

Abstract
This method works by partitioning the envelope of Gram-negative bacteria into total, inner, and outer-membrane (OM) fractions and concludes with assays to assess the purity of the bilayers. The OM has an increased overall density compared to the inner membrane, largely due to the presence of lipooligosaccharides (LOS) and lipopolysaccharides (LPS) within the outer leaflet. LOS and LPS molecules are amphipathic glycolipids that have a similar structure, which consists of a lipid-A disaccharolipid and core-oligosaccharide substituent. However, only LPS molecules are decorated with a third subunit known as the O-polysaccharide, or O-antigen. The type and amount of glycolipids present will impact an organism’s OM density. Therefore, we tested whether the membranes of bacteria with varied glycolipid content could be similarly isolated using our technique. For the LPS-producing organisms, Salmonella enterica serovar Typhimurium and Escherichia coli, the membranes were easily isolated and the LPS O-antigen moiety did not impact bilayer partitioning. Acinetobacter baumannii produces LOS molecules, which have a similar mass to O-antigen deficient LPS molecules; however, the membranes of these microbes could not initially be separated. We reasoned that the OM of A. baumannii was less dense than that of Enterobacteriaceae, so the sucrose gradient was adjusted and the membranes were isolated. The technique can therefore be adapted and modified for use with other organisms.
Keywords: membrane, inner membrane, outer membrane, lipopolysaccharide, lipooligosaccharide, capsule, bilayer, peptidoglycan, glycerophospholipid, Osborn-Munson, microbiology, bacteriology, biochemistry, chemistry, lipid, lipidomics, ISSUE #, isopycnic gradient
SUMMARY:
Gram-negative bacteria produce two spatially segregated membranes. The outer membrane is partitioned from the inner membrane by a periplasm and a peptidoglycan layer. The ability to isolate the dual bilayers of these microbes has been critical for understanding their physiology and pathogenesis.
INTRODUCTION:
Gram-negative bacteria produce two membranes that are separated by a periplasmic space and a peptidoglycan cell wall1. The inner membrane (IM) encases the cytosol and is a symmetric bilayer of phospholipids. Peptidoglycan protects against turgor pressure and provides the bacterium with a cell shape, and is attached to the outer membrane (OM) by lipoproteins2,3. The OM surrounds the periplasm and is predominantly asymmetric. The inner leaflet consists of phospholipids and the outer leaflet consists of glycolipids known as lipooligosaccharides (LOS) or lipopolysaccharides (LPS)4,5. The lipid asymmetry and the biochemistry of the LOS/ LPS molecules in the outer leaflet confer barrier properties to the cell surface that protect the bacterium against hazards in its environment6,7.
LPS molecules are comprised of three constituents: the lipid A disaccharolipid, the core oligosaccharide, and the O-polysaccharide or O-antigen. Lipid A is a multiply acylated disaccharolipid. Core-oligosaccharides consist of 10–15 sugars known as rough LPS or R-LPS. The core is subdivided into the inner region, composed of 2-keto-3-deoxy-D-manno-octulosonic acid (kdo) and one or more heptose residues, and an outer region that consists generally of hexoses (glucose or galactose) and heptoses, or acetamido sugars5. The outer core region is more variable in its components and structure than the inner core. In Salmonella spp., only one core structure has been described; however, in Escherichia coli there are five different core structures (designated K-12, R1, R2, R3, and R4)8. E. coli K-12 DH5α, which we use in this procedure carries a mutation that results in production R-LPS9. The R-LPS molecules lack the O-antigen moiety and have a similar molecular weight to LOS molecules.
The addition of O-antigen to R-LPS turns this molecule into smooth LPS, or S-LPS. The O-antigens are built from short 3–4 carbohydrate subunits and consist of multiple modalities with varying chain lengths10. Some LPS-producing bacteria, like Salmonella enterica serovar Typhimurium (S. Typhimurium), display a trimodal distribution of LPS molecules on their surface(10, 11). Very-long chain O-antigens can contain over one hundred subunits and weigh over one hundred kilodaltons. The O-antigens provide surface properties to the bacterium that are necessary to resist antibiotics, evade predation by bacteriophages, and cause disease.
Species of Campylobacter, Bordetella, Acinetobacter, Haemophilus, Neisseria and others generate LOS molecules instead of LPS molecules on their surface (12). LOS molecules consist of lipid A and core oligosaccharide, but lack the O-antigen. These types of Gram-negative bacteria modify their core oligosaccharides with additional sugars and combinations of sugars to alter surface properties (12). Both LOS and LPS-producing microbes derivatize the phosphates on lipid A and core molecules with cationic moieties7. These additions include phosphoethanolamine, galactosamine and aminoarabinose substitutions, which function by neutralizing anionic surface charge and thereby protecting against cationic antimicrobial peptides. Gram-negative bacteria also modify the core oligosaccharide structure with variable non-stoichiometric substitutions of sugars, or extra kdo molecules, and alter the number of acyl chains on lipid-A disaccharolipids7.
The ability to isolate the IM from the OM of Gram-negative bacteria has been instrumental for understanding the role of the cell envelope in antimicrobial resistance and disease pathogenesis11,12. Derivations of this approach have been used to deduce mechanisms of assembly, maintenance, and remodeling of the protein, phospholipid, and glycolipid constituents for the OM.
Our lab routinely performs bacterial lipidomic analyses to study protein-mediated lipid regulation and lipid function in a variety of Gram-negative species. The volumes used in the protocol reflect the routine use of this procedure to analyze non-radiolabeled phospholipids by thin-layer chromatography and liquid-chromatography tandem mass spectrometry13,14.
