

Cells allocate carbon resources using Kog1/Raptor mediated SNF1/AMPK activation.
Abstract
In changing environments, cells modulate resource budgeting through distinct metabolic routes to control growth. Accordingly, the TORC1 and SNF1/AMPK pathways operate contrastingly in nutrient replete or limited environments to maintain homeostasis. The functions of TORC1 under glucose and amino acid limitation are relatively unknown. We identified a modified form of the yeast TORC1 component Kog1/Raptor, which exhibits delayed growth exclusively during glucose and amino acid limitations. Using this, we found a necessary function for Kog1 in these conditions where TORC1 kinase activity is undetectable. Metabolic flux and transcriptome analysis revealed that Kog1 controls SNF1-dependent carbon flux apportioning between glutamate/amino acid biosynthesis and gluconeogenesis. Kog1 regulates SNF1/AMPK activity and outputs and mediates a rapamycin-independent activation of the SNF1 targets Mig1 and Cat8. This enables effective glucose derepression, gluconeogenesis activation, and carbon allocation through different pathways. Therefore, Kog1 centrally regulates metabolic homeostasis and carbon utilization during nutrient limitation by managing SNF1 activity.
INTRODUCTION
The effective utilization of available nutrients is of paramount importance for cell growth. In eukaryotes, this utilization is tightly controlled through the regulated outputs of multiple nutrient-responsive metabolic pathways (1, 2). This is particularly true for cells and sessile organisms that reside in fluctuating nutrient environments. In such environments, elaborate sensing and signaling mechanisms integrate nutrient inputs to appropriately control the outputs of carbon and nitrogen utilization. This thereby couples the metabolic state of the cell with growth outcomes (1). In all eukaryotes, the target of rapamycin complex 1 (TORC1) and the AMP (adenosine monophosphate)–activated kinase (AMPK) pathways are two important, evolutionarily conserved nutrient signaling pathways that control cellular metabolic states and regulate growth (3, 4).
The TORC1 is a metabolic regulator of growth and is activated by amino acids, glucose, and growth factors (3, 5). In general, TORC1 is active in nutrient-replete conditions. Upon activation, TORC1 increases anabolic processes including ribosome biogenesis, nucleotide synthesis, transcription, and translation and inhibits catabolic processes such as autophagy (3, 5). The TORC1 core components and its mechanism of action are well conserved across eukaryotes. In Saccharomyces cerevisiae, TORC1 has four major components—the Tor1/Tor2 kinases, Kog1, Lst8 (which are homologous, respectively, to mTOR, Raptor, and mLst8 in mammals), and Tco89 (3, 6, 7). TORC1 is inhibited by the macrolide drug rapamycin (6). When amino acids and other nutrients are abundantly available, the Tor1/Tor2 kinases are activated and phosphorylate downstream substrates that, in turn, control growth outcomes (3). Kog1 is a scaffolding protein that holds the TORC1 together and is critical for its amino acid–dependent activation (3, 6, 8). In contrast to the well-studied growth enabling roles of TORC1 in the presence of nutrients, little is known about the roles of TORC1 or its components when cells encounter nutrient-limited environments. Under these conditions, the kinase-dependent outputs of TORC1 are inhibited, and growth is reduced (3, 5, 9, 10). This has led to a view that the TORC1 is inactive in these environments.
In contrast to TORC1, AMPK responds to “energy stress” in eukaryotes and is active during glucose/energy limitation (4). The SNF1 (Sucrose NonFermenting 1) complex is the AMPK ortholog in budding yeast (4). In general, its activation increases catabolic processes such as the tricarboxylic acid (TCA) cycle, autophagy, and fatty acid oxidation (4, 11). SNF1 activates biomass building processes under nutrient limitation, such as gluconeogenesis and storage carbohydrate synthesis (4, 11). The SNF1 complex, similar to its mammalian counterpart AMPK, is a heterotrimeric complex consisting of a catalytic α subunit (Snf1), one of the three β subunits (Gal83, Sip1, and Sip2), and a regulatory γ subunit (Snf4) (4, 11). The SNF1 complex activity is induced by the specific phosphorylation of Thr210 (present on the catalytic α subunit) by upstream kinases (11–14). In addition, in mammalian cells, AMPK is regulated by the relative amounts of adenosine triphosphate [compared to adenosine di- or mono-phosphate (ADP/AMP)], while SNF1 responds to ADP levels (4, 15). While reports suggest that SNF1 might function in glucose-replete conditions (16–18), the roles of SNF1 in such nutrient-replete conditions remain unclear.
In general, the TORC1 and AMPK pathways are viewed as opposite ends of a spectrum, which, respectively, regulate anabolic and catabolic processes (19). Understanding the metabolic inputs and molecular mechanisms that regulate these contrasting roles of TORC1 and SNF1/AMPK activity remain major areas of research. In addition to their activation under opposing nutrient conditions, environmental stress inactivates TORC1 and activates SNF1/AMPK (4, 10, 13). This has logically resulted in questions on how these pathways interact with each other. Various reports show antagonistic roles of these two pathways under different conditions in budding yeast (19). Both pathways converge to conversely regulate downstream processes such as amino acid biosynthesis (20, 21), nitrogen catabolite repression (22), and autophagy (23, 24). Typically, high TORC1 correlates with low SNF1/AMPK activity and vice versa. Nevertheless, contradictory findings suggest a more complex cross-talk between these two pathways. A recent report suggests a role for Snf1 in the formation of inactive, TORC1 bodies under extreme starvation (25), yet this remains unresolved (26). A distinct study suggests a possible, rapamycin-dependent phosphorylation of Thr210 on Snf1 (27), which also remains unresolved (28, 29). It therefore appears that the interactions between these two pathways (in yeast and mammals) are more nuanced than commonly appreciated, particularly in complex nutrient environments (19, 30–32). Our comprehension of these opposite roles of TORC1 and SNF1/AMPK come primarily from studies where cells are either in nutrient-replete or starved conditions. The roles of TORC1 and SNF1/AMPK in fluctuating nutrient environments (where cells switch between different metabolic states to maintain homeostasis) are poorly understood. In addition, the functions of the TORC1 components when glucose and amino acids are not abundant remain unknown.
Here, we report an unexpected, essential role for Kog1 in controlling growth during glucose and amino acid limitation through the appropriate activation of SNF1. We identified a hypomorph of Kog1 (Kog1 with an N-terminal epitope tag), which showed a conditional growth defect only in glucose and amino acid limitation but no defects in Tor kinase activity in nutrient replete conditions. This hypomorph revealed a TORC1 kinase–independent role of Kog1 for cell growth under nutrient limitation. Kog1 is required to appropriately balance carbon flux toward amino acid biosynthesis and gluconeogenesis. As cells encounter glucose and amino acid limitations, Kog1 regulates the requisite transcriptional modulation of amino acid biosynthesis and carbohydrate metabolism genes (and their functional metabolic outputs) by tuning SNF1 activity. Kog1 enables the appropriate activation of the SNF1-dependent transcriptional regulators (Mig1 and Cat8). This Kog1-dependent function of SNF1 in regulating carbon flux toward amino acid biosynthesis is particularly critical for growth during glucose and amino acid limitation. These findings illustrate how a Kog1-SNF1 axis enables cells to judiciously balance carbon flux toward amino acid biosynthesis and gluconeogenesis, and control growth.
RESULTS
Kog1 is required for growth under glucose and amino acid limitation
TORC1 is a multiprotein complex that includes either of the Tor kinases (Tor1 or Tor2) assembled and held together by Kog1 (Fig. 1A). Kog1 enables the activation of the Tor kinase for growth during glucose and amino acid sufficiency. However, the roles of the TORC1 (including Kog1) in conditions where glucose and amino acids become limiting are largely unknown. This is because current studies have not been able to detect TORC1 kinase activity in these conditions (9, 10).
Fig. 1. Kog1 controls growth under glucose and amino acid limitation.
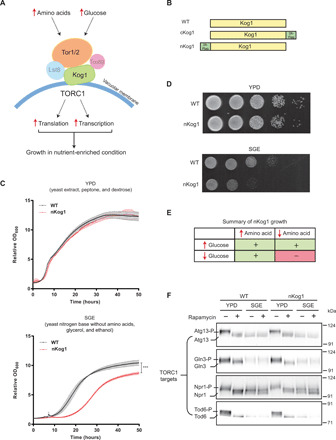
(A) A schematic showing components of the TORC1 pathway. Kog1 is a central scaffold that holds the different components of TORC1 together. (B) A schematic indicating the Kog1 strains used in this study. Kog1 was chromosomally tagged at C or N termini with a 3X-Flag epitope and named cKog1 and nKog1, respectively. WT is the unaltered, wild-type strain. (C and D) nKog1 cells show a growth defect in minimal medium with glycerol and ethanol as the carbon source (SGE). Growth curves and serial dilution spot assay for WT and nKog1 cells in YPD and SGE medium. Data are represented as means ± SD (n = 2). ***P < 0.0001 (unpaired Student’s t test). Also see fig. S1 (A to D) for growth in other conditions and fig. S1E for the growth of ∆kog1. (E) Summary of the comparative growth of nKog1 versus WT cells in different nutrient combinations (variables: glucose, glycerol/ethanol, free amino acids). “+” indicates normal growth and “−” indicates reduced growth compared to WT. (F) Canonical TOR kinase outputs are unaffected in nKog1. Western blots showing the electrophoretic mobility of Tor kinase targets—Atg13, Gln3, Npr1, and Tod6—in WT and nKog1 cells, in YPD, and after a 1-hour shift to SGE medium. Respective proteins were epitope tagged with hemagglutinin (HA) and detected using anti-HA antibodies. A representative blot (n = 2) is shown. Also see fig. S1 (F to I) for Kog1 protein, association of Kog1 with Tor kinase, and Sch9 electrophoretic mobility.
