

Abstract
Autophagy-associated genes have been identified as susceptible loci for inflammatory bowel disease. We investigated the role of a core autophagy factor, Atg5, in the development of dextran sodium sulfate (DSS)-induced colitis. Intestinal epithelial cell (IEC)-specific Atg5 gene deficient mice (Atg5ΔIEC mice) were generated by cross of Atg5-floxed mice (Atg5fl/fl) with transgenic mice expressing Cre-recombinase driven by the villin promotor. Mice were given three cycles of 1.5% DSS in drinking water for 5 days and regular water for 14 days over a 60-day period. The dysfunction of autophagy characterized by a marked accumulation of p62 protein, a substrate for autophagy degradation, was detected in epithelial cells in the non-inflamed and inflamed mucosa of inflammatory bowel disease patients. DSS-colitis was exacerbated in Atg5ΔIEC mice compared to control Atg5fl/fl mice. Phosphorylation of inositol-requiring transmembrane kinase/endonuclease1α (IRE1α), a sensor for endoplasmic reticulum stress, and c-Jun N-terminal kinase, a downstream target of IRE1α, were significantly enhanced in IECs in DSS-treated Atg5ΔIEC mice. Accumulation of phosphorylated IRE1α was enhanced by the treatment with chloroquine, an autophagy inhibitor. Apoptotic IECs were more abundant in DSS-treated Atg5ΔIEC mice. These findings suggest that Atg5 suppresses endoplasmic reticulum stress-induced apoptosis of IECs via the degradation of excess p-IRE1α.
Keywords: autophagy, IRE1α, IBD
Introduction
Inflammatory bowel disease (IBD), including Crohn’s disease (CD) and ulcerative colitis (UC), is characterized by chronic relapsing intestinal inflammation of unknown etiology.(1–3) Degradation and recycling of cellular components is critical for all eukaryotic cells for the maintenance of cellular homeostasis. Macroautophagy (hereafter referred to as autophagy) is an intracellular catabolic system which degrades large cytoplasmic components, including organelles, protein aggregates and intracellular pathogens, by sequestering these components in autophagosomes characterized by double membrane vesicles and delivering this cargo for lysosomal degradation.(4) Recent studies have revealed that autophagy selectively degrades unwanted components which are resistant to degradation by the ubiquitin-proteasome system.(5) In addition, autophagy regulates rapid cellular changes essential for development and differentiation.(6,7) Dysregulated autophagy is involved in the pathophysiology of various human disorders,(8) and genome-wide association studies have identified single nucleotide polymorphisms (SNPs) in the autophagy-related 16 like 1 (ATG16L1) gene as a susceptibility factor for CD.(9,10)
The endoplasmic reticulum (ER) is the cellular organelle that plays a critical role in protein folding and secretion.(11) In the ER lumen, proteins intended for secretion or display on the cell surface are folded into their proper conformation.(11) Protein folding in the ER is sometimes impaired under various conditions, leading to the accumulation of misfolded proteins (ER stress).(12) The misfolded proteins are harmful to cells and the unfolded protein response (UPR) mediates refolding them or targeting them for degradation.(11) The UPR is mediated by ER membrane-associated sensors such as inositol-requiring transmembrane kinase/endonuclease 1 (IRE1).(11,12) IRE1 stimulates a generation of transcription factor X box protein-1 (XBP-1), which activates several genes involved in the transport of misfolded proteins from the ER to cytosol and in ER-induced protein degradation.(13) XBP-1 deficiency in IECs promotes intestinal inflammation as consequence of increased ER stress, linking ER stress of IECs to gut inflammation.(14)
Recent studies have suggested an association of autophagy and ER stress in the pathophysiology of IBD.(15–17) For example, enhanced ER stress is observed in the Paneth cells of CD patients with SNP of the ATG16L1 gene.(18) The double knockout mice of ATG16L1 and XBP-1 develop spontaneous enterocolitis that resembles human IBD.(19) Mice with IEC specific ATG16L1 deletion caused accumulation of IRE1α in Paneth cells, resulting in CD-like ileitis.(20) However, it has not been fully elucidated how autophagy interacts with ER stress in the inflammatory conditions in the colon. Atg5 is a protein involved in the early stages of autophagosome formation.(21) In this study, we generated IEC specific Atg5 gene deficient mice and investigated the association between autophagy and ER stress using a dextran sodium sulfate (DSS)-induced colitis model.