The protocol begins by exposing a chilled suspension of Gram-negative bacteria to a high osmolar solution of sucrose and adding lysozyme to dissociate the OM from the underlying peptidoglycan layer (Fig. 1)12. EDTA is then added to facilitate penetration of the lysozyme, since divalent cation sequestration disrupts the lateral electrostatic bridging interactions between adjacent LOS/LPS molecules15. The original protocol from which ours has been adapted required the formation of spheroplasts, a Gram-negative bacterial cell form that consist of a plasma membrane and cytosol, but lacks the peptidoglycan layer and an OM. It is possible that spheroplasts are produced by our adapted method; however, the technique does not rely or intend on their formation for success. Instead, the lysozyme-EDTA treated bacteria are rapidly harvested by centrifugation and re-suspended in a sucrose solution of lesser concentration before pressurized lysis. The OMs that might have been released by forming spheroplasts should in theory be harvestable from the supernatants of the treated cells, but this approach is not detailed herein. Ultimately, the treated cells are subjected to conventional homogenization and lysis, which enhances the efficiency and reproducibility of the membrane separation procedure16.
Figure 1. A schematic depicting the Gram-negative bacterial membrane isolation procedure described in this methods article.

Shown is the procedure used for collecting bacterial cells and isolating the total, inner (IM), and outer membranes (OM). The approach relies upon the increased density of the OM bilayer for these microbes compared to density of the IM bilayer.
After lysis, the total membranes are collected by ultracentrifugation and applied to a discontinuous sucrose-density gradient to fractionate the IMs and OMs. The classical approach uses a more continuous gradient that consists of at least five different sucrose solutions11,12. The discontinuous gradient in our protocol consists of three sucrose solutions and partitions the bilayers into two distinct fractions17. The LOS and LPS molecules within the OMs of Gram-negative bacteria drive the envelope to partition into an upper brown low-density IM fraction and a lower white high-density OM fraction (Figs. 1 and 2).
Figure 2. Representative results for different Gram-negative species whose membranes were isolated using the standard and the modified sucrose density gradients described in this article.

Images of discontinuous sucrose density gradients post isopycnic centrifugation for A. wild-type Salmonella enterica serovar Typhimurium 14028s, B. galE-mutant S. Typhimurium LT2, which produces LPS molecules that are devoid of O-antigens, and C. Escherichia coli K-12 DH5α, which also produces LPS molecules that lack O-antigens. The inner membrane (IM) is separated from the outer membrane (OM) and localizes to the 20–53% sucrose interface as a brown material. The white OM layer localizes to the 53–73% sucrose interface due to the higher density of this fraction. D. The total membranes of the wild-type Acinetobacter baumanii 17978 did not separate using 20%/53%/73% w/v sucrose gradient, E. but did separate using the 20%/45%/73% w/v sucrose gradient.
Acinetobacter baumannii are important multidrug resistant human pathogens that produce LOS molecules in their OM and erect a cell envelope that is difficult to separate18,19. Recent work suggests that a derivation of the protocol we present here can be used to partition the bilayers of these organisms20. Therefore, we tested our protocol on A. baumannii 17978. Initially, the procedure was inadequate. However, we modified the sucrose concentration of the middle density solution and greatly improved separation (Fig. 2). An NADH dehydrogenase assay and a LOS/LPS extraction and detection procedure was used to confirm separation for A. baumannii, wild-type S. Typhimurium and two O-antigen deficient enterobacterial genotypes; namely, galE-mutant S. Typhimurium and a laboratory strain, E. coli DH5α (Fig. 3 and 4).
