

Abstract
Background
As a highly heterogeneous disease, lung cancer has a multitude of cellular components and patterns of gene expression which are not dependent on a single mutation or signaling pathway. Thus, using combined drugs to treat lung cancer may be a practical strategy.
Methods
The combined antitumor effects of HS‐10296, a third‐generation EGFR inhibitor targeting EGFR T790M mutation, with the multitargeted tyrosine kinase inhibitor (TKI) famitinib in non‐small cell lung cancer (NSCLC) were evaluated by in vitro methods such as cell proliferation, apoptosis, angiogenesis assays, and in vivo animal efficacy studies.
Results
Famitinib strengthened the effects of HS‐10296 on inhibiting proliferation and inducing apoptosis of NSCLC cells, possibly by synergistic inhibition of AKT and ERK phosphorylation. Meanwhile, HS‐10296 significantly potentiated the effects of famitinib on inhibiting the proliferation and migration of HUVEC, which may be through synergistic inhibition of ERK phosphorylation in HUVEC, suggesting that HS‐10296 may improve the inhibition of angiogenesis by famitinib. Moreover, combination of HS‐10296 and famitinib exerted synergistic antitumor activity in NCI‐H1975 and PC‐9 xenograft models, and this effect may be accomplished by synergistic inhibition of phosphorylation of AKT and ERK and tumor angiogenesis in tumor tissues.
Conclusions
Collectively, our results indicate that HS‐10296 and famitinib exhibit significant synergistic antitumor activity, suggesting that the third‐generation EGFR inhibitor combined with VEGFR inhibitor provides a promising strategy in the treatment of EGFR‐mutant NSCLC.
Keywords: EGFR‐mutant non‐small cell lung cancer, famitinib, HS‐10296, multi‐targeted tyrosine kinase inhibitor, third‐generation EGFR inhibitor
HS‐10296 combined with famitinib exhibits significant synergistic antitumor activity in EGFR‐mutant NSCLC cells. This synergistic effect may be accomplished by synergistic inhibition of phosphorylation of AKT and ERK and tumor angiogenesis in tumor tissues.

INTRODUCTION
Lung cancer has extremely high incidence and mortality rates worldwide 1 and approximately 80% of patients suffer from non‐small cell lung cancer (NSCLC). 2 For EGFR mutated NSCLC, the current standard first‐line treatment is EGFR tyrosine kinase inhibitors (EGFR‐TKIs), including first‐generation EGFR‐TKIs (e.g., gefitinib and erlotinib) and second‐generation EGFR‐TKIs (e.g., afatinib and dacomitinib). 3 While most patients acquire resistance after one to two years, approximately 50%–60% of cases are mediated by an acquired T790M mutation. 4 Therefore, the third‐generation EGFR‐TKIs (e.g., osimertinib) are designed to target the T790M resistance mutation. 5 A phase III study, the FLAURA trial, indicated that osimertinib was superior to gefitinib or erlotinib in untreated NSCLC patients with an activated EGFR mutation regarding progress free‐survival (PFS) (HR, 0.46; 95% CI: 0.37 to 0.57; p < 0.001; 18.9 vs. 10.2 months, respectively). 6 Although osimertinib has shown an excellent therapeutic effect on EGFR T790M mutated NSCLC, the development of acquired resistance to third‐generation EGFR‐TKI is still inevitable and the resistance mechanism is more complex. 4 Accordingly, new therapeutic strategies are urgently required.
HS‐10296 is an oral, potent, highly selective third generation EGFR‐TKI, targeting both EGFR‐sensitizing and T790M resistance mutations. HS‐10296 was approved by the National Medical Products Administration of China (NMPA) in 2020. A phase I study indicated that HS‐10296 resulted in an objective response rate (ORR) of 52%, a disease control rate (DCR) of 92%, and a median PFS of 11.0 months for advanced NSCLC patients, who acquired the T790M mutation. 7
Angiogenesis plays an essential role in tumor growth, proliferation and metastasis, thereby being the potential intervening target for cancer treatment. 8 For NSCLC, angiogenesis inhibitors mainly include monoclonal antibodies that block vascular endothelial growth factor (VEGF) binding and small molecule TKIs that inhibit the downstream VEGFR mediated signaling. 9 Famitinib is a novel oral multitarget RTK inhibitor against vascular endothelial growth factor receptor 2/3 (VEGFR2/3), stem cell factor receptor (C‐Kit), and platelet‐derived growth factor receptor (PDGFR), and has been shown to have promising antitumor activity in a range of solid tumors. 10 A phase I study indicated that famitinib plus docetaxel resulted in a partial response (PR) of 23.5% (4/17), stable disease (SD) of 64.7% (11/17), and median prolonged PFS of 4.35 months in famitinib 20 mg group for NSCLC patients, with lower and more manageable grade 3/4 toxicity events. 11 Preclinical and early clinical data (phase I and II trials) have shown that first‐generation EGFR‐TKIs combined with angiogenesis inhibitors are a potential strategy for the treatment of NSCLC. 12 A phase II clinical study indicated that bevacizumab plus erlotinib resulted in a significantly prolonged PFS in patients with EGFR mutation‐positive NSCLC compared to erlotinib monotherapy, with a median PFS (16.0 months vs 9.7 months, p = 0.0015). 13 Therefore, we aimed to investigate whether the combined therapy of HS‐10296, the third‐generation EGFR‐TKI, with famitinib could further improve the antitumor activity in EGFR‐mutant NSCLC compared to individual monotherapy and explore the possible mechanisms.
