

Autosomally replicating episomal DNA nanovectors allow clinical-scale CAR-T cell manufacturing.
Abstract
The compelling need to provide adoptive cell therapy (ACT) to an increasing number of oncology patients within a meaningful therapeutic window makes the development of an efficient, fast, versatile, and safe genetic tool for creating recombinant T cells indispensable. In this study, we used nonintegrating minimally sized DNA vectors with an enhanced capability of generating genetically modified cells, and we demonstrate that they can be efficiently used to engineer human T lymphocytes. This vector platform contains no viral components and is capable of replicating extrachromosomally in the nucleus of dividing cells, providing persistent transgene expression in human T cells without affecting their behavior and molecular integrity. We use this technology to provide a manufacturing protocol to quickly generate chimeric antigen receptor (CAR)–T cells at clinical scale in a closed system and demonstrate their enhanced anti-tumor activity in vitro and in vivo in comparison to previously described integrating vectors.
INTRODUCTION
Recombinant T cells that functionally express chimeric antigen receptors (CARs) for adoptive cell therapy (ACT) are an increasingly active field in oncology. CAR-T cell therapy has shown extraordinary efficacy in numerous clinical trials of hematological malignancies such as pediatric diffuse large B-cell lymphoma (DLBCL), and mantle cell lymphoma. Observed overall response rates of 81% in children and 83% in adults (1, 2) resulted in the clinical approval of the cell therapy–based therapeutics tisagenlecleucel (3), axicabtagene ciloleucel (4), and brexucabtagene autoleucel (5). These tremendously accelerated preclinical research activities have led toward more complex (6), controllable (7), and inducible (8) CAR constructs for the treatment of solid tumors as well as opening the fields of CAR applications up to autoinflammatory and cardiac diseases (9). However, despite the successes, substantial challenges remain, ranging from a long lead time and expensive manufacturing (10) to complicated vector-engineering, optimized gene expression and delivery, and reduced vector-mediated toxicities. Now, CAR-T cells are routinely produced using randomly integrating vectors such as gamma retroviruses (11) or transposons (12) that carry the inherent potential of genotoxic risk, which can be associated with costly long-term patient follow-up. CAR engineering by mRNA transfection might overcome this disadvantage, but this procedure is prohibitive for pharmaceutical manufacturing as it requires the production of several doses of vector per patient, due to transient protein expression (13).
As key players of the adaptive immune system, T cells are exquisitely sensitive to immunogenic exogenous DNA, and they rapidly lose functional capacity or induce apoptosis following transfection with typically used vectors. Here, we describe the development of nano-S/MARt (nS/MARt) vectors that are designed to provide stable expression while causing minimal disruption of T cell activity. This novel DNA vector platform is based on scaffold/matrix attachment region (S/MAR) motifs that can actively mediate the episomal maintenance and replication of DNA vectors in mitotically active cells (14). S/MARs are generally defined as the genomic DNA sequences that anchor chromatin to the nuclear matrix proteins during interphase (15). This binding mediates the generation of looped domains that contribute structurally to the tight packaging of the DNA into the nucleus and functionally to regulate of gene expression and replication of the genome (16). S/MARs have a size range of 60 to 5000 base pairs (bp) and are characterized by their AT-rich content, and although they are evolutionarily conserved across species, they do not show a consensus sequence. Through a process of iterative CpG depletion, selection marker minimalization, careful choice of the replication origin, empirical promoter design, and elimination of cryptic eukaryotic signals nS/MARt can be efficiently transfected into primary human T cells without toxicity. This novel DNA vector system is based on an antibiotic-free nanovector technology (17, 18); it contains no immunogenic and components and comprises only clinically approved sequences. It is easy, simple, versatile, and cost-efficient to produce. By combining our novel technology with well-established techniques such as flow electroporation and automated cell culture, we also describe a novel manufacturing protocol that allows the production of 6 × 108 recombinant T cells of which 55% stably express the novel CAR from 1 × 109 purified T cells, in only 5 days, an important step toward more sustainable and accessible ACTs.
RESULTS
Next-generation nS/MARt vectors efficiently modify human cells providing persistent transgene expression
The genetic composition of the S/MARt vectors described here was refined and optimized to produce a range of newly designed DNA vectors that can efficiently genetically modify primary T cells without any molecular or genetic impact (Fig. 1A). First, we modified pEPI (19) generating pS/MARt, a plasmid vector in which we reduced the size of the bacterial backbone from 3 to 1.5 kb by removing unnecessary DNA sequences and retaining only the bacterial origin of replication (Ori) and the bacterial antibiotic selection marker kanamycin (KanR). In the eukaryotic expression cassette, driven by the viral promoter cytomegalovirus (CMV), we coupled the expression of the selection marker puromycin (Puro) directly to the reporter gene green fluorescent protein (GFP) through the self-cleavage sequence P2A (20). The transcription of the expression cassette GFP-Puro terminates inside the S/MAR sequence isolated from the human interferon-β (IFN-β) cluster (IFN-β S/MAR), promoting its functionality that results in the extrachromosomal maintenance of the plasmid DNA (21). By introducing these changes, we generated a vector with improved capability in generating stably transfected clones in the human cell lines human embryonic kidney (HEK) 293T and HeLa (Fig. 1B). The vector pS/MARt was further refined into the minimally sized vector nS/MARt by retaining the expression cassette and exchanging the bacterial backbone containing the Ori and KanR for the antibiotic-free selection system described by Luke et al. (18). Stehle et al. (22) demonstrated that transcriptional activity through the S/MAR motif is essential for function. However, cellular mRNA transcripts that include stretches of AU-rich elements (AREs) are recognized and rapidly degraded by the human antigen R (23). As S/MAR motifs are stretches of genomic DNA enriched in adenine and thymidine, we introduced splicing sites to flank the IFN-β MAR generating the SP-nS/MARt-B vector that results in more stable transcripts that do not carry extraneous and antagonistic AREs derived from the unnecessary transcription of the S/MAR motif. The active splicing was demonstrated by Northern blot (fig. S1A) where the transcript length of the constructs generated from pS/MARt, nS/MARt, and SP-nS/MARt was investigated. As a last developmental step, we replaced the IFN-β S/MAR with a more compact version, isolated from the human apolipoprotein B (ApoB) gene cluster (24), which we also flanked with splicing sites. The nuclear matrix attachment sites found in specific AT-rich regions of genes predominantly comprise ATTA-ATTTA motifs, recognition sites of homeodomain proteins highly conserved in the Ori of yeast, viruses, mitochondria, chloroplasts, and mammalian Ori. The ApoB MAR is almost entirely composed of a contiguous stretch of 555 bp made of a mosaic of the TAAT, TAAAT, ATTA, ATTTTA, TAAAAT, and ATTTA motifs. The efficacy of establishment, measured as the number of colonies generated by each vector, demonstrated that the ultimate version, the SP-nS/MARt-A nanovector, was the most efficient, forming the highest number of colonies compared to previous generations of vectors both in HEK293T and HeLa cells (Fig. 1B). SP-nS/MARt-A not only generated the highest number of colonies but also provided the best transfection efficiency with the highest level of GFP expression [medium fluorescent intensity (MFI)] 24 hours after DNA delivery. The result was also confirmed when the cells were analyzed 30 days after DNA delivery (fig. S1B). In the modified cells, the S/MAR-based vectors’ episomal status was demonstrated by Southern blot (Fig. 1C), where the presence of single bands in the total DNA extracts of established cells showed the episomal forms of this class of vectors. The repetitive structure of the ApoB MAR sequence was fragmented to investigate whether the short motifs of this sequence were sufficient to sustain the vectors’ functionality. We generated seven different versions whose sequences varied by nucleotide composition, orientation, and length (fig. S1C) that were investigated for their capability to establish stable cells in HEK293T and HeLa. In both human cell lines, the wild-type full-length ApoB MAR sequence was found to be the most active. We then generated an SP-nS/MARt-A vector in which we swapped the CMV promoter for the human EF1α promoter (25), and we tested this construct in the human T cell cancer cell line Jurkat76 (J76) (26). We demonstrated that the vectors could be efficiently and consistently delivered by electroporation with more than 80% of the viable cells expressing the reporter gene (Fig. 1D). To isolate the cells in which the vector was actively maintained from the rest of the population, 15 days after delivery, GFP+ cells were isolated through fluorescence-activated cell sorting (FACS) and then further cultured for 360 days. We could successfully generate three independent cell lines that were monitored routinely for the percentage of GFP+ cells (GFP %) and intensity of expression (GFP MFI) (Fig. 1D), and we demonstrated that during this extended period, the GFP % and the GFP MFI remained stable. The plasmids’ episomal transmission was checked at day 360 through Southern blot (fig. S1D), and the copy number of ~1.71 copies per cell was determined (fig. S1E). In accordance with previous reports (19, 27, 28), vectors carrying S/MARs are replicated extrachromosomally in the nuclei of dividing cells where they establish at a low copy number. The persistent expression of the reporter gene GFP was also tested in primary human CD3+ cells for 27 days. CD3+ cells isolated from four different healthy donors were transfected with nS/MARt vectors carrying the IFN-β and the ApoB MAR compared to pEPI (Fig. 1E). The vector’s delivery by electroporation did not alter the CD4:CD8 ratio compared to mock electroporated cells (fig. S1F). However, 60% of the total cells transfected with pEPI showed no GFP expression 10 days after DNA delivery (fig. S1G), which was almost completely lost by day 27, while 40% of the cells transfected with SP-nS/MARt-A vectors remained GFP+. SP-nS/MARt-A displayed a significantly reduced percentage of GFP+ cells loss compared to pEPI and to nS/MARt vectors based on the IFN-β. Moreover, we demonstrated that the proliferation of T cells transfected with SP-nS/MARt-A vector was comparable to untreated CD3+ controls, indicating that there is a minimal impact of the DNA vectors and the DNA delivery system on cell proliferation (fig. S1H). The vectors’ episomal persistence in primary lymphocytes was demonstrated through rolling circle amplification (RCA) (Fig. 1F).
