

Abstract
Telomere erosion in cells with insufficient levels of the telomerase reverse transcriptase (TERT), contributes to age‐associated tissue dysfunction and senescence, and p53 plays a crucial role in this response. We undertook a genome‐wide CRISPR screen to identify gene deletions that sensitized p53‐positive human cells to telomerase inhibition. We uncovered a previously unannotated gene, C16ORF72, which we term Telomere Attrition and p53 Response 1 (TAPR1), that exhibited a synthetic‐sick relationship with TERT loss. A subsequent genome‐wide CRISPR screen in TAPR1‐disrupted cells reciprocally identified TERT as a sensitizing gene deletion. Cells lacking TAPR1 or TERT possessed elevated p53 levels and transcriptional signatures consistent with p53 upregulation. The elevated p53 response in TERT‐ or TAPR1‐deficient cells was exacerbated by treatment with the MDM2 inhibitor and p53 stabilizer nutlin‐3a and coincided with a further reduction in cell fitness. Importantly, the sensitivity to treatment with nutlin‐3a in TERT‐ or TAPR1‐deficient cells was rescued by loss of p53. These data suggest that TAPR1 buffers against the deleterious consequences of telomere erosion or DNA damage by constraining p53. These findings identify C16ORF72/TAPR1 as new regulator at the nexus of telomere integrity and p53 regulation.
Keywords: C16ORF72, CRISPR‐Cas9, genome‐wide screen, p53, synthetic‐sick‐lethal, telomerase inhibitor (BIBR1532), Telomere Attrition and P53 Response 1, telomere erosion
Telomere erosion leads to an upregulation of p53 that ultimately results in cell arrest or death. Via genome‐wide screens, we identified a previously unannotated gene, C16ORF72/TAPR1 (Telomere Attrition and P53 Response 1), that was synthetically sick with telomerase inhibition or TERT loss. TAPR1 deletion led to elevated p53 activation in response to DNA damage or telomere erosion, possibly via its interaction with the E3 ubiquitin ligase, HUWE1.
1. INTRODUCTION
Telomeres, which are the repetitive, G‐rich DNA sequences found at the ends of eukaryotic chromosomes, are a lynchpin of genome integrity. The loss of sufficient telomere reserves or end‐protection complexes elicits a DNA damage response that shares characteristics with the response to a double‐stranded DNA break (de Lange, 2018). In many organisms, this loss is prevented by the telomerase reverse transcriptase (TERT) and its associated telomerase RNA component (TERC), which add new telomeric DNA repeats to chromosome ends. The recruitment of telomerase to telomeres is highly regulated and depends on subunits of the telomere‐protective complex shelterin, POT1 and TPP1 (Aramburu et al., 2020). While TERT is expressed in stem/progenitor cells and in most cancers (Lorbeer & Hockemeyer, 2020), it is transcriptionally repressed in most adult human tissues (Roake & Artandi, 2020).
Primary human cells have a limited replicative capacity in culture, called cellular senescence or the Hayflick limit, which is tightly correlated with initial telomere length (Harley et al., 1994). When telomeres become sufficiently eroded, cells with functional p53 undergo induction of p21 and cell cycle arrest (Bunz et al., 1998; Herbig et al., 2004). Cells without functional p53 may temporarily avert senescence (called M1), but the eventual onset of telomere loss, fusions, or other genomic rearrangements lead to an M2 event called crisis (Harley et al., 1994). This crucial role of p53 in telomere‐induced or other types of senescence is context‐dependent and is influenced by mitochondrial activity, mTOR signaling and reactive oxygen species production (Maddocks & Vousden, 2011).
Genome‐wide screens in yeast have identified hundreds of genes that impact telomere length maintenance (Harari et al., 2020). Human gene networks that affect cellular senescence have also been identified in genome‐wide shRNA knockdown screens (Mazzucco et al., 2017; Wang et al., 2016). For example, the deubiquitinating enzyme USP28 is an important mediator that links p53 induction and the senescence‐associated secretory phenotype (Mazzucco et al., 2017) and plays a role in the response to DNA damage (Zhang et al., 2006). Despite these advances, the genetic landscape that helps human cells cope with eroded telomeres has remained elusive. Here, we exploited CRISPR‐Cas9 genome‐wide screens to identify genes that modulate cell fitness in the presence of critically eroded telomeres, which led to the discovery of a new modulator of the p53‐dependent response to telomere erosion, C16ORF72/TAPR1.
2. RESULTS
2.1. A genome‐wide CRISPR knockout screen identifies genetic dependencies of telomerase inhibition
Given the known importance of p53 in the response to critically eroded telomeres, we chose to conduct a genome‐wide CRISPR screen in the NALM‐6 pre‐B ALL cell line because it possesses wild‐type p53 and is well suited to large‐scale genetic screens owing to the ability to grow in suspension and a near‐diploid karyotype (Bertomeu et al., 2017). We first established that deletion of TERT in NALM‐6 cells led to an eventual loss of proliferative capacity and onset of caspase activation concomitant with critical telomere erosion (Figure S1a‐c). We chose to inhibit telomerase with the small molecule BIBR1532 in a chemical‐genetic screen as it induces telomere erosion in numerous cell models and its specificity is well established, including a co‐crystal structure with T. castaneum TERT (Bryan et al., 2015). In NALM‐6 cells, we established the IC50 for telomerase inhibition by BIBR1532 in cell extracts was 1.4 µM, with a growth inhibition IC50 of 30 µM in culture, consistent with concentrations described in other studies (Figure 1a‐b; see also Appendix S1). To facilitate the identification of CRISPR‐induced indels that either exacerbated or buffered against telomerase inhibition, we chose a BIBR1532 concentration of 20 µM (~IC30) to conduct the genome‐wide screen. We confirmed this BIBR1532 concentration was sufficient to elicit telomere erosion after 20 days of treatment relative to the vehicle control, 0.1% DMSO (v/v; Figure S1c).
FIGURE 1.
