

Abstract
The oxidation of tyrosine to form the neutral tyrosine radical via proton-coupled electron transfer is essential for a wide range of biological processes. The precise measurement of the proton-coupled redox potentials of tyrosine (Y) in complex protein environments is challenging mainly due to the highly oxidizing and reactive nature of the radical state. Herein a computational strategy is presented for predicting proton-coupled redox potentials in a protein environment. In this strategy, both the reduced Y-OH and oxidized Y-O• forms of tyrosine are sampled with molecular dynamics using a molecular mechanical force field. For a large number of conformations, a quantum mechanical/molecular mechanical (QM/MM) electrostatic embedding scheme is used to compute the free energy differences between the reduced and oxidized forms, including the zero-point energy and entropic contributions as well as the impact of the protein electrostatic environment. This strategy is applied to a series of fluorinated tyrosine derivatives embedded in a de novo α-helical protein denoted α3Y. The force fields for both the reduced and oxidized forms of these non-canonical fluorinated tyrosine residues are parameterized for general use. The calculated relative proton-coupled redox potentials agree with experimentally measured values with a mean unsigned error of 34 mV. Analysis of the simulations illustrate that hydrogen-bonding interactions between tyrosine and water increase the redox potentials by ~100–250 mV, with significant variations due to the fluctuating protein environment. This QM/MM approach enables the calculation of proton-coupled redox potentials of tyrosine and other residues such as tryptophan in a variety of protein systems.
Graphical Abstract
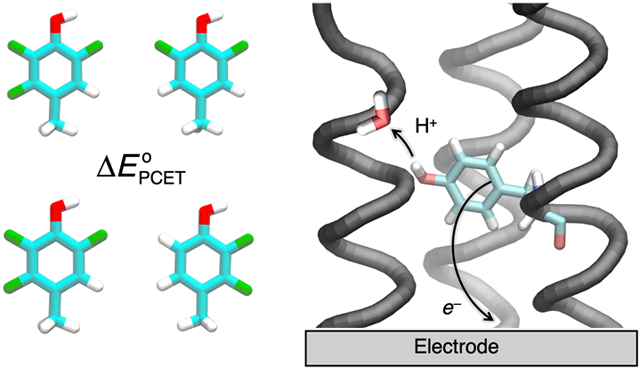
Introduction
Electron transfer (ET)1, 2 and proton-coupled electron transfer (PCET)3, 4 support the catalytic activities of a vast number of metabolic enzymes. These enzymes employ metallocofactors, organic molecules, and four types of amino acids to conduct the essential redox chemistry. In this context, tyrosine (Y), tryptophan (W), cysteine, and/or glycine protein residues serve as high potential one-electron redox mediators.5-17 The mechanistic properties of Y and W are particularly interesting because these residues can efficiently transfer highly oxidizing equivalents over many Ångströms.11, 12, 17-21 These amino acid based redox reactions are difficult to characterize for a number of reasons, including the very high reduction potentials (E°) involved, the reactive nature of the oxidized (radical) state, and the weaker optical features associated with amino acid radicals. Additionally, radical formation, transfer, and decay can be coupled to protein conformational changes occurring on a broad range of timescales.11, 22-24 For all of these reasons, detailed mechanistic information and thermodynamic information are challenging to obtain.
Site-specific incorporation of non-canonical amino acids (ncAAs) has emerged as a technique to investigate and tune protein function.25-27 In this technique, a protein residue of interest is replaced by a synthetic analogue with perturbed physiochemical and spectroscopic properties. Specifically, ncAAs have been employed to study catalytic mechanisms28, 29 and to probe mechanistically relevant redox-active tyrosines in E. coli ribonucleotide reductase,11, 30-36 BLUF domains,37, 38 and other protein systems.39-41 Enzymatic and spectroscopic studies of these ncAA mutant proteins have provided valuable insights. However, the interpretation of these types of studies would strongly benefit from accurate thermodynamic data for the ncAAs in the protein environment.
Given the challenges associated with studying redox-active amino acids experimentally, the development of predictive computational approaches that could provide this essential information is of great interest. A variety of methods have been developed for the computational prediction of redox potentials in proteins using quantum mechanical (QM) methods,42, 43 molecular mechanical (MM) force fields,44-46 or hybrid QM/MM approaches.47-49,50 These methods are often able to obtain redox potentials within 100 – 300 mV of the experimentally measured values.48 Recently a constant pH and redox potential molecular dynamics (MD) method was developed and used to compute proton-coupled redox potentials in several protein systems.51 This method performs conformational sampling with a MM force field and uses a Monte Carlo approach to attempt protonation and/or redox state changes while treating the solvent with a dielectric continuum model. Although the results are promising and provide useful insights, they highlight the challenges of using purely MM methods for redox potential calculations, as well as the need for careful benchmarking against experimental data.
Herein, we take advantage of the structurally and mechanistically well-defined α3X model protein system as a source of benchmarking data. The α3X system was specifically constructed to investigate Y/W-based proton-coupled redox processes. The single-stranded model proteins consist of three alpha helices (α3) with a single aromatic residue at interior position 32 (X32).52 Importantly, the α3 scaffold remains well-structured over a broad range of experimental conditions, supports reversible oxidation-reduction of X32, and facilitates protein film voltammetry studies.53-55 Thus, highly accurate and precise proton-coupled redox potentials (with errors ± 2-3 mV) are available for α3Y (X32 = Y) and a series of fluorotyrosine α3FnY variants (X32 = FnY where n = 2 or 3; see Figure 1). Because oxidation of tyrosine decreases its pKa by ~12 units,56 typically oxidation is accompanied by proton transfer from the phenol OH group to a nearby water molecule or protein residue via concerted PCET. Our previous MD simulations, combined with spectroscopic measurements, suggest that the proton acceptor for Y32 is a hydrogen-bonded water molecule, although a glutamate residue could not be ruled out.24 The small size and well-defined structure of the protein,22 as well as the precisely measured redox potentials,57 render the α3Y protein system ideally suited for benchmarking computational methods.
Figure 1.

PCET of the oxidative process in the α3Y protein, where the electron transfers to the electrode, and the proton acceptor is a water molecule, as suggested by prior simulations. X32 has been replaced by the depicted FnY residues55, 57 in previous experiments and in the present computational work.