Fig. 1. Optimized nS/MARt vectors efficiently modify cells in culture and provide prolonged transgene expression in primary CD3+ cells.
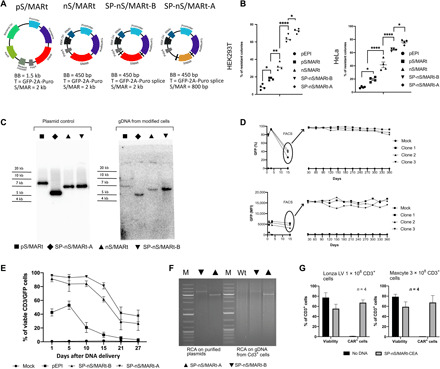
The vectors pS/MARt, nS/MARt, SP-nS/MARt-B, and SP-nS/MARt-A [(A) BB, bacterial backbone; T, transgene] were tested for their efficacy in generating stable clones with a colony-forming assay in HEK293T and HeLa (B). n = 4. In the graph, the line is at the median, and the analysis was performed with a one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test for multiple comparisons. *P < 0.05, **P < 0.01, and ****P < 0.0001. The episomal maintenance of the vectors was investigated by Southern blot (C) upon total genomic DNA (gDNA) extraction and digestion with the endonuclease Bam HI. The digested DNA was resolved on an agarose gel, transferred, and hybridized with a radioactive GFP probe. With SP-nS/MARt-EF1a, three independent J76-GFP stable cell lines were generated. The number of viable GFP+ and the medium fluorescent intensity (MFI) of cells were monitored, and 15 days after DNA delivery, GFP+ cells were isolated through FACS and further cultured for 360 days. GFP % and GFP MFI were monitored once per month (D). The GFP-prolonged expression from SP-nS/MARt vectors was investigated in primary human CD3+ cells isolated from four healthy volunteers. The number of CD3/GFP+ cells was monitored at days 1, 5, 10, 15, 21, and 27 after DNA delivery (E), and the episomal maintenance of the vectors was investigated by rolling circle amplification (RCA). The RCA reaction was digested with the endonuclease Bam HI and resolved on a 0.8% agarose gel [(F) M, molecular weight DNA marker; Wt, CD3+ cells unmodified]. SP-nS/MARt vectors expressing the CEA-CAR were optimized to be efficiently delivered with high viability into human CD3+ cells with two large-scale electroporation devices; the Lonza LV Unit and the MaxCyte ExPERT GTx platform (G).
To further investigate the possibility of using this novel DNA vector technology in clinical application, we replaced the GFP reporter gene in the SP-nS/MARt-A vector for the CAR against the human carcinoembryonic antigen (CEA) and the CMV promoter for the PGK (phosphoglycerate kinase) promoter generating the SP-nS/MARt-CEA vector. The efficiency of delivery and transgenic CAR expression was evaluated using two different large-scale electroporation devices: the Lonza LV Unit and the MaxCyte ExPERT GTx platform, with 1 × 108 and 3 × 108 CD3+ cells, respectively. With both devices, in comparison to the respective mock electroporated CD3+ cells, the delivery of the vector yielded cellular viability that ranged between 50 and 75% depending on the donor with the percentage of CAR+ T cells ranging from 65 to 80%, 24 hours after DNA delivery (Fig. 1G). These results demonstrated the minimal impact of this vector technology on primary T cells across electroporation devices, supporting the possibility of its use for the manufacturing of recombinant T cells for clinical applications.
nS/MARt vectors have minimal impact on human T cells and provide superior functional capability
To investigate the impact of the nS/MARt vectors on primary human T cells, isolated CD3+ cells manufactured with nS/MARt-CEA-CAR were compared with T cells transduced with lentivirus carrying the same expression cassette. Both approaches lead to similar percentages of CAR-T cells, while CAR-T cells generated with nS/MARt showed a greater median expression of the CAR (Fig. 2A). The modified T cells were then analyzed with the ESCAPE RNA sequencing (RNA-seq) proteogenomics (Proteona) platform (Fig. 2B). By single-cell RNA-seq (scRNA-seq), we showed that T cells electroporated with nS/MARt or transduced with lentivirus form separate clusters when compared to mock parental cells at the transcriptomic level. When the transcriptional profile of T cells transduced with lentiviral vectors was compared to unmodified parental cells, we could find 106 differentially expressed genes, with the most prominently regulated genes belonging to the IFIT (Interferon Induced proteins with Tetratricopeptide repeats) family or encoding for chemokines (Fig. 2, C and D). CAR expression was also negatively regulated in this subset of T cells. On the other hand, we could observe 61 differentially regulated genes upon gene delivery with nS/MARt (Fig. 2D); in this case, CAR mRNA expression was among the most up-regulated genes. Subsequent gene cluster analysis of the top 100 up-regulated genes in lentivirally transduced T cells among the cell subsets confirmed the distant relationship between nS/MARt and lentivirally modified cells. The minimal impact of the nS/MARt vectors was then investigated at the single-cell level where neither the cytokine IFN-β nor the genes that are typically up-regulated upon infection such as the cGAS (cyclic GMP-AMP synthase)–STING (stimulator of interferon) pathway appeared up-regulated (fig. S2A). This observation was then confirmed by enzyme-linked immunosorbent assay (ELISA) (fig. S2B), where we could not detect differences between cells electroporated with nS/MARt or without DNA, while those cells treated with a stimulatory synthetic double-stranded RNA (dsRNA) produced significant doses of IFN-β. These observations reflect the low immunogenic profile of the nS/MARt technology that presents a fully CpG-depleted backbone. In the nS/MARt-CEA vector, we observed 11 CpG motifs; seven are located internally within the PGK promoter, and four occur within the CEA-CAR cDNA. Furthermore, a closer relationship between n/SMARt− and unmodified T cells was observed. We performed a Gene Ontology (GO) term analysis of all up-regulated genes in both subsets of modified T cells and found several distinct gene sets that were significantly enriched. Transduced cells showed enrichment of gene sets involved in processing peptides through the major histocompatibility complex (MHC) class II pathway and associated with cell death (Fig. 2D). The concomitant down-regulation of the genes responsible for the IFN-β production (Fig. 2C) suggested that these cells acquired a nonphysiological phenotype, while in cells transfected with nS/MARt, we detected the up-regulation of 12 genes that are involved in processes initiated by the RNA polymerase II complex. In addition, a comparative gene set enrichment analysis (GSEA) applying the C7 immunologic gene sets panel of the Molecular Signature Database (MSigDB) on the scRNA-seq data revealed that T cells modified with nS/MARt vectors display a more naïve phenotype of CD8+ and CD4+ T cells than the lentivirally transduced counterparts.
Fig. 2. Single-cell RNA-seq (scRNA-seq) analysis of nS/MARt CAR-T cells.