We were interested in exploring the roles of Kog1 in nutrient-limited environments. Kog1 (and its mammalian ortholog Raptor) is commonly used to isolate the entire TORC1. Typically, either a C- or N-terminal epitope tag that is amenable to protein purification is incorporated into Kog1/Raptor, and the protein is easily isolated (6, 23, 33, 34). In this regard, to examine Kog1 in cells, a 3X-Flag epitope tag was incorporated within the endogenous chromosomal locus of Kog1 at either its C or N terminus. These were, respectively, named cKog1 and nKog1 (Fig. 1B), and this nomenclature is used henceforth. As a routine assessment, the growth of these epitope-tagged strains was tested in both liquid and solid medium. This was performed under four different nutrient conditions: (i) complex, rich medium with amino acids and glucose as the carbon source (YPD); (ii) complex, rich medium with amino acids and with glycerol and ethanol as the carbon source (YPGE); (iii) synthetic minimal medium without free amino acids, supplemented with ammonium sulfate and with glucose as the carbon source (SD); and (iv) synthetic minimal medium without amino acids, supplemented with ammonium sulfate and with glycerol and ethanol as the carbon source (SGE). Prototrophic strains were used, as these cells grow efficiently in minimal medium with no free amino acids supplemented, as long as a preferred nitrogen source (ammonium sulfate/chloride) is available. The cKog1 cells behaved in a manner indistinguishable from wild-type (WT) cells under all tested nutrient conditions (fig. S1A). Unexpectedly, nKog1 cells showed a very conditional growth defect. This strain exhibited reduced growth only in minimal medium with a nonfermentable (glycerol and ethanol) carbon source and no supplemented free amino acids, i.e., SGE medium (Fig. 1, C to E, and fig. S1, B and C). Notably, in standard complex growth medium with sufficient glucose and amino acids (which are conditions where the TORC1 kinase is highly active), no overt growth defect was observed (Fig. 1, C and D). This was intriguing and suggested a role for the N-terminal region of Kog1 for growth during glucose and amino acid limitation.
To independently validate this result and to ensure that it was not due to a feature of the 3X-Flag tag, a distinct, 9X-Myc epitope tag was similarly introduced at the N terminus of Kog1 (n-myc Kog1), and serial dilution–based growth assays in YPD, YPGE, SD, and SGE conditions were performed. A similar growth defect for this strain was observed only in minimal SGE medium (fig. S1D). This collectively suggested that a small addition to the N terminus of Kog1 results in a subtle, conditional growth phenotype. To rule out a general loss of Kog1 function in these cells, we compared this phenotype with the growth of cells lacking KOG1. For this, a single copy of the KOG1 gene was deleted in diploid cells, and this was followed by sporulation and tetrad dissection to generate haploid cells (of either WT or Δkog1 genotypes) (fig. S1E). Consistent with prior reports (6), the loss of KOG1 was lethal for cells (fig. S1E). This was in complete contrast with the conditional growth defect observed in nKog1 cells. These data therefore suggested a unique requirement of the N terminus of Kog1 for growth specifically under glucose and amino acid limitation.
Upon encountering nutrient limitations, cells rewire their metabolic state to effectively use any available nutrients for biomass and energy production. Defective metabolic switching will lead to delays or challenges in adapting to this specific condition. A more careful observation of cell growth in SGE medium (shown in Fig. 1C) revealed an increased lag phase in nKog1 cells. As opposed to an overt growth defect, this observation is instead consistent with an inability to adapt to growth under glucose and amino acid limitation. We first assessed whether this defect might be due to reduced amounts of the Kog1 protein in nKog1 cells, specifically under glucose and amino acid limitation. Notably, Kog1 protein amounts remained comparable and relatively unchanged in cKog1 and nKog1 cells (fig. S1, F and G). This ruled out the possibility that a conditional lack of Kog1 protein caused this growth/adaptation phenotype in nKog1 cells.
We had therefore serendipitously identified a conditional hypomorph of Kog1 (nKog1). Under glucose and amino acid limitation, a specific function of Kog1 had been altered in nKog1 cells. We therefore decided to use this hypomorph to uncover the role of Kog1 in mediating metabolic state switching when glucose and amino acids become limiting.
TORC1 kinase activity is undetectable under glucose and amino acid limitation
Since Kog1 is required for TORC1 activation, it is possible that the conditional growth defect observed in nKog1 cells is due to an altered association of Kog1 with the Tor kinase or decreased TORC1 kinase activity. However, TORC1 kinase is thought to be active only under glucose and amino acid sufficiency. nKog1 cells have no notable growth defects in nutrient-rich medium but exhibit a delayed adaptation to growth in glucose and amino acid limitation. These data diminish the possibility of inhibited TORC1 kinase activity in nKog1 cells. We further tested this by first assessing the association of Tor1 with Kog1 by immunopurifying Kog1 from either cKog1 or nKog1 cells. In both cells, Tor1 and Kog1 remained associated with each other regardless of nutrient medium (fig. S1H).
Next, we assessed TORC1 kinase output in different media. For this, we first examined Sch9 phosphorylation in WT cells growing in YPD and after a 1-hour shift to minimal SGE medium, with or without the TORC1 inhibitor rapamycin. Upon phosphorylation in a TORC1 kinase–dependent manner, Sch9 exhibits a reduced mobility in standard, reducing SDS–polyacrylamide gel electrophoresis (SDS-PAGE) (35). In YPD, the electrophoretic mobility of Sch9 was consistent with high TORC1 kinase activity in these conditions (as indicated with controls where rapamycin was used). Notably, Sch9 phosphorylation was undetectable in SGE medium (and similar to that of cells treated with rapamycin) (fig. S1I), suggesting low TORC1 kinase activity in SGE medium. Therefore, to more rigorously assess TORC1 kinase activity, we next compared the phosphorylation status of multiple, well-established TORC1 kinase targets—Atg13, Npr1, Gln3, and Tod6—in WT and nKog1 cells. These proteins also exhibit reduced electrophoretic mobility upon TORC1 kinase–dependent phosphorylation (23, 36–38). Similar to Sch9, the electrophoretic mobility of these proteins was consistent with high TORC1 kinase activity only in YPD medium and undetectable TORC1 kinase activity in SGE medium (Fig. 1F). Notably, the mobility of these proteins (and hence their phosphorylation status) was indistinguishable in nKog1 cells compared to WT (Fig. 1F). These findings show that (i) TORC1 kinase activity is not detectable in cells shifted from YPD medium to SGE medium and that (ii) TORC1 kinase outputs are indistinguishable in nKog1 and WT cells, regardless of nutrient conditions. These data collectively indicate that Kog1 has a distinct, putatively TORC1 kinase–independent function that is critical in conditions where glucose and amino acids are limiting.
Kog1 mediates the appropriate distribution of carbon between amino acid biosynthesis and gluconeogenesis
What role therefore might Kog1 perform in cells when they encounter glucose and amino acid limitations? We reasoned that an incomplete metabolic switch in SGE medium (in nKog1 cells) would limit the production of key metabolites required for growth under this specific condition. This putative metabolic bottleneck would thereby manifest as the observed growth defect. In nonfermentable medium (glycerol and ethanol as the carbon source), limited in free amino acids, the metabolic pathways necessary for appropriate cell growth are the following: (i) de novo amino acid biosynthesis, (ii) the TCA cycle, and (iii) gluconeogenesis. To identify the bottleneck, a targeted metabolomic approach was adopted to precisely pinpoint changes in these metabolic arms. The relative, steady-state amounts of metabolites in each of these pathways were compared 5 hours after WT or nKog1 cells were shifted to minimal SGE medium. Notably, steady-state amounts of amino acids were significantly higher in nKog1 cells (Fig. 2A and fig. S2A). The metabolites of the upper arm of the TCA cycle, particularly citrate/isocitrate and α-ketoglutarate levels, were also significantly higher in nKog1 cells (Fig. 2B). Here, it is important to recognize that α-ketoglutarate is the node of diversion toward glutamate and glutamine (and therefore all other amino acid) biosynthesis. Increased α-ketoglutarate, glutamate, and glutamine levels suggest that nKog1 cells might divert carbon toward amino acid biosynthesis (Fig. 2B). This also correlates with decreased steady-state amounts of succinate (Fig. 2B) and a corresponding increase in the amounts of other amino acids in nKog1 cells.
