

Abstract
Surface enhanced Raman scattering (SERS) spectra contain information on the chemical structure at nanoparticle surfaces through the position and alignment of molecules with the electromagnetic near field. Time dependent density functional theory (TDDFT) can provide the Raman tensors needed for a detailed interpretation of SERS spectra. Here the impact of molecular conformations on SERS spectra is considered. TDDFT calculations of the surfactant cetyltrimethylammonium bromide with five conformers produced more accurate unenhanced Raman spectra than a simple all-trans structure. The calculations and measurements also demonstrated a loss of structural information in the CH2/CH3 scissor vibration band at 1450 cm−1 in the SERS spectra. To study lipid bilayers, TDDFT calculations on conformers of methyl phosphorylcholine and cis-5-decene served as models for the symmetric choline stretch in the lipid headgroup and the C═C stretch in the acyl chains of 1,2-oleoyl-glycero-3-phosphocholine. Conformer considerations enabled a measurement of the distribution of double bond orientations with an order parameter of SC═C = 0.53.
Keywords: surface enhanced Raman scattering, Raman scattering, lipid membranes, gold nanorods, gold bipyramid, order parameter
Graphical Abstract
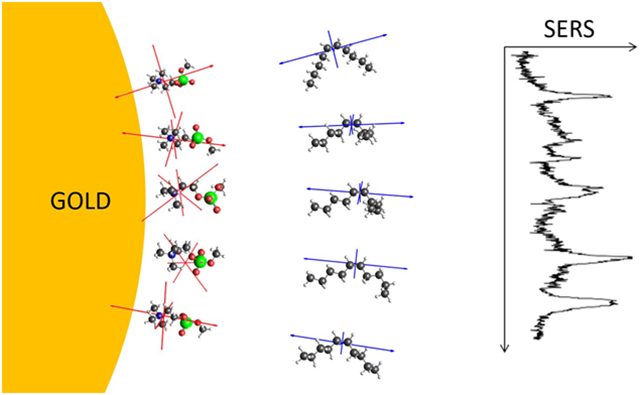
INTRODUCTION
Gold nanostructures concentrate light down to the nanometer scale due to resonant excitations of their free electrons called surface plasmons.1 The concentrated electromagnetic field greatly amplifies Raman scattered light for nearby molecules, referred to as surface enhanced Raman scattering (SERS).2,3 The gold nanostructure essentially acts as an antenna, both focusing light onto the molecule and broadcasting the Raman scattered signal with sufficient gain that vibrational spectra of single molecules can be recorded. Since SERS can detect minute quantities of small molecules based on inherent vibrations, it has been largely pursued as a technological platform for label-free biological and chemical sensing, with a focus on developing plasmonic substrates that provide large enhancements and high sensitivities.4
In addition to sensing, SERS spectra can be used to probe interfacial molecular structure and properties. One strategy is to apply insights from (unenhanced) Raman spectroscopy of molecules in solution to SERS spectra of molecules on surfaces. For example, self assembled monolayers (SAMS) that spontaneously form on gold and silver surfaces have been carefully studied by SERS. For alkanethiols, v(C-S) and v(C-C) peaks in solution indicate a mixture of trans and gauche conformations. SERS spectra for the same alkanethiols on gold show a shifted v(C-S) peak due to binding to the surface, and almost entirely trans conformation near the C-S bond, which indicates an ordered structure. Alkanethiol SAMs on roughened surfaces show more gauche character, indicating increased disorder.5 Studies of thioglycolic acid and 3-mercaptopropionic acid on gold and silver used SERS to detect the protonation state of the carboxyl group, and found it was correlated to molecular order through the trans and gauche character as described above.6,7 These spectral signatures of order in SAMs have also been used to indicate the insertion of molecules, such as glucose and ibuprofen.8,9
A more direct way to get structural information from SERS is to consider the relative intensities of the peaks since they depend on the alignment of each vibration’s Raman tensor with the enhanced field, as well as the vibration’s distance from the surface. This strategy has been applied to small molecules such as benzene on gold where the intensities of the stretching and ring C-H modes indicate the adsorbate orientation.10 The ratios of C-S, C-C, C-H, and pyridine ring vibrations in SAMs have been used to estimate the chain tilt relative to the surface.5,11 DNA strands have been designed with patterns of nucleic acids that encode the DNA surface tilt into the spectrum based on distance from the surface.12 However, these approaches are limited due to difficulty in assigning peaks and the lack of access to Raman tensor components. For small molecules the tensor directions can be deduced through symmetry, but larger, less symmetric molecules are more complicated. Some reports describe difficulties in getting structures from SERS and only use it in concert with other methods.13,14
We recently introduced a double-ratio method that finds molecular orientation and position from pairs of peaks in solution-phase SERS and Raman spectra, which we refer to as structural analysis by enhanced Raman scattering (SABERS).15 The ratios removed all experimental unknowns, and structural assignments were possible because Raman tensors were calculated with time-dependent density functional theory (TDDFT) and the electromagnetic near field was calculated with finite element method simulations (FEM). This combination of calculated polarizability tensors and near fields with spectral data has been used in a similar way to analyze structures of single molecules in tip enhanced Raman scattering (TERS). 16-18 While TERS provides much more structural information as a scanning microscopy, solution phase SERS can be applied to molecules free of a solid substrate.
Using the SABERS algorithm, the tilt of decyltrimethylammonium bromide on gold nanorods was found to be 27°, consistent with the reduced thickness observed for the longer chain cetyltrimethylammonium bromide (CTAB) bilayer from SANS and SAXS measurements.19 The acyl chain double bonds unsaturated lipid bilayers were found to be along the membrane normal and 13 Å from the choline nitrogen. The orientation and position of tryptophan in lipid bilayers was also measured. A second report described the orientation of the membrane probe di-4-ANEPPS in lipid bilayers by the same method.20 The Raman tensors for these results were calculated for simple all-trans hydrocarbon chains, but to achieve more accurate results, different molecular conformations should be considered. For example, in calculations of the Raman spectra of fatty acid methyl esters, the inclusion of multiple conformers improved the match to the experimental spectra in some cases.21-23 Here we consider the effects of different molecular conformations on structural interpretations of SERS spectra from surfactants and lipids on gold nanorods and pyramids.
MATERIALS AND METHODS
Gold Bipyramid and Nanorod Synthesis
Gold bipyramid synthesis followed a seed-mediated, cetyltrimethylammonium bromide (CTAB) directed growth method.24 To produce seeds particles, 10 mL of 0.2 M CTAB solution was heated to 60 °C, combined with 10 mL of 0.25 mM HAuCl4 solution, and allowed to mix under stirring for several minutes resulting in an orange solution. 0.6 mL of chilled (~3 °C) 0.1 M NaBH4 was prepared and added to the solution via a dropper. The solution turned from an orange to a light brown color, which indicates the formation of seed particles, after which the stirring was stopped and the stir bar was removed. The solution was then allowed to sit at 40 °C for five days in the dark, and then stored at room temperature also in the dark. This resulted in seeds with a useful shelf life of at least a month. For the growth solution, 10 mL of 0.1 M solution of CTAB was heated to 60 °C, and 0.3 mL of 10 mM solution of AgNO3 and 0.5 mL of 10 mM HAuCl4 were combined and allowed to heat for 20 minutes. Once the solution was well combined and heated thoroughly to 60 °C, then the solution was acidified by adding 0.2 mL of 0.1 M HCl and then mixed by inversion. This was followed by 80 μL of 100 mM solution of L-ascorbic acid which was mixed by inversion to reduce the HAuCl4. 60 μL of CTAB-capped seed solution was added to the vial and mixed. Then the solution was left on the hot plate covered for approximately 20 minutes to allow the bipyramids to grow.
Large diameter gold nanorods were synthesized following a seed mediated approach with CTAB and sodium oleate (NaOL) developed by Ye et al.25 Here it is scaled down to smaller batches. The growth solution is prepared by adding 50 mL of DI water to a round bottom flask and heating to 50 °C. 1.4 g of CTAB and 247 mg of NaOL powder were added and stirred as the solution cooled to 30 °C. Once the temperature is stabilized at 30 °C, 4.8 mL of 4 mM AgNO3 was added and left undisturbed at 30 °C. 50 mL 1 mM HAuCl4 was then added and medium stirred at 30 °C for 90 minutes. Next, 2.4 mL 3 M HCl is added and stirred slow for 15 minutes. Next, 0.25 mL 64 mM L-ascorbic acid is added and vigorously stirred for 30 seconds. Finally, 2 uL best seed solution is added and stirred for 30 seconds. The seed solutions were prepared separately by preparing four separate vials of 5 mL 0.5 mM HAuCl4 and 5 mL 200 mM CTAB. Since seed synthesis is not highly reproducible, several volumes (0.3, 0.6, 0.9, and 1.2 mL) of 10 mM NaBH4 were added to different vials and vigorously stirred for 120 seconds and then left to rest for 30 minutes. The seed solutions typically range from clear to dark brown. As reported in the original paper, the best seed solution is light brown and it is the one we select. The complete growth solution with seeds added is left to rest undisturbed at 30 °C.
