

Abstract
Our understanding of how the RAS protein family, and in particular mutant KRAS promote metabolic dysregulation in cancer cells has advanced significantly over the last decade. In this Review, we discuss the metabolic reprogramming mediated by oncogenic RAS in cancer, and elucidating the underlying mechanisms could translate to novel therapeutic opportunities to target metabolic vulnerabilities in RAS-driven cancers.
Keywords: KRAS, metabolism, autophagy, glutaminolysis, glycolysis, macropinocytosis, chemoresistance, ferroptosis, cancer therapeutics
Introduction
The RAS family proto-oncogenes KRAS, NRAS, and HRAS encode a group of small GTPases that are activated in response to growth factors and other extracellular stimuli and induce downstream signaling cascades, such as the MAPK pathway. When mutated, oncogenic RAS remains preferentially in the active GTP-bound state, whereas GTP hydrolysis by its GTPase function and enzymes such as GTPase-activating proteins, is compromised1,2. The resulting RAS-mediated signaling cascades drive tumor initiation, maintenance and progression by deregulating key cellular processes, for instance by increasing proliferation and suppressing apoptosis, but also by rewiring cellular metabolism and promoting alterations in the tumor microenvironment3.
Given that RAS dysregulation may cause aberrant cellular signaling and malignant transformation, the activation of the RAS signaling pathway is tightly controlled in normal cells1. However, RAS mutations and the resulting deregulated signaling events are responsible for one third of all human cancers4 (Figure 1). KRAS in particular, is among the most frequently mutated oncogenes in cancer and is commonly associated with therapeutic resistance and poor prognosis1,5,6. Being a critical cancer driver, RAS has been the focus of an intensive search for therapies7. However, no effective RAS inhibitor has been approved for clinical use to date. Recent preclinical and early clinical results on the efficacy of inhibitors against the KRAS G12C mutant8–10, sparked excitement in the scientific community. However, the initial enthusiasm has been somewhat tempered by work suggesting that acquired resistance may constrain the inhibitors’ efficacy, which indicates that combination therapies may be needed11–14.
Figure 1. Frequency and distribution of RAS mutations in human cancers.
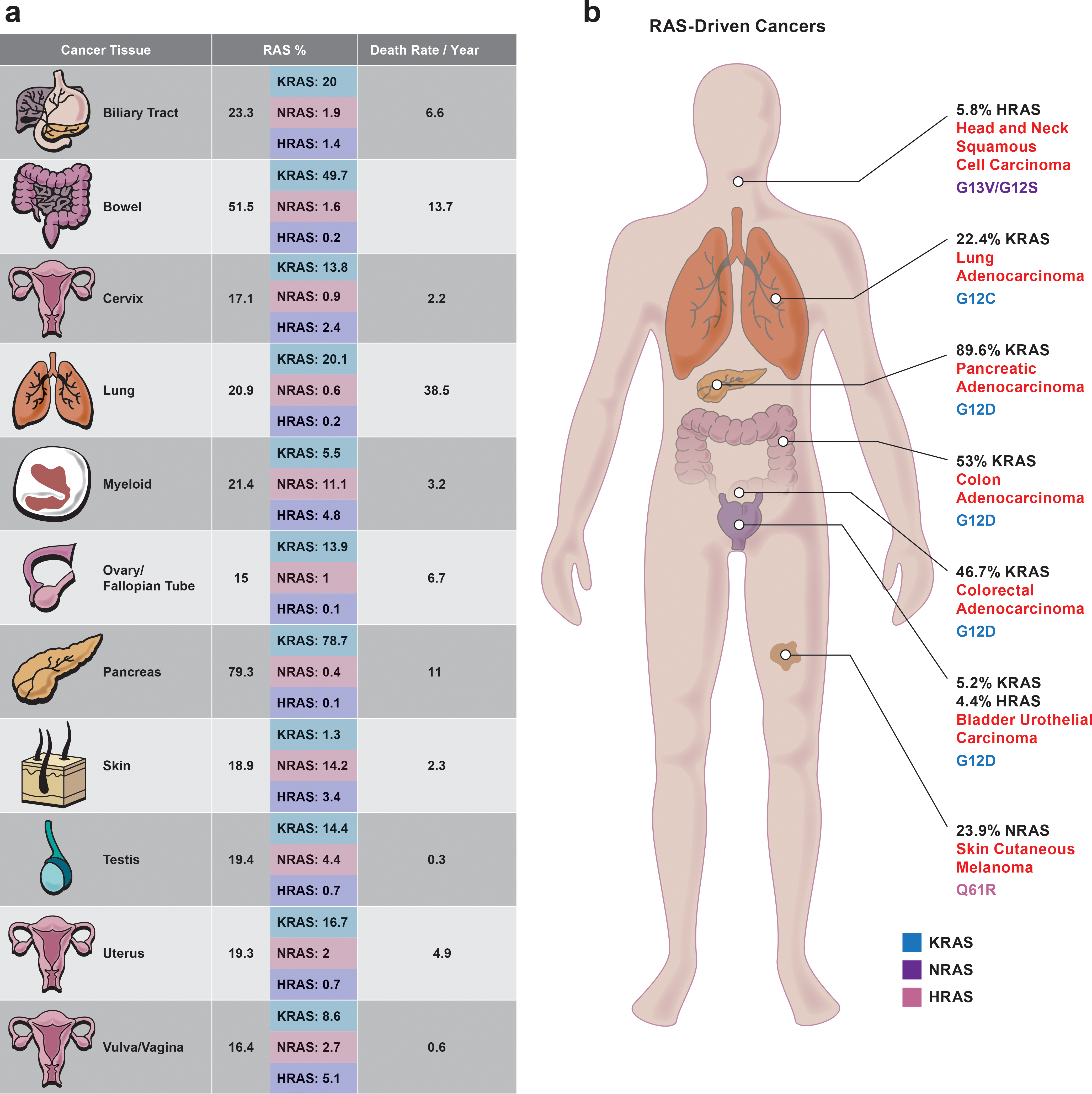
Human cancers differ by mutated RAS isoform, codon, and amino acid substitution. a. Distribution of RAS isoform (KRAS, NRAS, and HRAS) mutations across tumor types and frequency (%) of RAS mutations by isoform in each tumor type. Detailed information in Supplementary Table 1. b. Types of cancers commonly associated with RAS mutations. The frequency of the most commonly mutated RAS genes is listed by tumor type. For each tumor type, the amino acid substitutions that occur most frequently in the RAS isoform are shown. The color of the mutation refers to the mutated RAS gene (KRAS, blue; NRAS, purple; and HRAS, pink). Detailed information in Supplementary Tables 2 and 3. All human cancers that had a sample size greater than or equal to 300 and a total RAS mutation rate greater than or equal to 15% are listed from data resources. Death rate/ year (%) is based on the death rate per 100,000 men and women. Data collected from the National Cancer Institute SEER cancer statistics 2020 database.
The difficulty in targeting RAS has fueled a longstanding interest in identifying alternative approaches for treating RAS-driven cancers, efforts that have been supported by our increased understanding of RAS biology. It is now clear that the roles of oncogenic RAS extend far beyond its classic function of activating MAPK pathways. The links of RAS signaling to altered cellular metabolism are of particular interest in cancer research, given the potential to leverage RAS-related metabolic vulnerabilities to treat RAS-driven cancers. Here, we discuss the biology that connects RAS to metabolic dysregulation in cancer and evaluate the possibility of exploiting these connections for drug discovery and therapy.
RAS mutations in cancer
Cancers that harbor RAS mutations comprise a heterogeneous subset of all cancers, with the frequency of each mutant isoform and the specific mutation varying greatly across different cancer types (Fig. 1a and 1b). Most mutations of RAS family members occur at codons 12, 13, and 61, although the mutation frequency at each residue and isoform varies between cancer types that originate from different tissues. For instance, 22% of oncogenic mutations in lung adenocarcinoma (LUAD) occur in KRAS, whereas nearly 24% of such mutations in skin cutaneous melanoma occur in NRAS15 (Fig. 1b). The variation in substitution type is also striking. For instance, the G12C substitution is dominant in LUAD, whereas G12D is dominant in pancreatic adenocarcinoma (PDAC) (Fig. 1b). Overall, KRAS is mutated in many cancers (predominantly adenocarcinomas), whereas NRAS mutations are prominent in melanoma and myeloid cancer4,15,16 (Fig. 1). HRAS is mutated relatively infrequently, but when mutations do occur, they are primarily in bladder and head/neck squamous cell carcinoma. These observations indicate a fundamental difference in the biological effects of specific mutations on different RAS isoforms and in different tissues. Consequently, treatment efficacy cannot be extrapolated from one RAS-driven cancer to another, but rather therapeutic approaches must be tailored to the isoform, mutation and tissue. Altogether, despite a greater understanding of the RAS signaling cascade’s complexity, fundamental questions remain concerning the role of different oncogenic RAS mutations and isoforms on patients with cancer.
RAS and tumor metabolism
The reprogramming of cellular metabolism to support the energetic and biomass needs of uncontrolled proliferation, is a hallmark of cancer17. The use of FDG-PET imaging as a way to stage cancer18,19and of antimetabolites as chemotherapeutic agents20 in treating several cancers, further underscore the clinical importance of tumor metabolism21. The recognition that oncogenes, including RAS, can promote aerobic glycolysis, commonly known as the Warburg effect, and activate anabolic pathways 22,23, has increased efforts to understand the molecular underpinnings of altered metabolism in cancer24. In the subsequent sections we discuss the various manners in which oncogenic RAS reprograms metabolism, how these adaptations result in tumor-specific metabolic alterations that in turn modulate oncogenic signaling networks25,26, and how they may be targeted therapeutically. We focus primarily on KRAS, given the wealth of literature on this major oncogenic driver and note the roles of other isoforms where these are known.
Interplay between oncogenic RAS and glucose metabolism
Altered glucose metabolism, for example through the Warburg effect, is one of the most common metabolic changes differentiating normal and cancer cells. In addition to producing ATP, the breakdown of glucose through glycolysis produces metabolic intermediates, such as amino acids, and precursors for fatty acids and nucleotides that are required for cell growth and proliferation (Fig. 2a).
Figure 2. Oncogenic RAS-dependent control of cellular metabolism.