Fig. 2. Kog1 balances carbon flux between amino acid biosynthesis and gluconeogenesis.
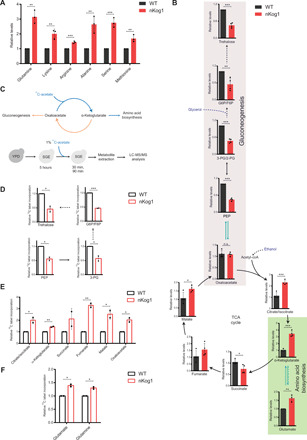
(A and B) Relative steady-state amounts of specific amino acids, gluconeogenic intermediates, and TCA cycle metabolites in WT and nKog1 cells after 5-hour shift to SGE medium. Data are represented as means ± SD (n = 3 for amino acids and n = 4 for gluconeogenic and TCA cycle intermediates). Also see fig. S2 (A to E). (C) Experimental design for 13C stable isotope label incorporation of metabolites using 13C-acetate in SGE media to estimate metabolic flux through the respective pathways. G6P, glucose-6-phosphate; F6P, fructose-6-phosphate; PEP, phosphoenolpyruvate; 3-PG, 3-phosphoglycerate, 2-PG, 2-phosphoglycerate; Acetyl-CoA, acetyl coenzyme A. (D) Relative 13C label incorporation in gluconeogenic intermediates after a 30-min pulse of 1% 13C-acetate after 5-hour shift to SGE media. Data are represented as means ± SD (n = 2). Also see fig. S2F. (E) Relative 13C label incorporation in TCA cycle intermediates in WT and nKog1 cells, measured after a 90-min pulse of 1% 13C-acetate after 5-hour shift to SGE media. Data are represented as means ± SD (n = 2). Also see fig. S2G. (F) Relative 13C label incorporation in glutamate and glutamine in WT and nKog1 cells, measured after a 90-min pulse of 1% 13C-acetate after 5-hour shift to SGE media. Data are represented as means ± SD (n = 2). For all panels, *P < 0.05, **P < 0.01, and ***P < 0.001; n.s., nonsignificant difference (unpaired Student’s t test).
Separately, gluconeogenesis is the other essential metabolic arm in glucose-limited SGE medium, and this pathway depends on sufficient oxaloacetate (which directly enters gluconeogenesis) (Fig. 2B). We therefore hypothesized that in case there was a carbon allocation imbalance, increased amino acid biosynthesis might lead to decreased gluconeogenesis. While testing this, we found that the steady-state levels of gluconeogenic intermediates were significantly reduced in nKog1 cells (Fig. 2B and fig. S2B). Trehalose, a storage carbohydrate and an end-point metabolite of gluconeogenesis (39), was notably reduced in nKog1 cells (Fig. 2B). At a later time point (18-hour shift to SGE medium), trehalose and glycogen (another gluconeogenic end-point metabolite) amounts were markedly reduced in nKog1 cells (fig. S2, C and D) and amino acid levels remained high (fig. S2E). These data suggest that in nKog1 cells, more carbon than required was routed toward amino acid biosynthesis, resulting in decreased gluconeogenesis.
To unambiguously test this possibility of altered carbon allocation, the relative metabolic flux through these pathways in WT and nKog1 cells were compared using a pulse label of an appropriate 13C-labeled carbon source. 13C label incorporation into gluconeogenic intermediates, TCA cycle intermediates, glutamate, and glutamine were measured after a pulse of 1% 13C2-acetate (Fig. 2C and Materials and Methods). Label incorporation into gluconeogenic intermediates was strongly reduced in nKog1 cells (Fig. 2D and fig. S2F). Contrastingly, label incorporation increased for α-ketoglutarate and oxaloacetate in nKog1 cells at 30 min (fig. S2G) and further increased at ~90 min for the TCA cycle metabolites as well as for glutamate and glutamine (Fig. 2, E and F). These data reveal a higher TCA cycle flux, coupled with increased amino acid synthesis and decreased gluconeogenesis in nKog1 cells. Overall, these results suggest that Kog1 maintains the balance of carbon flux toward amino acid biosynthesis or gluconeogenesis.
Kog1 temporally regulates amino acid biosynthetic and gluconeogenic gene transcripts
The metabolic characterization of nKog1 cells revealed a specific rerouting of carbon flux, resulting in a delayed adaptation to growth in medium lacking glucose and amino acids. We therefore asked whether the relative amounts of enzymes maintaining this flux were altered in nKog1 cells due to an altered transcriptional response when cells were shifted to glycerol/ethanol. For this, an RNA sequencing (RNA-seq)–based analysis was carried out for WT and nKog1 cells, comparing transcripts after 2- and 4-hour shift to minimal SGE medium (Fig. 3A). Transcript reads from biological replicates showed exceptional correlation across all conditions (correlation coefficient r2 ≥ 0.97) (fig. S3A and table S1). To define the WT transcriptional response for adapting to this nutrient environment, the differentially expressed transcripts in WT cells after 4-hour shift to SGE medium, relative to a 2-hour shift, were first identified. A cutoff of log2 fold change ≥ 1 (i.e., ∼2-fold change in expression) and a P value cutoff of ≤10−4 were set to consider differentially expressed genes for this analysis. A total of 494 genes were up-regulated, and 343 genes were down-regulated in WT cells in this duration (Fig. 3B and table S2). These genes were collectively designated as the differentially expressed transcripts occurring as cells metabolically adapt to SGE medium.
Fig. 3. Kog1 temporally regulates amino acid biosynthetic and gluconeogenic gene expression.
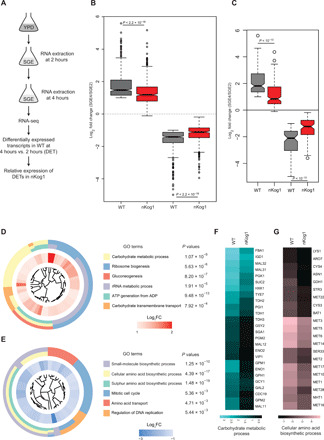
(A) Schematic showing the experimental design for comparative transcriptome analysis by RNA-seq. (B) Box plot showing global trends for up-regulated and down-regulated genes in WT and nKog1. Genes with ≥twofold change in mRNA expression (log2 fold change ≥ 1 and P value ≤ 10−4) after 4 hours in SGE, compared to 2-hour shift in WT cells, were classified as the differentially expressed transcripts (DETs). The corresponding fold changes of these DETs in nKog1 cells are shown, from identical experimental conditions. Also see fig. S3A. (C) Delayed up- or down-regulation of genes in nKog1. Box plot for transcripts (DETs) showing a differential expression in nKog1 cells with at least 1.5-fold difference (log2 fold change ≥ 0.6 and P ≤ 10−4) in relative expression compared to WT cells. (D and E) Gene Ontology (GO) mapping by biological processes for differentially expressed genes in nKog1 cells, in the up-regulated and down-regulated gene clusters, respectively. Enriched GO process terms along with their respective P values are shown. Inner circle shows expression (log2 fold) change for corresponding genes in nKog1 cells after a 4-hour shift to SGE, relative to 2-hour shift. FC, fold change. (F and G) Heatmaps showing relative changes in gene expression of transcripts in the most enriched GO processes, carbohydrate metabolic process and cellular amino acid biosynthetic process in WT and nKog1 cells. Also see fig. S3 (B to E).
For this set of transcripts, fold changes in expression after 4-hour shift to SGE medium, relative to the 2-hour shift, were then compared in nKog1 cells (Fig. 3B and table S2). Such an analysis would specifically indicate the nature of the global transcriptional response in nKog1 cells (compared to WT cells) during metabolic switching. For determining the differentially regulated transcripts in nKog1 cells, we then identified genes with at least 1.5-fold difference in their respective relative expression (after 4-hour shift relative to a 2-hour shift) in WT and nKog1 cells. 145 genes in the up-regulated cluster and 81 genes in the down-regulated cluster had significantly altered relative expression levels in nKog1 cells compared to WT (Fig. 3C and table S2). Notably, a clear trend was apparent. The transcripts that were up-regulated in WT cells showed a delayed induction in nKog1 cells. Similarly, the decreased transcripts in WT cells showed a delayed down-regulation in nKog1 cells (Fig. 3C). Therefore, a delayed transcriptional response for all these transcripts was observed in nKog1 cells (Fig. 3C).
Grouping these differentially transcribed genes using Gene Ontology (GO)–based analysis (GO Slim mapping by biological processes) revealed that a specific subset of metabolic genes was differentially regulated (had this delayed response) in nKog1 cells. The genes with delayed induction primarily grouped into carbohydrate metabolic processes as the most enriched GO process (Fig. 3D and table S3). Contrastingly, for the genes with a delayed down-regulation (i.e., higher in nKog1), the cellular amino acid biosynthetic processes were the most enriched (Fig. 3E and table S3).