Herein, we present a strategy for computing proton-coupled redox potentials and apply this approach to the α3Y protein system with the series of fluorinated tyrosines studied experimentally. Our computational strategy uses MM for conformational sampling and a QM/MM electrostatic embedding approach to compute the proton-coupled redox potentials for an ensemble of conformations. A comprehensive analysis of the results illustrates the importance of sampling hydrogen-bonding interactions between the tyrosine and water, as well as electronic effects due to the fluorine substituents. The development of a computational tool for the accurate and efficient prediction of proton-coupled redox potentials will help guide the design of new mechanistic probes or enzymes with enhanced reactivity.
Methods
Calculation of proton-coupled redox potentials
Under typical biological conditions, the oxidation of tyrosine (Y) occurs through a concerted PCET reaction, forming the neutral tyrosine radical and avoiding charged intermediates such as Y-OH+• or Y-O−. The PCET reaction associated with oxidation can be described as follows:
| (1) |
The relative proton-coupled redox potentials, ΔE°, with respect to a specified reference reaction were determined with the following expression:
| (2) |
where
| (3) |
Here ΔG° (YO• → YOH) is the free energy change for the reduction reaction, which is the reverse of Eq. (1). Thus, E° is a reduction potential but is referred to as a redox potential to signify the reversible reduction/oxidation process. The subscript “ref” denotes the reference reaction, and the value of is set to the experimentally determined value to obtain E° from Eq. (2). Using a reference reaction leads to cancellation effects that avoid introducing errors from the estimation of the solvation free energies of the proton and electron,58 as well as the value of the standard hydrogen electrode (SHE).59
All density functional theory (DFT) calculations in this paper were performed with the B3LYP-D3(BJ) functional60-62 and the 6-31+G(d,p) basis set63 using Q-Chem.64 This level of theory was found to produce redox potentials in reasonable agreement with experimental values. Prior to including the protein environment, we calculated the proton-coupled redox potentials of the fluorinated tyrosine side chains in the gas phase with phenol as the reference. For calculations in the protein environment, the reference was the α3Y protein with the unsubstituted tyrosine.
To calculate the proton-coupled redox potentials in the protein environment, we devised a QM/MM procedure with electrostatic embedding and applied it to an ensemble of conformations sampled from MD trajectories of both the reduced Y-OH and oxidized Y-O• states. The detailed workflow is shown in Scheme 1. For each conformation, the QM/MM interface in AMBER65 was used to automatically add a hydrogen link atom between the Cß and Cα carbons with the default charge redistribution scheme.66, 67 Adding the hydrogen link atom to the QM region is necessary to satisfy the valency of the carbon atom. The resulting QM region is then optimized in the field of the partial charges corresponding to the MM system. A frequency analysis was performed on the optimized geometry to ensure that it was a minimum in the field of MM partial charges and to compute the zero-point energy and entropic contributions. For the reduced (oxidized) state conformations, the same geometry optimization procedure was followed for the oxidized (reduced) state of tyrosine in the fixed protein environment. For the reduced Y-OH conformations, Y-O• was obtained by removing the hydrogen atom (i.e., deleting the hydroxyl proton and updating the multiplicity to reflect the loss of an electron that produces an unpaired electron). For the oxidized Y-O• conformations, a hydrogen atom was added to generate the reduced state. For each protein conformation, the free energy difference between the reduced and oxidized states was determined to compute the proton-coupled redox potential via Eq. (2). The conformations were sampled every 100 ps from a 1 μs trajectory for the reduced state and every 10 ps from a 1 ns trajectory for the oxidized state, which required less sampling for reasons discussed below. The total proton-coupled redox potential for a given system was determined as the average value over ~10,000 conformations sampled from each of the reduced and oxidized trajectories.
Scheme 1.
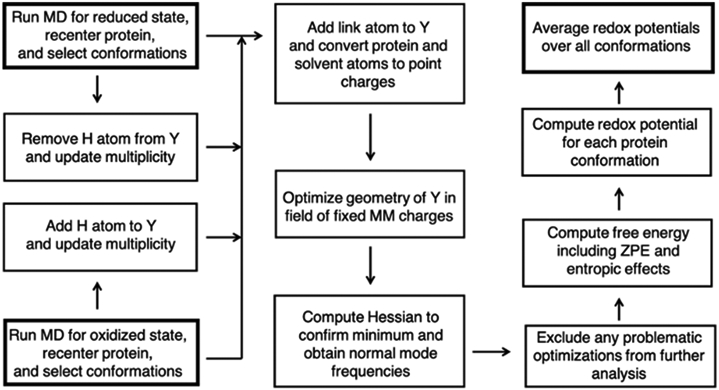
Computational workflow for calculating proton-coupled redox potentials. Bold boxes indicate the two possible starting points (left hand side) or the ending point (right hand side).
Parameterization of partial charges of fluorinated tyrosine
As the use of fluorinated and other non-canonical amino acids increases, the demand and interest in modeling systems containing these residues is rising. Unfortunately, parameters for fluorinated amino acids are not included in standard force fields. Although the Floudas group has developed FORCEFIELD_PTM68 and FORCEFIELD_NCAA69 to describe certain non-canonical amino acids, these parameters have not covered fluorinated amino acids. Fluoromethylated (-CF3) derivatives of branched chain apolar amino acids and relevant force field terms have been parameterized,70 but this effort has not been extended to tyrosine. We parameterized the non-canonical amino acids needed for this study, namely the series of fluorinated tyrosines shown in Fig. 1, in a manner that is compatible with the AMBER force field.