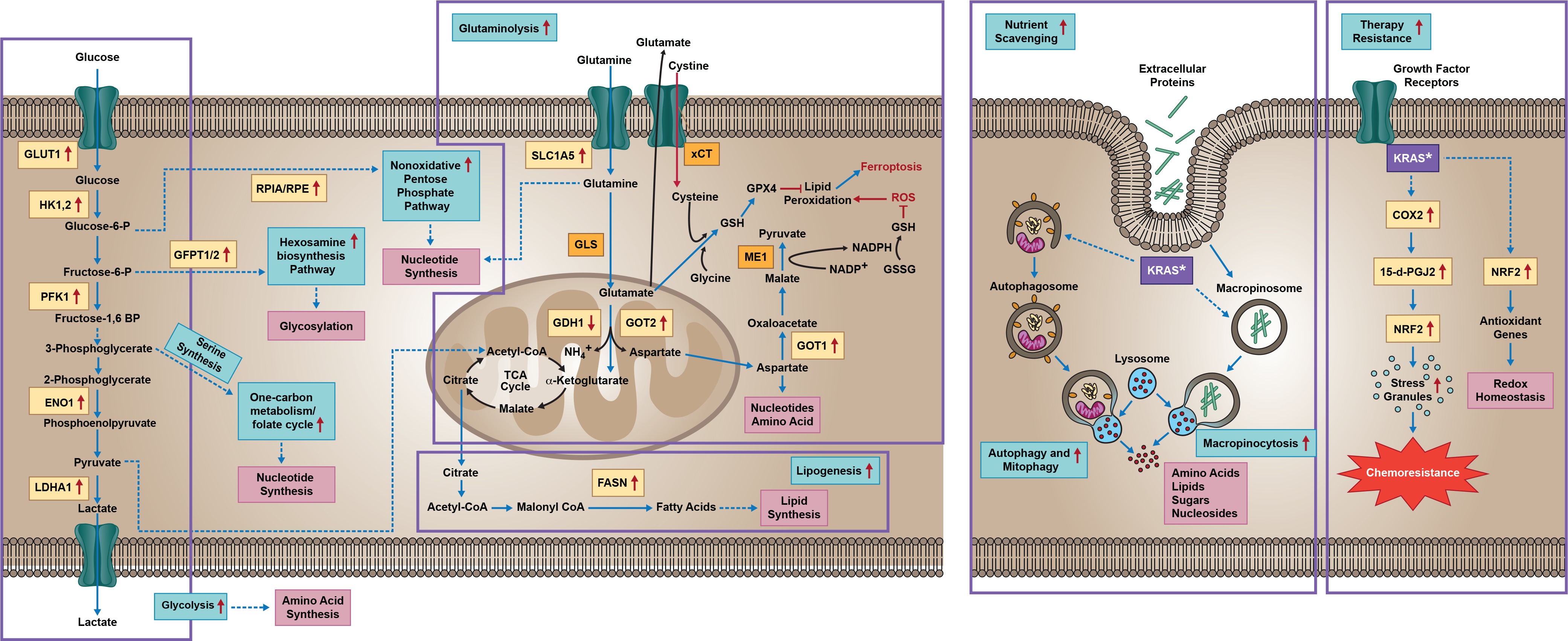
Mutant RAS deregulates key metabolic processes, including glutaminolysis, glycolysis, autophagy, and macropinocytosis. Oncogenic KRAS directs glucose metabolism into hexosamine biosynthetic pathways by upregulating several key glycolytic enzymes, and induces the nonoxidative pentose phosphate pathway to support increased nucleic acid biosynthesis. RAS-driven cancer cells alter glutaminolysis to support rewired metabolism. Altered glutaminolysis is a key feature of KRAS-dependent cancer cells. KRAS regulated glutamine metabolic rewiring influenced the TCA cycle, which is critical for nucleotide biosynthesis to support cell growth and survival. KRAS-driven tumors require glutaminolysis for redox balance. KRAS-mediated activated NRF2 is depicted as a key transcription factor that modulates redox homeostasis for the survival of cells under oxidative stress. Cells harboring mutant RAS are characterized by increased macropinocytosis, autophagy, and mitophagy, processes which help generate the energy and macromolecules needed for accelerated tumor proliferation. Mutant RAS also regulates stress granule formation, which helps mediate chemoresistance. Yellow box indicates RAS-dependent gene and/or protein expression, with arrows indicating increased or decreased expression. Purple box indicates oncogenic KRAS. GDH1: Glutamate DeHydrogenase 1; TCA: Tricarboxylic Acid; COX2: Cyclooxygenase 2; HK1,2: Hexokinase 1 and 2; GLUT1: Glucose Transporter-1; PFK1: Phosphofructokinase-1; ENO1: alpha-enolase-1; LDHA: Lactate Dehydrogenase; ME1: Malic Enzyme-1; ROS: Reactive Oxygen Species; GOT1,2: Glutamate Oxaloacetate Transaminase 1,2.
Mutant KRAS is involved in glucose metabolism in multiple ways. Gene expression and metabolic flux analyses have shown that it upregulates the expression of the GLUT1 glucose transporter to promote glucose uptake by cells, as well as inducing expression of hexokinase 1 and 2 (HK1 and HK2) rate-limiting enzymes of glycolysis, to increase glycolytic activity27–30 (Fig. 2a). One recent study reported the role of KRAS4A KRAS isoform in carbon metabolism through the direct regulation of the glycolytic enzyme HK1, which is of interest as it identifies the direct GTP-dependent regulation of a metabolic enzyme29 and further complicates the landscape of RAS mediated metabolism. Oncogenic KRAS also upregulates expression of other key glycolytic enzymes, such as PFK1, ENO1, and LDHA28,31–33, thereby promoting glycolytic flux and enabling the production of glycolytic intermediates that can be shunted into other anabolic pathways (Fig 2). KRAS also promotes the hexosamine biosynthesis pathway (HBP), which provides precursors for lipid and protein glycosylation34, and the non-oxidative arm of the pentose phosphate pathway (PPP)28, which supplies ribose, the backbone for nucleic acid production35 (Fig 2). KRAS regulates these pathways through MAPK-dependent signaling cascades, ultimately supporting cell survival and conferring a proliferative advantage on tumors28,36,37.
Highly glycolytic RAS-mutant cells have been found to be vulnerable to inhibition of the glycolytic enzymes glyceraldehyde 3-phosphate dehydrogenase (GAPDH) with Vitamin C, providing a mechanistic rationale for exploring the therapeutic use of this vitamin against KRAS or BRAF mutant colorectal cancer preclinical models 38. Additionally, oncogenic HRAS mediates enhanced glycolytic rates including increased glucose uptake, underscoring the fact that increased aerobic glycolysis is essential for RAS-mutant tumors to match energy production with the requirement for enhanced biosynthetic pathways39. Moreover, glycolytic KRAS-mutant cells produce increased amounts of potentially toxic byproducts of glycolysis such as methylglyoxal40. Methylglyoxal-mediated stress was shown to be involved in cancer progression41 and to be a potent activator of AKT signaling, suggesting that utilizing methylglyoxal scavengers in KRAS mutant colorectal cancer cells might be more effective when combined with AKT inhibitors40. Mutant KRAS has also been implicated in the induction of enzymes involved in the folate cycle42 and the aberrant activation of mTOR, a key regulator for both serine synthesis and the folate cycle43,44. Furthermore, the tumor suppressor LKB1, which activates the energy sensor and metabolic regulator AMPK, has been linked to serine metabolism and induction of tumorigenesis45. Of note, LKB1 loss is prevalent in KRAS mutant lung cancers46, indicating that oncogenic KRAS not only induces mTOR activity, but might also upregulate one-carbon metabolism by undermining AMPK’s inhibitory role in the folate cycle. However, systematic investigation is required to explore the role of oncogenic KRAS in one carbon metabolism in detail.
The interplay between oncogenic RAS and glycolysis provides a rationale for targeting glycolysis in RAS-driven cancers. A number of natural or synthetic products, including inhibitors of GLUT1–4, have been discovered over the years and validated through various pre-clinical cancer models before clinical trials47. A promising candidate is silybin, a natural flavonoid and potent inhibitor of GLUT1 and GLUT4, which was shown to be effective in phase 1 trial of prostate cancer with asymptotic liver toxicity as an adverse effect48. BAY-876, a potent GLUT1 inhibitor49, was separately shown as an effective candidate in pre-clinical setting 50. Several other compounds have been found to have inhibitory properties against glycolytic enzymes and some have been included in clinical trials47,51. Although preclinical studies support the effectiveness of these small molecule inhibitors, in-depth study is warranted to explore their true therapeutic and clinical potential. Additionally, toxicity and target specificity are a major concern for any drug and it is non-trivial to specifically inhibit these glycolytic enzymes while avoiding unwanted effects on normal cells. Further study is essential to identify potent inhibitors targeting glycolysis that would specifically impair RAS-driven cancer growth.
Oncogenic RAS in glutaminolysis and redox homeostasis
The nonessential amino acid glutamine is the most abundant amino acid in human sera and is necessary for cellular function and survival. The breakdown of glutamine through glutaminolysis gives rise to glutamate, a critical precursor of most other nonessential amino acids, including aspartate, alanine, arginine, and proline52. Thus, in addition to its central role in nucleotide and protein production, glutamine-derived carbon in the form of glutamate, can be an important anaplerotic substrate for the tricarboxylic acid (TCA) cycle (Fig. 2a). The process of anaplerosis replenishes metabolic intermediates removed from the TCA cycle, such as citrate, thereby increasing their availability for fatty acid and cholesterol biosynthesis53. Glutamine is also a major source of nitrogen for proliferating cells52,54.
Many tumors driven by oncogenic KRAS and its downstream effector, the transcription factor MYC, exhibit metabolic reprogramming to consume and rely more on glutamine for both catabolic to anabolic pathways55,56. Oncogenic KRAS elevates the gene expression of enzymes involved in glutaminolysis27. For instance, KRAS-dependent upregulation of Glutamate Oxaloacetate Transaminase 1,2. (GOT) expression in pancreatic cancer facilitates production of aspartate for nucleotide biosynthesis, and allows NADPH generation via ME157,58 (Fig 2). In addition to activating the GOT2/GOT1/Malic Enzyme 1 (ME1) pathway, oncogenic KRAS activates the NRF2 antioxidant system by inducing NRF2 expression59,60 and by constitutively activating the battery of genes controlled by NRF2 to maintain the redox balance and promote tumorigenesis61. The activation of NRF2 causes glutamine dependence in KRAS mutant lung and pancreatic cancers cells and preclinical mouse models60,62,63, and BRAF mutants can similarly activate NRF2 to promote reactive oxygen species (ROS) detoxification59. (Fig 2). Oncogenic KRAS maintains reduced glutathione pools by mediating GOT1,2/ME1 and NRF2-antioxidant pathways. However, KRAS has also been shown to promote cancer cell growth by stimulating alanine aminotransferase activity, leading to high levels of α-ketoglutarate for the TCA cycle and mitochondrial ROS generation, which was required for mutant KRAS-driven tumorigenesis in a mouse model of lung cancer64.