It was found that HS‐10296 combined with famitinib exerted significantly synergistic antitumor effects in NSCLC cells both in vitro and in vivo. These synergistic effects were possibly through the synergistic inhibition of AKT and ERK phosphorylation in tumor cells and synergistic inhibition of tumor angiogenesis in tumor tissues. Thus, we believe that the combination of HS‐10296 and famitinib is a promising strategy for the treatment of NSCLC harboring EGFR mutations.
METHODS
Materials
Famitinib Malate was provided by Jiangsu Hengrui Medicine Co., Ltd. HS‐10296 was obtained from Jiangsu Hansoh Pharmaceutical Co., Ltd. Sulforhodamine B (SRB) was purchased from Sigma‐Aldrich. Cell counting kit‐8 (CCK‐8) was purchased from Dalian Meilun Biotechnology Co., Ltd.
Antibodies against EGFR, phospho‐EGFR(Tyr1173), AKT, phospho‐AKT (Ser473), ERK1/2, phosphor‐ERK1/2 (Thr202/Tyr204), PARP, caspase‐3 and GAPDH were purchased from Cell Signaling Technology. Antibody against ERK1/2 was purchased from Santa Cruz Biotechnology. The anti‐CD31 antibody was purchased from Abcam.
Cell culture and treatment
NCI‐H1975 cell line was purchased from the American Type Culture Collection, NCI‐H292 cell line was obtained from the cell bank of the Chinese Academy of Sciences and PC‐9 cell line was provided by Shanghai Allist Pharmaceutical Technology Co., Ltd. Cells were cultured in RPMI‐1640 medium supplemented with 10% (vol/vol) FBS at 37°C in a humidified 5% CO2 atmosphere.
Cell proliferation assay
Cells were seeded in 96‐well plates overnight. After treatment with HS‐10296, famitinib alone or the combination at different concentrations for 72 h, cell proliferation was determined by sulforhodamine B (SRB) assay or CCK8 assay as previously described. 14 Combination drug index (CI) of HS‐10296 plus famitinb was calculated based on the median effect principle using CalcuSyn software. The CI < 1, CI = 1 and CI > 1 indicate the synergistic, additive and antagonistic effect, respectively.
Western blotting
After drug treatment, cells were lysed on ice for 5 min in sodium dodecyl sulfate (SDS) sample buffer. Cell lysates containing equal amounts of protein were applied on a 10% SDS‐PAGE and transferred to polyvinylidene fluoride (PVDF) membranes. Membranes were incubated with primary antibodies overnight at 4°C. After washing with TBST buffer three times, membranes were exposed to appropriate secondary antibodies for 2 h at room temperature. Immunoreactive proteins were visualized by using Western blot imaging Systems (Clinx Science Instruments) with Chemi Capture software.
Transwell migration assay
Cell migration was measured using a two‐chamber transwell system (8 μm pore size). Human umbilical vein endothelial cells (HUVEC) (1 × 105 cells) suspended in 100 μl of low‐serum (1% FBS) 1640‐RPMI medium were seeded in the upper chambers of the transwell. The vehicle, HS‐10296, famitinib alone or combination of HS‐10296 and famitinib were added to the upper chambers. Then, 600 μl of low‐serum 1640‐RPMI medium containing VEGF (50 ng/ml) and EGF (20 ng/ml) was added to the bottom chambers. Then, 12 h later, cells were fixed with 90% anhydrous ethanol for 20 min and stained with 0.5% crystal violet solution for 20 min. After washing with PBS, unmigrated cells in the upper chamber were scraped with a cotton swab. The migrated cells were photographed under an inverted microscope and three random fields were selected to count the number of cells migrated to the lower side.
