

Abstract
Hypochlorous acid (HOCl) is generated at a high concentration by activated neutrophils at sites of inflammation in a myeloperoxidase catalyzed reaction. The increased and sustained production of HOCl at inflammatory sites may lead to tissue injury and this process is believed to play an important role in the progression of several diseases like chronic inflammation, atherosclerosis and some types of cancers. We have examined the effect of HOCl on human red blood cells (RBCs) under in vitro conditions. Treatment of RBC with different concentrations of HOCl (0.05–2.5 mM) at 37°C resulted in decreased activities of major antioxidant enzymes while the antioxidant power of RBC was weakened, as shown by lowered metal-reducing and free radical quenching ability of HOCl treated cells. RBC plasma membrane redox system was also inhibited suggesting membrane damage. The enzymes of glucose metabolism were inhibited indicating deranged energy metabolism. Electron microscopic images showed gross morphological changes in HOCl treated RBC. These results show that HOCl causes major alterations in the cellular antioxidant defense system and inhibition of glycolytic pathways, which increase the susceptibility of RBC to oxidative damage.
Keywords: antioxidant power, HOCl, oxidative damage, PMRS, RBC, SEM
Graphical abstract
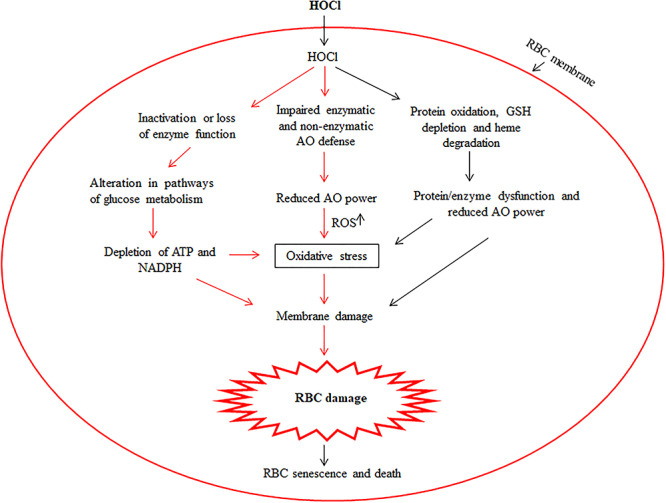
Introduction
Hypochlorous acid (HOCl) is generated by the myeloperoxidase catalyzed reaction between chloride ions and hydrogen peroxide. Activated phagocytes release myeloperoxidase during inflammation to produce substantial amounts of HOCl [1,2]. Under pathological conditions, HOCl/OCl− is found at concentrations up to 200 μM [3]. However, at sites of inflammation, it can be 3–8 mM [4]. HOCl is a highly reactive biological oxidant that has an important role in microbial killing, resulting in bactericidal action of phagocytic cells. It is, therefore, a critical component of host immune defence against invading bacteria, viruses and fungi. However, excessive or sustained production of HOCl may lead to tissue injury and this process is believed to play an important role in the progression of several diseases like chronic inflammation, atherosclerosis and some types of cancer [1,2]. Being a strong oxidant, HOCl has good antiseptic and germicidal properties [5] and is, therefore, used as a disinfectant of public water supplies. Sodium hypochlorite (0.1%) solution efficiently inactivated human coronaviruses (HCoV) similar to COVID-19, including SARS coronavirus, Middle East Respiratory Syndrome coronavirus and endemic HCoV, within 1 min on metal surfaces [6].
HOCl is a potent oxidant and highly reactive species. It reacts rapidly with most cellular components and biological molecules like lipids, proteins and DNA [1,7,8]. Due to their abundance, proteins are the major target and their reaction with HOCl results in fragmentation, dimerization and also modification of amino acid side chains [7,9]. Semi-stable chloramine intermediates are formed when HOCl reacts with lysine and histidine side chains of proteins [10]. HOCl reacts with the double bonds of unsaturated lipids to form α, β-chlorohydrin isomers that can cause cytotoxicity by affecting the cell membranes [11,12]. Thiols and thioesters react rapidly with HOCl and also reduced glutathione (GSH) and protein thiols (cysteine residues) are major targets of HOCl in biological systems [10]. HOCl exposure causes rapid depletion of GSH and inactivation of numerous thiol-dependent enzymes, which leads to cell dysfunction and death [2]. Recently, HOCl was shown to alter signalling pathways and confer insulin resistance to mice adipocytes [13]. HOCl was found to be genotoxic to lung epithelial cells at physiological concentrations [14].
There is great interest in the biochemistry of cell injury caused by oxidative stress, activated neutrophils and the role of damaging species involved in respiratory burst [15]. Blood represents a major target of xenobiotics and toxicants that enter by any route and red blood cell (RBC), being the most abundant cells not only in blood but the entire human body, are quickly exposed to their action. In addition to exogenous oxidative stress, RBCs are also exposed to neutrophil-derived oxidants at sites of inflammation. RBCs have, therefore, been used as the model for investigating molecular and cellular mechanisms of neutrophil-mediated cell damage [16]. RBCs are exposed to HOCl in the bloodstream during circulation, so studying their interaction is of significance in understanding the cellular effects of this oxidant. The results obtained with RBC can be extended and one can suggest similar cellular events produced by HOCl in other cell types. In addition, HOCl modified RBC can play an important role in the development of long-term vascular complications in inflammation, interact with neutrophils or other blood cells in circulation. We have investigated the cellular events, especially the antioxidant (AO) system and glycolytic pathways affected by the treatment of human RBC with HOCl. Our results showed for the first time that HOCl significantly decreased the activities of major AO defence and metabolic enzymes, inhibited the plasma membrane redox system (PMRS) and lowered the AO power of these specialized cells. This can exacerbate the oxidative stress condition leading to morphological changes in RBC.