Glutamate metabolism is also being investigated as a therapeutic target54. Although no clinical-grade inhibitors for the GOT/ME1 axis currently exist, the dependency of certain KRAS-driven cancers, such as pancreatic cancer60, lung cancer62, on glutamine could be exploited by targeting glutaminase-1 (GLS1), the enzyme that restricts glutamine’s conversion to glutamate and its anaplerotic entry into the TCA cycle. Limiting glutamine use combined with chemotherapy is a viable means to halt pancreatic tumor growth in preclinical mouse models and is not toxic to normal cells58,60,65. Separately, the loss of LKB1 in mutant KRAS non-small cell lung cancer preclinical mouse model was found to promote NRF2-dependent metabolic alterations that increased the tumor cells’ dependence on glutamine and created a vulnerability to glutaminase inhibition66. Additionally, mutations in the KEAP1 gene, which encodes a negative regulator of the NRF2, could point the way to treating lung adenocarcinoma, which is driven by oncogenic KRAS. Cells from advanced lung tumors with oncogenic KRAS and loss-of-function KEAP1 mutations were more dependent on increased amounts of glutamine than other cells, making them more susceptible to glutaminase inhibition62. KRAS activation is also commonly coupled with loss of LKB1 function. Co-occurrence of mutant KRAS and LKB1 deficiency in patients with lung cancer resulted in more aggressive tumors, higher frequency of metastasis, and therapy resistance67. This could be explained by the fact that loss of LKB1 sustains KRAS-mediated proliferation through autophagy and increased synthesis of essential macromolecules, even under nutrient-deprived conditions68. Moreover, in oncogenic-KRAS-driven lung adenocarcinoma, loss of LKB1 often induces KEAP1 activation69 and leads to metabolic alterations that could be counteracted by activation of NRF270, thereby maintaining redox homeostasis and fueling energy metabolism in a glutamine-dependent manner. Thus, cancer cells harboring KRAS, KEAP1 and LKB1 mutations may be more sensitive to glutaminase inhibition compared to normal counterparts66. Concurrent mutations in KRAS and LKB1 also confer vulnerabilities in pancreatic cancer, but the mechanisms are different from those in lung cancer45. In pancreatic cancer, such concurrent mutations support tumor growth by activating serine synthesis and increasing DNA methylation. Moreover, KRAS-driven lung adenocarcinomas with TP53 mutation induce immune cell production, while tumors with KEAP1 mutations rewire metabolism71. Exploiting these context-specific properties, either by depleting the immune cells in tumor tissues or by perturbing the altered metabolism, which could be effective in inhibiting tumor progression. This suggests that rather than considering a one-size-fits-all approach to therapy, individualized precision therapies based on co-occurring mutations could be more effective for patients with KRAS-driven cancers71. Thinking more broadly about the interplay of metabolism with RAS signaling, targeting glutamine metabolism has also been found to suppress acquired resistance to MAPK inhibitors in melanoma cells72. However, environmental factors may also come into play, as for instance, the availability of extracellular as discussed below, can influence the dependence of cancer cells on glutamine metabolism73. Consistently, not all KRAS tumors are sensitive to inhibition of glutamine metabolism in vivo74,75, indicating that a deeper understanding is required of the context in which KRAS-driven cancers would be most sensitive to agents that target this metabolic pathway.
xCT, the cystine/glutamate antiporter that exports glutamate to the extracellular space and imports cysteine into the cytosol for the production of the amino acid cysteine, has been shown to be essential for oncogenic-KRAS-mediated transformation and involved in intracellular redox balancing76. Cystine import is key to KRAS-driven PDAC cell survival as deprivation of cysteine or xCT inhibition were shown to undergo ferroptosis77, an iron-dependent form of programmed cell death characterized by a lethal buildup of lipid peroxides78. Moreover, NRF2 enhances xCT activity to mediate glutathione synthesis60,63,73 and also regulates glutathione peroxidase 4 (GPX4) activity, an enzyme that lies downstream of xCT and is involved in metabolic processing of ferroptosis78. In line with the known links of KRAS to NRF2, glutamine limitation was shown to induce pro-ferroptotic stimuli, including GPX4 inhibition, in KRAS mutant pancreatic cancer cells60, suggesting mutant RAS cancer cells displaying high levels of glutaminolysis might be more susceptible to ferroptosis. Although KRAS-driven pancreatic tumors depend on cysteine metabolism to prevent ROS-induced ferroptosis, making cysteine depletion a potentially useful clinical strategy77, it is unclear whether ferroptosis can be selectively activated in all RAS-driven tumors. In-depth studies of the roles of oncogenic KRAS in cysteine metabolism are needed to determine possible therapeutic approaches.
Lipid metabolism and fatty acid biosynthesis in RAS mutants
Lipids, including fatty acids, are an energy source in addition to glucose and glutamine, and proliferating cancer cells aberrantly activate lipid biosynthesis79. RAS-transformed cells depend on serum lipids for proliferation and survival80. Under metabolic stress, certain RAS-driven cancer cells stimulate lysophospholipid uptake and use it to support ATP production81. Oncogenic KRAS activates downstream signaling through AKT for the eventual activation of the ACLY enzyme, to enhance the conversion of citrate to acetyl-CoA, and increase de novo fatty acid and sterol biosynthesis82 (Fig. 2a). Furthermore, KRAS reprograms lipid homeostasis to support tumorigenesis by upregulating ACSL3, an enzyme involved in lipid synthesis83. In line with these findings, mutant KRAS drives a lipogenic gene-expression program to promote de novo lipogenesis and activates lipogenesis by inducing FASN expression, which can be exploited therapeutically84,85 (Fig 2). Inhibiting fatty acid oxidation in a mouse model of KRAS-driven pancreatic cancer was shown to reduces tumor recurrence86, suggesting potential therapeutic value in targeting lipid metabolism in RAS-driven cancers.
Recycling pathways and nutrient scavenging in RAS mutants
Oncogenic RAS-driven tumor cells develop distinct mechanisms to scavenge nutrients from extracellular sources and recycle intracellular fuel to provide metabolic flexibility and secure adequate nutrient availability (Fig. 2a). One such process is autophagy87, the regulated degradation and recycling of cellular components that is activated by starvation and stress, and provides energy and building blocks, such as amino acids, nucleotides, lipids, and sugars, necessary for cellular survival and organelle homeostasis. The role of autophagy in cancer is complex and context-dependent, but this process is known to be elevated in cancer cells harboring KRAS mutations and is required for tumor maintenance and cellular viability88,89. The nexus between oncogenic KRAS and autophagy is also sustained by increasing the glycolytic rate90 and supporting mitochondrial respiration91. In particular, basal autophagy has been shown to be elevated in KRAS-driven pancreatic cancers, where it provides nutrients to fuel the TCA cycle necessary for cell growth and survival88. Unlike normal cells, those harboring KRAS mutations upregulate basal autophagy by activating the MiT/TFE transcription program92. Separately, autophagic deficiency in KRAS- and BRAF-mutant cancers is known to enhance glutamine dependence, suggesting that autophagic protein degradation supplies cancer cells with certain amino acids required for metabolic pathways, including glutamine93. Blocking autophagy in a mutant-RAS setting can deplete glutamine and block fatty acid consumption, which compromises tumor growth. This concept further suggests that inhibiting downstream effectors of KRAS in the MAPK pathway can upregulate autophagic flux, potentially as a metabolic adaptation of compromised mitochondrial activity. Thus, combinatorial inhibition of MAPK effectors and autophagy was shown to reduce KRAS-driven tumor growth in preclinical mouse models of pancreatic cancer90,94,95.
Additionally, oncogenic KRAS upregulates mitophagy, a selective form of autophagy that clears damaged mitochondria and improves mitochondrial function under conditions of nutrient deficiency. Mutant KRAS stimulates a mitophagy receptor called NIX, leading to decreased mitochondrial metabolism and a shift toward glycolysis to stimulate cell proliferation and strengthen redox homeostasis96. Given these findings, it may be worth exploring mitophagy as a target for RAS-driven metabolic malignancies.
Although autophagy can produce diverse nutrients, it cannot increase the cell’s net biomass. To fuel elevated metabolic needs, KRAS-mutant tumors rely on lysosome-dependent macropinocytosis, the process in which cells non-specifically engulf material from the extracellular space93. For instance, RAS-stimulated macropinocytosis was shown to promote cellular uptake of extracellular albumin, followed by its degradation into amino acids (particularly glutamine) that could then enter the anaplerotic TCA cycle97 (Fig 2). Whereas oncogenic RAS enhances macropinocytosis, the process is initiated by growth factor–induced PI3K signaling98. In this context, the KRASG12R mutant was shown to be impaired in PI3K signaling and macropinocytosis, whereas the KRASG12D and G12V mutants relied on MYC to drive micropinocytosis in preclinical mouse models of pancreatic cancer99. These mutant-specific effects indicate that further exploration is needed to elucidate how macropinocytosis and KRAS activity are interlinked and whether such allele-specific nutrient supply is active in other tumor types.
Although there is no clinically-approved selective macropinocytosis inhibitor, EIPA, an inhibitor of Na+/H+ exchange, reportedly inhibits macropinocytosis and sensitizes KRAS mutants to the mTOR inhibitor rapamycin80,98. Moreover, the vacuolar ATPase, a transmembrane protein complex that transduces protons across cellular and organellar membranes, is essential for RAS-mediated macropinocytosis. HRASG12V or KRASG12V expression redistributed vacuolar ATPase from the cytoplasm to the plasma membrane of lung, pancreatic and colon cancer cells, raising the possibility that blocking macropinocytosis by targeting this complex may represent a new strategy to treat RAS-mutant cancers100.
Given that RAS-driven tumors activate nutrient-scavenging pathways, such as autophagy and micropinocytosis, targeting these processes represents an interesting therapeutic approach93, especially as non-cancer cells are less likely to rely on these metabolic alterations. For example, both autophagy and macropinocytosis involve the lysosome, suggesting that lysosome inhibitors may inhibit both these pathways to restrict mutant-RAS tumor growth, although this concept remains to be determined experimentally. In addition, further work will determine whether autophagy and/or macropinocytosis inhibition could be combined with established therapeutic approaches, such as chemotherapies.
Oncogenic RAS, metabolism and therapy resistance
Oncogenic KRAS mutations have been associated with reduced sensitivity to therapeutic agents101,102. For example, patients with mutant KRAS lung cancer had poor clinical outcomes following combined treatment with the EGFR inhibitor erlotinib and chemotherapy102. KRAS-dependent fibrosarcoma, colon and bladder cancer cell lines were also shown to become resistant to radiation therapy103,104. Both PI3K- and RAF-dependent, but MEK-independent signaling pathways have been suggested to underlie this KRAS-mediated radio-resistance in epithelial cells101. Consequently, targeting KRAS-mediated signaling can lead to the activation of compensatory pathways37,86,105 resulting in adaptive resistance to therapies. In line with this, most therapies induce ROS in cancer cells, with treatment-resistant tumors often developing ROS-inhibitory mechanisms106 or mechanisms that rely on ROS to sustain proliferation. For example, ROS generated through mitochondrial metabolism was shown to be required for KRAS-induced anchorage-independent growth and to be essential for cell proliferation and tumorigenesis in KRAS-driven mouse lung adenocarcinoma64. Separately, oncogenic KRAS was found to require ROS to promote the development of PDAC precursor lesions such as pancreatic intraepithelial neoplasia107. Moreover, oncogenic RAS-induced ROS was shown to be produced in a RAC1 and NADPH oxidase (Nox4)-dependent manner in zebrafish model system, leading to hyperproliferation and activation of DNA damage response pathways108.Thus, although mutant KRAS signaling reportedly leads to genotoxic stress stemming from ROS generation108, oncogenic KRAS can reprogram metabolism to favor glutathione biosynthesis and increase NADPH production. This may protect macromolecules from indiscriminate damage incurred by ROS (Fig. 2a). Various drugs with a direct or indirect effect on ROS metabolism are now in clinical trials testing whether targeting tumor cell antioxidant capacity is an effective therapy109. In summary, redox management may modulate tumor cell progression and therapeutic responses in RAS mutants.
