

Abstract
The abstract is published online only. If you did not include a short abstract for the online version when you submitted the manuscript, the first paragraph or the first 10 lines of the chapter will be displayed here. If possible, please provide us with an informative abstract.
It has been speculated for many years that heparanase plays an important role in the progression of cancer due largely to the finding that its expression is weak or absent in normal tissues but generally as tumors become more aggressive heparanase expression increases. However, it is only in the last decade or so that we have begun to understand the molecular mechanism behind the sinister role that heparanase plays in cancer. In this review, we describe the many functions of heparanase in promoting the growth, angiogenesis and metastasis of multiple myeloma, a devastating cancer that localizes predominantly within the bone marrow and spreads throughout the skeletal system devouring bone and ultimately leading to death of almost all patients diagnosed with this disease. We also explore recent discoveries related to how heparanase primes exosome biogenesis and how heparanase enhances myeloma tumor chemoresistance. Discovery of these multiple tumor-promoting pathways that are driven by heparanase identified the enzyme as an ideal target for therapy of applying a novel heparanase inhibitor recently tested in a Phase I trial in myeloma patients.
Keywords: Multiple myeloma, Exosomes, Chemoresistance, Syndecan-1, Proteases
1. Introduction
Multiple myeloma is a devastating cancer that is highly dependent on the bone marrow microenvironment for growth and survival [1]. Studies over the past two decades underscore the notion that the heparan sulfate degrading enzyme heparanase plays a major role in modulating the bone marrow microenvironment to support the progression and growth of multiple myeloma. Importantly, high heparanase activity in myeloma cells correlates with enhanced bone marrow angiogenesis, myeloma growth/metastasis and osteolytic bone disease (Table 1). A clinical trial testing anti-heparanase therapy in multiple myeloma patients was well-tolerated and showed some potential early signs of efficacy, emphasizing that targeting heparanase is a novel strategy for myeloma therapy [2]. Though heparanase has both enzymatic and non-enzymatic functions, much of its activity in myeloma is dependent on its enzymatic cleavage of heparan sulfate chains of syndecan-1 proteoglycan present on the surface of myeloma cells [3]. Heparanase mediated structural alterations of syndecan-1 leads to enhanced shedding of this proteoglycan from the surface of myeloma cells and high levels of shed syndecan-1 in myeloma patients are associated with poor prognosis and diminished overall survival [4, 5, 6]. Shed syndecan-1 plays diverse roles in the myeloma microenvironment, including shuttling of growth factors to both tumor and host cell surfaces and enhancing the formation of signaling complexes at the cell surface [7, 8, 9]. In addition, heparanase expression by myeloma cells enhances the activation of signaling pathways (ERK, p—38) and upregulation of multiple genes (VEGF, HGF, MMP-9) associated with enhancing myeloma progression [3, 10]. Recent studies also emphasize the involvement of heparanase in exosome secretion and chemoresistance in myeloma, highlighting its potential in mediating myeloma-host interactions and in dictating the response of myeloma cells to chemotherapeutic anti-myeloma drugs [9, 11, 10, 12] (Fig. 1). Importantly, heparanase levels were found to be elevated in myeloma cells from patients after the first and second rounds of chemotherapy, implying that heparanase is a highly desirable and druggable target for myeloma therapy [13]. This chapter focuses primarily on the diverse mechanisms employed by heparanase in the progression of myeloma including upregulation of multiple genes involved in remodeling of the tumor microenvironment, shedding of syndecan-1 from myeloma cell surface and formation of signaling complexes at the cell surface that are involved in tumor cell dissemination, bone damage, and angiogenesis.
Table 1.
Multi-functional role of heparanase in myeloma progression
| Impact | References |
|---|---|
| Promotes myeloma growth, proliferation and metastasis | [8] [54] [53] [44] [45] [61] [11] [14] |
| Promotes osteolysis | [54] [52] |
| Enhance endothelial cell invasion and angiogenesis | [56] [45] [8] |
| Promotes exosome biogenesis and exosome docking | [66, 71] [67] |
| Promotes syndecan-1 shedding and myeloma cell migration | [4] [8] |
| Regulates signaling and gene transcription | [22] [45] |
| Promotes drug resistance | [74] [13] [69] |
Fig. 1.
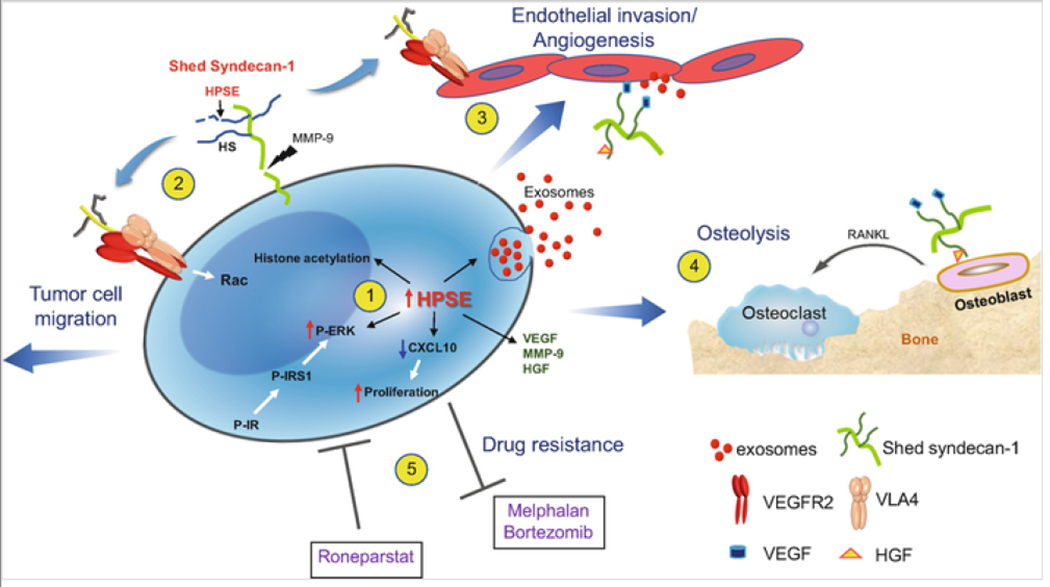
Heparanase triggers multiple pathways that drive myeloma progression. (1) Enhanced expression of heparanase by myeloma cells: augments gene transcription by enhancing acetylation of histones, stimulates exosome biogenesis by trimming the heparan sulfate chains of syndecan-1 thereby priming formation of the syndecan-syntenin-ALIX complex, downregulates CXCL10 causing increased tumor cell proliferation and activates ERK via the insulin signaling pathway resulting in enhanced expression of MMP-9 and VEGF. (2) Shedding of syndecan-1 from the myeloma surface is driven by the heparanase-mediated trimming of heparan sulfate and by the increase in MMP-9 secretion. The shed syndecan-1 complexes with VLA-4 and VEGFR2 on the tumor cell surface stimulating Rac signaling and resulting in cell migration/invasion. (3) Via the same mechanism as in tumor cells, shed syndecan-1 initiates Rac signaling in endothelial cells that promotes angiogenesis. Increased angiogenesis also occurs when angiogenic growth factors (VEGF, HGF) bound to shed syndecan-1 heparan sulfate chains activate receptors on endothelial cells and when exosomes bearing VEGF, HGF and heparanase cargo dock with endothelial cells. (4) Similarly, HGF bound to shed syndecan-1 activates the cMet receptor on osteoblasts that via an IL-11 feedback mechanism increases RANKL secretion leading to osteoclast activation and osteolysis. (5) Myeloma cells having elevated heparanase expression exhibit resistance to commonly used anti-myeloma drugs, including proteasome inhibitors (bortezomib, carfilzomib) and the alkylating agent melphalan. Conversely, exposure of cells to the heparanase inhibitor Roneparstat blocks the multiple pathways that are stimulated by heparanase (e.g., syndecan shedding, angiogenesis) resulting in decreased drug resistance and inhibition of myeloma growth in vivo
2. Heparanase Promotes Shedding of Syndecan-1 from the Myeloma Tumor Cell Surface
Shed syndecan-1 represents the soluble form of syndecan-1, containing intact heparan sulfate chains, that is released from the cell surface by proteolytic cleavage of the extracellular domain of its core protein [4]. Shed syndecan can either remain soluble or bind and accumulate on the cell surface or within the extracellular matrix [14, 15, 16]. Shed syndecan-1 is detected in a number of tumor types and in myeloma high levels of shed syndecan-1 in patient sera is an independent predictor of poor prognosis [5, 6]. This is consistent with the finding that enhanced expression of a soluble form of syndecan-1 by myeloma cells promotes tumor growth and metastasis in a mouse model [17, 18]. Importantly, heparanase upregulates both the expression of syndecan-1 and its shedding from the tumor cell surface [4, 19]. This is supported by the finding that silencing of heparanase gene expression in myeloma cells is associated with decreased levels of syndecan-1 shedding. Heparanase enzymatic activity is required (i.e., cleavage of heparan sulfate chains) for upregulation of both syndecan-1 expression and shedding because expression of enzymatically inactive form of heparanase failed to stimulate syndecan-1 expression and shedding [4]. Syndecan-1 shedding was also stimulated in myeloma cells after addition of recombinant active heparanase or bacterial heparitinase (heparinase III), indicating that cleavage of heparan sulfate chains by these enzymes .renders syndecan-1 core protein more susceptible to proteolysis by proteases that mediate syndecan-1 shedding. Further, it appears that heparanase plays a more direct role in facilitating syndecan-1 shedding by upregulating the expression of proteases (sheddases) that clip the syndecan-1 core protein in the extracellular region near the plasma membrane [20].
