

Abstract
Resting‐state functional connectivity in the human brain is heritable, and previous studies have investigated the genetic basis underlying functional connectivity. However, at present, the molecular mechanisms associated with functional network centrality are still largely unknown. In this study, functional networks were constructed, and the graph‐theory method was employed to calculate network centrality in 100 healthy young adults from the Human Connectome Project. Specifically, functional connectivity strength (FCS), also known as the “degree centrality” of weighted networks, is calculated to measure functional network centrality. A multivariate technique of partial least squares regression (PLSR) was then conducted to identify genes whose spatial expression profiles best predicted the FCS distribution. We found that FCS spatial distribution was significantly positively correlated with the expression of genes defined by the first PLSR component. The FCS‐related genes we identified were significantly enriched for ion channels, axon guidance, and synaptic transmission. Moreover, FCS‐related genes were preferentially expressed in cortical neurons and young adulthood and were enriched in numerous neurodegenerative and neuropsychiatric disorders. Furthermore, a series of validation and robustness analyses demonstrated the reliability of the results. Overall, our results suggest that the spatial distribution of FCS is modulated by the expression of a set of genes associated with ion channels, axon guidance, and synaptic transmission.
Keywords: Allen human brain atlas, functional connectivity, gene expression, resting‐state fMRI, transcriptome
This transcription‐neuroimaging association study identified that the expression profiles of a set of genes, especially those involved in processes related to ion channels, the axon guidance pathway and synaptic transmission, are related to FCS. These findings provide new insight into the biological substrates underlying intrinsic functional network centrality.
1. INTRODUCTION
Resting‐state refers to a state where people are in a calm and peaceful situation without external stimuli or explicit tasks (Sheline & Raichle, 2013). Although there is low physical energy expenditure when people are at rest, the brain at rest consumes ~80% of its energy supply, indicating a nontrivial role of the resting brain in cognitive functions (Raichle & Mintun, 2006). In recent decades, resting‐state functional magnetic resonance imaging (rs‐fMRI) has been widely used to explore blood oxygenation level‐dependent (BOLD) spontaneous fluctuations, partly due to its distinct advantages such as no potential performance confounding from a specific task, requiring the least amount of effort from the participants, and being relatively easy to implement in clinical populations (Greicius, 2008; Smith et al., 2013; Smith et al., 2013). Despite some concerns about the presence of uncontrolled brain signals from independent thoughts when subjects are in a resting state (Morcom & Fletcher, 2007) and that fMRI yields indirect measures of neuronal activity, fluctuations in BOLD signals appear to be a fundamental or intrinsic property of the brain that is found during sleep (Larson‐Prior et al., 2009) and general anesthesia (Vincent et al., 2007). As an important rs‐fMRI metric, resting‐state functional connectivity (rsFC) measures the temporal correlation of BOLD signals between spatially distinct brain regions and shows highly consistent patterns across human individuals (Yeo et al., 2011) and species (Laperchia et al., 2013), suggestive of a conserved genetic substrate.
Twin studies (Fornito et al., 2011; Glahn et al., 2010; van den Heuvel et al., 2013) and a genome‐wide association study (GWAS) (Elliott et al., 2018) have shown that rsFC in the human brain is moderate to highly heritable. Recent advances in comprehensive brain‐wide gene expression atlases such as the Allen Human Brain Atlas (AHBA) (Hawrylycz et al., 2012), have provided the possibility of linking spatial variations in gene expression to macroscopic neuroimaging phenotypes (Fornito, Arnatkeviciute, & Fulcher, 2019). Considering the highly conserved transcriptional architecture of the cerebral cortex across individuals (Hawrylycz et al., 2015), several transcription‐neuroimaging association studies have explored the molecular mechanisms underlying rsFC in healthy subjects (Anderson et al., 2018; Krienen, Yeo, Ge, Buckner, & Sherwood, 2016; Richiardi et al., 2015; Vertes et al., 2016) by correlating rsFC derived from living human brains to gene expression data derived from postmortem human brains. For example, a seminal study (Richiardi et al., 2015) applied independent component analysis (ICA) to build rsFC networks and focused on four rsFC networks, including the dorsal default‐mode network (DMN), salience network, sensorimotor network, and visuospatial network. They found that the transcriptome profile similarity with‐network was higher than that between different networks. In addition, Krienen et al. used a parcellation of 17 rsFC networks and found that laminar expression patterns of human supragranular enriched genes were associated with corticocortical network architecture (Krienen et al., 2016). Anderson and colleagues first identified genes with differential cortical network expression based on a parcellation of 7 rsFC networks and found strong correspondence with limbic and somato/motor cortico‐striatal functional networks (Anderson et al., 2018). Vertes et al. divided the brain into eight modules and examined the associations between transcription profiles associated with intra‐modular degree as well as inter‐modular degree and connection distance (Vertes et al., 2016). However, the abovementioned studies mainly assumed that the human brain consists of several independent networks/modules, and thus, such methods cannot fully delineate functional brain networks from a global perspective.
In contrast, network centrality measures show great promise for characterizing the complexity of the whole functional network (Zuo et al., 2012). Among the various centrality measures of the functional network, functional connectivity strength (FCS), also known as the “degree centrality” of weighted networks (Liang, Zou, He, & Yang, 2013; Wang et al., 2015), is a simple, direct, and commonly used measure and shows high test‐retest reliability (Wang et al., 2011). In addition, FCS has a close relationship with cerebral blood flow (Liang et al., 2013) and the cerebral metabolic rate of glucose (Tomasi & Volkow, 2011a), indicating that the FCS is biologically plausible. Furthermore, FCS abnormalities have been found in a variety of neurodegenerative and neuropsychiatric diseases such as Alzheimer's disease (AD) (Filippi et al., 2017), schizophrenia (SCZ) (Argyelan et al., 2014; Guo et al., 2015), bipolar disorder (BPD) (Argyelan et al., 2014) and autism spectrum disorder (ASD) (Khan et al., 2013). However, no transcription‐neuroimaging association study to date has investigated the genes linked with FCS. Considering that the human brain is a complex network and a vulnerable target of various neurodegenerative and neuropsychiatric conditions, an elaborate analysis of the association between FCS and gene expression is of clinical significance.
Inspired by previous work, in this exploratory study, we aimed to answer three questions: First, which genes are associated with FCS? Second, what are the biological functions of these genes? Third, in which cell types and developmental periods are these genes over‐represented? To this end, we performed a comprehensive transcriptome‐neuroimaging association to identify the molecular correlates of FCS. Specifically, we first computed the whole‐brain voxel‐wise FCS from high‐quality rs‐fMRI data from the Human Connectome Project (HCP) (Van Essen et al., 2013). Then, we extracted sample‐wise gene expression data across six postmortem adult human brains from AHBA. Subsequently, partial least squares regression (PLSR), a well‐established multivariate technique that generalizes and combines features from principal component analysis and multiple regression, was utilized to identify genes whose transcriptional profiles were significantly linked to the group mean FCS patterns. Finally, functional annotations of these FCS‐related genes may explain how these genes play crucial roles in regulating FCS using a suite of recently developed gene functional‐annotation tools. An overview of our study protocol is provided in Figure 1.
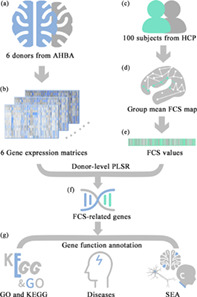
FIGURE 1.
The workflow of the study protocol. (a) Acquire the sample‐wise whole‐genomic transcriptomic profiles from the AHBA database; (b) Obtain the sample‐wise gene expression matrix of each donor; (c) Download rs‐fMRI data of 100 subjects from HCP; (d) Calculate group mean FCS map; (e) Obtain the sample‐wise group‐mean FCS values; (f) Identify FCS‐related genes by investigating correlations between cortical gene expression and FCS using PLSR; (g) Functional annotations for FCS‐related genes, including GO and KEGG enrichment analysis, disease enrichment analysis and specific expression analysis in three specific terms (cell types, brain regions, and developmental stages). Abbreviations: AHBA, Allen human brain atlas; FCS, functional connectivity strength; GO, gene ontology; HCP, human connectome project; KEGG, Kyoto encyclopedia of genes and genomes; PLSR, partial least squares regression; SEA, specific expression analysis
2. MATERIALS AND METHODS
2.1. Subjects, data acquisition, and preprocessing
The rs‐fMRI data of 100 subjects younger than 30 yo each with unique family IDs (50 males, mean age ± SD = 26.7 ± 2.3 years) were randomly chosen from publicly available HCP data (https://db.humanconnectome.org). We used the preprocessed imaging data based on an ICA‐fixed (FMRIB's ICA‐based X‐noisifier) pipeline (Salimi‐Khorshidi et al., 2014). For details of HCP rs‐fMRI data acquisition and denoised preprocessing, please refer to previous studies (Van Essen et al., 2012; Van Essen et al., 2013). Briefly, high‐resolution rs‐fMRI data (voxel size = 2 mm) of all participants were collected using a customized 3T Siemens Skyra scanner. The minimal preprocessing pipeline (Glasser et al., 2013) used to process the rs‐fMRI images included mainly gradient distortion correction, spatial distortion removal, rigid‐body head motion correction, fMRI data coregistering to the T1‐weighted anatomical scan, normalizing to Montreal Neurological Institute (MNI) space and high‐pass temporal filtering.
After the preprocessing steps of the ICA‐fixed pipeline, we further removed the linear detrend and regressed out several nuisance signals from the data, including Friston's 24 head‐motion parameters, global signal, white matter signal, cerebrospinal fluid signal, and “bad” time points with a frame‐wise displacement of head motion > 0.5 mm (Power, Barnes, Snyder, Schlaggar, & Petersen, 2013). Finally, temporal bandpass filtering (0.01–0.08 Hz) was performed to reduce the effects of low‐frequency drift and high‐frequency physiological noise.