For each fluorinated tyrosine, a tyrosine residue was constructed with AmberTools,71 the N- and C- termini were capped, and the relevant hydrogen atoms were changed to fluorines. The atom types and parameters from the general AMBER force field (GAFF)72 were used for the fluorine atoms, and the standard RESP73 procedure was followed to obtain the partial charges. Two conformers were generated with ϕ and ψ angles corresponding to a β-sheet and a right-handed α-helix, as given in Table II of Ref. 73. These structures were optimized at the Hartree-Fock level with the 6-31G* basis set74 to be consistent with the existing force field parameters. The atomic charges of the backbone (N, H, C, O) were fixed to the AMBER library charges for tyrosine during the multi-conformational RESP fit to be consistent with the ff14SB forcefield.75 A constraint on the blocking N-methyl and acetyl groups was applied to ensure that the overall charge on the tyrosine residue was zero. In addition to parameterizing the standard (reduced) state of each fluorinated tyrosine (Y-OH), we parameterized the neutral radical state (Y-O•) for each of them. The partial charges on the fluorine atoms derived within this work are comparable to the charges on the fluorine atoms parameterized for polar branched chain amino acids with CF3 groups.70 Additionally, these charges are similar to those found in the CHARMM cgenFF libraries.76 Although these charges have been found to be suitable for the purpose of this paper and are presumably appropriate for other uses, they have not been rigorously validated against experimental observables except for the redox potentials discussed herein. The partial charges and associated forcefield parameters adapted from GAFF for all of these non-canonical amino acids are reported in the SI (Tables S1-S5). The partial charges for the tyrosine radical are provided in our previous work.77
Computational details about molecular dynamics simulations
The starting structure for all of our MD simulations was the solution NMR structure for the α3Y protein (PDB code: 2MI7).22 As the NMR ensemble includes 32 structures, the last conformer was used as the starting point for each simulation. The structures of these different conformers are extremely similar, and our previous simulations starting from different conformers yielded similar results.24 Although the structures of the α3FnY variants have not been obtained, their global protein properties are nearly identical to those of α3Y.55, 57 Typically, fluorinated proteins show enhanced thermostability and minimal structural perturbations.78
For each α3FnY system studied, the non-canonical amino acid was introduced using the starting position of Y32 in the α3Y NMR structure and converting the relevant hydrogen atoms to fluorine atoms. Each initial protein structure was solvated with explicit TIP3P79 water in a periodic rectilinear box. The positive charge of the protein was neutralized with Cl− ions, followed by adding Na+ and Cl− ions to achieve a salt concentration of ~150 mM. The protonation states did not need to be determined, as the NMR structure contains hydrogen atoms. The MD simulations were performed using the AMBER 2018 software package80 with the AMBER ff14SB force field.75 The equilibration procedure and technical details are the same as those used in our previous studies of α3Y systems,24 and full details are given in the SI.
After equilibration, a trajectory was propagated for 1 μs for each reduced Y-OH system, and conformations were extracted every 100 ps over each 1 μs trajectory for computation of the proton-coupled redox potentials. Trajectories for the oxidized Y-O• systems were propagated for 100 ns, extracting conformations every 10 ps to compute the proton-coupled redox potentials. Less conformational sampling was required for the Y-O• systems because hydrogen-bonding interactions between the radical and water were not prevalent for any of the oxidized systems, as discussed further below. Moreover, we found that the proton-coupled redox potentials computed for the α3(2,3,5)F3Y-O• and α3(2,3,6)F3Y-O• systems using 10,000 conformations obtained from either the first or second 100 ns of a 200 ns trajectory are the same to within 5 mV for each of these two systems (Table S9). The simulations of the α3FnY proteins with the tyrosine in its standard (reduced) and neutral radical (oxidized) states were as stable as the wild-type simulations. The root-mean-square deviations (RMSDs) of the Cα atoms were ~1.1–1.8 Å for all systems studied (Table S6). Moreover, the fluorotyrosines exert minimal structural changes on the protein backbone (Figure 2).
Figure 2.

Backbones of representative equilibrated conformations of the α3FnY systems superimposed on a representative equilibrated conformation of the α3Y system with the tyrosine residues depicted explicitly.
The trajectories were visualized with the VMD program81 and analyzed with the CPPTRAJ program.82 The hydrogen-bonding interactions, inter-residue distances, and solvent occupancy were analyzed for all trajectories. Hydrogen bonds were defined to have a heavy-atom distance less than or equal to 3.0 Å and a donor-hydrogen-acceptor angle greater than or equal to 135 degrees.
Results and Discussion
Calculation of proton-coupled redox potentials in gas phase and protein
We computed the relative proton-coupled redox potentials in the gas phase with phenol as the reference. These computed relative redox potentials agree reasonably well with the values obtained experimentally in the protein environment (Table 1, ). The ordering of the redox potentials for the series of fluorinated tyrosines is preserved, with a mean unsigned error (MUE) of 20 mV for the series. We performed additional benchmarking with the ωB97XD83 functional and various basis sets (Table S7), yielding similar results with MUEs ranging from 16–20 mV. The agreement of the gas phase results with the experimental data obtained in the protein environment indicates that the effect of the protein environment is nearly uniform for all of the fluorinated tyrosines, as it does not significantly impact the relative values. If a non-canonical amino acid caused a structural rearrangement of the protein, this agreement would break down, and a gas phase calculation would not be able to account for these effects. However, the absolute values of the redox potentials for the tyrosines in the protein environment are shifted positively for the reduced (Y-OH) systems, mainly due to hydrogen-bonding interactions, as discussed below (Table S8). It is well established that the microenvironment can modulate the redox potential of protein-associated redox cofactors.e.g. 84-87 Our goal is to develop a computational strategy that would be reliable in these situations, although the protein environment appears to play a relatively uniform role in our specific application.
Table 1.
Experimental and Computed Relative Proton-Coupled Redox Potentials in the Gas Phase and Protein Environment.a
| System | b | c | d | e | f |
|---|---|---|---|---|---|
| α3(2,3,6)F3Y | 135 | 142 | 218 | 130 | 174 |
| α3(2,3)F2Y | 70 | 102 | 183 | −57 | 63 |
| α3(2,3,5)F3Y | 39 | 46 | 110 | −124 | −7 |
| α3Y | 0 | 0 | 0 | 0 | 0 |
| α3(3,5)F2Y | −25 | 10 | 33 | −77 | −22 |
| MUE | 20 | 81 | 87 | 34 |
All values are reported in mV relative to Y or α3Y in the associated column. The mean unsigned error (MUE) relative to experiment is given for each method. The standard deviations of the values computed in the protein environment values are given in Table S9 and are typically ~200 mV. The proton-coupled redox potentials were computed with the B3LYP-D3(BJ) functional. The values computed for the same set of conformations with the ωb97X-D functional are very similar and are given in Table S10.