Materials and Methods
Isolation of human RBC and treatment with HOCl
The study was approved by an institutional ethics committee that monitors research involving human subjects. Blood was taken from young (22–30 years), normal, non-smoking volunteers after getting their informed consent and used immediately. It was collected in heparinized tubes, centrifuged at 1000 × g for 10 min at 4°C in a clinical centrifuge and the supernatant containing plasma and buffy coat was removed. The RBC pellet was washed three times with phosphate-buffered saline (PBS) (0.9% NaCl in 10 mM sodium phosphate buffer, pH 7.4) and suspended in PBS to give a 20% cell suspension (hematocrit).
Sodium hypochlorite (NaOCl; Sigma-Aldrich, USA) was directly used as the source of HOCl [17,18]. HOCl concentration was determined from the absorbance at 292 nm (ε = 350 M−1 cm−1 [19]. RBCs (10% haematocrit) were incubated with different concentrations of HOCl (0.05–2.5 mM) at 37°C for 30 min. The HOCl reaction was quenched by the addition of methionine at five times the concentration of HOCl present in the samples [18]. Untreated RBC incubated at 37°C served as control. All samples were centrifuged at 1200 × g for 10 min at 4°C in a clinical centrifuge. The cell pellets were washed three times with PBS and RBC lysed with 10 volumes of hypotonic buffer (5 mM sodium phosphate buffer, pH 7.4) at 4°C for 2 h. The samples were centrifuged at 2000 × g for 10 min at 4°C and the supernatants (hemolysates) were quickly frozen in aliquots and used later.
Hemoglobin (Hb) was determined in hemolysates by the cyanomethemoglobin method using Drabkin’s reagent [20].
Nitrite and hydrogen peroxide levels
Nitrite concentration in hemolysates was determined using the Griess reagent [21]. H2O2 level was determined by using ferrous ammonium sulfate-xylenol orange (FOX reagent) in the presence of 100 mM sorbitol [22].
AO enzymes
Inhibition of the auto-oxidation of pyrogallol was used to assay Cu-Zn superoxide dismutase (SOD) [23] and catalase (CAT) activity was determined from the decrease in absorbance at 240 nm upon conversion of H2O2 to water [24]. Glutathione reductase (GR) was determined from the decrease in absorbance at 340 nm upon oxidation of NADPH to NADP+ when GSSG is reduced to GSH by the enzyme [25]. Glutathione-S-transferase (GST) was assayed using 1-chloro-2,4-dinitrobenzene as substrate [26] and thioredoxin reductase (TR) from the increase in the intensity of yellow colour formed upon reduction of 5,5′-dithiobisnotrobenzoic acid by NADPH [27]. Glutathione peroxidase (GPx) activity was determined from the oxidation of NADPH to NADP+ at 340 nm in presence of GSSG and GR [28].
AO power
The AO power/capacity of RBC was determined in hemolysates by several methods that measure the free-radical quenching (ABTS and DPPH assays) and metal-reducing [FRAP, K3Fe(CN)6, CUPRAC and phosphomolybdenum assays] ability of the cells. DPPH exists as a stable coloured radical that is quenched and decolourized by AOs in the sample; this quenching was monitored from the decrease in absorbance at 517 nm [29]. ABTS assay was done by following the reduction of coloured ABTS·+ cation radical to colourless form by AOs in hemolysates [30]. The reduction of Fe3+ to Fe2+ was determined by FRAP method and from the conversion of potassium ferricyanide to the ferrocyanide form [31, 32]. CUPRAC (cupric ion reducing AO capacity) assay was used to measure the reduction of cupric ions to cuprous form by hemolysates, exactly as described by Cekiç et al. [33]. The phosphomolybdenum assay measures the conversion of Mo6+ to Mo5+ which gives a green coloured complex whose absorbance was read at 695 nm [34].
Plasma membrane redox system
The RBC PMRS was determined from the reduction of extracellular ferricyanide by packed red blood cells (PRBCs) [35]; the ferrocyanide formed was measured by using 1,10-phenanthroline [36]. PMRS was also assayed by the recently developed ABTS·+ radical scavenging method [37,38]. Ascorbate free radical (AFR) reductase was assayed spectrophotometrically in hemolysates from the rate of NADH oxidation followed by absorbance at 340 nm [39].
Metabolic enzymes
Hexokinase activity was determined from the reduction in glucose concentration after its conversion to glucose 6-phosphate by the enzyme [40]. Lactate dehydrogenase (LDH) was assayed by following the conversion of NADH to NAD+ at 340 nm in presence of sodium pyruvate [41]. Glucose 6-phosphate dehydrogenase (G6PD) was assayed from the formation of NADPH at 340 nm in the presence of glucose 6-phosphate [42]. 5′-nucleotidase and AMP deaminase activities were determined from the release of inorganic phosphate and ammonia, respectively, from AMP [43,44].
Scanning electron microscopy
RBCs were fixed by adding 100 μl of each sample to 1 ml of 2.5% glutaraldehyde (in PBS) and then centrifuged at 1000 × g for 10 min. The cell pellets were washed two times with PBS and suspended in 0.5 ml PBS. Then, 20 μl of each sample was placed on siliconized glass coverslip, air-dried at room temperature, gold-coated for 3 min and examined by a scanning electron microscope (SEM) at 1000-fold magnification.
Statistical analysis
All data are expressed as mean ± standard error. Statistical evaluation was done by one-way analysis of variance in combination with post hoc test (Tukey Test) using GraphPad InStat 3.0. (USA). A probability level of P < 0.05 was selected as indicating statistical significance between the HOCl treated and control groups. All experiments were done in six different samples to document reproducibility.
Results
RBCs are abundant non-nucleated cells that lack protein-synthesizing machinery and represent a convenient model to examine the cellular effect of toxicants. In this work, we have studied the effect of HOCl on human RBC under in vitro conditions. RBCs were incubated at 37°C for 30 min with 0.05–2.5 mM HOCl. Since these concentrations are physiologically relevant and can be produced in vivo at the sites of inflammation [4]. Treatment of RBC with HOCl resulted in a decrease in nitrite and H2O2 concentrations upon incubation of RBC with HOCl (Fig. 1). H2O2 was 45% and nitrite was 13% of control values at 2.5 mM HOCl, the highest concentration used in this study.