A more in-depth analysis of these differentially transcribed groups of genes revealed a delayed induction of transcripts primarily involved in alternate carbon metabolism, specifically gluconeogenesis (which substantially overlaps with glycolysis) (Fig. 3F). However, the transcription of the uniquely gluconeogenic genes PCK1 and FBP1 were not affected (fig. S3B). In the cellular amino acid biosynthetic process group, transcripts of genes involved particularly in sulfur amino acid biosynthesis, along with the glutamate dehydrogenase gene Gdh1, showed a markedly delayed down-regulation in nKog1 cells (Fig. 3G and fig. S3C). The Gdh1 enzyme is positioned at a critical metabolic node. It is the major glutamate dehydrogenase in yeast and converts α-ketoglutarate to glutamate (40), thereby fueling amino acid biosynthesis. These transcriptional signatures were reflected at the protein level, as seen by comparative amounts of the Eno1 protein (significantly less in nKog1 cells, in SGE medium) (fig. S3D) and the Gdh1 protein (significantly high in nKog1 cells, in SGE medium) (fig. S3E). This transcriptional signature of nKog1 cells accurately correlates with the metabolic changes observed earlier, showing increased amino acid biosynthesis and decreased gluconeogenesis.
Therefore, Kog1 temporally regulates a precise transcriptional response required for cells to adapt to growth in minimal SGE medium. This transcriptional response enables cells to decrease the carbon allocation toward amino acid biosynthesis and to allow sufficient flux toward gluconeogenesis.
Kog1 controls SNF1/AMPK-dependent outputs during glucose and amino acid limitation
In nKog1 cells, an explicit metabolic rewiring including delay in gluconeogenic gene transcription results in reduced gluconeogenic flux. This underlies the bottleneck for growth. In this context, the SNF1 kinase complex (AMPK in mammals) is essential for growth specifically under glucose limitation (4, 11). This complex activates gluconeogenesis and other metabolic processes required to adapt to growth under such nutrient environments (Fig. 4A). We therefore wondered whether nKog1 might uniquely alter SNF1-dependent transcriptional outputs. To test this, we first compared our transcriptional dataset, with an available dataset of transcripts that are differentially regulated upon Snf1 kinase inhibition under glucose limitation (20). Although the experimental conditions used were different from the glucose and amino acid–limited conditions in this study, we observed a significant overlap (P < 10−4, Fisher’s exact test) between both these datasets (Fig. 4B and table S4). This alluded to a perturbation in SNF1-dependent transcriptional responses in nKog1 cells.
Fig. 4. Kog1 controls SNF1/AMPK dependent outputs in glucose and amino acid–limited conditions.
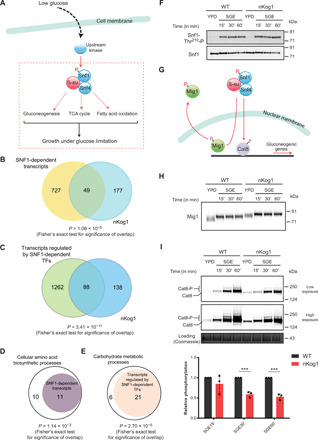
(A) Schematic illustrating the canonical outputs of SNF1/AMPK. In low glucose, Snf1 activation depends on T210 phosphorylation by upstream kinases. (B) Venn diagram depicting the overlap between SNF1-dependent transcripts (20) and differentially expressed genes in nKog1 cells. (C) Venn diagram depicting the overlap between transcripts regulated by SNF1-dependent transcription factors—Mig1, Cat8, Sip4, Rds2, and Adr1 (41)—and differentially expressed genes in nKog1 cells. Also see fig. S4A. (D and E) Plots showing a significant proportion of Kog1-dependent cellular amino acid biosynthetic processes and carbohydrate metabolic processes, genes that are known to be regulated by Snf1 and Snf1-dependent transcription factors, respectively. Also see fig. S4B. (F) Thr210 phosphorylation and levels of total Snf1 in WT and nKog1 cells in YPD and after shifting to SGE medium (time points indicated). A representative Western blot (n = 2) is shown. (G) Schematic showing downstream targets of SNF1 pathway, the Mig1 and Cat8 transcriptional regulators. (H) A representative Western blot (n = 3) for detection of Mig1 electrophoretic mobility, in WT and nKog1 cells in YPD and after shifting to SGE medium. (I) Representative Western blots and relative amounts of Cat8 phosphorylation in WT and nKog1 cells in YPD and after shifting to SGE medium. Data are represented as means ± SD (n = 3). ***P < 0.001 (unpaired Student’s t test). Also see fig. S4 (C to F).
To substantiate the nature of perturbation of SNF1-dependent outputs in nKog1 cells, we next carried out a more extensive comparison of our transcriptional dataset with those transcripts known to be regulated by SNF1-dependent transcription factors, Mig1, Cat8, Sip4, Rds2, and Adr1, irrespective of the nutrient condition (41). The SNF1-dependent regulation of these transcription factors is necessary for effective glucose derepression (11, 42). Through this analysis, a significant overlap (P < 10−10, Fisher’s exact test) was observed between Kog1-dependent transcripts and genes whose expression is regulated by SNF1-dependent transcription factors (Fig. 4C and table S4). 88 of the 226 differentially transcribed genes in the nKog1 dataset are known to be regulated by at least one of the SNF1-dependent transcription factors. Furthermore, this correlation was notable for the two main metabolic processes altered in nKog1. Approximately 52% (P < 10−3; Fisher’s exact test) of the genes of cellular amino acid biosynthetic process (that are induced in nKog1) and ~77% (P < 10−4; Fisher’s exact test) of the genes of carbohydrate metabolic processes (that are lower in nKog1) are known to be SNF1 dependent (Fig. 4, D and E). These data therefore suggested that Kog1 regulates the primary SNF1-dependent outputs.
We therefore next asked whether in nKog1 cells, direct biochemical readouts of SNF1 kinase activity were decreased. For this, we used two distinct outputs of SNF1 activity. One output is the phosphorylation of Thr210 present on Snf1, the α subunit (12, 13). This phosphorylation of T210 occurs during glucose starvation. In glucose-starved SGE medium, there was no obvious change in the relative phosphorylation of T210 of Snf1 in nKog1 cells compared to WT cells (Fig. 4F). These data suggest that the initial activation of Snf1 via the phosphorylation of T210 was likely intact in WT and nKog1 cells.
A second output of SNF1 activity, which more directly reflects the Snf1-dependent transcriptional changes observed, is the phosphorylation status of the transcription factors Mig1 and Cat8, the two major Snf1 kinase targets (Fig. 4G) (1, 11, 42, 43). A substantial fraction of Kog1-regulated transcripts and, particularly, almost all the carbohydrate metabolic processes genes are known to be Mig1 dependent (fig. S4, A and B, and table S4). The phosphorylation status of Mig1 and Cat8 was therefore examined. The SNF1-dependent phosphorylation of both these proteins correlates with their electrophoretic mobility (42, 43). For both Mig1 and Cat8, clearly altered phosphorylation of these proteins was observed in nKog1 cells (Fig. 4, H and I). When cells are shifted from glucose-rich medium to glucose-starved medium, Mig1 is inhibited by Snf1-dependent phosphorylation, and this is typically observed as a change in electrophoretic mobility on SDS-PAGE. Such an altered Mig1 mobility is seen in WT cells when shifted from rich YPD to minimal SGE medium (Fig. 4H). A glucose starvation–dependent change in Mig1 mobility was also observed in nKog1 cells (Fig. 4H), and this correlates with unimpaired Snf1-T210 phosphorylation in nKog1 cells. However, an additional, further reduced Mig1 mobility was observed in nKog1 cells, relative to WT (Fig. 4H). This therefore revealed that Mig1 function (as indicated by its electrophoretic mobility) is indeed altered in nKog1 cells.
Under glucose starvation, Snf1 regulates Cat8 function in two ways: (i) by its phosphorylation and (ii) by inducing its expression via Mig1 inhibition (11, 43). Notably, the phosphorylation-dependent mobility of Cat8 was significantly reduced in nKog1 cells in minimal SGE medium (Fig. 4I and fig. S4C). In addition, the total Cat8 protein amounts also decreased in nKog1 cells after 1-hour shift to SGE medium (fig. S4D), further correlating with decreased Mig1 inhibition.
These results collectively reveal that nKog1 cells have specifically decreased Snf1 kinase outputs when cells are shifted to SGE medium. While the glucose starvation–dependent T210 phosphorylation of Snf1 is not altered in nKog1 cells, a complete activation of Snf1 kinase does not occur in nKog1. This Snf1 function might therefore be required for the appropriate phosphorylation and corresponding inactivation and activation of Mig1 and Cat8, respectively. These data suggest that Kog1 promotes SNF1 function to appropriately inhibit Mig1 and activate Cat8. This Kog1-dependent activation of SNF1 alleviates glucose repression via a transcriptional response that enables cells to metabolically adapt to growth under glucose and amino acid limitation.