Materials and Methods
Tissue samples
Tissue samples were obtained by biopsy under colonoscopy and/or surgery from patients with UC (n = 6) and CD (n = 6). Normal colonic samples were obtained from surgically resected tissues of patients with colon cancer (n = 6). This project was approved by the ethics committee of the Shiga University of Medical Science, and written informed consent was obtained from all patients (Permit number: 27-27).
Animals and DSS-induced colitis
Atg5-floxed (Atg5fl/fl) mice were kindly provided by the RIKEN BRC through the National Bio-Resource Project of the MEXT, Japan.(22) Mice lacking the Atg5 gene in their IECs (Atg5ΔIEC) were generated by crossing Atg5fl/fl mice with transgenic mice that expressed Cre-recombinase driven by the villin promotor (Villin-Cre mice: Jackson Laboratory, Bar Harbor, ME). Mice were divided into 4 groups: Atg5fl/fl mice, Atg5ΔIEC mice, DSS-treated Atg5fl/fl mice (Atg5fl/fl DSS) and DSS-treated Atg5ΔIEC mice (Atg5ΔIEC DSS) (n = 10 mice/group). Mice were given three cycles of 1.5% DSS (molecular weight 36,000–50,000 Da; MP Biomedicals, Santa Ana, CA) in drinking water for 5 days and regular water for 14 days over a 60-day period prior to being euthanized.(23) This project was approved by the Research Center for Animal Life Science and Use Committee at the Shiga University of Medical Science (Permit number: 2016-4-6).
Assessment of DSS induced colitis
The severity of colitis was evaluated using the Disease Activity Index (DAI) based on a previous report.(24) The colon tissues were fixed with formalin, embedded in paraffin, and cut into 4 µm sections. Histological evaluations were performed according to a validated scoring system.(25)
Isolation of colonic epithelial cells
The isolation of colonic epithelial cells was performed according to a previously described method.(26) Briefly, the washed and inverted colons were incubated in Hanks’ balanced salt solution (HBSS) containing 2 mM EDTA at 37°C for 20 min with gentle shaking. Supernatants were centrifuged and pellets were resuspended in 40% percoll and centrifuged. The cells on the top layer were collected and used as colonic epithelial cells.
Extraction of mRNA and real-time polymerase chain reaction (PCR) analysis
Colonic epithelial mRNA was extracted using the TRIzol reagent (Invitrogen, Carlsbad, CA). Real-time PCR was performed using the LightCycler 480 System (Roche Applied Science, Penzberg, Germany) and SYBR Premix Ex TaqII (TAKARA, Otsu, Japan). The expression of each target gene was normalized with β-actin and expressed as a ratio relative to the control group. The PCR primers used in this study are listed in Supplemental Table 1*.
Extraction of protein and immunoblot analysis
The protein from colonic epithelial cells was extracted according to the method described previously.(27) Signal detection was performed using an ImageQuant LAS 4000 (GE Healthcare UK Ltd., Little Chalfont, UK). The antibodies used in this study are listed in Supplemental Table 2*.
Chloroquine treatment and TUNEL staining
Chloroquine (50 µg/g body weight) (Sigma-Aldrich, St. Louis, MO) or physiological saline was intraperitoneally administered 6 h before euthanasia on day 60. Perfusion fixation was performed using 4% paraformaldehyde (PFA), and frozen sections were prepared. TUNEL staining was performed according to the manufacturer’s protocol using a DeadEnd Fluorometric TUNEL System (Promega, Fitchburg, WI). TUNEL positive epithelial cells per 10 crypts were measured and compared between groups.(28)
Statistical analysis
The statistical significance of the differences was determined by one-way ANOVA with Bonferroni post hoc tests and Student’s t tests. P values less than 0.05 were considered to be statistically significant.
Results
The protein p62 is a selective substrate for autophagy degradation(29) and lack of autophagy leads to accumulation of p62. In order to investigate the activity of autophagy in the mucosa of IBD patients, we stained p62 protein. An enhanced accumulation of p62 protein was observed in the epithelial cells of the inactive and active mucosa of UC and CD patients compared to normal mucosa (Fig. 1), indicating an impaired autophagy flux in the colonic epithelial cells of IBD patients.
Fig. 1.

The expression of p62 protein in the epithelium of patients with IBD. Immunostaining for p62 was performed in normal mucosa and in the inactive and active mucosa of UC and CD patients. Pictures are shown from one of six independent samples with similar results. Original magnification: ×100.