Mutant KRAS tumor cells also shield themselves from the effects of stress stimuli and chemotherapy by promoting stress granule (SG) formation110 through the production of the 15-d-PGJ2 prostaglandin. 15-d-PGJ2 is in turn responsible for NRF2 activation111 and SG accumulation110, which could contribute to the ability of oncogenic KRAS cells to resist chemotherapy60,110 (Fig 2). However, glutamine restriction was shown to reduce the capacity of 15-d-PGJ2 to form SGs in chemotherapy-treated KRAS-driven PDAC cells60, thereby limiting the cytoprotection this pathway provides against chemotherapy-induced stress stimuli. This connection of the KRAS-NRF2-SG axis and glutaminolysis suggests a potential approach to counteract mutant KRAS-mediated drug resistance. However, given that the process of SG accumulation is incompletely understood, elucidating the precise underlying mechanisms and metabolic links in RAS-driven cancer will determine the feasibility of SG-based therapeutic strategies. Taken together, targeting RAS-dependent metabolic alterations might be useful to counteract drug resistance and inhibit tumor growth.
Targeting oncogenic RAS-related metabolism
As discussed in the previous sections, pleiotropic metabolic changes are among the primary events downstream of oncogenic KRAS expression (Fig. 3a), indicating that tumorigenesis progresses due to key oncogenic signaling that promotes metabolic adaptations to support proliferation112 and present therapeutic opportunities. More specifically, RAS-driven cancer cells appear to function optimally when nutrient supply is favorable, but undergo rapid bioenergetic collapse when starved of glucose or glutamine because their demands for energy cannot be met in the absence of sustained glycolysis or glutaminolysis, the major mechanisms that fuel energy production. The limited tolerance of malignant cells for metabolic imbalance creates a vulnerability that could be exploited with drugs targeting tumor metabolism. In this setting, conditions of limited nutrient availability would imbalance the ratio of energy produced per nutrient consumed, thereby leading to alterations in bioenergetic dynamics (Fig 3b).
Figure 3. Metabolic alterations and vulnerabilities of RAS-driven cancers.
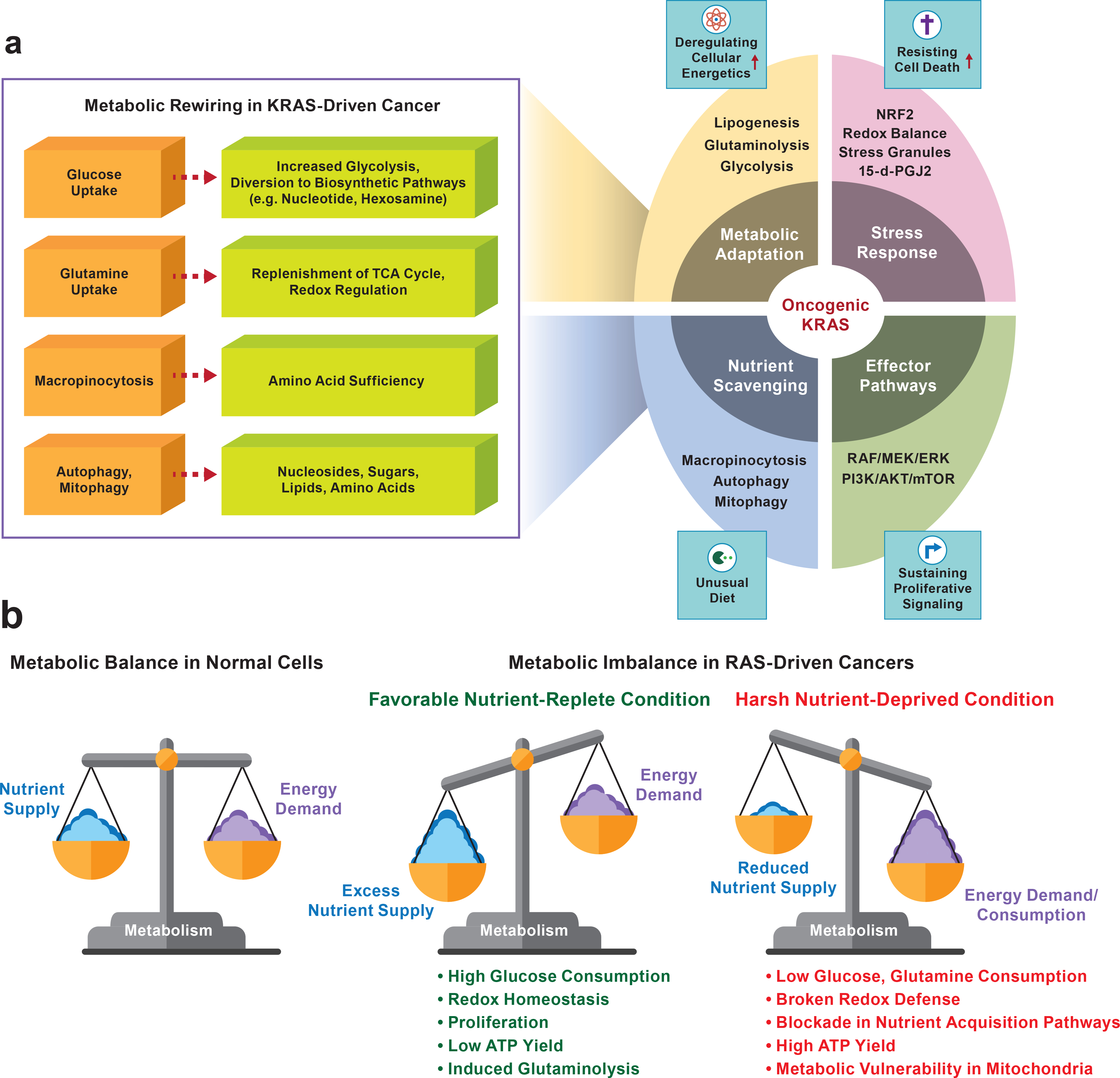
a. Schematic representation of the impact of oncogenic KRAS on cancer metabolism. Various metabolic pathways involving KRAS-mutants critical in cancer cell proliferation and cell survival. Increased glucose uptake, induced glutaminolysis, autophagy and micropinocytosis are involved in deregulating energetics and nutrient scavenging which results in RAS-driven cancer cells’ metabolic adaption for the benefit of cell growth. b. The delicate balance of nutrient supply and demand dynamics in RAS-driven cancers. In a balanced state, nutrient supply is sufficient to maintain energy demand (left). Excessive supply of nutrient availability, in the absence of a parallel increase in energy demand, represents a situation in which the energy required to satisfy energy demand is lower than the available energy (middle). A nutrient-deprived condition provokes metabolic inequity (energy supply < energy demand), leading to energetic stress and, ultimately, metabolic vulnerability (right). Nutrient-replete mutant RAS cells utilize rewired metabolism in their favor (middle panel). In the absence of glucose or glutamine, the metabolic vulnerabilities of mutant RAS cells intensify, leading to energetic stress and ultimately to cell death. Interventions that decrease nutrient consumption abolish redox defense and lead the cells to metabolic imbalance. This results in metabolic vulnerability, a potential therapeutic approach for RAS-driven cancer cells. (right panel).
Potential metabolic targets of signaling pathways for precision therapy have been documented previously22, and the advancing research on the roles of mutant RAS on cancer metabolism, either through direct effects on metabolic enzymes and pathways as discussed above, or through the actions of downstream RAS effectors29,113–117 provide many routes to explore therapeutic opportunities. For instance, detailed analysis of protein and genetic interactions in the RAS-driven pathway identified links between metabolic enzymes and oncogenic RAS, opening up a new avenue for potential RAS therapeutics114,116. Additionally, combining proximity-dependent proteomics with CRISPR screening identified a new set of functional RAS-associated proteins and several previously unrecognized direct RAS effectors, including metabolism-associated proteins, paving a way for the exploration of potential combinatorial therapies targeting the KRAS-effector pathway, RAS-mediated metabolic enzymes115, or other RAS-driven metabolic adaptations, including nutrient scavenging and stress response pathways (Fig 3a). As discussed further below, various signaling pathways often exist upstream metabolic processes to generate a common metabolic end product112 and surging evidence suggests that genetic alterations are associated with specific rewired oncogenic metabolic pathways118, which supports the idea of using several drugs to target metabolism for a particular disease. Combining agents to target complimentary metabolic pathways might be a suitable strategy for reducing the dose of individual drugs and eliminating unwanted toxicity levels in normal cells.
The successful development of potent inhibitors against KRASG12C, which have progressed to clinical trials8,9, now makes it possible to explore combinations of a RAS inhibitor with metabolism-focused treatment strategies. Indeed, one of the KRASG12C inhibitors, MRTX849, has revealed the potential resistance pathways that include the involvement of NRF2 in MRTX849 resistance, suggesting that a monotherapy approach might not work against RAS-driven cancers and that combinatorial therapies with mTOR or SHP2 or CDK4/6 inhibitors will be necessary9. As discussed, targeting metabolic enzymes has proven effective in some mutant KRAS cancer cell lines and mouse models from certain KRAS mutants60,62,65,90,119,120, and several other pharmacological inhibitors targeting dysregulated cancer metabolism are under development or in different preclinical stages21. If successful, such approaches could be combined with RAS inhibitors. Some metabolic pathway inhibitors, including against mTOR, have already been tested in combination with MRTX849 in a preclinical setting, with encouraging results12. In addition, several clinical trials that target metabolic dysregulations in mutant-RAS cancers are underway, including strategies against glutaminolysis and autophagy (Table 1).