Figure 1.
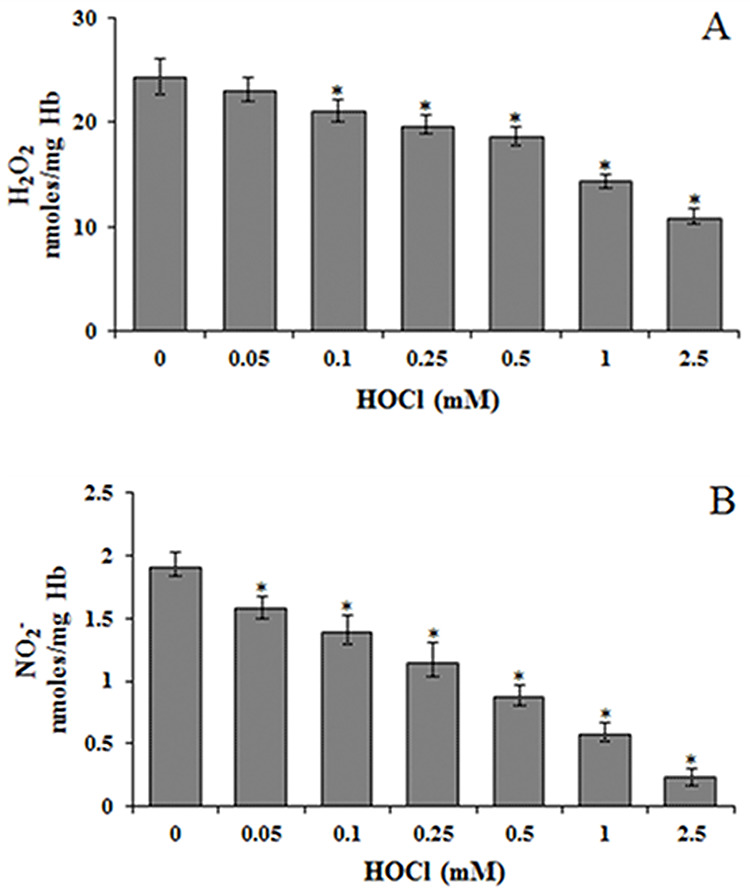
Effect of HOCl on (A) hydrogen peroxide (H2O2) and (B) nitrite (NO2−) levels in RBC. H2O2 and NO2− levels were determined in hemolysates from control and HOCl treated RBC. *Significantly different from control.
The specific activities of all major AO enzymes were greatly affected by HOCl. CAT, SOD, GST and GPx activities were increased while GR and TR decreased in HOCl treated RBC (Table 1). GR was the enzyme most affected by HOCl treatment. At 2.5 mM HOCl, it was 6-fold lower than the corresponding control value.
Table 1.
Effect of HOCl on some AO enzymes
| Control | 0.05 mM | 0.1 mM | 0.25 mM | 0.5 mM | 1.0 mM | 2.5 mM | |
|---|---|---|---|---|---|---|---|
| CAT | 20.43 ± 1.78 | 21.61 ± 2.85 | 23.78 ± 1.71a | 25.67 ± 3.10a | 28.71 ± 2.71a | 30.94 ± 3.81a | 35.29 ± 4.77a |
| SOD | 14.36 ± 1.01 | 15.87 ± 1.70 | 17.45 ± 1.60a | 19.21 ± 2.16a | 21.84 ± 2.59a | 23.23 ± 2.48a | 26.09 ± 3.51a |
| GPx | 90.61 ± 8.58 | 93.92 ± 7.33 | 100.01 ± 7.51a | 106.1 ± 6.76a | 116.13 ± 8.88a | 161.13 ± 11.09a | 210.58 ± 17.79a |
| GST | 14.37 ± 1.11 | 15.13 ± 1.52 | 16.58 ± 1.72a | 17.86 ± 1.91a | 20.16 ± 1.85a | 23.27 ± 1.97a | 26.41 ± 2.34a |
| GR | 5.44 ± 0.32 | 3.98 ± 0.41a | 2.95 ± 0.18a | 2.15 ± 0.17a | 1.66 ± 0.14a | 1.10 ± 0.10a | 0.93 ± 0.07a |
| TR | 29.09 ± 3.46 | 24.90 ± 1.44a | 22.80 ± 2.21a | 20.29 ± 1.76a | 19.9 ± 1.55a | 16.15 ± 1.25a | 14.50 ± 1.36a |
Specific activity of SOD is in units/mg Hb (one unit is the amount which causes 50% inhibition of pyogallol oxidation in a reaction volume of 3 ml). Specific activities of CAT, GPx, GST, GR and TR are in nmoles/min/mg Hb.
aSignificantly different from control.
The AO power of hemolysates was determined from metal (Fe3+, Cu2+, Mo6+) reducing and free radical quenching ability. HOCl treated RBC had lower FRAP values which were 30% less at the highest HOCl concentration used. Similarly, there was 39% reduction in Fe3+ reducing potential determined by the potassium ferricyanide method (Fig. 2A). The cupric reducing power was 40% while the reduction of Mo6+ to Mo5+ was 60% of the corresponding control values at 2.5 mM HOCl (Fig. 2B). The DPPH and ABTS assays showed lower free radical scavenging activity in HOCl-treated RBC (Fig. 3). Control samples showed 62% quenching of DPPH compared to 32% at 2.5 mM HOCl. Similarly, ABTS free radical scavenging was reduced to 52 μmoles trolox equivalent (TE) at 2.5 mM HOCl compared to 69 in control samples (Fig. 3).
Figure 2.