Atg5fl/fl and Atg5ΔIEC mice were normal in body weight gain and showed no symptoms of colitis (Fig. 2A and B). Colitis was induced by three cycles of administration of 1.5% DSS in Atg5fl/fl and Atg5ΔIEC mice. Body weight loss after each DSS treatment and disease activity index on day 60 were significantly higher in the Atg5ΔIEC DSS group than in the Atg5fl/fl DSS group (Fig. 2A and B). Moreover, colon length on day 60 was significantly shorter in the Atg5ΔIEC DSS group than in the Atg5fl/fl DSS group (Fig. 2C). Colon weight/length ratio, a marker of tissue edema, was significantly higher in the Atg5ΔIEC DSS group than in the Atg5fl/fl DSS group (Fig. 2D). In addition, histological scores on day 60 were significantly higher in the Atg5ΔIEC DSS group than in the Atg5fl/fl DSS group (Fig. 3A and B). Thus, Atg5ΔIEC mice were highly susceptible to DSS-colitis, indicating a protective role of Atg5-mediated autophagy in the colon.
Fig. 2.
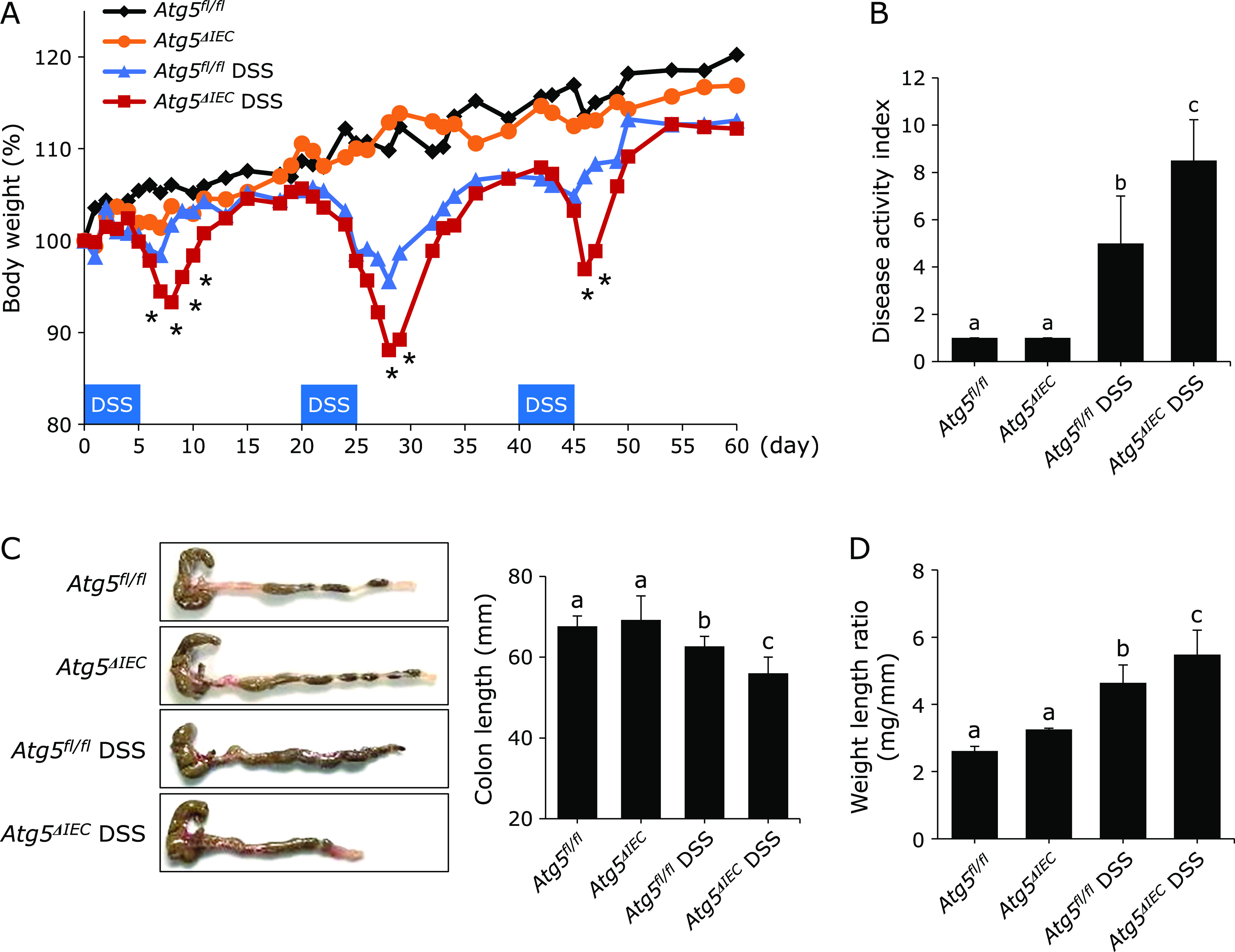
The deficiency of Atg5 increased susceptibility to DSS-induced colitis. IEC specific autophagy-deficient mice (Atg5ΔIEC) were generated by crossbreeding Atg5 floxed (Atg5fl/fl) mice with Villin-Cre mice. The experimental chronic colitis was induced by 3 five-day cycles of 1.5% w/v DSS treatment followed by 14 days of water. The mice were sacrificed on day 60. (A) Body weight. A statistically significant difference between the Atg5fl/fl DSS group and the Atg5ΔIEC DSS group was shown (*p<0.05). (B) Disease activity index. (C) Representative photographs of the colon. (D) Colonic weight per length on day 60. The data are expressed as means ± SEM (n = 10 mice/group). Values not sharing a letter are significantly different (p<0.05).