In vivo study
In vivo efficacy of HS‐10296, famitinib alone or cotreatment was measured by NCI‐H1975 and PC‐9 xenograft nude mice models. Female nude mice (BALB/c‐nude, 6–7‐week‐old) were purchased from Shanghai Lingchang Biotechnology Co. Ltd (Shanghai, China). Human tumor xenografts of NCI‐H1975 and PC‐9 cells were established by subcutaneously inoculating nude mice with cells. When the average tumor volume reached 100–150 mm3, the mice were randomly divided into control or treatment groups. Control groups were given the vehicle alone, and treatment groups received HS‐10296, famitiniib, or HS‐10296 plus famitinib daily by oral gavage. Tumor size and animal bodyweight were measured twice a week and tumor volume was calculated as (length × width2)/2. Tumor tissues were collected 10 h after the last dose. Tumor samples were analyzed by western blotting and immunohistochemistry. Animal experiments were conducted in accordance with the Institutional Animal Care and Use Committee guidelines of the Shanghai Institute of Materia Medica, Chinese Academy of Sciences.
Immunohistochemistry
Immunohistochemistry was conducted as previously described. 15 Images were captured by using Digital Slice Scanner. Microvessel density was obtained by counting the number of vessels in five random fields.
Data analysis
Data were analyzed with GraphPad Prism Version 7 software (GraphPad Software) and the results of repeated experiments were presented as mean ± standard deviation (SD). Half‐maximal inhibitory concentration (IC50) values and dose–response curves were obtained by nonlinear regression analysis. CI values were calculated using CalcuSyn software. A two‐tailed Student's t‐test was used to evaluate differences between groups and p‐value <0.05 was considered statistically significant.
RESULTS
Famitinib potentiated the activity of HS‐10296 on inhibiting cell proliferation and inducing cell apoptosis in NSCLC cells
We first examined the antiproliferation activity of HS‐10296 and famitinib alone on NSCLC cells with different EGFR genotype by SRB assay and the results are shown in Table 1. HS‐10296 strongly inhibited the proliferation of NCI‐H1975 cells (EGFR L858R/T790M mutation) and PC‐9 (EGFR ex19del), with the IC50 of 17.5 nM and 24.0 nM, respectively, while it inhibited the proliferation of NCI‐H292 (wild‐type EGFR) with much weaker activity (IC50 = 443.5 nM), indicating that as a third‐generation EGFR TKI, HS‐10296 mainly targets mutant EGFR. The activities of famitinib on inhibiting the proliferation of these three cells were almost the same, with the IC50 of about 3 μM (Table 1).
TABLE 1.
Antiproliferative effects of famitinib and HS‐10296 against NCI‐H1975, PC‐9 and NCI‐H292 cells
| Compound | IC50 (nM, mean ± SD) | ||
|---|---|---|---|
| NCI‐H1975 | PC‐9 | NCI‐H292 | |
| (L858R/T790M) | (ex19del) | (WT) | |
| Famitinib | 3816.7 ± 537.3 | 3590.0 ± 241.4 | 2956.5 ± 486.5 |
| HS‐10296 | 17.5 ± 3.8 | 24.0 ± 5.1 | 443.5 ± 28.9 |
Cells were treated with different concentrations of drugs for 72 h. Cell survival was analyzed by SRB assay. Data shown represent mean ± SD of three independent experiments.
When combined with HS‐10296, famitinib synergistically potentiated the activity of HS‐10296 on inhibiting the proliferation of four EGFR‐mutant NSCLC cells, with the CI values of 0.58 (NCI‐H1975), 0.53 (PC‐9), 0.57 (NCI‐H3255) and 0.73 (HCC827) (Figure 1(a), Figure S1). Moreover, the combination of HS‐10296 and famtinib induced more cleaved caspases 3 and poly‐(ADP‐ribose) polymerase (PARP), markers of cell apoptosis, than treatment with either drug alone in NCI‐H1975 and PC‐9 cells (Figure 1(b)), further indicating that the combination of HS‐10296 and famtinib shows the synergistic activity on inhibiting cell proliferation and inducing cell apoptosis in NSCLC cells.
FIGURE 1.