The duration of the ensuing synthesis must be carefully monitored. Monodisperse nanorods will grow up to a point, and then they become heterogenous. To monitor the synthesis, 2 mL of the growth solution is put in cuvette and its absorption spectrum is checked each minute at 30 °C. The growth is stopped once the absorption spectrum shows a high and narrow peak at about 785 nm. To step the reaction, the nanorods are centrifuged three times at 2000 rcf for 20 minutes and resuspended in 10 mM CTAB.
CTAB binding to the gold nanorods occurs through the strong adsorption of Br− to the gold surface, and electrostatic attraction of CTA+ ions to the Br− ions. A SERS peak for the Au-Br− bond at 180 cm−1 was originally detected in studies of CTAB in electrochemical cells, and was further analyzed in one of our previous publications, where we also show evidence for rapid exchange of surfactants on the surface with surfactants in solution.26,27
Lipid Preparation and Nanorod Encapsulation
1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-dioleoyl-sn-glycerol-3-phospho-(1’-rac-glycerol) (DOPG) in chloroform solution were purchased from Avanti Polar Lipids. Both were mixed to obtain a 9:1 molar ratio of DOPC:DOPG then dried in a rotating flask (30 rpm) under gentle Argon flow for several hours. The dried lipid sample was then rehydrated with deionized water to obtain a 10mg/mL total lipid concentration. To make sure the lipids fully detached from the flask wall, the solution was stirred for 30 min and sonicated for 15 min. If needed, additional sonication (no more than 30 min) was used to form SUVs until the solution became clear.
The nanorods are covered with a mixture of zwitterionic DOPC and anionic DOPG lipids. Encapsulation of the rods is possible because the positively charged choline headgroup of DOPC binds electrostatically to the Br− on the gold surface. DOPG has the same structure as DOPC except the positive choline headgroup is replaced by a neutral glycerol group. Lipid-encapsulated nanorods therefore have a net negative surface charge, preventing aggregation. The 9:1 ratio of DOPC:DOPG was previously found to minimize the fraction of DOPG while still preventing aggregation as detected by a large drop in the extinction of the nanorod solution.28
To encapsulate the nanorods in lipids, first the CTAB concentration of nanorod solution was lowered. 1.5 mL of nanorod solution was centrifuged at 1358 rcf for 10 min. The supernatant was removed and replaced by 1.5 mL of 2 mM CTAB. The solution was then vortexed for a few seconds and sonicated for 5 min. Second, the solution was centrifuged again at the same settings and the supernatant was removed and replaced by 0.5 mL of 10 mg/mL lipid solution. Again, the solution was vortexed for a few seconds and sonicated for 5 min. This step was repeated four more times for a total of five resuspensions in lipids.
Raman and SERS Measurements
All Raman and SERS measurements were recorded with a home-built microRaman system. The excitation source was a stabilized diode laser (Ondax RO-785) at 785 nm wavelength and 80 mW power. The beam was further filtered by a volume holographic grating (Ondax VHG), and the power was adjusted with a linear variable neutral density filter. The beam was brought into the imaging system via a dichroic mirror (Semrock LPD02-785RU-25x36x1.1). The beam was focused into the sample by a near-infrared corrected 20x/.45 NA microscope objective (Olympus LCPLN20XIR). The solution-phase samples were held in 1x1 mm glass capillaries with 0.2 mm thick walls (VitroCom, #8100) for optical access. The objective focussed the beam spot into the center of the capillary. Scattered light passed back through the objective, dichroic mirror, and through a blocking filter (Chroma RET792lp). A second near-infrared corrected objective, 5x/0.1 NA (Olympus LMPLN5XIR) focussed the beam onto the entrance slit of the spectrograph (Princeton Instruments IsoPlane SCT 320). The spectrum was recorded on a thermoelectrically cooled, open electrode CCD camera (Princeton Instruments Pixis 256E) at a resolution of 6 cm−1.
We have found solution-phase SERS measurements with static nanoparticle solutions to be unreliable in terms of stability and reproducibility. First, larger nanoparticles (> 100 nm) like those used here settle to the bottom of containers after several hours, altering the SERS signal with time. Even for brief exposure times, we find that optical forces and thermophoresis affect the concentration of nanoparticles in the beam spot in unpredictable ways.29 We have therefore constructed a cell that allows for continuous flow and mixing of the nanoparticle solutions so that long exposures can be recorded. A square 1 mm ID glass capillary was inserted into the conical vial, and connected to a syringe pump at the other end. The pump draws the solution up into the capillary above the optical axis of the objective lens. During the spectral acquisition, the solution was pumped up and down so that the volume exposed to the laser beam was repeatedly mixed against the bottom of the conical vial and replaced. The acquisition was typically 10 – 100 minutes and the pump rate was about 0.5 Hz, so samples were thoroughly mixed throughout the measurement. All SERS measurements presented in this paper were made using this flow system.
Even with continuous mixing, laser-induced heating at the nanorod surface could alter the membrane structure. In single particle trapping experiments, the surface temperature of plasmonic gold nanoparticles has been measured based on lipid phase transitions and on fluorescence polarization anisotropy. Using high numerical aperture objectives (NA = 1.25), temperature increases ranging from 1 to 100 K were observed, depending on excitation power and nanoparticle size.30,31 Our experiments use similar excitation laser power, but a much lower numerical aperture objective (NA = 0.45). Taking our lower irradiance into account, the surface heating is expected to be less than 10 K in our experiments.
Conformer Generation and TDDFT Calculations
Quantum chemical calculations were carried out with the Amsterdam Density Functional program from Software for Chemistry & Materials (ADF2018.105, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com).32-34 An all-trans CTAB structure, including the bromide counterion, was constructed and its geometry was optimized using the Becke-Perdew exchange-correlation potential under the generalized gradient approximation with a third order dispersion correction and Becke-Johnson damping (GGA:BP86-D3(BJ)). The calculation also used a quad zeta basis set with four polarization functions (QZ4P), no frozen core, no relativistic correction, and good numerical quality.35-38 The CTAB molecule was positioned to have the nitrogen atom at the origin and the centroid mass (calculated without the bromide ion) along the z-axis. Once the geometry was optimized an IR calculation was run with the same parameters to find the analytical normal mode frequencies. Finally, a Raman Range calculation was carried out for 1.58 eV excitation (785 nm) with two-point numerical differentiation of the polarizability tensor for a specific range of frequencies.39-44
The ADF program has RDKit functionality built into the software for conformer generation. 600 conformers of CTAB were generated using a 0.2 A RMS filter to remove duplicates. The software was unable to process the quaternary amine so the nitrogen atom was changed to carbon for the purpose of conformer generation. The bromide ion was also left out. The conformers were sorted by energy using a low level force field and the lowest energy structures were selected. For each selected structure, the quaternary amine was restored, the bromide ion was added, and the structure was aligned with the nitrogen at the origin and centroid on the z-axis. The structure was then further optimized by DFT and the Raman tensor calculated as described above for the all-trans CTAB molecule.
DOPC calculations were carried out on two fragments of the full molecule. For the symmetric stretch of the choline headgroup, methyl phosphorylcholine (MePC) was constructed, conformers were generated and optimized. The nitrogen atom was placed at the origin and the phosphorus atom along the z-axis. Raman tensor calculations were carried out as above. For the carbon double bonds of the fatty acid chains, the same procedure was applied to 5-decene with the carbon double bond at the origin and aligned to the z-axis.
Electromagnetic FEM Simulations
The electromagnetic near-field surrounding the gold bipyramids and nanorods were simulated with COMSOL Multiphysics 5.2 using the RF Module. The bipyramid was simulated as two circular cones placed back-to-back. The sharp tips of the cones were truncated and replaced with hemispheres. Nanorods were constructed with a circular cylinder and two hemispheres. Dimensions were set to match average values of TEM images of the bipyramids: the cone base diameter was set to 38 nm to match the bipyramid transverse diameter, and the tip-to-tip length was set to 113 nm to match the bipyramid length. The hemisphere diameter was set to 13 nm to match the bipyramid tip diameter. The nanorod structure was set to match in terms of cylinder diameter and length and tip radius. The dielectric permittivity of gold from Johnson and Christy was used.45 The bipyramid was embedded in a water dielectric and surrounded by a rectangular 190 x 190 x 510 nm perfectly matched layer (PML). An electromagnetic plane wave was incident and polarized along the bipyramid’s length. The simulated extinction curve was generated and compared to the experimental extinction spectrum. For a precise match, the bipyramid was lengthened to 114 nm. Using this optimized geometry, field values were generated throughout the volume at the excitation wavelength (785 nm) and red shifted Raman frequencies. The electric field versus distance from the bipyramid end at each frequency was exported for use in SABERS calculations. A similar procedure was used to generate the field at the tips of gold nanorods.