3. Heparanase Modulates the Expression of Proteases by Myeloma Cells
In myeloma cells, enhanced heparanase activity leads to increased MMP-9 expression and secretion, while silencing heparanase expression results in reduced MMP-9 activity [20]. In addition, levels of molecular determinants involved in the activation of MMP-9 such as urokinase plasminogen activator and urokinase-plasminogen activator receptor are also upregulated by heparanase expression in myeloma cells [20]. These findings have potential relevance in myeloma because inhibiting MMP-9 or uPA/uPAR interactions significantly reduce the shedding of syndecan-1 by myeloma cells. Though shedding of syndecan-1 has been attributed to several metalloproteinase enzymes such as MMP-9, MMP-1, MMP-14, MMP-16 and MMP-7, a role for uPA/uPAR as a sheddase and its correlation with heparanase expression revealed a novel mechanism underlying syndecan-1 shedding. However, it is likely that in myeloma cells, uPA does not directly cleave the syndecan-1 ectodomain at the cell surface, rather it activates the cascade that drives MMP-9 activation and subsequent syndecan-1 shedding. Also, it’s interesting that blocking MMP-9 or uPA/uPAR in cells expressing a low level of heparanase did not alter the constitutive level of shedding of syndecan-1 [20]. This suggests that upregulation of heparanase expression activates a shedding mechanism that is distinct from that mediating constitutive shedding. Despite the upregulation of MMP-9 by heparanase in myeloma tumor cells, in heparanase knockout mice, heparanase deficiency was accompanied by a marked elevation of MMP-9, MMP-2, and MMP-14, in an organ-dependent manner [21]. These findings suggest that the onset of heparanase expression marks a key defining event in mediating the induction and/or repression of protease gene depending on the biological setting.
Mechanistically, the upregulation of MMP-9 expression by heparanase in myeloma is mediated through the activation of ERK signaling [20]. In myeloma, heparanase-induced ERK activation is mediated through insulin receptor signaling. Heparanase promotes the phosphorylation of insulin receptors and enhances protein kinase C activity [22]. PKC activity, in turn, upregulates the levels of insulin receptor substrate-1 (IRS-1), the primary intracellular substrate for insulin receptor tyrosine kinase activity. IRS-1 plays a key role in transmitting signals from insulin and insulin-like growth factors. Tyrosine phosphorylation of insulin receptors induces the cytoplasmic binding of IRS-1 to these receptors which then undergo phosphorylation. This enables IRS-1 to activate ERK signaling [22]. Though ERK activation depends on the HS degrading activity of heparanase, the mechanism underlying this phenomenon is not clear. It is possible that trimming of heparan sulfate chains of syndecan-1 on the myeloma cell surface can trigger the clustering and activation of insulin receptors. This notion is supported by the findings that syndecan-1 couples with IGF-1 receptor on myeloma cells, and further the insulin receptor associates with IGF-1 receptors to form an insulin/IGF-1 hybrid receptor at the myeloma cell membrane [23]. This new insight into the mechanism of heparanase induced ERK activation provides a further understanding of how heparanase can impact myeloma progression. This is particularly relevant given the known effect of ERK activation in promoting myeloma cell proliferation, survival, drug resistance, and angiogenesis [24, 25].
4. Heparanase Regulates Gene Expression in Myeloma Cells by Altering Histone Acetylation
Although the tumor-promoting effects of heparanase can in part be attributed to its ability to remodel the extracellular matrix barrier by cleaving heparan sulfate chains, heparanase is also known to influence gene transcription. Elevation of heparanase levels in myeloma cells upregulates expression of multiple genes, including MMP-9, VEGF, and HGF, among others [3]. Heparanase is present and active in the nucleus where it could act locally to regulate gene expression [26]. Studies have shown that translocation of heparanase to the nucleus and degradation of nuclear HS chains regulates esophageal epithelial cell differentiation [27]. Further, the presence of heparanase in the nucleus can also regulate the activity of certain nuclear enzymes. For example, in breast cancer cells, the activity of topoisomerase I, an enzyme involved in DNA replication and transcription, is enhanced by nuclear translocation of heparanase [28]. In T lymphocytes, nuclear heparanase regulates the transcription of a group of inducible immune response genes by associating with euchromatin and controlling the pattern of histone 3 methylation [29]. Heparanase modifies the histone 3 methylation pattern by associating with the demethylase LSD1 and preventing recruitment of methylase MLL (Bhattacharya et al., Unpublished data). In some cancers, the distinct cellular localization of heparanase (either cytoplasmic or nuclear) may be of prognostic value [30, 31].
Acetylation by histone acetyltransferase (HAT) of N-terminal tails of histones, is a process that correlates with transcriptional activation [32, 33, 34, 35]. This process is balanced by selectively removing acetyl groups from histones by histone deacetylase (HDAC). A shift in the imbalance between HAT and HDAC activity modulates transcriptional activity and can lead to cell apoptosis, proliferation and malignancy [34]. Importantly, in addition to heparanase, there are numerous reports showing the presence of heparan sulfate proteoglycans in the nucleus of cells and gene repression due to the reduction in HAT activity mediated by heparan sulfate chains [36, 37, 38, 39, 40]. In a cell-free assay, exogenous heparin was demonstrated to block HAT activity, and further, the acetylation of histone H3 is reduced by 50% in pulmonary fibroblasts exposed to heparin [40]. In addition, a decrease in histone H3 acetylation is also observed in tumor cells that take up antiproliferative glycosaminoglycans [41]. Although the mechanism by which heparan sulfate chains inhibit HAT activity is unknown, the inhibitory activity is dependent upon heparan sulfate chain length and sulfation pattern, indicating that it is not a random inhibition but rather involves some degree of specificity [40, 41]. Regarding mechanisms by which negatively charged heparan sulfate blocks HAT activity, there are multiple possibilities including direct binding between HS and HAT and HS blocking of the acetylation sites on histones via binding to the positively charged lysine residues (acetylation sites in histones).