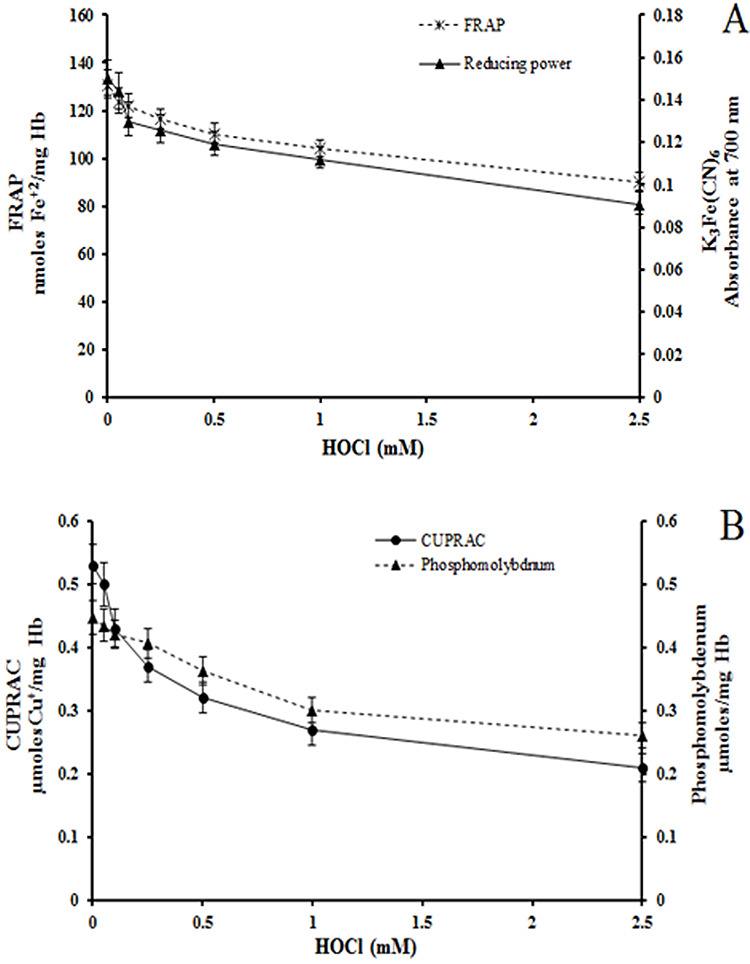
Effect of HOCl on metal reducing power of RBC. (A) FRAP and potassium ferrocyanide assays (B) CUPRAC and phosphomolybdenum assays. These assays were done using hemolysates from control and HOCl-treated RBC. Except for 0.05 mM HOCl, all other results were significantly different for HOCl treated RBCs as compared to control.
Figure 3.
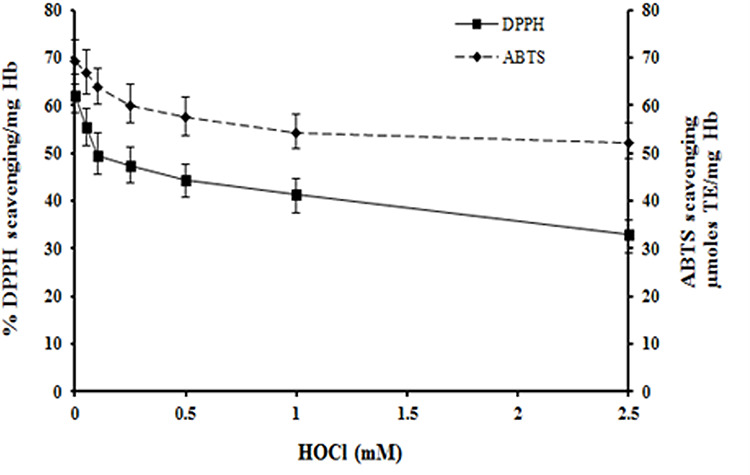
Effect of HOCl on DPPH and ABTS radical scavenging ability of RBC. The DPPH and ABTS assays were done using hemolysates from control and HOCl-treated RBC. TE: Trolox equivalent.
RBC membrane damage was monitored from PMRS activity. There was a dramatic HOCl dose-dependent decline in PMRS activity (Fig. 4). PMRS was reduced 6-fold (Fig. 4A) when assayed by ferricyanide reduction method and 3.3-fold by the more recent ABTS method (Fig. 4B) at 2.5 mM HOCl as compared to control values. This decline was accompanied by inhibition of AFR reductase activity which was 2.4-fold less at 2.5 mM HOCl (Fig. 4C).
Figure 4.
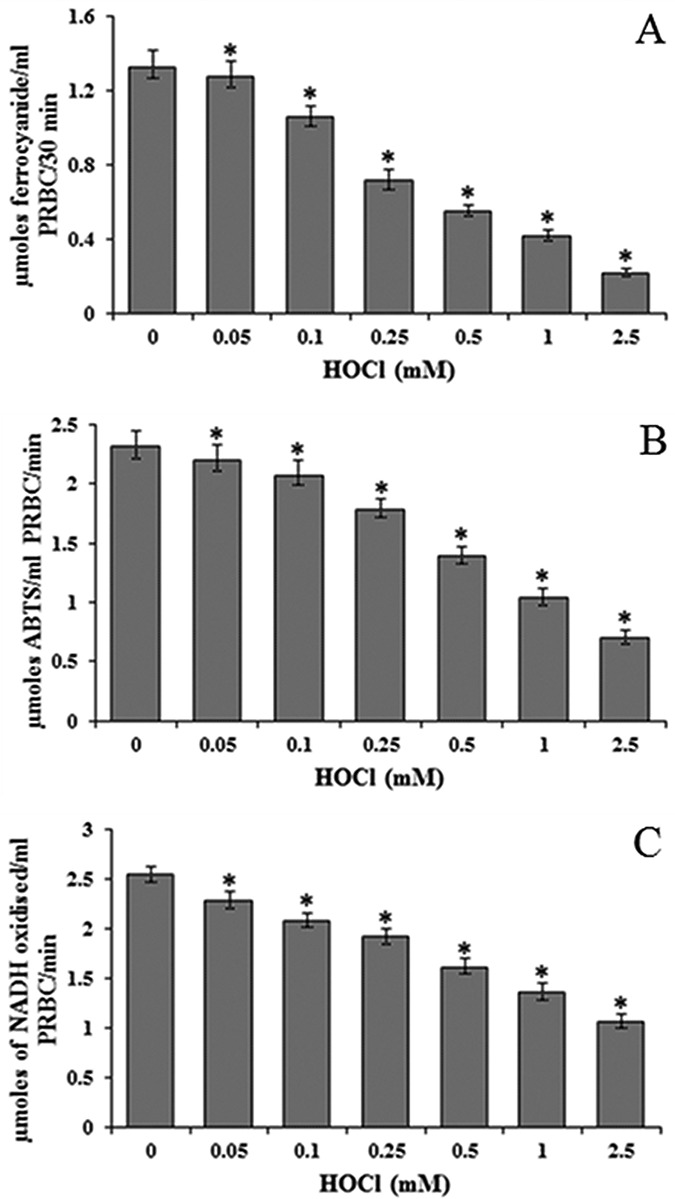
Effect of HOCl on PMRS of RBC determined by (A) ferricyanide and (B) ABTS* radical scavenging methods and (C) AFR reductase. *Significantly different from control. PRBC: packed red blood cells.