Fig. 3.

Histological evaluation of chronic DSS-induced colitis. (A) Histological pictures of the colonic tissue on day 60. Original magnification; ×200. (B) Histological score. The data are expressed as means ± SEM (n = 10 mice/group). Values not sharing a letter are significantly different (p<0.05).
It has recently been reported that autophagy in IECs suppresses excessive ER stress and controls intestinal inflammation.(19,20) Therefore, we hypothesized that the Atg5 deficiency enhanced ER stress in IECs, leading to the exacerbation of DSS-induced chronic colitis. To test this hypothesis, we examined the expression of IRE1α, which is a master regulator of the ER stress pathway. As shown in Fig. 4A, phosphorylated IRE1α was not detected in the absence of DSS treatment. DSS treatment induced an expression of phosphorylated IRE1α in the colonic epithelial cells of the Atg5fl/fl mice, and this was much enhanced in the Atg5ΔIEC mice. These results indicate that the Atg5 deficiency induced an enhancement of ER stress in colonic epithelial cells.
Fig. 4.
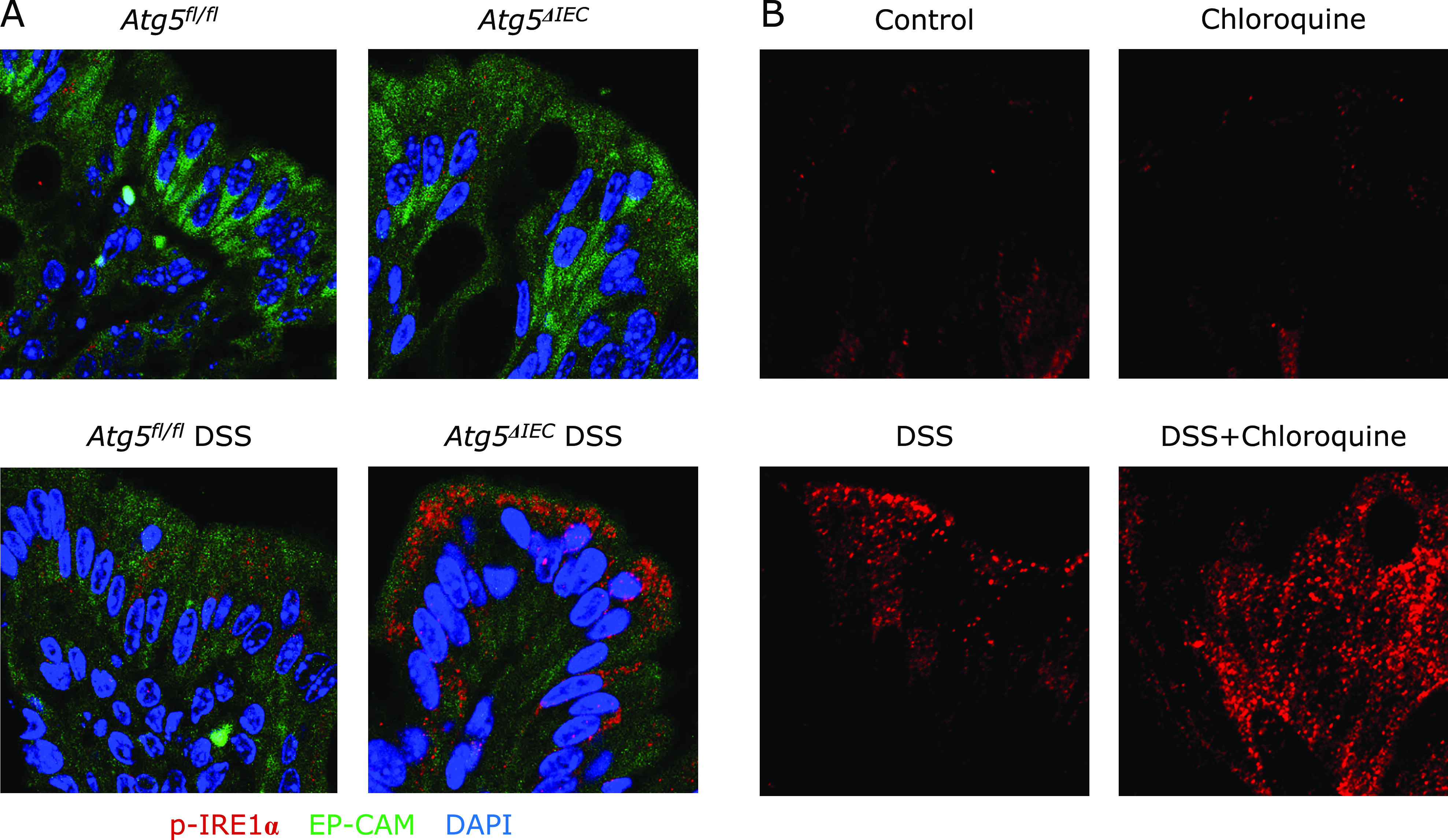
The deficiency of Atg5 enhanced an activation of IRE1α in IECs. (A) Immunofluorescence staining was used to evaluate expression of p-IRE1α (red fluorescence), EP-CAM (green fluorescence) and DAPI (blue fluorescence). Representative pictures are shown from one of four independent experiments with similar results. (B) Effects of chloroquine, an inhibitor of autophagy, in DSS-colitis developed in Atg5 normal mice. Chloroquine (50 µg/g body weight) (Sigma-Aldrich) or physiological saline was intraperitoneally administered 6 h before euthanasia on day 60.