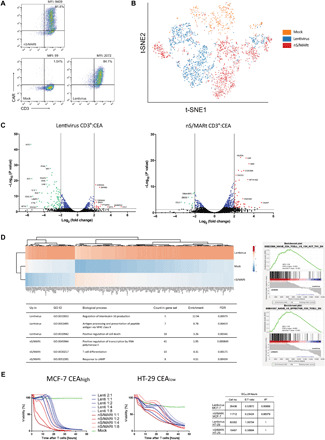
CEA-CAR T cells generated by electroporation with nS/MARt or by transduction with a vesicular stomatitis virus–pseudotyped lentivirus were analyzed by FACS for the expression of the transgene in comparison to mock cells (A). The modified cells were then analyzed with the ESCAPE RNA-seq proteogenomic platform (Proteona). t-SNE (t-distributed stochastic neighbour embedding) projection of RNA-seq reads are shown. Colors indicate clusters of cells identified by the transfection method (n = 2757 cells) (B). Volcano plots display the transcriptional profile of lentivirally transduced CAR-T cells and nS/MARt electroporated CAR-T cells versus mock T cells (C). The top 100 up-regulated genes in cells transduced with lentivirus underwent hierarchical cluster analysis in comparison with untransfected and electroporated cells. GO term analysis was performed on the full list of significantly up-regulated genes in each respective transfection method using GO terms and the PANTHER classification system. GSEA of all differentially expressed genes for each modified T cell approach reveals a more naïve T cell phenotype after nS/MARt transfection (D). The efficacy of tumor target killing was investigated in a real-time in vitro cytotoxicity assay. Target cells (2.5 × 104), either with a high (MCF-7) or a low (HT-29) expression of the target epitope CEA, were seeded at day 0, and the effector CEA-CAR T cells were added at day 1 at different effector-to-target ratios. The EC50 for effector CAR-T cells generated with the lentivirus and nS/MARt was estimated (E).
The efficacy of targeted tumor killing of CEA-CAR-T cells generated with nS/MARt technology was tested in vitro in a real-time cytotoxicity assay using the human cancer cell lines HT-29 and MCF-7 as targets and compared to T cells transduced with CEA lentivirus (Fig. 2E). For both tumor cell targets, regardless of high or low target antigen expression, we found nS/MARt-CEA-CAR-T cells to have a more efficient killing capability. When the target was MCF-7 cells, we showed that an effector to target ratio of 2.5:1 is necessary for lentivirally transduced T cells to match the functionality of 1:1 nS/MARt-CEA-CAR-T cells. The enhanced tumor killing efficacy was then confirmed when the target was HT-29 cells, where 2.7:1 cells generated with the virus were needed to match a 1:1 coculture with cells engineered with nS/MARt vectors. The calculated median effective concentration (EC50) values after 24 hours of cocultivation confirmed the observation. Following previous reports, we also confirmed that the cytolysis of the target cells was mediated by IFN-γ (fig. S3A) (29).
nS/MARt T cells mediate efficient tumor killing in vivo in three murine tumor xenograft models
To further demonstrate nS/MARt T cells’ functionality, we engineered human CD3+ cells to express anti–CEA-CARs, anti–NY-BR1–CARs, or anti–CD19-CARs, and we investigated the tumor killing capability of the modified CAR-T cells in xenografted mouse models. In the CEA mouse model, nS/MARt-CEA-CAR-T cells were compared against T cells transduced with a standard SIN (Self Inactivating)–lentiviral vector. Mice treated with nS/MARt-modified T cells showed a distinct reduction in the tumor size and delayed tumor growth (Fig 3A and fig. S3B) resulting in prolonged survival (Fig. 3B) in comparison to the groups treated with unmodified and lentivirally transduced CAR-T cells. However, a complete clearance of the tumor was not achieved in any of the groups, which was due to the loss of antigen expression in the target tumor cells (Fig. 3B), a common tumor escape mechanism during solid tumors treatment (30). In the tumors of mice treated with nS/MARt T cells, we could detect a higher tumor infiltration frequency (Fig 3C), while the engraftment of the CAR-T cells in the spleens was comparable in both approaches (Fig. 3D). An RCA test conducted on DNA extracted from the total blood, the spleens, and tumors of mice that received the therapy demonstrated the episomal maintenance of nS/MARt vectors in the genetically modified cells (Fig. 3F). The long-term antitumor activity of T cells modified with nS/MARt vectors was shown by coculturing splenocytes retrieved from treated mice and the cancer cell line MCF-7 (fig. S3D), in which a substantial activity from CAR-T cells was detected 40 days after treatment.
Fig. 3. In vivo targeted tumor killing.
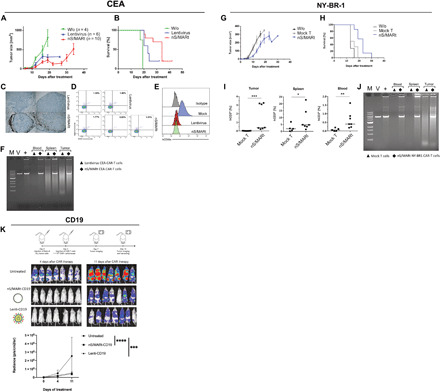
The efficacy of targeting tumor cells expressing the human epitopes CEA, NY-BR1, and CD19 was evaluated in preclinical models. For the CEA model, 1 × 106 cells were implanted subcutaneously in NSG mice, and on day 7, the mice were treated with mock, and CAR-T cells were generated with lentivirus and nS/MARt. Tumor growth (A) and mice survival (B) were monitored. nS/MARt CAR-T cells showed a higher tumor infiltration when compared to Lenti CAR-T cells (C), while the human T cells engraftment in both groups appeared similar (D). The outgrown tumors showed an antigen loss in the treated groups, while it remains expressed in the control group (E). CAR-T cells from the spleens, tumors, and total blood of the treated mice were isolated, and an RCA demonstrated the presence of the episomal plasmids [(F). M. DNA molecular marker; V, nS/MARt-CEA DNA restriction analysis; +, RCA on purified nS/MARt-CEA subsequentially digested with restriction enzymes]. Similarly, for the NY-BR1 model, cells expressing the epitope were xenografted into NSG mice. The efficacy of tumor killing was recorded as tumor growth (G) and survival (H). CAR-T cells in the tumors, spleens, and blood of treated mice were found 30 days after injection (I), and the episomal maintenance of the vector was demonstrated by RCA (J). Nalm-6 cells (1 × 105) modified to express the luciferase constitutively were injected into NSG mice by tail vein injection. Three days after tumor cell injection, 1 × 106 CD19-CAR-T cells generated with mock, nS/MARt, or lentivirus were administered. The tumor growth was monitored through bioluminescent imaging (K), as the radiance (p/s/cm2/sr), and the statistical analysis was performed with a one-way ANOVA followed by post hoc analysis (**P < 0.001 and ***P < 0.001).
Similarly, when we treated mice with the CAR against the breast cancer cells target NY-BR1 (31, 32), we demonstrated delayed tumor growth (Fig. 3G), alongside increased survival of the mice (Fig. 3H). Moreover, we could detect a significant percentage of nS/MARt T cells in the tumors, spleens, and blood of the treated animals (Fig. 3I) compared to mock control T cells. The extrachromosomal vector maintenance was also demonstrated by RCA in the analyzed tissues (Fig. 3J). Ultimately, we tested nS/MARt-CD19 CAR-T cells in the Nalm-6/CD19 model. Unmodified T cells, nS/MARt-CD19 CAR-T, and lentivirally produced CAR-T cells (Lenti-CD19) were injected into NSG (NOD.Cg-Prkdc Il2rg/SzJ) mice previously treated with Nalm-6 luciferase cells, and the efficacy of tumor killing was monitored through bioluminescent imaging, 4 and 11 days following CAR-T cell therapy (Fig. 3K). Mice treated with CD19-CAR-T cells significantly delayed tumor growth compared with the control group (Fig. 3K). However, no significant differences were observed between the groups that received CD19-CARs. Together, these results demonstrate that T cells modified with our novel DNA vector technology can target and kill tumor cells in vivo, with an efficacy that is higher, or at least comparable, to CAR-T cells modified with the state-of-the-art lentiviral approach.
