

Abstract
Objectives
Genetic variant classification is a challenge in rare adult‐onset disorders as in SCA‐PRKCG (prior spinocerebellar ataxia type 14) with mostly private conventional mutations and nonspecific phenotype. We here propose a refined approach for clinicogenetic diagnosis by including protein modeling and provide for confirmed SCA‐PRKCG a comprehensive phenotype description from a German multi‐center cohort, including standardized 3D MR imaging.
Methods
This cross‐sectional study prospectively obtained neurological, neuropsychological, and brain imaging data in 33 PRKCG variant carriers. Protein modeling was added as a classification criterion in variants of uncertain significance (VUS).
Results
Our sample included 25 cases confirmed as SCA‐PRKCG (14 variants, thereof seven novel variants) and eight carriers of variants assigned as VUS (four variants) or benign/likely benign (two variants). Phenotype in SCA‐PRKCG included slowly progressive ataxia (onset at 4–50 years), preceded in some by early‐onset nonprogressive symptoms. Ataxia was often combined with action myoclonus, dystonia, or mild cognitive‐affective disturbance. Inspection of brain MRI revealed nonprogressive cerebellar atrophy. As a novel finding, a previously not described T2 hyperintense dentate nucleus was seen in all SCA‐PRKCG cases but in none of the controls.
Interpretation
In this largest cohort to date, SCA‐PRKCG was characterized as a slowly progressive cerebellar syndrome with some clinical and imaging features suggestive of a developmental disorder. The observed non‐ataxia movement disorders and cognitive‐affective disturbance may well be attributed to cerebellar pathology. Protein modeling emerged as a valuable diagnostic tool for variant classification and the newly described T2 hyperintense dentate sign could serve as a supportive diagnostic marker of SCA‐PRKCG.
Introduction
Spinocerebellar ataxias (SCAs) denote rare autosomal‐dominant progressive ataxias and the most frequently diagnosed genotypes harbor trinucleotide‐repeat expansions. 1 , 2 In 2000, the first of now more than 20 conventional mutation SCA genotypes was identified in the protein kinase C gamma (PRKCG) gene 3 , 4 (MIM 176980) and termed SCA 14 5 or more recently SCA‐PRKCG. 6 Its prevalence estimates are continually rising and range from <1% to <6% in ataxia cohorts after repeat expansion SCAs have been excluded. 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14
The neuron‐specific gamma isoform of PRKC is most abundantly expressed in cerebellar Purkinje cells. 15 It regulates their dendritic growth and calcium permeability and the elimination of climbing fiber synapses. 16 , 17 It is yet unclear how different variants affect protein localization, aggregation, and kinase activity. 18 , 19 Histopathological reports in variants at residue H101 describe selective Purkinje cell loss in the cerebellar cortex without atrophy in neocortex or deep cerebellar nuclei. 20 , 21
SCA‐PRKCG diagnosis relies on genetic testing, however, the classification of variants according to current guidelines 22 , 23 is challenging. Some of the proposed criteria do not apply to this rare disorder: There is no established model of pathogenicity (criterion PS3) and mostly private mutations render population frequencies/novelty less informative (criterion PS4). Segregation analysis (supporting criterion PP1) is valuable but difficult to pursue outside the research context. Thus, novel PRKCG variants are often classified as of uncertain significance (VUS) and must be interpreted against clinical findings 24 . Conversely, informative phenotype description critically depends on correct genetic case ascertainment. Previous clinical descriptions (comprehensive list in Table S1) suggest a rather unspecific mildly progressive cerebellar ataxia of variable age of onset, in some cases with additional symptoms usually considered of extra‐cerebellar etiology.
To improve the clinicogenetic diagnosis of SCA‐PRKCG, this observational study investigates the SCA‐PRKCG phenotype in a multicenter cohort of PRKCG variant carriers with prospective and standardized data that is available to the clinician in an ataxia patient work‐up: clinical exam, brain structural MRI, and neuropsychologic and neuro‐ophthalmologic testing. 25 We add protein modeling as a supporting PP3‐criterion in the classification of pathogenic genetic variants and we explore phenotypic differences to nonconfirmed cases.
Instrumental gait analysis, 26 MR spectroscopy, 27 and detailed visual testing 28 from subcohorts are reported elsewhere.
Methods
The study included symptomatic subjects carrying a PRKCG variant considered of either pathogenic or uncertain significance, with referrals from five German university ataxia clinics. Exclusion criteria were any other disease involving the central nervous system, history of severe head trauma, severe psychiatric comorbidity, or contraindication for MRI investigation. A matched group of control subjects was included for the analysis of neuropsychology and brain imaging. Controls were assessed with identical protocol and had no history of neurologic or psychiatric disease, head trauma, no abnormal findings in neurological examination or contraindication for MRI investigation.
Subjects were investigated at one or both of the coordinating and neuroimaging centers (Berlin and Jülich). The study was approved by their respective Institutional Review Boards. Written informed consent was obtained from all participants.
All PRKCG variants were re‐evaluated by a geneticist (P. B.), first, according to current guidelines put forward by the American College of Medical Genetics and Genomics. 22 Minor allele frequency above 1% derived from published databases was set as stand‐alone evidence for “benign” variants. Second, a refined approach was applied that included results of protein modeling as a supporting criterion. This modeling evaluated the protein‐specific functional impact of a given variant (A. G.). Multitemplate homology modeling using the SwissModel webserver 29 was generated that covered the full PRKCG protein except for residues 0–35 (see Data S1). Within this model, two zinc‐binding cavities are formed by residues C49, C52, C77, and H74 (1st zinc‐binding site) and C85, C35, C69, and H36 (2nd zinc‐binding site).
The onset of ataxia was defined as onset of permanent gait ataxia. The clinical assessment comprised a structured medical history (including questions to capture history of seizures, myoclonus, dystonia, tremor, spasticity, cognitive or affective disturbance, pain, impairment of mobility and hand function), clinical examination, and application of clinical ratings of ataxia (SARA 30 , range 0–40) and nonataxia symptoms (INAS 31 , range 0–16). Comprehensive neuropsychological tests were applied (description and reference in Data S1) and validated screening for affective disturbance 32 (HADS) or cognitive impairment 33 (DemTect) performed using published cut‐offs.
The afferent visual pathway was assessed by functional testing (visual acuity) and retinal imaging (optical coherence tomography).
Brain MRI included 3D T1‐ and T2‐weighted sequences obtained at 3T (Magnetom Trio system, Siemens Healthineers, Germany).
Electrophysiology results and previously obtained routine brain MRI for longitudinal assessment were made available by patients and not part of the prospective protocol.
Further detail on methods is provided in Data S1.
Data processing and statistical analysis
PRKCG variants were checked against published reports and ordered by location to detect possible feature clusters (Tables 1 and 2, Table S1). Missing information was handled per item as indicated. Neuropsychological test results in confirmed SCA‐PRKCG were compared to results obtained in healthy controls matched for age, sex, education levels according to the International Standard of Education 34 and handedness according to Edinburgh Handedness Inventory. 35 Between‐group comparisons used t‐tests or Wilcoxon rank‐sum test as indicated in Table 4. In the case of between‐group difference in test results, correlations with ataxia ratings (SARA) and depression score (HADS‐D) were performed and, if significant, additional effects of age explored via partial correlations. Spearman or Pearson test was used as indicated in Table 5.
Table 1.
List of 20 PRKCG variants ordered by residue along with genetic classification by current (ACMG) guidelines and comprehensive classification decision which included results of protein modeling as supporting criterion, and CADD scores (not available for insertions or deletions as indicated).