Table 1:
Selected clinical studies with novel agents targeting metabolism in RAS-driven cancer
| Diseases | Biomarkers | Therapies | Phase | Study Identifier | Status |
|---|---|---|---|---|---|
| Lung Cancer: Small Cell or Squamous | HRAS, KRAS and NRAS mutations in Codons 12, 13, 61, 117, and 146 | Auranofin and Sirolimus | Phase 1/Phase 2 | NCT01737502 | Recruiting |
| Metastatic Pancreatic Carcinoma, Stage II, III, IV Pancreatic Cancer, Unresectable Pancreatic Carcinoma | MEK | Trametinib, Hydroxychloroquine | Phase 1 | NCT03825289 | Recruiting |
| Metastatic Pancreatic Adenocarcinoma, Stage IV Pancreatic Cancer | KRAS Mutation | Hydroxychloroquine, Binimetinib | Phase 1 | NCT04132505 | Recruiting |
| Lung Cancer: Non-Small Cell Lung Carcinoma | KRAS Mutation | CB-839, Docetaxel | Phase 1 | NCT02071862 | Completed |
| Colon Carcinoma | KRAS and BRAF mutation | Carbohydrate-Restricted Diet, Vitamin C Supplement | Phase 1/Phase 2 | NCT04035096 | Not yet recruiting |
| Metastatic Melanoma | NRAS Mutation | Trametinib, Hydroxychloroquine | Phase 1b/Phase 2 | NCT03979651 | Recruiting |
| Non-Small Cell Lung Carcinoma, Colorectal Carcinoma | KRAS Mutation | CB-839, Palbociclib | Phase 1/Phase 2 | NCT03965845 | Recruiting |
| Gastrointestinal Adenocarcinomas | MAPK mutations: KRAS, NRAS, HRAS, BRAF non-V600, MEK, and ERK mutations |
Ulixertinib, Hydroxychloroquine | Phase 1 | NCT04145297 | Recruiting |
| Pancreatic Cancer: Metastatic Adenocarcinoma | Mutant KRAS | Hydroxychloroquine, Gemcitabine | Phase 1/Phase 2 | NCT01506973 | Active, not recruiting |
| Non-squamous Cell Lung Cancer | Wild type and mutant KRAS | AZD2014, AZD6244 | Phase 1/Phase 2 | NCT02583542 | Active, not recruiting |
| Colon Cancer | KRAS, BRAF Mutation status | TVB-2640 | Phase 1 | NCT02980029 | Recruiting |
| Breast Cancer, Endometrial Cancer, Lung Cancer, Colorectal Cancer, Head and Neck Cancer | KRAS mutations | Serabelisib, Canagliflozin | Phase 1/Phase 2 | NCT04073680 | Not yet recruiting |
| Metastatic Colorectal Cancer | RAS Wild Type | CB-839, Panitumumab, Irinotecan | Phase 1/Phase 2 | NCT03263429 | Recruiting |
| Colorectal Cancer | RAS Wild Type | BAY94–9392, 11C-Glutamine | Phase 1 | NCT03275974 | Recruiting |
According to My Cancer Genome and ClinicalTrials.gov database
However, much work is still needed to fully explore the therapeutic potential of targeting altered metabolism in RAS-driven cancers. Among the complexities that require detailed study are the specific and/or tissue-dependent roles of different RAS mutations and isoforms. For example, different KRAS mutations likely have tissue-specific effects on metabolism; therefore, multiple strategies must be developed and matched to the RAS-mutant subsets. In line with this, there is considerable variation in glutamine dependencies between tissues based on their origin121–123, and various KRAS mutations can have different dependencies for as yet unclear reasons124. In vivo tumors also display variability in their glutamine dependencies compared to cell culture findings74, underscoring the importance of using appropriate model systems to draw firm conclusions. Further, various reports assessing the transcriptomic, proteomic, phosphor-proteomic and metabolic profiles of oncogenic RAS variants, including HRAS, NRAS and KRAS isoforms, indicate possible differences between their phenotypic effects 125–127. Nevertheless, the metabolic landscapes of HRAS- and NRAS-driven cancers remain less explored and it would be important to discover whether they resemble that of KRAS-driven tumors and whether different metabolic adaptations predominate in different tissue contexts for all RAS isoforms.
The use of metabolic therapies may have some advantages over other approaches. Such therapies could offer heightened specificity given that tumor cells appear to be more sensitive to metabolic inhibitors than their normal counterparts128. The success with chemotherapies that target metabolism129 intensifies hope that more metabolic therapies will ultimately reach the clinic. However, there are some limitations in targeting metabolism for therapeutic purposes130–132. Chief among them is the metabolic flexibility of cancer cells that can often switch their source of nutrients and energy and activate compensatory metabolic pathwasy for survival, when there is limitation in their favored metabolic pathways or they are deprived of preferred metabolic source75,133. This adaptive nature of cancer cells might limit the efficiency of targeting a single metabolic pathway for therapeutic purposes, a concern that could be addressed by combinatorial therapeutic approaches against multiple pathways, including known compensatory ones. Another major challenge in the development of drugs against cellular metabolism is the unwanted toxicity created by the effects of agents targeting metabolic enzymes in normal cells. Of particular concern is the dependency of immune cells on metabolic pathways134 similar to those utilized by tumor cells, which would make them vulnerable to the toxicity created by targeting metabolic pathways. Since affecting the metabolic processes of immune cells could potentially affect not only their anti-tumor activities but also the organism’s broader immune defenses, a detailed understanding of immunometabolism would be crucial in guiding the development and use of targeted therapies based on cancer metabolism. Although toxicity would limit the use of drugs in some cases, the fact that a therapeutic window may exist for many patients supports the notion that metabolic enzymes are attractive targets for cancer therapy. Better understanding of metabolic dependencies in specific tumor tissues, their links to different oncogenic alterations and signaling pathways and potential toxicities of targeted approaches is key for defining metabolism’s prospect for improving the therapeutic index.
Connecting RAS to other oncogenic drivers and metabolic pathways
Oncogene-directed metabolic alterations can have extensive impact, with multiple metabolic pathways being altered simultaneously in a single cancer type. Several oncogenes coordinate the transcriptional reprogramming that tumor cells need to thrive, with many cancer driver genes also perturbing metabolism135,136. Thus, to target oncogenic RAS and its downstream signaling and metabolic programs effectively, it is important to also understand how these are linked to other oncogenic drivers and pathways.
Analysis of data from the PanCancer Analysis of Whole Genomes Consortium samples135 on the KEGG pathway database137 depicts oncogenic driver genes involved in various metabolism processes in many cancer tissue types (Fig. 4a), with a gene-level analysis of each corresponding pathway unveiling driver genes that are co-altered in diverse tissue types (Fig. 4b). Future work should focus on investigating the potential crosstalk between RAS and commonly mutated driver genes known to be involved in metabolic and RAS signaling pathways in diverse tumor types, taking into account also that tumors from different tissues may display divergent metabolic phenotypes irrespective of their genomic profile. Network analysis of the several driver mutation genes, including KMT2D138, PIK3CA139, PTEN140, and IDH1141, that regulate signaling and metabolic pathways in KRAS-driven cancers, connects them to oncogenic RAS (Fig 4c). This analysis suggests a hypothetical view of a broader transcriptional and signaling circuit coordinated by RAS together with PI3KCA, NF1, and PTEN for the tight regulation of metabolic pathways. For example, KRAS could signal through P73142, or even c-Src143, to trigger PTEN, which may act through AKT to regulate KMT2D; through AP1143 to regulate KMT2C, or through p300144 to regulate the lipogenesis enzyme ACACA. KMT2D and KMT2C may in turn regulate c-MYC, SOX2, or CTNNB1 to control other metabolism driver genes, including GBA, IDH1, PTDSS1, POLE, and ACACA. Many of these circuits may feedback to NF1 to influence the RAS gene directly145 or to impact the RAS protein through a feedback loop from c-MYC146, SOX2147, or CTNNB1148. Alternatively, KRAS may directly trigger PIK3CA139, which impacts CTNNB1 for subsequent regulation of many metabolism driver genes. Various levels of interplay are known between the PI3K-AKT and KRAS-MAPK signaling cascades139, including for potential therapy. For example, using a combination of PI3K and MEK inhibitors has been shown to treat KRAS-driven lung cancers in mouse models149. However, detailed study is needed to understand precisely how these two pathways and the other factors depicted in this broader signaling circuit (Fig. 4c) coordinate with each other for the metabolic rewiring needed to sustain uncontrolled proliferation in cancer43. Such work will be instrumental for developing novel targeted therapies in RAS-driven cancers.
Figure 4. Oncogenic driver genes and their involvement in metabolic pathways.
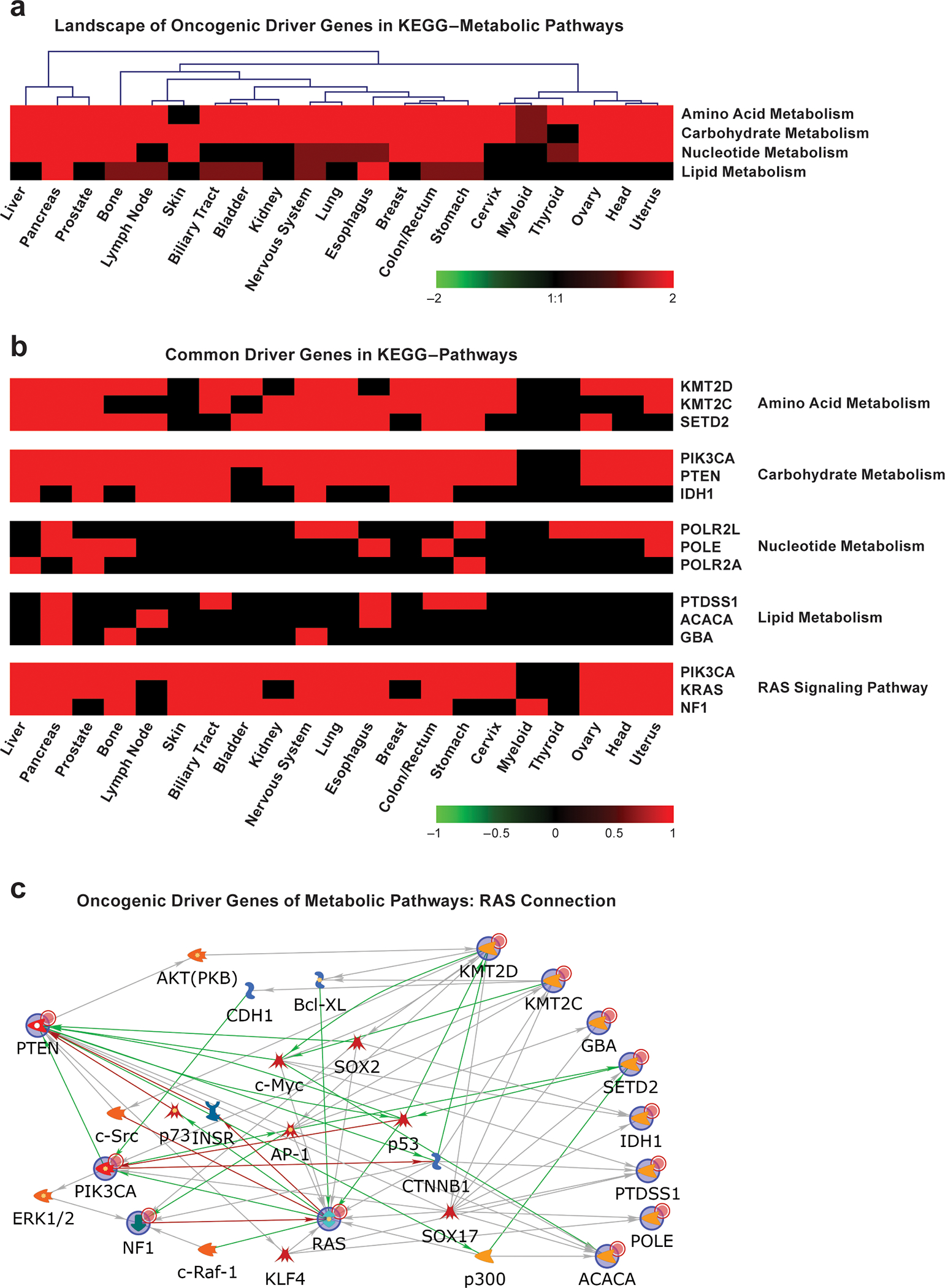
a. Pathway-level heatmap showing four KEGG composite metabolic pathways with the most hits of cancer driver mutation genes involved in various metabolic processes. Values are ListHits or numbers of cancer driver mutation genes from the tissue types involved in the composite metabolic pathways from the KEGG database. Red shows the number of driver genes from each tissue involved in each corresponding pathway. b. Gene-level heatmaps showing the three most common driver genes across tumor types for the KEGG metabolic pathways and the RAS signaling pathway. Red indicates an involved driver gene, while black indicates that the gene is not involved. c. Proposed RAS’ connection with oncogenic driver genes of metabolic pathways. KRAS, other top driver genes in the RAS pathway, and the top driver genes in KEGG metabolism pathways in (b) were used as seeds to retrieve their direct relations with each other or their indirect relations with other genes from the MetaCore™ database. Green lines indicate positive/activation relations, red lines indicate negative/inhibition relations, and gray lines indicate unspecified relations. The arrows indicate the relations’ directions. The nodes with blue circles are the original seed genes, whereas other genes were added based on evidence to help connect these genes, if needed. For further details on how analyses were performed see the Supplementary Note.