HS‐10296 combined with famitinib synergistically inhibits cell proliferation and induces apoptosis in NSCLC cell lines. (a) NCI‐H1975 and PC‐9 cells were seeded in 96 well plates overnight and then treated with different concentrations of HS‐10296 and famitinib alone or in combination. The cell viability was detected by SRB assay after treatment for 72 h (n = 3; error bars denote SD). The combination index (CI) was calculated with CalcuSyn software. (b) NCI‐H1975 and PC‐9 cells were treated with HS‐10296 and famitinib alone, or in combination at the indicated concentrations for 48 h. Total cell lysates were subjected to immunoblotting with anti‐PARP and anti‐caspase‐3 antibodies
Combination of HS‐10296 with famitinib synergistically inhibited EGFR downstream signaling pathway
To better understand the mechanism of synergistic inhibition on cell proliferation, we next measured the combined effect on EGFR and its downstream signaling pathways. As shown in Figure 2 and Figure S2, HS‐10296 alone significantly inhibited the phosphorylation of EGFR and its downstream AKT and ERK. Famitinib alone exhibited no effect; however, it greatly potentiated the inhibition of AKT and ERK phosphorylation by HS‐10296 in both NCI‐H1975 and PC‐9 cells. Considering that inhibition of AKT and ERK activities plays an essential role in the antitumor activity of HS‐10296, these results indicate that combination of HS‐10296 with famitinib synergistically inhibited AKT and ERK phosphorylation, thereby resulting in synergistic inhibition of cell proliferation.
FIGURE 2.
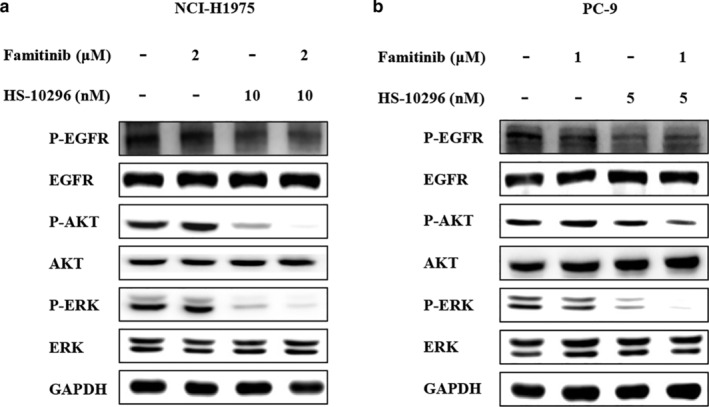
HS‐10296 combined with famitinib synergistically inhibits AKT and ERK phosphorylation in NSCLC cell lines. NCI‐H1975 (a) and PC‐9 (b) cells were treated with HS‐10296 and famitinib alone or in combination at indicated concentrations for 3 h. Total cell lysates were analyzed by Western blotting using the indicated antibodies
HS‐10296 potentiated the activity of famitinib on inhibiting cell proliferation and migration of HUVEC
The main antitumor mechanism of famitinib is inhibiting tumor angiogenesis through inhibition of multi‐target RTK containing VEGFR2 (KDR), C‐kit, PDGFR, 10 thus, to further investigate the combined antitumor effects of HS‐10296 with famitinib, it is necessary to determine the combined effects on tumor angiogenesis. Since HUVEC proliferation and migration in vitro are indicators of tumor angiogenesis, we first determined the combined effects on HUVEC proliferation and migration. Famitinib alone significantly inhibited the proliferation of HUVEC stimulated by VEGF and EGF with an IC50 value of 9.9 nM, while in the presence of 1 or 2 μM HS‐10296 with inhibition activity less than 20%, its IC50 values decreased to 3.2 and 1.7 nM, respectively (Figure 3(a)). We then investigated cell migration of HUVEC by transwell assay. As shown in Figure 3(b), famitinib alone (1 or 10 μM) significantly inhibited VEGF‐ and EGF‐stimulated HUVEC migration, as revealed by the number of cells migrated to the lower side of transwell chamber. In contrast, HS‐10296 only produced mild effects on the migration of HUVEC, but it greatly potentiated the inhibition of HUVEC migration by the combined treatment. Thus, these results collectively demonstrated that the combination of HS‐10296 with famitinib synergistically inhibited HUVEC proliferation and migration, probably resulting in inhibition of tumor angiogenesis. Moreover, these synergistic effects may result from synergistic inhibition of ERK (Figure 3(c)), which is important for HUVEC proliferation and migration, and for tumor angiogenesis. 16 , 17
FIGURE 3.