| PRKCG domain | PRKCG variants (all heterozygous) | n Subjects/families | Interpretation of protein modeling | Classification by current (ACMG) guidelines | Classification including protein modeling results | CADD PHRED score | CADD score |
|---|---|---|---|---|---|---|---|
| N‐terminal | c.68G > A, p. G23E | 3/2 | Not covered | VUS | Likely pathogenic 1 | 26.8 | 3.88 |
| c.70G > T, p. A24S | 1/1 | Not covered | VUS | Likely pathogenic 2 | 26.0 | 3.74 | |
| Regulatory domain C1 | c.146G > A, p. C49Y | 1/1 | 1st Zinc‐binding site probably disrupted | VUS | Likely pathogenic 3 | 27.7 | 4.00 |
| c.197G > A, p. C66Y | 5/2 | Pathogenic | Pathogenic 4 | 27.0 | 3.91 | ||
| c.207C > T, p. Cys69Cys | 2/1 | Benign | Benign | Benign 5 | 14.8 | 1.24 | |
| c.229T > A, p. C77S | 1/1 | 1st Zinc‐binding site probably disrupted | VUS | Likely pathogenic 3 | 25.2 | 3.57 | |
| c.244‐252delACCTTCGAG, p. T82_E84del | 2/1 | 2nd zinc‐binding site may be structurally affected | VUS | Likely pathogenic 6 | N/A | N/A | |
| c.338_340delTCT, p. F113_C114delinsC | 2/1 | Likely pathogenic | Likely pathogenic 7 | N/A | N/A | ||
| c.347A > C, p. H116P | 1/1 | Close to 2nd zinc‐binding site, may probably disrupt zinc binding | VUS | Likely pathogenic 8 | 27.0 | 3.91 | |
| c.353G > A, p. E118D | 2/1 | Pathogenic | Pathogenic 4 | 27.6 | 4.00 | ||
| c.367G > A, p. G123R | 1/1 | Likely pathogenic | Likely pathogenic 7 | 29.3 | 4.19 | ||
| c.368G > C, p. G123A | 1/1 | Change in local environment that may affect protein structure | VUS | Likely pathogenic 2 | 26.3 | 3.81 | |
| c.391T > C, p. C131R | 2/1 | Pathogenic | Pathogenic 4 | 28.6 | 4.10 | ||
| c.392G > C, p. C131S | 2/1 | Pathogenic | Pathogenic 4 | 25.9 | 3.73 | ||
| c.449G > A, p. C150Y | 1/1 | 2nd Zinc‐binding site probably disrupted | VUS | Likely pathogenic 2 | 29.7 | 4.23 | |
| Regulatory domain C2 | c.518T > G, p. I173S | 1/1 | Change to polar residue in conserved hydrophobic region may affect structure | VUS | VUS 9 | 25.2 | 3.55 |
| c638G > A, p. R213Q | 2/1 | Benign | Likely benign | Likely benign 9 | 23.2 | 2.84 | |
| c.768G > C, p. M256I | 1/1 | Near putative calcium‐binding site, but no change predicted in chemical properties | VUS | VUS 9 | 22.4 | 2.44 | |
| Kinase domain | c.1901G > A, p. R634H | 1/1 | Benign | VUS | VUS 9 | 24.8 | 3.44 |
| C‐terminal | c.2032C > G, p. P678A | 1/1 | Benign | VUS | VUS 9 | 23.2 | 2.85 |
Novel variants are written in bold. ACMG, American College of Medical Genetics and Genomics; CADD, combined annotation dependent depletion; PRKCG, protein kinase C gamma; VUS, variant of uncertain significance.
Based on ACMG variant classification (VUS), typical phenotype in two independent families within this study.
Based on ACMG variant classification (VUS), typical phenotype plus another SCA‐PRKCG patient with PRKCG missense variant at same residue.
Based on ACMG variant classification (VUS), typical phenotype plus abnormal PRKCG protein modeling.
Based on ACMG variant classification (pathogenic).
Based on ACMG variant classification (benign).
Based on ACMG variant classification (VUS), typical phenotype plus abnormal PRKCG protein modeling.
Based on ACMG variant classification (likely pathogenic).
Based on ACMG variant classification (VUS), typical phenotype in two families plus abnormal PRKCG protein modeling.
Based on ACMG variant classification (VUS), PRKCG protein modeling suggests functional consequence.
Based on ACMG variant classification (likely benign).
Based on ACMG variant classification (VUS); no further supportive evidence.
asterisk relates to explanation given in table caption, that CADD scores are not available for insertions or deletions.
Table 2.
Individual findings of selected outcomes in all 33 carriers of PRKCG variants, including four subjects with (likely) benign variants and four carriers of VUS.
| PRKCG variant | Disease onset | Clinical rating | Nonataxia movement disorder | Possible pyramidal | Possible peripheral | Cognitive/psychiatric screening | Nerve conduction studies abnormal | Brain MRI findings | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Domain | Variant classification including protein modeling results | Age at onset | disease duration (y) | SARA | INAS count | Myoclonus | Dystonia | Tremor | Increased tone/plantar extensor | Hyperreflexia | Areflexia (a) or mild pallhypesthesia (p) | Muscle atrophy | DemTect | HADS depression | HADS anxiety | Peripheral nerve | Tibial nerve somatosensory‐evoked potentials | Central motor conduction time | Cerebellar atrophy | Brainstem atrophy | T2 hyperintense dentate nucleus |
| N‐terminal | Likely pathogenic | 30 | 23 | 10 | 2 | no | yes | no | no | yes | no | no | 14 | 1 | 3 | no | no | no | 2 | no | yes |
| Likely pathogenic | 26 | 16 | 7.25 | 2 | yes | yes | no | no | no | no | no | 18 | 7 | 3 | no | no | no | 1 | no | yes | |
| Likely pathogenic | 35 | 20 | 8 | 2 | no | no | no | no | yes | no | no | 11 | 1 | 3 | yes | no | n.a. | 2 | no | yes | |
| Likely pathogenic | 30 | 21 | 7 | 1 | yes | no | no | no | no | no | no | 17 | 4 | 6 | no | no | n.a. | 1 | no | yes | |
| Regulatory domain C1 | Likely pathogenic | 37 | 17 | 15.5 | 2 | no | yes | no | no | no | a | no | 13 | 7 | 4 | no | yes | n.a. | 1 | no | yes |
| Pathogenic | 13 | 20 | 7 | 1 | yes | no | no | no | yes | no | no | 18 | 9 | 4 | no | no | no | 2 | no | yes | |
| Pathogenic | 48 | 14 | 25 | 2 | yes | no | no | no | no | no | no | 17 | 9 | 12 | yes | yes | no | 2 | no | yes | |
| Pathogenic | 50 | 15 | 6 | 3 | no | no | no | no | no | a, p | no | 13 | 4 | 5 | yes | yes | n.a. | 1 | no | yes | |
| Pathogenic | 33 | 4 | 8 | 0 | no | no | no | no | no | no | no | 18 | 9 | 6 | n.a. | n.a. | n.a. | 2 | no | yes | |
| Pathogenic | 48 | 4 | 15 | 2 | no | no | no | no | yes | no | no | 7 | 12 | 4 | n.a. | n.a. | n.a. | 1 | no | yes | |
| Benign | 40 | 27 | 15.5 | 5 | no | no | no | no | no | a, p | yes | 12 | 2 | 3 | n.a. | n.a. | n.a. | 1 | no | yes | |
| Benign | 38 | 9 | 2 | 3 | no | yes | no | no | no | a, p | no | 13 | 0 | 1 | n.a. | n.a. | n.a. | 1 | no | yes | |
| Likely pathogenic | 20 | 34 | 11.5 | 5 | no | no | no | no | no | p | yes | 14 | 7 | 5 | no | no | n.a. | 2 | no | yes | |
| Likely pathogenic | 36 | 34 | 12 | 2 | no | no | no | no | no | no | yes | 14 | 7 | 4 | no | yes | no | 2 | no | yes | |
| Likely pathogenic | 43 | 19 | 10 | 3 | no | no | no | no | yes | no | no | 12 | 6 | 1 | n.a. | n.a. | n.a. | 2 | no | yes | |
| Likely pathogenic | 47 | 11 | 12 | 2 | yes | yes | no | no | no | no | no | 13 | 8 | 10 | yes | no | no | 2 | no | yes | |
| Likely pathogenic | 20 | 11 | 4.5 | 0 | no | no | yes | no | no | no | no | 14 | 3 | n.a. | no | no | no | 2 | no | yes | |
| Likely pathogenic | 4 | 41 | 13 | 0 | no | no | no | no | no | no | no | 12 | 4 | 11 | yes | yes | no | 1 | no | yes | |
| Pathogenic | 45 | 11 | 12 | 2 | no | yes | no | no | no | no | yes | 14 | 9 | 8 | yes | n.a. | n.a. | 3 | no | yes | |
| Pathogenic | 50 | 3 | 5 | 0 | yes | no | no | no | no | no | no | 15 | 11 | 12 | n.a. | n.a. | n.a. | 2 | no | yes | |
| Likely pathogenic | 31 | 35 | 12.25 | 2 | no | no | no | no | no | no | no | 15 | n.a. | n.a. | n.a. | n.a. | n.a. | 2 | no | yes | |
| Likely pathogenic | 37 | 34 | 11 | 5 | yes | no | no | no | no | p | yes | n.a. | n.a. | n.a. | no | n.a. | n.a. | 2 | no | yes | |
| Pathogenic | 11 | 46 | 11 | 2 | yes | yes | yes | no | no | no | no | 15 | 12 | 2 | n.a. | n.a. | n.a. | 2 | no | yes | |
| Pathogenic | 29 | 2 | 7 | 2 | no | no | no | no | no | no | no | 10 | 3 | 6 | yes | no | n.a. | 1 | no | yes | |
| Pathogenic | 41 | 8 | 12.25 | 2 | yes | yes | yes | no | no | p | no | 12 | 4 | 4 | no | n.a. | n.a. | 2 | no | yes | |
| Pathogenic | 26 | 3 | 3 | 0 | yes | no | no | no | no | no | no | 18 | 9 | 6 | n.a. | n.a. | n.a. | 1 | no | yes | |
| Likely pathogenic | 20 | 29 | 5 | 1 | no | yes | no | no | no | no | no | 14 | 10 | 10 | yes | no | n.a. | 2 | no | yes | |
| Regulatory domain C2 | VUS | 44 | 9 | 7 | 0 | no | no | yes | no | no | no | no | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | 3 | no | yes |
| Likely benign | no ataxia | n.a. | 2 | 6 | yes | no | yes | no | no | a | yes | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | 0 | no | n.a. | |
| Likely benign | n.a. | 0 | 3 | no | no | no | no | no | p | yes | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | 0 | no | n.a. | ||
| VUS | 49 | 8 | 7.5 | 3 | no | yes | no | yes | no | no | no | 13 | n.a. | n.a. | no | yes | no | n.a.* | n.a.* | n.a.* | |
| Kinase domain | VUS | 46 | 6 | 12.5 | 1 | no | no | no | no | no | no | no | 13 | 6 | 3 | yes | yes | no | 2 | yes | no |
| C‐terminal | VUS | 47 | 4 | 7 | 2 | no | no | yes | no | no | no | no | 9 | 6 | 4 | n.a. | n.a. | n.a. | 2 | yes | no |
Subjects are ordered by location of variant (same order as Table 1).