Heparanase expression in myeloma cells significantly reduces the amount of syndecan-1 present in the nucleus of these cells [42]. This, in turn, is linked to high HAT activity in myeloma cells and elevated expression of genes that drive an aggressive disease [43]. The molecular mechanism by which heparanase regulates nuclear levels of syndecan-1 is not still clear. However, it’s possible that nuclear heparanase degrades syndecan-1 heparan sulfate chains, thereby eliminating syndecan-1 from the nucleus. In addition, it’s also possible that heparanase, via unknown mechanisms, can block the transport of syndecan-1 to the nucleus resulting in a significant reduction in total levels of nuclear syndecan-1. Together these findings reveal a novel and important function of heparanase in regulating myeloma cell behavior via upregulating HAT activity and gene transcription.
Heparanase is present and active in plasma isolated from the bone marrow of myeloma patients and studies have shown that both myeloma cells and stromal cells in the bone marrow can express heparanase [44, 19]. The finding that exogenous heparanase can cause upregulation of HAT activity by myeloma cells demonstrates an important and novel mechanism whereby gene expression can be regulated by cross talk between cells within a tumor. Heparanase released from either myeloma cells or host cells could alter HAT activity and gene expression in adjacent tumor or host cells not expressing heparanase. This may be particularly important in cancers such as myeloma, which are highly dependent on the tumor microenvironment for their survival.
5. How Does Heparanase Promote, Myeloma Growth, Metastasis, Angiogenesis and Osteolysis?
5.1. Down-Regulation of CXCL10 Cytokine
Using a Tet-on system, the mechanisms underlying the pro-tumorigenic activity of heparanase were explored in myeloma. Induction of heparanase expression in myeloma cells by doxycycline increased the colony number and size in soft agar and tumor growth in vivo. As determined by gene array profiling, the induction of heparanase was associated with downregulation of cytokine CXCL10 [45]. Overexpression of CXCL10 in heparanase-high myeloma cells results in significantly fewer and smaller colonies in soft agar compared to control cells, clearly demonstrating that CXCL10 suppresses myeloma proliferation [46]. Silencing of the CXCL10 gene or addition of CXCL10 neutralizing antibody enhanced cell proliferation and colony formation, further supporting the notion that CXCL10 can attenuate myeloma cell proliferation. Importantly, CXCL10 gene silencing resulted in tumor xenografts that were larger than control myeloma tumors, while overexpression of CXCL10 or its injection into tumor-bearing mice resulted in a marked decrease in tumor development. CXCL10 has multiple functions in inhibiting myeloma progression. It directly inhibits myeloma cell proliferation, endothelial cell proliferation and angiogenesis, and it attracts anti-tumor immune cells [46]. CXCL10 is an interferon-inducible chemokine with potent chemotactic activity on activated effector T cells and other leukocytes that express the CXCL10 G protein-coupled receptor CXCR3. Consistent with this function, an increase in infiltration of cytotoxic NK and T cells was observed in myeloma tumor-bearing mice administered with CXCL10-Ig fusion protein [46].
5.2. Upregulation of HGF Expression and Activity
HGF is among the most upregulated genes in multiple myeloma, and elevated levels of HGF in myeloma are associated with poor prognosis [47, 48]. Myeloma cells produce HGF and express its receptor c-met [49]. HGF upregulation in the bone marrow microenvironment of multiple myeloma is associated with lytic bone disease [50]. HGF has a heparin binding domain and thus binds to heparan sulfate proteoglycans, in addition to binding to its c-met receptor [51]. In myeloma, HGF binds to syndecan-1 on the surface of myeloma cells and when syndecan-1 is shed from the surface, the syndecan-1/HGF complex can potentiate signaling via the c-met receptor present on distal cells [51, 52]. Apparently, heparanase expression in myeloma cells fuels this signaling pathway by increasing syndecan-1 shedding and by enhancing the expression of HGF. However, upregulation of HGF expression is not dependent on heparanase enzymatic activity [52].
Heparanase-induced HGF secretion by myeloma cells activates c-met signaling in osteoblasts leading to an increase in IL-11 secretion [52, 50]. IL-11, via a feedback loop, stimulates osteoblasts to produce RANKL, a key factor that drives osteolytic bone disease in myeloma [52]. Studies utilizing SCID-hu and SCID-tibia animal models of myeloma demonstrated that myeloma tumors, growing in bone and expressing high heparanase, increase both local and systemic bone damage as compared to control myeloma tumors expressing much lower levels of heparanase [53, 54]. This enhanced bone damage caused by heparanase appears linked to upregulation of RANKL by myeloma cells or via indirect impact on osteoblasts through the HGF/-IL-11 axis. The latter is supported by the finding that heparanase expression dramatically stimulates osteolysis in distal bones prior to the arrival of tumor cells at those sites [54]. This finding underscores the possibility that heparanase-induced production of HGF and soluble syndecan-1 by myeloma cells act upon distal osteoblasts to produce RANKL and subsequent osteolysis. Another possible impact of heparanase-mediated HGF signaling could be in mechanisms leading to minimal residual disease, a precursor to patient relapse and eventual death. Shed syndecan-1 that is known to accumulate within the bone marrow of myeloma patients likely facilitates the accumulation of a reservoir of HGF that is available for the growth of myeloma cells that escape therapy, thereby contributing to myeloma relapse [16]. The ability of heparanase to enhance syndecan-1 shedding and the downstream effect of shed syndecan-1 in regulating the activity of HGF and other heparin-binding growth factors are likely crucial promoters of myeloma progression.
5.3. Enhanced Angiogenesis and Polarized Migration of Myeloma Cells
Multiple myeloma is a plasma cell dyscrasia characterized by multiple lytic lesions at the time of diagnosis. There is continuous spread or dissemination of tumor cells from the original site of tumor development to multiple sites in the bone marrow niche. Heparanase promotes both bone marrow angiogenesis and metastasis by altering the structure and function of heparan sulfate proteoglycans and contributing to tumor-mediated remodeling of both cell surfaces and the extracellular matrix [44, 55, 53]. These actions dynamically impact multiple regulatory pathways, most notably by augmenting the bioavailability of growth factors and cytokines bound to heparan sulfate chains. Using myeloma and endothelial cell models, the novel roles of heparanase in promoting metastasis and angiogenesis in myeloma have been determined. Surprisingly, key to these mechanisms is heparanase induced shedding of syndecan-1 [4, 8]. Both the heparan sulfate chains and the core protein of shed syndecan-1, through different mechanisms, participate in promoting myeloma cell invasion and endothelial cell angiogenesis.
Upregulation of VEGF Expression and Endothelial Invasion
High heparanase activity in the plasma harvested from the bone marrow of myeloma patients is associated with elevated microvessel density [44]. Heparanase, in addition to enhancing syndecan-1 shedding, upregulates VEGF expression and secretion by myeloma cells [56]. VEGF binds to heparan sulfate chains of shed syndecan-1 present in the conditioned medium of myeloma cells and when incubated with endothelial cells, this complex stimulates ERK signaling leading to enhanced endothelial invasion and angiogenesis [56]. Prior removal of the VEGF/syndecan-1 complex from the conditioned medium, either by treating with heparinase III, a bacterial enzyme that degrades heparan sulfate chains, or by immunoprecipitation using anti-syndecan-1 antibody, abolishes the activation of ERK signaling and subsequent invasion of endothelial cells. It is important to note that immunoprecipitation of shed syndecan-1 from the conditioned medium captures only the intact ectodomain core protein containing heparan sulfate chains, however, the heparan sulfate fragments generated by heparanase action remain in the medium. Surprisingly, these fragments which also have bound VEGF fail to enhance invasion of the endothelial cells [56]. Thus, it appeared in these initial studies that the key mechanism by which heparanase promoted angiogenesis was by the upregulation of syndecan-1 shedding. The presence of shed syndecan-1 extends the range of proteoglycan function beyond that at the cell surface within the tumor microenvironment. Conceivably, shed syndecan-1 which is known to enter the circulation, could, with its bound VEGF travel to distal sites and initiate angiogenesis that supports the establishment of metastatic lesions [57].