SABERS Calculation
SABERS is based on a double ratio of SERS and Raman peaks that eliminates experimental unknowns such as instrument sensitivities and numbers of molecules sampled. For each peak, 1 and 2, the SERS intensity is divided by the Raman intensity, and then those values are divided to calculate the combined experimental ratio R1-2. See the Supporting Information for a derivation that shows that R1-2 can be calculated with the electromagnetic near field from FEM calculations and Raman tensors for each vibrational mode from TDDFT. For peaks each composed of a single vibrational mode, for a single molecular position, and for a single conformational structure, the combined ratio is:
| (1) |
where S1, U1, S2, and U2 are the calculated SERS and unenhanced Raman signals for modes 1 and 2. The ratio is calculated for all molecular polar and roll angles. Note that the expression we use for the unenhanced Raman value (U) is invariant with respect to orientation, so it does not depend on polar or roll angle. See the Supporting Information for details.
To consider the effects of having (1) multiple vibrational modes that contribute to an experimental peak, (2) multiple molecular positions in the structure (such as upper and lower leaflet), and (3) multiple molecular conformations, R1-2 is calculated with sums of the Raman and SERS signals:
| (2) |
where the indices p, c, and m represent the positions, conformers, and vibration modes, respectively. Note that each peak, 1 and 2, has a distinct set of indices. The resulting maps of R1-2 versus the polar and roll angle are compared to the experimental value to find which molecular orientations are consistent with the measurements.
For CTAB, five conformers are included each in two positions (upper and lower leaflet). One calculated vibrational mode is used for the 760 cm−1 peak and 24 modes are used for the 1450 cm−1 peak. The R1-2 value is calculated for each possible polar and roll angle of the molecule to create the maps shown below. Note that here the “position” index does not indicate the various polar and roll angles calculated, it refers to the upper and lower leaflets. We follow the same procedure for DOPC, with one mode assigned to the 1650 cm−1 carbon double bond stretch, and one mode for the 720 cm−1 symmetric headgroup stretch.
As described below, for the DOPC result we also considered a distribution of polar angles to represent disorder. R1-2 therefore depends on the distribution’s mean polar angle θ0 and standard deviation σ. For each polar angle θo, we sum the signal for all roll angles since our double bond fragment is axially symmetric. We sum all of the modes, conformers, and positions as described above. Finally, we sum for the distribution of polar angles θ with a normalized solid angle gaussian weighting factor:
| (3) |
where
| (4) |
The resulting maps of R1-2 versus the polar angle and standard deviation of the polar angle distribution are compared to the experimental value.
RESULTS AND DISCUSSION
Raman and SERS Intensities
It seems reasonable to assume that any spectrum recorded from resonant plasmonic structures will be SERS spectrum. Although unenhanced Raman signals may contribute, SERS should be orders of magnitude stronger given the large enhancements at electromagnetic hotspots.46-48 This assumption likely holds for SERS substrates, but for solution phase SERS from colloidal suspensions of nanoparticles, it may not. The enhancements at the open ends of nanoparticles are several orders of magnitude lower than hotspots found on substrates.27 Also, if the molecules that stabilize nanoparticle suspensions remain in the solution at mM concentrations, their large numbers counteract the enhancement effect, and the Raman signal may be comparable or even larger than the SERS signal.
Since SABERS relies on accurate Raman and SERS intensities, we distinguished the two signals by recording spectra at decreasing nanoparticle concentration, holding all other parameters constant. Using the open flow system described above, we took spectra at an initial nanoparticle concentration in 10 mM CTAB or DOPC solution and then diluted the nanoparticles with the same solution to 80%, 60%, 40%, and 20% of the original nanoparticle concentration. As displayed in Figure 1B and 1D, the resulting spectral peak intensities create a line with a slope that is proportional to the SERS intensity and a vertical intercept that is proportional to the unenhanced Raman intensity. Note that the absorption correction described in the Supporting Information is required to get an accurate line. If uncorrected for absorption, the intensities are underestimated at high nanorod concentration. The slopes of the lines in Figure 1B and 1D were used for SERS intensities of the relevant peaks for CTAB and DOPC. Although the Raman signals could be taken from the vertical intercept, dilution curves were also created for pure CTAB and DOPC solutions (diluted with water) for a more accurate measurement of the Raman intensity (displayed in Figure 1A and 1C).
Figure 1.
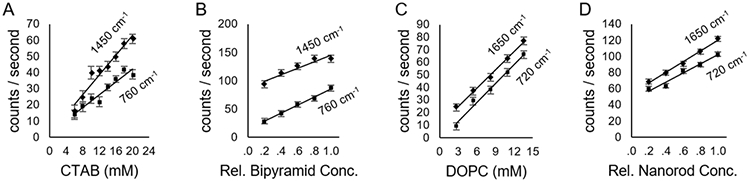
Raman and SERS intensities. Peak intensity dilution curves for (A) CTAB Raman spectra, (B) CTAB SERS spectra, (C) DOPC Raman spectra, and (D) DOPC SERS spectra. The peaks are identified on the plot and discussed in the main text. The slopes of these lines are used as SERS and Raman intensities.
CTAB Conformers
In our previous structural analysis of the surfactant layer, we compared the CH2/CH3 scissor mode of the hydrocarbon chain and the symmetric stretch of the N+(CH3)3 headgroup.15 The Raman tensors were calculated by TDDFT, yielding a Raman spectrum that closely matched the experimental spectrum. The symmetric stretch and scissor mode tensors were identified and used in the ratiometric SABERS method, which concluded that the surfactant formed a tilted bilayer. This result was in agreement with SANS and SAXS studies which found a thinner layer than expected but interpreted it as an effect of interdigitation.19 More recently, the surfactant layer on gold nanoparticles has been simulated with molecular dynamics.49-53 The simulations found that CTAB adsorbs as bilayers under some conditions and adsorbs as surfactant micelles under others. However, in either case, it is clear that the surfactant chains undergo rapid conformational fluctuations whether in solution or adsorbed to the gold surface. Since conformation should affect the identity and alignment of the vibrational modes and Raman tensors, we have studied its effect on structural analysis of SERS spectra.
Conformers of the CTAB molecule were generated with RDKit, software that applies torsion angles to rotatable bonds based on statistics from structural databases, and then carries out a low level structural refinement.54 The resulting lowest energy conformers were further structurally optimized by TDDFT, and their Raman tensors were calculated. Figure 2A-E displays the structures and eigenvectors of the Raman tensors that correspond to the CH2/CH3 scissoring band at 1450 cm−1. Figure 3 compares the calculated Raman spectrum of CTAB for a straight hydrocarbon chain to the average of the four conformers from Figure 2 B-E, and to the experimental Raman and SERS spectra. The spectral region assignments described here (III - VII) follow a recent review of the Raman spectra of lipids and other biomembrane components.55
Figure 2.

CTAB conformer structures and Raman tensors. The left structure (A) is a straight hydrocarbon chain, and the four on the right (B-E) are conformers generated by RDKit and further optimized by DFT. Eigenvectors of the 24 Raman tensors that correspond to the broad band at 1450 cm−1 are displayed.
Figure 3.
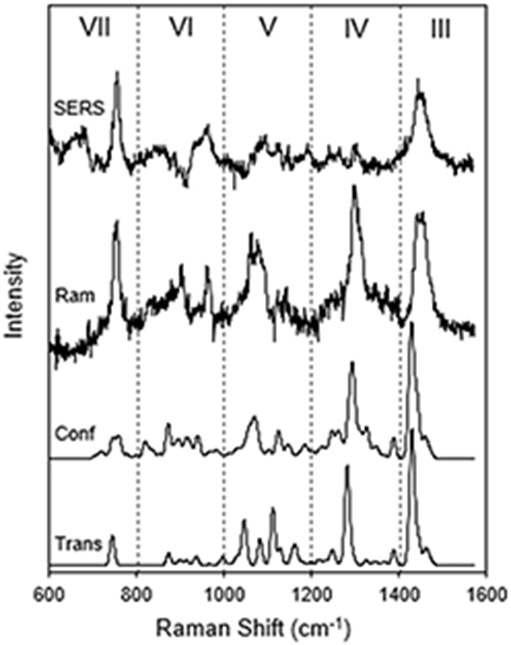
The experimental and calculated Raman and SERS spectra of CTAB. The experimental spectra (top) were recorded from a 10 mM CTAB micelle solution with (SERS) and without (Ram) gold bipyramids. The TDDFT calculated spectra for a straight hydrocarbon chain (Trans) and an average of four conformers (Conf) are shown. Peaks in the five spectral regions correspond to the symmetric N+(CH3)3 stretch (VII), skeletal C-C modes (V), CH2 twist modes (IV), and CH2 and CH3 scissor modes (III).