2.2. Whole‐brain FCS analysis
Pearson's correlation coefficients were computed between the time series of each pair of voxels, resulting in a whole‐brain rsFC matrix for each subject. To exclude spurious correlations from nongray matter voxels, the computation was constrained within a gray matter mask by thresholding (a threshold of > 0.2) a prior gray matter probability map in SPM12 (https://www.fil.ion.ucl.ac.uk/spm/software/spm12/). Of note, we also set a correlation coefficient of r > .2 to eliminate weak correlations possibly arising from noise signals and negative correlations caused by global signal removal (Wang et al., 2011). Subsequently, we calculated the FCS as the sum of the rsFC values between a given voxel and all other voxels for each subject. Finally, we averaged whole‐brain FCS maps of 100 subjects into a group mean FCS map.
2.3. Gene expression data
The human microarray‐based gene expression data were downloaded from AHBA (http://human.brain-map.org) (Hawrylycz et al., 2012), which were taken from six postmortem adult brains (1 Hispanic, 2 African‐American and 3 Caucasian; aged 24–57 years, one female and 5 males). The demographic information of each donor is shown in Table S1. Specifically, AHBA offers comprehensive coverage of nearly the entire brain, consisting of normalized expression data of 20,737 genes with unique Entrez IDs detected by 58,692 probes taken from 3,702 spatially distinct tissue samples. Since the AHBA‐provided coordinates were calculated by affine registering donors' T1‐weighted anatomical images to the MNI space, a more accurate nonlinear coregistration consisting of four steps (translation, rigid body, affine, and nonlinear deformation field) generated by Gorgolewski et al. (https://github.com/chrisgorgo/alleninf), which was performed using Advanced Normalization Tools (Avants et al., 2011), was used in this study. Therefore, spatial correspondence between gene expression and neuroimaging phenotypes could be established in MNI space.
The expression level of each gene from each sample of each donor was extracted from the AHBA database (the expression level of each gene from all probes was averaged if there were multiple probes for the same gene). Taking the MNI coordinates of each sample as the center, a 4‐mm radius (two times the voxel size) sphere was drawn, and the mean FCS value of the sphere was computed as the FCS value of the sample. Given the gross dissimilarities in the gene expression profiles of the cerebral cortex, subcortex, and cerebellum (Kang et al., 2011), we restricted our analyses to a mask of the cerebral cortex generated from the Brainnetome Atlas (Fan et al., 2016). Finally, a total of 1,765 samples of the cerebral cortex were included in the subsequent analyses (Table S1). The gene expression levels and FCS values were z‐score normalized by subtracting the mean and dividing by the SD across all the samples.
2.4. Donor‐level PLSR analyses
PLSR, a data reduction and regression technique, was used to explore genes significantly associated with FCS. PLSR is a widely used multivariate method to identify relationships between multiple predictor and response variables, with the goal of predicting response variables from predictors. Here, the predictor variables were the z‐score normalized matrix of the sample‐wise mean gene expression level of all probes from each donor ([donor ID: sample size × gene number]: H0351.1009: 178 × 20,737, H0351.1012: 258 × 20,737, H0351.1015: 225 × 207,37, H0351.1016: 255 × 20,737, H0351.2001: 474 × 20,737, and H0351.2002: 375 × 20,737). In addition, the response variables were the z‐score normalized vectors of the sample‐wise group mean FCS values (178 × 1, 258 × 1, 225 × 1, 255 × 1, 474 × 1, and 375 × 1). The PLSR components are ranked by the explained variances between predictor and response variables, and thus, the first several PLSR components provide the optimal low‐dimensional representation of the covariance of the higher dimensional data matrices (Abdi & Williams, 2013). In the current study, permutation tests were used to test the statistical significance of the first five PLSR components of each donor‐level model. To do this, we resampled the data (i.e., AHBA samples) without replacement within each PLSR model 1,000 times and counted the number of times of the explained variances which were higher than the real explained variances. The p‐values with statistical significance were calculated by dividing by the total number of permutation tests (1,000 times). Moreover, bootstrap tests were utilized to evaluate the significance of genes contributing to components. After obtaining real PLSR weights for each gene on each PLSR component, we resampled the data with replacement 1,000 times to reperform PLSR analysis at the donor level by bootstrap tests and generated the weights of each gene. The standard errors were assessed by bootstrapping, and z‐values (PLSR weights divided by their standard errors) were transformed to p‐values to evaluate the contribution of each gene to each PLSR component. We used the Benjamini‐Hochberg false discovery rate (BH‐FDR) correction (q < 0.05) to define the significance threshold. PLSR models were constructed for each donor to remove the bias of transcriptomic variations among the six donors. Only genes whose transcriptional profiles were significantly associated with FCS patterns in all six donors were defined as FCS‐related genes and were used for the following functional annotation analyses.
2.5. Gene functional annotations
Gene ontology (GO) (Ashburner et al., 2000) and the Kyoto encyclopedia of genes and genomes (KEGG) (Kanehisa & Goto, 2000) imbedded in the Enrichr tool (http://amp.pharm.mssm.edu/Enrichr/) (Chen et al., 2013; Kuleshov et al., 2016) were used for functional annotations of the FCS‐related genes (with both positive and negative weights). GO was used to enrich these genes for specific molecular functions, biological processes and cellular components, and KEGG was used to identify related biological pathways.
In addition, Enrichr and ToppGene (https://toppgene.cchmc.org/) (Dougherty, Schmidt, Nakajima, & Heintz, 2010) were applied to enrich the FCS‐related genes for specific neurodegenerative and neuropsychiatric diseases.
Furthermore, specific expression analysis (SEA) (Xu, Wells, O'Brien, Nehorai, & Dougherty, 2014) (http://genetics.wustl.edu/jdlab/csea-tool-2/) was utilized to evaluate whether these FCS‐related genes were over‐represented in three specific terms (cell types, brain regions and developmental stages) with a specificity index probability (pSI) (to assess how genes are more enriched in the specific terms relative to others). Specifically, cell‐type transcriptome data were collected from the translational profiles of 25 central nervous system cell populations in mice, including cerebellar (1–9), spinal cord (10), striatal and basal forebrain (11–14), brainstem (15), and cortical (16–25) cell types (Dougherty et al., 2010). The brain development transcriptional profiles were acquired from the BrainSpan Atlas of the Developing Human Brain (http://www.brainspan.org/), which was generated across 13 developmental stages in 8–16 brain structures.
All enrichment analyses were performed using Fisher's exact tests. BH‐FDR correction for multiple comparisons (q < 0.05) was applied for gene functional annotations.
2.6. Robustness analyses
To exclude the potential influence of different preprocessing approaches for neuroimaging and gene expression data on our main findings, we performed a series of robustness analyses.
2.6.1. The effect of correlation thresholds
In the main analysis, a single correlation threshold of r > .2 was used during the FCS calculation. To assess the effects of different thresholds on our results, we recomputed the FCS using two other thresholds (r > .1 and r > .3) and then reperformed donor‐level PLSR analyses.
2.6.2. Global signal regression effect
The use of global signal regression (GSR) is a debate in the field of rs‐fMRI preprocessing (Liu, Nalci, & Falahpour, 2017). We recalculated FCS (r > .2) without GSR and reperformed donor‐level PLSR analyses to compare with our main results (r > .2 with GSR).
2.6.3. Independent imaging data set
We applied another 100 HCP subjects with unique family IDs to test the stability of our main results, and after removing those with the same family IDs from the main analysis, 88 subjects were kept (44 males, mean age ± SD = 25.76 ± 2.70 years). The sex and age of the subjects were matched in the two neuroimaging data sets. We reperformed all the analyses in the same manner as mentioned above based on FCS values calculated by this independent imaging data set and gene expression values extracted from AHBA.
2.6.4. Individual FCS map level PLSR analyses
In view of individual differences in FCS patterns (Mueller et al., 2013), we repeated donor‐level PLSR analyses with the FCS pattern of each subject rather than the group mean FCS map, which resulted in a total of 600 (100 subjects × 6 donors) PLSR analyses. For each donor, the significant genes obtained with each subject overlapped together, and genes with repetition rates greater than 95% were retained. Finally, the FCS‐related genes were defined as the genes kept in all six donors.
2.6.5. Different preprocessing approaches for gene expression data
The preprocessing approaches for gene expression data are highly inconsistent across studies (Hawrylycz et al., 2015; Krienen et al., 2016; Romero‐Garcia et al., 2018). We used a newly proposed pipeline (Arnatkevic Iute, Fulcher, & Fornito, 2019) to repreprocess the gene expression data to validate our findings (please see the Supplementary Materials for details).
2.6.6. Remove the effect of the DMN
In consideration of the high correspondence of the final computed grand‐average FCS map with the DMN (i.e., higher FCS values, see Figure 2 for details), the results may be biased by the expression gradients between DMN regions and non‐DMN regions. Therefore, we divided the group mean FCS map into two parts, DMN regions, and non‐DMN regions, based on the DMN mask embedded in the GIFT software (https://trendscenter.org/software/gift/). Then, donor‐level PLSR analyses were carried out within the samples in DMN regions and non‐DMN regions, respectively.