Large-scale GMP-compatible production of nS/MARt T cells
For translation into a clinically relevant application of nS/MARt T cells, a manufacturing protocol that allows the generation of clinical-grade recombinant T cells was developed (Fig. 4). For this, we coupled the CliniMACS Prodigy device (Miltenyi), a fully automated and closed system for the isolation and culturing of primary human CD3+, with the GTx ExPERT (MaxCyte) extra large-scale electroporation platform. With this protocol, we aimed to generate recombinant T cells in less than 7 days from isolating the sample. The nS/MARt-CEA-CAR vector was used to simulate the production of a clinical-grade product. The process started with a leukapheresis product containing 10.35 × 109 blood cells from which 1 × 109 T cells were isolated by the CliniMACS Prodigy using the implemented TCT (T Cell Transduction) process and GMP (Good Manufacturing Practice)–grade CD8/CD4 microbeads. At this stage, a sample was taken from the culture and kept for further analysis serving as a negative control for downstream experiments. The cells were cultured for 3 days in TransAct, interleukin-7 (IL-7), and IL-15 in TexMACS medium following the enhanced feeding protocol of the device and using a TS520 tubing set. Continuous monitoring was performed using the inbuilt microscope (fig. S4), and T cell activation was confirmed. On day 4, activated CD3+ cells were transferred into the HyClone electroporation buffer (MaxCyte) using the automated final formulation procedure. Electroporation of the DNA was performed in the GTx ExPERT platform using the CL-2 processing assembly and the “T Cell Program 3” as pulsing code with nS/MARt-CEA-CAR (125 μg/ml) for CD3+ cells (1 × 108/ml). Shortly after electroporation, the collection bag of the MaxCyte CL-2 processing assembly was reconnected to the CliniMACS Prodigy, and the cells were transferred to the culturing chamber. Twenty-four hours after DNA delivery, a sample was taken for analysis, and this showed a robust CAR expression with a viability of 72%. The CAR-T cells’ functionality was subsequently tested in an in vitro real-time killing assay using the HT-29 human cancer cell line as a tumor target. The CAR-T cells were shown to kill the tumor cells at different ratios in compared to unmodified CD3+ cells (mock T). Moreover, CAR-T cells produced IFN-γ in response to the tumor cell target. Using this novel protocol, we were able to generate 3.6 × 108 functional CAR-T cells within 5 days.
Fig. 4. GMP-compatible, large-scale, CAR-T cell manufacturing using nS/MARt vectors.
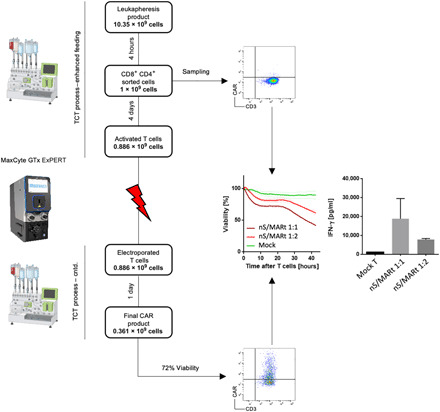
For the generation of recombinant T cells at a clinical scale, a manufacturing protocol was developed. The process started with a leukapheresis product that was subjected to CD3+ cell isolation with the TCT process in the CliniMACS Prodigy. Isolated CD3+ cells were then cultured in the presence of TransAct, IL-7, and IL-15 for 3 days when the cells were counted and transferred into the electroporation bag of the MaxCyte ExPERT GTx device. The electroporation was conducted with 1 × 108 cell/ml and DNA (125 μg/ml). Shortly after electroporation, the cells were retransferred into the culturing chamber of the CliniMACS Prodigy and fed with medium supplemented with IL-7 and IL-15 for 1 day, when the cells were harvested and analyzed for the CAR expression. Their capability of killing tumor cells was tested in an in vitro killing assay, in which the IFN-γ production was also assessed.
DISCUSSION
Despite the enthusiasm generated by the U.S. Food and Drug Administration’s approval of several cellular-based therapies, the long lead time of the manufacturing and their high cost severely limit the application of ACT products to only severe cases for which there are typically limited therapeutic possibilities. We describe here an economical and rapid clinical scale nonviral manufacturing protocol for generating CAR-T cells. We developed minimally sized nS/MARt DNA nanovectors that were specifically refined and optimized for their low toxicity (Fig. 2C and fig. S2, A and B) and capacity to generate genetically modified human T cells efficiently. Nonviral approaches for the genetic engineering of human cells have been extensively described, and several clinical trials based on CRISPR-Cas9 or transposons are underway. However, in both cases, the modification of the lymphocytes requires the integration of the transgene into the cellular genome. By systematically refining the composition of DNA vectors, we have enhanced their capacity to modify cells persistently. We improved the vectors’ stability and reduced their toxic burden at the DNA level by removing extraneous bacterial sequences, depleting the CpG motifs and incorporating an alternative S/MAR enriched with a core of the element’s distinguishing A and T motifs. The vectors were also refined at the RNA level by designing the S/MAR motif to be spliced out from the vectors’ mRNA, which leads to improved expression and more stable transcripts (Fig. 1). We showed that DNA nanovectors induce no genotoxicity; they do not integrate and instead replicate autonomously and extrachromosomally in the nuclei of cells, avoiding the inherent risk of integrative mutagenesis (Fig. 1F), while standard non-SMAR minicircles are lost during cell proliferation (33). Episomal vectors present then the big advantage of not being limited in the cargo capacity, as demonstrated by Hibbit et al. (34), in which a 135-kb genetic locus was isolated, cloned, and delivered. nS/MARt vectors comprise only mammalian sequences for their replication, maintenance, and transgene expression (35) without the requirement of additional elements (36). Compared to previously described SMAR vectors (19, 27, 37), nS/MARt plasmids do not require a mandatory 2-week period of drug selection pressure to drive episomal replication. After an initial period of establishment, the vectors are retained long term in cells at a low copy number. This initial phase lasts for approximately 14 days in the rapidly dividing cell lines such as Jurkat cells, but it is dependent on the cell type. A spontaneous decrease of genetically modified T cells is observed at ~21 days after DNA delivery, which can be considered a desirable safety feature during clinical applications where only a subpopulation of transfected cells will be retained in the body. By high-resolution scRNA-seq, we demonstrate that these DNA vectors do not cause molecular, genetic, or vector-mediated damage to the recipient cell. Populations of CAR-T cells generated with DNA nanovectors are unperturbed, and their RNA expression profiles cluster with unmodified cells, whereas those transduced with lentivirus form distinct dysregulated clusters despite having comparable levels of CAR expression (Fig. 2, A and D). Notably, there are specific gene families that are perturbed by lentiviral transduction that affect the capacity of the CAR-T cell to function. These include the anti-infection IFIT gene family that acts as regulators of antiviral responses including regulation of T cell activation genes such as IFN-β and RANTES (38, 39). The improved functionality of these nanovector-modified cells is reflected in more effective targeted killing of human cancer cells in culture and xenografted mouse models than those engineered with the current state-of-the-art integrative lentiviruses (Fig. 3). A major challenge in translating cellular immunotherapy from preclinical studies to applicable products involves the manufacturing process. Depending on the tumor entity, treatment protocol, and bodyweight of the patient, the number of CAR-T cells required ranges from 1 × 108 to 5 × 109 per treatment. In a GMP environment, this is time and cost intensive. The manufacturing of tisagenlecleucel, for instance, is estimated to take 3 to 4 weeks including a double shipment of frozen cells (40). Lentiviral production is expensive, batches are small, and their storage capacity is limited (41). In contrast, we estimate that only 1.5 mg of nanovector is required to manufacture a single batch of CAR-T cells. This DNA can be simply and economically prepared and efficiently stored providing a marked improvement upon existing platforms. Assuming a lentiviral vector is readily available, a decentralized on-site production using semiautomated manufacturing plants still projects a production time of 12 to 14 days (10). Harnessing the nanovector technology with a refined combination of the high-capacity MaxCyte ExPERT GTx platform for electroporation and the CliniMACS Prodigy system for automated cell sorting and culture, we describe a novel manufacturing protocol that allows the production of clinically relevant 1.6 × 108 recombinant T cells in only 5 days (Fig. 4). All consumables and materials used in this process are fully GMP-compliant, and the workflow is done with tubing sets and bags in a closed system. Further developments in our protocols and an extended capacity of the culturing system will be needed to increase cells’ production. With these refinements, we believe that our method of immune cell modification can serve as a new standard for the clinical manufacturing of cellular immunotherapies, whether it is CAR or T cell receptor engraftment in T or natural killer cells. In conclusion, the clinical application of genetically modified cells demands the highest levels of vector efficiency with minimal toxicity. The capability to introduce functional CARs into naïve human T cells currently represents a promising therapeutic strategy for treating a range of cancers. The development of safer, more efficient, easier, and economically prepared, persistently expressing genetic vectors is, therefore, a principle strategic task and is the crucial prerequisite for its extended implementation and successful clinical application. Permanently maintained, episomal, and autonomously replicating DNA nanovectors provide the most suitable method of achieving this. With this shortened protocol associated with the extra-efficient DNA production by fermentation (42), we will be able to produce the materials to treat over 1000 patients from a single batch of vector while halving the time necessary to manufacture the T cell product for patients, a noteworthy improvement upon existing platforms.