Kog1 regulates SNF1-dependent outputs irrespective of glucose and amino acid availability
The data presented thus far indicate that Kog1 mediates glucose derepression by controlling SNF1 function. This will balance the amount of carbon allocated to flow from α-ketoglutarate toward amino acid synthesis. In high glucose, yeast cells are glycolytic, and gluconeogenesis is not required. However, the metabolic arm from α-ketoglutarate and toward glutamate/amino acid synthesis will still function to maintain amino acid balance (Fig. 5A). Therefore, it is possible that this SNF1 function (regulating carbon flux toward amino acid biosynthesis) remains active even in glucose-replete conditions. Such a role for SNF1 in glucose-replete conditions has not been explored, although subtle requirements of SNF1 for cell growth have been observed in glucose-replete conditions (16–18). Any such function might go undetected in YPD (complex, high glucose) medium at least at the level of growth, as cells are already highly glycolytic and amino acids are not limiting. As shown earlier, in nKog1 cells, no overt growth defects were observed in complex, glucose-replete medium, and TORC1 kinase activity appears unperturbed. However, a difference in Mig1 mobility in nKog1 cells was observed even in YPD medium compared to WT cells (Fig. 4H). Similarly, for Cat8 phosphorylation–dependent electrophoretic mobility, a subtle difference was noticed, where only one protein band was observed in nKog1 cells instead of two bands seen in WT (Fig. 4I and fig. S4E). In addition, the total Cat8 protein amounts were higher in YPD in nKog1 cells (fig. S4F), suggesting altered Mig1 inhibition.
Fig. 5. Kog1 regulates Snf1-dependent outputs and carbon homeostasis irrespective of glucose and amino acid availability.
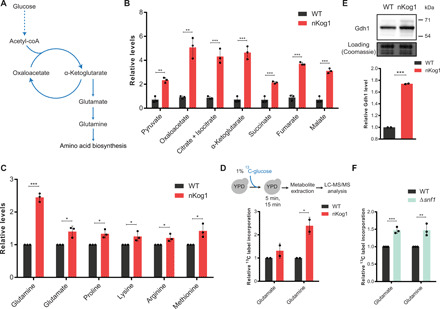
(A) Schematic showing amino acid biosynthesis from glucose via α-ketoglutarate. (B) Relative steady-state amounts of TCA cycle intermediates measured by LC-MS/MS (liquid chromatography–tandem mass spectrometry) in WT and nKog1 cells grown in YPD. Data are represented as means ± SD (n = 3). (C) Relative steady-state amounts of specific amino acids measured using LC-MS/MS in WT and nKog1 cells grown in YPD. Data are represented as means ± SD (n = 3). (D) Relative 13C label incorporation into glutamate and glutamine, measured in WT and nKog1 cells after a 15-min pulse of 1% 13C-glucose in YPD. Data are represented as means ± SD (n = 2). Also see fig. S5A. (E) A representative Western blot and relative amounts of Gdh1 protein in YPD in WT and nKog1 cells. Gdh1 was epitope tagged with HA and detected using anti-HA antibody. Data are represented as means ± SD (n = 2). (F) Relative 13C label incorporation into glutamate and glutamine measured in WT and Δsnf1 cells after a 15-min pulse of 1% 13C-glucose in YPD. Data are represented as means ± SD (n = 3). Also see fig. S5B. For all panels, *P < 0.05, **P < 0.01, and ***P < 0.001 (unpaired Student’s t test).
Hence, we asked whether the functional outputs of SNF1 are altered in YPD medium in nKog1 cells. In other words, does an imbalance in carbon flux exist in nKog1 cells even in complex medium with high glucose? To answer this, TCA cycle intermediates and amino acids were measured in cells growing in YPD. Notably, these intermediates and metabolites were significantly higher in nKog1 cells compared to WT cells (Fig. 5, B and C). Next, the carbon flux toward amino acid biosynthesis was estimated in these cells by pulsing 13C6-glucose into cells growing in YPD and estimating the relative carbon flux toward glutamate and glutamine biosynthesis (Fig. 5D and Materials and Methods). Here, increased label incorporation into glutamate and glutamine was observed in nKog1 cells (Fig. 5D and fig. S5A). This correlated with increased Gdh1 protein amounts in nKog1 cells in YPD medium (Fig. 5E). These data suggest that Kog1 is required to balance carbon flux toward glutamate/glutamine (which will increase amino acid biosynthesis), even when glucose and amino acids are not limiting.
We next assessed whether this carbon flux balance is regulated by SNF1 in WT cells, when glucose is abundant. For this, a 13C6-glucose pulse-labeling experiment in ∆snf1 cells growing in YPD was carried out, similar to that shown in Fig. 5D for nKog1. Significantly higher label incorporation into glutamate and glutamine was observed in ∆snf1 cells after the label pulse (Fig. 5F and fig. S5B). Thus, the loss of Snf1 effectively phenocopies nKog1 cells in YPD medium. These data reveal that SNF1 activity is required to maintain carbon flux balance.
Collectively, Kog1 controls appropriate SNF1 activation. Through this, cells can apportion carbon flux toward amino acid biosynthesis. This is irrespective of glucose availability, although overt growth defects will only be observed in low glucose and amino acid conditions. This is because in glucose limitation, the SNF1 function in increasing carbon flux toward gluconeogenesis becomes essential for growth.
Kog1 controls Snf1 activation to regulate growth under nutrient limitation
We asked whether Kog1 might interact with Snf1. For this, Snf1 was immunopurified and assessed for an association with Kog1. A small fraction of Kog1 consistently coimmunoprecipitated with Snf1, irrespective of the medium that cells are growing in (Fig. 6, A and B, and fig. S6A). This suggests that a fraction of the SNF1 complex associates with a fraction of Kog1 in vivo. We also assessed whether in nKog1 cells, this Snf1-Kog1 interaction is perturbed. However, the Snf1-Kog1 interaction persisted in nKog1 cells (Fig. 6C). This therefore suggests that the N terminus of Kog1 is important not for the interaction of Snf1 with Kog1 but possibly in regulating the ability of Snf1 to phosphorylate its targets.
Fig. 6. Kog1 controls Snf1 activation to regulate growth under nutrient limitation.
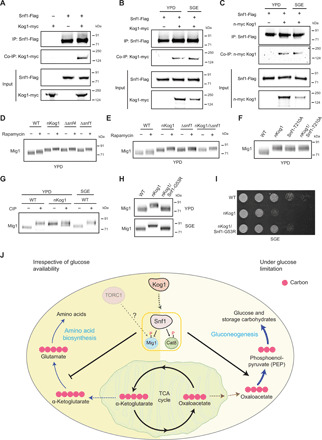
(A to C) Snf1 interacts with Kog1. Flag-tagged Snf1 was immunoprecipitated from cells in YPD or after a 1-hour shift to SGE medium and eluted, and coimmunoprecipitated myc-tagged Kog1 was detected. A representative image (n = 2) is shown. Also see fig. S6A. IP, immunoprecipitation. (D and E) A representative Western blot (n = 3) depicting the electrophoretic mobility of Mig1 in WT, nKog1, Δsnf4, Δsnf1, and nKog1/Δsnf4 cells before and after rapamycin treatment in YPD. Also see fig. S6 (B and C). (F) A representative Western blot (n = 2) depicting the electrophoretic mobility of Mig1 in WT, nKog1, Snf1-T210A, and nKog1/Snf1-T210A cells in YPD medium. (G) Cell lysates from WT and nKog1 cells in the indicated conditions were treated with phosphatase (CIP), and the electrophoretic mobility of Mig1 was assessed. A representative image (n = 3) is shown. Also see fig. S6D. (H) A representative Western blot (n = 2) depicting the electrophoretic mobility of Mig1 in WT, nKog1, and nKog1/Snf1-G53R cells in YPD and after a 30-min shift to SGE medium. (I) Comparative growth of WT, nKog1, and nKog1/Snf1-G53R cells in SGE medium. Also see fig. S6H. (J) A mechanistic model proposing Kog1-dependent regulation of SNF1. Kog1 controls SNF1 activation irrespective of nutrient availability, and reduces carbon flux toward amino acid biosynthesis. Upon glucose limitation, the Kog1-dependent activation of SNF1 (and phosphorylation of Mig1 and Cat8) regulates growth by appropriately inducing gluconeogenesis and increasing carbon flux toward the synthesis of gluconeogenic intermediates and sugars.