Conclusion and future perspectives
Although substantial progress has been made in unraveling the role of oncogenic RAS in metabolic pathways, many open questions remain about the links between RAS biology and metabolic dysregulation. For instance, the complex interplay between KRAS isoforms, oncogenic RAS alleles, tissues of tumor origin, and metabolic alterations is not well understood. The crucial question of whether RAS-mediated altered metabolic pathways are common to all RAS-driven cancers remains unanswered. Moreover, the connections between RAS and other oncogenic drivers, and the signaling and metabolic processes they each control also require attention. The elucidation of these events should also take into account the fact that metabolic phenotype is not uniform across different tumor types, and variability also exists between different tumors of the same type. A deeper understanding of these complexities, combined with the renewed excitement around targeting oncogenic RAS, will pave the way for the development of well-tolerated and effective therapies for patients with RAS-driven cancer.
Data Availability Statement
For figure 1 and Supplementary Tables 1, 2, 3, genome-wide cancer mutation data were compiled from databases and public resources, including AACR Genie (Release 6.1-public)150, COSMIC (v90)151, and cBioPortal152,153, TCGA Research Network: https://www.cancer.gov/tcga and NCI’s Genomic Data Commons (GDC)154, that are openly accessible to the public and are cited in the manuscript. The datasets derived from these resources that support the analyses and discussion presented in this article are available in the cited references. For figure 4a–b, previously published cancer driver mutation data were acquired from Ref135,136 and the International Cancer Genome Consortium/The Cancer Genome Atlas via controlled access through rigorous application and are available from these resources.
Code availability statement
For figure 4, Driver mutation genes were applied to an in-house pathway pattern extraction pipeline (PPEP) tool described in Ref155 and implemented in customized R scripts (www.r-project.org). PPEP and corresponding databases (WPS version 2) can be downloaded from WPS homepage. This tool represents a pathway-based platform for discovery integration to maximize analysis power. The tool is freely available at http://www.abcc.ncifcrf.gov/wps/wps_index.php156 or can be available on request from the correspondence author.
Supplementary Material
Acknowledgements
We thank Dr. Ming Yi, Frederick National Laboratory for Cancer Research (FNL), for helping search cancer databases for RAS-mutant samples and assisting with driver gene data preparation. We are grateful to Dr. Dwight V. Nissley, NCI RAS Initiative, FNL, for critical comments and thoughtful suggestions on the manuscript. We also thank FNL’s Scientific Publications, Graphics and Media for their technical assistance. This work was supported by grants from NCI, HHS-National Institutes of Health, under Contract No. HHSN261200800001E. Research in FM lab is supported by the National Cancer Institute of the National Institutes of Health under Award Number R35CA197709. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services (HHS), nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Competing Interests
The authors are aware of no direct conflicts with the topic of the paper; however, M.G.V.H. is a Scientic Advisory Board member for Agios Pharmaceuticals, Aeglea Biotherapetics, iTeos Therapeutics, Faeth Therapeutics, and Auron Therapeutics. F.M. is a consultant for the following companies: Amgen; Daiichi Ltd.; Ideaya Biosciences; Kura Oncology; Leidos Biomedical Research, Inc.; PellePharm; Pfizer Inc.; PMV Pharma and Quanta Therapeutics. F.M. is a consultant and co-founder for the following companies (with ownership interest including stock options): BridgeBio; DNAtrix Inc.; Olema Pharmaceuticals, Inc.; and Quartz. F.M. is the scientific director of the National Cancer Institute (NCI) RAS Initiative at the Frederick National Laboratory for Cancer Research/Leidos Biomedical Research, Inc. None of these affiliations represents a conflict of interest with respect to this manuscript.
REFERENCES:
- 1.Simanshu DK, Nissley DV & McCormick F RAS Proteins and Their Regulators in Human Disease. Cell 170, 17–33, doi: 10.1016/j.cell.2017.06.009 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stalnecker CA & Der CJ RAS, wanted dead or alive: Advances in targeting RAS mutant cancers. Sci Signal 13, doi: 10.1126/scisignal.aay6013 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pylayeva-Gupta Y, Grabocka E & Bar-Sagi D RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 11, 761–774, doi: 10.1038/nrc3106 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prior IA, Lewis PD & Mattos C A comprehensive survey of Ras mutations in cancer. Cancer Res 72, 2457–2467, doi: 10.1158/0008-5472.CAN-11-2612 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vogelstein B et al. Cancer genome landscapes. Science 339, 1546–1558, doi: 10.1126/science.1235122 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haigis KM KRAS Alleles: The Devil Is in the Detail. Trends Cancer 3, 686–697, doi: 10.1016/j.trecan.2017.08.006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cox AD, Fesik SW, Kimmelman AC, Luo J & Der CJ Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov 13, 828–851, doi: 10.1038/nrd4389 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Canon J et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223, doi: 10.1038/s41586-019-1694-1 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Hallin J et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov 10, 54–71, doi: 10.1158/2159-8290.CD-19-1167 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Janes MR et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 172, 578–589 e517, doi: 10.1016/j.cell.2018.01.006 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Hata AN & Shaw AT Resistance looms for KRAS(G12C) inhibitors. Nat Med 26, 169–170, doi: 10.1038/s41591-020-0765-z (2020). [DOI] [PubMed] [Google Scholar]
- 12.Bar-Sagi D, Knelson EH & Sequist LV A bright future for KRAS inhibitors. Nature Cancer 1, 25–27, doi: 10.1038/s43018-019-0016-8 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Fedele C et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J Exp Med 218, doi: 10.1084/jem.20201414 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ryan MB et al. Vertical Pathway Inhibition Overcomes Adaptive Feedback Resistance to KRAS(G12C) Inhibition. Clin Cancer Res 26, 1633–1643, doi: 10.1158/1078-0432.CCR-19-3523 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prior IA, Hood FE & Hartley JL The Frequency of Ras Mutations in Cancer. Cancer Res 80, 2969–2974, doi: 10.1158/0008-5472.CAN-19-3682 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moore AR, Rosenberg SC, McCormick F & Malek S RAS-targeted therapies: is the undruggable drugged? Nat Rev Drug Discov 19, 533–552, doi: 10.1038/s41573-020-0068-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674, doi: 10.1016/j.cell.2011.02.013 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Kelloff GJ et al. Progress and promise of FDG-PET imaging for cancer patient management and oncologic drug development. Clin Cancer Res 11, 2785–2808, doi: 10.1158/1078-0432.CCR-04-2626 (2005). [DOI] [PubMed] [Google Scholar]
- 19.d’Amico A Review of clinical practice utility of positron emission tomography with 18F-fluorodeoxyglucose in assessing tumour response to therapy. Radiol Med 120, 345–351, doi: 10.1007/s11547-014-0446-4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peters GJ et al. Basis for effective combination cancer chemotherapy with antimetabolites. Pharmacol Ther 87, 227–253, doi: 10.1016/s0163-7258(00)00086-3 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Luengo A, Gui DY & Vander Heiden MG Targeting Metabolism for Cancer Therapy. Cell Chem Biol 24, 1161–1180, doi: 10.1016/j.chembiol.2017.08.028 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagarajan A, Malvi P & Wajapeyee N Oncogene-directed alterations in cancer cell metabolism. Trends Cancer 2, 365–377, doi: 10.1016/j.trecan.2016.06.002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levine AJ & Puzio-Kuter AM The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 330, 1340–1344, doi: 10.1126/science.1193494 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Pavlova NN & Thompson CB The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23, 27–47, doi: 10.1016/j.cmet.2015.12.006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mayers JR et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 353, 1161–1165, doi: 10.1126/science.aaf5171 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuneva MO et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab 15, 157–170, doi: 10.1016/j.cmet.2011.12.015 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaglio D et al. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol 7, 523, doi: 10.1038/msb.2011.56 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ying H et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670, doi: 10.1016/j.cell.2012.01.058 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amendola CR et al. KRAS4A directly regulates hexokinase 1. Nature 576, 482–486, doi: 10.1038/s41586-019-1832-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yun J et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 325, 1555–1559, doi: 10.1126/science.1174229 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kole HK, Resnick RJ, Van Doren M & Racker E Regulation of 6-phosphofructo-1-kinase activity in ras-transformed rat-1 fibroblasts. Arch Biochem Biophys 286, 586–590, doi: 10.1016/0003-9861(91)90084-v (1991). [DOI] [PubMed] [Google Scholar]
- 32.Racker E, Resnick RJ & Feldman R Glycolysis and methylaminoisobutyrate uptake in rat-1 cells transfected with ras or myc oncogenes. Proc Natl Acad Sci U S A 82, 3535–3538, doi: 10.1073/pnas.82.11.3535 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chun SY et al. Oncogenic KRAS modulates mitochondrial metabolism in human colon cancer cells by inducing HIF-1alpha and HIF-2alpha target genes. Mol Cancer 9, 293, doi: 10.1186/1476-4598-9-293 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slawson C, Copeland RJ & Hart GW O-GlcNAc signaling: a metabolic link between diabetes and cancer? Trends Biochem Sci 35, 547–555, doi: 10.1016/j.tibs.2010.04.005 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stincone A et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc 90, 927–963, doi: 10.1111/brv.12140 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taparra K et al. O-GlcNAcylation is required for mutant KRAS-induced lung tumorigenesis. J Clin Invest 128, 4924–4937, doi: 10.1172/JCI94844 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Santana-Codina N et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat Commun 9, 4945, doi: 10.1038/s41467-018-07472-8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yun J et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 350, 1391–1396, doi: 10.1126/science.aaa5004 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lock R et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell 22, 165–178, doi: 10.1091/mbc.E10-06-0500 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bellier J et al. Methylglyoxal Scavengers Resensitize KRAS-Mutated Colorectal Tumors to Cetuximab. Cell Rep 30, 1400–1416 e1406, doi: 10.1016/j.celrep.2020.01.012 (2020). [DOI] [PubMed] [Google Scholar]