The experimental values were obtained from Ref. 57.
These values were computed for the substituted tyrosine sidechains in the gas phase relative to phenol, which represents Y.
These values were obtained by averaging over all conformations with Y-OH hydrogen bonded to at least one water molecule among 10,000 conformations equally distributed along the 1 μs trajectory with Y-OH for each system.
These values were obtained by averaging over 10,000 conformations equally distributed along the 100 ns trajectory with Y-O• for each system.
These values were obtained by averaging the previous two columns corresponding to Y-OH and Y-O• for each system.
The proton-coupled redox potentials were computed in the protein environment using the QM/MM approach described above. The MD conformational sampling was performed with both the reduced tyrosine (Y-OH) and the oxidized tyrosine (Y-O•) with the corresponding relative redox potentials denoted and , respectively. For the calculation of given in Table 1, only the conformations with Y-OH hydrogen bonded to at least one water molecule were included for reasons that will be discussed below. The degree of hydrogen bonding of Y-O• to water in the trajectories used to compute was minimal due to the absence of the polar hydroxyl group. As shown in Table 1, is greater than for the fluorinated tyrosine systems, mainly due to the more extensive hydrogen-bonding interactions with Y-OH than Y-O• and the greater impact of hydrogen bonding on the fluorinated species than on the canonical tyrosine. The average of the results from sampling with Y-OH and Y-O• to model the reversible PCET process leads to a MUE of 34 mV compared to experiment. This level of agreement is considered to be state-of-the-art among current computational methods.
A challenge that arises in this application is that the differences between the redox potentials in this series are relatively small and are difficult to capture when sampling over a large ensemble of protein conformations. To elucidate these local protein environments, we analyzed the hydrogen-bonding interactions of the tyrosine for each 1 μs trajectory used to compute the proton-coupled redox potentials (Table 2). As shown in our previous work, the degree of hydrogen bonding of Y-OH to water and the protein is sensitive to the force field, and extensive sampling is required to converge the hydrogen-bonding percentages. 24
Table 2.
Hydrogen-Bonding Interactions Involving Y for MD Trajectories of α3Y and α3FnY Systemsa
| System | V9:O | E13:Oε1, ε2 | L58:O | L12:O | WAT:O |
|---|---|---|---|---|---|
| α3(2,3,6)F3Y | ND | <1% | 28.1% | 1.2% | 58.2% |
| α3(2,3)F2Y | ND | ND | 23.1% | 17.9% | 27.5% |
| α3(2,3,5)F3Y | ND | ND | 26.2% | 17.3% | 17.4% |
| α3Y | 54.1% | 24.0% | 5.12% | ND | 38.2% |
| α3(3,5)F2Y | ND | ND | 27.2% | 3.1% | 5.1% |
Each value is the percentage of conformations with Y-OH hydrogen bonded to the specified atom over a 1 μs trajectory. ND denotes trajectories where this interaction was not detected. Hydrogen-bonding percentages involving water may reflect contributions from multiple water molecules and a mixture of water molecules accepting or donating a hydrogen bond to Y, with a full breakdown provided in Table S11.
The hydrogen-bonding results are only given for the trajectories with the reduced tyrosine (Y-OH) because the oxidized tyrosine (Y-O•) did not exhibit hydrogen-bonding interactions for more than 1% of the trajectory except for α3Y-O•, which exhibited hydrogen-bonding interactions with water for 3% of the 100 ns trajectory. Propagation of the α3Y-O• system for a longer 1 μs trajectory resulted in a similar hydrogen-bonding interaction percentage of 6%. Previous experimental and computational studies have shown that the phenoxyl radical in solution is able to form a relatively strong hydrogen-bonding interaction with water.88-90 To test our force field parameters, we simulated an individual tyrosyl radical in aqueous solution and found that such hydrogen-bonding interactions persisted for the majority of a 100 ns trajectory (see SI). This test implies that the low prevalence of hydrogen-bonding interactions in the α3Y-O• systems arises from the hydrophobic protein environment. As discussed for the α3Y-OH systems, however, the α3Y-O• hydrogen-bonding interactions are also sensitive to the force field and degree of conformational sampling.
Although Y-OH can serve as a hydrogen bond donor or acceptor to water, typically it serves as a hydrogen bond donor in this system (Table S11). Moreover, when Y-OH serves as a hydrogen bond acceptor to water, it is almost always simultaneously serving as a hydrogen bond donor to a protein residue, such as E13. Thus, virtually all of the conformations with Y-OH hydrogen bonded to water are hydrogen bond donors. These conformations are expected to be most relevant to proton-coupled oxidation because the proton must be transferred to a molecule such as water or E13. To maintain consistency among the systems given the challenges in conformational sampling, only those conformations with Y-OH hydrogen bonded to water are included in the calculation of . The results obtained including all conformations are given in Table S9 and, as expected, do not provide quite the same level of quantitative accuracy.
Given the impact of hydrogen-bonding interactions on the proton-coupled redox potentials, we analyzed the hydrogen-bonding interactions within the protein environment. This analysis focused on the distance between the Y32 hydroxyl oxygen and the oxygen of the closest water molecule because of the postulated role of water as the proton acceptor, the increase in the redox potential observed upon hydrogen bonding to water, and the nearly bimodal distributions of these distances (Figure 3).
Figure 3.

Distribution of hydrogen-bonding O–O distances between Y-OH and the closest water molecule in the reduced (Y-OH) trajectories for the five systems studied.
We further scrutinized the α3(2,3,6)F3Y system because it exhibits the greatest population of conformations with a hydrogen-bonded water molecule. Figure 4 illustrates that the population of hydrogen-bonded conformations (i.e., shorter Y-OH—OH2 distances) exhibits proton-coupled redox potentials that are ~100–200 mV greater than those corresponding to the more diffuse population of conformations without this hydrogen-bonding interaction (i.e., larger distances). Although the fluctuations within each population are significant, this increase in the proton-coupled redox potential is consistent with the 4–6 kcal/mol interaction energy of phenol forming a hydrogen bond with water,88 given that the Y-OH typically donates a hydrogen bond to water, and this interaction is not possible for the Y-O• radical.
Figure 4.