Region III
The broad band at 1450 cm−1 is typically assigned to CH2 and CH3 scissor modes of a hydrocarbon chain. The TDDFT analysis finds that 24 normal mode vibrations make up the band for CTAB. Most of them are scissor-like vibrations of CH2 groups on the chain, with a few localized on the CH3 groups at the ends of the CTAB molecule. Although slightly red shifted and more narrow, the calculated peaks match the experimental Raman peak well. The conformer-averaged spectrum has a larger width than the straight hydrocarbon chain, so it is a moderately improved match to the experimental peak.
Region IV
The band at 1300 cm−1 is due to CH2 twisting deformations. TDDFT analysis reveals 9 normal modes that are composed almost entirely of CH2 twisting motion along the hydrocarbon chain. There is minimal motion of the terminal CH3 groups. There are some CH2 wag modes, but they are calculated to have negligible amplitude. The conformer-averaged band is slightly broadened and blue-shifted relative to the straight chain calculation, and it more accurately captures the broadening at the base of the experimental Raman peak due to weak modes, making it a better match.
Region V
This region has contributions from the skeletal C-C stretching modes of the hydrocarbon chain. It is highly sensitive to conformer structure and can be used to detect the phase of lipid bilayer membranes. For the TDDFT calculations on CTAB, the averaging of only four conformers makes a remarkable improvement in matching the experimental Raman peak (relative to the calculation for the straight hydrocarbon chain).
Region VI
This region contains more skeletal C-C modes, in addition to the asymmetric N+(CH3)3 stretch vibrations. It is not highly dependent on conformation and has some potentially assignable peaks that match the experimental Raman spectrum. The straight chain and conformer averaged spectra are very similar.
Region VII
The peak at 760 cm−1 in this region is the symmetric N+(CH3)3 headgroup stretch. The calculated results are somewhat surprising. The experimental peak is sharp as though it is due to a single, well defined mode independent of conformation. The straight chain calculation identifies such a single mode at the correct Raman shift. However, the conformers split the symmetric N+(CH3)3 stretch into multiple modes that include vibrations of different parts of the hydrocarbon chain. These modes occur at varied Raman shifts, resulting in a broadened peak. The calculated variations among the conformers therefore do not appear to correspond to reality given the sharp experimental peak. These variations could stem from the TDDFT calculation overestimating the coupling between the trimethylammonium headgroup and the hydrocarbon chain.
The above results show that considering conformers in TDDFT Raman calculations creates moderate improvements in some bands (regions III and IV), significantly clarifies the interpretation of some bands (region V), and can even lower the accuracy in some cases (region VII). However, the effects of conformers on the Raman spectra and on structural information in SERS are not necessarily correlated. The calculated Raman spectra in Figure 3 used an expression that averages all molecular orientations relative to the incoming electric field direction (polarization).56 This matches the Raman experiment in which scattering comes from solution-phase CTAB micelles with no preferred molecular orientation. However, in solution-phase SERS there is an orientational dependence. Although solution-phase nanorods and bipyramids take random orientations with respect to the laser polarization, the enhanced near field remains approximately normal to the nanoparticle surface, and the SERS signal depends on the alignment between the molecule and that near field. For example, averaging conformers only slightly improves the CH2/CH3 scissor mode spectrum at 1450 cm−1, but the effect on structural information is significant if one considers that the SERS signal is a dot product between a vertical electric field and the tensors in Figure 2A-E. The straight chain has large tensor components along the vertical direction, while the conformers have much less alignment since they take more random directions, guided by the hydrocarbon chain. Also consider the symmetric N+(CH3)3 stretch at 760 cm−1. Although the conformers artificially shift the peak to degrade the match to the experimental Raman spectrum, the mode is highly symmetric so the effect on the SERS signal is minimal.
For a structural analysis of the CTAB layer, consider the SERS spectrum in Figure 3. Two peaks were compared: the symmetric N+(CH3)3 stretch at 760 cm−1 and the CH2/CH3 scissoring band at 1450 cm−1. In SABERS we create a ratio between the SERS and Raman intensities of each of these peaks, and then compare it with a calculated ratio at various molecular angles and positions. The experimental value is R1450-760 = 0.5 +/− 0.3 based on the slopes in Figure 1 A-D. Calculated values of R1450-760 included sums over positions, conformers, and modes as described in the Methods section. Two positions were summed: the upper leaflet and the lower leaflet of a putative bilayer. 24 modes were used for the 1450 cm−1 peak and 1 for the HG peak. To create maps for comparison, R1450-760 was calculated for various polar angles (relative to the membrane normal) and roll angles (around the long axis of the molecule). To generate the maps in Figure 4A-E, the Raman tensors for the CH2/CH3 scissor modes were rotated along the polar and roll angles, while the tensor for the symmetric N+(CH3)3 stretch was averaged over all angles since the modes are highly symmetric. Maps were calculated for the straight hydrocarbon (Figure 4A) chain and the four conformers (Figure 4 B-E).
Figure 4.

SABERS ratio maps for the symmetric N+(CH3)3 peak and CH2/CH3 scissor band of CTAB. The left map (A) is calculated for a straight hydrocarbon chain. The right maps (B-E) are calculated for the conformers displayed in Figure 2 B-E, respectively. The same polar and roll axis scales and R color scale apply to all maps.
The maps in Figure 4 A-E reveal that using a conformer average smooths the map to remove much of the potential for structural analysis. The experimental value matches the straight hydrocarbon chain map for a range of polar angles between 30 and 60 degrees, depending on the roll angle. The conformer-averaged map has much less variation, and the experimental value matches at almost all polar and roll angles. This illustrates the loss of structural information in the Raman tensors due to conformational variation. Note that while the conformer-averaged map provides no resolution on the CTAB orientation, the values match the experimental value within the uncertainty, indicating that our measurements and analysis are accurate.
Lipid Order Parameter
The CTAB layer on gold nanoparticles can be displaced by phosphocholine lipids due to the similar chemical structure and electrostatics of their headgroups.28,57,57-65 Several lines of evidence suggest that the lipids form a bilayer structure that is electrostatically bound to the gold nanoparticle surface. Our previous SABERS analysis between vibrations of the headgroup choline and the fatty acid double bond was consistent with a bilayer structure with respect to orientation and spacing.15 Here we present new data and consider variations in the double bond angle due to conformational flexibility.
Experimental Raman and SERS spectra were recorded with 9:1 mixtures of DOPC and DOPG, the latter added to provide sufficient surface charge for colloidal stability. Both lipid species are unsaturated with a double bond in each acyl chain. Figure 5 presents lipid Raman and SERS spectra that contributed to Figure 1C and 1D. Here we analyze the symmetric N+(CH3)3 stretch of the choline group at 720 cm−1 and the C═C stretch at 1650 cm−1. DOPC is a rather large molecule for a TDDFT calculation and, furthermore, running the full molecule is unnecessary considering the local nature of the two vibrations of interest. Instead, MePC and 5-decene were used as models of the lipid headgroup and acyl chain double bond. Region VII of Figure 5 displays a five-conformer MePC average spectrum. All conformers have a well-defined symmetric N+(CH3)3 stretch near 720 cm−1, matching the experimental spectra. The other nearby calculated peaks are MePC vibrations that are not relevant to the lipid spectrum. Region II contains the C═C stretch of the acyl chains. The calculated 5-decene spectrum only contains the C═C stretch in this region, and a five-conformer average is presented.
Figure 5.