MATERIALS AND METHODS
DNA vectors
pEPI-GFP vector was provided by C. Hagedorn (43). In pS/MARt-CMV-GFP, the bacterial backbone was swapped for a smaller one composed of the high copy number ColE1/pMB1/pBR322/pUC Ori and the KanR gene encoding for the aminoglycoside phosphotransferase from Tn5. In pS/MARt, the expression of the reporter gene GFP was coupled with the expression of the Puro resistance by the self-cleavage 2A peptide from porcine teschovirus-1 polyprotein. nS/MARt was obtained by exchanging the bacterial backbone containing a bacterial Ori and the antibiotic section marker, for the antibiotic-free nanobackbone described by Luke et al. (18) without changing the other functional features of the plasmid. SP-nS/MARt-B was created by introducing a splicing donor and a splicing acceptor site before and after the IFN-β S/MAR sequence, respectively. In nS/MARt-A, the S/MAR isolated from the IFN-β cluster (19) was replaced by the ApoB S/MAR (24) without altering the other functional features of the plasmid. SP-nS/MARt-EF1a was generated by replacing the CMV promoter of nS/MARt-A with the EF1a.
SP-nS/MARt-CEA was created by introducing the coding sequence for the CEA-CAR (SCA431scFv-hFc-CD28-CD3z-OX40), a gift of H. Abken (44), into the SP-nS/MARt-A plasmid, removing the GFP-2A-Puro expression cassette. The human PGK promoter was chosen for the expression of the CAR instead of the viral CMV. SP-nS/MARt-CD19-CAR and SP-nS/MARt–NY-BR1–CAR were created in a similar way replacing the GFP-2A-Puro cDNA for the CD19-CAR (FMC63scFv-CD8BBz) (45), and the NY-BR1 CAR was generated by replacing the binding moiety of the CEA-CAR (SCA431scFv) with the one having specificity for NY-BR1.
The lentiviral vector expressing the CEA-CAR was generated by introducing the expression cassette into the pRRLSIN.cPPT.PGK-GFP.WPRE plasmid, a gift from D. Trono (Addgene plasmid no. 12252), replacing the GFP expression cassette. Similarly, the CD19-CAR cDNA was cloned under the control of the human PGK promoter in the pRRLSIN vector.
ApoB MAR variants
The ApoB 3′ S/MAR (NG_042877.1) was amplified by polymerase chain reaction (PCR) with the forward (CTTCTCCACTCCTGGCAGG) and reverse (ATTTAAGTAACAGAATAAAAATGGAAACGGAGAAATTATGG) primers generating an 854-bp construct containing the highly repetitive structure and 251 bp at the 5′ end and 83 bp at the 3′ end. This construct was used to generate the nS/MARt-A vector. nS/MARt-ApoBcore was made by cloning only the central part of the sequence containing the mosaic of TAAT, TAAAT, ATTA, ATTTTA, TAAAAT, and ATTTA motifs. nS/MARt-ApoBD was found to be spontaneously generated from bacteria of the strain Stellar Competent Cells (Takara), and it showed the presence of a 383-bp ApoB MAR mapping at the 5′ end of the sequence. The core sequence was then split into two subfragments, Frag1 (277 bp) and Frag2 (248 bp) based on the motif’s enrichment. The core sequence is almost exclusively composed by A and T, and in the nS/MARt-ApoBS, we introduced through gene synthesis either a G or a C in the motifs that were not TAAT, TAAAT, ATTA, ATTTTA, TAAAAT, or ATTTA, while for the nS/MARt-ApoLscramble, we generated a random synthetic sequence by assembling the core motifs. For the generation of the nS/MARt-ApoB complement, we generated the reverse complement of the 555-bp core sequence.
Cell culture
HEK293T, HeLa, MCF-7, and HT-29 were maintained in Dulbecco’s modified Eagle’s medium (Life Technologies), while J76 and Nalm-6 were cultured in RPMI 1640 (Life Technologies) at 37°C in a 5% CO2 humidified incubator. All growth media were supplemented with penicillin (100 U/ml) and streptomycin (100 mg/ml; Life Technologies) and 10% fetal calf serum (FCS) (Gibco). Primary human CD3+ cells for small-scale and animal experiments were isolated from healthy volunteer donors using the human Pan T cell isolation kit II (Miltenyi Biotec), activated by the use of the human T Cell TransAct (Miltenyi Biotec) kit and subsequently cultured in TexMACS (Miltenyi Biotec) medium supplemented with IL-7 and IL-15, both at the final concentration of 0.1 mg/ml (Miltenyi Biotec). Concentration of primary CD3+ culture was kept at 1 × 106 cells/ml per cm2. For large-scale production, a leukapheresis product was obtained from the Heidelberg University Hospital Blood Bank (IKTZ gGmbH), which was donated by a random volunteer. T cell isolation, activation, and cultivation were performed in the CliniMACS Prodigy device using the TS520 tubing set, CliniMACS buffer (supplemented with human serum albumin), CliniMACS CD4 reagent, CliniMACS CD8 reagent, GMP-TransAct, GMP-TexMACS medium, GMP–IL-7, and GMP–IL-15 (all Miltenyi Biotec) according to the manufacturer’s instructions. Process steering was done by the internal software of the device, the process “TCT-enhanced feeding” was chosen.
Cell line transfection
HEK293T and HeLas were transfected with jetPEI DNA Transfection Reagent (Polyplus) following the manufacturer’s protocol for the delivery of DNA to adherent cells. J76 cells were transfected with the Lonza 4D Nucleofector using 2 × 106 cells, 1 mg of DNA, the SE solution, and the CL-120 pulse code. Before transfection, the cells were washed with phosphate-buffered saline (PBS) and resuspended into 20 ml of supplemented SE solution. After the electroporation, the cells were transferred into a 24-well plate.
Lentivirus production
A second-generation packaging system was used to generate lentiviral particles. Packaging plasmids pMD2.G and pCMVR8.74 were a gift from D. Trono (Addgene plasmid nos. 12259 and 22036). HEK293T cells were seeded onto 15-cm plates 2 days before transfection (6 × 106 cells per plate, five plates comprise one stack). Transfection of cells was done using 7.5 mM PEI in 150 mM NaCl and with equimolar amounts of plasmids (per stack: pRRLSIN, 112.5 μg; pMD2.G, 39.5 μg; pCMVR8.74, 73 μg). One day after transfection, the medium was replaced with 14 ml of RPMI 1640 (without phenol red and without FCS) per plate. Two days after transfection, culture supernatant (70 ml per stack) was harvested, filtered (45-μm polyvinylidene difluoride filter membranes), and concentrated using sterilized Centricon Plus-70 Ultracel PL-100 devices (Merck). Lentiviral particles from one production were pooled and stored at −80°C. Determination of infectious titer was calculated as described previously (46) by FACS measurement of CAR expression, respectively.
Primary cell transfection and transduction
Primary human CD3+ cells were transfected through electroporation either with the Neon electroporator (Life Technologies), the Lonza 4D Nucleofector device, or a MaxCyte GTx platform. Small-scale electroporation with the Neon device was performed after 2 days of activation. CD3+ cells (5 × 106) were resuspended in 100 μl of buffer T containing 15 μg of DNA and collected into a 100-μl electroporation tip. Pulsing conditions were set to 2200 V, 20 ms, and 1 pulse. Immediately after pulsing, cells were transferred to one well of a six-well plate containing prewarmed RPMI 1640 medium (without phenol red, without FCS, and without cytokines). Four hours after electroporation, cells were washed and resuspended in cultivation medium (TexMACS IL-7/15). For the Lonza device, 1 × 108 cells/ml were transfected with DNA (100 μg/ml) in the P3 solution using the FI-115 pulse code. In the MaxCyte GTx device, 1 × 108 cells/ml were transfected with DNA (125 μg/ml). Shortly after the electroporation, the cells were transferred into a culture vessel at the density of 1 × 106cell/ml in an unsupplemented TexMACS medium. Six hours after transfection, IL-7 (0.1 mg/ml) and IL-15 (0.1 mg/ml) were added. Transduction of primary CD3+ cells was done by incubation with concentrated lentiviral supernatant at day 2 after activation with a multiplicity of infection of 5 in TexMACS medium containing IL-7 and IL-15.
FACS analysis
For FACS analysis, HEK293T and HeLa were detached from their culturing dishes with Trypsin, washed three times in cold PBS, and resuspended in PBS containing 1% fetal bovine serum (FBS). Before flow cytometry analysis (LSR Fortessa, BD Sciences), the viability staining was performed by adding 4′,6-diamidino-2-phenylindole (DAPI) as a live/dead marker. Jurkat cells and primary human CD3+ were harvested from their culture vessel, washed three times in PBS, resuspended into PBS and 1% FBS, and stained with DAPI before analysis. When needed, a 30-min staining with antibodies against human CD3 (UCHT1, allophycocyanin; BD Biosciences), human CD4 (RPA-T4, fluorescein isothiocyanate; Leinco Technologies), human CD8 (SK1, phycoerythrin/cyanine7; BioLegend) was performed before adding the live/dead marker. The analysis of the data was operated with the FlowJo software, which was also used to measure the median fluorescent intensity of the populations established with the vectors expressing the reporter gene GFP. The expression of the CEA-CAR was detected with the anti-human FCg antibody (polyclonal and R-PE conjugated; Jackson ImmunoResearch), while biotinylated human CD19 (Acro Biosystems) followed by Streptavidin–Alexa Fluor 488 (Life Technologies) was used for the detection of the expression of the CD19-CAR.