HS‐10296 combined with famitinib synergistically inhibits HUVEC proliferation and migration stimulated with VEGF and EGF. (a) HUVECs were treated with different concentrations of HS‐10296 and famitinib alone or in combination for 72 h, and cell viability was determined by CCK8 assay (n = 3; error bars denote SD). (b) HUVECs were treated with HS‐10296 (2 μM), famitinib (10 μM or 1 μM), HS‐10296 plus famitinib (2 μM + 10 μM or 2 μM +1 μM), and VEGF (50 ng/ml) and EGF (20 ng/ml) for 12 h. then, HUVEC migration was measured by transwell migration assay. The migrated cells from three random fields of vision (scale bar, 100 μm) were counted and analyzed using a two‐tailed Student's t‐test. **p < 0.01, ***p < 0.001. (c) Serum starved HUVECs were treated with HS‐10296, famitinib alone or in combination at indicated concentrations for 1.5 h. VEGF (50 ng/ml) and EGF (20 ng/ml) was added 5 min before harvest. Cells were harvested and lysed, and total cell lysates were analyzed by Western blotting using the indicated antibodies. Abbreviations: HS: HS‐10296, Fa, famitinib
Combination of HS‐10296 with famitinib exerts synergistic antitumor efficacy in NSCLC xenografts
Given the synergistic effects observed in vitro, we next investigated the antitumor efficacy of combined treatment with HS‐10296 and famitinib in NSCLC in vivo. Nude mice bearing tumor NCI‐H1975 and PC‐9 xenografts were treated with HS‐10296 or famitinib alone or both (Figure 4(a)). HS‐10296 combined with famitinib produced enhanced antitumor efficacy in NCI‐H1975 xenograft model compared with each single treatment (p < 0.05). Moreover, partial tumor regression was observed in four of eight tumors in cotreatment groups (HS‐10296 5 mg/kg + famitinib 10 mg/kg). Similar results were also observed in PC‐9 xenograft model, for HS‐10296 combined with famitinib producing an antitumor efficacy that was significantly superior to single treatment (p < 0.01) and partial tumor regression was observed in three of eight tumors in cotreatment groups (HS‐10296 3 mg/kg + famitinib 10 mg/kg). In addition, the combination of HS‐10296 with famitinib was generally well tolerated and no significant bodyweight loss was observed during the course of the experiment in all groups (Figure 4(a)).
FIGURE 4.

Combined HS‐10296 and famitinib exerts synergistic antitumor efficacy in NSCLC xenografts. (a) NCI‐H1975 and PC‐9 tumor‐bearing mice received vehicle, HS‐10296 and famitinib alone or in combination. Tumor volumes were measured and bodyweights were determined. Tumor volumes and bodyweights are presented as means ± SD. ** p < 0.01 versus the 10 mg/kg famitinib group; ## p < 0.01 versus the 3 mg/kg HS‐10296 group; $ p < 0.05 versus the 5 mg/kg HS‐10296 group; $$ p < 0.01 versus the 1 mg/kg HS‐10296 group. (b) Tumor vascularization was significantly suppressed according to CD31 staining. MVD in the vehicle, HS‐10296 (5 mg/kg), famitinib (10 mg/kg), and HS‐10296 plus famitinib (5 + 10 mg/kg) groups were measured by immunohistochemistry. Scale bar 100 μm. *p < 0.05, **p < 0.01. (c) Mice were sacrificed 10 h after the last treatment with vehicle, 3 mg/kg HS‐10296, 10 mg/kg famitinib, or HS‐10296 plus famitinib, and tumors were removed and analyzed by Western blotting
To better understand the mechanism of synergistic effects in vivo, the tumor xenografts were lysed and the EGFR signaling pathway was analyzed. As a result, the levels of AKT and ERK phosphorylation were remarkably decreased in the coadministration group compared with single treatment groups (Figure 4(c)). Moreover, the results from immunohistochemistry staining demonstrated that CD31 staining in co‐administration group was much weaker than single treatment groups (Figure 4(b)). Knowing that the less CD31 staining means the less microvascular density (MVD), these results suggest that the combination of HS‐10296 with famitinib synergistically inhibits tumor angiogenesis. Taken together, consistent with the results in vitro, the data in vivo indicate that combination of HS‐10296 with famitinib produces synergistic antitumor activity in vivo, probably through synergistic inhibition of AKT and ERK phosphorylation and tumor angiogenesis.