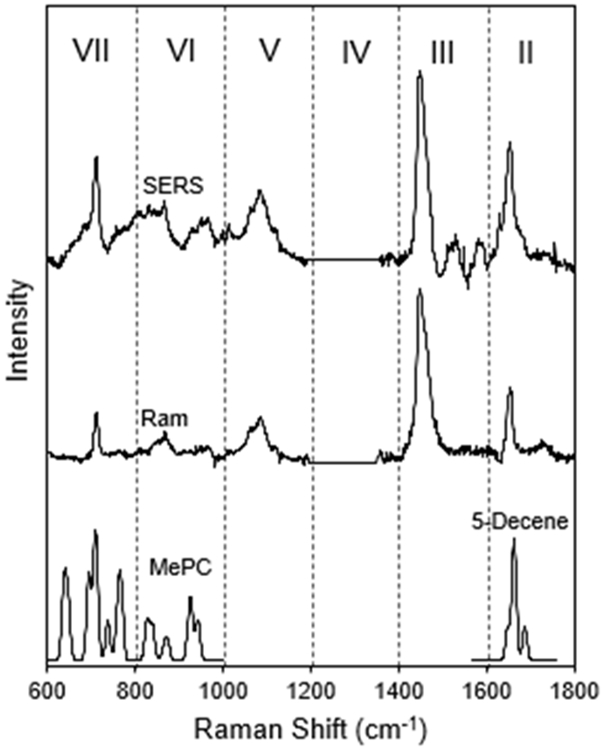
The experimental and calculated Raman and SERS spectra of phospholipids. The experimental spectra (top) were recorded from a 10 mg/mL small unilamellar vesicle solution with (SERS) and without (Ram) gold bipyramids. The TDDFT calculated spectra for MePC and 5-decene are also displayed. Region VII contains the symmetric N+(CH3)3 stretch and Region II contains the C═C stretch modes
The TDDFT calculations on lipid fragments were not intended to produce a close match to the experimental Raman spectrum as was the case for CTAB. For the lipid analysis presented here, they provide Raman tensors for two isolated vibrational peaks, and how those tensors are affected by local changes in conformation. Figure 6 displays the conformer structures and Raman tensor eigenvectors that correspond to the symmetric N+(CH3)3 stretch (MePC) and C═C stretch (5-decene). For MePC, the nitrogen and phosphate atoms were aligned with the z-axis (membrane normal). Although this does not represent the actual lipid headgroup structure, the MePC orientation is largely irrelevant in our analysis since the N+(CH3)3 stretch is highly symmetric. The MePC headgroup mode is therefore used as a reference value in SABERS. For 5-decene, the double bond was aligned with the z-axis. Figure 6 illustrates the small effect of conformational fluctuations of nearby atoms on the Raman tensor for the C═C stretch at 1650 cm−1.
Figure 6.
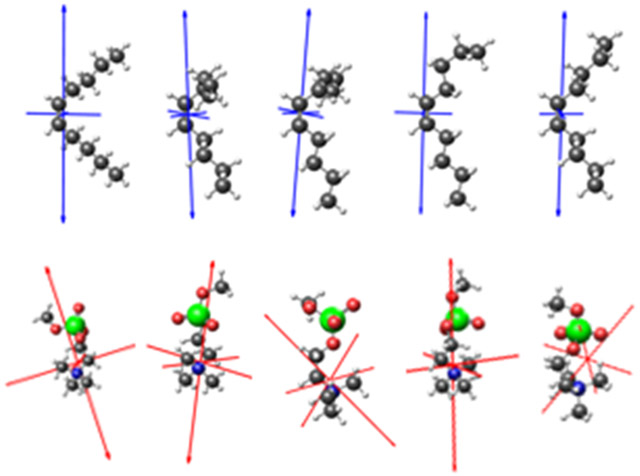
Lipid fragment conformer structures and Raman tensors. 5-decene structures are shown on the top row with the eigenvectors of the Raman tensor for the C═C stretching mode in blue. MePC structures are on the bottom row with eigenvectors for the Raman tensor for the symmetric stretch in red. These conformers were averaged to produce the calculated spectra in Figure 5.
The experimental ratio from the data in Figure 1 A-D for the two lipid modes is R1650-720 = 0.9 +/− 0.3. The calculated map for different orientations of the C═C bond is shown in Figure 7A. It includes the 5 conformers in Figure 6, each in two positions (upper and lower leaflet). Each peak was represented by a single vibrational mode rather than a band of modes as for the CH2/CH3 scissor band of CTAB at 1450 cm−1. According to the map, a vertically oriented double bond would result in R = 1.5, and the value decreases as the polar angle increases. This matches expectations from Figure 6, where an increased polar angle would result in a smaller dot product between the C═C tensors and the membrane normal. The best match between the calculated map and experimental value occurs where the double bonds have a polar angle of 40°. Such a highly tilted acyl chain suggests an altered bilayer structure on the gold nanoparticle surface or perhaps inaccurate tensors from TDDFT. However, a third possibility is that disorder in the acyl chains affects the measurement.
Figure 7.

Calculated maps of the SERS and Raman intensity ratio, R, for the symmetric N+(CH3)3 stretch at 720 cm−1 and the C═C stretch at 1650 cm−1. A) A plot of the ratio for different polar and roll angles of the C═C bond. B) A plot of the ratio for the different polar angles (θ) and normal distribution width (σ). C) The polar angle distribution of C═C bonds.
The structures in Figure 6 only probe the effect of local conformational variation on the direction of the Raman tensor relative to the C═C bond, but fluctuations of the entire lipid molecule create a distribution of C═C bond orientations. We therefore generated a map where each point represents a normal distribution of polar angles with the axes set to the average polar angle (θ) and standard deviation (σ). This distribution map is displayed in Figure 7B. For an average C═C bond direction normal to the membrane (θ = 0°), the experimental ratio matches the map for a distribution with a standard deviation of 27°. The resulting distribution of C═C polar angles is plotted in Figure 7C.
Membrane structure is often characterized with order parameters of specific atoms and bonds relative to the membrane normal. Here an order parameter was calculated for the C═C bond using the distribution in Figure 7C according to the the second-order Legendre polynomial:
| (5) |
which yields SC═C = 0.53. Unfortunately, there are no published values of this order parameter for a direct comparison. Deuterium NMR can measure the order parameters of C-D bonds (SCD), which are related to the segmented order parameter (Smol), at specific locations along the acyl chains. For a saturated lipid (dipalmitoylphosphatidylcholine, DPPC) Smol is found to decrease along the chain with values ranging from 0.47 to 0.21.66,67 For unsaturated chains (1-palmitoyl-2-oleoyl-glycero-3-phosphocholine, POPC) there is a significant dip in Smol near the double bond.68 However, measurements there are influenced by the bond geometry as well as disorder, so comparison to our value of SC═C above may not be meaningful.67
Molecular dynamics (MD) simulations provide a direct view of the trajectories of all atoms in a lipid membrane. MD acts as a computational microscope that has yielded significant insights into membrane phases and transitions, membrane diffusion, protein crowding, small molecule permeation, and other aspects of complex biomembranes.69,70 To validate MD algorithms and sampling methods, order parameters can be extracted from the results and compared to NMR measurements. MD simulations of unsaturated lipid membranes find similar order parameter values to NMR, but the C═C bond angle distribution in particular has not been described.71-74 In one case, the average chain orientation for a DOPC bilayer was extracted and found to be 34°, in reasonable agreement with the distribution in Figure 7C.73 MD could act as a source of more realistic conformers than the random generation used for this study. Ultimately, ab initio molecular dynamics (AIMD) will yield more realistic Raman tensors, but is still under development for large molecules like surfactants and lipids.75-77
CONCLUSION
Both Raman and SERS spectra contain structural information, but it can be difficult to extract without a precise knowledge of the connection between spectral peaks and vibrational motions. TDDFT can calculate Raman spectra of large molecules, but the spectra depend on the detailed conformational structure. Here we show that averaging TDDFT results for several randomly generated conformers of CTAB significantly improves the match to the experimental Raman spectrum. This leads to better identification of spectral peaks for further analysis. However, for SERS spectra, where the signal depends on the orientation of the Raman tensor, conformational variations can lead to a loss of structural information, demonstrated here for a vibration band in CTAB. For lipids, conformational variation in nearby atoms had a minor impact on the mode tensors investigated, but variations in bond angles due to fluctuations of the entire molecule could be characterized. These results show that conformational variations can have a significant impact on SERS spectra and should be considered for detailed interpretations.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge the Welch Foundation (grant C-1761), the National Science Foundation (award number 1709084), and the National Institutes of Health grant 1SC2GM118273-01A1 from the National Institute of General Medical Sciences.
Footnotes
Supporting Information
The supporting information section contains a detailed description of the spectral data processing, including the background subtraction and correction for absorption by the sample. It also has a derivation of the SABERS ratio and how it depends only on Raman tensors and electromagnetic field intensities. Finally, it contains TEM images of the gold bipyramids and nanorods.