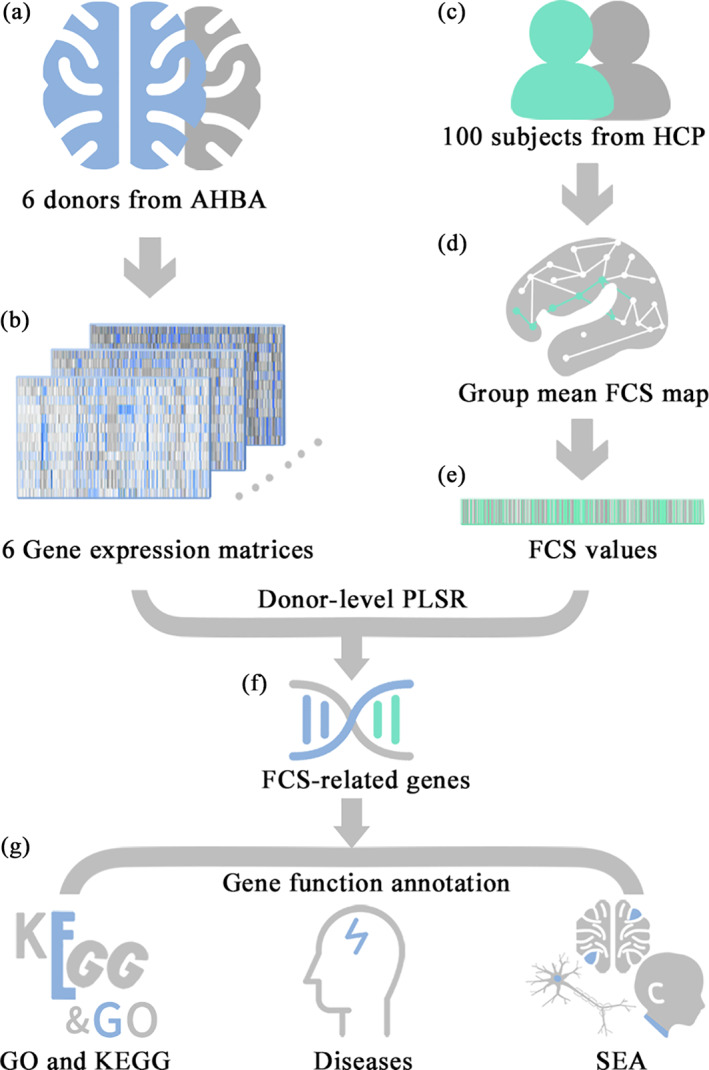
FIGURE 2.
The spatial distribution pattern of the group mean FCS pattern in healthy young adults. The voxel‐wise group mean FCS spatial distribution pattern of the cerebral cortex in healthy young adults (a correlation threshold of r > .2 with GSR) shows that the regions with higher FCS are primarily located in the DMN (mainly involving the PCC/PCU, inferior parietal lobule, medial prefrontal cortex) and in the DLPFC and the lateral temporal, parietal, and visual cortices. The color bar represents the z‐scored FCS values. Of note, only positive FCS values (i.e., higher FCS values) are shown here for visualization purpose. Abbreviations: L, left; R, right
2.6.7. Parcellation effect
To investigate the effect of parcellation on the results, we performed transcriptome‐neuroimaging association analyses at the regional level rather than at the voxel level. We assigned the cerebral cortical brain samples into the Brodmann atlas (Brodmann, 1909) according to the MNI coordinate of each sample. We assigned a total of 2,273 samples (H0351.1009: 243 samples, left hemisphere; H0351.1012: 318 samples, left hemisphere; H0351.1015: 294 samples, left hemisphere; H0351.1016: 312 samples, left hemisphere; H0351.2001: 581 samples, both hemispheres; H0351.2002: 525 samples, both hemispheres) to cerebral cortex regions of the Brodmann Atlas.
The voxel‐wise group mean FCS map was calculated with the same methods mentioned in the main analyses. After sample assignment, region‐wise FCS values were obtained by averaging a set of FCS values of samples in the same region, and a region‐wise expression matrix was extracted in the same way. The gene expression levels and FCS values were z‐score normalized by subtracting the mean and dividing by the standard deviation across all the regions at the donor level.
Donor‐level region‐wise PLSR analyses were carried out with permutation and bootstrap tests to identify components and genes that correlated with FCS. The predictor variables were as follows: H0351.1009: 35 × 20,737, H0351.1012: 39 × 20,737, H0351.1015: 37 × 20,737, H0351.1016: 41 × 20,737, H0351.2001: 76 × 20,737, and H0351.2002: 74 × 20,737. In addition, the response variables were the z‐score normalized vectors of the region‐wise group mean FCS values (H0351.1009: 35 × 1, H0351.1012: 39 × 1, H0351.1015: 37 × 1, H0351.1016: 41 × 1, H0351.2001: 76 × 1, and H0351.2002: 74 × 1). Finally, only genes significantly associated with FCS in six PLSR models were considered FCS‐related genes.
2.6.8. Comparison with the prior publications
We compared the FCS‐related genes with the gene lists obtained by correlating the rsFC network with gene expression in healthy subjects (Anderson et al., 2018; Krienen et al., 2016; Richiardi et al., 2015; Vertes et al., 2016) by using permutation tests. Specifically, we selected 1,518 genes (the number of FCS‐related genes was 1,518, please see details in the RESULTS section) from 20,737 genes randomly 1,000 times and obtained the overlapping genes by intersecting them with the results of the studies mentioned above. We counted the number of overlapping genes more than the real overlapping genes, and the p‐values with statistical significance were calculated by dividing by the total number of permutation tests (1,000 times).
2.7. Validation analyses
We performed validation analyses using independent imaging data sets and Brainspan expression data. Notably, the independent imaging data were the same as the data set in subsection 2.6.3.
Brainspan is a transcriptome atlas designed as a foundational resource for studying transcriptional mechanisms involved in human brain development (Miller et al., 2014). The data were generated across 13 developmental stages in 8–16 brain structures. The normalized expression data of 20,563 genes from seven donors aged 20–40 in the adult stage were extracted, which contained 11 cerebral regions (each region consists of one or more Brodmann areas) in both hemispheres (https://help.brain‐map.org/display/devhumanbrain/Documentation). The average expression level of samples in the same area was taken as the expression level of the area. Then, functional connectivity networks were constructed based on the Brodmann atlas, and mean FCS values in each of the 11 cerebral regions were calculated.
The gene expression levels and FCS values were z‐score transformed across all the samples before PLSR analyses. Here, the independent variables were the gene expression levels of the cortical regions (22 regions × 20,563 genes), and the dependent variables were the FCS values of the 22 regions. After PLSR analyses, functional annotations were reperformed with FCS‐related genes in the exact same way as mentioned above.
3. RESULTS
3.1. Whole‐brain voxel‐wise FCS pattern
As illustrated in Figure 2, the regions with higher FCS values were predominantly located in the DMN (involving mainly the posterior cingulate cortex/precuneus [PCC/PCU], inferior parietal lobule, medial prefrontal cortex) and dorsolateral prefrontal cortex (DLPFC), lateral temporal, parietal, and visual cortices, which is highly in line with the findings of previous studies (Bokde et al., 2006; Liang et al., 2013; Liu et al., 2015; Tomasi & Volkow, 2011a; Tomasi & Volkow, 2011b).
3.2. PLSR characterization
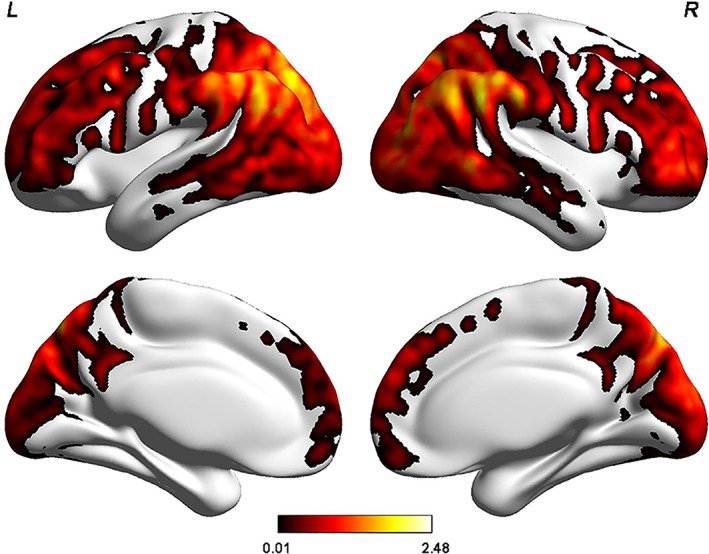
As shown in Table S2 and Figure 3, the permutation tests revealed that only the scores of the first PLSR component (PLSR1) were significantly positively associated with the FCS in all six donors (all ps < .001). A total of 1,518 genes were repeatedly identified to make significant contributions to PLSR1 (BH‐FDR q < 0.05) in all six donors (Figure S1). Thus, these genes were considered FCS‐related genes and were used for the following gene functional annotation analyses.
FIGURE 3.