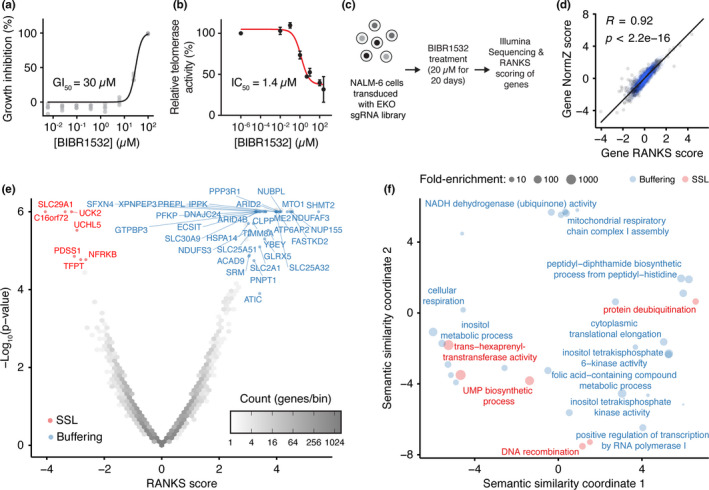
Genome‐wide CRISPR knockout screen identifies chemical‐genetic interactions with telomerase inhibition by BIBR1532. (a) Growth inhibition of NALM‐6 cells upon treatment with the indicated concentrations of BIBR1532 for 72 h (n = 4). (b) Inhibition of telomerase activity in NALM‐6 cell lysates by BIBR1532 measured by qTRAP (n = 3). (c) Genome‐wide CRISPR knockout screen schematic and genetic interaction identification using the RANKS algorithm. (d) Pearson correlation between the chemical‐genetic interaction for each gene with BIBR1532 (20 days, 20 µM) as analyzed by the RANKS or DrugZ algorithms. (e) Volcano plot showing the RANKS scores from each gene treated with BIBR1532 (20 µM) relative to the negative log‐transformed p‐value. Shades of gray in each hexagonal bin represent gene count and synthetic‐sick/lethal (SSL) chemical‐genetic interactions are labeled in red (RANKS < −2 & FDR < 0.05 for visualization purpose) while buffering interactions are labeled in blue (RANKS > 3 & FDR < 0.05 for visualization purpose). (f) Gene ontology (GO) term enrichment in the list of buffering (RANKS > 2 & FDR < 0.1, shown in blue) or SSL (RANKS < −2 & FDR < 0.1, shown in red) hits. The position of GO terms represents their semantic similarity and a subset is labeled to aid visualization
The genome‐wide screen was carried out using the previously described extended knockout (EKO) pooled CRISPR‐Cas9 sgRNA library transduced into an inducible Cas9 NALM‐6 cell clone (Benslimane et al., 2020; Bertomeu et al., 2017) treated with either 0.1% DMSO (v/v) or 20 µM BIBR1532 for 20 days (Figure 1c‐d). Several gene deletions were identified that either buffered cells against (synthetic‐rescue) or sensitized (synthetic‐sick‐lethal, SSL) to treatment with BIBR1532. Of the SSL interactions, we identified processes involved in pyrimidine salvage (e.g., UCK2), de novo pyrimidine synthesis (e.g. DHODH, PDSS1, PDSS2), and components of the INO80 chromatin remodeling complex (INO80E, TFPT, NFRKB, UCHL5; Figure 1e‐f, Table S1). These SSL relationships were recapitulated in cells deleted for the telomerase reverse transcriptase, TERT (TERT KO; Figure S1d‐g) using chemical inhibitors of the INO80 complex subunit UCHL5 (NSC‐687852 and WP‐1130), the nucleotide transporter SLC29A1 (NBMPR), and DHODH (atovaquone and brequinar; Figure S1h‐i). Given the known role of DHODH in pyrimidine biosynthesis (Evans & Guy, 2004), we tested if the reduced cell fitness observed in TERT KO cells treated with DHODH inhibitors could be rescued by nucleoside supplementation, and found that addition of exogenous nucleosides partially or completely rescued the sensitivity of TERT KO cells to atovaquone or brequinar, respectively (Figure S1j). These results are consistent with the known sensitivity of cells without telomerase to nucleotide pool homeostasis and replicative stress (Matmati et al., 2020), and with a previously reported role of INO80 in telomere replication, recombination and length homeostasis (Hu et al., 2013; Min et al., 2013; Morrison & Shen, 2009). Thus, the telomerase inhibitor BIBR1532 identified several genes whose function is known to be important to cell fitness when telomeres become eroded.
2.2. Genetic validation of a synthetic‐sick‐lethal interaction with TAPR1 (C16ORF72) in cells lacking the telomerase reverse transcriptase
One of the SSL interactions with BIBR1532 treatment was an unnamed gene, C16ORF72 (Figure 1e, Table S1), hereafter referred to as TAPR1 for Telomere Attrition and P53 Response 1. This gene encodes a predicted protein of 275 amino acids of unknown function that was also recently identified in a high‐throughput genetic screen as SSL with ATR inhibition (Hustedt et al., 2019). We disrupted TAPR1 in NALM‐6 cells with two different sgRNAs (targeting exons 1 and 2) and isolated clones disrupted for TAPR1 (TAPR1 KO; Figure 2a; see Appendix S1 for details). Competitive growth experiments to assess the relative fitness of TAPR1‐deleted NALM‐6 cells versus NALM‐6 cells deleted for both TAPR1 and TERT revealed a statistically significant decrease in cell fitness in cells lacking TAPR1 and TERT (Figure 2b‐c, S2a,b). These data show that TAPR1 exhibits a SSL interaction with cells in which telomerase function has been compromised.
FIGURE 2.
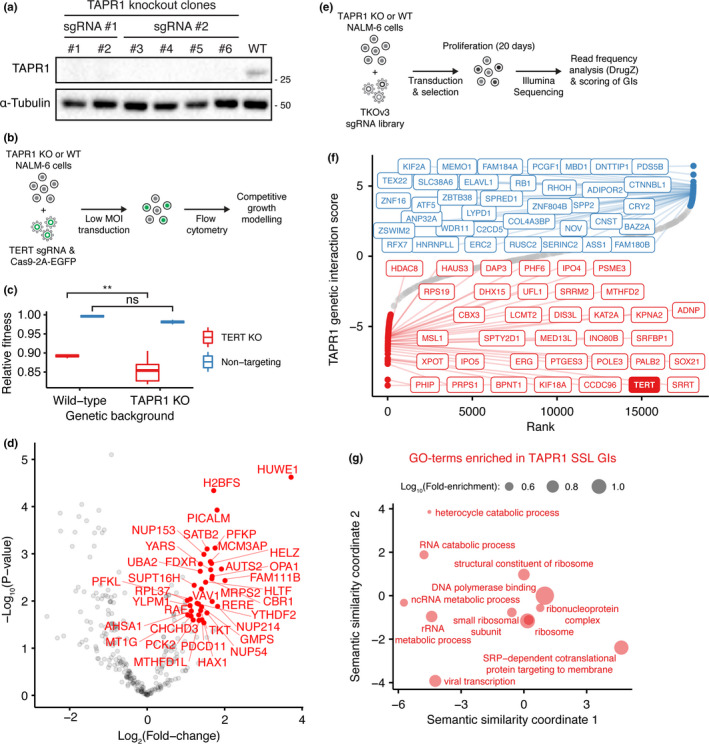
Analysis of TAPR1/TAPR1 protein and genetic interactions. (a) NALM‐6 lysates from clonal TAPR1‐disrupted (TAPR1 KO) or wild‐type NALM‐6 cells were blotted against TAPR1 and α‐tubulin (1 representative blot of 2 independent replicates). (b) Schematic of competitive growth assays used to query the genetic interaction between TAPR1 and TERT. (c) Relative fitness of TERT‐disrupted (TERT KO) or non‐targeting control in wild‐type or TAPR1‐deleted NALM‐6 cell background (n ≥ 3). (d) Volcano plot showing TAPR1 protein–protein interactions measured by BioID. Proteins with a peptide count fold‐change higher than 2 and a FDR lower than 0.1 are labeled in red (n ≥ 3). (e) Schematic of genome‐wide CRISPR screen in TAPR1‐deficient cells and genetic interaction scores. (f) Ranked TAPR1 genetic interaction scores (see Appendix S1 for details). The top 1% SSL and buffering interactions (with the top 0.2% interactions labeled) are shown in red and in blue respectively. (g) Gene ontology (GO) term enrichment in the list of SSL genetic interactions with TAPR1. The position of GO terms represents their semantic similarity and a subset is labeled to aid visualization
2.3. Identification of physical and genetic interaction partners of TAPR1