REFERENCES
- (1).Schuller JA; Barnard ES; Cai W; Jun YC; White JS; Brongersma ML Plasmonics for Extreme Light Concentration and Manipulation. Nat. Mater 2010, 9 (3), 193–204. 10.1038/nmat2630. [DOI] [PubMed] [Google Scholar]
- (2).Pérez-Jiménez AI; Lyu D; Lu Z; Liu G; Ren B Surface-Enhanced Raman Spectroscopy: Benefits, Trade-Offs and Future Developments. Chem. Sci 2020, 11 (18), 4563–4577. 10.1039/D0SC00809E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bell SEJ; Charron G; Cortés E; Kneipp J; Chapelle M. L. de la; Langer J; Procházka M; Tran V; Schlücker S Towards Reliable and Quantitative Surface-Enhanced Raman Scattering (SERS): From Key Parameters to Good Analytical Practice. Angew. Chem. Int. Ed 2020, 59 (14), 5454–5462. 10.1002/anie.201908154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Golightly RS; Doering WE; Natan MJ Surface-Enhanced Raman Spectroscopy and Homeland Security: A Perfect Match? ACS Nano 2009, 3 (10), 2859–2869. 10.1021/nn9013593. [DOI] [PubMed] [Google Scholar]
- (5).Bryant M; Pemberton J Surface Raman-Scattering of Self-Assembled Monolayers Formed from 1-Alkanethiols - Behavior of Films at Au and Comparison to Films at Ag. J. Am. Chem. Soc 1991, 113 (22), 8284–8293. 10.1021/ja00022a014. [DOI] [Google Scholar]
- (6).Meuse CW; Niaura G; Lewis ML; Plant AL Assessing the Molecular Structure of Alkanethiol Monolayers in Hybrid Bilayer Membranes with Vibrational Spectroscopies. Langmuir 1998, 14 (7), 1604–1611. 10.1021/la9700679. [DOI] [Google Scholar]
- (7).Królikowska A; Kudelski A; Michota A; Bukowska J SERS Studies on the Structure of Thioglycolic Acid Monolayers on Silver and Gold. Surf. Sci 2003, 532–535, 227–232. 10.1016/S0039-6028(03)00094-3. [DOI] [Google Scholar]
- (8).Levin CS; Kundu J; Janesko BG; Scuseria GE; Raphael RM; Halas NJ Interactions of Ibuprofen with Hybrid Lipid Bilayers Probed by Complementary Surface-Enhanced Vibrational Spectroscopies. J. Phys. Chem. B 2008, 112 (45), 14168–14175. 10.1021/jp804374e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Rycenga M; McLellan JM; Xia Y A SERS Study of the Molecular Structure of Alkanethiol Monolayers on Ag Nanocubes in the Presence of Aqueous Glucose. Chem. Phys. Lett 2008, 463 (1), 166–171. 10.1016/j.cplett.2008.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Gao Xiaoping.; Davies JP; Weaver MJ Test of Surface Selection Rules for Surface-Enhanced Raman Scattering: The Orientation of Adsorbed Benzene and Monosubstituted Benzenes on Gold. J. Phys. Chem 1990, 94 (17), 6858–6864. 10.1021/j100380a059. [DOI] [Google Scholar]
- (11).Matulaitienė I; Kuodis Z; Matijoška A; Eicher-Lorka O; Niaura G SERS of the Positive Charge Bearing Pyridinium Ring Terminated Self-Assembled Monolayers: Structure and Bonding Spectral Markers. J. Phys. Chem. C 2015, 119 (47), 26481–26492. 10.1021/acs.jpcc.5b07687. [DOI] [Google Scholar]
- (12).Barhoumi A; Zhang D; Halas NJ Correlation of Molecular Orientation and Packing Density in a DsDNA Self-Assembled Monolayer Observable with Surface-Enhanced Raman Spectroscopy. J. Am. Chem. Soc 2008, 130 (43), 14040–14041. 10.1021/ja804367c. [DOI] [PubMed] [Google Scholar]
- (13).Caldwell W; Campbell D; Chen K; Herr B; Mirkin C; Malik A; Durbin M; Dutta P; Huang K A Highly Ordered Self-Assembled Monolayer Film of an Azobenzenealkanethiol on Au(111) - Electrochemical Properties and Structural Characterization by Synchrotron in-Plane X-Ray-Diffraction, Atomic-Force Microscopy, and Surface-Enhanced Raman-Spectroscopy. J. Am. Chem. Soc 1995, 117 (22), 6071–6082. 10.1021/ja00127a021. [DOI] [Google Scholar]
- (14).Seballos L; Olson TY; Zhang JZ Effects of Chromophore Orientation and Molecule Conformation on Surface-Enhanced Raman Scattering Studied with Alkanoic Acids and Colloidal Silver Nanoparticles. J. Chem. Phys 2006, 125 (23), 234706. 10.1063/1.2404648. [DOI] [PubMed] [Google Scholar]
- (15).Matthews JR; Shirazinejad CR; Isakson GA; Demers SME; Hafner JH Structural Analysis by Enhanced Raman Scattering. Nano Lett. 2017, 17 (4), 2172–2177. 10.1021/acs.nanolett.6b04509. [DOI] [PubMed] [Google Scholar]
- (16).Lee J; Tallarida N; Rios L; Ara Apkarian V The Raman Spectrum of a Single Molecule on an Electrochemically Etched Silver Tip. Appl. Spectrosc 2020, 74 (11), 1414–1422. 10.1177/0003702820949274. [DOI] [PubMed] [Google Scholar]
- (17).El-Khoury PZ; Aprà E Spatially Resolved Mapping of Three-Dimensional Molecular Orientations with ~2 Nm Spatial Resolution through Tip-Enhanced Raman Scattering. J. Phys. Chem. C 2020, 124 (31), 17211–17217. 10.1021/acs.jpcc.0c04263. [DOI] [Google Scholar]
- (18).Liu P; Chen X; Ye H; Jensen L Resolving Molecular Structures with High-Resolution Tip-Enhanced Raman Scattering Images. ACS Nano 2019, 13 (8), 9342–9351. 10.1021/acsnano.9b03980. [DOI] [PubMed] [Google Scholar]
- (19).Gómez-Graña S; Hubert F; Testard F; Guerrero-Martínez A; Grillo I; Liz-Marzán LM; Spalla O Surfactant (Bi)Layers on Gold Nanorods. Langmuir 2012, 28 (2), 1453–1459. 10.1021/la203451p. [DOI] [PubMed] [Google Scholar]
- (20).Hughes HJ; Demers SME; Zhang A; Hafner JH The Orientation of a Membrane Probe from Structural Analysis by Enhanced Raman Scattering. Biochim. Biophys. Acta BBA - Biomembr 2020, 1862 (2), 183109. 10.1016/j.bbamem.2019.183109. [DOI] [PubMed] [Google Scholar]
- (21).Oakes RE; Renwick Beattie J; Moss BW; Bell SEJ Conformations, Vibrational Frequencies and Raman Intensities of Short-Chain Fatty Acid Methyl Esters Using DFT with 6-31G(d) and Sadlej PVTZ Basis Sets. J. Mol. Struct. THEOCHEM 2002, 586 (1), 91–110. 10.1016/S0166-1280(02)00064-7. [DOI] [Google Scholar]
- (22).Oakes RE; Renwick Beattie J; Moss BW; Bell SEJ DFT Studies of Long-Chain FAMEs: Theoretical Justification for Determining Chain Length and Unsaturation from Experimental Raman Spectra. J. Mol. Struct. THEOCHEM 2003, 626 (1), 27–45. 10.1016/S0166-1280(02)00670-X. [DOI] [Google Scholar]
- (23).Miranda AM; Castilho-Almeida EW; Martins Ferreira EH; Moreira GF; Achete CA; Armond RASZ; Dos Santos HF; Jorio A Line Shape Analysis of the Raman Spectra from Pure and Mixed Biofuels Esters Compounds. Fuel 2014, 115, 118–125. 10.1016/j.fuel.2013.06.038. [DOI] [Google Scholar]
- (24).Navarro JRG; Manchon D; Lerouge F; Cottancin E; Lermé J; Bonnet C; Chaput F; Mosset A; Pellarin M; Parola S Synthesis, Electron Tomography and Single-Particle Optical Response of Twisted Gold Nano-Bipyramids. Nanotechnology 2012, 23 (14), 145707. 10.1088/0957-4484/23/14/145707. [DOI] [PubMed] [Google Scholar]