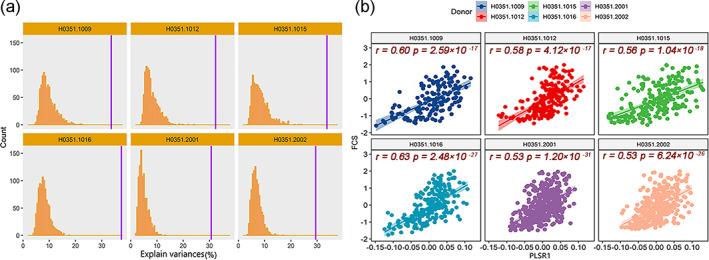
Results derived from PLSR analyses. (a) Permutation tests revealed that only the PLSR1 scores explain a significant amount of the variance in the FCS in all six donors (explained variances at least > 29%). Purple vertical lines represent the real explained variances in each donor, and bar plots indicate the distribution of random explained variance. (b) All donors show significantly positive correlations between PLSR1 scores (the liner combinations of the expression matrix) and group‐mean FCS values in each donor, with Pearson's correlation coefficients ranging from 0.53 to 0.63. Abbreviations: PLSR1, the first component of partial least squares regression
3.3. Gene enrichments
In the GO enrichment analysis, FCS‐related genes were enriched for the biological processes of metal ion transport (corrected q = 1.85 × 10−2) and the molecular functions of voltage‐gated‐like ion channels, especially potassium (corrected q = 1.53 × 10−4) and sodium (corrected q = 5.46 × 10−3) channels (Figure 4a). In the KEGG pathway analysis, FCS‐related genes were enriched for the calcium signaling pathway (corrected q = 1.99 × 10−5), axon guidance pathway (corrected q = 5.32 × 10−4) and synaptic transmission processes such as dopaminergic synaptic transmission (corrected q = 5.32 × 10−4), long‐term potentiation (corrected q = 5.32 × 10−4), long‐term depression (corrected q = 2.79 × 10−4) and glutamatergic synaptic transmission (corrected q = 2.79 × 10−3) (Figure 4b).
FIGURE 4.
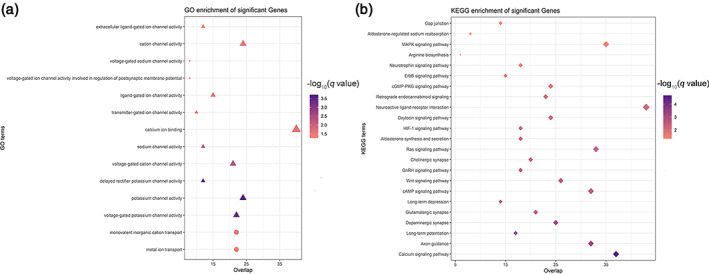
Biological function analysis for FCS‐related genes. The GO (a) and KEGG (b) enrichment analyses show that FCS‐related genes are significantly enriched for ion channels, axon guidance pathway, and synaptic transmission processes. The color bars represent the ‐log10 (q value) of BH‐FDR correction. The size of the circles (biological processes), triangles (molecular functions), and rhombus (KEGG terms) represents the gene number hit on the terms. Abbreviations: GO, gene ontology; KEGG, Kyoto encyclopedia of genes and genomes
In the disease‐related enrichment analyses, FCS‐related genes were enriched for several common neurodegenerative and neuropsychiatric disorders and were differentially expressed in patients with these disorders such as AD, ASD, BPD, and SCZ (Figure 5a). Moreover, both upregulated (Figure 5b) and downregulated (Figure 5c) genes were found in patients with these disorders rather than healthy populations.
FIGURE 5.
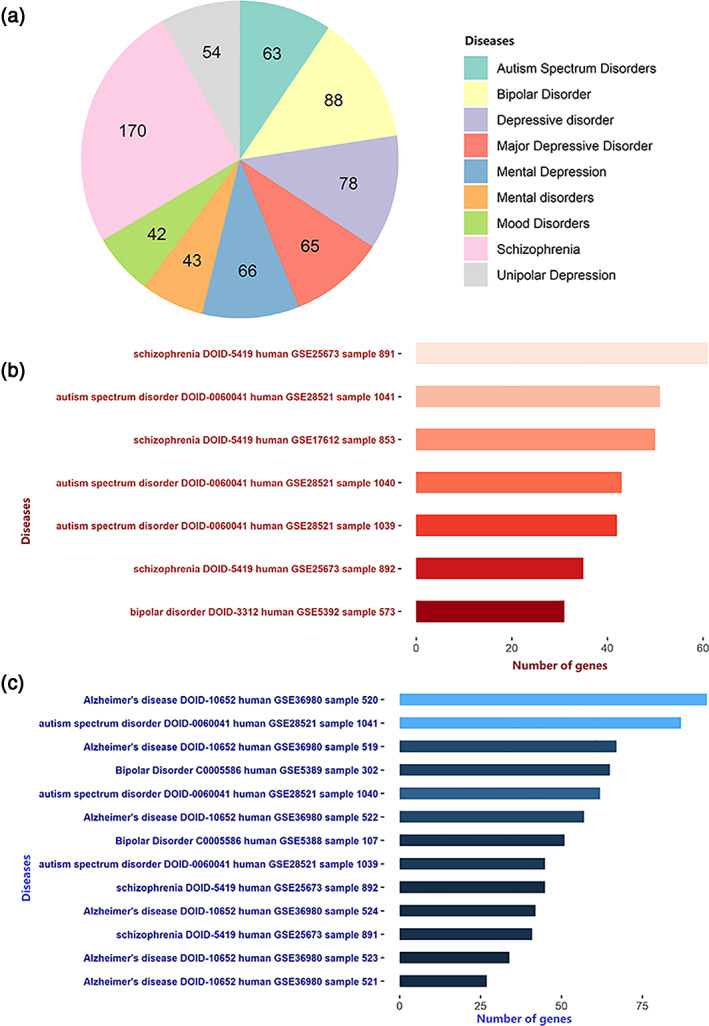
Disease enrichment analysis for the FCS‐related genes. (a) ToppGene shows the number of FCS‐related genes associated with neurodegenerative and neuropsychiatric disorders. Enrichr shows that FCS‐related genes are enriched for genes with different expression patterns in patients with neurodegenerative and neuropsychiatric diseases; the red and blue bars indicate upregulated (b) and downregulated (c) genes, respectively. The colors indicate the significances of enrichment. These data are obtained from Gene Expression Omnibus (GEO) data sets, and the text on the left shows the name of the disease, disease ID, species, GEO Series (GSE) ID, and sample ID for each term
In the human brain, FCS‐related genes were preferentially expressed during young adulthood (corrected q = 2.541 × 10−4, pSI = .0001), particularly in the cerebral cortex (corrected q = 0.03, pSI = .0001). In mice, 1130 homologs of the 1518 FCS‐related genes were primarily overexpressed in neurons, including layer 5b corticospinal and corticopontine pyramidal neurons (corrected q = 1.151 × 10−4, pSI = .05) and layer six corticothalamic pyramidal neurons (corrected q = 2.366 × 10‐4, pSI = .05) (Figure 6 and Tables [Link], [Link]).
FIGURE 6.
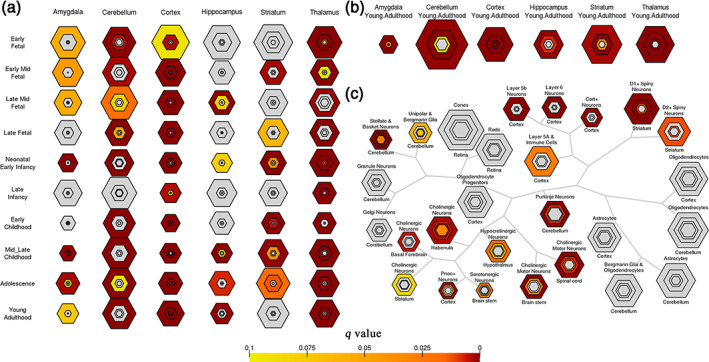
Specific expression analyses of the FCS‐related genes. (a) Development SEA indicates that the FCS‐related genes show the most significant enrichment during young adulthood (corrected q = 2.541×10−4, pSI = .0001). (b) Brain region SEA indicates that FCS‐related genes preferentially express in the cerebral cortex (corrected q = 0.03, pSI = .0001). (c) Cell type SEA indicates that FCS‐related genes have higher expression levels in the neurons of several brain regions. The sizes of the bullseyes are scaled to the numbers of enriched genes at different thresholds (i.e., pSI = .05 [outermost], .01 [outer], .001 [inner], and .0001 [innermost]). The bullseyes are color‐coded according to the q values (BH‐FDR correction)
3.4. Robustness analyses
3.4.1. The effect of correlation thresholds
The correlation threshold effect was tested by recomputing the FCS maps with two other thresholds (r > .1 and r > .3, Figure S2a,b) and reperforming PLSR analyses. We calculated Spearman's rank correlation coefficient between the group mean FCS maps to evaluate the effect of correlation thresholds on the FCS pattern both in the voxel and sample levels (Figures S3a,b and S4a,b). The results of PLSR analyses based on the two FCS maps with different thresholds were highly similar to our main findings (r > .2). Specifically, only the PLSR1 scores were significantly positively correlated with the FCS maps. The number of significant FCS‐related genes, the genes overlapping with those in our main findings, and the variance in the FCS patterns explained by the PLSR1 scores are shown in Tables S6–S8.
3.4.2. The GSR effect
We recomputed the FCS maps (r > .2) without GSR (Figure S2c) and reperformed the PLSR analysis. Spearman's rank correlation showed that FCS maps were significantly positively correlated both in the voxel and sample levels (Figures S3c and S4c). The PLSR results were also highly similar to our main findings. Specifically, only the PLSR1 scores were significantly positively correlated with the FCS maps. The number of significant FCS‐related genes, the genes overlapping with those in our main findings and the variance in the FCS patterns explained by the PLSR1 scores are shown in Tables S6 and S9.