Activation of VEGFR2 Downstream of Heparanase Activity Promotes Polarized Migration of Myeloma Cells and Angiogenesis
Though the above-described roles of shed syndecan-1 are mediated through heparan sulfate binding to VEGF, the role of the syndecan-1 core protein ectodomain in stimulating myeloma migration and angiogenesis was recently discovered. Myeloma cells expressing high levels of heparanase form a highly polarized morphology on fibronectin or VCAM, two ligands enriched in the bone marrow that are recognized by VLA integrin expressed by myeloma cells [8]. However, myeloma cells expressing a low level of heparanase or an enzymatically inactive form of heparanase failed to polarize. Interestingly, it was discovered that when syndecan-1 is shed from the myeloma surface, it exposes a cryptic juxtamembrane site on the syndecan-1 core protein that binds VEGFR2 and VLA-4 (α4β1 integrin). This coupling of VEGFR2 (that is aberrantly expressed on myeloma tumor cells) to the integrin on the surface of myeloma cells reorients VLA-4 from the uropod (trailing edge of the cell) to the leading edge of the cell and also activates VEGFR2 leading to Rac signaling [8]. These events trigger polarized migration of myeloma cells. Interestingly, it was found that the same molecular mechanisms drive endothelial tube formation, thereby revealing a new mechanism of heparanase activity in driving angiogenesis.
Similar to the interaction of shed syndecan-1 with VEGFR2 and VLA-4, the coupling of syndecan-1 with other integrins also occur, leading to activation of tyrosine kinases (IGF-1R, HER2, EGFR) in other types of cancer [58, 59, 60]. The signaling mechanism involving shed syndecan-1/VGFR2/VLA4 in myeloma cell migration and angiogenesis is highly dependent on heparanase stimulation of syndecan-1 shedding as the initiating step, thereby identifying an important role for this enzyme in myeloma progression. This role of heparanase was confirmed using a heparanase inhibitor Roneparstat (Noseda and Barbieri, in this volume), a chemically modified anticoagulant heparin derivative, that diminishes syndecan-1 shedding and subsequent myeloma cell invasion and angiogenesis [8, 61]. It is important to note that an active motif in shed syndecan-1 is responsible for promoting the invasive phenotype in myeloma cells by coupling VEGFR2 to VLA4 [8]. This active site, amino acid 210 to 236 of the syndecan-1 ectodomain, is fully functional only when syndecan-1 is shed from the myeloma cell surface. Its binding to VEGFR2 or VLA4 can be mimicked by short peptides, called synstatins (SSTNs), encompassing part of this sequence that acts to competitively inhibit this mechanism. Peptides that bind only VLA integrin (SSTN 210–233) or VEGFR2 (214–240) blocks myeloma cell invasion and angiogenesis due to their inhibition of the coupling of VGFR2 and VLA-4 [8]. The inhibitory synstatins are likely to show promise against myeloma extravasation and spread [62].
It is well documented that heparanase enhances both tumor metastasis and angiogenesis. Many of the known mechanisms of heparanase activity likely contribute to these processes in various ways. However, to our knowledge, the finding that heparanase induces shedding of syndecan-1 leading to the downstream activation of Rac is the first demonstration that, by this single mechanism, both metastasis and angiogenesis are stimulated.
6. Impact of Heparanase on Exosome Biogenesis by Myeloma Cells and on Exosome Docking with Target Cells
6.1. Exosome Biogenesis
Exosomes are best defined as extracellular vesicles that are released from cells upon fusion of endocytic multivesicular bodies with the plasma membrane [63, 64]. This liberates the vesicles contained within the multivesicular body into the extracellular milieu. Once released these vesicles are referred to as exosomes. Exosomes are composed of a vast array of cellular molecules, most prominently proteins and nucleic acids. Once exosomes dock with either adjacent or distal cells they can reprogram these recipient cells. Although essentially, all cells can secrete exosomes, cancer cell secretion of exosomes is elevated, and these exosomes can play important roles in promoting tumor progression and metastasis [65]. Heparanase plays an important role in exosome biogenesis by participating in activation of the syndecan-syntenin-ALIX complex [66, 67, 68]. Briefly, trimming of heparan sulfate chains of syndecan-1 by heparanase facilitates the binding of the syndecan cytoplasmic domain via syntenin to the syntenin-ALIX complex. This leads to recruitment of a larger complex of proteins known as the endosomal sorting complex required for transport (ESCRT). This complex activates the budding and scission process that generates the intraluminal vesicles (David and Zimmermann, in this volume). Utilizing myeloma cells, it was discovered that high heparanase expression dramatically enhances cellular production of exosomes [66]. Heparanase enzymatic activity is required for enhanced exosome biogenesis because enzymatically inactive heparanase, even when present in high levels, does not substantially increase exosome biogenesis. Addition of recombinant heparanase to myeloma cells expressing a low level of the enzyme also enhances exosome biogenesis, indicating that heparanase released into the tumor microenvironment can aid in driving exosome secretion within the bone marrow [66]. Importantly, heparanase has been shown to be present in its soluble and active form within plasma harvested from the bone marrow of myeloma patients [44].
Heparanase also regulates the protein cargo of myeloma-derived exosomes which is evident from the fact that exosomes from heparanase-high myeloma cells contain high levels of syndecan-1, VEGF and HGF in their cargo compared to exosomes from heparanase-low cells [66]. The difference in exosome cargo due to heparanase expression, in fact, reflects on the impact of exosomes on tumor and host cell behavior. For example, exosomes from myeloma cells expressing high levels of heparanase stimulated myeloma cell spreading on fibronectin, and endothelial cell invasion through Matrigel matrix better than exosomes from myeloma cells expressing low levels of heparanase [66]. Further analysis of exosome secreted by myeloma cells revealed that heparanase is present as cargo on the surface of these exosomes and is available to degrade heparan sulfate within the extracellular matrix [66, 69]. Moreover, this exosomal heparanase can be delivered to cells within the tumor microenvironment and perhaps distally to other parts of the body [69]. Because of the known role of heparanase in promoting angiogenesis and metastasis, exosomes bearing heparanase may play a role in establishing niches to which tumor cells eventually home and grow.
6.2. Docking of Exosomes with Target Cells
The functional effects of exosomes rely on their interaction with, and subsequent delivery of cargo to, target cells. Heparan sulfate chains on the surface of target cells function as receptors for exosomes and can also assist in the internalization of exosomes [70]. A role for heparanase in exosome uptake remained unknown until recently when it was revealed that in cells expressing a high level of heparanase, the exosomes secreted contained abundant fibronectin on the exosome surface [71]. Fibronectin binds to heparan sulfate via a strong and well-characterized heparin/heparan sulfate binding domain. This region of the fibronectin protein, designated as the Hep-II domain, is located within the C-terminal repeat units 12–14 of fibronectin [72]. Exosomes from heparanase-high cells interacted with target cells much better than did the exosomes from heparanase-low cells pointing to the fact that the levels of fibronectin on exosomes correlates with exosome ability to interact with target cells [71]. Mechanistically, it was demonstrated that fibronectin is bound to heparan sulfate on the surface of exosomes and facilitates exosome interaction with heparan sulfate chains present on the surface of target cells. Therefore, cell surface heparan sulfate proteoglycans such as syndecan-1 play a dual role in the interaction between exosomes and cells. Heparan sulfate on the exosome surface binds fibronectin, and subsequently, when the exosome encounters a target cell, it binds via fibronectin to heparan sulfate on that cell. Because heparan sulfate is ubiquitously expressed on cell surfaces, the mechanism described here may be a general mechanism of exosomes binding to most cells and is likely not mediating targeting of exosomes to specific cell types. However, some specificity of exosome binding to cells could be conferred through the structure of cell surface heparan sulfate chains. For example, cells lacking heparan sulfate 2-O- or -N-sulfation exhibited reduced exosome binding compared to cells containing these structures [70]. Cell surface heparan sulfate proteoglycans, after binding to exosomes, are internalized by cells and thus function as internalizing receptors for exosomes, rather than just cell surface attachment sites [70]. Even though the mechanism by which exosomes release their cargo within cells is not clear, once internalized, exosomes can fuse with the delimiting membrane of the endocytic compartment of target cells to deliver the cargo [73].