PRKCG protein kinase C gamma; VUS variant of uncertain significance.
n.a. not assessed.
yes/no refers to symptom, sign or abnormal finding present:
MRI cerebellar atrophy rated by inspection as (0) none, (1) mild, (2) moderate and (3) severe.
MRI results: only report of routine MRI available.
Table 4.
Results of neuropsychological testing performed in 23 confirmed SCA‐PRKCG (13 females; age 49 ± 11 years) and 23 age‐ and sex‐matched controls (13 females; age 49 ± 11 years) along with statistics for group comparison (t‐test or *Wilcoxon rank‐sum (WRS) test).
| Domain | Specific skill | Test acronym |
n SCA‐PRKCG/control |
Mean/median | SD/SE | T/U * | P‐value |
|---|---|---|---|---|---|---|---|
| Attention | Selective attention | TAP‐Flexibility |
22 22 |
783.5 605.7 |
247.8 191.6 |
2.7 | 0.011 |
| Inhibition | TAP‐Go/NoGo |
22 23 |
550.1 506.3 |
58.7 48.2 |
2.7 | 0.010 | |
| Processing speed | TAP‐Alertness* |
22 23 |
298.5 246 |
12.1 13.6 |
135* | 0.007* | |
| Executive functioning | Affinity of interference | FWIT* |
23 23 |
30.7 27.6 |
5 2.1 |
207* | 0.207* |
| Interhemispheric motor inhibition | COMO* |
23 23 |
4.6 0 |
0.7 0.7 |
122.5* | 0.001* | |
| Visuospatial mental rotation | LPS 50 + subtest 7 |
23 22 |
11.8 18.3 |
4.3 9 |
‐3.1 | 0.004 | |
| Language | Vocabulary | MWT‐B* |
23 23 |
28 29 |
1 0.8 |
213* | 0.254* |
| Phonemic verbal fluency | RWT phon. * |
23 22 |
21 19.5 |
1.2 1.1 |
242* | 0.802* | |
| Semantic verbal fluency | RWT sem. |
23 22 |
24.6 25.6 |
5.7 5.3 |
‐0.6 | 0.553 | |
| Memory | Figural memory | ROCFT learning* |
23 23 |
18.5 23 |
1.5 1.3 |
192* | 0.111* |
| ROCFT delayed* |
23 23 |
18 22 |
1.5 1.5 |
211.5* | 0.244* | ||
| Visual spatial working memory | CBT* |
23 23 |
10 10 |
0.3 0.4 |
207.5* | 0.196* | |
| Verbal episodic memory | VLMT learning* |
23 23 |
59 57 |
1.8 1.6 |
260* | 0.921* | |
| VLMT delayed* |
23 23 |
13 12 |
0.5 0.5 |
226* | 0.391* | ||
| Verbal working memory | Digit‐span test* |
23 23 |
11 12 |
0.3 0.5 |
185* | 0.077* | |
| Perception | Emotional perception | FEFA |
22 22 |
42.5 43.1 |
3.3 2.8 |
‐0.6 | 0.524 |
Groups did not differ regarding education according to the International Standard of Education or handedness according to Edinburgh Handedness Inventory. For test descriptions and references see Table S1.
Table 5.
Correlation of neuropsychological test results – performed only for those tests that indicated group differences, see Table 4 – to ataxia severity, depressive symptoms, and age, using Spearman’s rho or Pearson’s r as indicated with results.
| Domain | Test acronym | Zero‐order correlations SCA‐PRKCG/control | Partial correlations SCA‐PRKCG | ||
|---|---|---|---|---|---|
| Ataxia (SARA) | Depression symptoms (HADS‐D) | Age | Ataxia (SARA) controlled for age | ||
| Attention | TAP Flexibility |
ρ = 0.43 P = 0.044 – |
ρ = 0.01 P = 0.950 ρ = 0.11 P = 0.639 |
r = 0.41 P = 0.060 r = 0.58 P = 0.004 |
ρ = 0.24 P = 0.298 |
| TAP Go/NoGo |
ρ = 0.33 P = 0.133 – |
ρ = −0.15 P = 0.509 ρ = 0.18 P = 0.416 |
r = 0.64 P = 0.001 r = 0.64 P = 0.001 |
– | |
| TAP Alertness |
ρ = 0.20 P = 0.380 – |
ρ = 0‐.38 P = 0.085 ρ = 0.08 P = 0.710 |
ρ = 0.52 P = 0.013 ρ = 0.38 P = 0.071 |
– | |
| Executive functioning | COMO |
ρ = 0.33 P = 0.129 – |
ρ = 0.03 P = 0.909 ρ = 0‐.04 P = 0.849 |
ρ = 0‐.07 P = 0.755 ρ = 0.34 P = 0.110 |
– |
| LPS 50 + subtest 7 |
ρ = 0‐.55 P = 0.008 – |
ρ = 0.21 P = 0.358 ρ = 0.11 P = 0.619 |
r = −0.46 P = 0.026 r = −0.46 P = 0.032 |
ρ = −0.21 P = 0.351 |
|
Results of visual pathway assessment (H. Z., T. O.) and brain MRI (M. Sch., S. G.) were each independently inspected and interpreted by two experienced raters. Interpretation of imaging results included a comparison to healthy controls with groups matched for age and sex. The results of electrophysiological testing were reviewed by examiners of the respective centers.
Results
Genetic findings
We investigated 33 subjects (22 families) with 20 PRKCG variants, thereof 11 novel variants (Fig. 1).
Figure 1.
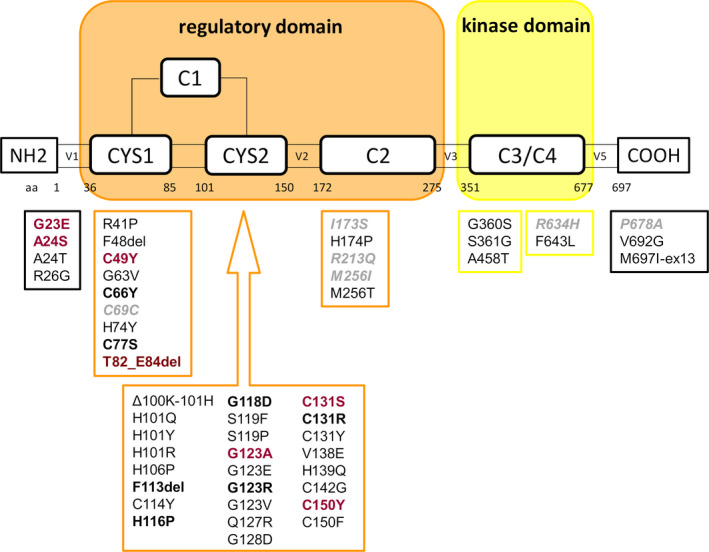
Overview of all PRKCG variants published to date. Variants included in this case series are marked in bold, novel (likely) pathogenic variants are marked in red. Variants of uncertain significance or likely benign variants of our cohort are marked in gray italic.
All participants were of Caucasian ethnicity. The genetic re‐evaluation according to current guidelines suggested (likely) pathogenicity in only 6/20 variants and (likely) benign variants in 2/20, whereas VUS was assigned in 12/20 (this included nine novel variants and three variants with suggested pathogenicity in previous reports (p. C77S, p. H116P, p. I173S, see Table S1). Of note, five novel variants were in residues previously published as disease‐causing (p. A24S, p. G123A, p. G131S, p. C150Y, p. M256I).