Since TORC1 kinase outputs were unaffected in nKog1 cells, these data indicate that Kog1 controls proper Snf1 activation independent of TORC1 kinase activity. We therefore reassessed whether Kog1-dependent regulation of SNF1 outputs is TORC1 kinase independent. We compared the electrophoretic mobility of Mig1 in rapamycin-treated cells (where TORC1 kinase activity is inhibited) with nKog1 cells, all growing in YPD medium. The mobility of Mig1 in nKog1 and rapamycin-treated cells were clearly distinct (Fig. 6D). This is consistent with the data presented earlier and indicates that Kog1 regulates Mig1 phosphorylation independent of TORC1 kinase activity. Next, we compared Mig1 phosphorylation/electrophoretic mobility in nKog1, ∆snf4 (SNF4 is the γ regulatory subunit of the SNF1 kinase complex), ∆snf1, and WT cells (Fig. 6D). Two observations are clear: First, nKog1, ∆snf4, and ∆snf1 phenocopy each other and show identical Mig1 electrophoretic mobility (Fig. 6D). This electrophoretic mobility of Mig1 is distinct from that seen in WT cells. Second, adding rapamycin reduces the mobility of Mig1 in WT cells. This trend is fully recapitulated in nKog1, ∆snf4, and ∆snf1 cells, reiterating a TORC1 kinase–independent regulation of Mig1 mobility in this condition (Fig. 6D). Further, nKog1/Δsnf1, nKog1/Δsnf4, and Δsnf1/Δsnf4 cells phenocopied nKog1 cells with respect to Mig1 electrophoretic mobility (Fig. 6E and fig. S6B). This is consistent with Kog1 and Snf1 both functioning in the same signaling pathway to control Mig1 phosphorylation and mobility. We assessed whether Mig1 phosphorylation and electrophoretic mobility is rapamycin dependent in SGE medium and found no change in Mig1 mobility upon rapamycin treatment in WT and nKog1 cells (fig. S6C). Collectively, these data reveal that Kog1 functions together with Snf1, and independent of TORC1 kinase activity, to control Mig1 phosphorylation.
We further assessed the importance of Snf1 kinase activity in regulating Mig1 phosphorylation/mobility. T210 phosphorylation is the dominant mode of Snf1 kinase activation under glucose limitation. Basal T210 phosphorylation is correlated with Snf1 kinase activity even under high-glucose condition (17). Hence, we generated Snf1-T210A mutants in WT and nKog1 cells and examined Mig1 mobility in these cells in YPD medium. Snf1-T210A cells will have low kinase activity. Notably, Mig1 mobility in Snf1-T210A and nKog1/Snf1-T210A cells were similar to nKog1 cells (Fig. 6F). These data therefore show that reduced Snf1 kinase activity decreases Mig1 mobility even under high-glucose conditions.
Therefore, we addressed the nature of Snf1-dependent regulation of Mig1 mobility putatively by decreasing Mig1 phosphorylation. We treated cell lysates with alkaline phosphatase and monitored Mig1 electrophoretic mobility in WT and nKog1 cells. Mig1 mobility in WT cells, in YPD and in SGE medium, was reduced upon phosphatase treatment, phenocopying nKog1 cells (Fig. 6G). Forms of dephosphomimic mutants of Mig1 are known to show reduced electrophoretic mobility (42). Therefore, this suggests that Mig1 is dephosphorylated in nKog1 cells. Decreased Snf1 function in nKog1 cells leads to decreased Mig1 phosphorylation. Consistent with this result suggesting reduced Snf1 kinase activity as the cause of decreased Mig1 phosphorylation, the loss of Reg1 [the regulatory subunit of Glc7 phosphatase regulating Mig1 phosphorylation (44)] had no additional effect on Mig1 mobility in this context (fig. S6D). Collectively, these data strongly suggest that Snf1 kinase activity and outputs are decreased in nKog1 cells.
We next asked whether Snf1 is up or downstream of Kog1 function. Under extreme starvation, Kog1 can form Kog1 bodies. Kog1 body formation requires the low-complexity prion-like domains of Kog1—PrD1, PrD2, and Snf1-dependent phosphorylation of serine residues (S491 and S494) on Kog1 (25). We hence asked whether these Snf1-dependent phosphorylation sites of Kog1 are important for growth in SGE medium. Using CRISPR-based approaches, we generated cells where the glutamines in the PrD regions of Kog1 and the Snf1-dependent phosphorylation sites (S491 and S494) are mutated to alanines. We assayed the growth of these cells in minimal medium (fig. S6E). Notably, these strains of Kog1 did not show any growth defect in SGE medium and fully phenocopied WT cells (fig. S6E). This therefore rules out the possibility of decreased Snf1-dependent Kog1 phosphorylation being the cause of growth defect in nKog1 cells. Collectively, these data suggest a role of Snf1 downstream of Kog1.
To establish whether that is the case, we created a hyperactive version of Snf1 (45), in nKog1 cells, using a single-point mutation on Snf1 (G53R). In nKog1/Snf1-G53R cells, we first assessed Mig1 electrophoretic mobility. These cells showed similar Mig1 electrophoretic mobility to that of WT cells in both YPD and SGE medium (Fig. 6H), revealing a rescue of Snf1-dependent Mig1 phosphorylation. Last, we compared the growth of WT, nKog1, and nKog1/Snf1-G53R cells in SGE medium. The growth defect originally observed in nKog1 cells (Fig. 1D) is rescued in nKog1/Snf1-G53R (Fig. 6I). Collectively, these data strongly suggest that Snf1 function is downstream of Kog1 and that Kog1 is required for appropriate Snf1 activation.
Overall, these results reveal a previously unidentified role for Kog1 in maintaining metabolic homeostasis by distinctly regulating SNF1 activation. This allows cells to temporally regulate the activity and expression of SNF1-dependent genes, which is critical for cells to appropriately allocate carbon toward amino acid biosynthesis and gluconeogenesis to balance carbon flux.
DISCUSSION
Cell growth is a collective output of the interconnected biochemical reactions within. Accordingly, to grow effectively in a particular nutrient environment, cells balance the allocations of carbon and nitrogen through distinct, integrated metabolic pathways. Under glucose limitation, carbon flux through gluconeogenesis allows cells to synthesize critical sugars, nucleotides, and storage carbohydrates (39). In such conditions, the catabolism of amino acids also contributes toward the gluconeogenic flux (46). However, in conditions where amino acids are also limiting, cells apportion carbon flux between two metabolic pathways: gluconeogenesis and amino acid biosynthesis. This balance becomes critical, since cell growth depends on protein and nucleotide synthesis, both of which require amino acids as building blocks. In contexts where both glucose and amino acids become limiting, we found how Kog1 (“kontroller of growth 1”) controls growth by mediating this carbon flux apportioning.
Collectively, our data support a model where Kog1 controls carbon flux homeostasis via moderating SNF1 activity (Fig. 6J). This Kog1-dependent SNF1 activation, and the subsequent phosphorylation of its downstream transcription factors (Mig1 and Cat8), initiates a response required to apportion carbon between gluconeogenesis and amino acid biosynthesis (Fig. 6J). This carbon flux balance is particularly important for growth under glucose and amino acid limitation (Fig. 6J). Our discovery hinged on the serendipitous identification of a hypomorphic mutant of Kog1 (nKog1). Introducing an N-terminal epitope tag on Kog1 results in reduced growth specifically under glucose and amino acid limitation (Fig. 1). This hypomorph showed no overt defects in growth or TORC1 kinase activity in glucose and amino acid sufficiency. This unexpected observation allowed us to investigate regulatory roles that might be independent of this dominant function of Kog1. TORC1 kinase has no observable activity in glucose and amino acid limitation (Fig. 1), suggesting a TORC1 kinase–independent role for Kog1. However, since growth directly correlates with an increase in biomass, the hypomorph revealed that Kog1 regulates biomass production by maintaining metabolic homeostasis during nutrient limitations. nKog1 cells exhibited a precise metabolic rewiring, where the balance of carbon flux toward amino acid biosynthesis increased and gluconeogenesis decreased (Fig. 2 and fig. S2). This rewiring depends on a clear-cut transcriptional program. In WT cells, gluconeogenic genes are up-regulated, with a concurrent down-regulation of amino acid biosynthetic genes during this period of adaptation to growth under glucose and amino acid limitation. This response is delayed in nKog1 cells (Fig. 3). When glucose becomes limiting, the SNF1/AMPK pathway mediates the adaptation for this condition, by activating the transcription of gluconeogenic genes and repressing amino acid biosynthetic genes (4, 11, 14, 20). Since the complete loss of Kog1 is lethal (fig. S1E) (6) and Δsnf1 cells do not grow under glucose limitation (47), neither Δkog1 nor Δsnf1 cells can be used to probe any coupling between these two pathways. The chance identification of this hypomorphic mutant of Kog1 (nKog1) therefore became invaluable for this study. Using this hypomorph, we identified that a component of SNF1 kinase activity depends upon Kog1, and nKog1 cells exhibit reduced Snf1 kinase outputs. This is observed in the functional transcriptional outputs and the phosphorylation/electrophoretic mobility of the major downstream targets of Snf1, Mig1, and Cat8 (Fig. 4). SNF1 can associate with Kog1. Notably, while a component of Mig1 regulation is rapamycin/TORC1 kinase dependent (Fig. 6), this arm of regulation is intact in nKog1 cells. In this scenario and specifically under nutrient limitation, the phosphorylation of Mig1 is SNF1 complex and Kog1 dependent and is likely TORC1 kinase independent (Fig. 6). In nKog1 cells, SNF1 activation (and Mig1 phosphorylation) is downstream of Kog1, and a hyperactive Snf1 allele rescues SNF1-dependent function and cell growth in nKog1 cells. These data therefore reveal an integrative response wherein Kog1 activates SNF1 function. This Kog1-SNF1 axis is critical when glucose and amino acids are limiting and where the careful management of carbon flux is required. This remains operational even when nutrients are abundant and where cells have to more subtly prevent carbon flux imbalances to manage growth.