Together, these studies underscore the importance of the heparanase/syndecan axis in regulating the biology of exosomes and show that their impact is not restricted solely to exosome biogenesis and cargo content but also impact exosome-target cell interaction, a key step in the ability of exosomes to regulate cell behavior. These findings not only support a role for heparanase in regulating exosome action in myeloma but also expose multiple ways by which exosome-cell interactions can be therapeutically targeted in patients. Importantly, a fully sulfated 12-mer heparin mimetic, and heparin-derived heparanase inhibitor, Roneparstat, are both capable of inhibiting exosome binding to cells [71]. Since both the 12-mer mimetic and Roneparstat lack anti-coagulant activity, these compounds could potentially be delivered to patients to block exosome uptake by target cells, thereby diminishing the biological impact of exosomes in disease settings.
7. Heparanase Modulates Sensitivity of Myeloma Cells to Therapy
Gene expression profiling of myeloma cells from patients demonstrated that following high dose chemotherapy, the cells present upon tumor relapse exhibited a high level of heparanase expression [74, 13]. These data are clinically relevant because heparanase expression within the bone marrow microenvironment of newly diagnosed myeloma patients treated with chemotherapy and stem cell transplantation is associated with shorter survival [19]. Also, elevated heparanase level is associated with myeloma cell resistance to bortezomib and melphalan, two drugs widely-utilized for anti-myeloma therapy [74]. Mechanistically, heparanase promotes drug resistance by activating ERK signaling, and this signaling pathway requires enzyme activity of heparanase [74]. Heparanase thus plays an important role in determining the outcome of anti-myeloma therapy. Usage of inhibitors of heparanase such as Roneparstat, in combination with drugs like melphalan, can, therefore, enhance the efficacy of melphalan even against highly aggressive myeloma [74]. Moreover, in a model of dexamethasone resistant multiple myeloma, the combination of Roneparstat with dexamethasone inhibited tumor growth [61]. All these findings point to the fact that heparanase inhibitors can be potential drugs to target minimal residual disease in myeloma patients, because inhibition of heparanase may interfere with the reestablishment of a tumor-promoting microenvironment, thereby preventing relapse. Combining anti-heparanase therapy with standard chemotherapy drugs may prevent myeloma relapse and improve patient outcome.
8. Heparanase Inhibitor for Myeloma Therapy
Heparanase impacts multiple regulatory pathways within the myeloma microenvironment that together drive myeloma growth, dissemination, angiogenesis, osteolysis, and chemoresistance (Table 1, Fig. 1). Heparanase, therefore, plays an important role in the pathogenesis of multiple myeloma, and its inhibition will disrupt the myeloma microenvironment leading to diminished myeloma growth. These results prompted a first in man, multicenter phase I clinical study of Roneparstat in advanced heavily pretreated refractory myeloma patients who had exhausted currently available anti-myeloma therapies [2]. The drug was well tolerated and in some patients showed early signs of efficacy. Roneparstat is composed of 100% N-acetylated and glycol-split heparin. It is a potent inhibitor of heparanase enzyme activity (IC50 = 3 nM) that is devoid of any significant anticoagulant activity [75, 76] (Noseda & Barbieri; Cassinelli, Torri and Naggi, in this volume). The impact of Roneparstat has been tested in vivo using different models of myeloma where human myeloma tumor cells were injected either subcutaneously, into fragments of human bone implanted in mice, or intravenously into the mouse tail vein [74, 61]. Roneparstat significantly inhibited growth, angiogenesis and bone metastasis of myeloma tumors in these models. Analysis of myeloma tumors from animals treated with Roneparstat demonstrated that these tumors have diminished levels of VEGF, HGF and MMP-9, reduced angiogenesis and reduced levels of shed syndecan-1 compared to animals treated with vehicle [61]. This highlights that the mechanism of action of Roneparstat is consistent with it having anti-heparanase activity in vivo. Further, using an in vivo model of disseminated myeloma, where myeloma cells expressing a high level of heparanase home and grow exclusively in bone, Roneparstat in combination with either bortezomib or melphalan, significantly decreased both the number of animals with detectable tumors and tumor burden compared to animals treated with either of these drugs alone [74]. The ability of Roneparstat to dramatically reduce tumor growth in bone when used in combination with either bortezomib or melphalan indicates that blocking heparanase diminishes drug resistance in myeloma.
9. Concluding Remarks
Over the last two decades, heparanase has been shown to be involved in many important steps necessary for the progression of multiple myeloma (Fig. 1). Based on the copious evidence demonstrating the role of heparanase in myeloma growth, metastasis, angiogenesis, exosome biogenesis, and chemoresistance, heparanase can be defined as a multifunctional protein whose activity fuels the aggressive progression of myeloma. Surprisingly, many of the downstream impacts of heparanase are dependent on its ability to enhance the shedding of syndecan-1. Of note is the novel role of shed syndecan-1 in activating VEGFR2, by coupling VEGFR2 with VLA-4 thereby initiating downstream signaling pathways that trigger polarized migration of myeloma cells and endothelial cells. It will be important to determine if this mechanism is at play in other types of cancer. Considering the fact that much of the heparanase function in myeloma progression is dependent on its enzymatic cleavage of heparan sulfate chains, and that heparanase is the only known mammalian endoglycosidase that cleaves heparan sulfate chains and is not expressed abundantly in normal tissue, this enzyme presents an ideal pharmaceutical target for myeloma and other cancers.
Acknowledgments
This study was supported by research grants awarded to R.S. (CA138340 and CA211752) by the National Institutes of Health. The authors thank the many members of the Sanderson’s laboratory who have contributed over the years to much of the knowledge reviewed in this chapter.