3.4.3. Independent imaging data set
We replicated our main findings in another independent neuroimaging data set with 88 subjects and recomputed the FCS maps with r > .2 and GSR. The group mean FCS patterns are shown in Figure S2d. Spearman's rank correlation was utilized to evaluate the relationship between the group mean FCS maps of our main results and independent imaging data sets. We found that the two group mean FCS maps were significantly positively correlated both in the voxel and sample levels (Figures S3d and S4d). The permutation tests revealed that only the PLSR1 scores explained a significant amount of the variance in the FCS in all six donors (all ps < .001, Table S10). Additionally, the PLSR1 scores were positively correlated with the sample‐wise group mean z‐scored FCS values of each donor, with Pearson's correlation coefficients ranging from 0.52 to 0.63 (Figure S5). We identified 1,603 FCS‐related genes (Figure S6), of which 1,449 genes (95%) overlapped with those in our main findings. Gene enrichment analyses and SEA revealed that the 1,603 FCS‐related genes had highly similar functions to those in our main results (Figures S7–S9).
3.4.4. Individual FCS map level PLSR analyses
After 600 PLSR analyses, 113 FCS‐related genes were found, and all these genes were in total in our main results.
3.4.5. The new preprocessing pipeline for gene expression data
According to the newly proposed pipeline for preprocessing gene expression data, probe‐to‐gene reannotations resulted in 47,795 probes and 20,919 unique genes. After all the preprocessing procedures, 10,185 genes and 1,122 cortical samples were left for the PLSR analyses. We found that the gene expression profiles of the first two components (PLSR1 and PLSR2) significantly predicted the FCS pattern (r > .2 with GSR) with explanatory variances of > 26.25%, and > 15.12%, respectively. From these two components, we found that 1,121 genes (74%) overlapped with those in our main findings.
3.4.6. Remove the effect of the DMN
Seventy‐six and 1591 genes were significantly related to the FCS patterns of DMN and non‐DMN regions, respectively. Most of those genes overlapped with the 1518 FCS‐related genes, especially the genes related to the FCS patterns of the non‐DMN (63.2% [48/76] and 85.3% [1357/1591]), indicating that our transcriptome‐neuroimaging association analysis was not driven by the gene expression gradient between the DMN and non‐DMN regions.
3.4.7. Parcellation effect
We obtained 1,685 FCS‐related genes by using the Brodmann Atlas, 693 of which overlapped with 1,518 FCS‐related genes found in the main results. The genes were enriched in potassium ion transport biological process, voltage‐gated cation channel activity and voltage‐gated cation channel activity molecular function, and in KEGG analysis, the genes were enriched in pathways similar to the main results.
3.4.8. Comparison with the prior publications
We found significant overlap between the 1,518 FCS‐related genes and the results in prior publications (permutation test, all ps < .05), which indicated the reliability of our results (Table S11).
3.5. Validation analyses
By using independent imaging data and the Brainspan atlas, we found 651 FCS‐related genes overlapping with the main results. Gene functional annotations revealed that the FCS‐related genes had highly similar functions to those in our main results. In the GO enrichment analysis, these genes were enriched for the biological processes of metal ion transport (corrected q = 2.50 × 10−2), neuronal action potential (corrected q = 1.49 × 10−2) and monovalent inorganic cation transport (corrected q = 1.18 × 10−2), and some ion channel activities of molecular functions such as voltage‐gated sodium channel activity (corrected q = 2.24 × 10−3) and voltage‐gated potassium channel activity (corrected q = 5.15 × 10−3). In the KEGG pathway analysis, similar terms of the main results were also validated. Moreover, in the disease‐related enrichment analyses, these genes were enriched for several common neurodegenerative and neuropsychiatric disorders and were differentially expressed in patients with these disorders such as AD, ASD, BPD, and SCZ. Furthermore, both upregulated and downregulated genes were found in patients with these disorders rather than healthy populations.
4. DISCUSSION
This transcriptome‐neuroimaging association study investigated spatial correlations between gene transcriptional profiles and resting‐state functional network centrality in healthy young adults. Specifically, we identified a set of genes correlated with FCS using PLSR. The FCS‐related genes were enriched for ion channels, axon guidance pathways, and synaptic transmission processes. These genes were preferentially expressed in cortical neurons in young adulthood and were associated with common neurodegenerative and neuropsychiatric disorders. Our results showed high robustness and large reproducibility by using a series of robustness analyses and validation analyses.
In total, 1,518 FCS‐related genes were enriched for ion channels, especially voltage‐gated potassium and sodium channels, and calcium signaling pathways. Ion channels are vital for neuronal functions, triggering nerve impulses, and neurotransmitter release (Kumar, Kumar, Jha, Jha, & Ambasta, 2016). Mutations in ion channel‐related genes are associated with diverse neuropsychiatric disorders in humans (Kim, 2014; Pietrobon, 2002). Voltage‐gated potassium channels are widely present in the central nervous system (CNS) and are responsible for neuronal excitability (Shah & Aizenman, 2014). Abnormally high or low neuronal excitability can lead to various neurodegenerative and neuropsychiatric disorders. For example, abnormally low neuronal excitability can be seen in major depressive disorder (MDD) and AD; in contrast, attention deficit/hyperactivity disorder (ADHD), anxiety, BPD, and SCZ show abnormally high neuronal excitability. Similarly, voltage‐gated sodium channels are also broadly present in the CNS (Eijkelkamp et al., 2012) and are linked to neuronal excitability, with their dysfunction being implicated in brain disorders such as SCZ (Rees et al., 2019), AD (Verret et al., 2012) and ASD (Weiss et al., 2003). Moreover, the calcium signaling pathway also plays a pivotal role in neurodegenerative and neuropsychiatric disorders. For example, in AD, Aβ aggregation can increase neuronal cytosolic calcium concentrations and further trigger synaptic dysfunction and neurodegeneration (Alzheimer's Association Calcium Hypothesis Workgroup, 2017). Furthermore, GWAS have indicated that genetic variations associated with the calcium signaling pathway can increase the risk of developing five major psychiatric disorders: ASD, ADHD, BPD, SCZ, and MDD (Cross‐Disorder Group of the Psychiatric Genomics Consortium, 2013). FCS‐related genes were also enriched for axon guidance pathways. Directing axon growth and elongation during development and regulating the structural plasticity of synaptic connections in adults rely heavily on axon guidance pathway proteins encoded by axon guidance genes. Common variants of axon guidance pathway genes (Hollingworth et al., 2011) or alterations in their expression profiles have been found to contribute to the pathogenesis of AD (Antonell et al., 2013; Simon et al., 2009) and ASD (Melin et al., 2006; Suda et al., 2011). The role of these genes in FCS abnormalities in these disorders needs to be further elucidated.
FCS‐related genes were also enriched for synaptic transmission and plasticity. Synaptic transmission is important for information exchange between neurons, which depends on neurotransmitter systems. Changes in the synaptic plasticity of dopamine neurons may affect dopamine release (Slifstein et al., 2015). Dopamine is one of the most important neuromodulators regulating circuit function and plasticity (Tritsch & Sabatini, 2012), including neurotransmitter release, postsynaptic sensitivity to neurotransmitters, and the membrane excitability of pre‐ and postsynaptic cells. Dopaminergic neurotransmission dysfunction has been found to be involved in the pathogenesis of several psychiatric and neurological disorders (Ko & Strafella, 2012) such as MDD (Pogarell et al., 2006), AD (Nobili et al., 2017), and SCZ (Chuhma, Mingote, Kalmbach, Yetnikoff, & Rayport, 2017). Glutamate is one of the major excitatory neurotransmitters in the mammalian CNS and is involved in numerous neuronal functions, including synaptic transmission (Pittenger, Bloch, & Williams, 2011). This role is consistent with previous findings that mutations and expression abnormalities in genes encoding glutamate and glutamatergic receptors are associated with ASD (Leblond et al., 2014; Rabaneda, Robles‐Lanuza, Nieto‐Gonzalez, & Scholl, 2014), ADHD (Miladinovic, Nashed, & Singh, 2015) and SCZ (Zhou et al., 2016). Long‐term potentiation and long‐term depression are two forms of synaptic plasticity and are substrates for learning and memory in the brain (Anggono & Huganir, 2012; Collingridge, Peineau, Howland, & Wang, 2010; Pignatelli & Bonci, 2015). Thus, the genes associated with synaptic plasticity have been linked to many brain disorders.
FCS‐related genes were preferentially expressed during young adulthood, which is the late developmental stage of the cerebral cortex and a period in which the risk for many neuropsychiatric disorders increased. Brain tissue expresses more alternatively spliced transcripts than other tissues at this stage (Yeo, Holste, Kreiman, & Burge, 2004), which is one of the biological mechanisms for generating the complexity and function of the transcriptome in the brain and is involved in numerous neuropsychiatric disorders (Licatalosi & Darnell, 2006). This theory is supported by the findings of the disease‐related enrichment analysis in that these genes were enriched in several neuropsychiatric disorders (such as SCZ, MDD, and BPD), with a higher incidence at this stage (Argyelan et al., 2014; Filippi et al., 2017; Guo et al., 2015; Khan et al., 2013).
Several limitations need to be considered when interpreting our results. First, the gene expression data and neuroimaging data were not collected from the same participants. However, comparative studies have shown a high degree of conservation in overall gene expression across human populations (Stranger et al., 2007), and the resulting FCS‐related genes should be considered those with high conservation across subjects. Second, the gene expression data were derived from six adult brains from donors of different ages, ethnicities, sexes, medical histories, and causes of death, which may have led to variance in transcriptome profiles and could have further influenced our results. Third, transcriptional activity in the AHBA was measured by microarray (the relative levels of gene expression were measured in a tissue sample), which is limited to known gene sequences and is prone to background noise. In addition, the expression levels of the bulk tissue samples might be influenced by the cellular composition. These factors might confound the transcription‐neuroimaging associations. Fourth, the different preprocessing procedures for the gene expression data may have impacted the resulting FCS‐related genes, and although the proposed pipeline has several merits, only including a single cerebral hemisphere may introduce biases from functional connectivity and gene expression asymmetries across the two hemispheres (Hawrylycz et al., 2012; Pletikos et al., 2014; Sun et al., 2005). Therefore, we did not use the newly proposed preprocessing pipeline for our main analysis. In addition, the reliability of our findings is supported by the 74% overlap in the FCS‐related genes with the two pipelines for preprocessing gene expression data. Fifth, the sample sizes of image data in our study were small due to the heavy calculation burden (voxel size: 2 × 2 × 2 mm3). Our results are preliminary, and a larger sample is needed to verify this association in the future. Sixth, although there is an improved gene set enrichment analysis method (https://doi.org/10.1101/2020.04.24.058958), the codes in this paper now only support univariate analysis (e.g., Pearson or Spearman analysis) and cover only biological processes of GO terms. In the future, we will test the PLSR results using this method. Finally, although we identified a set of genes associated with FCS, this relationship is correlational rather than causative. Our results need to be validated by animal experiments in the future.