When protein modeling and evaluation of clinical findings from this and previous reports were considered in the second step of variant classification, a genetic diagnosis of SCA‐PRKCG was assigned to 14/20 variants (25 subjects/16 families), including seven novel likely pathogenic variants (Table 1, Fig. 1). All these variants were located in the N‐terminal or C1 regulatory domain. Results of structural modeling clearly supported a pathogenic relevance in five variants classified as VUS by current guidelines, as they were likely to impose critical changes at zinc‐binding sites. In two other variants, possibly deleterious conformational changes were assigned due to changes in local structural properties (p. G123A) or change from hydrophobic to polar residue (p. I173S). In three other VUS and two benign/likely benign variants, no relevant effects were predicted on protein structure or function, whereas the two N‐terminal variants were not covered by the model. Four remaining VUS were located in the C2 regulatory, kinase, or C‐terminal domain. Two variants were classified as (likely) benign despite one (p. C69C) located within the mutational hotspot/1st zinc‐binding site.
Family history was negative or not informative in only 3/25 SCA‐PRKCG subjects – thereof one singular index case – but in 3/4 VUS carriers and 2/4 carriers of benign/likely benign PRKCG variants.
Phenotype in SCA‐PRKCG
An excerpt of individual findings in 33 subjects (whether confirmed SCA‐PRKCG or benign/VUS) is presented in Table 2, whereas Table 3 summarizes the SCA‐PRKCG phenotype based on 25 subjects confirmed as SCA‐PRKCG as annotated in Table 2.
Table 3.
Summary of clinical findings in 25 cases of confirmed SCA‐PRKCG given as proportion (%) of sample with specific findings, ordered by possible structural attribution.
| Structure | System | Sign | Observed or reported (% of sample) | n = 25 unless stated otherwise |
|---|---|---|---|---|
| Cerebellum | Cerebellar ataxia (SARA ratings > 0) | Gait ataxia | 25 (100) | |
| Stance ataxia | 21 (84) | |||
| Dysarthria | 23 (92) | |||
| Limb ataxia | 25 (100) | |||
| Cerebellar oculomotor signs | Saccadic pursuit | 25 (100) | ||
| Saccadic dysmetria | 24 (96) | |||
| Gaze‐evoked nystagmus | 15 (60) | |||
| Non‐ataxia movement disorder, observed or reported | Myoclonus | 10 (40) | ||
| Dystonia | 8 (32) | |||
| Tremor | 3 (12) | |||
|
Other symptoms or signs of suspected cerebellar attribution |
Diplopia | 11 (44) | ||
| Dysphagia | 12 (48) | |||
| Mild cognitive impairment by clinical suspicion or subjective complaint | 11 (44) | |||
| Cognitive screening test positive | 6 (25) | (n = 24) | ||
| Brainstem |
Brainstem oulomotor signs |
Ophthalmoparesis | 0 | |
| Slowing of saccades | 0 | |||
| Retina/optic nerve | Symptoms or signs of retinal/optic nerve involvement | Reduced visual acuity (monocular) | 0 | (n = 13) |
| Optical coherence tomography pRNFL reduction | 0 | (n = 13) | ||
| Spinal tract | Symptoms or signs of pyramidal involvement | Hyperreflexia | 5 (20) | |
| Spasticity | 0 | |||
| Plantar extensor | 0 | |||
| Electrophysiology: CMCT abnormal | 0 | (n = 8) | ||
| Symptoms or signs of spinal or peripheral involvement | Fasciculations | 5 (20) | ||
| Muscle atrophy | 4 (16) | |||
| Pareses | 3 (12) | |||
| Reduced vibration sense (ankle) | 4 (16) | (n = 25) | ||
| Electrophysiology: mild neuropathy | 8 (44) | (n = 18) | ||
| Electrophysiology: SSEP abnormal | 5 (33) | (n = 15) | ||
| Undefined | Symptoms of unclear attribution | Depression/anxiety screening test positive | 11 (48) | (n = 23) |
| Depression/anxiety clinically relevant | 5 (22) | (n = 23) | ||
| Cramps or sensation of muscle stiffness | 10 (40) | |||
| Pain in legs or lower back unpexplained otherwise | 5 (20) |
CMCT, central motor conduction time; PRKCG, protein kinase C gamma; pRNFL, peripapillary retinal nerve fiber layer; SARA, scale for the assessment and rating of ataxia; SSEP, somatosensory‐evoked potentials; VUS, variant of uncertain significance.
The confirmed SCA‐PRKCG had a mean age of 38 (SD 13.4) years and a disease duration of 19 (SD 12.5) years. Patients featured mild to moderate ataxia (SARA < 20) in all but one patient (score 25) presenting with additional myoclonus. Three patients reported permanent use of walking aids and none were wheel‐chair dependent. INAS count indicated up to five nonataxia signs per patient (none in five patients). Myoclonus involved trunk or hands in most cases and stimulus sensitivity was observed in one patient. Mild focal dystonia was often reported as action‐induced or task‐specific. Although hyperreflexia was noted in five and a sensation of leg stiffness was reported by two subjects, no spasticity or extensor plantar response was observed. Five subjects reported persistent bone or muscle pain located in the legs or back that increased with exertion without other identifiable cause. Fasciculations or mild to moderate muscle atrophy affected proximal or distal muscle groups with mild to moderate weakness in three subjects.
There was clinical suspicion or subjective complaint of mild cognitive dysfunction in almost half of the patients, whereas DemTect indicated mild cognitive impairment in only five subjects. Screening tests indicated dementia in one subject but coincident with relevant depressive symptoms.
Neuropsychological test results indicated lower performance compared to healthy controls in mainly two domains: attentional functions and executive function (Table 4). Results of SCA‐PRKCG in respective tests were unrelated to depressive symptoms (HADS‐D) while an association with ataxia severity (SARA) was seen for visuospatial mental rotation and selective attention (Table 5). When effects of age were taken into account using partial correlations, the associations with ataxia scores were no longer significant (Fig. S1).
Assessments of the visual pathway did not indicate pathology of the optic nerve (see Ihl et al. 28 for detail).
Electroneurographic signs of mild axonal or mixed neuropathy of single nerves were seen in some subjects but did not qualify for a diagnosis of polyneuropathy. Of note, findings were normal in three of four patients who featured reduced vibration sense. Central motor conduction time was normal in all eight subjects with reports available.
Symptom onset and progression
Due to cross‐sectional study design, the information in this section relies on patient report. The onset of gait ataxia varied between 4 and 50 years of age (mean/SD 38/13). In two very mildly affected subjects (SARA score 3 and 7), one subjectively unaware of ataxia, limb ataxia of the legs was more prominent than gait/stance ataxia. Disease manifestation coincided with giving birth to the 2nd child in one subject.
Several subjects reported possible early manifestations: minor difficulty with locomotor coordination since childhood (four patients, combination with early learning deficits in one), childhood‐onset, nonprogressive slurring of speech (two patients), and reading–writing difficulties (one patient).
The onset of dysarthria was mostly close to or even coincident with the onset of gait ataxia, whereas (mild) dysphagia started later in the disease course. The onset of impaired hand coordination was on average >10 years after the onset of gait ataxia. Early mild writing difficulties before the onset of gait ataxia in one subject were likely attributable to task‐specific dystonia. The onset of myoclonus remained unresolved as it often went unrecognized by patients themselves.
Progression of ataxia was slow (SARA annual progression rate 0.99 ± 1.01 pt/year, estimated as SARA scores by disease duration). In the subject with the most severe ataxia (SARA 25), valproate 900mg/day almost completely resolved the action‐induced truncal myoclonus with subsequent sustained SARA improvement by five points. Results of earlier neuropsychological testing, available in one patient, indicated only a mild decrease in tests of attention and semantic verbal fluency over a period of 8 years but no impairment in semantic or verbal episodic memory.
Brain imaging
There was no atrophy of cerebrum, brainstem, or cervical spinal cord, but cerebellar atrophy was seen in all SCA‐PRKCG subjects (Table 2), particularly pronounced in anterior lobe and upper vermis and including middle or superior cerebellar peduncles in three and two subjects, respectively.
A peculiar symmetrical hyperintensity of the dentate nucleus on T2‐weighted images was unequivocally seen in all SCA‐PRKCG subjects but none of the healthy controls. It extended from the dentate nucleus toward the superior cerebellar peduncle, whereas in healthy subjects the dentate nucleus was generally hypointense, presenting only a central clear‐cut hyperintense spot in some cases resembling dilated perivascular space. The T2‐hyperintense signal of the dentate nucleus had a hypointense correlate in T1‐weighted images (Fig. 2). The detection of the T2‐hyperintense dentate sign was improved by (para)coronar angulation of images along the superior cerebellar peduncles.
Figure 2.