Our data expand known roles for Kog1 in metabolic regulation. Kog1/Raptor has largely been studied as an essential part of TORC1 in the context of Tor kinase activity (6, 8, 33). The N terminus of Kog1 has characteristic disordered regions along with the conserved RNC domain (Raptor-like N-terminal–conserved domain). This domain helps provide substrates to TORC1 for phosphorylation, in a rapamycin-dependent manner (8). Most rapamycin-dependent phenotypes are similar to that of Kog1/Raptor-depleted cells (6, 33, 48, 49). However, emerging data hints at more nuanced roles for both TORC1 and Kog1. This includes TORC1-dependent functions in contexts where TORC1 kinase activity may not be pertinent, and such roles can no longer be ignored (34, 50, 51). TORC1 activation is tightly linked to its localization on the vacuole (3, 52, 53). Contrastingly, distinct localizations of a fraction of TORC1 components, notably Kog1, outside the vacuole are known (7, 48, 54). Recent studies unambiguously conform the existence of pools of TORC1 within the cell (51, 55). These localizations are linked to different TORC1 kinase outputs, observed either as the phosphorylation of specific downstream targets or rapamycin-resistant phosphorylation of select targets (51, 55). It now appears plausible that distinct pools of Kog1 exist, and such a pool of Kog1 is required for complete SNF1 activation. Consistent with this idea of a rapamycin-resistant pool of Kog1, nKog1 cells exhibit normal TORC1 kinase activity and rapamycin inhibition, and only a fraction of Kog1 coimmunoprecipitates with SNF1 (Fig. 6). In nKog1 cells, the N-terminal region of Kog1 reduces the ability of SNF1 to phosphorylate its major downstream targets, thereby mediating a global metabolic state shift. Since both Snf1 and Kog1 are constitutively expressed and interact even in complex, glucose-replete medium (Fig. 6), these data support a required, Kog1-dependent SNF1 activation irrespective of the nutrients available (as observed in Fig. 5). Hence, Kog1 becomes an important regulator of growth during both the availability and the absence of nutrients.
Our data reveal an important role for SNF1/AMPK in always balancing carbon allocations through different pathways, depending on the needs of the cell. This is via the nuanced control of SNF1 function by Kog1. SNF1 has been extensively studied for its roles in maintaining energy balance under glucose limitation (4, 11, 14, 15). Snf1 is activated via the phosphorylation of Thr210, which is critical for complete Snf1 function (12, 13). However, multiple reports suggest additional functions of Snf1 (beyond only the need for T210 phosphorylation), where growth, cell cycle defects, and the drug sensitivity of Δsnf1 cells are partly rescued by T210A mutant forms of Snf1 (18, 56, 57). This indicates a complex, as yet poorly studied control of SNF1 activity. This might include alternate mechanisms such as the phosphorylation of a distinct Ser214 residue on the Snf1 α subunit or the SUMOylation and ubiquitination of Snf1 (58–60). The function of T210 phosphorylation in complex rich medium is also poorly studied (61). Our results suggest that while T210-P–dependent activation of Snf1 kinase activity is required for effective Mig1 phosphorylation/inhibition, Kog1 mediates SNF1 kinase activation and subsequent downstream regulation of global metabolic outputs even in complex medium with high glucose, irrespective of T210 phosphorylation. The role of Snf1-T210–dependent Mig1 phosphorylation in rich complex medium itself has not been reported previously. However, it is known that when cells in high glucose encounter low-glucose environments, the Snf1-T210–dependent phosphorylation of Mig1 causes its delocalization from nucleus to cytosol (44, 62). This relieves Cat8 repression and further induces gluconeogenesis (43). However, the regulation of Mig1 and Cat8 itself appears to have more layers and depends on the existing nutrient/environmental stress (12, 43, 44). Even in high glucose, Mig1 interacts with Snf1 (62). The inhibition of Snf1 kinase activity (as observed by using an analog-sensitive mutant of Snf1) leads to a dispersed localization of Mig1, with Mig1 being present in both cytosol and nucleus (44). Consistent with this observation, we observe distinct Cat8 regulation in nKog1 cells, where Snf1 activity is perturbed (Fig. 4 and fig. S4). In high glucose, Cat8 amounts are high, presumably due to ineffective Mig1 inhibition resulting from altered Mig1 localization in cytosol. Contrastingly, in low glucose, Cat8 is repressed, likely due to residual nuclear localization. Cat8 is extensively phosphorylated depending on the carbon source provided, and the loss of Mig1 (relieving the repression of Cat8) increases the nonphosphorylated population of Cat8 (43). We noted an equivalent response for Cat8 induction in nKog1 cells in high glucose, observed as a single band corresponding to the nonphosphorylated form of Cat8 (Fig. 4). This indicates ineffective Mig1 inhibition. In low glucose, the Snf1-dependent Cat8 phosphorylation leads to gluconeogenic induction, including the expression of the PCK1 and FBP1 genes (11, 43). The expression of these two gluconeogenic genes correlate with T210 phosphorylation of Snf1 (12, 58). While T210 phosphorylation is unhindered in nKog1 cells, the full transcriptional response that leads to the regulated expression of genes shared between glycolysis and gluconeogenesis requires Kog1-dependent SNF1 activation. The laws of mass action dictate that these common genes will function predominantly in gluconeogenesis when glucose is limiting. In addition, this allocation of carbon toward gluconeogenesis must reciprocally affect amino acid biosynthesis. Largely overlooked studies suggest roles of SNF1 in repressing amino acid synthesis, indicating that SNF1 might reduce carbon flux toward amino acid biosynthesis in a coordinated manner (16, 20, 63). Together, our data when contextualized suggest two critical points. First, there is an unhindered SNF1-dependent acute/short-term adaptation response in nKog1 cells (including T210 phosphorylation), where the induction of PCK1, FBP1, and amino acid biosynthetic genes occurs. Second, and importantly, the Kog1-dependent SNF1 activation mediates a longer-term adaptation. This is required to induce the shared gluconeogenic/glycolytic genes and concurrently downregulate amino acid biosynthetic genes. This metabolic rewiring appropriately balances carbon allocations toward amino acid biosynthesis and gluconeogenesis to maintain growth under nutrient limitation. Therefore, SNF1 enforces a metabolic switch that enables growth throughout nutrient limitation. This SNF1-dependent carbon allocation is required to maintain metabolic homeostasis even in abundance of nutrients. Similar to nKog1 cells, Δsnf1 cells in high glucose show increased accumulation of amino acids and TCA cycle intermediates (16, 63). Collectively, these data clarify the role of SNF1 as a global metabolic regulator, with distinct functions irrespective of nutrient condition and energy limitation.
Inadequate carbon appropriations in a cell will imbalance metabolic homeostasis. This causes functional deficiencies in a cell, which manifest as subtle growth defects that become apparent only in challenging nutrient environments (64). In challenging environments, the starting carbon backbones are allocated distinctly through central and peripheral pathways of carbon, amino acid, and nucleotide metabolism. This hinges on a critical node where carbon flux is apportioned toward amino acid biosynthesis—the conversion of α-ketoglutarate to glutamate. One possible role for maintaining this carbon flux via the TCA cycle, even under conditions of glucose and amino acid sufficiency, might be in nitrogen cycling (65). Upon amino acid limitation, the conversion of α-ketoglutarate to glutamate is essential for nitrogen/ammonia assimilation. Glutamate facilitates further ammonia assimilation by providing the backbone for glutamine synthesis. Approximately 80 to 85% of the cellular nitrogen are incorporated via glutamate, and the rest through glutamine, by transaminase reactions (65). Both glutamate and glutamine donate nitrogen toward the synthesis of all other amino acids. Excess ammonia must therefore be removed by assimilating it into glutamate and glutamine, which can then be either used or secreted out (66, 67). The conversion of α-ketoglutarate to glutamate would therefore be critical during amino acid limitations. This would have subtle but important homeostatic roles in maintaining the ammonia–glutamate–amino acid balance even under conditions where glucose and free amino acids are abundant. As central growth regulators, Kog1 and SNF1 therefore must function in concert to regulate this pivotal anabolic node for maintaining carbon flux homeostasis. This regulation will be critical for cells to appropriately modulate growth outcomes in fluctuating nutrient environments.