Cell line establishment
Seventy-two hours after DNA delivery, transfected HEK293T and HeLa were transferred into a 10-cm2 dish containing growth medium supplemented with Puro (0.5 mg/ml, PanReac Applichem). After 3 days, the culture was refreshed by the addition of growing medium with Puro. The cells were then grown for at least 30 days in the absence of selection, and the established cell populations were expanded. Cells transfected with the pEPI vector were cultured in the presence of the antibiotic selection G418 (1 mg/ml, Roth). Upon DNA delivery, J76 cells were grown in absence of selection for 15 days; afterward, GFP+ cells were FACS-sorted (FACSAria, BD) and further expanded.
Colony-forming assay
To determine replicating episomal establishment efficiency of the different plasmids, colony-forming assays were performed (47). Twenty-four hours after DNA delivery efficiency was determined by flow cytometry, and cells expressing the reporter gene were FACS-sorted (FACSAria, BD). One hundred and 1000 GFP+ cells were seeded into a 150-mm cell culture dish in the presence of G418 or Puro, respectively. After 14 days of selection, antibiotic-resistant clones were fixed with 1% formaldehyde/PBS for 15 min at room temperature (RT) and stained with 0.5% crystal violet/25% methanol for 10 min at RT. The plates were then rinsed with water, and the colonies were counted. The establishment efficiency of a respective vector was calculated using the ratio of resistant colonies to seeded cells.
Southern blot
For DNA analysis, total DNA was extracted using the DNA Blood and Tissue Extraction (Qiagen) and quantified using a NanoDrop 2000c (Thermo Fisher Scientific) spectrophotometer. For Southern analysis, total DNA (10 to 15 mg) was digested overnight with Bam HI mixed with 10X Loading Dye and separated slowly on 0.8% agarose gel at 20 mV overnight. The gel was immersed in 0.25 M HCl for 10 min and incubated twice for 15 min in depurination buffer followed by a 15-min incubation in neutralization buffer. The gel was supported on a layer of Whatman 3MM paper with a tank containing 20× SSC nucleic acid transfer buffer. A Hybond-XL nylon membrane from Amersham Bioscience was soaked with buffer and placed on top of the gel, taking care to remove any bubbles. Once the paper towel was positioned, a weight was balanced on top, and the apparatus was left overnight to allow transfer to be completed. The following day, the apparatus was disassembled, and the nylon membrane was exposed to ultraviolet radiation for 1 min to cross-link the DNA to the membrane permanently. The GFP gene was used to generate DNA fragments that were labeled with 32P (Prime-It II Random Primer Labeling kit, Agilent Technologies) and used as a probe. The hybridization was performed in Church buffer at 65°C for 16 hours.
Rolling circle amplification
For the detection of episomally maintained DNA vectors, the TempliPhi Amplification Kit (GE Healthcare) was used following the manufacturer’s protocol. Total DNA was extracted with the DNA Blood and Tissue Extraction kit (Qiagen) and quantified using a NanoDrop 2000c (Thermo Fisher Scientific) spectrophotometer. Total DNA (20 ng) was used for each reaction. The samples were first denatured at 95°C for 3 min and quickly transferred into ice. The master mix provided in the kit was added, and the reaction was then incubated for 18 hours at 30°C. After a 2-min incubation at 65°C, the reaction was cooled to 37°C and digested with Bam HI (Thermo Fisher Scientific) for 30 min. The restriction pattern was resolved on a 0.8% agarose gel.
Quantitative PCR and copy number calculation
Quantitative PCR analysis was performed using a Light Cycler 96 instrument (Roche) and the QuantiTect SYBR Green PCR Kit (Qiagen). The reaction volume was 25 μl containing 0.3 mM of each primer. PCR reactions were performed at 35 cycles in a three-step cycling program. The reaction steps are summarized: heat inactivation, 95°C for 900 s; denaturation, 94°C for 15 s; annealing, 60°C for 25 s; extension, 72°C for 30 s for 35 cycles. Then, the melting curve was generated through the incubation of the reaction for 10 s at 95°C, 65°C for 60 s, and 97°C for 1 s before the instrument was cooled down to 4°C for storage. Plasmid copies were quantified using a single-copy housekeeping gene [glyceraldehyde-3-phosphate dehydrogenase: GCAAATCAAAGCCCTGGGACTA (forward) and AGGGCAGGAGTAAAGGTCAGAA (reverse); product of 145 bp], compared to the reporter gene GFP [CTTCCTGCACGCCATCAACAACG (forward) and GATGATCTTGTCGGTGAAGATCACG (reverse); product of 181 bp]. The specificity of the PCR reactions was determined by electrophoresis on a 0.8% agarose gel.
Single-cell protein and gene expression
To evaluate the impact of gene and protein expression across the different transfection methods, transfected cells were cryopreserved in 10% dimethyl sulfoxide and shipped to Proteona for their enhanced single-cell analysis with protein expression (ESCAPE) RNA-seq analysis. The method uses a panel of DNA-barcoded antibodies to stain cells before running those cells through a scRNA-seq protocol (10X Genomics, 3′ RNA-seq, v2). The cells were stained with CAR-T Panel v1 (Proteona) containing 49 antibodies related to T cell biology. Each set of cells was additionally stained with a unique anti-CD45 to hash the samples, and after staining, cells were combined into one lane of the 10X system. Sequencing was performed on an Illumina HiSeq, and resulting data were analyzed using Cell Ranger and the Loupe Browser (10X Genomics). Principal components analysis and heatmap visualization of significantly up-regulated genes were performed using the online tool ClustVis (48). In the heatmap, both rows and columns were clustered using Euclidean distance and average linkage. GO enrichment analysis of genes was conducted online (www.geneontology.org) by applying the PANTHER classification system (49). GSEA was performed for lentivirus and nS/MARt modified T cells with the gene lists derived from scRNA-seq results against a panel of gene sets obtained from the MSigDB (C7 immunologic gene sets collection). Analysis was performed using the desktop GSEA (v4.1.0). To identify gene sets enriched in significantly differentially expressed genes [false discovery rate (FDR) < 0.05] between CAR-T cells, we ran the GSEA software.
Cell proliferation assay
To evaluate the proliferation rate of cells transfected with nS/MARt vectors compared to mock-electroporated T cells, an assay with the intercalating dye CellTrace Violet (Thermo Fisher Scientific) was performed. The CellTrace Violet (CTV) Proliferation Kit is used for in vitro and in vivo labeling of cells to trace multiple generations using dye dilution by flow cytometry. Cells (1 × 106/ml) were resuspended in PBS and stained with CTV to a final concentration of 5 mM for 20 min at 37°C in the dark. Supplemented culture medium was then added, and the mixture was centrifuged for 10 min at 200g. The cell pellet was washed twice with PBS and 5% FCS before the cells were placed into an appropriate cell culture vessel with culture medium supplemented with cytokines. Directly after the staining, a fraction of the cells treated with CTV was used to detect generation 0 at the cytometer (detection channel: 405 to 450/50). For the proliferation assay of electroporated CD3+ cells with nS/MARt, CD3+ cells were isolated and activated for 2 days in the presence of TransAct (Miltenyi) following the manufacturer’s protocol and IL-7 and IL-15. The cells were then stained with CTV and subsequentially electroporated. One hour, 3 days, and 5 days after electroporation, cells were recorded (detection channel: 405 to 450/50) using the 7AAD (7-Aminoactinomycin D) as live/dead marker.
In vitro cytotoxicity and T cell activation assays
Real-time cytotoxicity assays were performed by impedance measurement using the xCELLigence RTCA SP device (ACEA Biosciences). Target cells were preseeded on an E-Plate 96 in triplicates (2.5 × 104 cells per well) for 24 hours, and impedance was measured every 5 min and plotted as cell index. CAR-T effector cells were added in serial dilution starting with 1 × 105 CAR+ cells per well (equals an effector-target cell ratio of 2:1), and the cell indices were normalized. The viability of target cells was calculated on the basis of the measured cell indices compared to untreated target cells as a reference and plotted as mean viability ± SD over time. The EC50 values of effector cell number at 24 hours were calculated by the RTCA software using the sigmoidal dose response (variable slope) algorithm. Forty-eight hours after CAR-T cell coculture, 30 μl of assay supernatant per well was taken, and IFN-γ concentration in the supernatant was measured using the BD OptEIA Human IFN-γ ELISA kit (BD Biosciences).