To examine physical interactors of TAPR1, we performed protein proximity labeling (BioID) in NALM‐6 cells using TAPR1 as a bait (Figure 2d, see Appendix S1). The top TAPR1‐interactor was the E3 ligase HUWE1, which is known to mediate the ubiquitin‐dependent degradation of p53, MYC, and other substrates (Giles & Grill, 2020; Gong et al., 2020; Figure 2d, Table S2). Other TAPR1 interactors identified in the BioID analysis included proteins involved in proteostasis, the mRNA export machinery, and the nuclear pore, such as MCM3AP, NUP214, NUP153, NUP54, and RAE1 (Figures 2d, S2c, Table S2). To identify TAPR1 genetic interactions, we performed a genome‐wide CRISPR screen in NALM‐6 cells disrupted for TAPR1 and compared the cell fitness profiles to wild‐type NALM‐6 cells (Figures 2e, S2d). TERT was among the top SSL genetic interactions with TAPR1 along with ACD, a gene that encodes the telomerase recruitment factor TPP1 (Figure 2f, S2d). GO‐term analysis of the list of TAPR1 SSL genetic interactions revealed enrichment of genes involved ribosome biogenesis (Figure 2g, Table S3, see also Appendix S1). We compared the TAPR1‐/‐ genome‐wide screen data with TAPR1 co‐dependencies in 769 different cell lines (Meyers et al., 2017; www.depmap.com). Genetic co‐dependencies with TAPR1 were enriched in genes involved in ribosome biogenesis, replicative senescence, and p53 signal transduction in response to DNA damage (Figure S2e‐f). For example, genes with the highest co‐dependency included negative regulators of p53 activity such as HUWE1, MDM2, MDM4, USP7, and PPM1D. Genes with a negative co‐dependency included positive p53 effectors such as TP53 itself, TP53BP1, USP28, ATM, CHEK2, and CDKN1A (p21). Several of these gene deletions also exhibited a buffering or SSL interaction with TAPR1 loss, consistent with a role of these gene products as effectors or attenuators of p53, respectively (Figure S3). These data reveal that TAPR1 exhibits a wide array of physical and genetic interactions that suggest a role in the p53 response. Importantly, our unbiased genome‐wide screen data indicate that a major genetic vulnerability of TAPR1‐deficient cells is the loss of telomere homeostasis.
2.4. The transcriptome of cells lacking TAPR1 reveals signatures consistent with p53 signaling
To further probe the potential relationship between TAPR1 and p53, we used RNA‐seq to assess the transcriptional response to deletion of either TAPR1 or TERT compared with non‐targeted control cells (Figure 3a, Table S4). Of the gene networks upregulated in TAPR1‐deficient cells, we identified processes regulated by p53, as well as the response to proteotoxic stress via ribosome biogenesis (rRNA cleavage, SSU, and LSU assembly) and chaperone‐mediated protein folding (HSP90AA1, HSP90B1, HSPA1A, HSPA5, HSPA8, HSPE1, HSPH1, DNAJA1; Figure 3b‐c, Table S4). An analysis of the transcriptome of TERT KO NALM‐6 cells similarly detected an upregulation of p53‐regulated genes (Figure S4a‐b, Table S4), with a statistically significant overlap between the TERT KO and TAPR1 KO RNA‐seq datasets (Figure 3d‐e; see Appendix S1 for details). This upregulation of p53 transcriptional targets in TERT KO cells was accompanied by an increase in p53 levels (Figure S4c). These results suggest that TAPR1 deficiency is associated with an upregulation of p53 and p53‐regulated genes, a subset of which are also observed in TERT‐deficient cells with eroded telomeres.
FIGURE 3.
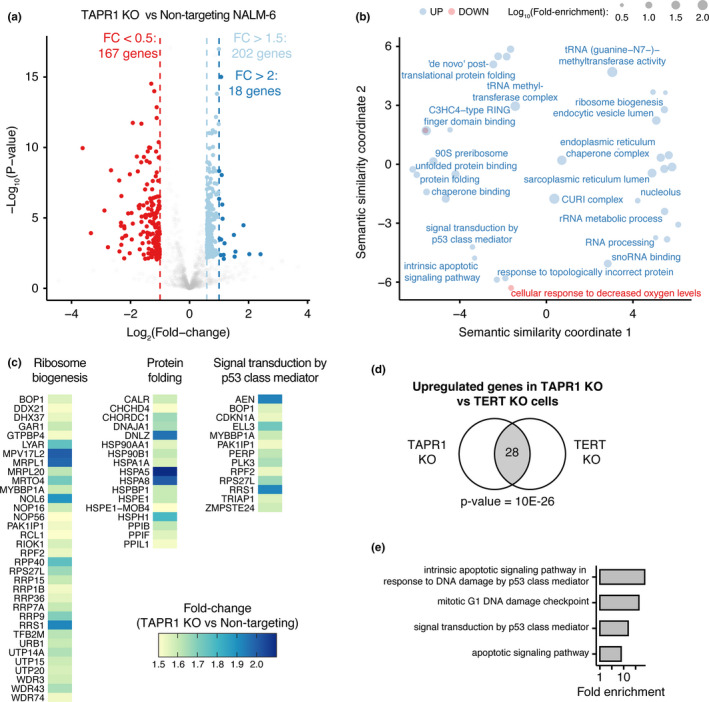
The transcriptome of cells lacking TAPR1 exhibits upregulation of p53 signaling. (a) Volcano plot showing transcriptome changes in TAPR1‐disrupted (TAPR1 KO) NALM‐6 cells relative to non‐targeting controls, with differentially expressed genes (FDR < 0.05) shown for the fold‐change thresholds indicated (n ≥ 3). (b) Gene ontology (GO) term enrichment in the list of upregulated (fold change >1.5, shown in blue) or downregulated (fold change <0.5, shown in red) genes in TAPR1 KO NALM‐6 cells. The position of GO terms represents their semantic similarity and a subset is labeled to aid visualization. (c) Heatmap showing the fold change of upregulated genes within the indicated enriched GO terms. (d) Upregulated genes in cells lacking TERT (TERT KO) or TAPR1 (TAPR1 KO) were used to calculate the statistical significance of the overlap (shown as number of genes in common in the gray‐shaded area) between the indicated lists of genes using the hypergeometric test. (e) GO‐term enrichment in the list of overlapping upregulated genes in NALM‐6 cells deleted for TERT or TAPR1
2.5. TAPR1 modulates p53‐mediated growth arrest
The upregulation of p53‐responsive genes in cells lacking TERT or TAPR1 suggested that TAPR1 may be required to attenuate the p53 response as telomeres become eroded. To test this hypothesis, we examined if the stabilization of p53 had an adverse effect on NALM‐6 TERT KO cell fitness. We first confirmed that nutlin‐3a, which inhibits the interaction of p53 with MDM2 and thereby stabilizes p53 (Secchiero et al., 2011), led to the expected upregulation of p53‐dependent genes such as BAX, CDKN1, and MDM2 (Figure S4d). We next tested the impact of nutlin‐3a treatment on the fitness of TERT KO cells, and observed that nutlin‐3a elicited a reduction in cell fitness compared with wild‐type cells (Figure S4e). This impact of nutlin‐3a on cell fitness in TERT KO cells was TP53‐dependent, since deletion of TP53 rescued the reduction in cell fitness (Figure S4e). These data show that NALM‐6 cells exhibited a p53‐dependent reduction in cell fitness upon TERT loss.