Figure 3. Representative results for the NADH dehydrogenase assay to test outer membrane (OM) purity.
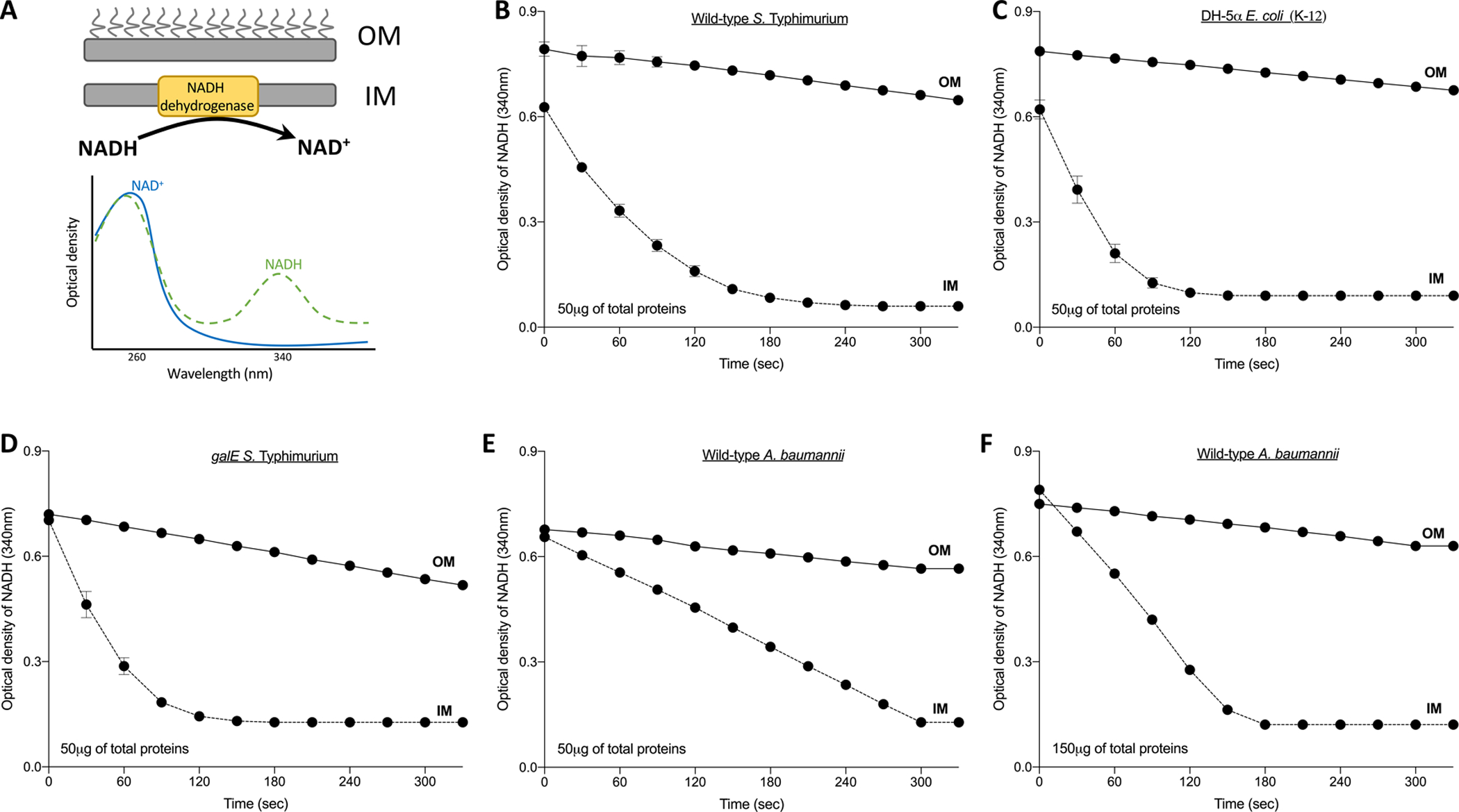
The presence of the enzyme, NADH-dehydrogenase, was tested in inner (IM) and OM samples to test the efficiency of the separation. A. The oxidation of NADH to NAD is catalyzed by an enzyme located in the bacterial IM. The reaction substrate (NADH) has a maximum absorbance at 340 nm; therefore, a decrease in optical density at this wavelength is indicative of the presence of the enzyme in the sample. The IMs and OMs were measured for B. wild-type S. Typhimurium, C. E. coli K-12 DH5α and D. galE-mutant S. Typhimurium. These membranes were isolated using an isopycnic sucrose density gradient of 20%/53%/73% w/v sucrose. For A. baumannii, the membranes were isolated using a gradient of 20%/45%/73% w/v sucrose. The NADH assay to test the purity of membranes was done using E. 50 μg for the enterobacterial organisms and F. 150μg of total proteins for A. baumannii. A higher concentration of protein was added in F., since the curve for E. suggested that the relative levels of NADH dehydrogenase compared to total protein were less for A. baumannii than for S. Typhimurium and E. coli.
Figure 4. Representative results for the LPS and LOS extraction and visualization procedure to test inner membrane (IM) purity.

The volume of membrane sample corresponding to 50 μg of total protein was used to extract LPS from the IM and outer membrane (OM) of S. Typhimurium and E. coli DH5-alpha. The volume of membrane sample corresponding to 300 μg of total protein was used to extract LPS from the membranes of A. baumannii. The volumes were normalized to 100 μl with endotoxin-free water and treated with Proteinase K. LPS or LOS were extracted by hot-phenol extraction (1:1 water:phenol) and 21 μl of the extracts were loaded onto a 4–20% gradient polyacrylamide gel and visualized by PRO-Q Emerald 300 staining to assess the cross contamination of IM fractions with OM materials.
The intent of this work is to supply a streamlined approach for reproducibly isolating the membranes of Gram-negative bacteria. The protocol can be used to study many types of membrane-associated molecules for these microbes.
PROTOCOL:
1. GENERAL REAGENTS AND MEDIA PREPARATION FOR MEMBRANE EXTRACTION
1.1 Bacterial growth media: Prepare and sterilize 1 L of broth media in a thoroughly cleaned and autoclaved 2 L flask.
1.2. General resuspension buffer (1 M Tris Buffer pH 7.5; 50 mL): Dissolve 6.05 g of Tris base in 30 mL of H2O. Adjust pH to 7.5 with 5 M HCl. Adjust final volume to 50 mL with ultrapure H2O.
1.3. Master stock of divalent cation chelation solution (0.5 M EDTA pH 8; 100 mL): Add 18.6 g of disodium ethylene tetraacetate·2H2O to 80 mL of H2O. Stir vigorously and adjust pH to 8.0 with NaOH. Adjust final volume to 100 mL with ultrapure H2O.
Note: The disodium salt of EDTA will not dissolve until the pH of the solution is adjusted to ~8.0 by the addition of NaOH.
1.4. Osmotic buffer A (0.5 M sucrose, 10 mM Tris pH 7.5; 1 L): Weigh 171.15 g of sucrose and transfer to a 1 L cylinder. Add 10 mL of 1 M Tris pH 7.5. Adjust to a final volume of 1 L with ultrapure H2O. Store at 4 °C.
1.5. Lysozyme (10mg/mL; 5 mL): Weigh 50 mg of (chicken egg-white) lysozyme and dissolve in 5 mL ultrapure H2O. Store at 4 °C.
1.6. Diluted divalent cation chelation solution (1.5 mM EDTA; 500 mL): Add 1.5 mL of 0.5 M EDTA (Step 1.3) to 497.5 mL ultrapure H2O. Store at 4 °C.
1.7. Osmotic buffer B (0.2 M sucrose, 10 mM Tris pH 7.5; 2 L): Weigh 136.8 g of sucrose and transfer to a 2 L cylinder. Add 20 mL of 1 M Tris pH 7.5. Adjust final volume to 2 L with ultrapure H2O. Store at 4 °C.
1.8. Nuclease co-factor (1 M MgCl2; 10 mL): Dissolve 2.03 g of MgCl2·6H2O in 8 mL of ultrapure H2O. Adjust volume to 10 mL. Store at room temperature.