Two-dimensional contour plot of the relative proton-coupled redox potential versus distance between the tyrosine hydroxyl oxygen and the closest water molecule for the α3(2,3,6)F3Y system. These results were obtained from the 1 μs trajectories with Y-OH, and the redox potentials are given relative to the average proton-coupled redox potential computed for α3Y. The color bar represents density, and the bins used to generate the underlying histogram were 0.2 Å and 100 mV for distance and redox potential, respectively. The top black and white dashed line passes through the average proton-coupled redox potential for the conformations with Y-OH hydrogen bonded to at least one water molecule, while the bottom dashed line passes through the average value for the conformations without such hydrogen bonds.
This analysis indicates that the relatively large fluctuations observed in the computed proton-coupled redox potentials arise from the variations in hydrogen-bonding interactions, as well as other environmental factors within the protein. Given the sensitivity of the proton-coupled redox potential to hydrogen-bonding interactions, as well as the challenges inherent to sampling these interactions sufficiently even over 1 μs, including only the conformations with Y-OH hydrogen bonded to a water molecule ensures a degree of consistency. Furthermore, the proton-coupled oxidation of tyrosine requires proton transfer to a water molecule or other residue, suggesting that these conformations are most relevant to the experimental measurements.
Conclusions
This paper presents a QM/MM strategy for calculating proton-coupled redox potentials in proteins. This strategy entails conformational sampling of both the Y-OH and Y-O• forms of tyrosine with a molecular mechanical force field in conjunction with a QM/MM electrostatic embedding scheme to compute the free energy differences for a large number of conformations. To allow the investigation of non-canonical tyrosine amino acids, we derived partial charges for the Y-OH and Y-O• forms of a series of fluorinated tyrosines. The QM/MM scheme involves geometry optimizations and free energy calculations of these Y-OH and Y-O• residues in the electrostatic field of the protein. This scheme can be used in conjunction with any QM/MM method, such as ONIOM or other embedding approaches.91, 92 We applied this strategy to a series of systems that each involve a tyrosine or fluorotyrosine in the α3Y protein. Analysis of the results indicates that hydrogen-bonding interactions between tyrosine and water increase the proton-coupled redox potential by 100–250 mV, although significant fluctuations arise from the varying protein environment. This strategy produces relative proton-coupled redox potentials with a mean unsigned error of less than 40 mV relative to experimental measurements for these systems. However, the quantitative results are sensitive to the force field, conformational sampling, and the treatment of hydrogen-bonding interactions. Further benchmarking is required to determine if this accuracy will be attained for a wider range of protein systems.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health Grant Number GM056207 (S.H-S.) and GM79190 (C.T). C.R.R is supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE1752134 and in part by the National Institutes of Health (5T32GM06754 3-12). Computational resources of the Yale Center for Research Computing (YCRC) were used for processing trajectories. C.R.R thanks Alexander Soudackov for technical assistance and helpful discussions.
Footnotes
Supporting Information
Computational details, forcefield parameters for fluorinated tyrosine residues, and additional analysis of proton-coupled redox potentials computed in the protein environment.
References
- 1.Page CC; Moser CC; Chen X; Dutton PL Natural Engineering Principles of Electron Tunnelling in Biological Oxidation-Reduction. Nature 1999, 402, 47–52. [DOI] [PubMed] [Google Scholar]
- 2.Winkler JR; Gray HB Long-Range Electron Tunneling. J. Am. Chem. Soc 2014, 136, 2930–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reece SY; Nocera DG Proton-Coupled Electron Transfer in Biology: Results from Synergistic Studies in Natural and Model Systems. Annu. Rev. Biochem 2009, 78, 673–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hammes-Schiffer S Proton-Coupled Electron Transfer: Moving Together and Charging Forward. J. Am. Chem. Soc 2015, 137, 8860–8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uhlin U; Eklund H Structure of Ribonucleotide Reductase Protein R1. Nature 1994, 370, 533–539. [DOI] [PubMed] [Google Scholar]
- 6.Tommos C; Babcock GT Proton and Hydrogen Currents in Photosynthetic Water Oxidation. Biochim. Biophys. Acta 2000, 1458, 199–219. [DOI] [PubMed] [Google Scholar]
- 7.Stubbe J; Nocera DG; Yee CS; Chang MCY Radical Initiation in the Class I Ribonucleotide Reductase: Long-Range Proton-Coupled Electron Transfer? Chem. Rev 2003, 103, 2167–2202. [DOI] [PubMed] [Google Scholar]