In keeping with the transcriptional upregulation of p53‐dependent targets, we observed an increase in p53 protein levels in TAPR1‐deficient cells relative to wild‐type cells that was also apparent after nutlin‐3a treatment (Figure 4a). We also observed a decrease in relative fitness of TAPR1‐deleted cells upon treatment with nutlin‐3a or doxorubicin, a topoisomerase II poison that activates p53 in response to DNA damage (Figure 4b). These treatments led to a statistically significant upregulation of the p53 transcriptional target CDKN1A (Figure 4c‐d). These data suggested that the increased sensitivity of TAPR1‐deficient cells to nutlin‐3a and doxorubicin could be due to a p53‐mediated elevation of CDKN1A. We therefore tested whether disruption of TP53 would rescue the sensitivity of TAPR1‐deficient cells to nutlin‐3a. Competitive growth assays in cells disrupted for TAPR1 or TP53 alone, or both genes deleted together (Figure 4e, see Appendix S1), revealed that the deletion of TP53 was epistatic to deletion of TAPR1 with respect to nutlin3a sensitivity (Figure 4f; compare first and third lower graphs). These data are consistent with a role of TAPR1 in buffering against the deleterious effect on cell fitness when p53 is activated by eroded telomeres, DNA damage or MDM2 inhibition.
FIGURE 4.
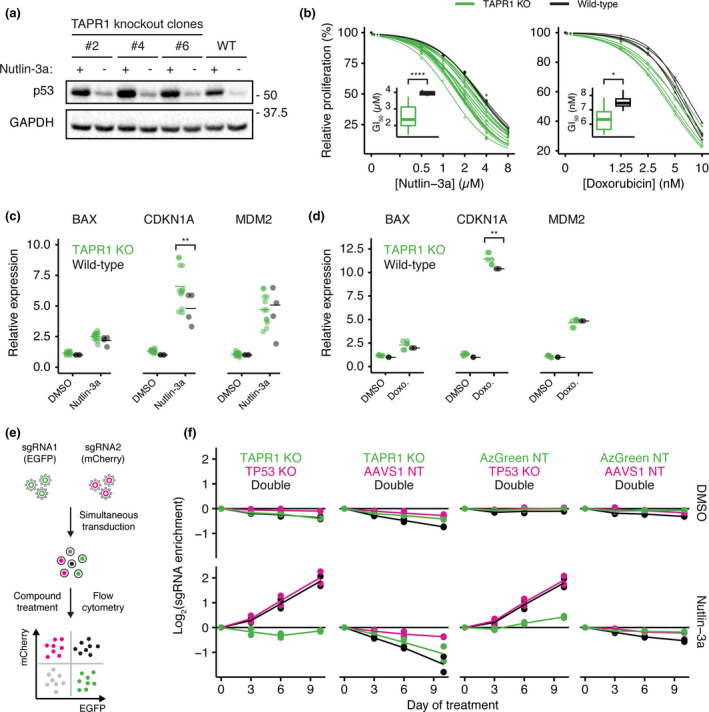
The impact of TAPR1 loss on cell fitness is TP53‐dependent. (a) NALM‐6 lysates from clonal TAPR1‐disrupted (TAPR1 knockout) or wild‐type (WT) NALM‐6 cells treated with nutlin‐3a (2 µM, 4 h) were blotted against p53 and GAPDH (1 representative blot of 3 independent replicates). (b) Relative proliferation of TAPR1‐disrupted (TAPR1 KO) or wild‐type cells treated with the indicated concentrations of nutlin‐3a or doxorubicin for 72 h. Dose–response curves were fitted and the GI50 concentration is shown as inset plots (n ≥ 3). (c) Relative expression of the indicated transcripts in wild‐type or TAPR1 KO cells treated with 2 µM nutlin‐3a or 0.1% (v/v) DMSO for 4 h (n ≥ 4). (d) Relative expression of the indicated transcripts in wild‐type or TAPR1 KO cells treated with 0.5 µM doxorubicin (Doxo.) or 0.1% (v/v) DMSO for 4 h (n ≥ 2). (e) Competitive growth assay schematic for NALM‐6 cells transduced with non‐targeting sgRNAs and sgRNAs targeting TAPR1 and TP53. (f) sgRNA enrichment in NALM‐6 cells treated with 2 µM nutlin‐3a or 0.1% (v/v) DMSO shown for the indicated TAPR1/TP53 sgRNA combinations (n = 2)
3. DISCUSSION
We conducted a genome‐wide screen using CRISPR‐Cas9 in the p53‐positive cancer cell line, NALM‐6, for gene deletions that sensitized cells to telomere erosion. We uncovered a previously unannotated gene, C16ORF72, which we named TAPR1 (Telomere Attrition and P53 Response 1) as it exhibits a strong synthetic genetic interaction with telomerase inhibition or deletion of TERT, and appears to taper the response to p53 upon loss of telomere integrity.
Prior to this study, C16ORF72/TAPR1 was identified as a hit in human genome‐wide CRISPR‐Cas9 chemogenetic screens for sensitizers to ATR kinase inhibition or hydroxyurea (Hustedt et al., 2019; Benslimane et al., 2020). C16ORF72/TAPR1 is also a target of miR‐134, a microRNA associated with tumorigenesis and chemo‐resistance (Shuang et al., 2015), and is associated with rare de novo CNVs in autism spectrum disorder (ASD) and schizophrenia (Levinson et al., 2011; Sanders et al., 2011). In our study, TAPR1 was identified as an interactor with HUWE1, and C16ORF72/TAPR1 also interacts with HUWE1 in previous high‐throughput mass spectrometry surveys (Hein et al., 2015; Thompson et al., 2014). This HUWE1‐TAPR1 interaction may be biologically relevant, especially as p53 is ubiquitinated by HUWE1 (Giles & Grill, 2020; Gong et al., 2020). This interaction could be mediated by a domain of unknown function within TAPR1 (DUF4588), as the S. cerevisiae homologue of HUWE1 (Tom1) interacts with a protein of unknown function called YJR056C that contains the same DUF4588 domain (Sung et al., 2016). Tom1 and HUWE1 are involved in the degradation of ribosomal (ERISQ pathway) and non‐ribosomal proteins (Sung et al., 2016; Xu et al., 2016), and in the regulation of genes involved in ribosomal biogenesis (Gomez‐Herreros et al., 2017). HUWE1 is also implicated in the p53 response to proteotoxic stress caused by the imbalance between ribosomal protein (RP) and ribosomal RNA (rRNA) production (Hipp et al., 2019). We found several genes involved in proteostasis that were upregulated in TAPR1‐deficient cells, including components of the CURI complex that coordinate the HSF1‐dependent response to the imbalance between RPs and rRNA, subunits of the ribosomal SSU and LSU processosome, and other transcriptional targets of HSF1 (Figure 3a, Table S4). Future studies will determine if TAPR1 functions with HUWE1 in these ubiquitin‐mediated processes, and their relationship to the response to telomere erosion and DNA damage.