HOCl treatment also resulted in lowered activities of hexokinase and G6PD, enzymes of glycolysis and hexose monophosphate shunt pathway, respectively. LDH, an enzyme of anaerobic glycolysis, was increased in RBC treated with HOCl (Table 2). The activity of 5′-nucleotidase, an enzyme involved in nucleotide metabolism in RBC, was lowered. However, AMP deaminase, which is involved in purine metabolism, was greatly increased in hemolysates from HOCl treated RBC. There was 2.7-fold increase in AMP deaminase activity at 2.5 mM HOCl when compared to the control (Table 2).
Table 2.
Effect of HOCl on some metabolic enzymes
| Control | 0.05 mM | 0.1 mM | 0.25 mM | 0.5 mM | 1.0 mM | 2.5 mM | |
|---|---|---|---|---|---|---|---|
| Hexokinase | 32.36 ± 3.01 | 27.94 ± 2.24a | 24.54 ± 2.13a | 21.72 ± 2.08a | 19.11 ± 1.43a | 14.81 ± 1.29a | 9.39 ± 1.03a |
| LDH | 46.63 ± 4.93 | 49.16 ± 4.74 | 52.23 ± 4.91a | 56.22 ± 5.18a | 58.66 ± 4.18a | 62.18 ± 5.14a | 66.86 ± 4.74a |
| G6PD | 3.08 ± 0.26 | 1.87 ± 0.15a | 1.51 ± 0.13a | 1.13 ± 0.10a | 0.76 ± 0.09a | 0.59 ± 0.05a | 0.19 ± 0.03a |
| 5′-Nucleotidase | 0.56 ± 0.03 | 0.45 ± 0.04a | 0.44 ± 0.04a | 0.39 ± 0.03a | 0.34 ± 0.04a | 0.29 ± 0.02a | 0.25 ± 0.02a |
| AMP deaminase | 0.98 ± 0.13 | 1.10 ± 0.10a | 1.14 ± 0.12a | 1.28 ± 0.10a | 1.34 ± 0.14a | 2.16 ± 0.20a | 2.64 ± 0.25a |
LDH and G6PD are in in nmoles/min/mg Hb, and hexokinase is in nmoles/hr/mg Hb while AMP- deaminase and 5′-nucleotidase are in units/min/mg Hb.
aSignificantly different from control.
The SEM images of HOCl treated RBC showed progressive cell damage in an HOCl concentration-dependent manner (Fig. 5). The control (untreated) RBC showed normal biconcave disc-shaped morphology (Fig. 5A). RBC treated with 0.25 mM HOCl showed mild anisopoikilocytosis, formation of echinocytes (black arrows), acanthocytes (white arrows) and stomatocytes (deep depressions on the membranes) (Fig. 5B). Incubation of RBC with 1.0 mM HOCl led to the formation of anisopoikilocytosis, echinocytes, fragmented (white arrows) and teardrop cells (light-coloured cells shown in black arrows) and many abnormally shaped cells (Fig. 5C). RBC incubated with 2.5 mM HOCl exhibited even more extensive cell damage and predominantly showed crenation of cells and severe anisopoikilocytosis (Fig. 5D). The white arrows represent some stomatocytes entangled in the aggregates.
Figure 5.
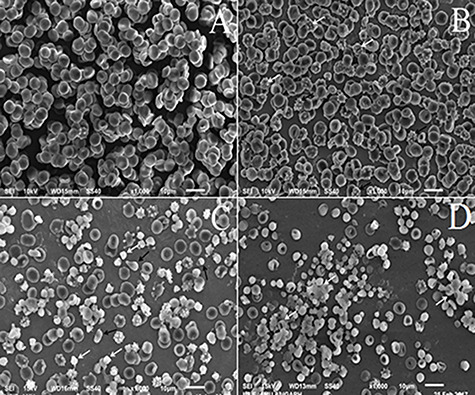
SEM images of control and HOCl treated RBC. (A) untreated RBC (control); RBC treated with (B) 0.25 mM (C) 1.0 mM and (D) 2.5 mM HOCl. Magnification is ×1000.
Discussion
HOCl is generated by neutrophils using the enzyme myeloperoxidase. The reactions of HOCl with cell constituents (proteins, lipids and DNA) are well investigated but the molecular mechanism of cytotoxic action of HOCl is still not well known. Circulating blood comes in contact with various tissues, including the sites of inflammation and tissue injury, where it can be exposed to high HOCl concentrations. This can result in HOCl-induced changes/damage in the RBC. Previous studies have shown that HOCl causes red cell lysis, depletion of GSH levels and protein oxidation [8,17,45]. In agreement with these results, we also found that HOCl caused concentration-dependent hemolysis, lowering of GSH levels and increase in protein carbonyl content in RBC (data not shown). The HOCl-induced modification of proteins can alter their structure and function while GSH depletion will affect the redox status of the cell.