- (25).Ye X; Zheng C; Chen J; Gao Y; Murray CB Using Binary Surfactant Mixtures To Simultaneously Improve the Dimensional Tunability and Monodispersity in the Seeded Growth of Gold Nanorods. Nano Lett. 2013, 13 (2), 765–771. 10.1021/nl304478h. [DOI] [PubMed] [Google Scholar]
- (26).Koglin E; Tarazona A; Kreisig S; Schwuger MJ In-Situ Investigations of Coadsorbed Cationic Surfactants on Charged Surfaces: A SERS Microprobe Study. Colloids Surf. Physicochem. Eng. Asp 1997, 123–124, 523–542. 10.1016/S0927-7757(96)03803-4. [DOI] [Google Scholar]
- (27).Lee S; Anderson LJE; Payne CM; Hafner JH Structural Transition in the Surfactant Layer That Surrounds Gold Nanorods as Observed by Analytical Surface-Enhanced Raman Spectroscopy. Langmuir 2011, 27 (24), 14748–14756. 10.1021/la202918n. [DOI] [PubMed] [Google Scholar]
- (28).Matthews JR; Payne CM; Hafner JH Analysis of Phospholipid Bilayers on Gold Nanorods by Plasmon Resonance Sensing and Surface-Enhanced Raman Scattering. Langmuir 2015, 31 (36), 9893–9900. 10.1021/acs.langmuir.5b01203. [DOI] [PubMed] [Google Scholar]
- (29).Makihara T; Demers SME; Cole LED; Zhang A; Hafner JH Thermophoresis of Gold Nanorods from Surface Enhanced Raman Scattering and Real-Time Rayleigh Scattering in Solution. Anal. Methods 2019, 11 (18), 2482–2488. 10.1039/C9AY00104B. [DOI] [Google Scholar]
- (30).Bendix PM; Reihani SNS; Oddershede LB Direct Measurements of Heating by Electromagnetically Trapped Gold Nanoparticles on Supported Lipid Bilayers. ACS Nano 2010, 4 (4), 2256–2262. 10.1021/nn901751w. [DOI] [PubMed] [Google Scholar]
- (31).Baffou G; Kreuzer MP; Kulzer F; Quidant R Temperature Mapping near Plasmonic Nanostructures Using Fluorescence Polarization Anisotropy. Opt. Express 2009, 17 (5), 3291–3298. 10.1364/OE.17.003291. [DOI] [PubMed] [Google Scholar]
- (32).Baerends EJ;Ziegler T;Atkins AJ;Autschbach J;Bashford D;Baseggio O;Berces A;Bickelhaupt FM;Bo C;Boerritger PM;Cavallo L;Daul C;Chong DP;Chulhai DV;Deng L;Dickson RM;Dieterich JM;Ellis DE;van Faassen M;Ghysels A;Giammona A;van Gisbergen SJA;Goez A;Gotz AW;Gusarov S;Harris FE;van den Hoek P;Hu Z;Jacob CR;Jacobsen H;Jensen L;Joubert L;Kaminski JW;van Kessel G;Konig C;Kootstra F;Kovalenko A;Krykunov M;van Lenthe E;McCormack DA;Michalak A;Mitoraj M;Morton SM;Neugebauer J;Nicu VP;Noodleman L;Osinga VP;Patchkovskii S;Pavanello M;Peeples CA;Philipsen PHT;Post D;Pye CC;Ramanantoanina H;Ramos P;Ravenek W;Rodriguez JI;Ros P;Ruger R;Schipper PRT;Schluns D;van Schoot H;Schreckenbach G;Seldenthuis JS;Seth M;Snijders JG;Sol\à Miquel;Stener M;Swart M;Swerhone D;te Velde G;Tognetti V;Vernooijs P;Versluis L;Visscher L;Visser O;Wang F;Wesolowski TA;van Wezenbeek EM;Wiesenekker G;Wolff SK;Woo TK; Yakovlev AL ADF; SCM: Amsterdam, The Netherlands. [Google Scholar]
- (33).Fonseca Guerra C; Snijders JG; te Velde G; Baerends EJ Towards an Order-N DFT Method. Theor. Chem. Acc 1998, 99 (6), 391–403. 10.1007/s002140050353. [DOI] [Google Scholar]
- (34).Velde G. te; Bickelhaupt FM; Baerends EJ; Guerra CF; Gisbergen S. J. A. van; Snijders JG; Ziegler T Chemistry with ADF. J. Comput. Chem 2001, 22 (9), 931–967. 10.1002/jcc.1056. [DOI] [Google Scholar]
- (35).van Lenthe E; Baerends EJ Optimized Slater-Type Basis Sets for the Elements 1–118. J. Comput. Chem 2003, 24 (9), 1142–1156. 10.1002/jcc.10255. [DOI] [PubMed] [Google Scholar]
- (36).van Lenthe E; Ehlers A; Baerends E-J Geometry Optimizations in the Zero Order Regular Approximation for Relativistic Effects. J. Chem. Phys 1999, 110 (18), 8943–8953. 10.1063/1.478813. [DOI] [Google Scholar]
- (37).van Lenthe E; Baerends EJ; Snijders JG Relativistic Regular Two-component Hamiltonians. J. Chem. Phys 1993, 99 (6), 4597–4610. 10.1063/1.466059. [DOI] [Google Scholar]
- (38).van Lenthe E; Baerends EJ; Snijders JG Relativistic Total Energy Using Regular Approximations. J. Chem. Phys 1994, 101 (11), 9783–9792. 10.1063/1.467943. [DOI] [Google Scholar]
- (39).Fan L; Ziegler T Application of Density Functional Theory to Infrared Absorption Intensity Calculations on Main Group Molecules. J. Chem. Phys 1992, 96 (12), 9005–9012. 10.1063/1.462258. [DOI] [Google Scholar]
- (40).Fan L; Ziegler T Application of Density Functional Theory to Infrared Absorption Intensity Calculations on Transition-Metal Carbonyls. J. Phys. Chem 1992, 96 (17), 6937–6941. 10.1021/j100196a016. [DOI] [Google Scholar]
- (41).van Gisbergen SJA; Snijders JG; Baerends EJ A Density Functional Theory Study of Frequency-dependent Polarizabilities and Van Der Waals Dispersion Coefficients for Polyatomic Molecules. J. Chem. Phys 1995, 103 (21), 9347–9354. 10.1063/1.469994. [DOI] [Google Scholar]
- (42).van Gisbergen SJA; Snijders JG; Baerends EJ Application of Time-Dependent Density Functional Response Theory to Raman Scattering. Chem. Phys. Lett 1996, 259 (5), 599–604. 10.1016/0009-2614(96)00858-5. [DOI] [Google Scholar]
- (43).van Gisbergen SJA; Snijders JG; Baerends EJ Implementation of Time-Dependent Density Functional Response Equations. Comput. Phys. Commun 1999, 118 (2), 119–138. 10.1016/S0010-4655(99)00187-3. [DOI] [Google Scholar]
- (44).Jensen L; Autschbach J; Krykunov M; Schatz GC Resonance Vibrational Raman Optical Activity: A Time-Dependent Density Functional Theory Approach. J. Chem. Phys 2007, 127 (13), 134101. 10.1063/1.2768533. [DOI] [PubMed] [Google Scholar]
- (45).Johnson PB; Christy RW Optical Constants of the Noble Metals. Phys. Rev. B 1972, 6 (12), 4370–4379. 10.1103/PhysRevB.6.4370. [DOI] [Google Scholar]
- (46).Kneipp K; Wang Y; Kneipp H; Perelman LT; Itzkan I; Dasari RR; Feld MS Single Molecule Detection Using Surface-Enhanced Raman Scattering (SERS). Phys. Rev. Lett 1997, 78 (9), 1667–1670. 10.1103/PhysRevLett.78.1667. [DOI] [Google Scholar]
- (47).Nie S; Emory SR Probing Single Molecules and Single Nanoparticles by Surface-Enhanced Raman Scattering. Science 1997, 275 (5303), 1102–1106. 10.1126/science.275.5303.1102. [DOI] [PubMed] [Google Scholar]
- (48).Doering WE; Nie S Single-Molecule and Single-Nanoparticle SERS: Examining the Roles of Surface Active Sites and Chemical Enhancement. J. Phys. Chem. B 2002, 106 (2), 311–317. 10.1021/jp011730b. [DOI] [Google Scholar]
- (49).da Silva JA; Meneghetti MR New Aspects of the Gold Nanorod Formation Mechanism via Seed-Mediated Methods Revealed by Molecular Dynamics Simulations. Langmuir 2018, 34 (1), 366–375. 10.1021/acs.langmuir.7b03703. [DOI] [PubMed] [Google Scholar]
- (50).González-Rubio G; Díaz-Núñez P; Rivera A; Prada A; Tardajos G; González-Izquierdo J; Bañares L; Llombart P; Macdowell LG; Palafox MA; Liz-Marzán LM; Peña-Rodríguez O; Guerrero-Martínez A Femtosecond Laser Reshaping Yields Gold Nanorods with Ultranarrow Surface Plasmon Resonances. Science 2017, 358 (6363), 640–644. 10.1126/science.aan8478. [DOI] [PubMed] [Google Scholar]