To confirm a contribution of autophagy to induction of phosphorylated (p)-IRE1α in DSS mice, we used chloroquine. Chloroquine has been reported to affect the endo-lysosomal system and impair the basal autophagic flux by decreasing autophagosome-lysosome fusion and lysosomal protein degradation.(30) This leads to intracellular accumulation of proteins which should be disassembled by autophagy. In this study, DSS colitis was induced in the mice bearing normal Atg5 gene (the Atg5fl/fl mice) (Fig. 4B). Chloroquine itself did not stimulate p-IRE1α expression. DSS-treatment induced phosphorylated IRE1α, and this was markedly enhanced by chloroquine treatment. This finding suggests that autophagy might mediate degradation of p-IRE1α in DSS colitis.
It has been reported that phosphorylation of IRE1α leads to activation of c-Jun N-terminal kinase (JNK)(31) which is a down-stream target of IRE1α and plays a critical role in various cellular responses. As shown in Fig. 5A and B, immunoblot analysis of proteins isolated from colonic epithelial cells revealed that the phosphorylation of IRE1α was significantly enhanced in the Atg5ΔIEC DSS group as compared to in the Atg5fl/fl DSS group, and these responses were accompanied by a significant enhancement of phosphorylation of c-JUN (Fig. 5A and C). These results indicate that IEC-specific deficiency of autophagy enhanced the activation of IRE1α-JNK pathway in colonic epithelial cells.
Fig. 5.

Immunoblot for phosphorylated (p)-IRE1α, IRE1α, p-JNK and JNK in the isolated colonic epithelial cells. (A) Total protein was isolated from colonic epithelial cells. GAPDH was used as a loading control. The pictures are representative of four independent experiments. Relative expression of p-IRE1α (B) and p-JNK (C). The data are expressed as means ± SEM (n = 4 mice/group). Values not sharing a letter are significantly different (p<0.05).