- 8.Tsai AL; Kulmacz RJ Prostaglandin H Synthase: Resolved and Unresolved Mechanistic Issues. Arch. Biochem. Biophys 2010, 493, 103–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Craciun S; Balskus EP Microbial Conversion of Choline to Trimethylamine Requires a Glycyl Radical Enzyme. Proc. Natl. Acad. Sci 2012, 109, 21307–21312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davidson VL; Wilmot CM Posttranslational Biosynthesis of the Protein-Derived Cofactor Tryptophan Tryptophylquinone. Annu. Rev. Biochem 2013, 82, 531–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Minnihan EC; Nocera DG; Stubbe J Reversible, Long-Range Radical Transfer in E. coli Class Ia Ribonucleotide Reductase. Acc. Chem. Res 2013, 46, 2524–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Z; Tan C; Guo X; Li J; Wang L; Sancar A; Zhong D Determining Complete Electron Flow in the Cofactor Photoreduction of Oxidized Photolyase. Proc. Natl. Acad. Sci 2013, 110, 12966–12971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broderick JB; Duffus BR; Duschene KS; Shepard EM Radical S-Adenosylmethionine Enzymes. Chem. Rev 2014, 114, 4229–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Backman LRF; Funk MA; Dawson CD; Drennan CL New Tricks for the Glycyl Radical Enzyme Family. Crit. Rev. Biochem. Mol 2017, 52, 674–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blomberg MR Mechanism of Oxygen Reduction in Cytochrome c Oxidase and the Role of the Active Site Tyrosine. Biochemistry 2016, 55, 489–500. [DOI] [PubMed] [Google Scholar]
- 16.Wikström M; Krab K; Sharma V Oxygen Activation and Energy Conservation by Cytochrome c Oxidase. Chem. Rev 2018, 118, 2469–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gray HB; Winkler JR Living with Oxygen. Acc. Chem. Res 2018, 51, 1850–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Acebes S; Ruiz-Dueñas FJ; Toubes M; Sáez-Jiménez V; Pérez-Boada M; Lucas MF; Martínez AT; Guallar V Mapping the Long-Range Electron Transfer Route in Ligninolytic Peroxidases. J. Phys. Chem. B 2017, 121, 3946–3954. [DOI] [PubMed] [Google Scholar]
- 19.Brettel K; Byrdin M Reaction Mechanisms of DNA Photolyase. Curr. Opin. Struct. Biol 2010, 20, 693–701. [DOI] [PubMed] [Google Scholar]
- 20.Chaves I; Pokorny R; Byrdin M; Hoang N; Ritz T; Brettel K; Essen L-O; Horst G. T. J. v. d.; Batschauer A; Ahmad M The Cryptochromes: Blue Light Photoreceptors in Plants and Animals. Annu. Rev. Plant Biol 2011, 62, 335–364. [DOI] [PubMed] [Google Scholar]
- 21.Kang G; Taguchi AT; Stubbe J; Drennan CL Structure of a Trapped Radical Transfer Pathway Within a Ribonucleotide Reductase Holocomplex. Science 2020, 368, 424–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glover SD; Jorge C; Liang L; Valentine KG; Hammarstrom L; Tommos C Photochemical Tyrosine Oxidation in the Structurally Well-Defined Alpha3Y Protein: Proton-Coupled Electron Transfer and a Long-Lived Tyrosine Radical. J. Am. Chem. Soc 2014, 136, 14039–14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glover SD; Tyburski R; Liang L; Tommos C; Hammarstrom L Pourbaix Diagram, Proton-Coupled Electron Transfer, and Decay Kinetics of a Protein Tryptophan Radical: Comparing the Redox Properties of W32(*) and Y32(*) Generated Inside the Structurally Characterized Alpha3W and Alpha3Y Proteins. J. Am. Chem. Soc 2018, 140, 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nilsen-Moe A; Reinhardt CR; Glover SD; Liang L; Hammes-Schiffer S; Hammarström L; Tommos C Proton-Coupled Electron Transfer from Tyrosine in the Interior of a De Novo Protein: Mechanisms and Primary Proton Acceptor. J. Am. Chem. Soc 2020, 142, 11550–11559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Young DD; Schultz PG Playing with the Molecules of Life. ACS Chem. Biol 2018, 13, 854–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chin JW Expanding and Reprogramming the Genetic Code. Nature 2017, 550, 53–60. [DOI] [PubMed] [Google Scholar]
- 27.Wang L, Engineering the Genetic Code in Cells and Animals: Biological Considerations and Impacts. Acc. Chem. Res 2017, 50, 2767–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu Y; Cui C; Wang J; Lu Y Biosynthetic Approach to Modeling and Understanding Metalloproteins Using Unnatural Amino Acids. Sci. China Chem 2017, 60, 188–200. [Google Scholar]
- 29.Mirts EN; Bhagi-Damodaran A; Lu Y Understanding and Modulating Metalloenzymes with Unnatural Amino Acids, Non-Native Metal Ions, and Non-Native Metallocofactors. Acc. Chem. Res 2019, 52, 935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seyedsayamdost MR; Reece SY; Nocera DG; Stubbe J Mono-, Di-, Tri-, and Tetra-Substituted Fluorotyrosines: New Probes for Enzymes That Use Tyrosyl Radicals in Catalysis. J. Am. Chem. Soc 2006, 128, 1569–1579. [DOI] [PubMed] [Google Scholar]
- 31.Seyedsayamdost MR; Stubbe J Site-Specific Replacement of Y356 with 3,4-Dihydroxyphenylalanine in the β2 Subunit of E. coli Ribonucleotide Reductase. J. Am. Chem. Soc 2006, 128, 2522–2523. [DOI] [PubMed] [Google Scholar]
- 32.Seyedsayamdost MR; Stubbe J Replacement of Y730 and Y731 in the Alpha2 Subunit of Escherichia coli Ribonucleotide Reductase with 3-Aminotyrosine Using an Evolved Suppressor tRNA/tRNA-Synthetase Pair. Methods Enzymol. 2009, 462, 45–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seyedsayamdost MR; Yee CS; Reece SY; Nocera DG; Stubbe J pH Rate Profiles of FnY356–R2s (n = 2, 3, 4) in Escherichia coli Ribonucleotide Reductase: Evidence that Y356 Is a Redox-Active Amino Acid along the Radical Propagation Pathway. J. Am. Chem. Soc 2006, 128, 1562–1568. [DOI] [PubMed] [Google Scholar]
