some of the CCR5-expressing T-cell subsets, which suggest that these populations of tumor infiltrating lymphocytes might not be able to control tumor growth because they are exhausted. T-cell exhaustion post-ASCT has been noted by us and others1,3,7,16 and has been reversed in vivo by use of checkpoint inhibitors, such as anti-TIGIT antibodies.17
Several of the identified subsets were senescent. Immunosenescent T cells are characterized by a decrease in surface CD27 and CD28 costimulating molecules and increase of KLRG1 and in some cases CD57+.18 They have been shown to have immunoregulatory properties and to suppress effective antitumor immune responses.19 It has been demonstrated that therapies such as ASCT7,16 and MM itself can induce T-cell immunosenescence20 and that higher levels of senescent T cells are associated with worse outcomes at diagnosis in MM.7 Our study suggests that the same is true after ASCT. In fact, existing maintenance approaches, such as lenalidomide, skew the T-cell repertoire toward a less terminally differentiated phenotype.21 These immunosenescent subsets also increase in triple refractory patients but not during earlier relapses,22 which could be a result of clonal T-cell expansion induced by CD38 monoclonal antibodies.23 The concurrent increase in the naive T-cell pool in these patients suggests that immunomodulation early post-ASCT using agents that are able to skew a residual naive T-cell pool toward an early effector phenotype, such as CD38 antibodies, perhaps in combination with anti-TIGIT agents to “revive” exhausted subsets could be a reasonable therapeutic strategy.
We noted that the NK-cell pool post-ASCT recovered in a homogenous way with the exception of male patients having more CD16− NK cells. CD16 is a key receptor mediating antibody-dependent cell cytotoxicity, which is a major mechanism of antitumor efficacy by therapeutic antibodies to tumor antigens, including elotuzumab.24 Our findings suggest that elotuzumab efficacy early after ASCT might be decreased in male patients. Our differences compared with other studies, that identified differences in NK-cell subsets in the peripheral blood of patients with MRD− CR,3 may have to do with differences in the markers tested (limited NK-cell–specific markers in our panel), sample source (marrow vs peripheral blood), and patient cohort characteristics. Similar to others,2 we noted less robust B-cell immune reconstitution in patients with inferior outcomes. Interestingly, patients not achieving CR had higher levels of exhausted T cells. More exhausted T cells in patients at higher risk for relapse has been reported in the past.1 These cells in our study expressed CD27 and CD28 but also some markers of senescence such as KLRG1 but not CD57. It is unclear if these cells represent progenitor or terminally exhausted T cells25 and to what extent they could be modulated by checkpoint inhibition. Finally, we did not observe any differences in the cytokine compartment across the various immune groups or when performing prespecified comparisons according to clinical characteristics. This suggests that the humoral iTME compartment post-ASCT might be too homogenous to detect differences in this cohort possibly as a result of common humoral factors driving immune and hematopoietic recovery early post-ASCT.
Our study has several limitations. Most of our patients were male, and we did not validate our findings in a separate cohort, so generalizability is limited. We were restricted by the number of patients with all available samples and enough clinical follow-up for this study. MRD status was only available for half of the patients in CR. Therefore, we could not accurately assess differences between MRD+ and MRD− CRs. Several (40%) patients did not receive maintenance, which reflects differences in practice patterns for patients diagnosed at an earlier period. However, receiving maintenance was not an independent predictor of outcomes in this cohort. We used cryopreserved samples, and therefore, myeloid populations could not be adequately assessed, because several key populations, such as myeloid derived suppressor cells, are lost after cryopreservation,26 whereas T-cell loss is thought to be stochastic. For this reason, we did not include markers for myeloid populations and focused on the lymphoid compartment.
In summary, here we define major immune reconstitution patterns early after ASCT using novel multiparametric approaches that can help deepen our understanding of the state of the iTME in this setting.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Michelle Manske for her help with some of the experiments performed in this study. The visual abstract was created using biorender.com.
This work was supported by an Eagles 5th District Cancer Telethon–Cancer Research Fund, the Mayo Clinic Myeloma SPORE P50 National Institutes of Health, National Cancer Institute grant (CA186781-03) a Mayo Clinic Benefactor Career Development Award, and a Mayo Clinic Transplant Center Scholar Award.
Footnotes
For original data, please contact the corresponding author at kourelis.taxiarchis@mayo.edu.
Authorship
Contribution: T.V.K. provided the concept and design; H.P., M.G., E.I.A., S.K., and T.V.K. provided the study materials of the patients; H.P., M.G., E.I.A., S.K., and T.V.K. collected and assembled the data; T.V.K. and H.P. analyzed and interpreted the data; H.P., M.G., E.I.A., S.K., and T.V.K. wrote and had final approval of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Taxiarchis Kourelis, Division of Hematology, Mayo Clinic, 200 First St SW, Rochester, MN 55905; e-mail: kourelis.taxiarchis@mayo.edu.