Our choice of a p53‐positive cell line for the CRISPR‐Cas9 screen enabled the identification of TAPR1 as a gene whose function enables NALM‐6 cells to better cope with telomere loss via the attenuation of p53 activation. This result is supported by our unbiased genome‐wide screen data that identified deletion of TERT or ACD (encoding TPP1) as SSL with TAPR1 loss. The role of TAPR1 may not be limited to a telomere‐induced DNA damage response (DDR), as TAPR1‐deficient cells were sensitive to nutlin‐3a, doxorubicin, and ATR inhibition (Hustedt et al., 2019; this study), and HUWE1 is also involved in the DDR (Hall et al., 2007; Mandemaker et al., 2017; Myant et al., 2017 Guo, 2020 #299). Intriguingly, the profile of genetic interactions also hint that TAPR1 might link proteotoxic stress and genome/telomere integrity, especially as aneuploidy is known to induce proteotoxic stress due to gene copy number imbalance (Brennan et al., 2019; Ohashi et al., 2015; Oromendia et al., 2012).
Cells must optimize the p53 response to satisfy two countervailing forces: too little p53 activity allows the propagation of damaged genomes that sets cells on the road to tumorigenesis, whereas too much p53 activity can drive cells into premature senescence (Wu & Prives, 2018). Because p53 lies at the nexus of cancer and aging, its appropriate regulation necessitates many context‐specific checks and balances that shape its overall activity (Sharpless & DePinho, 2002). Notably, a gradient of p53 activity appears to govern stem cell fitness in part through cell non‐autonomous competition effects that are manifest at lower levels of p53 activation (Bondar & Medzhitov, 2010). Our identification of TAPR1 as an attenuator of p53 in the context of low level genomic damage caused by eroded telomeres adds a new element to the p53 regulatory network (Figure 5). Given the role of p53 activation as a key mediator of senescence (Wu & Prives, 2018), it will be important to determine how TAPR1 expression is regulated in primary cells as telomeres erode, and if TAPR1 serves to protract the entry into senescence in normal cells and/or tissues by counterbalancing p53 activation. In mice with eroded telomeres, p53 deficiency initially rescues the adverse effects of telomere dysfunction on proliferation but then promotes subsequent tumor initiation (Chin et al., 1999). TAPR1 may thus play a role in delaying cellular senescence on the one hand and suppressing tumorigenesis or apoptosis in response to genotoxic stress on the other hand. Regardless of its specific physiological roles, TAPR1 represents a previously unappreciated genetic modulator of the delicate equilibrium that governs p53 activity. Future work will illuminate the precise genetic and cellular contexts in which TAPR1 is important for p53 function and other biological responses to stress, and how these functions impinge on cancer and aging.
FIGURE 5.
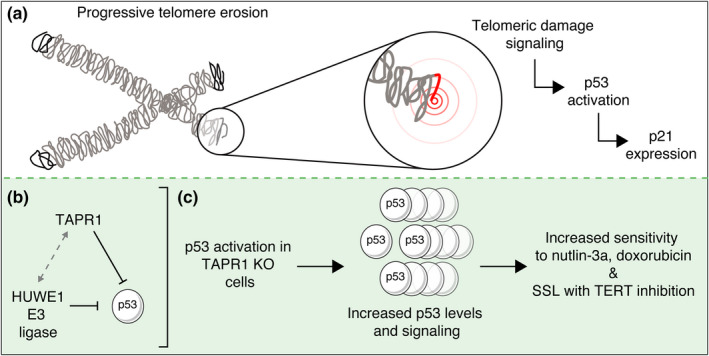
Model of TAPR1 modulation of p53 signaling in response to telomere erosion. (a) In the absence of sufficient telomere‐replenishing activity (e.g. telomerase inhibition), telomeres progressively erode and eventually induce a DNA damage response, resulting in p53 activation and induction of transcriptional targets such as CDKN1a/p21. (b) TAPR1 attenuates p53 activation in the pre‐B cell line NALM‐6. Proximity labeling identified HUWE1, an E3 ubiquitin ligase that targets p53 for degradation, as an interaction partner with TAPR1. Whether TAPR1 attenuates p53 in a HUWE1‐dependent manner has not yet been determined. (c) Deletion of TAPR1 leads to excessive p53 induction and increased sensitivity of cells treated with the MDM2 inhibitor nutlin‐3a or the DNA damaging agent doxorubicin, and a synthetic/sick/lethal (SSL) phenotype in cells either deleted for telomerase (TERT) or treated with the telomerase inhibitor BIBR1532. TAPR1 may also limit p53 activation in other circumstances including DNA damage, senescence and cancer
4. EXPERIMENTAL PROCEDURES
4.1. Cell culture
NALM‐6 cells are an immortalized cell line that was established from the peripheral blood of a man, 19 years of age, with acute lymphoblastic leukemia (see Key Resources in Appendix S1) and was provided by the laboratory of Steve Elledge. Cells were propagated in 10% FBS (v/v) RPMI 1640 medium and 37°C, and sub‐cultured every 2–3 days. HEK293 T cells (for lentiviral packaging) were propagated in 10% FBS (v/v) DMEM medium at 5% (v/v) CO2 and 37°C, and sub‐cultured every 2–3 days. Parental and modified (e.g. knockout) cell lines used for this study were tested for mycoplasma contamination by standard multiplex PCR. The genotype of knockout lines (populations or clonal isolates) were confirmed by indel sequencing (see Appendix S1). Further details on generation of knockout lines, proliferation assays and competitive growth modeling are provided in Appendix S1.
4.2. Genome‐wide CRISPR screens
The CRISPR knockout screen to identify chemical‐genetic interactions with BIBR1532 was performed as previously described with the following changes (Benslimane et al., 2020). A NALM‐6 clone with inducible Cas9 expression previously transduced with the EKO library was treated with Doxycycline (2 µg/mL) for 8 days to induce Cas9 expression and knockout generation followed by treatment (28 million cells per treatment, corresponding to 100 cells/sgRNA) with 20 µM BIBR1532 or 0.1% (v/v) DMSO for 20 days. For the genome‐wide CRISPR screen in TAPR1‐deficient cells, wild‐type NALM‐6 cells or two independent clones of TAPR1‐disrupted NALM‐6 cells were transduced with the TKOv3 sgRNA library as previously described (Benslimane et al., 2020; Hart et al., 2017). Briefly, 120 million cells were transduced at a MOI of 0.5, corresponding to a coverage of 800 cells/sgRNA. Two days after infection, cells were selected with puromycin (0.5 µg/mL) for 4 days. After selection, 36 million cells from each of the cell populations harboring the TKOv3 libraries proliferated for an additional 15 days (with sub‐culturing every 3 days). After the sgRNA library outgrowth period (for both genome‐wide screens performed), cells were collected, genomic DNA extracted and sgRNA sequences were PCR‐amplified followed by Illumina sequencing (Illumina NextSeq500) at the Genomic Platform at Institute for Research in Immunology and Cancer (IRIC). Reads were aligned using Bowtie2.2.5 in the forward direction only (‐‐norc option) with otherwise default parameters and total read counts per sgRNA tabulated to obtain sgRNA frequencies. For the DMSO controls, sgRNA sequencing reads from multiple DMSO‐treated samples were pooled as background, to decrease noise. Chemical‐genetic interactions were scored relative to DMSO using the previously published RANKS algorithm (Bertomeu et al., 2017), which estimates the p‐values for the fold change between individual sgRNAs to control sgRNAs (non‐targeting sgRNAs in the EKO library) or the DrugZ algorithm (see Appendix S1, Key Resources). The TAPR1 genetic interaction data was analyzed using the DrugZ algorithm by comparing the sgRNA read frequency in each TAPR1‐deleted NALM‐6 clone against the summed reads from two replicates in a wild‐type NALM‐6 background. The NormZ scores for each clone were subsequently summed to generate a TAPR1 genetic interaction (GI) score to highlight genes that show a SSL or buffering phenotype in both TAPR1‐deleted clones.