IFN-β releasing assay
The production of IFN-β from transfected cells with nS/MARt vectors and transduced with lentivirus was evaluated through LumiKine Xpress hIFN-β 2.0 ELISA (InvivoGen) following the manufacturer’s guidelines. The assay was evaluated using synthetically produced dsRNA as a positive control, and the data were interpolated using GraphPad Prism 9.
Animal experiments
CEA-CAR treatment
Six- to 8-week-old NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ mice received a subcutaneous injection of 2 × 106 HT-29 tumor cells in 100 μl of PBS on day 0. On day 7 after tumor injection and when a palpable tumor was established, a single intravenous injection of 2 × 105 CAR+ T cells per mouse in 100 μl of PBS was administered through the tail vein. Tumor size was measured every third day in three dimensions using a caliper, and tumor volume was calculated by the respective formula for equipotential ellipsoids.
NY-BR1-CAR treatment
Six- to 8-week-old NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ mice received a subcutaneous injection of 2 × 106 BOSC23-NY-BR1 tumor cells in 100 μl of PBS on day 0. On day 7 after tumor injection and when a palpable tumor was established, a single intravenous injection of 1 × 106 CAR+ T cells per mouse in 100 μl of PBS was administered through the tail vein. Tumor size was measured every third day in three dimensions using a caliper, and tumor volume was calculated by the respective formula for equipotential ellipsoids.
CD19-CAR treatment
Six- to 8-week-old NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ mice received an intravenous injection of 2 × 105 NALM6-luc tumor cells in 100 μl of PBS on day 0. Tumor burden was assessed by measurement of luciferase activity (intraperitoneal injected d-luciferin, 15 ng/ml, 200 μl) in anesthetized mice twice a week using the IVIS 200 system (Xenogen). On day 7 after tumor injection, a single intravenous injection of 1 × 106 CAR+ T cells per mouse in 100 μl of PBS was administered through the tail vein. All animal experiments were performed following the local animal welfare guidance and with permission of the regional authorities (Regierungspräsidium Karlsruhe, reference no. 35-9185.81/G-199/16).
Immunohistochemistry
Tumor samples from xenotransplantation and CAR treatment experiments were explanted at day 40 after tumor injection, immediately embedded in Tissue-Tek O.C.T. compound (Sakura), and frozen at −80°C. Cryosections of 10-μm thickness were prepared on SuperFrost Plus slides (Thermo Fisher Scientific), and fully automated staining was performed on the Bond-MAX device (Leica). Staining for CD3 was done using the NCL-L-CD3-565 reagent (clone LN10, Leica) followed by polymer-DAB detection and hematoxylin counterstain. Images were acquired and analyzed with the BZ-9000 microscope and ImageViewer software (Keyence).
Statistical analysis
The data were analyzed with the software GraphPad Prism 9. The number of replicates, the statistical tests, and the number of groups are specified for each experiment.
Mouse strains
NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ mice for xenotransplantation assays were purchased from the Jackson Laboratory, and continuous breeding was conducted under a licensed agreement in DKFZ’s (Deutsches Krebsforschungszentrum/German Cancer Research Center) animal facility.
Acknowledgments
We thank J. Williams for support with the production of nS/MARt vectors, J. Scolnick for the support during the scRNA-seq, N. Codeluppi for helping during the experimental phase, and S. Niesik for the production of the dsRNA. We also thank the Flow Cytometry Core Facility of the DKFZ for the support provided during FACS sorting. Funding: This work was funded by the Deutsches Krebsforschungs Zentrum in Heidelberg, “CEECINST/00091/2018” (to M.P.C), the Elevator Pitch Programme of the National Center for Tumor Diseases in Heidelberg, and the NCT 3.0 Proof of Concept Program. Author contributions: M.B. performed the experiments, analyzed the data, and wrote the manuscript. A.D.R., A.B., and A.T. performed the experiments. M.P.C. performed the experiments and wrote the manuscript. A.S. supervised the single-cell analysis. I.Z. and D.J. supervised the project. P.S. performed the experiments, analyzed the data, wrote the manuscript, and supervised the project. R.P.H. supervised the project and wrote the manuscript. Competing interests: M.B. and R.P.H. are inventors on a patent application related to this work filed by the Deutsches Krebsforschungszentrum and Nature Technology Corporation (no. WO2019057774A1, filed 19 September 2018, published 28 March 2019). M.B. and R.P.H. are also inventors on another patent application filed by the Deutsches Krebsforschungszentrum and Nature Technology Corporation (no. WO2019060253A1, filed 17 September 2018, published 28 March 2019). A.S. has commercial interests in Proteona. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/16/eabf1333/DC1
REFERENCES AND NOTES
- 1.Zheng P.-P., Kros J. M., Li J., Approved CAR T cell therapies: Ice bucket challenges on glaring safety risks and long-term impacts. Drug Discov. Today 23, 1175–1182 (2018). [DOI] [PubMed] [Google Scholar]
- 2.June C. H., Sadelain M., Chimeric antigen receptor therapy. N. Engl. J. Med. 379, 64–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maude S. L., Tisagenlecleucel in pediatric patients with acute lymphoblastic leukemia. Clin. Adv. Hematol. Oncol. 16, 664–666 (2018). [PubMed] [Google Scholar]
- 4.Neelapu S. S., Locke F. L., Bartlett N. L., Lekakis L. J., Miklos D. B., Jacobson C. A., Braunschweig I., Oluwole O. O., Siddiqi T., Lin Y., Timmerman J. M., Stiff P. J., Friedberg J. W., Flinn I. W., Goy A., Hill B. T., Smith M. R., Deol A., Farooq U., Sweeney P. M., Munoz J., Avivi I., Castro J. E., Westin J. R., Chavez J. C., Ghobadi A., Komanduri K. V., Levy R., Jacobsen E. D., Witzig T. E., Reagan P., Bot A., Rossi J., Navale L., Jiang Y., Aycock J., Elias M., Chang D., Wiezorek J., Go W. Y., Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 377, 2531–2544 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang M., Munoz J., Goy A., Locke F. L., Jacobson C. A., Hill B. T., Timmerman J. M., Holmes H., Jaglowski S., Flinn I. W., McSweeney P. A., Miklos D. B., Pagel J. M., Kersten M. J., Milpied N., Fung H., Topp M. S., Houot R., Beitinjaneh A., Peng W., Zheng L., Rossi J. M., Jain R. K., Rao A. V., Reagan P. M., KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 382, 1331–1342 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cho J. H., Collins J. J., Wong W. W., Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell 173, 1426–1438.e11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu C.-Y., Roybal K. T., Puchner E. M., Onuffer J., Lim W. A., Remote control of therapeutic T cells through a small molecule–gated chimeric receptor. Science 350, aab4077 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y., Di S., Shi B., Zhang H., Wang Y., Wu X., Luo H., Wang H., Li Z., Jiang H., Armored inducible expression of IL-12 enhances antitumor activity of Glypican-3–targeted chimeric antigen receptor–engineered T cells in hepatocellular carcinoma. J. Immunol. 203, 198–207 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Visan I., CAR T cells for heart disease. Nat. Immunol. 20, 1414 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Ran T., Eichmüller S. B., Schmidt P., Schlander M., Cost of decentralized CAR T-cell production in an academic nonprofit setting. Int. J. Cancer 147, 3438–3445 (2020). [DOI] [PubMed] [Google Scholar]
- 11.Casati A., Varghaei-Nahvi A., Feldman S. A., Assenmacher M., Rosenberg S. A., Dudley M. E., Scheffold A., Clinical-scale selection and viral transduction of human naïve and central memory CD8+ T cells for adoptive cell therapy of cancer patients. Cancer Immunol. Immunother. 62, 1563–1573 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh H., Moyes J. S. E., Huls M. H., Cooper L. J. N., Manufacture of T cells using the Sleeping Beauty system to enforce expression of a CD19-specific chimeric antigen receptor. Cancer Gene Ther. 22, 95–100 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Beatty G. L., Haas A. R., Maus M. V., Torigian D. A., Soulen M. C., Plesa G., Chew A., Zhao Y., Levine B. L., Albelda S. M., Kalos M., June C. H., Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2, 112–120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bozza M., Green E. W., Espinet E., De Roia A., Klein C., Vogel V., Offringa R., Williams J. A., Sprick M., Harbottle R. P., Novel non-integrating DNA Nano-S/MAR vectors restore gene function in isogenic patient-derived pancreatic tumor models. Mol. Ther. Methods Clin. Dev. 17, 957–968 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mirkovitch J., Mirault M. E., Laemmli U. K., Organization of the higher-order chromatin loop: Specific DNA attachment sites on nuclear scaffold. Cell 39, 223–232 (1984). [DOI] [PubMed] [Google Scholar]