It is known that apoptosis of colonic epithelial cells contributes to the severity of DSS-induced colitis.(32,33) As shown in Fig. 6A, TUNEL-positive colonic epithelial cells were more abundant in the Atg5ΔIEC DSS mice than in the Atg5fl/fl DSS mice. The number of apoptosis cells/10 crypts was significantly higher in the Atg5ΔIEC DSS mice than in the Atg5fl/fl DSS mice (Fig. 6B). These results were also observed in the samples from mice of a similar degree of histological score (Fig. 6C). The number of TUNEL positive epithelial cells in the samples with a histological score between 3 to 6 points was significantly higher in the Atg5ΔIEC DSS group than in the Atg5fl/fl DSS group (Fig. 6D). These results indicate that Atg5 suppresses apoptosis of colonic epithelial cells in DSS-induced colitis.
Fig. 6.

The deficiency of Atg5 increased the apoptosis of colonic epithelial cells. (A) TUNEL staining in the colon sections. (B) Number of TUNEL positive epithelial cells per 10 crypts. The data are expressed as means ± SEM (n = 10 mice/group). Values not sharing a letter are significantly different (p<0.05). (C) Correlation between number of TUNEL positive epithelial cells and histological score. (D) Number of TUNEL positive epithelial cells per 10 crypts in the comparison using samples with a similar degree of histological score (from 3 to 6 points). The data are expressed as means ± SEM. **p<0.01.
Discussion
Previous studies have revealed that autophagy and ER stress play important roles in the pathophysiology of IBD.(34,35) However, the mechanistic relationship between autophagy and ER stress has not been fully elucidated. In the present study, we demonstrated that Atg5 in colonic epithelial cells plays a protective role in the development of DSS-induced colitis by regulating excessive ER stress via inhibition of the IRE1α-JNK pathway.
Genome-wide association studies identified autophagy-related susceptibility gene loci for IBD such as IRGM, ATG16L1 and NOD2,(34,36,37) suggesting the involvement of autophagy dysfunction in the pathophysiology of IBD. In this study, we examined the autophagy activity in the inflamed mucosa of IBD patients by p62 immunostaining. The p62 protein is degraded by autophagy and its expression has a negative correlation with the autophagy activity.(38) A marked accumulation of p62 was observed in the colonic epithelial cells of non-inflamed and inflamed mucosa of UC and CD patients, suggesting an impaired autophagy response in the colonic epithelial cells of IBD patients. Although impaired autophagy response has been mainly implicated to the pathophysiology of CD,(16) our observation suggest that autophagy response is also defective in UC patients. Furthermore, these changes were unrelated to the severity of inflammation, suggesting that autophagy dysfunction may be a fundamental etiologic factor of IBD.
This study indicated that IEC-specific Atg5 deletion aggravated DSS-induced colitis, suggesting a protective role of epithelial Atg5. In addition, IEC-specific Atg5 deletion was accompanied by an increased number of epithelial apoptotic cells. This was not a consequence of inflammation, since the number of apoptotic cells was significantly elevated in the Atg5ΔIEC mice compared to the Atg5fl/fl mice with similar disease activity. Pott et al.(39) recently reported a similar observation that chronic colitis induced by infection of Helicobacter hepaticus in IEC-specific Atg16l1-deficient mice exhibited increased apoptosis of colonic epithelial cells. These findings suggest that the dysfunction of autophagy might predispose IECs to apoptosis during mucosal inflammation via an intrinsic mechanism. Epithelial cell apoptosis results in a blockade of the epithelial barrier and leads to easy access of luminal antigens and bacteria to immune cells in the lamina propria and leads to the aggravation of colitis.
Along with the studies of autophagy-associated susceptible genes, genome-wide association studies have identified a strong association of IBD with ER stress/UPR genes,(16) suggesting a linkage of dysfunction of autophagy and ER stress in the pathophysiology of IBD. IRE1α, which is one of the ER stress sensors, exists as a monomer in a steady state. Upon sensing ER stress, IRE1α is activated by autophosphorylation leading to the formation of dimers and oligomers.(40) In a steady state, monomeric IRE1α is degraded via a ubiquitin ligase complex,(41) but the ER stress-induced molecular clusters of p-IRE1α are degraded by autophagy.(20,42,43) In order to improve accumulated ER stress, the UPR induce autophagy to degrade misfolded proteins.(16) ER stress is harmful in any cells when the UPR or autophagy does not work normally. In this study, marked accumulation of p-IRE1α in colonic epithelial cells was found in Atg5ΔIEC DSS mice. Inhibition of autophagy by chloroquine induced strong intracellular accumulation of IRE1α in DSS colitis in Atg5 normal mice. These results suggest that autophagy mediates degradation of ER stress-induced phosphorylation of IRE1α in IECs in DSS colitis. In addition, we found that autophagy deficiency in IECs was accompanied by an enhancement of JNK activation and apoptosis of colonic epithelial cells. Since excessive ER stress has been reported to induce apoptosis via the IRE1α-JNK pathway in various cell types,(44) these findings suggest that autophagy in IECs suppresses epithelial apoptosis via inhibition of the IRE1α-JNK pathway leading to protection against chronic colitis.