- 41.Bellahcene A, Nokin MJ, Castronovo V & Schalkwijk C Methylglyoxal-derived stress: An emerging biological factor involved in the onset and progression of cancer. Semin Cancer Biol 49, 64–74, doi: 10.1016/j.semcancer.2017.05.010 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Moran DM et al. KRAS mutation status is associated with enhanced dependency on folate metabolism pathways in non-small cell lung cancer cells. Mol Cancer Ther 13, 1611–1624, doi: 10.1158/1535-7163.MCT-13-0649 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Gomes AP & Blenis J A nexus for cellular homeostasis: the interplay between metabolic and signal transduction pathways. Curr Opin Biotechnol 34, 110–117, doi: 10.1016/j.copbio.2014.12.007 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM & Manning BD mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 351, 728–733, doi: 10.1126/science.aad0489 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kottakis F et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature 539, 390–395, doi: 10.1038/nature20132 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Calles A et al. Immunohistochemical Loss of LKB1 Is a Biomarker for More Aggressive Biology in KRAS-Mutant Lung Adenocarcinoma. Clin Cancer Res 21, 2851–2860, doi: 10.1158/1078-0432.CCR-14-3112 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Lv J, Wang J, Chang S, Liu M & Pang X The greedy nature of mutant RAS: a boon for drug discovery targeting cancer metabolism? Acta Biochim Biophys Sin (Shanghai) 48, 17–26, doi: 10.1093/abbs/gmv102 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Flaig TW et al. A phase I and pharmacokinetic study of silybin-phytosome in prostate cancer patients. Invest New Drugs 25, 139–146, doi: 10.1007/s10637-006-9019-2 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Siebeneicher H et al. Identification and Optimization of the First Highly Selective GLUT1 Inhibitor BAY-876. ChemMedChem 11, 2261–2271, doi: 10.1002/cmdc.201600276 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma Y et al. Ovarian Cancer Relies on Glucose Transporter 1 to Fuel Glycolysis and Growth: Anti-Tumor Activity of BAY-876. Cancers (Basel) 11, doi: 10.3390/cancers11010033 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van der Mijn JC, Panka DJ, Geissler AK, Verheul HM & Mier JW Novel drugs that target the metabolic reprogramming in renal cell cancer. Cancer Metab 4, 14, doi: 10.1186/s40170-016-0154-8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang L, Venneti S & Nagrath D Glutaminolysis: A Hallmark of Cancer Metabolism. Annu Rev Biomed Eng 19, 163–194, doi: 10.1146/annurev-bioeng-071516-044546 (2017). [DOI] [PubMed] [Google Scholar]
- 53.DeBerardinis RJ & Cheng T Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 29, 313–324, doi: 10.1038/onc.2009.358 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoo HC, Yu YC, Sung Y & Han JM Glutamine reliance in cell metabolism. Exp Mol Med 52, 1496–1516, doi: 10.1038/s12276-020-00504-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Toda K et al. Clinical Role of ASCT2 (SLC1A5) in KRAS-Mutated Colorectal Cancer. Int J Mol Sci 18, doi: 10.3390/ijms18081632 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dejure FR & Eilers M MYC and tumor metabolism: chicken and egg. EMBO J 36, 3409–3420, doi: 10.15252/embj.201796438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Son J et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105, doi: 10.1038/nature12040 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mukhopadhyay S, Saqcena M & Foster DA Synthetic lethality in KRas-driven cancer cells created by glutamine deprivation. Oncoscience 2, 807–808, doi: 10.18632/oncoscience.253 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.DeNicola GM et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109, doi: 10.1038/nature10189 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mukhopadhyay S et al. Undermining Glutaminolysis Bolsters Chemotherapy While NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res, doi: 10.1158/0008-5472.CAN-19-1363 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chio IIC et al. NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell 166, 963–976, doi: 10.1016/j.cell.2016.06.056 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Romero R et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med 23, 1362–1368, doi: 10.1038/nm.4407 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sayin VI et al. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. Elife 6, doi: 10.7554/eLife.28083 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weinberg F et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A 107, 8788–8793, doi: 10.1073/pnas.1003428107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saqcena M et al. Blocking anaplerotic entry of glutamine into the TCA cycle sensitizes K-Ras mutant cancer cells to cytotoxic drugs. Oncogene 34, 2672–2680, doi: 10.1038/onc.2014.207 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Galan-Cobo A et al. LKB1 and KEAP1/NRF2 Pathways Cooperatively Promote Metabolic Reprogramming with Enhanced Glutamine Dependence in KRAS-Mutant Lung Adenocarcinoma. Cancer Res 79, 3251–3267, doi: 10.1158/0008-5472.CAN-18-3527 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Skoulidis F & Heymach JV Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat Rev Cancer 19, 495–509, doi: 10.1038/s41568-019-0179-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Faubert B, Solmonson A & DeBerardinis RJ Metabolic reprogramming and cancer progression. Science 368, doi: 10.1126/science.aaw5473 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kaufman JM et al. LKB1 Loss induces characteristic patterns of gene expression in human tumors associated with NRF2 activation and attenuation of PI3K-AKT. J Thorac Oncol 9, 794–804, doi: 10.1097/JTO.0000000000000173 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li F et al. LKB1 Inactivation Elicits a Redox Imbalance to Modulate Non-small Cell Lung Cancer Plasticity and Therapeutic Response. Cancer Cell 27, 698–711, doi: 10.1016/j.ccell.2015.04.001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Best SA et al. Distinct initiating events underpin the immune and metabolic heterogeneity of KRAS-mutant lung adenocarcinoma. Nat Commun 10, 4190, doi: 10.1038/s41467-019-12164-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hernandez-Davies JE et al. Vemurafenib resistance reprograms melanoma cells towards glutamine dependence. J Transl Med 13, 210, doi: 10.1186/s12967-015-0581-2 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Muir A et al. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife 6, doi: 10.7554/eLife.27713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Davidson SM et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab 23, 517–528, doi: 10.1016/j.cmet.2016.01.007 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Biancur DE et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nature Communications 8, 15965, doi: 10.1038/ncomms15965 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lim JKM et al. Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc Natl Acad Sci U S A 116, 9433–9442, doi: 10.1073/pnas.1821323116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Badgley MA et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85–89, doi: 10.1126/science.aaw9872 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stockwell BR et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171, 273–285, doi: 10.1016/j.cell.2017.09.021 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.DeBerardinis RJ & Chandel NS Fundamentals of cancer metabolism. Sci Adv 2, e1600200, doi: 10.1126/sciadv.1600200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Salloum D, Mukhopadhyay S, Tung K, Polonetskaya A & Foster DA Mutant ras elevates dependence on serum lipids and creates a synthetic lethality for rapamycin. Mol Cancer Ther 13, 733–741, doi: 10.1158/1535-7163.MCT-13-0762 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kamphorst JJ et al. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci U S A 110, 8882–8887, doi: 10.1073/pnas.1307237110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Carrer A et al. Acetyl-CoA Metabolism Supports Multistep Pancreatic Tumorigenesis. Cancer Discov 9, 416–435, doi: 10.1158/2159-8290.CD-18-0567 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Padanad MS et al. Fatty Acid Oxidation Mediated by Acyl-CoA Synthetase Long Chain 3 Is Required for Mutant KRAS Lung Tumorigenesis. Cell Rep 16, 1614–1628, doi: 10.1016/j.celrep.2016.07.009 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Singh A et al. De novo lipogenesis represents a therapeutic target in mutant Kras non-small cell lung cancer. FASEB J, fj201800204, doi: 10.1096/fj.201800204 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gouw AM et al. Oncogene KRAS activates fatty acid synthase, resulting in specific ERK and lipid signatures associated with lung adenocarcinoma. Proc Natl Acad Sci U S A 114, 4300–4305, doi: 10.1073/pnas.1617709114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Viale A et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514, 628–632, doi: 10.1038/nature13611 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Amaravadi RK, Kimmelman AC & Debnath J Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov 9, 1167–1181, doi: 10.1158/2159-8290.CD-19-0292 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Guo JY et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 25, 460–470, doi: 10.1101/gad.2016311 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Guo JY et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev 27, 1447–1461, doi: 10.1101/gad.219642.113 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bryant KL et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med 25, 628–640, doi: 10.1038/s41591-019-0368-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Guo JY & White E Autophagy is required for mitochondrial function, lipid metabolism, growth, and fate of KRAS(G12D)-driven lung tumors. Autophagy 9, 1636–1638, doi: 10.4161/auto.26123 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Perera RM et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 524, 361–365, doi: 10.1038/nature14587 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.White E Exploiting the bad eating habits of Ras-driven cancers. Genes Dev 27, 2065–2071, doi: 10.1101/gad.228122.113 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kinsey CG et al. Protective autophagy elicited by RAF-->MEK-->ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med 25, 620–627, doi: 10.1038/s41591-019-0367-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee CS et al. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc Natl Acad Sci U S A 116, 4508–4517, doi: 10.1073/pnas.1817494116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Humpton TJ et al. Oncogenic KRAS Induces NIX-Mediated Mitophagy to Promote Pancreatic Cancer. Cancer Discov 9, 1268–1287, doi: 10.1158/2159-8290.CD-18-1409 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Commisso C et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637, doi: 10.1038/nature12138 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang Y & Commisso C Macropinocytosis in Cancer: A Complex Signaling Network. Trends Cancer 5, 332–334, doi: 10.1016/j.trecan.2019.04.002 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hobbs GA et al. Atypical KRAS(G12R) Mutant Is Impaired in PI3K Signaling and Macropinocytosis in Pancreatic Cancer. Cancer Discov 10, 104–123, doi: 10.1158/2159-8290.CD-19-1006 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ramirez C, Hauser AD, Vucic EA & Bar-Sagi D Plasma membrane V-ATPase controls oncogenic RAS-induced macropinocytosis. Nature 576, 477–481, doi: 10.1038/s41586-019-1831-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Grana TM, Rusyn EV, Zhou H, Sartor CI & Cox AD Ras mediates radioresistance through both phosphatidylinositol 3-kinase-dependent and Raf-dependent but mitogen-activated protein kinase/extracellular signal-regulated kinase kinase-independent signaling pathways. Cancer Res 62, 4142–4150 (2002). [PubMed] [Google Scholar]