- 16.Bode J., Goetze S., Heng H., Krawetz S. A., Benham C., From DNA structure to gene expression: Mediators of nuclear compartmentalization and dynamics. Chromosome Res. 11, 435–445 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Luke J. M., Vincent J. M., Du S. X., Gerdemann U., Leen A. M., Whalen R. G., Hodgson C. P., Williams J. A., Improved antibiotic-free plasmid vector design by incorporation of transient expression enhancers. Gene Ther. 18, 334–343 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luke J. M., Carnes A. E., Williams J. A., Development of antibiotic-free selection system for safer DNA vaccination. Methods Mol. Biol. 1143, 91–111 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Piechaczek C., Fetzer C., Baiker A., Bode J., Lipps H. J., A vector based on the SV40 origin of replication and chromosomal S/MARs replicates episomally in CHO cells. Nucleic Acids Res. 27, 426–428 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim J. H., Lee S.-R., Li L.-H., Park H.-J., Park J.-H., Lee K. Y., Kim M.-K., Shin B. A., Choi S.-Y., High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLOS ONE 6, e18556 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jenke A. C., Stehle I. M., Herrmann F., Eisenberger T., Baiker A., Bode J., Fackelmayer F. O., Lipps H. J., Nuclear scaffold/matrix attached region modules linked to a transcription unit are sufficient for replication and maintenance of a mammalian episome. Proc. Natl. Acad. Sci. U.S.A. 101, 11322–11327 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stehle I. M., Postberg J., Rupprecht S., Cremer T., Jackson D. A., Lipps H. J., Establishment and mitotic stability of an extra-chromosomal mammalian replicon. BMC Cell Biol. 8, 33 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ripin N., Boudet J., Duszczyk M. M., Hinniger A., Faller M., Krepl M., Gadi A., Schneider R. J., Šponer J., Meisner-Kober N. C., Allain F. H.-T., Molecular basis for AU-rich element recognition and dimerization by the HuR C-terminal RRM. Proc. Natl. Acad. Sci. U.S.A. 116, 2935–2944 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boulikas T., Homeotic protein binding sites, origins of replication, and nuclear matrix anchorage sites share the ATTA and ATTTA motifs. J. Cell. Biochem. 50, 111–123 (1992). [DOI] [PubMed] [Google Scholar]
- 25.Wakabayashi-Ito N., Nagata S., Characterization of the regulatory elements in the promoter of the human elongation factor-1 alpha gene. J. Biol. Chem. 269, 29831–29837 (1994). [PubMed] [Google Scholar]
- 26.Heemskerk M. H. M., Hoogeboom M., de Paus R. A., Kester M. G. D., van der Hoorn M. A., Goulmy E., Willemze R., Falkenburg J. H. F., Redirection of antileukemic reactivity of peripheral T lymphocytes using gene transfer of minor histocompatibility antigen HA-2-specific T-cell receptor complexes expressing a conserved alpha joining region. Blood. 102, 3530–3540 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Hagedorn C., Antoniou M. N., Lipps H. J., Genomic cis-acting sequences improve expression and establishment of a nonviral vector. Mol. Ther. Nucleic Acids 2, e118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong S.-P., Harbottle R. P., Genetic modification of dividing cells using episomally maintained S/MAR DNA vectors. Mol. Ther. Nucleic Acids 2, e115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tau G. Z., Cowan S. N., Weisburg J., Braunstein N. S., Rothman P. B., Regulation of IFN-γ signaling is essential for the cytotoxic activity of CD8+ T cells. J. Immunol. 167, 5574–5582 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Rourke D. M., Nasrallah M. P., Desai A., Melenhorst J. J., Mansfield K., Morrissette J. J. D., Martinez-Lage M., Brem S., Maloney E., Shen A., Isaacs R., Mohan S., Plesa G., Lacey S. F., Navenot J.-M., Zheng Z., Levine B. L., Okada H., June C. H., Brogdon J. L., Maus M. V., A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 9, eaaa0984 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seil I., Frei C., Sültmann H., Knauer S. K., Engels K., Jäger E., Zatloukal K., Pfreundschuh M., Knuth A., Tseng-Chen Y., Jungbluth A. A., Stauber R. H., Jäger D., The differentiation antigen NY-BR-1 is a potential target for antibody-based therapies in breast cancer. Int. J. Cancer 120, 2635–2642 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Jäger D., Filonenko V., Gout I., Frosina D., Eastlake-Wade S., Castelli S., Varga Z., Moch H., Chen Y.-T., Busam K. J., Seil I., Old L. J., Nissan A., Frei C., Gure A. O., Knuth A., Jungbluth A. A., NY-BR-1 is a differentiation antigen of the mammary gland. Appl. Immunohistochem. Mol. Morphol. 15, 77–83 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Cheng C., Tang N., Li J., Cao S., Zhang T., Wei X., Wang H., Bacteria-free minicircle DNA system to generate integration-free CAR-T cells. J. Med. Genet. 56, 10–17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Hibbitt O. C., Harbottle R. P., Waddington S. N., Bursill C. A., Coutelle C., Channon K. M., Wade-Martins R., Delivery and long-term expression of a 135 kb LDLR genomic DNA locus in vivo by hydrodynamic tail vein injection. J. Gene Med. 9, 488–497 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Tessadori F., Zeng K., Manders E., Riool M., Jackson D., van Driel R., Stable S/MAR-based episomal vectors are regulated at the chromatin level. Chromosome Res. 18, 757–775 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stavrou E. F., Lazaris V. M., Giannakopoulos A., Papapetrou E., Spyridonidis A., Zoumbos N. C., Gkountis A., Athanassiadou A., The β-globin replicator greatly enhances the potential of S/MAR based episomal vectors for gene transfer into human haematopoietic progenitor cells. Sci. Rep. 7, 40673 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baiker A., Maercker C., Piechaczek C., Schmidt S. B., Bode J., Benham C., Lipps H. J., Mitotic stability of an episomal vector containing a human scaffold/matrix-attached region is provided by association with nuclear matrix. Nat. Cell Biol. 2, 182–184 (2000). [DOI] [PubMed] [Google Scholar]
- 38.Liu X.-Y., Chen W., Wei B., Shan Y.-F., Wang C., IFN-induced TPR protein IFIT3 potentiates antiviral signaling by bridging MAVS and TBK1. J. Immunol. 187, 2559–2568 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Fensterl V., Sen G. C., Interferon-induced Ifit proteins: Their role in viral pathogenesis. J. Virol. 89, 2462–2468 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tyagarajan S., Spencer T., Smith J., Optimizing CAR-T cell manufacturing processes during pivotal clinical trials. Mol. Ther. Methods Clin. Dev. 16, 136–144 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schweizer M., Merten O.-W., Large-scale production means for the manufacturing of lentiviral vectors. Curr. Gene Ther. 10, 474–486 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Carnes A. E., Hodgson C. P., Luke J. M., Vincent J. M., Williams J. A., Plasmid DNA production combining antibiotic-free selection, inducible high yield fermentation, and novel autolytic purification. Biotechnol. Bioeng. 104, 505–515 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Hagedorn C., Gogol-Döring A., Schreiber S., Epplen J. T., Lipps H. J., Genome-wide profiling of S/MAR-based replicon contact sites. Nucleic Acids Res. 45, 7841–7854 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hombach A. A., Heiders J., Foppe M., Chmielewski M., Abken H., OX40 costimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4+ T cells. Onco. Targets. Ther. 1, 458–466 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maude S. L., Frey N., Shaw P. A., Aplenc R., Barrett D. M., Bunin N. J., Chew A., Gonzalez V. E., Zheng Z., Lacey S. F., Mahnke Y. D., Melenhorst J. J., Rheingold S. R., Shen A., Teachey D. T., Levine B. L., June C. H., Porter D. L., Grupp S. A., Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 371, 1507–1517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barde I., Salmon P., Trono D., Production and titration of lentiviral vectors. Curr. Protoc. Neurosci. Chapter 4, Unit 4 21 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Hagedorn C., Baiker A., Postberg J., Ehrhardt A., Lipps H. J., A colony-forming assay for determining the establishment efficiency of S/MAR-containing nonviral episomal expression vectors. Cold Spring Harb. Protoc. 2012, 706–708 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Metsalu T., Vilo J., ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 43, W566–W570 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mi H., Muruganujan A., Casagrande J. T., Thomas P. D., Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 8, 1551–1566 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/16/eabf1333/DC1