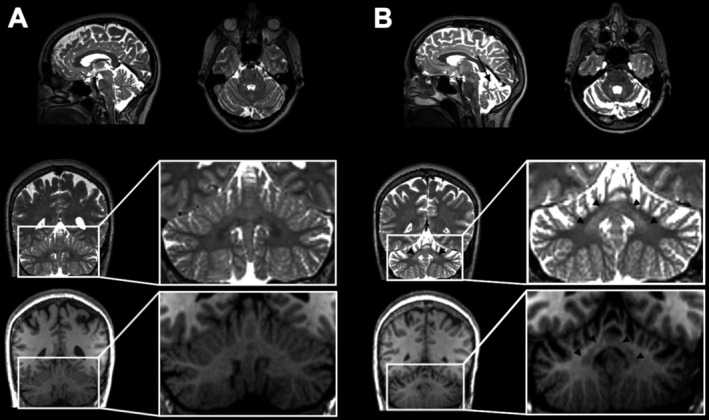
Example of MRI findings, specifically T2 signal of the dentate nuclei, in (A) a healthy subject aged 54 years and (B) a subject with SCA‐PRKCG aged 53 years with 23 years since disease onset. As seen in all confirmed SCA‐PRKCG of our cohort, there was a peculiar homogeneous T2 hyperintensity of the dentate nucleus (arrow head coronar image) that was not seen in any of the age‐ and gender‐matched control subjects. This hyperintensity had a hypointense correlate in T1‐weighted images. Further, as in all SCA‐PRKCG, the patient featured cerebellar atrophy most pronounced in upper vermis (arrow sagittal image) and anterior lobe (arrow axial image).
Both, the cerebellar atrophy and T2‐hyperintense dentate sign, were clearly observed also in two subjects with clinically incipient manifestation. Cerebellar atrophy even preceded clinical manifestation by 8 years in one of the three subjects with preceding routine clinical MRI available. By inspection, no obvious progression of atrophy was seen over periods of 8 to 17 years (Fig. 3). Volume loss could not be quantified since prior routine MRI was not obtained in 3D and slicing did not allow a statement on dentate signal alterations.
Figure 3.
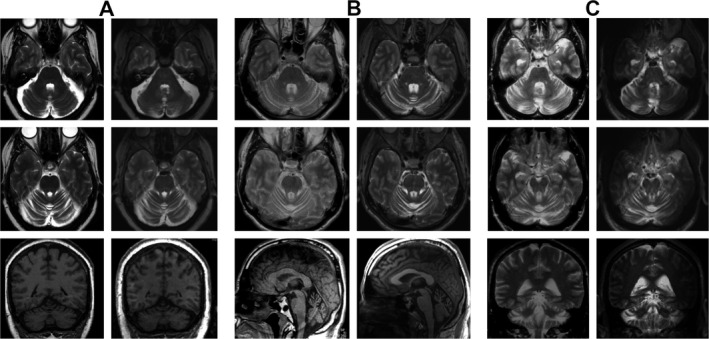
Evolution of MRI findings over eight (A), 12 (B) and 17 (C) years in three clinically manifest confirmed SCA‐PRKCG subjects who made prior clinical MR imaging available (age at second scan 57 (A), 37 (B) and 56 (C) years). Most recent imaging is presented on the right hand columns, previous MRI presented on the left for each case. In case B, the first scan was obtained for other symptoms than ataxia, that is, in the premanifest stage.
Clinical and imaging findings in VUS/(likely) benign cases
The four subjects classified as VUS had nominally older age (mean 46.5 years) and shorter disease duration (mean 6.8 years) than those confirmed as SCA‐PRKCG. Nonataxia movement disorder was seen in three of four carriers of VUS and disturbed memory was reported by two (Table 2). Signs of spasticity were reported in one subject (p. M256I) despite normal central motor conduction times and slowed saccades and horizontal ophthalmoparesis were seen in another subject (p. P678A).
In one parent‐offspring pair of a likely benign variant (p. R213Q), no signs of ataxia were observed but myoclonus, resting tremor, mild muscle atrophy, and weakness in the index case. The other family carrying a benign variant (p. C69C) presented with slowly progressive ataxia, areflexia, mild muscle atrophy (1), focal dystonia (1), and moderate to severe sensory disturbance.
Structural brain MRI in carriers of VUS showed extracerebellar pathology in three of four cases with brainstem atrophy (p. R634H and p. P678A), whole‐brain atrophy (p. M256I and p. P678A), or hyperintense middle cerebellar peduncle (p. R634H). Such features were not observed in any SCA‐PRKCG subject. Furthermore, no T2 hyperintense dentate sign was seen in two cases (only report of routine MRI was available for p. M256I carrier). In both carriers of variant p. R213Q, brain MRI was unremarkable without cerebellar atrophy. However, both carriers of variant p. C69C and a singular carrier of VUS (p. I173S) had imaging findings compatible with SCA‐PRKCG, including the hyperintense T2 dentate sign.
Discussion
As a main result, we describe a refined variant classification for the clinico‐genetic diagnosis of SCA‐PRKCG and summarize the SCA‐PRKCG phenotype from prospective investigation of clinical, neuropsychological, and imaging findings in the largest cohort to date. The novel brain MRI finding of T2 hyperintense/T1 hypointense dentate nuclei was shared by all confirmed SCA‐PRKCG and may serve as a supportive marker for PRKCG variant classification.
Clinical findings support a variable combination of three motor symptoms: (1) mild to moderate cerebellar ataxia, (2) multifocal action myoclonus, and (3) task‐specific or cervical dystonia (including dystonic tremor). The age of ataxia onset had a remarkably wide range apparently unrelated to phenotype or progression. The onset of ataxia related to childbirth in one of our subjects was also described in other PRKCG variants 11 , 36 and in SCA‐ATXN10 37 but possible mechanisms of aggravation remain speculative. Long‐standing mild or nonprogressive symptoms of walking, speech, or learning dysfunction were reported by 8/25 subjects and also noted in previous reports, 7 , 10 , 38 , 39 suggestive of an early developmental component.
Action myoclonus in SCA‐PRKCG may aggravate ataxia or be even mistaken for ataxia in commonly used motor coordination tests and obviously interfered with SARA rating in one of our subjects. Such interference is known from early‐onset ataxias. 40 Further, history taking for myoclonus required specific enquiry, for example, for “muscle jerks at rest or action like you would sometimes experience falling asleep,” as most subjects did not complain of jerks spontaneously, even if clinically observed. Action as a trigger argues for a cortical origin, 41 further supported by the previous notion of negative myoclonus, 42 response to valproate 5 and this first description of stimulus‐sensitivity in one patient. Dystonia was mild or intermittent in our study though disabling predominant myoclonus‐dystonia has been described in SCA‐PRKCG. 42 The observed head and upper limb tremor was difficult to classify; classification in previous reports included tremulous dystonia, rhythmic myoclonus, or segmental myorhythmia (Table S1).
The nonataxia movement disorders observed in SCA‐PRKCG have been referred to as extracerebellar signs in previous reports and are not (yet) considered part of the clinical cerebellar syndrome. 43 However, converging arguments attribute them to cerebellar pathology. For myoclonus, the coincidence with symptoms of ataxia has long been described 44 For dystonia, an ataxia to dystonia continuum was suggested from animal models 45 related to the irregularity of excitatory outflow from deep cerebellar nuclei impacting on cerebral cortical functions. 46 , 47 Consistently, a previous SCA‐PRKCG series 36 revealed changes in intracortical inhibition similar to those reported in cortical myoclonus or DYT‐TOR1A/DYT1 carriers and clinical signs of reduced interhemispheric motor inhibition (contralateral movement test, Table 4) were observed in our study. In sum, nonataxia movement disorders in SCA‐PRKCG may be related to a distinct cerebellar pathology that SCA‐PRKCG possibly shares with other movement disorders: a pure Purkinje cell dysfunction/loss in coincidence with structurally intact but disinhibited deep cerebellar nuclei. This pattern is in line with a recent histopathological report of SCA‐PRKCG (p. H101Q). 21
Aside from hyperreflexia, there were no other signs of pyramidal affection and motor‐evoked potentials were normal as in all previous reports (one previous report of abnormal central motor conduction times 48 was found unremarkable later‐on, personal communication D. T.). The etiology of muscle atrophy/pareses, fasciculations, mild sensory symptoms, and pain remains unclear. Electrophysiological findings here and in other studies do not support large‐fiber neuropathy as a feature of SCA‐PRKCG (severe axonal neuropathy has hitherto been described in only one singular index case (p. A458T) 14 ). PRKCG expression in dorsal horn and nucleus gracilis 49 may be of relevance and requires further investigations of spinal structures in SCA‐PRKCG.