Despite the extensive work done on HOCl, there are no reports of its effect on AO enzymes and the AO power of exposed cells. Cellular AO defence mechanisms involve several essential enzymes. These enzymes form a major defence and protect the cell against damage by reactive oxygen species (ROS) and free radicals, which are frequently induced by toxicants. The primary defence enzymes are SOD, CAT and GPx. SOD catalyses the dismutation of superoxide radical (O2←) to H2O2 which is then converted to water and molecular oxygen by CAT [46]. GPx is also involved in the detoxification of peroxides, including H2O2 and organic peroxides. The marked increase in SOD, CAT and GPx activities in HOCl treated RBC is probably an adaptive response of the system to counter the damaging effects of HOCl-induced oxidative stress. The increased activities of CAT and GPx must have contributed to the observed decline in cellular H2O2 concentration. Another possibility is the consumption of H2O2 due to a Fenton type reaction leading to the generation of hydroxyl radicals (OH) [46,47]. Furthermore, the greatly reduced nitrite levels might reflect direct scavenging of HOCl by this inorganic anion. Nitrite accumulates at sites of chronic inflammation where HOCl is overproduced and has been proposed to be cytoprotective against HOCl-induced injury [48].
The AO power of control and HOCl-treated RBC was determined next since it has not been reported before in any system. Several assays were used due to the complexity of AOs present in the cells and reflect the electron-donating ability of the cells. In these assays, the donated electrons quench stable coloured free radicals (DPPH· and ABTS·+) or reduce metal ions (Fe+3, Cu+2 and Mo+6) to lower oxidation states. The results of all these assays clearly showed that HOCl treatment impaired the AO power resulting in greatly lowered free radical quenching and metal-reducing ability of the RBC. The lowered AO power upon exposure to HOCl can have a major deleterious effect on the cell since it will greatly reduce the ability of the RBC to quench free radicals and ROS and, therefore, exacerbate the damaging effects of oxidative stress condition induced by HOCl. This decline in the AO power/capacity may be due to the depletion of GSH because of its rapid reaction with HOCl [17,49]. Thus the insufficient cellular AO defence of HOCl-treated RBC will be unable to cope with the damaging effects of this oxidizing agent.
The two major pathways of glucose metabolism in RBC, glycolysis and hexose monophosphate shunt were inhibited when monitored from the activities of their enzymes. In the absence of the citric acid cycle in RBC, glycolysis is the only metabolic pathway that generates ATP. Inhibition of hexokinase will slow down glycolysis and deplete cellular ATP levels. The activity of LDH, the terminal enzyme of anaerobic glycolysis which is responsible for the regeneration of NAD+ from NADH, was increased. LDH is bound to the membrane on the inner side and its activation suggests that it may have been released due to perturbation of plasma membrane integrity induced by HOCl [50]. It could also be a cellular response to ensure that enough NAD+ is available for glycolysis to proceed. G6PD is the first enzyme of hexose monophosphate shunt that generates NADPH, the major cellular reductant of RBC. G6PD activity was dramatically inhibited by HOCl. This will greatly reduce NADPH levels, which in turn, will affect the AO defence enzymes GR and TR. NADPH is used by GR for the regeneration of GSH from GSSG. Low NADPH levels will result in reduced activity of GR and less regeneration of GSH from its oxidized form. NADPH is also used by TR which maintains the sulfhydryl groups of proteins in the reduced state, especially under conditions of oxidative stress. Thus, reduced levels of NADPH, due to inhibition of G6PD, will result in lowered activities of both GR and TR. Importantly, the activity of AMP deaminase, an enzyme of purine metabolism, was increased almost 3-fold. AMP deaminase is considered an important marker of RBC oxidative stress [51] and its enhanced activity is suggestive of increased generation of ROS in the cell.
Membrane damage also led to inhibition of PMRS and AFR reductase. The PMRS transfers electrons to extracellular acceptors. Higher PMRS activity is correlated with increased ability to combat oxidative stress [52]. Since RBCs are constantly exposed to oxidative stress, attenuation of ROS induced damage is one of the main roles of PMRS. The PMRS and AFR reductase are important not only in maintaining the redox status of the cell but also the extracellular levels of ascorbate, a major AO of plasma [37]. Their inhibition will result in lowered AO power not only of RBC but also of whole blood.
The electron microscopic images of HOCl treated RBC show extensive cellular damage and morphological changes in an HOCl dose-dependent manner. These images provide physical evidence and confirmation of cell damage and toxicity exerted by this oxidant. SEM studies greatly support the results of biochemical studies described above.
The above results on the effects of HOCl on RBC should be seen in the context of RBC being present in blood along with plasma which is a complex mixture of proteins, cells, nutrients, amino acids, AOs, metal ions, hormones, etc. The presence of these components is going to affect the reaction of HOCl with RBC. The reaction of HOCl with plasma proteins, thiols and lipids is already reported [53,54]. The presence of water-soluble (ascorbic acid, uric acid) and insoluble (alpha-tocopherol, ubiquinone) AOs, in addition to AO enzymes (CAT, SOD, GPx), in plasma is also going to significantly lower the effect of HOCl on RBC. However, the use of isolated RBC offers a clear advantage in that represents a cellular system that has a well-developed endogenous AO system to combat the effects of extracellular and intracellular oxidants and can rapidly yield results on the toxic effects of a compound.
Conclusions
HOCl decreases the enzymatic AO defence and power. The PMRS and the enzymes of major pathways of glucose metabolism are inhibited. Thus, HOCl generated from MPO at sites of inflammation may lead to extensive cell damage to the RBC. Our results demonstrate therefore that the inhibition of AO enzymes and glycolytic pathways by HOCl can be one of the reasons for perpetuating the oxidative stress induced by this oxidant. This increased destruction/damage of RBC at the site of inflammation can lead to hemolytic anemia and red cell senescence.
Acknowledgements
We are thankful to Ms. Rubina Hameed for help in the EM studies. We are also grateful to Fariheen Aisha Ansari and Shaikh Nisar Ali for their help in this study. This work was funded by a grant from CST-UP. Financial support to the department from the University Grants Commission (SAP-DRS-III scheme) and Department of Science and Technology (DST-FIST), New Delhi, is gratefully acknowledged. I.Q.T. is the recipient of a UGC Non-NET fellowship from Aligarh Muslim University.