1.9. Nuclease solution cocktail including RNase and DNase enzymes (see table of reagents). Store at −20 °C.
TABLE OF MATERIALS:
| Name | Company | Catalog Number | Comments |
|---|---|---|---|
| 1 L Centrifuge bottles, PC/PPCO, super speed, with sealing cap, Nalgene | VWR | 525–0466 | |
| 1 L Pyrex Media Storage Bottle with High Temperature Cap | VWR | 10416–312 | |
| 2,000 mL Erlenmeyer Flask, Narrow Mouth | VWR | 10545–844 | |
| 4–20% mini PROTEAN Precast Protein Gels, 12 well | BIORAD | 4561095 | |
| 4x Laemmli Sample Buffer 10 mL | BIORAD | 1610747 | |
| 50 mL sterile polypropylene centrifuge tubes | VWR | 89049–174 | |
| 7 mL Dounce Tissue Homogenizer with Two Glass Pestles | VWR | 71000–518 | |
| 70 mL polycarbonate bottle assembly | Beckman Coulter Life Sciences | 355622 | |
| Agar Powder | VWR | A10752 | |
| B10P Benchtop pH Meter with pH Probe | VWR | 89231–664 | |
| Barnstead GenPure xCAD Plus UV/UF - TOC (bench version) | ThermoFisher Scientific | 50136146 | |
| Benzonase Nuclease | MilliporeSigma | 70746–3 | |
| EDTA disodium salt dehydrate 99.0–101.0%, crystals, ultrapure Bioreagent Molecular biology grade, J.T. Baker | VWR | 4040–01 | |
| EmulsiFlex-C3 | Avestin, Inc. | ||
| Fiberlite F13–14 × 50cy Fixed Angle Rotor | ThermoFisher Scientific | 75006526 | |
| Hydrochloric acid 6.0 N | VWR | BDH7204 | |
| IBI Scientific Orbital Platform Shaker | Fischer Scientific | 15-453-211 | |
| LB Broth Miller | VWR | 214906 | |
| Lysozyme, Egg White, Ultra Pure Grade | VWR | VWRV0063 | |
| Magnesium chloride hexahydrate | Sigma Aldrich | M2670 | |
| NADH | Sigma Aldrich | 10107735001 | |
| Optima XPN-80 - IVD | Beckman Coulter Life Sciences | A99839 | |
| Pharmco Products PURE ALCOHOL 200 PROOF GL 4/CS | Fischer Scientific | NC1624582 | |
| Phenol Solution, Equilibrated with 10 mM Tris HCI, pH 8.0, 1 mM EDTA, BioReagent, for molecular biology | Sigma - Millipore | P4557 | |
| Pierce Coomassie Plus (Bradford) Assay Kit | Thermofisher | 23238 | |
| Pro-Q emerald 300 Lipopolysaccharide Gel Stain Kit | Thermofisher | P20495 | |
| Proteacease −50, EDTA free | G Biosciences | 786–334 | |
| Proteinase K, Molecular Biology Grade | New England Biolabs | P8107S | |
| Sodium hydroxide >99.99% | VWR | AA45780–22 | |
| Sorval RC 6 Plus Centrifuge | ThermoFisher Scientific | 36-101-0816 | |
| Sucrose | MilliporeSigma | SX1075–3 | |
| SW 41 Ti Swinging-Bucket Rotor | Beckman Coulter Life Sciences | 331362 | |
| Tris(hydroxymethyl)aminomethane (TRIS, Trometamol) ≥99.9% (dried basis), ultrapure Bioreagent Molecular biology grade, J.T. Baker | VWR | JT4109–6 | |
| Type 45 Ti Fixed-Angle Titanium Rotor | Beckman Coulter Life Sciences | 339160 | |
| Ultra-Clear Tube, 14 × 89mm | Beckman Coulter Life Sciences | 344059 | |
| Vortex-Genie 2 | VWR | 102091–234 |
1.10. Protease inhibitor cocktail (see table of reagents). Store at 4 °C.
1.11. Low-density isopycnic sucrose gradient solution (20% w/v sucrose, 1 mM EDTA, 1 mM Tris pH 7.5 Solution; 100 mL): Weigh 20 g of sucrose and transfer to a 200 mL cylinder. Add 100 μL of 1 M Tris Buffer pH 7.5 and 200 μL of 0.5 M EDTA pH 8. Adjust final volume to 100 mL with ultrapure H2O. Store at room temperature.
1.12. Medium-density isopycnic sucrose gradient solution (53 % w/v sucrose, 1 mM EDTA, 1mM Tris pH 7.5 Solution; 100 mL): Weigh 53 g of sucrose and transfer to a 200 mL graduated cylinder. Add 100 μL of 1 M Tris Buffer pH 7.5 and 200 μL of 0.5 M EDTA pH 8. Adjust final volume to 100 mL with ultrapure H2O. Store at room temperature.
NOTE: it is very important to prepare this solution in a graduated cylinder to ensure accuracy due to the high percentage of sucrose. Add a magnetic stir bar and stir until the sucrose is completely dissolved in solution. This process may take several hours.
1.13. High-density isopycnic sucrose gradient solution (73 % w/v sucrose, 1 mM EDTA, 1 mM Tris pH 7.5 Solution; 100 mL): Weigh 73 g of sucrose and transfer to a 200 mL graduated cylinder. Add 100 μL of 1 M Tris Buffer pH 7.5 and 200 μL of 0.5 M EDTA pH 8. Adjust final volume to 100 mL with ultrapure H2O. Store at room temperature.
Note: it is very important to prepare this solution in a graduated cylinder to ensure accuracy due to the high percentage of sucrose. Add a magnetic stir bar and stir until the sucrose is completely dissolved. This process may take several hours.