- 34.Yokoyama K; Uhlin U; Stubbe J Site-Specific Incorporation of 3-Nitrotyrosine as a Probe of pKa Perturbation of Redox-Active Tyrosines in Ribonucleotide Reductase. J. Am. Chem. Soc 2010, 132, 8385–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Minnihan EC; Seyedsayamdost MR; Uhlin U; Stubbe J Kinetics of Radical Intermediate Formation and Deoxynucleotide Production in 3-Aminotyrosine-Substituted Escherichia coli Ribonucleotide Reductases. J. Am. Chem. Soc 2011, 133, 9430–9440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seyedsayamdost MR; Yee CS; Stubbe J Use of 2,3,5-F3Y-β2 and 3-NH2Y-α2 To Study Proton-Coupled Electron Transfer in Escherichia coli Ribonucleotide Reductase. Biochemistry 2011, 50, 1403–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gil AA; Haigney A; Laptenok SP; Brust R; Lukacs A; Iuliano JN; Jeng J; Melief EH; Zhao R-K; Yoon E; et al. Mechanism of the AppABLUF Photocycle Probed by Site-Specific Incorporation of Fluorotyrosine Residues: Effect of the Y21 pKa on the Forward and Reverse Ground-State Reactions. J. Am. Chem. Soc 2016, 138, 926–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gil AA; Laptenok SP; Iuliano JN; Lukacs A; Verma A; Hall CR; Yoon GE; Brust R; Greetham GM; Towrie M; et al. Photoactivation of the BLUF Protein PixD Probed by the Site-Specific Incorporation of Fluorotyrosine Residues. J. Am. Chem. Soc 2017, 139, 14638–14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rappaport F; Boussac A; Force DA; Peloquin J; Brynda M; Sugiura M; Un S; Britt RD; Diner BA robing the Coupling Between Proton and Electron Transfer in Photosystem II Core Complexes Containing a 3-Fluorotyrosine. J. Am. Chem. Soc 2009, 131, 4425–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ayyadurai N; Prabhu NS; Deepankumar K; Kim A; Lee SG; Yun H Biosynthetic Substitution of Tyrosine in Green Fluorescent Protein with its Surrogate Fluorotyrosine in Escherichia coli. Biotechnol. Lett 2011, 33, 2201–2207. [DOI] [PubMed] [Google Scholar]
- 41.Yee EF; Dzikovski B; Crane BR Tuning Radical Relay Residues by Proton Management Rescues Protein Electron Hopping. J. Am. Chem. Soc 2019, 141, 17571–17587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ashizawa R; Noguchi T Effects of Hydrogen Bonding Interactions on the Redox Potential and Molecular Vibrations of Plastoquinone as Studied using Density Functional Theory Calculations. Phys. Chem. Chem. Phys 2014, 16, 11864–11876. [DOI] [PubMed] [Google Scholar]
- 43.Blomberg MRA Active Site Midpoint Potentials in Different Cytochrome c Oxidase Families: A Computational Comparison. Biochemistry 2019, 58, 2028–2038. [DOI] [PubMed] [Google Scholar]
- 44.Cruzeiro VWD; Amaral MS; Roitberg AE Redox potential Replica Exchange Molecular Dynamics at Constant pH in AMBER: Implementation and Validation. J. Chem. Phys 2018, 149, 072338. [DOI] [PubMed] [Google Scholar]
- 45.Breuer M; Zarzycki P; Blumberger J; Rosso KM Thermodynamics of Electron Flow in the Bacterial Deca-heme Cytochrome MtrF. J. Am. Chem. Soc 2012, 134, 9868–9871. [DOI] [PubMed] [Google Scholar]
- 46.Li W; Baldus IB; Gräter F Redox Potentials of Protein Disulfide Bonds from Free-Energy Calculations. J. Phys. Chem. B 2015, 119, 5386–5391. [DOI] [PubMed] [Google Scholar]
- 47.Shen L; Zeng X; Hu H; Hu X; Yang W Accurate Quantum Mechanical/Molecular Mechanical Calculations of Reduction Potentials in Azurin Variants. J. Chem. Theory Comput 2018, 14, 4948–4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blumberger J Free Energies for Biological Electron Transfer from QM/MM Calculation: Method, Application and Critical Assessment. Phys. Chem. Chem. Phys 2008, 10, 5651–5667. [DOI] [PubMed] [Google Scholar]
- 49.Kamerlin SC; Haranczyk M; Warshel A Progress in Ab Initio QM/MM Free-Energy Simulations of Electrostatic Energies in Proteins: Accelerated QM/MM Studies of pKa, Redox Reactions and Solvation Free Energies. J. Phys. Chem. B 2009, 113, 1253–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Datta SN; Sudhamsu J; Pandey A Theoretical Determination of the Standard Reduction Potential of Plastocyanin in Vitro. J. Phys. Chem. B 2004, 108, 8007–8016. [Google Scholar]
- 51.Cruzeiro VWD; Feliciano GT; Roitberg AE Exploring Coupled Redox and pH Processes with a Force-Field-Based Approach: Applications to Five Different Systems. J. Am. Chem. Soc 2020, 142, 3823–3835. [DOI] [PubMed] [Google Scholar]
- 52.Tommos C; Skalicky JJ; Pilloud DL; Wand AJ; Dutton PL De Novo Proteins as Models of Radical Enzymes. Biochemistry 1999, 38, 9495–9507. [DOI] [PubMed] [Google Scholar]
- 53.Martínez-Rivera MC; Berry BW; Valentine KG; Westerlund K; Hay S; Tommos C Electrochemical and Structural Properties of a Protein System Designed To Generate Tyrosine Pourbaix Diagrams. J. Am. Chem. Soc 2011, 133, 17786–17795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berry BW; Martínez-Rivera MC; Tommos C Reversible Voltammograms and a Pourbaix Diagram for a Protein Tyrosine Radical. Proc. Natl. Acad. Sci 2012, 109, 9739–9743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ravichandran KR; Liang L; Stubbe J; Tommos C Formal Reduction Potential of 3,5-Difluorotyrosine in a Structured Protein: Insight into Multistep Radical Transfer. Biochemistry 2013, 52, 8907–8915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dixon WT; Murphy D Determination of the Acidity Constants of Some Phenol Radical Cations by Means of Electron Spin Resonance. J. Chem. Soc., Faraday Trans. 2 1976, 72, 1221–1230. [Google Scholar]
- 57.Ravichandran KR; Zong AB; Taguchi AT; Nocera DG; Stubbe J; Tommos C Formal Reduction Potentials of Difluorotyrosine and Trifluorotyrosine Protein Residues: Defining the Thermodynamics of Multistep Radical Transfer. J. Am. Chem. Soc 2017, 139, 2994–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Solis BH; Hammes-Schiffer S Proton-Coupled Electron Transfer in Molecular Electrocatalysis: Theoretical Methods and Design Principles. Inorg. Chem 2014, 53, 6427–6443. [DOI] [PubMed] [Google Scholar]
- 59.Marenich AV; Cramer CJ; Truhlar DG Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [DOI] [PubMed] [Google Scholar]
- 60.Becke AD Density - Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- 61.Lee C; Yang W; Parr RG Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B Condens. Matter 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- 62.Johnson ER; Becke AD A Post-Hartree-Fock Model of Intermolecular Interactions: Inclusion of Higher-Order Corrections. J. Chem. Phys 2006, 124, 174104. [DOI] [PubMed] [Google Scholar]