- (51).Llombart P; Palafox MA; MacDowell LG; Noya EG Structural Transitions and Bilayer Formation of CTAB Aggregates. Colloids Surf. Physicochem. Eng. Asp 2019, 580, 123730. 10.1016/j.colsurfa.2019.123730. [DOI] [Google Scholar]
- (52).Meena SK; Celiksoy S; Schäfer P; Henkel A; Sönnichsen C; Sulpizi M The Role of Halide Ions in the Anisotropic Growth of Gold Nanoparticles: A Microscopic, Atomistic Perspective. Phys. Chem. Chem. Phys 2016, 18 (19), 13246–13254. 10.1039/C6CP01076H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Meena SK; Sulpizi M Understanding the Microscopic Origin of Gold Nanoparticle Anisotropic Growth from Molecular Dynamics Simulations. Langmuir 2013, 29 (48), 14954–14961. 10.1021/la403843n. [DOI] [PubMed] [Google Scholar]
- (54).Seballos L; Olson TY; Zhang JZ Effects of Chromophore Orientation and Molecule Conformation on Surface-Enhanced Raman Scattering Studied with Alkanoic Acids and Colloidal Silver Nanoparticles. J. Chem. Phys 2006, 125 (23), 234706. 10.1063/1.2404648. [DOI] [PubMed] [Google Scholar]
- (55).Czamara K; Majzner K; Pacia MZ; Kochan K; Kaczor A; Baranska M Raman Spectroscopy of Lipids: A Review. J. Raman Spectrosc 2015, 46 (1), 4–20. 10.1002/jrs.4607. [DOI] [Google Scholar]
- (56).Long DA The Raman Effect: A Unified Treatment of the Theory of Raman Scattering by Molecules, 1st Edition.; Wiley: Chichester ; New York, 2001. [Google Scholar]
- (57).Lee Y; Jang J; Yoon J; Choi J-W; Choi I; Kang T Phase Transfer-Driven Rapid and Complete Ligand Exchange for Molecular Assembly of Phospholipid Bilayers on Aqueous Gold Nanocrystals. Chem. Commun 2019, 55 (22), 3195–3198. 10.1039/C8CC10037C. [DOI] [PubMed] [Google Scholar]
- (58).Takahashi H; Niidome Y; Niidome T; Kaneko K; Kawasaki H; Yamada S Modification of Gold Nanorods Using Phosphatidylcholine to Reduce Cytotoxicity. Langmuir 2006, 22 (1), 2–5. 10.1021/la0520029. [DOI] [PubMed] [Google Scholar]
- (59).Lee SE; Sasaki DY; Perroud TD; Yoo D; Patel KD; Lee LP Biologically Functional Cationic Phospholipid–Gold Nanoplasmonic Carriers of RNA. J. Am. Chem. Soc 2009, 131 (39), 14066–14074. 10.1021/ja904326j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Orendorff CJ; Alam TM; Sasaki DY; Bunker BC; Voigt JA Phospholipid–Gold Nanorod Composites. ACS Nano 2009, 3 (4), 971–983. 10.1021/nn900037k. [DOI] [PubMed] [Google Scholar]
- (61).Kah JCY; Zubieta A; Saavedra RA; Hamad-Schifferli K Stability of Gold Nanorods Passivated with Amphiphilic Ligands. Langmuir 2012, 28 (24), 8834–8844. 10.1021/la3000944. [DOI] [PubMed] [Google Scholar]
- (62).Su X; Wang Y; Wang W; Sun K; Chen L Phospholipid Encapsulated AuNR@Ag/Au Nanosphere SERS Tags with Environmental Stimulus Responsive Signal Property. ACS Appl. Mater. Interfaces 2016, 8 (16), 10201–10211. 10.1021/acsami.6b01523. [DOI] [PubMed] [Google Scholar]
- (63).Santhosh PB; Thomas N; Sudhakar S; Chadha A; Mani E Phospholipid Stabilized Gold Nanorods: Towards Improved Colloidal Stability and Biocompatibility. Phys. Chem. Chem. Phys 2017, 19 (28), 18494–18504. 10.1039/C7CP03403B. [DOI] [PubMed] [Google Scholar]
- (64).Luchini A; D’Errico G; Leone S; Vaezi Z; Bortolotti A; Stella L; Vitiello G; Paduano L Structural Organization of Lipid-Functionalized-Au Nanoparticles. Colloids Surf. B Biointerfaces 2018, 168, 2–9. 10.1016/j.colsurfb.2018.04.044. [DOI] [PubMed] [Google Scholar]
- (65).Bruzas I; Brinson BE; Gorunmez Z; Lum W; Ringe E; Sagle L Surface-Enhanced Raman Spectroscopy of Fluid-Supported Lipid Bilayers. ACS Appl. Mater. Interfaces 2019, 11 (36), 33442–33451. 10.1021/acsami.9b09988. [DOI] [PubMed] [Google Scholar]
- (66).Seelig J; Niederberger W Two Pictures of a Lipid Bilayer. Comparison between Deuterium Label and Spin-Label Experiments. Biochemistry 1974, 13 (8), 1585–1588. 10.1021/bi00705a005. [DOI] [PubMed] [Google Scholar]
- (67).Seelig A; Seelig J Effect of a Single Cis Double Bond on the Structure of a Phospholipid Bilayer. Biochemistry 1977, 16 (1), 45–50. 10.1021/bi00620a008. [DOI] [PubMed] [Google Scholar]
- (68).Seelig J; Waespe-Sarcevic N Molecular Order in Cis and Trans Unsaturated Phospholipid Bilayers. Biochemistry 1978, 17 (16), 3310–3315. 10.1021/bi00609a021. [DOI] [PubMed] [Google Scholar]
- (69).Enkavi G; Javanainen M; Kulig W; Róg T; Vattulainen I Multiscale Simulations of Biological Membranes: The Challenge To Understand Biological Phenomena in a Living Substance. Chem. Rev 2019, 119 (9), 5607–5774. 10.1021/acs.chemrev.8b00538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Marrink SJ; Corradi V; Souza PCT; Ingólfsson HI; Tieleman DP; Sansom MSP Computational Modeling of Realistic Cell Membranes. Chem. Rev 2019, 119 (9), 6184–6226. 10.1021/acs.chemrev.8b00460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Balali-Mood K; Harroun TA; Bradshaw JP Molecular Dynamics Simulations of a Mixed DOPC/DOPG Bilayer. Eur. Phys. J. E 2003, 12 (1), 135–140. 10.1140/epjed/e2003-01-031-3. [DOI] [PubMed] [Google Scholar]
- (72).Klauda JB; Venable RM; Freites JA; O’Connor JW; Tobias DJ; Mondragon-Ramirez C; Vorobyov I; MacKerell AD; Pastor RW Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114 (23), 7830–7843. 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Reddy AS; Warshaviak DT; Chachisvilis M Effect of Membrane Tension on the Physical Properties of DOPC Lipid Bilayer Membrane. Biochim. Biophys. Acta BBA - Biomembr 2012, 1818 (9), 2271–2281. 10.1016/j.bbamem.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Piggot TJ; Allison JR; Sessions RB; Essex JW On the Calculation of Acyl Chain Order Parameters from Lipid Simulations. J. Chem. Theory Comput 2017, 13 (11), 5683–5696. 10.1021/acs.jctc.7b00643. [DOI] [PubMed] [Google Scholar]
- (75).Thomas M; Brehm M; Fligg R; Vöhringer P; Kirchner B Computing Vibrational Spectra from Ab Initio Molecular Dynamics. Phys. Chem. Chem. Phys 2013, 15 (18), 6608–6622. 10.1039/C3CP44302G. [DOI] [PubMed] [Google Scholar]
- (76).Luber S; Iannuzzi M; Hutter J Raman Spectra from Ab Initio Molecular Dynamics and Its Application to Liquid S-Methyloxirane. J. Chem. Phys 2014, 141 (9), 094503. 10.1063/1.4894425. [DOI] [PubMed] [Google Scholar]
- (77).Aprà E; Bhattarai A; Baxter E; Wang S; Johnson GE; Govind N; El-Khoury PZ Simplified Ab Initio Molecular Dynamics-Based Raman Spectral Simulations. Appl. Spectrosc 2020, 74 (11), 1350–1357. 10.1177/0003702820923392. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.