- 102.Eberhard DA et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol 23, 5900–5909, doi: 10.1200/JCO.2005.02.857 (2005). [DOI] [PubMed] [Google Scholar]
- 103.Miller AC, Kariko K, Myers CE, Clark EP & Samid D Increased radioresistance of EJras-transformed human osteosarcoma cells and its modulation by lovastatin, an inhibitor of p21ras isoprenylation. Int J Cancer 53, 302–307, doi: 10.1002/ijc.2910530222 (1993). [DOI] [PubMed] [Google Scholar]
- 104.Bernhard EJ et al. Direct evidence for the contribution of activated N-ras and K-ras oncogenes to increased intrinsic radiation resistance in human tumor cell lines. Cancer Res 60, 6597–6600 (2000). [PubMed] [Google Scholar]
- 105.Muzumdar MD et al. Survival of pancreatic cancer cells lacking KRAS function. Nat Commun 8, 1090, doi: 10.1038/s41467-017-00942-5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Diehn M et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458, 780–783, doi: 10.1038/nature07733 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Liou GY et al. Mutant KRas-Induced Mitochondrial Oxidative Stress in Acinar Cells Upregulates EGFR Signaling to Drive Formation of Pancreatic Precancerous Lesions. Cell Rep 14, 2325–2336, doi: 10.1016/j.celrep.2016.02.029 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ogrunc M et al. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ 21, 998–1012, doi: 10.1038/cdd.2014.16 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chio IIC & Tuveson DA ROS in Cancer: The Burning Question. Trends Mol Med 23, 411–429, doi: 10.1016/j.molmed.2017.03.004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Grabocka E & Bar-Sagi D Mutant KRAS Enhances Tumor Cell Fitness by Upregulating Stress Granules. Cell 167, 1803–1813 e1812, doi: 10.1016/j.cell.2016.11.035 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kansanen E, Kivela AM & Levonen AL Regulation of Nrf2-dependent gene expression by 15-deoxy-Delta12,14-prostaglandin J2. Free Radic Biol Med 47, 1310–1317, doi: 10.1016/j.freeradbiomed.2009.06.030 (2009). [DOI] [PubMed] [Google Scholar]
- 112.Hsu PP & Sabatini DM Cancer cell metabolism: Warburg and beyond. Cell 134, 703–707, doi: 10.1016/j.cell.2008.08.021 (2008). [DOI] [PubMed] [Google Scholar]
- 113.Adhikari H & Counter CM Interrogating the protein interactomes of RAS isoforms identifies PIP5K1A as a KRAS-specific vulnerability. Nat Commun 9, 3646, doi: 10.1038/s41467-018-05692-6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kelly MR et al. Combined Proteomic and Genetic Interaction Mapping Reveals New RAS Effector Pathways and Susceptibilities. Cancer Discov 10, 1950–1967, doi: 10.1158/2159-8290.CD-19-1274 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kovalski JR et al. The Functional Proximal Proteome of Oncogenic Ras Includes mTORC2. Mol Cell 73, 830–844 e812, doi: 10.1016/j.molcel.2018.12.001 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Beganton B et al. Proximal Protein Interaction Landscape of RAS Paralogs. Cancers (Basel) 12, doi: 10.3390/cancers12113326 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mukhopadhyay S et al. Reciprocal regulation of AMP-activated protein kinase and phospholipase D. J Biol Chem 290, 6986–6993, doi: 10.1074/jbc.M114.622571 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.DeBerardinis RJ, Lum JJ, Hatzivassiliou G & Thompson CB The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7, 11–20, doi: 10.1016/j.cmet.2007.10.002 (2008). [DOI] [PubMed] [Google Scholar]
- 119.Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, Sotgia F & Lisanti MP Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol 14, 11–31, doi: 10.1038/nrclinonc.2016.60 (2017). [DOI] [PubMed] [Google Scholar]
- 120.Biancur DE et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat Commun 8, 15965, doi: 10.1038/ncomms15965 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.van den Heuvel AP, Jing J, Wooster RF & Bachman KE Analysis of glutamine dependency in non-small cell lung cancer: GLS1 splice variant GAC is essential for cancer cell growth. Cancer Biol Ther 13, 1185–1194, doi: 10.4161/cbt.21348 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kung HN, Marks JR & Chi JT Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia. PLoS Genet 7, e1002229, doi: 10.1371/journal.pgen.1002229 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nelson BS et al. Tissue of origin dictates GOT1 dependence and confers synthetic lethality to radiotherapy. Cancer Metab 8, 1, doi: 10.1186/s40170-019-0202-2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Brunelli L, Caiola E, Marabese M, Broggini M & Pastorelli R Capturing the metabolomic diversity of KRAS mutants in non-small-cell lung cancer cells. Oncotarget 5, 4722–4731, doi: 10.18632/oncotarget.1958 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Stolze B, Reinhart S, Bulllinger L, Frohling S & Scholl C Comparative analysis of KRAS codon 12, 13, 18, 61, and 117 mutations using human MCF10A isogenic cell lines. Sci Rep 5, 8535, doi: 10.1038/srep08535 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Santra T et al. An Integrated Global Analysis of Compartmentalized HRAS Signaling. Cell Rep 26, 3100–3115 e3107, doi: 10.1016/j.celrep.2019.02.038 (2019). [DOI] [PubMed] [Google Scholar]
- 127.Brunelli L, Caiola E, Marabese M, Broggini M & Pastorelli R Comparative metabolomics profiling of isogenic KRAS wild type and mutant NSCLC cells in vitro and in vivo. Sci Rep 6, 28398, doi: 10.1038/srep28398 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Moreno-Sanchez R, Rodriguez-Enriquez S, Marin-Hernandez A & Saavedra E Energy metabolism in tumor cells. FEBS J 274, 1393–1418, doi: 10.1111/j.1742-4658.2007.05686.x (2007). [DOI] [PubMed] [Google Scholar]
- 129.DeVita VT Jr. & Chu E A history of cancer chemotherapy. Cancer Res 68, 8643–8653, doi: 10.1158/0008-5472.CAN-07-6611 (2008). [DOI] [PubMed] [Google Scholar]
- 130.Vander Heiden MG Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov 10, 671–684, doi: 10.1038/nrd3504 (2011). [DOI] [PubMed] [Google Scholar]
- 131.Vander Heiden MG & DeBerardinis RJ Understanding the Intersections between Metabolism and Cancer Biology. Cell 168, 657–669, doi: 10.1016/j.cell.2016.12.039 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Vander Heiden MG Exploiting tumor metabolism: challenges for clinical translation. J Clin Invest 123, 3648–3651, doi: 10.1172/JCI72391 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lue HW et al. Metabolic reprogramming ensures cancer cell survival despite oncogenic signaling blockade. Genes Dev 31, 2067–2084, doi: 10.1101/gad.305292.117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Loftus RM & Finlay DK Immunometabolism: Cellular Metabolism Turns Immune Regulator. J Biol Chem 291, 1–10, doi: 10.1074/jbc.R115.693903 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sabarinathan R et al. , doi: 10.1101/190330 (2017). [DOI] [Google Scholar]
- 136.Consortium, I. T. P.-C. A. o. W. G. Pan-cancer analysis of whole genomes. Nature 578, 82–93, doi: 10.1038/s41586-020-1969-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kanehisa M, Goto S, Furumichi M, Tanabe M & Hirakawa M KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res 38, D355–360, doi: 10.1093/nar/gkp896 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Alam H et al. KMT2D Deficiency Impairs Super-Enhancers to Confer a Glycolytic Vulnerability in Lung Cancer. Cancer Cell 37, 599–617 e597, doi: 10.1016/j.ccell.2020.03.005 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Castellano E & Downward J RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2, 261–274, doi: 10.1177/1947601911408079 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nogueira C et al. Cooperative interactions of PTEN deficiency and RAS activation in melanoma metastasis. Oncogene 29, 6222–6232, doi: 10.1038/onc.2010.349 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wakimoto H et al. Targetable signaling pathway mutations are associated with malignant phenotype in IDH-mutant gliomas. Clin Cancer Res 20, 2898–2909, doi: 10.1158/1078-0432.CCR-13-3052 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fernandez-Garcia B et al. p73 cooperates with Ras in the activation of MAP kinase signaling cascade. Cell Death Differ 14, 254–265, doi: 10.1038/sj.cdd.4401945 (2007). [DOI] [PubMed] [Google Scholar]
- 143.Dias Carvalho P et al. KRAS Oncogenic Signaling Extends beyond Cancer Cells to Orchestrate the Microenvironment. Cancer Res 78, 7–14, doi: 10.1158/0008-5472.CAN-17-2084 (2018). [DOI] [PubMed] [Google Scholar]
- 144.Lo Re AE et al. Novel AKT1-GLI3-VMP1 pathway mediates KRAS oncogene-induced autophagy in cancer cells. J Biol Chem 287, 25325–25334, doi: 10.1074/jbc.M112.370809 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cichowski K & Jacks T NF1 tumor suppressor gene function: narrowing the GAP. Cell 104, 593–604, doi: 10.1016/s0092-8674(01)00245-8 (2001). [DOI] [PubMed] [Google Scholar]
- 146.Dang CV MYC on the path to cancer. Cell 149, 22–35, doi: 10.1016/j.cell.2012.03.003 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhang S, Xiong X & Sun Y Functional characterization of SOX2 as an anticancer target. Signal Transduct Target Ther 5, 135, doi: 10.1038/s41392-020-00242-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jeong WJ, Ro EJ & Choi KY Interaction between Wnt/beta-catenin and RAS-ERK pathways and an anticancer strategy via degradations of beta-catenin and RAS by targeting the Wnt/beta-catenin pathway. NPJ Precis Oncol 2, 5, doi: 10.1038/s41698-018-0049-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Engelman JA et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 14, 1351–1356, doi: 10.1038/nm.1890 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Consortium, A. P. G. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov 7, 818–831, doi: 10.1158/2159-8290.CD-17-0151 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tate JG et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Research 47, D941–D947, doi: 10.1093/nar/gky1015 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cerami E et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404, doi: 10.1158/2159-8290.CD-12-0095 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gao J et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1, doi: 10.1126/scisignal.2004088 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Grossman RL et al. Toward a Shared Vision for Cancer Genomic Data. N Engl J Med 375, 1109–1112, doi: 10.1056/NEJMp1607591 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Yi M, Mudunuri U, Che A & Stephens RM Seeking unique and common biological themes in multiple gene lists or datasets: pathway pattern extraction pipeline for pathway-level comparative analysis. BMC Bioinformatics 10, 200, doi: 10.1186/1471-2105-10-200 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yi M, Horton JD, Cohen JC, Hobbs HH & Stephens RM WholePathwayScope: a comprehensive pathway-based analysis tool for high-throughput data. BMC Bioinformatics 7, 30, doi: 10.1186/1471-2105-7-30 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
For figure 1 and Supplementary Tables 1, 2, 3, genome-wide cancer mutation data were compiled from databases and public resources, including AACR Genie (Release 6.1-public)150, COSMIC (v90)151, and cBioPortal152,153, TCGA Research Network: https://www.cancer.gov/tcga and NCI’s Genomic Data Commons (GDC)154, that are openly accessible to the public and are cited in the manuscript. The datasets derived from these resources that support the analyses and discussion presented in this article are available in the cited references. For figure 4a–b, previously published cancer driver mutation data were acquired from Ref135,136 and the International Cancer Genome Consortium/The Cancer Genome Atlas via controlled access through rigorous application and are available from these resources.