Contributor Information
Irfan Qadir Tantry, Department of Biochemistry, Faculty of Life Sciences, Aligarh Muslim University, Aligarh 202002, India; Department of Biochemistry, J.N. Medical College, Aligarh Muslim University, Aligarh 202002, India.
Asif Ali, Department of Biochemistry, J.N. Medical College, Aligarh Muslim University, Aligarh 202002, India.
Riaz Mahmood, Department of Biochemistry, Faculty of Life Sciences, Aligarh Muslim University, Aligarh 202002, India.
Funding
This work was funded by a grant from Council of Science and Technology, UP. India (Grant No. 2123).
Conflict of interest statement
None declared.
References
- 1. Davies MJ, Hawkins CL, Pattison DIet al. Mammalian heme peroxidases: from molecular mechanisms to health implications. Antioxid Redox Signal 2008;10:1199–234. [DOI] [PubMed] [Google Scholar]
- 2. Rayner BS, Love DT, Hawkins CL. Comparative reactivity of myeloperoxidase-derived oxidants with mammalian cells. Free Radic Biol Med 2014;71:240–55. [DOI] [PubMed] [Google Scholar]
- 3. Favero TG, Colter D, Hooper PFet al. Hypochlorous acid inhibits Ca (2+)-ATPase from skeletal muscle sarcoplasmic reticulum. J Appl Physiol 1998;84:425–30. [DOI] [PubMed] [Google Scholar]
- 4. Guo Y, Krumwiede M, White JCet al. HOCl effects on the tight junctions of rabbit tracheal epithelium. Am J Physiol 1996;270:L224–31. [DOI] [PubMed] [Google Scholar]
- 5. Sakarya S, Gunay N, Karakulak Met al. Hypochlorous acid: an ideal wound care agent with powerful microbicidal antibiofilm and wound healing potency. Wound 2014;26:342–50. [PubMed] [Google Scholar]
- 6. Kampf G, Todt D, Pfaender S, Steinmann E. Persistence of coronaviruses on inanimate surfaces and their inactivation with biocidal agents. J Hosp Infect 2020;104:246–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hawkins CL. Hypochlorous acid-mediated modification of proteins and its consequences. Essays Biochem 2020;64:75–86. [DOI] [PubMed] [Google Scholar]
- 8. Pattison DI, Davies MJ. Reactions of myeloperoxidase-derived oxidants with biological substrates: gaining chemical insight into human inflammatory diseases. Curr Med Chem 2006;13:3271–90. [DOI] [PubMed] [Google Scholar]
- 9. Imlay JA. Pathways of oxidative damage. Annu Rev Microbiol 2003;57:395–418. [DOI] [PubMed] [Google Scholar]
- 10. Hawkins CL, Pattison DI, Davies MJ. Hypochlorite-induced oxidation of amino acids, peptides and proteins. Amino Acids 2003;25:259–74. [DOI] [PubMed] [Google Scholar]
- 11. Spalteholz H, Panasenko OM, Arnhold J. Formation of reactive halide species by myeloperoxidase and eosinophil peroxidase. Arch Biochem Biophys 2006;445:225–34. [DOI] [PubMed] [Google Scholar]
- 12. Robaszkiewicz A, Bartosz G, Pitt ARet al. HOCl-modified phosphatidylcholines induce apoptosis and redox imbalance in HUVEC-ST cells. Arch Biochem Biophys 2014;548:1–10. [DOI] [PubMed] [Google Scholar]
- 13. Zhou J, Wang Q, Ding Yet al. Hypochlorous acid via peroxynitrite activates protein kinase Cθ and insulin resistance in adipocytes. J Mol Endocrinol 2015;54:25–37. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14. Gungor N, Knaapen AM, Munnia Aet al. Genotoxic effects of neutrophils and hypochlorous acid. Mutagenesis 2010;25:149–54. [DOI] [PubMed] [Google Scholar]
- 15. Weiss SJ. Tissue destruction by neutrophils. N Engl J Med 1989;320:365–76. [DOI] [PubMed] [Google Scholar]
- 16. Vissers MCM, Carr AC, Chapman ALP. Comparison of human red cell lysis by hypochlorous and hypobromous acids: insights into the mechanism of lysis. Biochem J 1998;330:131–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vissers MCM, Winterbourn CC. Oxidation of intracellular glutathione after exposure of human red blood cells to hypochlorous acid. Biochem J 1995;307:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marsche G, Furtmuller PG, Obinger Cet al. Hypochlorite-modified high-density lipoprotein acts as a sink for myeloperoxidase in vitro. Cardiovasc Res 2008;79:187–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Morris JC. The acid ionization constant of HOCl from 5° to 35°. J Phys Chem 1966;70:3798–805. [Google Scholar]
- 20. Acker JP, Croteau IM, Yi QL. An analysis of the bias in red blood cell hemolysis measurement using several analytical approaches. Clin Chim Acta 2012;413:1746–52. [DOI] [PubMed] [Google Scholar]
- 21. Miranda KM, Espey MG, Wink DA. A rapid, simple spectrophotometric method for simultaneous detection of nitrate and nitrite. Nitric Oxide 2001;5:62–71. [DOI] [PubMed] [Google Scholar]
- 22. Gay C, Gebicki JM. A critical evaluation of the effect of sorbitol on the ferric-xylenol orange hydroperoxide assay. Anal Biochem 2000;248:217–20. [DOI] [PubMed] [Google Scholar]
- 23. Marklund S, Marklund G. Involvement of the superoxide anion radical in the autooxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur J Biochem 1974;47:469–74. [DOI] [PubMed] [Google Scholar]
- 24. Aebi H. Catalase in vitro. Methods Enzymol 1984;105:121–6. [DOI] [PubMed] [Google Scholar]
- 25. Carlberg I, Mannervik B. Glutathione reductase. Methods Enzymol 1985;113:484–90. [DOI] [PubMed] [Google Scholar]