References
- 1.Marino S, & Roodman GD (2018). Multiple myeloma and bone: The fatal interaction. Cold Spring Harbor Perspectives in Medicine, 8(8). 10.1101/cshperspect.a031286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galli M, Chatterjee M, Grasso M, Specchia G, Magen H, Einsele H, Celeghini I, Barbieri P, Paoletti D, Pace S, Sanderson RD, Rambaldi A, & Nagler A (2018). Phase I study of the heparanase inhibitor roneparstat: An innovative approach for ultiple myeloma therapy. Haematologica, 103(10), e469–e472. 10.3324/haematol.2017.182865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramani VC, Purushothaman A, Stewart MD, Thompson CA, Vlodavsky I, Au JL, & Sanderson RD (2013). The heparanase/syndecan-1 axis in cancer: Mechanisms and therapies. The FEBS Journal, 280(10), 2294–2306. 10.1111/febs.12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang Y, Macleod V, Miao HQ, Theus A, Zhan F, Shaughnessy JD Jr., Sawyer J, Li JP, Zcharia E, Vlodavsky I, & Sanderson RD (2007). Heparanase enhances syndecan-1 shedding: A novel mechanism for stimulation of tumor growth and metastasis. The Journal of Biological Chemistry, 282(18), 13326–13333. [DOI] [PubMed] [Google Scholar]
- 5.Lovell R, Dunn JA, Begum G, Barth NJ, Plant T, Moss PA, Drayson MT, Pratt G, & Working Party on Leukaemia in Adults of the National Cancer Research Institute Haematological Oncology Clinical Studies G. (2005). Soluble syndecan-1 level at diagnosis is an independent prognostic factor in multiple myeloma and the extent of fall from diagnosis to plateau predicts for overall survival. British Journal of Haematology, 130(4), 542–548. 10.1111/j.1365-2141.2005.05647.x. [DOI] [PubMed] [Google Scholar]
- 6.Seidel C, Sundan A, Hjorth M, Turesson I, Dahl IM, Abildgaard N, Waage A, & Borset M (2000). Serum syndecan-1: A new independent prognostic marker in multiple myeloma. Blood, 95(2), 388–392. [PubMed] [Google Scholar]
- 7.Stewart MD, Ramani VC, & Sanderson RD (2015). Shed Syndecan-1 Translocates to the nucleus of cells delivering growth factors and inhibiting histone acetylation: A NOVEL MECHANISM OF TUMOR-HOST CROSS-TALK. The Journal of Biological Chemistry, 290(2), 941–949. 10.1074/jbc.M114.608455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jung O, Trapp-Stamborski V, Purushothaman A, Jin H, Wang H, Sanderson RD, & Rapraeger AC (2016). Heparanase-induced shedding of syndecan-1/CD138 in myeloma and endothelial cells activates VEGFR2 and an invasive phenotype: Prevention by novel synstatins. Oncogene, 5, e202. 10.1038/oncsis.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Couchman JR, Multhaupt H, & Sanderson RD (2016). Recent insights into cell surface Heparan sulphate proteoglycans and Cancer. F1000Res, 5. 10.12688/f1000research.8543.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vlodavsky I, Singh P, Boyango I, Gutter-Kapon L, Elkin M, Sanderson RD, & Ilan N (2016). Heparanase: From basic research to therapeutic applications in cancer and inflammation. Drug Resistance Updates, 29, 54–75. 10.1016/j.drup.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramani VC, Zhan F, He J, Barbieri P, Noseda A, Tricot G, & Sanderson RD (2016). Targeting heparanase overcomes chemoresistance and diminishes relapse in myeloma. Oncotarget, 7, 1598–1607. 10.18632/oncotarget.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Franqui-Machin R, Hao M, Bai H, Gu Z, Zhan X, Habelhah H, Jethava Y, Qiu L, Frech I, Tricot G, & Zhan F (2018). Destabilizing NEK2 overcomes resistance to proteasome inhibition in multiple myeloma. The Journal of Clinical Investigation, 128(7), 2877–2893. 10.1172/JCI98765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramani VC, Vlodavsky I, Ng M, Zhang Y, Barbieri P, Noseda A, & Sanderson RD (2016). Chemotherapy induces expression and release of heparanase leading to changes associated with an aggressive tumor phenotype. Matrix Biology, 55, 22–34. 10.1016/j.matbio.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Y, MacLeod V, Dai Y, Khotskaya-Sample Y, Shriver Z, Venkataraman G, Sasisekharan R, Naggi A, Torri G , Casu B, Vlodavsky I, Suva LJ, Epstein J, Yaccoby S, Shaughnessy JD Jr., Barlogie B, & Sanderson RD (2007). The syndecan-1 heparan sulfate proteoglycan is a viable target for myeloma therapy. Blood, 110(6), 2041–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanderson RD, & Yang Y (2008). Syndecan-1: A dynamic regulator of the myeloma microenvironment. Clinical & Experimental Metastasis, 25(2), 149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bayer-Gamer IB, Sanderson RD, Dhodapkar MV, Owens RB, & Wilson CS (2001). Syndecan-1 (CD138) immunoreactivity in bone marrow biopsies of multiple myeloma: Shed syndecan-1 accumulates in fibrotic regions. Modern Pathology, 14(10), 1052–1058. [DOI] [PubMed] [Google Scholar]
- 17.Sanderson RD, Yang Y, Suva LJ, & Kelly T (2004). Heparan sulfate proteoglycans and heparanase--partners in osteolytic tumor growth and metastasis. Matrix Biology, 23(6), 341–352. [DOI] [PubMed] [Google Scholar]
- 18.Yang Y, Yaccoby S, Liu W, Langford JK, Pumphrey CY, Theus A, Epstein J, & Sanderson RD (2002). Soluble syndecan-1 promotes growth of myeloma tumors in vivo. Blood, 100(2), 610–617. [DOI] [PubMed] [Google Scholar]
- 19.Mahtouk K, Hose D, Raynaud P, Hundemer M, Jourdan M, Jourdan E, Pantesco V, Baudard M, De Vos J, Larroque M, Moehler T, Rossi JF, Reme T, Goldschmidt H, & Klein B (2007). Heparanase influences expression and shedding of syndecan-1, and its expression by the bone marrow environment is a bad prognostic factor in multiple myeloma. Blood, 109(11), 4914–4923. 10.1182/blood-2006-08-043232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Purushothaman A, Chen L, Yang Y, & Sanderson RD (2008). Heparanase stimulation of protease expression implicates it as a master regulator of the aggressive tumor phenotype in myeloma. The Journal of Biological Chemistry, 283(47), 32628–32636. 10.1074/jbc.M806266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zcharia E, Jia J, Zhang X, Baraz L, Lindahl U, Peretz T, Vlodavsky I, & Li JP (2009). Newly generated heparanase knock-out mice unravel co-regulation of heparanase and matrix metalloproteinases. PLoS One, 4(4), e5181. 10.1371/journal.pone.0005181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Purushothaman A, Babitz SK, & Sanderson RD (2012). Heparanase enhances the insulin receptor signaling pathway to activate extracellular signal-regulated kinase in multiple myeloma. The Journal of Biological Chemistry, 287(49), 41288–41296. 10.1074/jbc.M112.391417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sprynski AC, Hose D, Kassambara A, Vincent L, Jourdan M, Rossi JF, Goldschmidt H, & Klein B (2010). Insulin is a potent myeloma cell growth factor through insulin/IGF-1 hybrid receptor activation. Leukemia, 24(11), 1940–1950. 10.1038/leu.2010.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hideshima T, Bergsagel PL, Kuehl WM, & Anderson KC (2004). Advances in biology of multiple myeloma: Clinical applications. Blood, 104(3), 607–618. 10.1182/blood-2004-01-0037. [DOI] [PubMed] [Google Scholar]