References
- 1.Chung DJ, Pronschinske KB, Shyer JA, et al. T-cell exhaustion in multiple myeloma relapse after autotransplant: optimal timing of immunotherapy. Cancer Immunol Res. 2016;4(1):61-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ho CM, McCarthy PL, Wallace PK, et al. Immune signatures associated with improved progression-free and overall survival for myeloma patients treated with AHSCT. Blood Adv. 2017;1(15):1056-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhutani M, Foureau D, Zhang Q, et al. Peripheral immunotype correlates with minimal residual disease status and is modulated by immunomodulatory drugs in multiple myeloma. Biol Blood Marrow Transplant. 2019;25(3):459-465. [DOI] [PubMed] [Google Scholar]
- 4.Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17(8):e328-e346. [DOI] [PubMed] [Google Scholar]
- 5.Maiolino A, Biasoli I, Lima J, Portugal AC, Pulcheri W, Nucci M. Engraftment syndrome following autologous hematopoietic stem cell transplantation: definition of diagnostic criteria. Bone Marrow Transplant. 2003;31(5):393-397. [DOI] [PubMed] [Google Scholar]
- 6.Van Gassen S, Callebaut B, Van Helden MJ, et al. FlowSOM: using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A. 2015;87(7):636-645. [DOI] [PubMed] [Google Scholar]
- 7.Kourelis TV, Villasboas JC, Jessen E, et al. Mass cytometry dissects T cell heterogeneity in the immune tumor microenvironment of common dysproteinemias at diagnosis and after first line therapies. Blood Cancer J. 2019;9(9):72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amir ED, Lee B, Badoual P, et al. Development of a comprehensive antibody staining database using a standardized analytics pipeline. Front Immunol. 2019;10:1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amir AD, Davis KL, Tadmor MD, et al. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol. 2013;31(6):545-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steele NG, Carpenter ES, Kemp SB, et al. Multimodal mapping of the tumor and peripheral blood immune landscape in human pancreatic cancer. Nat Cancer. 2020;1(11):1097-1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castellino F, Huang AY, Altan-Bonnet G, Stoll S, Scheinecker C, Germain RN. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature. 2006;440(7086):890-895. [DOI] [PubMed] [Google Scholar]
- 12.González-Martín A, Gómez L, Lustgarten J, Mira E, Mañes S. Maximal T cell-mediated antitumor responses rely upon CCR5 expression in both CD4(+) and CD8(+) T cells. Cancer Res. 2011;71(16):5455-5466. [DOI] [PubMed] [Google Scholar]
- 13.Vallet S, Pozzi S, Patel K, et al. A novel role for CCL3 (MIP-1α) in myeloma-induced bone disease via osteocalcin downregulation and inhibition of osteoblast function. Leukemia. 2011;25(7):1174-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cózar JM, Canton J, Tallada M, et al. Analysis of NK cells and chemokine receptors in tumor infiltrating CD4 T lymphocytes in human renal carcinomas. Cancer Immunol Immunother. 2005;54(9):858-866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zimmermann T, Moehler M, Gockel I, et al. Low expression of chemokine receptor CCR5 in human colorectal cancer correlates with lymphatic dissemination and reduced CD8+ T-cell infiltration. Int J Colorectal Dis. 2010;25(4):417-424. [DOI] [PubMed] [Google Scholar]
- 16.Lucas F, Pennell M, Huang Y, et al. T cell transcriptional profiling and immunophenotyping uncover LAG3 as a potential significant target of immune modulation in multiple myeloma. Biol Blood Marrow Transplant. 2020;26(1):7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Minnie SA, Kuns RD, Gartlan KH, et al. Myeloma escape after stem cell transplantation is a consequence of T-cell exhaustion and is prevented by TIGIT blockade [published correction appears in Blood. 2019;134(21):1878]. Blood. 2018;132(16):1675-1688. [DOI] [PubMed] [Google Scholar]
- 18.Liu X, Hoft DF, Peng G. Senescent T cells within suppressive tumor microenvironments: emerging target for tumor immunotherapy. J Clin Invest. 2020;130(3):1073-1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Montes CL, Chapoval AI, Nelson J, et al. Tumor-induced senescent T cells with suppressor function: a potential form of tumor immune evasion. Cancer Res. 2008;68(3):870-879. [DOI] [PubMed] [Google Scholar]
- 20.Suen H, Brown R, Yang S, et al. Multiple myeloma causes clonal T-cell immunosenescence: identification of potential novel targets for promoting tumour immunity and implications for checkpoint blockade. Leukemia. 2016;30(8):1716-1724. [DOI] [PubMed] [Google Scholar]
- 21.Fostier K, Caers J, Meuleman N, et al. Impact of lenalidomide maintenance on the immune environment of multiple myeloma patients with low tumor burden after autologous stem cell transplantation. Oncotarget. 2018;9(29):20476-20489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Visram A, Dasari S, Anderson E, Kumar SK, Kourelis TV. Describing the cellular and humoral immune tumor microenvironment and malignant transcriptome across the multiple myeloma disease spectrum, in American Society of Hematology Annual Meeting. 2020, Blood. p. 39-40.
- 23.Krejcik J, Casneuf T, Nijhof IS, et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood. 2016;128(3):384-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balasa B, Yun R, Belmar NA, et al. Elotuzumab enhances natural killer cell activation and myeloma cell killing through interleukin-2 and TNF-α pathways. Cancer Immunol Immunother. 2015;64(1):61-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller BC, Sen DR, Al Abosy R, et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. 2019;20(3):326-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kadić E, Moniz RJ, Huo Y, Chi A, Kariv I. Effect of cryopreservation on delineation of immune cell subpopulations in tumor specimens as determinated by multiparametric single cell mass cytometry analysis. BMC Immunol. 2017;18(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.