5. CONCLUSIONS
This transcription‐neuroimaging association study identified that the expression profiles of a set of genes, especially those involved in processes related to ion channels, the axon guidance pathway, and synaptic transmission, are related to FCS. These findings provide new insight into the biological substrates underlying intrinsic functional network centrality.
CONFLICT OF INTEREST
All authors report no biomedical financial interests or potential conflicts of interest.
Supporting information
Table S1. AHBA donors' demographic information
Table S2. The explained variances of the FCS patterns by the PLSR1‐5 scores
Table S3. Brain region and development specific expression analysis.
Table S4. Brain region specific analysis
Table S5. Cell type specific expression analysis result
ACKNOWLEDGMENTS
This work was supported by National Key Research and Development Program of China (2018YFC1314300), Natural Science Foundation of China (Grant No. 81871431, 82030053, 82001796, and 82072001), and Natural Science Foundation of Tianjin Municipal Science and Technology Commission (Grant No. 18JCYBJC26300 and 18JCQNJC10900).
Zhu D, Yuan T, Gao J, et al. Correlation between cortical gene expression and resting‐state functional network centrality in healthy young adults. Hum Brain Mapp. 2021;42:2236–2249. 10.1002/hbm.25362
Funding information National Key Research and Development Program of China, Grant/Award Number: 2018YFC1314300; National Natural Science Foundation of China, Grant/Award Numbers: 82030053, 81871431, 82001796, 82072001; Natural Science Foundation of Tianjin Municipal Science and Technology Commission, Grant/Award Numbers: 18JCQNJC10900, 18JCYBJC26300
Contributor Information
Feng Liu, Email: fengliu@tijmu.edu.cn.
Junping Wang, Email: wangjunping_tj@163.com.
DATA AVAILABILITY STATEMENT
The rs‐fMRI data were requested and downloaded from publicly available HCP data (https://db.humanconnectome.org). The human microarray‐based gene expression data were downloaded from AHBA (http://human.brain‐map.org). All codes included in this study are available upon request by contact with the corresponding author.
REFERENCES
- Abdi, H. , & Williams, L. J. (2013). Partial least squares methods: Partial least squares correlation and partial least square regression. Methods in Molecular Biology, 930, 549–579. 10.1007/978-1-62703-059-5_23 [DOI] [PubMed] [Google Scholar]
- Alzheimer's Association Calcium Hypothesis Workgroup . (2017). Calcium Hypothesis of Alzheimer's disease and brain aging: Aframework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement, 13, 178–182.e117. 10.1016/j.jalz.2016.12.006 [DOI] [PubMed] [Google Scholar]
- Anderson, K. M. , Krienen, F. M. , Choi, E. Y. , Reinen, J. M. , Yeo, B. T. T. , & Holmes, A. J. (2018). Gene expression links functional networks across cortex and striatum. Nature Communications, 9, 1428. 10.1038/s41467-018-03811-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anggono, V. , & Huganir, R. L. (2012). Regulation of AMPA receptor trafficking and synaptic plasticity. Current Opinion in Neurobiology, 22, 461–469. 10.1016/j.conb.2011.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonell, A. , Llado, A. , Altirriba, J. , Botta‐Orfila, T. , Balasa, M. , Fernandez, M. , … Molinuevo, J. L. (2013). A preliminary study of the whole‐genome expression profile of sporadic and monogenic early‐onset Alzheimer's disease. Neurobiology of Aging, 34, 1772–1778. 10.1016/j.neurobiolaging.2012.12.026 [DOI] [PubMed] [Google Scholar]
- Argyelan, M. , Ikuta, T. , DeRosse, P. , Braga, R. J. , Burdick, K. E. , John, M. , … Szeszko, P. R. (2014). Resting‐state fMRI connectivity impairment in schizophrenia and bipolar disorder. Schizophrenia Bulletin, 40, 100–110. 10.1093/schbul/sbt092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnatkevic Iute, A. , Fulcher, B. D. , & Fornito, A. (2019). A practical guide to linking brain‐wide gene expression and neuroimaging data. NeuroImage, 189, 353–367. 10.1016/j.neuroimage.2019.01.011 [DOI] [PubMed] [Google Scholar]
- Ashburner, M. , Ball, C. A. , Blake, J. A. , Botstein, D. , Butler, H. , Cherry, J. M. , … Sherlock, G. (2000). Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nature Genetics, 25, 25–29. 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avants, B. B. , Tustison, N. J. , Song, G. , Cook, P. A. , Klein, A. , & Gee, J. C. (2011). A reproducible evaluation of ANTs similarity metric performance in brain image registration. NeuroImage, 54, 2033–2044. 10.1016/j.neuroimage.2010.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokde, A. L. W. , Lopez‐Bayo, P. , Meindl, T. , Pechler, S. , Born, C. , Faltraco, F. , … Hampel, H. (2006). Functional connectivity of the fusiform gyrus during a face‐matching task in subjects with mild cognitive impairment. Brain, 129, 1113–1124. 10.1093/brain/awl051 [DOI] [PubMed] [Google Scholar]
- Brodmann, K. (1909). Vergleichende Lokalisationslehre der Grosshirnrinde in ihren Prinzipien dargestellt auf Grund des Zellenbaues: Barth.
- Chen, E. Y. , Tan, C. M. , Kou, Y. , Duan, Q. , Wang, Z. , Meirelles, G. V. , … Ma'ayan, A. (2013). Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics, 14, 128. 10.1186/1471-2105-14-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuhma, N. , Mingote, S. , Kalmbach, A. , Yetnikoff, L. , & Rayport, S. (2017). Heterogeneity in dopamine neuron synaptic actions across the striatum and its relevance for schizophrenia. Biological Psychiatry, 81, 43–51. 10.1016/j.biopsych.2016.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge, G. L. , Peineau, S. , Howland, J. G. , & Wang, Y. T. (2010). Long‐term depression in the CNS. Nature Reviews. Neuroscience, 11, 459–473. 10.1038/nrn2867 [DOI] [PubMed] [Google Scholar]
- Cross‐Disorder Group of the Psychiatric Genomics . (2013). Identification of risk loci with shared effects on five major psychiatric disorders: A genome‐wide analysis. Lancet, 381, 1371–1379. 10.1016/S0140-6736(12)62129-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty, J. D. , Schmidt, E. F. , Nakajima, M. , & Heintz, N. (2010). Analytical approaches to RNA profiling data for the identification of genes enriched in specific cells. Nucleic Acids Research, 38, 4218–4230. 10.1093/nar/gkq130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijkelkamp, N. , Linley, J. E. , Baker, M. D. , Minett, M. S. , Cregg, R. , Werdehausen, R. , … Wood, J. N. (2012). Neurological perspectives on voltage‐gated sodium channels. Brain, 135, 2585–2612. 10.1093/brain/aws225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott, L. T. , Sharp, K. , Alfaro‐Almagro, F. , Shi, S. , Miller, K. L. , Douaud, G. , … Smith, S. M. (2018). Genome‐wide association studies of brain imaging phenotypes in UK Biobank. Nature, 562, 210–216. 10.1038/s41586-018-0571-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, L. , Li, H. , Zhuo, J. , Zhang, Y. , Wang, J. , Chen, L. , … Jiang, T. (2016). The human Brainnetome atlas: A new brain atlas based on connectional architecture. Cerebral Cortex, 26, 3508–3526. 10.1093/cercor/bhw157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippi, M. , Basaia, S. , Canu, E. , Imperiale, F. , Meani, A. , Caso, F. , … Agosta, F. (2017). Brain network connectivity differs in early‐onset neurodegenerative dementia. Neurology, 89, 1764–1772. 10.1212/wnl.0000000000004577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornito, A. , Arnatkeviciute, A. , & Fulcher, B. D. (2019). Bridging the gap between connectome and transcriptome. Trends in Cognitive Sciences, 23, 34–50. 10.1016/j.tics.2018.10.005 [DOI] [PubMed] [Google Scholar]
- Fornito, A. , Zalesky, A. , Bassett, D. S. , Meunier, D. , Ellison‐Wright, I. , Yucel, M. , … Bullmore, E. T. (2011). Genetic influences on cost‐efficient organization of human cortical functional networks. The Journal of Neuroscience, 31, 3261–3270. 10.1523/JNEUROSCI.4858-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glahn, D. C. , Winkler, A. M. , Kochunov, P. , Almasy, L. , Duggirala, R. , Carless, M. A. , … Blangero, J. (2010). Genetic control over the resting brain. Proceedings of the National Academy of Sciences of the United States of America, 107, 1223–1228. 10.1073/pnas.0909969107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasser, M. F. , Sotiropoulos, S. N. , Wilson, J. A. , Coalson, T. S. , Fischl, B. , Andersson, J. L. , … Jenkinson, M. (2013). The minimal preprocessing pipelines for the Human Connectome Project. NeuroImage, 80, 105–124. 10.1016/j.neuroimage.2013.04.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greicius, M. (2008). Resting‐state functional connectivity in neuropsychiatric disorders. Current Opinion in Neurology, 21, 424–430. 10.1097/WCO.0b013e328306f2c5 [DOI] [PubMed] [Google Scholar]
- Guo, W. , Liu, F. , Xiao, C. , Liu, J. , Yu, M. , Zhang, Z. , … Zhao, J. (2015). Increased short‐range and long‐range functional connectivity in first‐episode, medication‐naive schizophrenia at rest. Schizophrenia Research, 166, 144–150. 10.1016/j.schres.2015.04.034 [DOI] [PubMed] [Google Scholar]