1.14. Isolated-membrane-storage buffer (10 mM Tris Buffer pH 7.5; 1 L): Add 1 mL of 1 M Tris Buffer pH 7.5 to a 1 L flask and adjust the final volume to 1 L with ultrapure H2O.
1.15 β-Nicotinamide adenine dinucleotide (NADH) (10 mg/mL solution). Resuspend 10 mg of NADH in ultrapure H2O. Prepare fresh stocks, weekly. Store at −20 °C.
1.16 Phenol solution. Equilibrated with 10 mM Tris HCl, pH 8.0, 1 mM EDTA, for molecular biology (see table of reagents). Store the solution at 4 °C.
1.17 Pro-Q™ Emerald 300 Lipopolysaccharide Gel Stain Kit (see table of reagents).
1.18. Bradford Reagent (see table of reagents)
Note: All solutions and media should be prepared within the week of performing the assay to ensure consistent results.
2. PREPARATION OF BACTERIA FOR MEMBRANE EXTRACTION
2.1. Streak the bacteria from frozen glycerol stocks onto fresh agar plates. Store the plates at 4 °C once colonies develop. Inoculate a single colony into a 5 mL tube filled with broth media and culture the bacteria as desired overnight.
2.2. Back-dilute the overnight bacterial culture into 1 L of preferred broth media and culture the bacteria until the desired optical density is achieved.
Note: Inoculating a single bacterial colony into 1 L of broth media is recommended for mutant genotypes that are prone to suppressing growth phenotypes, but some Gram-negative bacteria simply grow slower than others. If it is not possible to achieve a sufficient culture density by single-colony inoculation, back diluting an overnight culture into 1 L of media is one strategy to synchronize growth. Bacterial-membrane composition varies depending upon the growth phase of the culture (logarithmic vs stationary phase)13. Growth curves measuring the change in optical density for the bacterial cultures as a function of time should be performed with all strains to correlate culture density with growth phase.
2.3. Set the flasks containing the broth cultures on ice. Read the optical density at 600 nm (OD600) and calculate the volume of culture that is equivalent to between 6.0 and 8.0×1011 bacterial colony forming units (cfu). For S. Typhimurium, this corresponds to 1 L of culture at an OD600 of between 0.6–0.8, since an OD600 of 1.0 is equal to roughly 1.0×109 cfu/mL. Add this volume to a centrifuge tube and ensure that the remaining cultures stay on ice until they are to be used.
2.4. Pellet the bacteria by centrifugation at 4 °C at 7,000–10,000 × g in a fixed angle high-speed centrifuge for 10 minutes.
Note: Pre-cool and maintain the centrifuges at a low temperature. Maintain the samples on ice during the entire procedure.
2.5 Decant and discard the supernatant carefully.
Note: The pellet can be flash frozen and/or stored at −80 °C if the membrane fractions are not going to be extracted immediately. However, it is recommended to proceed directly with plasmolysis on the same day the cells are harvested, and is especially recommended for non-enterobacterial species.
3. DISSOCIATION OF THE OUTER MEMBRANE AND PLASMOLYSIS.
3.1 Thaw the cell pellets on ice if previously stored at −80 °C and retain the samples on ice for the remainder of the procedure. Resuspend each cell pellet within the centrifuge tube in 12.5 mL of buffer A. Add a magnetic stir bar to the suspension of cells.
3.2. Add 180 μl of 10 mg/mL lysozyme (final concentration of 144 μg/mL) to each cell resuspension. Keep the samples on ice while stirring for 2 min.
3.3. Add 12.5 mL of 1.5 mM EDTA solution to each cell resuspension and continue stirring on ice for an additional 7 min.
3.4. Decant the suspension into a 50 mL conical tube and centrifuge at 9000–11000 × g for 10 min at 4 °C.
3.5. Discard supernatants into a biohazard waste container and retain the pellets on ice.
3.6. Add 25 mL of buffer B to the cell pellet
3.7. Add 55 μl of 1 M MgCl2, 1 μl of RNase/DNase nuclease reagent (to avoid viscosity problems associated with bacteria undergoing plasmolysis prior to homogenization), and 1 μl of protease inhibitor cocktail to the volume of buffer B that sits atop the cell pellet
3.8. Resuspend the pellet in the buffer B mixture. Vigorously pipet and vortex until observing a homogenous solution.
Note: It is very important to have a homogenous solution before proceeding to Step 4 of this protocol. The resuspended cells should have a viscous cake-batter like appearance and consistency.
3.9. Vortex each sample for 15 seconds. Retain pellets on ice and proceed to Step 4.
4. PRESSURIZED HOMOGENIZATION AND LYSIS
Note: Several methods can be used for lysis. Sonication is not ideal due to the generation of heat. Osmotic lysis can be achieved, but is often inefficient. Therefore, we recommend high-pressure lysis. High pressure lysis can be achieved using a variety of instruments. We suggest homogenization machines, such as the French Press or the Emusliflex. We work with many types of Gram-negative bacteria whose response to high osmolar sucrose solutions varies. The high-pressure homogenization step improves efficiency, reproducibility, and yield.
4.1 Prechill the French Press cell at 4 °C or insert the metal coil from the Emulsiflex machine on ice.
4.2 Pour the sample into the French-pressure cell or the Emulsiflex-sample cylinder and bring the cell under the desired homogenization pressure (10,000 psi should be adequate when using a French Press or 20,000 psi when using an Emulsiflex).
4.3 Adjust the outlet flow rate to approximately one drop per second while maintaining the pressure if utilizing a French Press.
4.4 Collect the cell lysate in 50 mL conical tubes while keeping samples on ice.
4.5 Repeat steps 4.2–4.4 three to five times to achieve complete lysis, typically indicated by gradual increase in transparency of sample.
Note: The sample chamber should be washed and equilibrated with Buffer B in between samples.