The results of neuropsychological testing were compatible with previously described cognitive features of cerebellar pathology. 50 Longitudinal data of neuropsychological testing available in one of our subjects indicated mild progression of cognitive dysfunction, in line with few previous descriptions. 7 , 51 , 52 Few reports of overt dementia were all in SCA‐PRKCG with long‐standing disease (Table S1) or probable comorbidity. In one report, marked cognitive decline coincided with hearing loss, diabetes, and epilepsy, suggestive of other pathology. 53 A role of (physiologically weak) neocortical expression of mutant PRKCG is not excluded, but dementia of rather subcortical type, normal structural MRI, MR spectroscopy, 27 and histopathology of cerebral cortex 20 , 21 argue against it.
Standardized structural brain MRI confirmed pure cerebellar atrophy, predominantly of vermis and anterior lobe. This may precede clinical manifestation and disclose carrier status in premanifest stages as in other SCAs. 54 Atrophy was nonprogressive in serial MRI of three cases spanning up to 17 years and may thus be interpreted as a maldevelopmental or early degenerative change that occurs independent of the manifestation or progression of ataxia. Of note, cerebellar (cortical) atrophy in absence of ataxia has been reported in other movement disorders. 55 , 56 The clinical manifestation of SCA‐PRKCG may thus be more related to dysfunctional cerebellar signaling than to cerebellar structural change, whereas the early developmental or even congenital cerebellar atrophy/hypoplasia may explain early nonprogressive subtle clinical signs. Both hypotheses await further exploration in longitudinal, histopathological, and functional studies.
The finding of symmetrically T2 hyperintense/T1 hypointense dentate nuclei was consistently seen in all 25 SCA‐PRKCG cases irrespective of time since onset. We were unable to relate this finding to previous reports, as these displayed only sagittal view images. This sign was not seen in any of our healthy controls in whom a physiological decrease of T2 signal in the dentate nucleus is expected throughout the lifespan. 57 Brain T2 hyperintensity with corresponding T1 hypointensity has been proposed to indicate myelin degradation. 58 In SCA‐PRKCG, the extension of this signal along the superior cerebellar peduncles suggests that this may also affect the dentato‐thalamic efferents. The T2 hyperintense dentate sign was not observed in two VUS carriers who showed instead clinical and imaging features not seen in any confirmed SCA‐PRKCG case. However, it was present in two related carriers of a benign variant (p. C69C) and in one VUS carrier (p. I173S). As all three shared a phenotype compatible with SCA‐PRKCG this would rather support pathogenicity in the latter and should stimulate further (e.g., intronic) genetic investigation of PRKCG in the other family. Specificity and histopathological correlates of this novel sign are yet unknown. There have been reports of altered dentate signals in T2 weighted or FLAIR sequences in different movement disorders, usually as part of a more widespread pattern of imaging abnormalities. 59 Signal alterations confined to the dentate nucleus have recently been described in few genetic (ataxia) movement disorders 60 , 61 , 62 but have not been systematically investigated and T2 hyperintensity of the dentate nucleus is currently not considered a characteristic imaging finding in neurodegenerative ataxia. 63 , 64 Although it is possibly not specific, our results suggest the T2 hyperintense dentate sign as a supporting criterion for PRKCG variant classification in cases with typical phenotype. Its absence, as well as the presence of atypical clinical findings (e.g., brainstem or pyramidal affection, early cognitive decline, retinal atrophy), or extracerebellar pathology on brain MRI may contribute to exclude PRKCG variants as causative. This should then stimulate further investigation into alternative causes or even genetic comorbidity.
The validity of this clinicogenetic description is strengthened by the use of standardized phenotype assessment applied in a prospective manner and a standardized refined procedure of variant classification. This is expected to reduce reporting bias for phenotypic features often seen with retrospective studies and to reduce misclassifications of pathogenicity. Some previous descriptions of SCA‐PRKCG were published before the consensus guidelines on variant interpretation, the application of which, in fact, led to re‐assignment as VUS in some (Table 1). All (likely) pathogenic variants in this study were within N‐terminal or C1 regulatory domain. Conclusions on the (rarer) kinase domain mutations could thus only rely on literature review (Table S1) which did not convincingly reveal distinctive features. 18 The comprehensive variant classification proposed here clearly increased diagnostic yield by inclusion of protein modeling results. Their interpretation weighs the structural and functional consequences of each variant on PRKCG function. Such a protein‐specific approach can be considered more specific than generic pathogenicity prediction tools that are largely based on evolutionary conservation. Segregation analysis may have added certainty but we decided to systematically not consider such information, as its unavailability reflects the prevalent clinical reality. Clearly, most valid claims of pathogenicity would require functional study in a valid disease model, which has not yet been established for SCA‐PRKCG. Thus, this refined classification approach may be generalizable to assign pathogenicity to missense variants in the case of other very rare, multi‐allelic adult‐onset disorders, in a gene with low tolerance to variability and in the absence of reliable biomarkers, functional models, or a highly specific phenotype. It should be noted, that the interpretation of both, genetic variants and protein modeling results, requires relevant expertise but would be feasible in the context of emerging research networks for rare diseases.
Conflict of Interest
Dr. Schmitz‐Hübsch reports honoraria from Biogen and Bayer AG outside the submitted work. Dr. Lux, Dr. Bauer, E. Schlapakow, Dr. Greschus, Dr. Scheel, Dr. Gärtner, Dr. Gras, Dr. Timmann, Dr. Synofzik, Dr. Giorgetti, Dr. Carloni, Dr. Shah, Dr. Schöls, Dr. Kopp, Dr. Bußenius, T. Oberwahrenbrock, Dr. Pfueller, Ms. Grosch, Dr. Amunts, Dr. Doss, M. Rönnefarth, and Dr. Minnerop have nothing to disclose. Dr. Brandt is cofounder and shareholder of medical technology companies Motognosis GmbH, Germany, and Nocturne GmbH, Germany, outside the submitted work. Dr. Kirlangic reports a position at the Gegenbauer Services GmbH, and a patent DE102016214575 with Volkswagen Aktiengesellschaft, outside the submitted work. Dr. Zimmermann reports grants from Novartis, outside the submitted work. E. Kadas is cofounder of Nocturne GmbH, Germany, outside the submitted work. Dr. Endres reports ME reports grants from Bayer and fees paid to the institution from Bayer, Boehringer Ingelheim, BMS, Daiichi Sankyo, Amgen, GSK, Sanofi, Covidien, Novartis, Pfizer, all outside the submitted work. Dr. Paul reports receives honoraria for lecturing, and travel expenses for attending meetings from Guthy Jackson Foundation, Sanofi Genzyme, Novartis, Alexion, Viela Bio, Roche, UCB, Mitsubishi Tanabe and Celgene. His research is funded by the German Ministry for Education and Research (BMBF), Deutsche Forschungsgemeinschaft (DFG), Einstein Foundation, Guthy Jackson Charitable Foundation, EU FP7 Framework Program, Biogen, Genzyme, Merck Serono, Novartis, Bayer, Teva, Alexion, Roche, Parexel, and Almirall. All funding is outside the submitted work.
Author Contributions
T. S‐H. contributed to the conception and design of the study, acquisition and analysis of data, and drafting the manuscript and tables. S. L. contributed to the conception and design of the study (especially neuropsychology and imaging), analysis of data, and drafting parts of the manuscript and figures. P. B. contributed to study design, provided genetic analysis and interpretation, and co‐drafted the manuscript and tables. A. U. B. contributed to the conception and design of the study, acquisition, and analysis of data and revising the manuscript for intellectual content. E. S. contributed to the conception of the study, acquisition of data, and revising the manuscript. S. G. provided acquisition and interpretation of imaging data, co‐drafted the manuscript and figures. M. Sch. contributed to the acquisition and analysis of imaging data and revised the manuscript. H. G. contributed to the acquisition and analysis of data and revised the manuscript. M. E. K. contributed to the acquisition of data (particularly motor function and brain imaging), and editing and revising the manuscript for intellectual content. V. G. contributed to the conception and design of the MRI protocol, supported the acquisition of data, and revised the manuscript. D. T. contributed to the interpretation of clinical data and MRI data, and revised the manuscript for intellectual content. M. Sy. contributed to the discussion of the study concept, contributed to the interpretation of clinical and genetic findings, and revised the manuscript for intellectual content. A. G. contributed to the conception and design of the study, calculation, and analysis of protein modeling, and drafting the manuscript. P. C. contributed to the discussion of the study concept and revised the manuscript for intellectual content. N. J. S. contributed to the conception and design of the MRI protocol; revision of the manuscript. L. S. contributed to the discussion of the study concept, contributed to the interpretation of clinical and genetic findings, and revised the manuscript for intellectual content. U. K. contributed to the discussion of study concept, to data acquisition and analysis (especially neuropsychology) and revised manuscript for intellectual content. L. B. contributed to the data analysis of neuropsychological statistics and drafting part of the figures and tables. T. O. contributed to data acquisition and interpretation (specifically the assessment of afferent visual pathway), revised manuscript for intellectual content. H. Z. contributed to data acquisition and interpretation (specifically assessment of afferent visual pathway), revised manuscript for intellectual content. C. P. contributed to coordinate study visits, to data acquisition and interpretation, and revised the manuscript for intellectual content. E. M. K. contributed to interpretation (specifically assessment of afferent visual pathway), and revised the manuscript for intellectual content. M. R. contributed to the acquisition and analysis of patients’ and clinical data as well as revising the manuscript. A. S. G. contributed to the acquisition and analysis of data as well as revising the manuscript. M. E. involved in the discussion of study concept and revision of the manuscript for intellectual content. K. A. contributed to the conception and design of the study and revision of the manuscript for intellectual content. F. P. contributed to the conception and design of the study and revision of the manuscript for intellectual content. S. D. contributed to the conception and design of the study, acquisition and analysis of data, and revising the manuscript draft for intellectual content. M. M. contributed to the conception and design of the study, acquisition and analysis of data, and drafting the manuscript and figures.