DISCUSSION
As a highly heterogeneous disease, lung cancer has a multitude of cellular components and patterns of gene expression, which are not dependent on a single mutation or signaling pathway. 2 Thus, using combined drugs to treat lung cancer may be a practical strategy. 18 Herein, in this report, the combination of third‐generation EGFR inhibitor HS‐10296 and multi‐targeted RTK inhibitor famitinib exerts synergistic antitumor effects on EGFR‐mutant NSCLC cells both in vitro and in vivo, and this synergistic antitumor effect is accomplished by synergistic inhibition of phosphorylation of AKT and ERK in tumor cells and tumor angiogenesis. This report may provide a promising strategy in the treatment of EGFR‐mutant NSCLC.
A previous study reported that VEGFR2 inhibitor apatinib enhanced antitumor activity of third‐generation EGFR‐TKI osimertinib on T790M positive lung adenocarcinoma, but the exact mechanism of synergistic effects has not been further explored. 3 Our study shows that the synergistic effects result from synergistic inhibition of EGFR downstream pathways in tumor cells and inhibition of tumor angiogenesis, both of which are essential for tumor growth. The combination of VEGF monoclonal antibody bevacizumab with the first‐generation EGFR inhibitor erlotinib has been previously reported to exhibit synergistic antitumor activity in NSCLC tumors with VEGF overexpression. 19 However, bevacizumab failed to potentiate the antiproliferation activity of erlotinib in NSCLC cell lines in vitro. 19 Thus, the mechanism in our study is distinct from that of combination of erlotinib and VEGF monoclonal antibody. Since VEGFR2/KDR and c‐kit were undetectable in the NSCLC cells used in our study, this synergistic inhibition of AKT and ERK phosphorylation may be due to inhibition of another tyrosine kinase by famitinib, which may also play a role in AKT and ERK activation. In this study, not only dual inhibition of EGFR and KDR, but also inhibition of other tyrosine kinases, may have contributed to the synergistic antitumor effects and is therefore worthy of further exploration.
The major consequence of dual inhibition of EGFR and KDR underlying synergistic effects is inhibiting tumor angiogenesis. 20 The pathways of EGFR signaling and VEGFR transduction are tightly interconnected. 21 VEGF is downregulated by EGFR inhibition and overactive VEGF pathway independent of EGFR plays a role in resistance to EGFR TKIs. 19 In this regard, it is easy to understand that dual inhibition of EGFR and VEGFR signal pathways by respective inhibitors benefit each other, since inhibition of EGFR pathway decreases VEGF production and the inhibition of VEGFR inhibits tumor angiogenesis, thus leading to tumor growth inhibition. In addition, we found that without altering VEGF production, HS‐10296 potentiated the activity of famitinib on inhibiting HUVEC proliferation and migration, which may be through synergistic inhibition of ERK phosphorylation. Thus, the impact of EGFR inhibitors on VEGFR signal pathway may be not only on reducing VEGF production in tumor tissues, but also on enhancing the inhibition of KDR inhibitors on VEGFR signal pathway in endothelial cells.
Acquired resistance of the third‐generation EGFR inhibitor is still inevitable, and it is also important to search novel approaches to overcome this resistance. 4 , 22 Considering that famitinib mainly potentiated the antitumor activity of HS‐10296 through inhibiting tumor angiogenesis, regardless of EGFR mutation or gene phenotype in tumor cells, it is reasonable that famitinib combined with HS‐10296 may be an efficient strategy to overcome HS‐10286 resistance. In addition, knowing that VEGF/VEGFR signal pathway is an essential mechanism of resistance to EGFR inhibitor, 23 , 24 the combination approach may also an effective way to prevent drug resistance to EGFR inhibitors.
Although this combination therapy exhibited synergistic effects, the study still has some limitations. Our study highlighted that the synergistic antitumor effect was accomplished by synergistic inhibition of phosphorylation of AKT and ERK in tumor cells and tissues. However, it remains unclear which upstream target associated with AKT and ERK activation is inhibited by famitinib in NSCLC cells in vitro. Considering that famitinib is a multi‐targeted tyrosine kinase inhibitor, other potential targets which contribute to synergistic antitumor effects should be explored further. In addition, it should be noted that this combination therapy is effective in EGFR mutated NSCLC, which has only been examined in preclinical studies. The long‐term effect of the combination of VEGFR inhibitors and third‐generation EGFR inhibitors requires further input.
In summary, we found that the combination of HS‐10296 and famatinib exerted significant synergistic antitumor activity in NSCLC cell lines. Currently, first‐generation EGFR‐TKIs in combination with VEGF/VEGFR pathway inhibition are being investigated in clinical trials. We believe that the combination of VEGFR inhibitors with third‐generation EGFR inhibitors may gain significant benefits in clinical trials and is a novel and effective strategy to treat patients with EGFR‐mutant NSCLC.