4.6 Keep lysed cells on ice.
5. TOTAL MEMBRANE FRACTIONATION
5.1. Centrifuge the lysed bacterial samples at 6169 × g for 10 min at 4 °C to pellet remaining intact cell material. (e.g. unlysed bacterial cells)
5.2. Distribute the remaining portion of the supernatant which now contains the homogenized membranes into a polycarbonate bottle for ultracentrifugation.
CAUTION: If needed, cell samples can be balanced by diluting with buffer B.
5.3. Ultracentrifuge the cell lysates at 184,500 × g for at least 1 h, at 4 °C. This step can be performed overnight without affecting the quality of the membranes.
5.4. Discard remaining supernatant present in the ultracentrifuge tube and retain membrane pellets on ice. (Fig. 1)
5.5 Resuspend the membrane pellets in 1 mL of the low-density isopycnic-sucrose gradient solution using a glass-dounce homogenizer. Use a glass-Pasteur pipette to transfer the sample homogenate to a 1.5 mL microcentrifuge tube and retain on ice.
NOTE: If only total membrane composition analysis is desired, substitute 1 mL of low-density isopycnic-sucrose gradient solution for 1 mL of isolated-membrane storage buffer. Step 5.5 is the endpoint of isolation if only total-bacterial membrane samples are desired. Store samples at −20 °C until further downstream analysis is required.
6. DENSITY GRADIENT ULTRACENTRIFUGATION TO SEPARATE THE DUAL MEMBRANES
6.1 Gather the appropriate number of 13 mL polypropylene or ultra-clear open-top tubes specified for a swinging bucket rotor and ultracentrifuge.
6.2. Hold the tube in a slightly tilted position and prepare the sucrose gradient by slowly adding sucrose solutions from higher density to lower density in the following order:
- 2 mL of 73% w/v sucrose, 1 mM EDTA, 1 mM Tris pH 7.5
- 4 mL of 53% w/v sucrose, 1 mM EDTA, 1 mM Tris pH 7.5
- Next, add the total membrane fraction (1 mL), which has been resuspended in the 20% w/v sucrose solution (step 5.5). Avoid mixing the membrane fraction with the sucrose solution that lies beneath it. Divisions should be visible between each of these layers. Finally, fill the tube with the low-density isopycnic-sucrose gradient solution (approx. 6 mL). Polypropylene and ultra-clear open-top tubes should be filled as full as possible (2 or 3 mm from the tube top) for tube support.
Adjustment for Acinetobacter baumannii 17978
An adjusted sucrose gradient can be adapted for use with different bacterial specimens. For A. baumannii, the following sucrose gradient afforded more complete separation of the membranes (Fig. 2).
- 2 mL of 73% w/v sucrose, 1 mM EDTA, 1 mM Tris pH 7.5
- 4 mL of 45% w/v sucrose, 1 mM EDTA, 1 mM Tris pH 7.5
- Next, add the total membrane fraction (1 mL), which has been resuspended in the 20% w/v sucrose solution (step 5.5). Avoid mixing the membrane fraction with the sucrose solution that lies beneath it. Divisions should be visible between each of these layers. Finally, fill the tube with low-density isopycnic-sucrose gradient solution (approx. 6 mL). Polypropylene and ultra-clear open-top tubes should be filled as full as possible (2 or 3 mm from the tube top) for tube support.
6.3. Ultracentrifuge the samples using a swinging-bucket rotor at 288,000 × g and 4 °C overnight.
Note: For the volumes used in the previous steps we recommend 16 h< centrifugation time < 23 h.
HARVESTING AND WASHING THE MEMBRANES TO REMOVE THE SUCROSE
6.4. Cut the end of a P1000 pipette tip about 5 mm from the point. Remove the upper-brown IM layer using the pipette. Transfer the IM fraction into a polycarbonate bottle for ultracentrifugation.
6.5. Leave about 2 mL of the sucrose solution above the 53–73% interface to ensure that the lower white OM is not cross contaminated with the IM fraction. Repeat the pipetting procedure from step 6.4 for the OM fraction (Fig. 1).
Note: the membranes can also be collected by puncturing the centrifuge tubes at the bottom using a needle and collecting the membranes as fractions dropwise.
6.6. Fill the remaining void of the ultracentrifuge tube with isolated-membrane storage buffer, and mix by inversion or pipetting. Retain the samples on ice.
6.7. Collect the now washed and isolated membranes by ultracentrifugation at 184500 × g for 1 h and 4 °C.
6.8. Discard the supernatant and resuspend the membranes by dounce-homogenization. Add 500–1000 μl of storage buffer. Collect samples in 2 mL microcentrifuge tubes.
6.9. Store the bacterial membrane samples at −20 °C.
7. CONFIRMING SEPARATION OF THE BILAYERS
Note: Incomplete separation of the bilayers can occur due to technical error or the unique cell envelope composition of some species. To confirm separation, we advise using two independent assays to quantify the degree of cross contamination between the bilayers. The first assay detects the enzymatic activity of NADH dehydrogenase, which exists exclusively in the IM. The second assay detects the presence of LOS or LPS, which predominantly exists in the OM.
Confirming that the OMs are isolated from the IMs
NADH oxidation assay
7.1 Measure the protein concentration in each isolated membrane fraction using a Bradford protein assay.
7.2. Add the volume of sample corresponding to between 50 and 500 μg of protein to an empty 2 mL microcentrifuge tube. The concentration will vary depending upon the species, but 50 μg is typically sufficient for Enterobacteriaceae. Add the appropriate volume of 10 mM Tris-buffer to achieve a total volume of 990 μl. Transfer the content to a cuvette for spectrophotometry.