- 63.Clark T; Chandrasekhar J; Spitznagel GW; Schleyer PVR Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. III. The 3-21+G Basis Set For First-Row Elements, Li–F. J. Comput. Chem 1983, 4, 294–301. [Google Scholar]
- 64.Shao Y; Gan Z; Epifanovsky E; Gilbert ATB; Wormit M; Kussmann J; Lange AW; Behn A; Deng J; Feng X; et al. Advances in Molecular Quantum Chemistry Contained in the Q-Chem 4 Program Package. Mol. Phys 2015, 113, 184–215. [Google Scholar]
- 65.Götz AW; Clark MA; Walker RC An Extensible Interface for QM/MM Molecular Dynamics Simulations with AMBER. J Comput. Chem 2014, 35, 95–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Walker RC; Crowley MF; Case DA The Implementation of a Fast and Accurate QM/MM Potential Method in Amber. J. Comput. Chem 2008, 29, 1019–1031. [DOI] [PubMed] [Google Scholar]
- 67.König PH; Hoffmann M; Frauenheim T; Cui Q A Critical Evaluation of Different QM/MM Frontier Treatments with SCC-DFTB as the QM method. J. Phys. Chem. B 2005, 109, 9082–9095. [DOI] [PubMed] [Google Scholar]
- 68.Khoury GA; Thompson JP; Smadbeck J; Kieslich CA; Floudas CA Forcefield_PTM: Ab Initio Charge and AMBER Forcefield Parameters for Frequently Occurring Post-Translational Modifications. J. Chem. Theory Comput 2013, 9, 5653–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khoury GA; Smadbeck J; Tamamis P; Vandris AC; Kieslich CA; Floudas CA Forcefield_NCAA: Ab Initio Charge Parameters to Aid in the Discovery and Design of Therapeutic Proteins and Peptides with Unnatural Amino Acids and Their Application to Complement Inhibitors of the Compstatin Family. ACS Synth. Biol 2014, 3, 855–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Robalo JR; Huhmann S; Koksch B; Vila Verde A The Multiple Origins of the Hydrophobicity of Fluorinated Apolar Amino Acids. Chem 2017, 3, 881–897. [Google Scholar]
- 71.Case DA; Cheatham TE; Darden T; Gohlke H; Luo R; Merz KM Jr; Onufriev A; Simmerling C; Wang B; Woods RJ The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and Testing of a General Amber Force Field. J. Comput. Chem 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- 73.Cieplak P; Cornell WD; Bayly C; Kollman PA Application of the Multimolecule and Multiconformational RESP Methodology to Biopolymers: Charge Derivation for DNA, RNA, and Proteins. J. Comput. Chem 1995, 16, 1357–1377. [Google Scholar]
- 74.Hehre WJ; Ditchfield R; Pople JA Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys 1972, 56, 2257–2261. [Google Scholar]
- 75.Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vanommeslaeghe K; Hatcher E; Acharya C; Kundu S; Zhong S; Shim J; Darian E; Guvench O; Lopes P; Vorobyov I; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem 2010, 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reinhardt CR; Li P; Kang G; Stubbe J; Drennan CL; Hammes-Schiffer S Conformational Motions and Water Networks at the α/β Interface in E. coli Ribonucleotide Reductase. J. Am. Chem. Soc 2020, 142, 13768–13778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marsh EN Fluorinated Proteins: From Design and Synthesis to Structure and Stability. Acc. Chem. Res 2014, 47, 2878–2886. [DOI] [PubMed] [Google Scholar]
- 79.Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- 80.Case DA; Ben-Shalom IY; Brozell SR; Cerutti DS; T.E. Cheatham I; Cruzeiro VWD; Darden TA; Duke RE; Ghoreishi D; Gilson MK; et al. AMBER 2018, University of California, San Francisco: 2018. [Google Scholar]
- 81.Humphrey W; Dalke A; Schulten K VMD: Visual Molecular Dynamics. J. Mol. Graph 1996, 14, 33–38, 27-38. [DOI] [PubMed] [Google Scholar]
- 82.Roe DR; Cheatham TE PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput 2013, 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- 83.Chai J-D; Head-Gordon M Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys 2008, 10, 6615–6620. [DOI] [PubMed] [Google Scholar]
- 84.Reedy CJ; Gibney BR Heme Protein Assemblies. Chem. Rev 2004, 104, 617–650. [DOI] [PubMed] [Google Scholar]
- 85.Yanagisawa S; Banfield MJ; Dennison C The Role of Hydrogen Bonding at the Active Site of a Cupredoxin: The Phe114Pro Azurin Variant. Biochemistry 2006, 45, 8812–8822. [DOI] [PubMed] [Google Scholar]
- 86.Ishikita H; Loll B; Biesiadka J; Saenger W; Knapp E-W Redox Potentials of Chlorophylls in the Photosystem II Reaction Center. Biochemistry 2005, 44, 4118–4124. [DOI] [PubMed] [Google Scholar]
- 87.Ishikita H; Knapp EW Function of Redox-Active Tyrosine in Photosystem II. Biophys. J 2006, 90, 3886–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Scheiner S; Kar T; Pattanayak J Comparison of Various Types of Hydrogen Bonds Involving Aromatic Amino Acids. J. Am. Chem. Soc 2002, 124, 13257–13264. [DOI] [PubMed] [Google Scholar]
- 89.Crespo-Otero R; Bravo-Rodriguez K; Roy S; Benighaus T; Thiel W; Sander W; Sánchez-García E Interactions of Aromatic Radicals with Water. ChemPhysChem 2013, 14, 805–811. [DOI] [PubMed] [Google Scholar]
- 90.Sander W; Roy S; Polyak I; Ramirez-Anguita JM; Sanchez-Garcia E The Phenoxyl Radical–Water Complex—A Matrix Isolation and Computational Study. J. Am. Chem. Soc 2012, 134, 8222–8230. [DOI] [PubMed] [Google Scholar]
- 91.Chung LW; Sameera WMC; Ramozzi R; Page AJ; Hatanaka M; Petrova GP; Harris TV; Li X; Ke Z; Liu F; et al. The ONIOM Method and Its Applications. Chem. Rev 2015, 115, 5678–5796. [DOI] [PubMed] [Google Scholar]
- 92.Friesner RA; Guallar V Ab Initio Quantum Chemical and Mixed Quantum Mechanics/Molecular Mechanics (QM/MM) Methods for Studying Enzymatic Catalysis. Annu. Rev. Phys. Chem 2005, 56, 389–427. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.