4.3. Transcriptome analysis by RNA‐Seq
RNA from 1 million clonal NALM‐6 cells of the indicated genotypes was extracted using the QIAGEN Mini RNeasy kit according to the manufacturer's protocol. Sample purity was assessed by nanodrop using 260/280 nm and 260/230 nm ratios. Total RNA was quantified by QuBit (ABI) and 1 µg of total RNA was used for library preparation. RNA quality control was assessed with the Bioanalyzer RNA 6000 Nano assay on the 2100 Bioanalyzer system (Agilent technologies) and all samples had a RIN of 10. Library preparation was performed with the KAPA mRNAseq Hyperprep kit (KAPA, #KK8581). Libraries were quantified by QuBit and BioAnalyzer and diluted to 10 nM before normalization by qPCR using the KAPA library quantification kit (KAPA; #KK4973). Libraries were then pooled to equimolar concentration. Sequencing was performed with the Illumina Nextseq500 on half a flowcell of Nextseq 75 cycles High Output v2 using 2.8 pM of the pooled libraries. Around 20 million single‐end PF reads were generated per sample. Analysis of RNA‐seq reads is described further in Appendix S1.
4.4. Gene list enrichment and network analysis
Statistical significance of the overlap between gene lists was calculated using the hypergeometric test in R. Gene ontology term enrichment was calculated with the “gprofiler2” package in R where the gene lists were considered an unordered query and with subsequent filtering for GO terms (GO:BP, GO:CC, GO:MF as data sources) that contain <1000 terms with an adjusted p‐value below .05 (see Appendix S1, Key Resources). Further filtering of the enriched GO terms was performed to remove redundant GO terms that share a high semantic similarity using the REVIGO tool (http://revigo.irb.hr/) to aid with visualization. The network of TAPR1 co‐dependencies was built using the Avana CRISPR dataset (Public release 20Q3). This dataset is available as a matrix of normalized dependency scores that represents the effect of a CRISPR knockout in a given cell line. By calculating a correlation matrix using the Pearson method, co‐dependencies with a given gene knockout (most highly correlated or anti‐correlated genes) can be retrieved. The top 100 co‐dependencies with TAPR1 (absolute Pearson correlation coefficient) as well as their respective top 10 co‐dependencies were combined (as nodes, with edges representing the co‐dependency). The network shown in Figure S3 was built with isolated nodes (i.e. genes with only 1 co‐dependency) removed for visual simplicity. The network was built and visualized as an undirected force‐directed layout (with minimal manual adjustments were made to improve legibility) using the “network”, “ggnetwork,” and “ggplot2” packages in R. The nodes in the network are color‐coded based on the score in the TAPR1 genetic interaction screen.
4.5. Quantification and statistical analysis
Unless otherwise indicated, statistical analyses were performed on PRISM 8 (www.graphpad.com). Statistical significance was carried out with a Student t test (2 groups), or with ANOVA (more than 2 groups) using the Sidak or Tukey correction for multiple comparisons.
CONFLICT OF INTEREST
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Y.B., M.T., and L.H. conceptualized the study. Y.B., M.S.‐O., J.C.‐H., T.B., D.H. C.H., and E.B. carried out the methodology and investigation. Y.B. and L.H. wrote the manuscript with editorial input from all authors. P.T., M.T., and L.H. acquired funding and supervised the study.
Supporting information
Appendix S1
Table S1
Table S2
Table S3
Table S4
ACKNOWLEDGEMENTS
The authors thank past and present members of the laboratory, and IRIC for providing FACS, Illumina sequencing, RNA‐seq, and mass spectrometry services. We thank Steve Elledge for providing NALM‐6 cells, Jason Moffat for the TKOv3 CRISPR library, Dan Durocher and Daniel Schramek for sharing unpublished findings, and Anne‐Claude Gingras and Brian Raught for providing the miniTURBO plasmids and sharing unpublished data. We thank Tsz‐Wai Chu for assistance with the Telomere Restriction Fragment protocol, and Corinne St‐Denis for technical assistance. This research was supported by the Canadian Institutes for Health Research (367427 to L.H.; FDN‐167277 to M.T.), and a Genomics Technology Platform award from Genome Canada and Genome Quebec (M.T. and P.T.). Salaries were supported by the Fonds de Recherche du Quebec – Nature et Technologie (FRQ‐NT) and the Cole Foundation (Y.B), an Institute for Data Valorisation (IVADO) postdoctoral fellowship (J. C.‐H.) and a Canada Research Chair in Systems and Synthetic Biology (M.T.).
DATA AVAILABILITY STATEMENT
All unique/stable reagents generated in this study will be made available upon reasonable request. Depending on the reagent, a completed materials transfer agreement may also be required. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lea Harrington ( lea.harrington@umontreal.ca ). CRISPR screen HTS sequencing and RNA‐Seq data are uploaded and available at NCBI GEO https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE160869. The mass spectrometry proteomics data (BioID) have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD022128 and 10.6019/PXD022128 (see also Appendix S1, Key Resources).