JNK activation in Atg5ΔIEC DSS mice plays another role in inflammatory response. It has been reported that ER stress-induced IRE1α activation stimulates JNK activation and recruits TNF receptor-associated factor 2 (TRAF2) to trigger NF-κB signaling.(16) Since NF-κB is a central transcription factor in immune and inflammatory responses such as cytokine secretion and free radical production,(45) aggravation of DSS colitis in Atg5ΔIEC mice might be mediated by ER stress-associated activation of NF-κB pathway. This possibility is supported by a recent report that ER stress increased inflammation and IL-6 production.(46)
In conclusion, Atg5 in IECs directly protects against ER stress-induced apoptosis via the IRE1α-JNK pathway during chronic colitis. Our observation indicates that the autophagy and ER stress/UPR pathways are closely linked and contributes to the maintenance of intestinal homeostasis. Therefore, the development of therapeutic drugs targeting autophagy and UPR may open new avenues for the future treatment of IBD.
Acknowledgments
This study was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan [18K07909 (AN), 18K08241 (SK), 16K09308 (OI), 16K9846 (MK), 18H02862 (HM), and 18K08002 (AA)], a grant for the Intractable Diseases from the Ministry of Health, Labour and Welfare of Japan [067 (AA)], a grant from the Practical Research Project for Rare/Intractable Diseases from the Japan Agency for Medical Research and Development, AMED [15AeK0109047h0002 (AA)] and a grant from the Smoking Research Foundation [1848 (AA)]. We also thank Noboru Mizushima (Tokyo University) for providing Atg5-floxed mice.
Conflict of Interest
No potential conflicts of interest were disclosed.
Supplementary Material
References
- 1.Verstockt B, Ferrante M, Vermeire S, Van Assche G. New treatment options for inflammatory bowel diseases. J Gastroenterol 2018; 53: 585–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol 2010; 28: 573–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Podolsky DK. Inflammatory bowel disease. N Engl J Med 2002; 347: 417–429. [DOI] [PubMed] [Google Scholar]
- 4.Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J Cell Biol 1992; 119: 301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baxt LA, Xavier RJ. Role of autophagy in the maintenance of intestinal homeostasis. Gastroenterology 2015; 149: 553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldman SJ, Zhang Y, Jin S. Autophagic degradation of mitochondria in white adipose tissue differentiation. Antioxid Redox Signal 2011; 14: 1971–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mortensen M, Ferguson DJ, Edelmann M, et al. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci U S A 2010; 107: 832–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang P, Mizushima N. Autophagy and human diseases. Cell Res 2014; 24: 69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet 2007; 39: 596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cadwell K. Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis. Nat Rev Immunol 2016; 16: 661–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest 2005; 115: 2656–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007; 8: 519–529. [DOI] [PubMed] [Google Scholar]
- 13.Rao RV, Bredesen DE. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr Opin Cell Biol 2004; 16: 653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaser A, Lee AH, Franke A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008; 134: 743–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao SS. Epithelial ER stress in Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis 2016; 22: 984–993. [DOI] [PubMed] [Google Scholar]
- 16.Hooper KM, Barlow PG, Henderson P, Stevens C. Interactions between autophagy and the unfolded protein response: implications for inflammatory bowel disease. Inflamm Bowel Dis 2019; 25: 661–671. [DOI] [PubMed] [Google Scholar]
- 17.Sugiyama T, Sasaki M, Nakagawa S, et al. The association among enterobacterial flora, dietary factors, and prognosis in patients with ulcerative colitis. J Clin Biochem Nutr 2020; 66: 152–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deuring JJ, Fuhler GM, Konstantinov SR, et al. Genomic ATG16L1 risk allele-restricted Paneth cell ER stress in quiescent Crohn’s disease. Gut 2014; 63: 1081–1091. [DOI] [PubMed] [Google Scholar]
- 19.Adolph TE, Tomczak MF, Niederreiter L, et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013; 503: 272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tschurtschenthaler M, Adolph TE, Ashcroft JW, et al. Defective ATG16L1-mediated removal of IRE1α drives Crohn’s disease-like ileitis. J Exp Med 2017; 214: 401–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Codogno P, Meijer AJ. Atg5: more than an autophagy factor. Nat Cell Biol 2006; 8: 1045–1047. [DOI] [PubMed] [Google Scholar]