CONFLICT OF INTEREST
The authors declare that there are no conflicts of interest.
Supporting information
Appendix S1. Supporting Information
Figure S1. HS‐10296 combined with famitinib synergistically inhibits cell proliferation in EGFR‐mutant NSCLC cell lines. NCI‐H3255 and HCC827 cells were seeded in 96 well plates overnight and then treated with different concentrations of HS‐10296 and famitinib alone or in combination. The cell viability was detected by SRB assay after treatment for 72 h (n = 2; error bars denote SD). The combination index (CI) was calculated by the CalcuSyn software.
Figure S2. HS‐10296 combined with famitinib significantly inhibits AKT and ERK phosphorylation in PC‐9 cell. PC‐9 cell was treated with HS‐10296 and famitinib alone or in combination at indicated concentrations for 3 h. Total cell lysates were analyzed by Western blotting using the indicated antibodies.
ACKNOWLEDGMENTS
This research was supported by grants from the Science and Technology Commission of Shanghai Municipality (№ 18DZ2293200) and the Yunnan Province Sciences and Technology plan (№ 2017ZF010).
Zhang M, Quan H, Fu L, Li Y, Fu H, Lou L. Third‐generation EGFR inhibitor HS‐10296 in combination with famitinib, a multi‐targeted tyrosine kinase inhibitor, exerts synergistic antitumor effects through enhanced inhibition of downstream signaling in EGFR‐mutant non‐small cell lung cancer cells. Thorac Cancer. 2021;12:1210–1218. 10.1111/1759-7714.13902
Funding information The Science and Technology Commission of Shanghai Municipality, Grant/Award Number: № 18DZ2293200; The Yunnan Province Sciences and Technology plan, Grant/Award Number: № 2017ZF010
REFERENCES
- 1. Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Piñeros M, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144(8):1941–53. [DOI] [PubMed] [Google Scholar]
- 2. Testa U, Castelli G, Pelosi E. Lung cancers: molecular characterization, clonal heterogeneity and evolution and cancer stem cells. Cancers (Basel). 2018;10(8):248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu Y, Xiong Z‐C, Sun X, Sun L, Zhang SL, Ma JT, et al. Impact of apatinib in combination with osimertinib on EGFR T790M‐positive lung adenocarcinoma. Transl Cancer Res. 2019;8(5):2151–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nagano T, Tachihara M, Nishimura Y. Mechanism of resistance to epidermal growth factor receptor‐tyrosine kinase inhibitors and a potential treatment strategy. Cell. 2018;7(11):212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M‐mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4(9):1046–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR‐mutated advanced non‐small‐cell lung cancer. N Engl J Med. 2018;378(2):113–25. [DOI] [PubMed] [Google Scholar]
- 7. Chih‐Hsin Yang J, Camidge DR, Yang CT, et al. Safety, efficacy and pharmacokinetics of almonertinib (HS‐10296) in pretreated patients with EGFR‐mutated advanced NSCLC: a multicenter, open‐label phase I trial. J Thorac Oncol. 2020;15(12):1907–1918. [DOI] [PubMed] [Google Scholar]
- 8. Aggarwal C, Somaiah N, Simon G. Antiangiogenic agents in the management of non‐small cell lung cancer: where do we stand now and where are we headed? Cancer Biol Ther. 2012;13(5):247–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hall RD, Le TM, Haggstrom DE, Gentzler RD. Angiogenesis inhibition as a therapeutic strategy in non‐small cell lung cancer (NSCLC). Transl Lung Cancer Res. 2015;4(5):515–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ge S, Zhang Q, He Q, Zou J, Liu X, Li N, et al. Famitinib exerted powerful antitumor activity in human gastric cancer cells and xenografts. Oncol Lett. 2016;12(3):1763–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhou C, Ren S, Su C, Wu F. A phase 1b study of famitinib plus docetaxel as 2nd line setting in patients with metastatic non‐squamous non‐small cell lung cancer and EGFR wild type. J Clin Oncol. 2016;34(15_suppl):e20603. [Google Scholar]