Supporting information
Table S1. Content summary and full reference list of all available published clinicogenetic descriptions of SCA‐PRKCG ordered by location of variant from lower to higher number of amino acid residue.
Data S1. Details on methods (genetic classification, protein modeling, assessment protocols and test references (Table S2)) and additional results (Fig. S1: effects of age on cognitive test results).
Table S2. Description and reference of neuropsychological tests and screening instruments applied in this study.
Acknowledgments
We are grateful for the support by Heidi Mellenthin, Kerstin Jütten, and Leonora Zange in data acquisition and Graham Cooper for proof‐reading the final version of the manuscript as a native speaker. We greatly appreciate the willingness and efforts taken by all participants to support research by their participation in this study. We acknowledge public funding from DFG under Germany´s Excellence Strategy – EXC‐2049 – 390688087, BMBF, DZNE, DZHK, EU, Corona Foundation, and Fondation Leducq for M. E. and the Deutsche Forschungsgemeinschaft (DFG), NeuroCure Cluster of Excellence grant number EXC 257 to F. P., and grant 779257 “Solve‐RD” from the EU Horizon 2020 program to M. Sy. Open Access funding enabled and organized by ProjektDEAL
Funding Information
We acknowledge public funding from DFG under Germany´s Excellence Strategy – EXC‐2049 – 390688087, BMBF, DZNE, DZHK, EU, Corona Foundation, and Fondation Leducq for M. E. and the Deutsche Forschungsgemeinschaft (DFG), NeuroCure Cluster of Excellence grant number EXC 257 to F. P., and grant 779257 “Solve‐RD” from the EU Horizon 2020 program to M. Sy.
Funding Statement
This work was funded by Deutsche Forschungsgemeinschaft grant 390688087; NeuroCure Cluster of Excellence grant EXC 257; EU Horizon 2020 grant 779257.
Contributor Information
Tanja Schmitz‐Hübsch, Email: tanja.schmitz-huebsch@charite.de.
Martina Minnerop, Email: m.minnerop@fz-juelich.de.
References
- 1. Klockgether T. Update on degenerative ataxias. Curr Opin Neurol 2011;24:339–345. [DOI] [PubMed] [Google Scholar]
- 2. Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol 2010;9:885–894. [DOI] [PubMed] [Google Scholar]
- 3. Chen DH, Brkanac Z, Verlinde CL, et al. Missense mutations in the regulatory domain of PKC gamma: a new mechanism for dominant nonepisodic cerebellar ataxia. Am J Hum Genet 2003;72:839–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yabe I, Sasaki H, Chen DH, et al. Spinocerebellar ataxia type 14 caused by a mutation in protein kinase C gamma. Arch Neurol 2003;60:1749–1751. [DOI] [PubMed] [Google Scholar]
- 5. Yamashita I, Sasaki H, Yabe I, et al. A novel locus for dominant cerebellar ataxia (SCA14) maps to a 10.2‐cM interval flanked by D19S206 and D19S605 on chromosome 19q13.4‐qter. Ann Neurol 2000;48:156–163. [DOI] [PubMed] [Google Scholar]
- 6. Marras C, Lang A, van de Warrenburg BP, et al. Nomenclature of genetic movement disorders: recommendations of the international Parkinson and movement disorder society task force. Mov Disord 2016;31:436–457. [DOI] [PubMed] [Google Scholar]
- 7. Koht J, Stevanin G, Durr A, et al. SCA14 in Norway, two families with autosomal dominant cerebellar ataxia and a novel mutation in the PRKCG gene. Acta Neurol Scand 2012;125:116–122. [DOI] [PubMed] [Google Scholar]
- 8. Alonso I, Costa C, Gomes A, et al. A novel H101Q mutation causes PKCgamma loss in spinocerebellar ataxia type 14. J Hum Genet 2005;50:523–529. [DOI] [PubMed] [Google Scholar]
- 9. Dalski A, Mitulla B, Burk K, et al. Mutation of the highly conserved cysteine residue 131 of the SCA14 associated PRKCG gene in a family with slow progressive cerebellar ataxia. J Neurol 2006;253:1111–1112. [DOI] [PubMed] [Google Scholar]
- 10. Hiramoto K, Kawakami H, Inoue K, et al. Identification of a new family of spinocerebellar ataxia type 14 in the Japanese spinocerebellar ataxia population by the screening of PRKCG exon 4. Mov Disord 2006;21:1355–1360. [DOI] [PubMed] [Google Scholar]
- 11. Klebe S, Durr A, Rentschler A, et al. New mutations in protein kinase Cgamma associated with spinocerebellar ataxia type 14. Ann Neurol 2005;58:720–729. [DOI] [PubMed] [Google Scholar]
- 12. Sailer A, Scholz SW, Gibbs JR, et al. Exome sequencing in an SCA14 family demonstrates its utility in diagnosing heterogeneous diseases. Neurology 2012;79:127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van de Warrenburg BP, Verbeek DS, Piersma SJ, et al. Identification of a novel SCA14 mutation in a Dutch autosomal dominant cerebellar ataxia family. Neurology 2003;61:1760–1765. [DOI] [PubMed] [Google Scholar]
- 14. Chelban V, Wiethoff S, Fabian‐Jessing BK, et al. Genotype‐phenotype correlations, dystonia and disease progression in spinocerebellar ataxia type 14. Mov Disord 2018;33:1119–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jarius S, Wildemann B. 'Medusa head ataxia': the expanding spectrum of Purkinje cell antibodies in autoimmune cerebellar ataxia. Part 2: Anti‐PKC‐gamma, anti‐GluR‐delta2, anti‐Ca/ARHGAP26 and anti‐VGCC. J Neuroinflammation 2015;12:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Asai H, Hirano M, Shimada K, et al. Protein kinase C gamma, a protein causative for dominant ataxia, negatively regulates nuclear import of recessive‐ataxia‐related aprataxin. Hum Mol Genet 2009;18:3533–3543. [DOI] [PubMed] [Google Scholar]
- 17. Adachi N, Kobayashi T, Takahashi H, et al. Enzymological analysis of mutant protein kinase Cgamma causing spinocerebellar ataxia type 14 and dysfunction in Ca2+ homeostasis. J Biol Chem 2008;283:19854–19863. [DOI] [PubMed] [Google Scholar]
- 18. Shimobayashi E, Kapfhammer JP. Increased biological activity of protein Kinase C gamma is not required in Spinocerebellar ataxia 14. Mol Brain 2017;10:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Verbeek DS, Goedhart J, Bruinsma L, et al. PKC gamma mutations in spinocerebellar ataxia type 14 affect C1 domain accessibility and kinase activity leading to aberrant MAPK signaling. J Cell Sci 2008;121(Pt 14):2339–2349. [DOI] [PubMed] [Google Scholar]
- 20. Brkanac Z, Bylenok L, Fernandez M, et al. A new dominant spinocerebellar ataxia linked to chromosome 19q13.4‐qter. Arch Neurol 2002;59:1291–1295. [DOI] [PubMed] [Google Scholar]
- 21. Wong MMK, Hoekstra SD, Vowles J, et al. Neurodegeneration in SCA14 is associated with increased PKCgamma kinase activity, mislocalization and aggregation. Acta Neuropathol Commun 2018;6:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Olgiati S, Quadri M, Bonifati V. Genetics of movement disorders in the next‐generation sequencing era. Mov Disord 2016;31:458–470. [DOI] [PubMed] [Google Scholar]
- 24. Coutelier M, Hammer MB, Stevanin G, et al. Efficacy of exome‐targeted capture sequencing to detect mutations in known cerebellar ataxia genes. JAMA Neurol 2018;75:591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van de Warrenburg BP, van Gaalen J, Boesch S, et al. EFNS/ENS Consensus on the diagnosis and management of chronic ataxias in adulthood. Eur J Neurol 2014;21:552–562. [DOI] [PubMed] [Google Scholar]
- 26. Schmitz‐Hubsch T, Brandt AU, Pfueller C, et al. Accuracy and repeatability of two methods of gait analysis ‐ GaitRite und Mobility Lab ‐ in subjects with cerebellar ataxia. Gait Posture 2016;48:194–201. [DOI] [PubMed] [Google Scholar]
- 27. Doss S, Rinnenthal JL, Schmitz‐Hubsch T, et al. Cerebellar neurochemical alterations in spinocerebellar ataxia type 14 appear to include glutathione deficiency. J Neurol 2015;262:1927–1935. [DOI] [PubMed] [Google Scholar]
- 28. Ihl T, Kadas EM, Oberwahrenbrock T, et al. Investigation of visual system involvement in spinocerebellar ataxia type 14. Cerebellum 2020;19:469–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Biasini M, Bienert S, Waterhouse A, et al. SWISS‐MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 2014;42(W1):W252–W258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schmitz‐Hubsch T, du Montcel ST, Baliko L, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 2006;66:1717–1720. [DOI] [PubMed] [Google Scholar]
- 31. Jacobi H, Rakowicz M, Rola R, et al. Inventory of Non‐Ataxia Signs (INAS): validation of a new clinical assessment instrument. Cerebellum 2013;12:418–428. [DOI] [PubMed] [Google Scholar]
- 32. Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatr Scand 1983;67:361–370. [DOI] [PubMed] [Google Scholar]
- 33. Kalbe E, Kessler J, Calabrese P, et al. DemTect: a new, sensitive cognitive screening test to support the diagnosis of mild cognitive impairment and early dementia. Int J Geriatr Psychiatry 2004;19:136–143. [DOI] [PubMed] [Google Scholar]
- 34. UNESCO‐United Nations Educational SaCO. International Standard Classification of Education – ISCED. 1997.