- 26. Habig WH, Pabst MJ, Jokoby WB. Glutathione-S-transferases. J Biol Chem 1974;249:7130–9. [PubMed] [Google Scholar]
- 27. Tamura T, Stadtman TC. A new selenoprotein from human lung adenocarcinoma cells: purification, properties and thioredoxin reductase activity. Proc Natl Acad Sci USA 1996;93:1006–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Flohe L, Gunzler W. Assays of glutathione peroxidase. Methods Enzymol 1984;105:114–21. [DOI] [PubMed] [Google Scholar]
- 29. Mishra K, Ojha H, Chaudhury NK. Estimation of antiradical properties of antioxidants using DPPH assay: A critical review and results. Food Chem 2012;130:1036–43. [Google Scholar]
- 30. Re R, Pellegrini N, Proteggente Aet al. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic Biol Med 1999;26:1231–7. [DOI] [PubMed] [Google Scholar]
- 31. Benzie IFF, Stain JJ. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: the FRAP assay. Anal Biochem 1996;239:70–6. [DOI] [PubMed] [Google Scholar]
- 32. Yen GC, Chen HY. Antioxidant activity of various tea extracts in relation to their antimutagenicity. J Agric Food Chem 1995;43:27–32. [Google Scholar]
- 33. Çekiç SD, Kara N, Tütem Eet al. Protein–incorporated serum total antioxidant capacity measurement by a modified CUPRAC (CUPRIC reducing antioxidant capacity) method. Anal Lett 2012;45:754–63. [Google Scholar]
- 34. Prieto P, Pineda M, Aguilar M. Spectrophotometric quantitation of antioxidant capacity through the formation of a phosphomolydenum complex: specific application to the determination of vitamin E. Anal Biochem 1999;269:337–41. [DOI] [PubMed] [Google Scholar]
- 35. Rizvi SI, Srivastava N. Erythrocyte plasma membrane redox system in first degree relatives of type 2 diabetic patients. Int J Diabetes Mellit 2010;2:119–21. [Google Scholar]
- 36. Avron M, Shavit N. A sensitive and simple method for determination of ferrocyanide. Anal Biochem 1963;6:549–54. [DOI] [PubMed] [Google Scholar]
- 37. Sadowska-Bartosz I, Bartosz G. Effect of 3-bromopyruvic acid on human erythrocyte antioxidant defense system. Cell Biol Int 2013;37:1285–90. [DOI] [PubMed] [Google Scholar]
- 38. Sadowska-Woda I, Bartosz G. How do erythrocytes contribute to the ABTS* scavenging capacity of blood? Free Radic Res 2013;47:35–43. [DOI] [PubMed] [Google Scholar]
- 39. May JM, Qu ZC, Cobb CE. Human erythrocyte recycling of ascorbic acid: relative contributions from the ascorbate free radical and dehydroascorbic acid. J Biol Chem 2004;279:14975–82. [DOI] [PubMed] [Google Scholar]
- 40. Crane RK, Sols A. The association of hexokinase with particulate fractions of brain and other tissue homogenates. J Biol Chem 1953;203:273–92. [PubMed] [Google Scholar]
- 41. Khundmiri SJ, Asghar M, Khan Fet al. Effect of ischemia and reperfusion on enzymes of carbohydrate metabolism in rat kidney. J Nephrol 2004;17:377–83. [PubMed] [Google Scholar]
- 42. Shonk CC, Boxer GE. Enzyme patterns in human tissues. I. Methods for the determination of glycolytic enzymes. Cancer Res 1964;24:709–21. [PubMed] [Google Scholar]
- 43. Heppel PA, Hilmore RJ. Purification and properties of 5-nucleotidase. J Biol Chem 1951;88:665–76. [PubMed] [Google Scholar]
- 44. Pedersen RC, Berry AJ. Sensitive, optimized assay for serum AMP deaminase. Clin Chem 1977;23:1726–33. [PubMed] [Google Scholar]
- 45. Carr AC, Vissers MCM, Domigan NMet al. Modification of red cell membrane lipids by hypochlorous acid and haemolysis by preformed lipid chlorohydrins. Redox Rep 1997;3:263–71. [DOI] [PubMed] [Google Scholar]
- 46. Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine, 4th edn. New York: Oxford University Press, 2007. [Google Scholar]
- 47. Powers SK, Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev 2008;88:1243–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Whiteman M, Hooper DC, Scott GSet al. Inhibition of hypochlorous acid-induced cellular toxicity by nitrite. Proc Natl Acad Sci USA 2002;99:12061–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pullar JM, Winterbourn CC, Vissers MCM. Loss of GSH and thiol enzymes in endothelial cells exposed to sublethal concentrations of hypochlorous acid. Am J Physiol 1999;277:H1505–12. [DOI] [PubMed] [Google Scholar]
- 50. Schraufstatter IU, Browne K, Harris Aet al. Mechanisms of hypochlorite injury of target cells. J Clin Invest 1990;85:554–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tavazzi B, Amorini AM, Fazzina Get al. Oxidative stress induces impairment of human erythrocytes energy metabolism through the oxygen radical mediated direct activation of AMP-deaminase. J Biol Chem 2001;276:48083–92. [DOI] [PubMed] [Google Scholar]
- 52. Kennett EC, Kuchel PW. Redox reactions and electron transfer across the red cell membrane. IUBMB Life 2003;55:375–85. [DOI] [PubMed] [Google Scholar]
- 53. Hawkins CL, Davies MJ. Hypochlorite-induced oxidation of proteins in plasma: formation of chloramines and nitrogen-centred radicals and their role in protein fragmentation. Biochem J 1999;340:539–48. [PMC free article] [PubMed] [Google Scholar]
- 54. Pattison DI, Hawkins CL, Davies MJ. What are the plasma targets of the oxidant hypochlorous acid? A kinetic modelling approach. Chem Res Toxicol 2009;22:807–17. [DOI] [PubMed] [Google Scholar]