- 25.Kim K, Kong SY, Fulciniti M, Li X, Song W, Nahar S, Burger P, Rumizen MJ, Podar K, Chauhan D, Hideshima T, Munshi NC, Richardson P, Clark A, Ogden J, Goutopoulos A, Rastelli L, Anderson KC, & Tai YT (2010). Blockade of the MEK/ERK signalling cascade by AS703026, a novel selective MEK1/2 inhibitor, induces pleiotropic anti-myeloma activity in vitro and in vivo. British Journal of Haematology, 149(4), 537–549. 10.1111/j.1365-2141.2010.08127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schubert SY, Ilan N, Shushy M, Ben-Izhak O, Vlodavsky I, & Goldshmidt O (2004). Human heparanase nuclear localization and enzymatic activity. Laboratory Investigation, 84(5), 535–544. [DOI] [PubMed] [Google Scholar]
- 27.Kobayashi M, Naomoto Y, Nobuhisa T, Okawa T, Takaoka M, Shirakawa Y, Yamatsuji T, Matsuoka J, Mizushima T, Matsuura H, Nakajima M, Nakagawa H, Rustgi A, & Tanaka N (2006). Heparanase regulates esophageal keratinocyte differentiation through nuclear translocation and heparan sulfate cleavage. Differentiation, 74(5), 235–243. [DOI] [PubMed] [Google Scholar]
- 28.Zhang L, Sullivan P, Suyama J, & Marchetti D (2010). Epidermal growth factor-induced heparanase nucleolar localization augments DNA topoisomerase I activity in brain metastatic breast cancer. Molecular Cancer Research, 8(2), 278–290. 10.1158/1541-7786.MCR-09-0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He YQ, Sutcliffe EL, Bunting KL, Li J, Goodall KJ, Poon IK, Hulett MD, Freeman C, Zafar A, McInnes RL, Taya T, Parish CR, & Rao S (2012). The endoglycosidase heparanase enters the nucleus of T lymphocytes and modulates H3 methylation at actively transcribed genes via the interplay with key chromatin modifying enzymes. Transcription, 3(3), 130–145. 10.4161/trns.19998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doweck I, Kaplan-Cohen V, Naroditsky I, Sabo E, Ilan N, & Vlodavsky I (2006). Heparanase localization and expression by head and neck cancer: Correlation with tumor progression and patient survival. Neoplasia, 8(12), 1055—1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohkawa T, Naomoto Y, Takaoka M, Nobuhisa T, Noma K, Motoki T, Murata T, Uetsuka H, Kobayashi M, Shirakawa Y, Yamatsuji T, Matsubara N, Matsuoka J, Haisa M, Gunduz M, Tsujigiwa H, Nagatsuka H, Hosokawa M, Nakajima M, & Tanaka N (2004). Localization of heparanase in esophageal cancer cells: Respective roles in prognosis and differentiation. Laboratory Investigation, 84(10), 1289–1304. 10.1038/labinvest.3700159. [DOI] [PubMed] [Google Scholar]
- 32.Hebbes TR, Thorne AW, & Crane-Robinson C (1988). A direct link between core histone acetylation and transcriptionally active chromatin. The EMBO Journal, 7(5), 1395–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turner BM, & O’Neill LP (1995). Histone acetylation in chromatin and chromosomes. Seminars in Cell Biology, 6(4), 229–236. [DOI] [PubMed] [Google Scholar]
- 34.Roth SY, Denu JM, Allis CD (2001) Histone acetyltransferases. Annual Review of Biochemistry 70:81–120. doi:70/1/81 [pii] 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 35.Loidl P (1994). Histone acetylation: Facts and questions. Chromosoma, 103(7), 441–449. [DOI] [PubMed] [Google Scholar]
- 36.Ishihara M, Fedarko NS, & Conrad HE (1986). Transport of heparan sulfate into the nuclei of hepatocytes. The Journal of Biological Chemistry, 261(29), 13575–13580. [PubMed] [Google Scholar]
- 37.Richardson TP, Trinkaus-Randall V, & Nugent MA (2001). Regulation of heparan sulfate proteoglycan nuclear localization by fibronectin. Journal of Cell Science, 114(Pt 9), 1613–1623. [DOI] [PubMed] [Google Scholar]
- 38.Hsia E, Richardson TP, & Nugent MA (2003). Nuclear localization of basic fibroblast growth factor is mediated by heparan sulfate proteoglycans through protein kinase C signaling. Journal of Cellular Biochemistry, 88(6), 1214–1225. [DOI] [PubMed] [Google Scholar]
- 39.Brockstedt U, Dobra K, Nurminen M, & Hjerpe A (2002). Immunoreactivity to cell surface syndecans in cytoplasm and nucleus: Tubulin-dependent rearrangements. Experimental Cell Research, 274(2), 235–245. 10.1006/excr.2002.5477. [DOI] [PubMed] [Google Scholar]
- 40.Buczek-Thomas JA, Hsia E, Rich CB, Foster JA, & Nugent MA (2008). Inhibition of histone acetyltransferase by glycosaminoglycans. Journal of Cellular Biochemistry, 105(1), 108–120. 10.1002/jcb.21803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nilsson U, Johnsson R, Fransson LA, Ellervik U, & Mani K (2010). Attenuation of tumor growth by formation of antiproliferative glycosaminoglycans correlates with low acetylation of histone H3. Cancer Research, 70(9), 3771–3779. 10.1158/0008-5472.CAN-09-4331. [DOI] [PubMed] [Google Scholar]
- 42.Chen L, & Sanderson RD (2009). Heparanase regulates levels of syndecan-1 in the nucleus. PLoS One, 4(3), e4947. 10.1371/journal.pone.0004947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Purushothaman A, Hurst DR, Pisano C, Mizumoto S, Sugahara K, & Sanderson RD (2011). Heparanasemediated loss of nuclear syndecan-1 enhances histone acetyltransferase (HAT) activity to promote expression of genes that drive an aggressive tumor phenotype. The Journal of Biological Chemistry, 286(35), 30377–30383. 10.1074/jbc.M111.254789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kelly T, Miao HQ, Yang Y, Navarro E, Kussie P, Huang Y, MacLeod V, Casciano J, Joseph L, Zhan F, Zangari M, Barlogie B, Shaughnessy J, & Sanderson RD (2003). High heparanase activity in multiple myeloma is associated with elevated microvessel density. Cancer Research, 63(24), 8749–8756. [PubMed] [Google Scholar]
- 45.Barash U, Zohar Y, Wildbaum G, Beider K, Nagler A, Karin N, Ilan N, & Vlodavsky I (2014). Heparanase enhances myeloma progression via CXCL10 downregulation. Leukemia, 28, 2178–2187. 10.1038/leu.2014.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barash U, Zohar Y, Wildbaum G, Beider K, Nagler A, Karin N, Ilan N, & Vlodavsky I (2014). Heparanase enhances myeloma progression via CXCL10 downregulation. Leukemia, 28(11), 2178–2187. 10.1038/leu.2014.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seidel C, Borset M, Turesson I, Abildgaard N, Sundan A, & Waage A (1998). Elevated serum concentrations of hepatocyte growth factor in patients with multiple myeloma. The Nordic Myeloma Study Group. Blood, 91(3), 806–812. [PubMed] [Google Scholar]
- 48.Zhan F, Hardin J, Kordsmeier B, Bumm K, Zheng M, Tian E, Sanderson R, Yang Y, Wilson C, Zangari M, Anaissie E, Morris C, Muwalla F, van Rhee F, Fassas A, Crowley J, Tricot G, Barlogie B, & Shaughnessy J Jr. (2002). Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood, 99(5), 1745–1757. [DOI] [PubMed] [Google Scholar]
- 49.Borset M, Hjorth-Hansen H, Seidel C, Sundan A, & Waage A (1996). Hepatocyte growth factor and its receptor cmet in multiple myeloma. Blood, 88(10), 3998–4004. [PubMed] [Google Scholar]
- 50.Hjertner O, Torgersen ML, Seidel C, Hjorth-Hansen H, Waage A, Borset M, & Sundan A (1999). Hepatocyte growth factor (HGF) induces interleukin-11 secretion from osteoblasts: A possible role for HGF in myeloma-associated osteolytic bone disease. Blood, 94(11), 3883–3888. [PubMed] [Google Scholar]
- 51.Seidel C, Borset M, Hjertner O, Cao D, Abildgaard N, Hjorth-Hansen H, Sanderson RD, Waage A, & Sundan A (2000). High levels of soluble syndecan-1 in myeloma-derived bone marrow: Modulation of hepatocyte growth factor activity. Blood, 96(9), 3139–3146. [PubMed] [Google Scholar]