- 35. Oldfield RC. The assessment and analysis of handedness: the Edinburgh inventory. Neuropsychologia 1971;9:97–113. [DOI] [PubMed] [Google Scholar]
- 36. Ganos C, Zittel S, Minnerop M, et al. Clinical and neurophysiological profile of four German families with spinocerebellar ataxia type 14. Cerebellum 2014;13:89–96. [DOI] [PubMed] [Google Scholar]
- 37. Silva GV, Bonilha P, Moro A, et al. Spinocerebellar ataxias type 3 and 10: onset and progression of ataxia during pregnancy and puerperium. Parkinsonism Relat Disord 2018;52:119–120. [DOI] [PubMed] [Google Scholar]
- 38. Nolte D, Landendinger M, Schmitt E, Muller U. Spinocerebellar ataxia 14: novel mutation in exon 2 of PRKCG in a German family. Mov Disord 2007;22:265–267. [DOI] [PubMed] [Google Scholar]
- 39. Vlak MH, Sinke RJ, Rabelink GM, et al. Novel PRKCG/SCA14 mutation in a Dutch spinocerebellar ataxia family: expanding the phenotype. Mov Disord 2006;21:1025–1028. [DOI] [PubMed] [Google Scholar]
- 40. Lawerman TF, Brandsma R, Verbeek RJ, et al. Construct validity and reliability of the SARA gait and posture sub‐scale in early onset ataxia. Front Hum Neurosci 2017;11:605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zutt R, Elting JW, van der Hoeven JH, et al. Myoclonus subtypes in tertiary referral center. Cortical myoclonus and functional jerks are common. Clin Neurophysiol 2017;128:253–259. [DOI] [PubMed] [Google Scholar]
- 42. Visser JE, Bloem BR, van de Warrenburg BP. PRKCG mutation (SCA‐14) causing a Ramsay Hunt phenotype. Mov Disord 2007;22:1024–1026. [DOI] [PubMed] [Google Scholar]
- 43. Bodranghien F, Bastian A, Casali C, et al. Consensus paper: revisiting the symptoms and signs of cerebellar syndrome. Cerebellum 2016;15:369–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lance JW. Action myoclonus, Ramsay Hunt syndrome, and other cerebellar myoclonic syndromes. Adv Neurol 1986;43:33–55. [PubMed] [Google Scholar]
- 45. Shakkottai VG, Batla A, Bhatia K, et al. Current opinions and areas of consensus on the role of the cerebellum in dystonia. Cerebellum 2017;16:577–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kaji R, Bhatia K, Graybiel AM. Pathogenesis of dystonia: is it of cerebellar or basal ganglia origin? J Neurol Neurosurg Psychiatry 2018;89:488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jinnah HA, Neychev V, Hess EJ. The anatomical basis for dystonia: the motor network model. Tremor Other Hyperkinet Mov 2017;7:506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wieczorek S, Arning L, Gizewski ER, et al. Benign SCA14 phenotype in a German patient associated with a missense mutation in exon 3 of the PRKCG gene. Mov Disord 2007;22:2135–2136. [DOI] [PubMed] [Google Scholar]
- 49. Hughes AS, Averill S, King VR, et al. Neurochemical characterization of neuronal populations expressing protein kinase C gamma isoform in the spinal cord and gracile nucleus of the rat. Neuroscience 2008;153:507–517. [DOI] [PubMed] [Google Scholar]
- 50. Timmann D, Daum I. Cerebellar contributions to cognitive functions: a progress report after two decades of research. Cerebellum 2007;6:159–162. [DOI] [PubMed] [Google Scholar]
- 51. Stevanin G, Hahn V, Lohmann E, et al. Mutation in the catalytic domain of protein kinase C gamma and extension of the phenotype associated with spinocerebellar ataxia type 14. Arch Neurol 2004;61:1242–1248. [DOI] [PubMed] [Google Scholar]
- 52. Wedding IM, Koht J, Dietrichs E, et al. Cognition is only minimally impaired in Spinocerebellar ataxia type 14 (SCA14): a neuropsychological study of ten Norwegian subjects compared to intrafamilial controls and population norm. BMC Neurol 2013;13:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shirafuji T, Shimazaki H, Miyagi T, et al. Spinocerebellar ataxia type 14 caused by a nonsense mutation in the PRKCG gene. Mol Cell Neurosci 2019;98:46–53. [DOI] [PubMed] [Google Scholar]
- 54. Jacobi H, Reetz K, du Montcel ST, et al. Biological and clinical characteristics of individuals at risk for spinocerebellar ataxia types 1, 2, 3, and 6 in the longitudinal RISCA study: analysis of baseline data. Lancet Neurol 2013;12:650–658. [DOI] [PubMed] [Google Scholar]
- 55. Le Ber I, Clot F, Vercueil L, et al. Predominant dystonia with marked cerebellar atrophy: a rare phenotype in familial dystonia. Neurology 2006;67:1769–1773. [DOI] [PubMed] [Google Scholar]
- 56. Tijssen MA, Thom M, Ellison DW, et al. Cortical myoclonus and cerebellar pathology. Neurology 2000;54:1350–1356. [DOI] [PubMed] [Google Scholar]
- 57. Maschke M, Weber J, Dimitrova A, et al. Age‐related changes of the dentate nuclei in normal adults as revealed by 3D fast low angle shot (FLASH) echo sequence magnetic resonance imaging. J Neurol 2004;251:740–746. [DOI] [PubMed] [Google Scholar]
- 58. Schiffmann R, van der Knaap MS. Invited article: an MRI‐based approach to the diagnosis of white matter disorders. Neurology 2009;72:750–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bond KM, Brinjikji W, Eckel LJ, et al. Dentate update: imaging features of entities that affect the dentate nucleus. AJNR Am J Neuroradiol 2017;38:1467–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ronsin S, Hannoun S, Thobois S, et al. A new MRI marker of ataxia with oculomotor apraxia. Eur J Radiol 2019;110:187–192. [DOI] [PubMed] [Google Scholar]
- 61. Hewamadduma CA, Hoggard N, O'Malley R, et al. Novel genotype‐phenotype and MRI correlations in a large cohort of patients with SPG7 mutations. Neurol Genet 2018;4:e279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cocozza S, Pontillo G, De Michele G, et al. The "crab sign": an imaging feature of spinocerebellar ataxia type 48. Neuroradiology 2020;62:1095–1103. [DOI] [PubMed] [Google Scholar]
- 63. Baldarcara L, Currie S, Hadjivassiliou M, et al. Consensus paper: radiological biomarkers of cerebellar diseases. Cerebellum 2015;14:175–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Khadilkar S, Jaggi S, Patel B, et al. A practical approach to diseases affecting dentate nuclei. Clin Radiol 2016;71:107–119. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Content summary and full reference list of all available published clinicogenetic descriptions of SCA‐PRKCG ordered by location of variant from lower to higher number of amino acid residue.
Data S1. Details on methods (genetic classification, protein modeling, assessment protocols and test references (Table S2)) and additional results (Fig. S1: effects of age on cognitive test results).
Table S2. Description and reference of neuropsychological tests and screening instruments applied in this study.