REFERENCES
- Aramburu, T. , Plucinsky, S. , & Skordalakes, E. (2020). POT1‐TPP1 telomere length regulation and disease. Computational and Structural Biotechnology Journal, 18, 1939–1946. 10.1016/j.csbj.2020.06.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benslimane, Y. , Bertomeu, T. , Coulombe‐Huntington, J. , McQuaid, M. , Sanchez‐Osuna, M. , Papadopoli, D. , Avizonis, D. , Russo, M. S. T. , Huard, C. , Topisirovic, I. , Wurtele, H. , Tyers, M. , & Harrington, L. (2020). Genome‐wide screens reveal that resveratrol induces replicative stress in human cells. Molecular Cell, 79(5), 846–856.e8. 10.1016/j.molcel.2020.07.010 [DOI] [PubMed] [Google Scholar]
- Bertomeu, T. , Coulombe‐Huntington, J. , Chatr‐Aryamontri, A. , Bourdages, K. G. , Coyaud, E. , Raught, B. , Xia, Y. , & Tyers, M. (2017). A high‐resolution genome‐wide CRISPR/Cas9 viability screen reveals structural features and contextual diversity of the human cell‐essential proteome. Molecular and Cellular Biology, 38(1), e00302‐17. 10.1128/MCB.00302-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondar, T. , & Medzhitov, R. (2010). p53‐mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell, 6(4), 309–322. 10.1016/j.stem.2010.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan, C. M. , Vaites, L. P. , Wells, J. N. , Santaguida, S. , Paulo, J. A. , Storchova, Z. , Harper, J. W. , Marsh, J. A. , & Amon, A. (2019). Protein aggregation mediates stoichiometry of protein complexes in aneuploid cells. Genes & Development, 33(15–16), 1031–1047. 10.1101/gad.327494.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan, C. , Rice, C. , Hoffman, H. , Harkisheimer, M. , Sweeney, M. , & Skordalakes, E. (2015). Structural basis of telomerase inhibition by the highly specific BIBR1532. Structure, 23(10), 1934–1942. 10.1016/j.str.2015.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunz, F. , Dutriaux, A. , Lengauer, C. , Waldman, T. , Zhou, S. , Brown, J. P. , Sedivy, J. M. , Kinzler, K. W. , & Vogelstein, B. (1998). Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science, 282(5393), 1497–1501. 10.1126/science.282.5393.1497 [DOI] [PubMed] [Google Scholar]
- Chin, L. , Artandi, S. E. , Shen, Q. , Tam, A. , Lee, S. L. , Gottlieb, G. J. , Greider, C. W. , & DePinho, R. A. (1999). p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell, 97(4), 527–538. 10.1016/s0092-8674(00)80762-x [DOI] [PubMed] [Google Scholar]
- de Lange, T. (2018). Shelterin‐mediated telomere protection. Annual Review of Genetics, 52, 223–247. 10.1146/annurev-genet-032918-021921 [DOI] [PubMed] [Google Scholar]
- Evans, D. R. , & Guy, H. I. (2004). Mammalian pyrimidine biosynthesis: Fresh insights into an ancient pathway. Journal of Biological Chemistry, 279(32), 33035–33038. 10.1074/jbc.R400007200 [DOI] [PubMed] [Google Scholar]
- Giles, A. C. , & Grill, B. (2020). Roles of the HUWE1 ubiquitin ligase in nervous system development, function and disease. Neural Development, 15(1), 6. 10.1186/s13064-020-00143-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Herreros, F. , Margaritis, T. , Rodriguez‐Galan, O. , Pelechano, V. , Begley, V. , Millan‐Zambrano, G. , Morillo‐Huesca, M. , Munoz‐Centeno, M. C. , Perez‐Ortin, J. E. , de la Cruz, J. , Holstege, F. C. P. , & Chavez, S. (2017). The ribosome assembly gene network is controlled by the feedback regulation of transcription elongation. Nucleic Acids Research, 45(16), 9302–9318. 10.1093/nar/gkx529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, X. , Du, D. , Deng, Y. , Zhou, Y. , Sun, L. , & Yuan, S. (2020). The structure and regulation of the E3 ubiquitin ligase HUWE1 and its biological functions in cancer. Investigational New Drugs, 38(2), 515–524. 10.1007/s10637-020-00894-6 [DOI] [PubMed] [Google Scholar]
- Hall, J. R. , Kow, E. , Nevis, K. R. , Lu, C. K. , Luce, K. S. , Zhong, Q. , & Cook, J. G. (2007). Cdc6 stability is regulated by the Huwe1 ubiquitin ligase after DNA damage. Molecular Biology of the Cell, 18(9), 3340–3350. 10.1091/mbc.e07-02-0173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harari, Y. , Gershon, L. , Alonso‐Perez, E. , Klein, S. , Berneman, Y. , Choudhari, K. , Singh, P. , Sau, S. , Liefshitz, B. , & Kupiec, M. (2020). Telomeres and stress in yeast cells: When genes and environment interact. Fungal Biology, 124(5), 311–315. 10.1016/j.funbio.2019.09.003 [DOI] [PubMed] [Google Scholar]
- Harley, C. B. , Kim, N. W. , Prowse, K. R. , Weinrich, S. L. , Hirsch, K. S. , West, M. D. , Bacchetti, S. , Hirte, H. W. , Counter, C. M. , Greider, C. W. , Piatyszek, M. A. , Wright, W. E. , & Shay, J. W. (1994). Telomerase, cell immortality, and cancer. Cold Spring Harbor Symposia on Quantitative Biology, 59, 307–315. 10.1101/sqb.1994.059.01.035 [DOI] [PubMed] [Google Scholar]
- Hart, T. , Tong, A. H. Y. , Chan, K. , Van Leeuwen, J. , Seetharaman, A. , Aregger, M. , Chandrashekhar, M. , Hustedt, N. , Seth, S. , Noonan, A. , Habsid, A. , Sizova, O. , Nedyalkova, L. , Climie, R. , Tworzyanski, L. , Lawson, K. , Sartori, M. A. , Alibeh, S. , Tieu, D. , … Moffat, J. (2017). Evaluation and design of genome‐wide CRISPR/SpCas9 knockout screens. G3: Genes – Genomes ‐ Genetics, 7(8), 2719–2727. 10.1534/g3.117.041277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein, M. Y. , Hubner, N. C. , Poser, I. , Cox, J. , Nagaraj, N. , Toyoda, Y. , Gak, I. A. , Weisswange, I. , Mansfeld, J. , Buchholz, F. , Hyman, A. A. , & Mann, M. (2015). A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell, 163(3), 712–723. 10.1016/j.cell.2015.09.053 [DOI] [PubMed] [Google Scholar]
- Herbig, U. , Jobling, W. A. , Chen, B. P. , Chen, D. J. , & Sedivy, J. M. (2004). Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Molecular Cell, 14(4), 501–513. 10.1016/s1097-2765(04)00256-4 [DOI] [PubMed] [Google Scholar]
- Hipp, M. S. , Kasturi, P. , & Hartl, F. U. (2019). The proteostasis network and its decline in ageing. Nature Reviews Molecular Cell Biology, 20(7), 421–435. 10.1038/s41580-019-0101-y [DOI] [PubMed] [Google Scholar]
- Hu, Y. , Tang, H. B. , Liu, N. N. , Tong, X. J. , Dang, W. , Duan, Y. M. , Fu, X. H. , Zhang, Y. , Peng, J. , Meng, F. L. , & Zhou, J. Q. (2013). Telomerase‐null survivor screening identifies novel telomere recombination regulators. PLoS Genetics, 9(1), e1003208. 10.1371/journal.pgen.1003208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hustedt, N. , Alvarez‐Quilon, A. , McEwan, A. , Yuan, J. Y. , Cho, T. , Koob, L. , Hart, T. , & Durocher, D. (2019). A consensus set of genetic vulnerabilities to ATR inhibition. Open Biology, 9(9), 190156. 10.1098/rsob.190156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinson, D. F. , Duan, J. , Oh, S. , Wang, K. , Sanders, A. R. , Shi, J. , Zhang, N. , Mowry, B. J. , Olincy, A. , Amin, F. , Cloninger, C. R. , Silverman, J. M. , Buccola, N. G. , Byerley, W. F. , Black, D. W. , Kendler, K. S. , Freedman, R. , Dudbridge, F. , Pe'er, I. , … Gejman, P. V. (2011). Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. American Journal of Psychiatry, 168(3), 302–316. 10.1176/appi.ajp.2010.10060876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorbeer, F. K. , & Hockemeyer, D. (2020). TERT promoter mutations and telomeres during tumorigenesis. Current Opinion in Genetics & Development, 60, 56–62. 10.1016/j.gde.2020.02.001 [DOI] [PubMed] [Google Scholar]