7.3. Add 10 μL of a 10 mg/mL solution of NADH to the sample and measure the initial absorbance at 340 nm.
7.4. Continue measuring the absorbance every 30 seconds for 5 minutes.
7.5. Compile the data and graph the change in Absorbance (y-axis) vs the change in Time (x-axis) for each membrane fraction (Fig. 3).
Confirming that the IMs are isolated from the OMs
LPS/LOS extraction and visualization in a gel.
7.5. Add the volume of sample corresponding to between 50 and 500 μg of protein to an empty 2 mL microcentrifuge tube. The concentration will vary depending upon the bacterial species, but 50 μg is typically sufficient for Enterobacteriaceae. Fill to a final volume of 100 μL with phosphate buffered saline, which is hereafter referred to as the aqueous phase.
7.7. Add 5 μL of Proteinase K (stock 800 U/ml) to the aqueous phase and incubate overnight at 59° C.
7.8. Warm a 10 mL aliquot of Tris-saturated Phenol for 10 min at 68° C.
7.9. Spin down the aqueous phase samples that were treated with the Proteinase K and add the “hot” Tris-saturated Phenol at a 1:1 ratio with the aqueous phase, vortex vigorously and incubate at 68° C for 10 min.
7.10. Transfer the now milky-white samples from 68° C to an ice-water bath and incubate for 10 min.
7.11. Centrifuge the samples at 2100 × g for 10 min at 4° C.
7.12. Transfer the upper aqueous phase to a new tube and store the tube at −20° C if the sample is not to be used immediately.
7.13. Dilute samples in SDS-loading buffer and load the wells of a 4–20 % Tris-glycine gradient gel.
7.14. Electrophorese for 45 min or until the dye-front is at the bottom of the gel.
7.15. Stain the gel using the PRO-Q Emerald 300 LPS staining kit following manufacturer’s instructions.
REPRESENTATIVE RESULTS:
This technique provides an effective means to isolate the IMs and OMs for Gram-negative bacteria. An outline of the entire procedure is illustrated (Fig. 1). In general, the normalization of cultures to an OD600 of 0.6–0.8 in 1 L of media, or harvesting between 6.0 and 8.0×1011 bacterial cells will ensure that the appropriate amount of membrane material is collected for subsequent separation.
Upon lysing the bacteria and ultracentrifuging the lysate, a sticky brown total membrane pellet will be visible at the bottom of the ultracentrifuge tube. After scraping, dounce-homogenizing, and ultracentrifuging the membrane over the discontinuous sucrose density gradient, the IM and OM should be separated as depicted (Fig. 2). We found that the 20%/53%/73% w/v sucrose-density gradient was insufficient to partition the envelope of A. baumannii, while a 20%/45%/73% w/v gradient was sufficient (Fig. 2).
Various analytical methods can be used to assess the quality and purity of each membrane fraction. NADH-dehydrogenase is an inner membrane enzyme that catalyzes NADH oxidation to NAD (Fig. 3). Given its cellular localization, it can be used to determine cross contamination between IMs and OMs. According to the absorbance spectra of both molecules, NAD and NADH each have a peak absorbance at 260 nm, while only NADH has a maximum absorbance at 340 nm. Thus, a decrease in the absorbance at 340 nm would be indicative of oxidation of NADH to NAD and therefore the presence of the enzyme in the sample. If the membranes separate properly, this change in absorbance should only occur in IM samples (Fig. 3). A decrease in absorbance in OM samples would indicate cross contamination with IM materials. For A. baumannii, three times the amount of membrane, or 150 μg of protein equivalents, was needed to demonstrate similar levels of NADH dehydrogenase activity to what was measured for the enterobacterial strains (Fig. 3). Therefore, it is possible that the levels of NADH oxidase are lower for A. baumannii or that the specific activity of the enzyme is decreased.
The outer leaflet of the OM is mainly composed of LOS or LPS molecules. Therefore, the extraction of LPS / LOS from the IM and OM samples with subsequent electrophoresis and visualization by PRO-Q Emerald 300 staining will reflect the enrichment of these structures in the OM compared to the IM fractions. Synthesis of LOS and LPS molecules begins in the cytoplasm and is completed at the surface of the IM5. LOS and LPS structures are unidirectionally transported to the OM and inserted into the outer leaflet. Since biosynthesis involves precursor attachment to the IM, a faint banding pattern is always observed for the IM fractions. However, the intensity of the molecules in the OM fraction is much greater than in the IM fraction, due to the enrichment of the LOS/ LPS structures (Fig. 4). The amount of membrane obtained was measured by determining the protein concentration in the suspension. Six times the amount of membrane was necessary to extract and detect LOS from A. baumannii compared to the amount necessary to detect LPS molecules from the enteric organisms (Fig. 4). We reason that this might reflect a decreased level of LOS molecules in the OM for these organisms compared to protein levels, but have not pursued this hypothesis in detail.
DISCUSSION:
This method will continue to aid researchers in understanding the role of the cell envelope in bacterial physiology and pathogenesis. Following the sequential ultracentrifugation steps a purified total, inner, and OM fraction can be obtained. These membranes can be assayed in isolation to test hypotheses related to membrane protein localization and function, transport and trafficking across the periplasm, and the composition of the individual bilayers under various environmental conditions. Biological studies exploring the involvement of individual OM components in pathogenesis, such LOS/ LPS and OM proteins, can also be conducted in animal and cellular models using isolated membrane fractions collected by this technique.