- Hawrylycz, M. , Miller, J. A. , Menon, V. , Feng, D. , Dolbeare, T. , Guillozet‐Bongaarts, A. L. , … Lein, E. (2015). Canonical genetic signatures of the adult human brain. Nature Neuroscience, 18, 1832–1844. 10.1038/nn.4171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawrylycz, M. J. , Lein, E. S. , Guillozet‐Bongaarts, A. L. , Shen, E. H. , Ng, L. , Miller, J. A. , … Jones, A. R. (2012). An anatomically comprehensive atlas of the adult human brain transcriptome. Nature, 489, 391–399. 10.1038/nature11405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingworth, P. , Harold, D. , Sims, R. , Gerrish, A. , Lambert, J. C. , Carrasquillo, M. M. , … Williams, J. (2011). Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nature Genetics, 43, 429–435. 10.1038/ng.803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa, M. , & Goto, S. (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Research, 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, H. J. , Kawasawa, Y. I. , Cheng, F. , Zhu, Y. , Xu, X. , Li, M. , … Sestan, N. (2011). Spatio‐temporal transcriptome of the human brain. Nature, 478, 483–489. 10.1038/nature10523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, S. , Gramfort, A. , Shetty, N. R. , Kitzbichler, M. G. , Ganesan, S. , Moran, J. M. , … Kenet, T. (2013). Local and long‐range functional connectivity is reduced in concert in autism spectrum disorders. Proceedings of the National Academy of Sciences of the United States of America, 110, 3107–3112. 10.1073/pnas.1214533110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. B. (2014). Channelopathies. Korean Journal of Pediatrics, 57, 1–18. 10.3345/kjp.2014.57.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko, J. H. , & Strafella, A. P. (2012). Dopaminergic neurotransmission in the human brain: New lessons from perturbation and imaging. The Neuroscientist, 18, 149–168. 10.1177/1073858411401413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krienen, F. M. , Yeo, B. T. , Ge, T. , Buckner, R. L. , & Sherwood, C. C. (2016). Transcriptional profiles of supragranular‐enriched genes associate with corticocortical network architecture in the human brain. Proceedings of the National Academy of Sciences of the United States of America, 113, E469–E478. 10.1073/pnas.1510903113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov, M. V. , Jones, M. R. , Rouillard, A. D. , Fernandez, N. F. , Duan, Q. , Wang, Z. , … Ma'ayan, A. (2016). Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Research, 44, W90–W97. 10.1093/nar/gkw377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, P. , Kumar, D. , Jha, S. K. , Jha, N. K. , & Ambasta, R. K. (2016). Ion Channels in Neurological Disorders. Advances in Protein Chemistry and Structural Biology, 103, 97–136. 10.1016/bs.apcsb.2015.10.006 [DOI] [PubMed] [Google Scholar]
- Laperchia, C. , Allegra Mascaro, A. L. , Sacconi, L. , Andrioli, A. , Matte, A. , De Franceschi, L. , … Pavone, F. S. (2013). Two‐photon microscopy imaging of thy1GFP‐M transgenic mice: A novel animal model to investigate brain dendritic cell subsets in vivo. PLoS One, 8, e56144. 10.1371/journal.pone.0056144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson‐Prior, L. J. , Zempel, J. M. , Nolan, T. S. , Prior, F. W. , Snyder, A. Z. , & Raichle, M. E. (2009). Cortical network functional connectivity in the descent to sleep. Proceedings of the National Academy of Sciences of the United States of America, 106, 4489–4494. 10.1073/pnas.0900924106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblond, C. S. , Nava, C. , Polge, A. , Gauthier, J. , Huguet, G. , Lumbroso, S. , … Bourgeron, T. (2014). Meta‐analysis of SHANK Mutations in Autism Spectrum Disorders: A gradient of severity in cognitive impairments. PLoS Genetics, 10, e1004580. 10.1371/journal.pgen.1004580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, X. , Zou, Q. , He, Y. , & Yang, Y. (2013). Coupling of functional connectivity and regional cerebral blood flow reveals a physiological basis for network hubs of the human brain. Proceedings of the National Academy of Sciences of the United States of America, 110, 1929–1934. 10.1073/pnas.1214900110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi, D. D. , & Darnell, R. B. (2006). Splicing regulation in neurologic disease. Neuron, 52, 93–101. 10.1016/j.neuron.2006.09.017 [DOI] [PubMed] [Google Scholar]
- Liu, F. , Zhu, C. , Wang, Y. , Guo, W. , Li, M. , Wang, W. , … Chen, H. (2015). Disrupted cortical hubs in functional brain networks in social anxiety disorder. Clinical Neurophysiology, 126, 1711–1716. 10.1016/j.clinph.2014.11.014 [DOI] [PubMed] [Google Scholar]
- Liu, T. T. , Nalci, A. , & Falahpour, M. (2017). The global signal in fMRI: Nuisance or information? NeuroImage, 150, 213–229. 10.1016/j.neuroimage.2017.02.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melin, M. , Carlsson, B. , Anckarsater, H. , Rastam, M. , Betancur, C. , Isaksson, A. , … Dahl, N. (2006). Constitutional downregulation of SEMA5A expression in autism. Neuropsychobiology, 54, 64–69. 10.1159/000096040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miladinovic, T. , Nashed, M. G. , & Singh, G. (2015). Overview of glutamatergic dysregulation in central pathologies. Biomolecules, 5, 3112–3141. 10.3390/biom5043112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, J. A. , Ding, S.‐L. , Sunkin, S. M. , Smith, K. A. , Ng, L. , Szafer, A. , … Lein, E. S. (2014). Transcriptional landscape of the prenatal human brain. Nature, 508, 199–206. 10.1038/nature13185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morcom, A. M. , & Fletcher, P. C. (2007). Does the brain have a baseline? Why we should be resisting a rest. NeuroImage, 37, 1073–1082. 10.1016/j.neuroimage.2007.06.019 [DOI] [PubMed] [Google Scholar]
- Mueller, S. , Wang, D. , Fox, M. D. , Yeo, B. T. , Sepulcre, J. , Sabuncu, M. R. , … Liu, H. (2013). Individual variability in functional connectivity architecture of the human brain. Neuron, 77, 586–595. 10.1016/j.neuron.2012.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobili, A. , Latagliata, E. C. , Viscomi, M. T. , Cavallucci, V. , Cutuli, D. , Giacovazzo, G. , … D'Amelio, M. (2017). Dopamine neuronal loss contributes to memory and reward dysfunction in a model of Alzheimer's disease. Nature Communications, 8, 14727. 10.1038/ncomms14727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrobon, D. (2002). Calcium channels and channelopathies of the central nervous system. Molecular Neurobiology, 25, 31–50. 10.1385/mn:25:1:031 [DOI] [PubMed] [Google Scholar]
- Pignatelli, M. , & Bonci, A. (2015). Role of dopamine neurons in reward and aversion: A synaptic plasticity perspective. Neuron, 86, 1145–1157. 10.1016/j.neuron.2015.04.015 [DOI] [PubMed] [Google Scholar]
- Pittenger, C. , Bloch, M. H. , & Williams, K. (2011). Glutamate abnormalities in obsessive compulsive disorder: Neurobiology, pathophysiology, and treatment. Pharmacology & Therapeutics, 132, 314–332. 10.1016/j.pharmthera.2011.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletikos, M. , Sousa, A. M. , Sedmak, G. , Meyer, K. A. , Zhu, Y. , Cheng, F. , … Sestan, N. (2014). Temporal specification and bilaterality of human neocortical topographic gene expression. Neuron, 81, 321–332. 10.1016/j.neuron.2013.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogarell, O. , Koch, W. , Popperl, G. , Tatsch, K. , Jakob, F. , Zwanzger, P. , … Padberg, F. (2006). Striatal dopamine release after prefrontal repetitive transcranial magnetic stimulation in major depression: Preliminary results of a dynamic [123I] IBZM SPECT study. Journal of Psychiatric Research, 40, 307–314. 10.1016/j.jpsychires.2005.09.001 [DOI] [PubMed] [Google Scholar]
- Power, J. D. , Barnes, K. A. , Snyder, A. Z. , Schlaggar, B. L. , & Petersen, S. E. (2013). Steps toward optimizing motion artifact removal in functional connectivity MRI; a reply to Carp. NeuroImage, 76, 439–441. 10.1016/j.neuroimage.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabaneda, L. G. , Robles‐Lanuza, E. , Nieto‐Gonzalez, J. L. , & Scholl, F. G. (2014). Neurexin dysfunction in adult neurons results in autistic‐like behavior in mice. Cell Reports, 8, 338–346. 10.1016/j.celrep.2014.06.022 [DOI] [PubMed] [Google Scholar]
- Raichle, M. E. , & Mintun, M. A. (2006). Brain work and brain imaging. Annual Review of Neuroscience, 29, 449–476. 10.1146/annurev.neuro.29.051605.112819 [DOI] [PubMed] [Google Scholar]