- 52.Ramani VC, Yang Y, Ren Y, Nan L, & Sanderson RD (2011). Heparanase plays a dual role in driving hepatocyte growth factor (HGF) signaling by enhancing HGF expression and activity. The Journal of Biological Chemistry, 286(8), 6490–6499. 10.1074/jbc.M110.183277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang Y, Macleod V, Bendre M, Huang Y, Theus AM, Miao HQ, Kussie P, Yaccoby S, Epstein J, Suva LJ, Kelly T, & Sanderson RD (2005). Heparanase promotes the spontaneous metastasis of myeloma cells to bone. Blood, 105(3), 1303–1309. [DOI] [PubMed] [Google Scholar]
- 54.Yang Y, Ren Y, Ramani VC, Nan L, Suva LJ, & Sanderson RD (2010). Heparanase enhances local and systemic osteolysis in multiple myeloma by upregulating the expression and secretion of RANKL. Cancer Research, 70(21), 8329–8338. 10.1158/0008-5472.CAN-10-2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Masola V, Beilin G, Gambaro G, & Onisto M (2018). Heparanase: A multitasking protein involved in extracellular matrix (ECM) Remodeling and intracellular events. Cell, 7(12). 10.3390/cells7120236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Purushothaman A, Uyama T, Kobayashi F, Yamada S, Sugahara K, Rapraeger AC, & Sanderson RD (2010). Heparanase-enhanced shedding of syndecan-1 by myeloma cells promotes endothelial invasion and angiogenesis. Blood, 115(12), 2449–2457. 10.1182/blood-2009-07-234757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nurcombe V, Cool SM (2007) Heparan sulfate control of proliferation and differentiation in the stem cell niche. Critical Reviews in Eukaryotic Gene Expression 17 (2): 159—171. doi:31c5e2d111e631ad,4e9eaa466db6cc30 [pii]. [DOI] [PubMed] [Google Scholar]
- 58.Beauvais DM, Rapraeger AC (2010) Syndecan-1 couples the insulin-like growth factor-1 receptor to inside-out integrin activation. Journal of Cell Science 123 (Pt 21):3796–3807. doi: 10.1242/jcs.067645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rapraeger AC, Ell BJ, Roy M, Li X, Morrison OR, Thomas GM, & Beauvais DM (2013). Vascular endothelial-cadherin stimulates syndecan-1-coupled insulin-like growth factor-1 receptor and cross-talk between alphaVbeta3 integrin and vascular endothelial growth factor receptor 2 at the onset of endothelial cell dissemination during angiogenesis. The FEBS Journal, 280(10), 2194–2206. 10.1111/febs.12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang H, Jin H, & Rapraeger AC (2015). Syndecan-1 and Syndecan-4 Capture epidermal growth factor receptor family members and the alpha3beta1 integrin via binding sites in their Ectodomains: NOVEL SYNSTATINS PREVENT KINASE CAPTURE AND INHIBIT alpha6beta4-INTEGRIN-DEPENDENT EPITHELIAL CELL MOTILITY. The Journal of Biological Chemistry, 290(43). 26103–26113. 10.1074/jbc.M115.679084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ritchie JP, Ramani VC, Ren Y, Naggi A, Torri G, Casu B, Penco S, Pisano C, Carminati P, Tortoreto M, Zunino F, Vlodavsky I, Sanderson RD, & Yang Y (2011). SST0001, a chemically modified heparin, inhibits myeloma growth and angiogenesis via disruption of the heparanase/syndecan-1 axis. Clinical Cancer Research, 17(6), 1382–1393. 10.1158/1078-0432.CCR-10-2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beauvais DM, Jung O, Yang Y, Sanderson RD, & Rapraeger AC (2016). Syndecan-1 (CD138) suppresses apoptosis in multiple myeloma by activating IGF1 receptor: Prevention by SynstatinIGF1R inhibits tumor growth. Cancer Research, 76(17), 4981–4993. 10.1158/0008-5472.CAN-16-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thery C, Zitvogel L, & Amigorena S (2002). Exosomes: Composition, biogenesis and function. Nature Reviews. Immunology, 2(8), 569–579. 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- 64.Sanderson RD, Bandari SK, & Vlodavsky I (2017). Proteases and glycosidases on the surface of exosomes: Newly discovered mechanisms for extracellular remodeling. Matrix Biology, 10.1016/j.matbio.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wortzel I, Dror S, Kenific CM, & Lyden D (2019). Exosome-mediated metastasis: Communication from a distance. Developmental Cell, 49(3), 347–360. 10.1016/j.devcel.2019.04.011. [DOI] [PubMed] [Google Scholar]
- 66.Thompson CA, Purushothaman A, Ramani VC, Vlodavsky I, & Sanderson RD (2013). Heparanase regulates secretion, composition, and function of tumor cell-derived exosomes. The Journal of Biological Chemistry, 288(14), 10093–10099. 10.1074/jbc.C112.444562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roucourt B, Meeussen S, Bao J, Zimmermann P, & David G (2015). Heparanase activates the syndecan-syntenin-ALIX exosome pathway. Cell Research, 25(4), 412–428. 10.1038/cr.2015.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.David G, & Zimmermann P (2016). Heparanase tailors syndecan for exosome production. Molecular & Cellular Oncology, 3(3), e1047556. 10.1080/23723556.2015.1047556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bandari SK, Purushothaman A, Ramani VC, Brinkley GJ, Chandrashekar DS, Varambally S, Mobley JA, Zhang Y, Brown EE, Vlodavsky I, & Sanderson RD (2018). Chemotherapy induces secretion of exosomes loaded with heparanase that degrades extracellular matrix and impacts tumor and host cell behavior. Matrix Biology, 65, 104–118. 10.1016/j.matbio.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Christianson HC, Svensson KJ, van Kuppevelt TH, Li JP, & Belting M (2013). Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proceedings of the National Academy of Sciences of the United States of America, 110(43), 17380–17385. 10.1073/pnas.1304266110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Purushothaman A, Bandari SK, Liu J, Mobley JA, Brown EE, & Sanderson RD (2016). Fibronectin on the surface of myeloma cell-derived Exosomes mediates exosome-cell interactions. The Journal of Biological Chemistry, 291(4), 1652–1663. 10.1074/jbc.M115.686295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carpentier M, Denys A, Allain F, & Vergoten G (2014). Molecular docking of heparin oligosaccharides with hep-II heparin-binding domain of fibronectin reveals an interplay between the different positions of sulfate groups. Glycoconjugate Journal, 31(2), 161–169. 10.1007/sl0719-013-9512-8. [DOI] [PubMed] [Google Scholar]
- 73.Raposo G, & Stoorvogel W (2013). Extracellular vesicles: Exosomes, microvesicles, and friends. The Journal of Cell Biology, 200(4), 373–383. 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ramani VC, Zhan F, He J, Barbieri P, Noseda A, Tricot G, & Sanderson RD (2016). Targeting heparanase overcomes chemoresistance and diminishes relapse in myeloma. Oncotarget, 7(2), 1598–1607. 10.18632/oncotarget.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Naggi A, Casu B, Perez M, Torri G, Cassinelli G, Penco S, Pisano C, Giannini G, Ishai-Michaeli R, & Vlodavsky I (2005). Modulation of the heparanase-inhibiting activity of heparin through selective desulfation, graded Nacetylation, and glycol splitting. The Journal of Biological Chemistry, 280(13), 12103–12113. [DOI] [PubMed] [Google Scholar]
- 76.Pala D, Rivara S, Mor M, Milazzo FM, Roscilli G, Pavoni E, & Giannini G (2016). Kinetic analysis and molecular modeling of the inhibition mechanism of roneparstat (SST0001) on human heparanase. Glycobiology, 26(6), 640–654. 10.1093/glycob/cww003. [DOI] [PMC free article] [PubMed] [Google Scholar]