Our procedure has been optimized for use with Enterobacteriaceae, specifically S. Typhimurium, which produces LPS molecules that contain O-antigens of variable chain length. This protocol also works for the model bacterium, E. coli K-12, which has lost the genetic ability to synthesize O-antigens. Using wild type and O-antigen deficient S. Typhimurium 14028s and E. coli K-12 strain DH5α, we show that the ability to separate the membranes for these microbes is not substantially influenced by the presence of the O-antigens. However, to separate the envelope of the LOS-producing bacterium, A. baumannii 17978, we had to reduce the concentration of the middle-density sucrose solution in the discontinuous gradient to isolate the bilayers (Fig. 2). In particular, shifting the concentration of the middle-density solution from 53 to 45 % was sufficient to allow the OM to partition to the 45–73 % interface in the adapted gradient. When using the 53–73% gradient for A. baumannii, the majority of the OM material was often observed slightly below the IM fraction at the 20–53% interface (Fig. 2). Sparse OM material was present at the 53–73% interface for A. baumannii. These results suggested that the 20%/53%/73% gradient is inadequate for separating the bilayers of A. baumannii under these conditions.
In summary, adjustments can be made to the density gradient to accommodate organisms with varied OM-glycolipid content and level, and the approach can be adapted for other Gram-negative bacteria.
ACKNOWLEDGMENTS:
This work was funded by P20GM10344 and R01AI139248 awarded to Z. D. Dalebroux.
Footnotes
DISCLOSURES:
No conflicts of interest declared.
REFERENCES
- 1.Silhavy TJ, Kahne D & Walker S The bacterial cell envelope. Cold Spring Harb Perspect Biol 2, a000414, doi: 10.1101/cshperspect.a000414 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Egan AJ & Vollmer W The physiology of bacterial cell division. Ann N Y Acad Sci 1277, 8–28, doi: 10.1111/j.1749-6632.2012.06818.x (2013). [DOI] [PubMed] [Google Scholar]
- 3.Pazos M, Peters K & Vollmer W Robust peptidoglycan growth by dynamic and variable multi-protein complexes. Curr Opin Microbiol 36, 55–61, doi: 10.1016/j.mib.2017.01.006 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Raetz CR Enzymology, genetics, and regulation of membrane phospholipid synthesis in Escherichia coli. Microbiol Rev 42, 614–659 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whitfield C & Trent MS Biosynthesis and export of bacterial lipopolysaccharides. Annu Rev Biochem 83, 99–128, doi: 10.1146/annurev-biochem-060713-035600 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Needham BD & Trent MS Fortifying the barrier: the impact of lipid A remodelling on bacterial pathogenesis. Nat Rev Microbiol 11, 467–481, doi: 10.1038/nrmicro3047 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simpson BW & Trent MS Pushing the envelope: LPS modifications and their consequences. Nature Reviews Microbiology 17, 403–416, doi: 10.1038/s41579-019-0201-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ebbensgaard A, Mordhorst H, Aarestrup FM & Hansen EB The Role of Outer Membrane Proteins and Lipopolysaccharides for the Sensitivity of Escherichia coli to Antimicrobial Peptides. Front Microbiol 9, 2153, doi: 10.3389/fmicb.2018.02153 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu D & Reeves PR Escherichia coli K12 regains its O antigen. Microbiology 140 ( Pt 1), 49–57, doi: 10.1099/13500872-140-1-49 (1994). [DOI] [PubMed] [Google Scholar]
- 10.Kalynych S, Morona R & Cygler M Progress in understanding the assembly process of bacterial O-antigen. FEMS Microbiol Rev 38, 1048–1065, doi: 10.1111/1574-6976.12070 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Osborn MJ, Gander JE, Parisi E & Carson J Mechanism of assembly of the outer membrane of Salmonella typhimurium. Isolation and characterization of cytoplasmic and outer membrane. J Biol Chem 247, 3962–3972 (1972). [PubMed] [Google Scholar]
- 12.Osborn MJ & Munson R Separation of the inner (cytoplasmic) and outer membranes of Gram-negative bacteria. Methods Enzymol 31, 642–653, doi: 10.1016/0076-6879(74)31070-1 (1974). [DOI] [PubMed] [Google Scholar]
- 13.Cian MB, Giordano NP, Masilamani R, Minor KE & Dalebroux ZD Salmonella enterica serovar Typhimurium use PbgA/YejM to regulate lipopolysaccharide assembly during bacteremia. Infect Immun, doi: 10.1128/IAI.00758-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Masilamani R, Cian MB & Dalebroux ZD Salmonella Tol-Pal Reduces Outer Membrane Glycerophospholipid Levels for Envelope Homeostasis and Survival during Bacteremia. Infect Immun 86, doi: 10.1128/IAI.00173-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nikaido H Outer Membrane of Salmonella-Typhimurium Transmembrane Diffusion of Some Hydrophobic Substances. Biochimica Et Biophysica Acta 433, 118–132, doi:Doi 10.1016/0005-2736(76)90182-6 (1976). [DOI] [PubMed] [Google Scholar]
- 16.Dalebroux ZD, Matamouros S, Whittington D, Bishop RE & Miller SI PhoPQ regulates acidic glycerophospholipid content of the Salmonella Typhimurium outer membrane. Proc Natl Acad Sci U S A 111, 1963–1968, doi: 10.1073/pnas.1316901111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Castanie-Cornet MP, Cam K & Jacq A RcsF is an outer membrane lipoprotein involved in the RcsCDB phosphorelay signaling pathway in Escherichia coli. J Bacteriol 188, 4264–4270, doi: 10.1128/JB.00004-06 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thorne KJ, Thornley MJ & Glauert AM Chemical analysis of the outer membrane and other layers of the cell envelope of Acinetobacter sp. J Bacteriol 116, 410–417 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geisinger E, Huo W, Hernandez-Bird J & Isberg RR Acinetobacter baumannii: Envelope Determinants That Control Drug Resistance, Virulence, and Surface Variability. Annu Rev Microbiol 73, 481–506, doi: 10.1146/annurev-micro-020518-115714 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Kamischke C et al. The Acinetobacter baumannii Mla system and glycerophospholipid transport to the outer membrane. Elife 8, doi: 10.7554/eLife.40171 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]