- 12. Chen Z, Wei J, Ma X, Yu J. Efficacy of EGFR‐TKIs with or without angiogenesis inhibitors in advanced non‐small‐cell lung cancer: a systematic review and meta‐analysis. J Cancer. 2020;11(3):686–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seto T, Kato T, Nishio M, Goto K, Atagi S, Hosomi Y, et al. Erlotinib alone or with bevacizumab as first‐line therapy in patients with advanced non‐squamous non‐small‐cell lung cancer harbouring EGFR mutations (JO25567): an open‐label, randomised, multicentre, phase 2 study. Lancet Oncol. 2014;15(11):1236–44. [DOI] [PubMed] [Google Scholar]
- 14. Zhao H, Quan H, Xie C, Xu Y, Xie F, Hu Y, et al. YHHU0895, a novel synthetic small‐molecule microtubule‐destabilizing agent, effectively overcomes P‐glycoprotein‐mediated tumor multidrug resistance. Cancer Lett. 2012;314(1):54–62. [DOI] [PubMed] [Google Scholar]
- 15. Wang L, Xu Y, Fu L, Li Y, Lou L. (5R)‐5‐hydroxytriptolide (LLDT‐8), a novel immunosuppressant in clinical trials, exhibits potent antitumor activity via transcription inhibition. Cancer Lett. 2012;324(1):75–82. [DOI] [PubMed] [Google Scholar]
- 16. Nieminen T, Toivanen PI, Rintanen N, Heikura T, Jauhiainen S, Airenne KJ, et al. The impact of the receptor binding profiles of the vascular endothelial growth factors on their angiogenic features. Biochim Biophys Acta. 2014;1840(1):454–63. [DOI] [PubMed] [Google Scholar]
- 17. Sadremomtaz A, Mansouri K, Alemzadeh G, Safa M, Rastaghi AE, Asghari SM. Dual blockade of VEGFR1 and VEGFR2 by a novel peptide abrogates VEGF‐driven angiogenesis, tumor growth, and metastasis through PI3K/AKT and MAPK/ERK1/2 pathway. Biochim Biophys Acta Gen Subj. 2018;1862(12):2688–700. [DOI] [PubMed] [Google Scholar]
- 18. Naumov GN, Nilsson MB, Cascone T, Briggs A, Straume O, Akslen LA, et al. Combined vascular endothelial growth factor receptor and epidermal growth factor receptor (EGFR) blockade inhibits tumor growth in xenograft models of EGFR inhibitor resistance. Clin Cancer Res. 2009;15(10):3484–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li H, Takayama K, Wang S, Shiraishi Y, Gotanda K, Harada T, et al. Addition of bevacizumab enhances antitumor activity of erlotinib against non‐small cell lung cancer xenografts depending on VEGF expression. Cancer Chemother Pharmacol. 2014;74(6):1297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Masuda C, Yanagisawa M, Yorozu K, Kurasawa M, Furugaki K, Ishikura N, et al. Bevacizumab counteracts VEGF‐dependent resistance to erlotinib in an EGFR‐mutated NSCLC xenograft model. Int J Oncol. 2017;51(2):425–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pennell NA, Lynch TJ Jr. Combined inhibition of the VEGFR and EGFR signaling pathways in the treatment of NSCLC. Oncologist. 2009;14(4):399–411. [DOI] [PubMed] [Google Scholar]
- 22. Chong CR, Jänne PA. The quest to overcome resistance to EGFR‐targeted therapies in cancer. Nat Med. 2013;19(11):1389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Perdrizet K, Leighl NB. The role of angiogenesis inhibitors in the era of immune checkpoint inhibitors and targeted therapy in metastatic non‐small cell lung cancer. Curr Treat Options Oncol. 2019;20(3):21. [DOI] [PubMed] [Google Scholar]
- 24. Seki N, Natsume M, Ochiai R, Haruyama T, Ishihara M, Fukasawa Y, et al. Promising combination therapy with Bevacizumab and Erlotinib in an EGFR‐mutated NSCLC patient with MET amplification who showed intrinsic resistance to initial EGFR‐TKI therapy. Case Rep Oncol. 2019;12(1):91–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information
Figure S1. HS‐10296 combined with famitinib synergistically inhibits cell proliferation in EGFR‐mutant NSCLC cell lines. NCI‐H3255 and HCC827 cells were seeded in 96 well plates overnight and then treated with different concentrations of HS‐10296 and famitinib alone or in combination. The cell viability was detected by SRB assay after treatment for 72 h (n = 2; error bars denote SD). The combination index (CI) was calculated by the CalcuSyn software.
Figure S2. HS‐10296 combined with famitinib significantly inhibits AKT and ERK phosphorylation in PC‐9 cell. PC‐9 cell was treated with HS‐10296 and famitinib alone or in combination at indicated concentrations for 3 h. Total cell lysates were analyzed by Western blotting using the indicated antibodies.