- Rees, E. , Carrera, N. , Morgan, J. , Hambridge, K. , Escott‐Price, V. , Pocklington, A. J. , … O'Donovan, M. C. (2019). Targeted sequencing of 10,198 samples confirms abnormalities in neuronal activity and implicates voltage‐gated sodium channels in schizophrenia pathogenesis. Biological Psychiatry, 85, 554–562. 10.1016/j.biopsych.2018.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richiardi, J. , Altmann, A. , Milazzo, A. C. , Chang, C. , Chakravarty, M. M. , Banaschewski, T. , … Greicius, M. D. (2015). Brain networks. Correlated gene expression supports synchronous activity in brain networks. Science, 348, 1241–1244. 10.1126/science.1255905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero‐Garcia, R. , Whitaker, K. J. , Vasa, F. , Seidlitz, J. , Shinn, M. , Fonagy, P. , … Vertes, P. E. (2018). Structural covariance networks are coupled to expression of genes enriched in supragranular layers of the human cortex. NeuroImage, 171, 256–267. 10.1016/j.neuroimage.2017.12.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salimi‐Khorshidi, G. , Douaud, G. , Beckmann, C. F. , Glasser, M. F. , Griffanti, L. , & Smith, S. M. (2014). Automatic denoising of functional MRI data: Combining independent component analysis and hierarchical fusion of classifiers. NeuroImage, 90, 449–468. 10.1016/j.neuroimage.2013.11.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah, N. H. , & Aizenman, E. (2014). Voltage‐gated potassium channels at the crossroads of neuronal function, ischemic tolerance, and neurodegeneration. Translational Stroke Research, 5, 38–58. 10.1007/s12975-013-0297-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline, Y. I. , & Raichle, M. E. (2013). Resting state functional connectivity in preclinical Alzheimer's disease. Biological Psychiatry, 74, 340–347. 10.1016/j.biopsych.2012.11.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon, A. M. , de Maturana, R. L. , Ricobaraza, A. , Escribano, L. , Schiapparelli, L. , Cuadrado‐Tejedor, M. , … Frechilla, D. (2009). Early changes in hippocampal Eph receptors precede the onset of memory decline in mouse models of Alzheimer's disease. Journal of Alzheimer's Disease, 17, 773–786. 10.3233/jad-2009-1096 [DOI] [PubMed] [Google Scholar]
- Slifstein, M. , van de Giessen, E. , Van Snellenberg, J. , Thompson, J. L. , Narendran, R. , Gil, R. , … Abi‐Dargham, A. (2015). Deficits in prefrontal cortical and extrastriatal dopamine release in schizophrenia: A positron emission tomographic functional magnetic resonance imaging study. JAMA Psychiatry, 72, 316–324. 10.1001/jamapsychiatry.2014.2414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, S. M. , Beckmann, C. F. , Andersson, J. , Auerbach, E. J. , Bijsterbosch, J. , Douaud, G. , … Consortium, W. U.‐M. H. (2013). Resting‐state fMRI in the Human Connectome Project. NeuroImage, 80, 144–168. 10.1016/j.neuroimage.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, S. M. , Vidaurre, D. , Beckmann, C. F. , Glasser, M. F. , Jenkinson, M. , Miller, K. L. , … Van Essen, D. C. (2013). Functional connectomics from resting‐state fMRI. Trends in Cognitive Sciences, 17, 666–682. 10.1016/j.tics.2013.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger, B. E. , Nica, A. C. , Forrest, M. S. , Dimas, A. , Bird, C. P. , Beazley, C. , … Dermitzakis, E. T. (2007). Population genomics of human gene expression. Nature Genetics, 39, 1217–1224. 10.1038/ng2142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda, S. , Iwata, K. , Shimmura, C. , Kameno, Y. , Anitha, A. , Thanseem, I. , … Mori, N. (2011). Decreased expression of axon‐guidance receptors in the anterior cingulate cortex in autism. Molecular Autism, 2, 14. 10.1186/2040-2392-2-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, T. , Patoine, C. , Abu‐Khalil, A. , Visvader, J. , Sum, E. , Cherry, T. J. , … Walsh, C. A. (2005). Early asymmetry of gene transcription in embryonic human left and right cerebral cortex. Science, 308, 1794–1798. 10.1126/science.1110324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasi, D. , & Volkow, N. D. (2011a). Association between functional connectivity hubs and brain networks. Cerebral Cortex, 21, 2003–2013. 10.1093/cercor/bhq268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasi, D. , & Volkow, N. D. (2011b). Functional connectivity hubs in the human brain. NeuroImage, 57, 908–917. 10.1016/j.neuroimage.2011.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch, N. X. , & Sabatini, B. L. (2012). Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron, 76, 33–50. 10.1016/j.neuron.2012.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Heuvel, M. P. , van Soelen, I. L. , Stam, C. J. , Kahn, R. S. , Boomsma, D. I. , & Hulshoff Pol, H. E. (2013). Genetic control of functional brain network efficiency in children. European Neuropsychopharmacology, 23, 19–23. 10.1016/j.euroneuro.2012.06.007 [DOI] [PubMed] [Google Scholar]
- Van Essen, D. C. , Smith, S. M. , Barch, D. M. , Behrens, T. E. , Yacoub, E. , Ugurbil, K. , & Consortium, W. U.‐M. H. (2013). The WU‐minn human connectome project: An overview. NeuroImage, 80, 62–79. 10.1016/j.neuroimage.2013.05.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Essen, D. C. , Ugurbil, K. , Auerbach, E. , Barch, D. , Behrens, T. E. , Bucholz, R. , … Consortium, W. U.‐M. H. (2012). The human connectome project: A data acquisition perspective. NeuroImage, 62, 2222–2231. 10.1016/j.neuroimage.2012.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verret, L. , Mann, E. O. , Hang, G. B. , Barth, A. M. , Cobos, I. , Ho, K. , … Palop, J. J. (2012). Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell, 149, 708–721. 10.1016/j.cell.2012.02.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertes, P. E. , Rittman, T. , Whitaker, E. J. , Romero‐Garcia, R. , Vasa, F. , Kitzbichler, M. G. , … Bullmore, E. T. (2016). Gene transcription profiles associated with inter‐modular hubs and connection distance in human functional magnetic resonance imaging networks. Philosophical Transactions of the Royal Society of London, Series B, Biological Sciences, 371. 10.1098/rstb.2015.0362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent, J. L. , Patel, G. H. , Fox, M. D. , Snyder, A. Z. , Baker, J. T. , Van Essen, D. C. , … Raichle, M. E. (2007). Intrinsic functional architecture in the anaesthetized monkey brain. Nature, 447, 83–86. 10.1038/nature05758 [DOI] [PubMed] [Google Scholar]
- Wang, J. H. , Zuo, X. N. , Gohel, S. , Milham, M. P. , Biswal, B. B. , & He, Y. (2011). Graph theoretical analysis of functional brain networks: Test‐retest evaluation on short‐ and long‐term resting‐state functional MRI data. PLoS One, 6, e21976. 10.1371/journal.pone.0021976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Xia, M. , Li, K. , Zeng, Y. , Su, Y. , Dai, W. , … Si, T. (2015). The effects of antidepressant treatment on resting‐state functional brain networks in patients with major depressive disorder. Human Brain Mapping, 36, 768–778. 10.1002/hbm.22663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss, L. A. , Escayg, A. , Kearney, J. A. , Trudeau, M. , MacDonald, B. T. , Mori, M. , … Meisler, M. H. (2003). Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Molecular Psychiatry, 8, 186–194. 10.1038/sj.mp.4001241 [DOI] [PubMed] [Google Scholar]
- Xu, X. , Wells, A. B. , O'Brien, D. R. , Nehorai, A. , & Dougherty, J. D. (2014). Cell type‐specific expression analysis to identify putative cellular mechanisms for neurogenetic disorders. The Journal of Neuroscience, 34, 1420–1431. 10.1523/jneurosci.4488-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo, B. T. , Krienen, F. M. , Sepulcre, J. , Sabuncu, M. R. , Lashkari, D. , Hollinshead, M. , … Buckner, R. L. (2011). The organization of the human cerebral cortex estimated by intrinsic functional connectivity. Journal of Neurophysiology, 106, 1125–1165. 10.1152/jn.00338.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo, G. , Holste, D. , Kreiman, G. , & Burge, C. B. (2004). Variation in alternative splicing across human tissues. Genome Biology, 5, R74. 10.1186/gb-2004-5-10-r74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y. , Kaiser, T. , Monteiro, P. , Zhang, X. , Van der Goes, M. S. , Wang, D. , … Feng, G. (2016). Mice with Shank3 Mutations Associated with ASD and Schizophrenia Display Both Shared and Distinct Defects. Neuron, 89, 147–162. 10.1016/j.neuron.2015.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo, X. N. , Ehmke, R. , Mennes, M. , Imperati, D. , Castellanos, F. X. , Sporns, O. , & Milham, M. P. (2012). Network centrality in the human functional connectome. Cerebral Cortex, 22, 1862–1875. 10.1093/cercor/bhr269 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. AHBA donors' demographic information
Table S2. The explained variances of the FCS patterns by the PLSR1‐5 scores
Table S3. Brain region and development specific expression analysis.
Table S4. Brain region specific analysis
Table S5. Cell type specific expression analysis result
Data Availability Statement
The rs‐fMRI data were requested and downloaded from publicly available HCP data (https://db.humanconnectome.org). The human microarray‐based gene expression data were downloaded from AHBA (http://human.brain‐map.org). All codes included in this study are available upon request by contact with the corresponding author.