- Maddocks, O. D. , & Vousden, K. H. (2011). Metabolic regulation by p53. Journal of Molecular Medicine (Berlin), 89(3), 237–245. 10.1007/s00109-011-0735-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandemaker, I. K. , van Cuijk, L. , Janssens, R. C. , Lans, H. , Bezstarosti, K. , Hoeijmakers, J. H. , Demmers, J. A. , Vermeulen, W. , & Marteijn, J. A. (2017). DNA damage‐induced histone H1 ubiquitylation is mediated by HUWE1 and stimulates the RNF8‐RNF168 pathway. Scientific Reports, 7(1), 15353. 10.1038/s41598-017-15194-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matmati, S. , Lambert, S. , Geli, V. , & Coulon, S. (2020). Telomerase repairs collapsed replication forks at telomeres. Cell Reports, 30(10), 3312–3322 e3313. 10.1016/j.celrep.2020.02.065 [DOI] [PubMed] [Google Scholar]
- Mazzucco, A. E. , Smogorzewska, A. , Kang, C. , Luo, J. , Schlabach, M. R. , Xu, Q. , Patel, R. , & Elledge, S. J. (2017). Genetic interrogation of replicative senescence uncovers a dual role for USP28 in coordinating the p53 and GATA4 branches of the senescence program. Genes & Development, 31(19), 1933–1938. 10.1101/gad.304857.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers, R. M. , Bryan, J. G. , McFarland, J. M. , Weir, B. A. , Sizemore, A. E. , Xu, H. , Dharia, N. V. , Montgomery, P. G. , Cowley, G. S. , Pantel, S. , Goodale, A. , Lee, Y. , Ali, L. D. , Jiang, G. , Lubonja, R. , Harrington, W. F. , Strickland, M. , Wu, T. , Hawes, D. C. , … Tsherniak, A. (2017). Computational correction of copy number effect improves specificity of CRISPR‐Cas9 essentiality screens in cancer cells. Nature Genetics, 49(12), 1779–1784. 10.1038/ng.3984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min, J. N. , Tian, Y. , Xiao, Y. , Wu, L. , Li, L. , & Chang, S. (2013). The mINO80 chromatin remodeling complex is required for efficient telomere replication and maintenance of genome stability. Cell Research, 23(12), 1396–1413. 10.1038/cr.2013.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison, A. J. , & Shen, X. (2009). Chromatin remodelling beyond transcription: the INO80 and SWR1 complexes. Nature Reviews Molecular Cell Biology, 10(6), 373–384. 10.1038/nrm2693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myant, K. B. , Cammareri, P. , Hodder, M. C. , Wills, J. , Von Kriegsheim, A. , Gyorffy, B. , Rashid, M. , Polo, S. , Maspero, E. , Vaughan, L. , Gurung, B. , Barry, E. , Malliri, A. , Camargo, F. , Adams, D. J. , Iavarone, A. , Lasorella, A. , & Sansom, O. J. (2017). HUWE1 is a critical colonic tumour suppressor gene that prevents MYC signalling, DNA damage accumulation and tumour initiation. EMBO Molecular Medicine, 9(2), 181–197. 10.15252/emmm.201606684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi, A. , Ohori, M. , Iwai, K. , Nakayama, Y. , Nambu, T. , Morishita, D. , Kawamoto, T. , Miyamoto, M. , Hirayama, T. , Okaniwa, M. , Banno, H. , Ishikawa, T. , Kandori, H. , & Iwata, K. (2015). Aneuploidy generates proteotoxic stress and DNA damage concurrently with p53‐mediated post‐mitotic apoptosis in SAC‐impaired cells. Nature Communications, 6, 7668. 10.1038/ncomms8668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oromendia, A. B. , Dodgson, S. E. , & Amon, A. (2012). Aneuploidy causes proteotoxic stress in yeast. Genes & Development, 26(24), 2696–2708. 10.1101/gad.207407.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roake, C. M. , & Artandi, S. E. (2020). Regulation of human telomerase in homeostasis and disease. Nature Reviews Molecular Cell Biology, 21(7), 384–397. 10.1038/s41580-020-0234-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders, S. J. , Ercan‐Sencicek, A. G. , Hus, V. , Luo, R. , Murtha, M. T. , Moreno‐De‐Luca, D. , Chu, S. H. , Moreau, M. P. , Gupta, A. R. , Thomson, S. A. , Mason, C. E. , Bilguvar, K. , Celestino‐Soper, P. B. S. , Choi, M. , Crawford, E. L. , Davis, L. , Davis Wright, N. R. , Dhodapkar, R. M. , DiCola, M. , … State, M. W. (2011). Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with Autism. Neuron, 70(5), 863–885. 10.1016/j.neuron.2011.05.00 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secchiero, P. , Bosco, R. , Celeghini, C. , & Zauli, G. (2011). Recent advances in the therapeutic perspectives of Nutlin‐3. Current Pharmaceutical Design, 17(6), 569–577. 10.2174/138161211795222586 [DOI] [PubMed] [Google Scholar]
- Sharpless, N. E. , & DePinho, R. A. (2002). p53: good cop/bad cop. Cell, 110(1), 9–12. 10.1016/s0092-8674(02)00818-8 [DOI] [PubMed] [Google Scholar]
- Shuang, T. , Wang, M. , & Chang, S. (2015). Hybrid‐polymerase chain reaction to identify novel target genes of miR‐134 in paclitaxel resistant human ovarian carcinoma cells. Oncology Letters, 9(6), 2910–2916. 10.3892/ol.2015.3137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung, M. K. , Porras‐Yakushi, T. R. , Reitsma, J. M. , Huber, F. M. , Sweredoski, M. J. , Hoelz, A. , Hess, S. , & Deshaies, R. J. (2016). A conserved quality‐control pathway that mediates degradation of unassembled ribosomal proteins. Elife, 5, e19105. 10.7554/eLife.19105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J. W. , Nagel, J. , Hoving, S. , Gerrits, B. , Bauer, A. , Thomas, J. R. , Kirschner, M. W. , Schirle, M. , & Luchansky, S. J. (2014). Quantitative Lys–Gly‐Gly (diGly) proteomics coupled with inducible RNAi reveals ubiquitin‐mediated proteolysis of DNA damage‐inducible transcript 4 (DDIT4) by the E3 ligase HUWE1. Journal of Biological Chemistry, 289(42), 28942–28955. 10.1074/jbc.M114.573352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Xu, Q. , Sack, L. , Kang, C. , & Elledge, S. J. (2016). A gain‐of‐function senescence bypass screen identifies the homeobox transcription factor DLX2 as a regulator of ATM‐p53 signaling. Genes & Development, 30(3), 293–306. 10.1101/gad.271445.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, D. , & Prives, C. (2018). Relevance of the p53‐MDM2 axis to aging. Cell Death and Differentiation, 25(1), 169–179. 10.1038/cdd.2017.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Y. , Anderson, D. E. , & Ye, Y. (2016). The HECT domain ubiquitin ligase HUWE1 targets unassembled soluble proteins for degradation. Cell Discovery, 2, 16040. 10.1038/celldisc.2016.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, D. , Zaugg, K. , Mak, T. W. , & Elledge, S. J. (2006). A role for the deubiquitinating enzyme USP28 in control of the DNA‐damage response. Cell, 126(3), 529–542. 10.1016/j.cell.2006.06.039 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Table S1
Table S2
Table S3
Table S4
Data Availability Statement
All unique/stable reagents generated in this study will be made available upon reasonable request. Depending on the reagent, a completed materials transfer agreement may also be required. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lea Harrington ( lea.harrington@umontreal.ca ). CRISPR screen HTS sequencing and RNA‐Seq data are uploaded and available at NCBI GEO https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE160869. The mass spectrometry proteomics data (BioID) have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD022128 and 10.6019/PXD022128 (see also Appendix S1, Key Resources).