- 22.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15: 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang YR, Kim DH, Seo YK, et al. Elevated O-GlcNAcylation promotes colonic inflammation and tumorigenesis by modulating NF-κB signaling. Oncotarget 2015; 6: 12529–12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berberat PO, YI AR, Yamashita K, et al. Heme oxygenase-1-generated biliverdin ameliorates experimental murine colitis. Inflamm Bowel Dis 2005; 11: 350–359. [DOI] [PubMed] [Google Scholar]
- 25.Obermeier F, Kojouharoff G, Hans W, Schölmerich J, Gross V, Falk W. Interferon-gamma (IFN-γ)- and tumour necrosis factor (TNF)-induced nitric oxide as toxic effector molecule in chronic dextran sulphate sodium (DSS)-induced colitis in mice. Clin Exp Immunol 1999; 116: 238–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singh N, Gurav A, Sivaprakasam S, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014; 40: 128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nishida A, Hidaka K, Kanda T, et al. Increased expression of interleukin-36, a member of the interleukin-1 cytokine family, in inflammatory bowel disease. Inflamm Bowel Dis 2016; 22: 303–314. [DOI] [PubMed] [Google Scholar]
- 28.Hino K, Saito A, Asada R, Kanemoto S, Imaizumi K. Increased susceptibility to dextran sulfate sodium-induced colitis in the endoplasmic reticulum stress transducer OASIS deficient mice. PLoS One 2014; 9: e88048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bjørkøy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171: 603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mauthe M, Orhon I, Rocchi C, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018; 14: 1435–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000; 287: 664–666. [DOI] [PubMed] [Google Scholar]
- 32.Qiu W, Wu B, Wang X, et al. PUMA-mediated intestinal epithelial apoptosis contributes to ulcerative colitis in humans and mice. J Clin Invest 2011; 121: 1722–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frey MR, Edelblum KL, Mullane MT, Liang D, Polk DB. The ErbB4 growth factor receptor is required for colon epithelial cell survival in the presence of TNF. Gastroenterology 2009; 136: 217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patel KK, Stappenbeck TS. Autophagy and intestinal homeostasis. Annu Rev Physiol 2013; 75: 241–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 2013; 13: 722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaser A, Blumberg RS. Autophagy, microbial sensing, endoplasmic reticulum stress, and epithelial function in inflammatory bowel disease. Gastroenterology 2011; 140: 1738–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cadwell K, Patel KK, Komatsu M, Virgin HW 4th, Stappenbeck TS. A common role for Atg16L1, Atg5 and Atg7 in small intestinal Paneth cells and Crohn disease. Autophagy 2009; 5: 250–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007; 131: 1149–1163. [DOI] [PubMed] [Google Scholar]
- 39.Pott J, Kabat AM, Maloy KJ. Intestinal epithelial cell autophagy is required to protect against TNF-induced apoptosis during chronic colitis in mice. Cell Host Microbe 2018; 23: 191–202.e4. [DOI] [PubMed] [Google Scholar]
- 40.Credle JJ, Finer-Moore JS, Papa FR, Stroud RM, Walter P. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc Natl Acad Sci U S A 2005; 102: 18773–18784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun S, Shi G, Sha H, et al. IRE1α is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat Cell Biol 2015; 17: 1546–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghosh R, Wang L, Wang ES, et al. Allosteric inhibition of the IRE1α RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014; 158: 534–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Korennykh A, Walter P. Structural basis of the unfolded protein response. Annu Rev Cell Dev Biol 2012; 28: 251–277. [DOI] [PubMed] [Google Scholar]
- 44.Wu L, Liu X, Wang L, et al. Exendin-4 protects HUVECs from tunicamycin-induced apoptosis via inhibiting the IRE1a/JNK/caspase-3 pathway. Endocrine 2017; 55: 764–772. [DOI] [PubMed] [Google Scholar]
- 45.Li Q, Verma IM. NF-κB regulation in the immune system. Nat Rev Immunol 2002; 2: 725–734. [DOI] [PubMed] [Google Scholar]
- 46.Keestra-Gounder AM, Byndloss MX, Seyffert N, et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature 2016; 532: 394–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.