MATERIALS AND METHODS
Yeast strains used
All strains were made in S. cerevisiae CEN.PK prototrophic, haploid “a” background (68) unless otherwise indicated. C- and N-terminal epitope-tagged and knockout strains at the endogenous locus were generated using standard polymerase chain reaction (PCR) gene tagging and deletion technique with appropriate flanking sequences to amplify drug resistance cassettes and replacing the target genomic sequence by homologous recombination. For N-terminal tagging, the drug resistance cassettes including LoxP recombination sites were removed using Cre recombinase. Strains were then sporulated, and tetrads were dissected to obtain haploid cells of the required genotype. All strains were verified by PCRs, serial dilution–based growth assays, and Western blotting for validating the appropriate insertion of an epitope tag. To make ∆kog1 cells, a CEN.PK diploid “a/α” background was used, single-copy (heterozygous) deletions were obtained, and haploid strains with the gene deletion were obtained after sporulation and tetrad dissection (Singer SporePlay dissection microscope). Mating types were confirmed by standard methods. Wherever necessary, DNA sequencing of the appropriate region was done to confirm the strains. Strains used in this study are listed in table S5.
CRISPR-based mutagenesis
Guide RNA (gRNA) was cloned into a plasmid containing gRNA scaffold with Bpl I restriction sites using standard cloning procedures. Respective strains to be mutated were transformed first with plasmid constitutively expressing Cas9. Cells positive for Cas9 plasmid expression were transformed with the gRNA plasmid and homology repair (HR) fragment and plated on YPD plates. For Kog1 PrDm1 and PrDm2 mutants, the PrD regions to be mutated were synthesized (Thermo Fisher Scientific/GeneArt). For Kog1 S491A and S494A mutation, site-directed mutagenesis PCR was used to amplify mutated sequence and used as HR fragment. Clones were then sequenced to confirm the mutation. The gRNA and oligonucleotides used as HR fragment are listed in table S6.
Media used
YPD (1% yeast extract, 2% peptone, and 2% glucose), SD (yeast nitrogen base without amino acids with 2% glucose), YPGE (1% yeast extract, 2% peptone, 1% glycerol, and 2% ethanol), SGE (yeast nitrogen base without amino acids with 1% glycerol and 2% ethanol). For solid agar plates, 2% granulated agar was added. Glucose was filter sterilized and added to the medium.
Yeast growth details
A primary culture of respective strain was set up in 4 ml of YPD medium and incubated overnight at 30°C/250 rpm. This primary culture was used to set up a secondary culture at a starting optical density at 600 nm (OD600) of ~0.2 in YPD. For changing media, after the cells reached an OD600 of ~1.0/ml (mid-log phase), they were collected (centrifuging at 1500g for 2 min at room temperature), washed in water, and resuspended in the desired media. The culture was incubated at 30°C/250 rpm for the indicated time. Rapamycin treatment was done by incubating with 200 nM rapamycin (Selleck Chemicals, S1039) for 20 min at 30°C/250 rpm.
Serial dilution “spot” growth assay
At an OD600 of ~1.0/ml (mid-log phase) in YPD, cells were collected and washed. Serial dilutions were made (OD600 of 1.0, 0.1, 0.01, 0.001, and 0.0001). 5 μl of the solution was spotted on respective agar plates, incubated at 30°C, and growth monitored.
Liquid growth curves
Primary cultures were collected and washed with water, inoculated at OD600 ~ 0.15 in 500 μl of respective medium in a 48-well plate, in biological replicates. The 48-well plate was incubated in a plate reader (Tecan, Infinite F200 PRO) at 30°C/200 rpm for 48 hours. OD600 was measured every 15 min, and values were normalized to the starting OD600 for respective strains and plotted (GraphPad Prism 6). Statistical significance was calculated using unpaired Student’s t test.
Sporulation and tetrad dissection
Respective strains were cultured in YPD overnight. 200 μl of the primary culture was collected, washed, resuspended in sporulation medium (3 g of potassium acetate and 0.2 g of raffinose in 1 liter of water), and incubated at 30°C for 2 to 5 days (until sporulation). Spores were resuspended in 50 μl of Zymolyase (0.5 mg/ml in 1 M sorbitol) and incubated at 30°C/~10 min, tetrads were dissected, and spores were scored for their mating type.
Glycogen and trehalose measurements
Ten OD600 of cells were collected and washed. Cell pellets were resuspended in 250 μl of 0.25 M Na2CO3 and incubated at 98°C/4 hours. 150 μl of 1 M acetic acid and 600 μl of 0.2 M CH3COONa were added to the solution to bring its pH to 5.2. For glycogen measurements, amyloglucosidase (1 U/ml; Sigma-Aldrich, 10115) was added to the suspension and incubated at 57°C overnight with gentle rotation. For trehalose measurements, the solution was incubated with trehalase (0.025 U/ml; Sigma-Aldrich, T8778) at 37°C overnight. The overnight samples were spun at maximum speed, and supernatants were transferred to a fresh tube. The amount of glucose in the supernatant was measured using a glucose assay kit (Sigma-Aldrich, GAGO20). The experiment was performed with >3 biological replicates, and observed values were plotted (GraphPad Prism 6). Statistical significance was calculated using unpaired Student’s t test.
Metabolite extraction and metabolites analysis using liquid chromatography–tandem mass spectrometry
Metabolite quenching and extraction was performed following protocols described in (69). Unless indicated, ~10 OD600 of cells were used in each experiment. HPLC separation was performed using Synergi 4-μm Fusion-RP 80 Å (150 × 4.6 mm) LC column (Phenomenex, 00F-4424-E0). Solvents used for amino acids and TCA intermediates detection are the following: 0.1% formic acid in water (solvent A) and 0.1% formic acid in methanol (solvent B). Solvents used for sugar phosphates detection are the following: 5 mM ammonium acetate in water (solvent A) and 5 mM ammonium acetate in acetonitrile (solvent B). For TCA cycle intermediates, metabolites were extracted from ~3 OD600 cells and derivatized as described earlier (69). Mass spectrometer used is ABSciex QTRAP 6500 with Waters Acquity UPLC system. Detection for TCA intermediates and amino acids was done in positive polarity mode. For sugar phosphates, detection was done in negative polarity mode. Mass spectrometry data were acquired using Analyst 1.6.2 software (Sciex). For analysis, MultiQuant version 3.0.1 and PeakView version 2.0 were used. Analyzed data were normalized and plotted. Statistical significance was calculated using unpaired Student’s t test.
13C carbon flux measurement using liquid chromatography–tandem mass spectrometry
For carbon flux toward amino acid biosynthesis in SGE medium, cells were cultured in YPD medium, shifted to SGE medium for 5 hours and then pulsed with 1% 13C2-acetate (Cambridge Isotope Laboratories, CLM-440). Metabolite extraction was performed as described previously (69) from ~3 OD600 of cells after 30 and 90 min of labeled acetate addition. For 13C6-glucose labeling, cells were grown in YPD with 1% unlabeled glucose until mid-log phase (~4 hours), and a pulse of 1% 13C6-glucose (Cambridge Isotope Laboratories, CLM-1396) was provided. Metabolite extraction was performed after 5- and 15-min pulsing. Samples were dissolved in respective solvents and detected as described earlier. Parent and product ions used for label detection are given in table S7. Total 13C label incorporation was calculated as the sum of all individual 13C-labeled intermediates detected for a specific metabolite (table S7). Relative label incorporation was calculated by normalizing with respective WT values in each set and plotted. Statistical significance was calculated using unpaired Student’s t test.
Protein extraction and Western blotting for detection
Cells were collected and processed for protein analysis as described earlier (64). Briefly, ~10 OD600 of cells were used for protein extraction and Western blotting. Cells were collected after adding 10% trichloroacetic acid. Subsequently, cells were processed and proteins were extracted as described (64). For Sch9, the NTCB (2-Nitro-5-thiocyanatobenzoic acid) cleavage assay (to assess phosphorylation status) was performed as described in (35). Phosphatase treatment was performed as described in (37). Total protein concentration was estimated using BCA (bicinchoninic acid) protein assay kit (G-Biosciences, 786-570). Total protein amounts were normalized, and 5 to 15 μl of the same were loaded on the gel. Different SDS-PAGE gels used for the mobility shift assays are (i) Atg13 and Tod6, 7% SDS-PAGE with TGS buffer (25 mM tris, 0.192 M glycine, and 0.1% SDS); (ii) Snf1, 10% SDS-PAGE gel with TGS buffer; (iii) Npr1, Gln3, and Cat8, 3 to 8% tris-acetate gel (Invitrogen, EA0375BOX) with tris-acetate buffer (50 mM tris, 50 mM tricine, and 0.1% SDS); (iv) Kog1, Gdh1, Mig1, and Eno1, 4 to 12% bis-tris gel (Invitrogen, NP0336BOX) with MOPS buffer (50 mM tris, 50 mM MOPS, 0.1% SDS, and 1 mM EDTA); and (v) Sch9, 10% SDS-PAGE with TGS buffer. Protein transfer: 0.45 μm of nitrocellulose membrane (GE Healthcare, 10600003) in transfer buffer (25 mM tris, 0.192 M glycine, and 20% methanol). Antibodies used are as follows: anti-HA mouse (1:2000; Sigma-Aldrich, 11583816001), anti-Flag mouse (1:2000; Sigma-Aldrich, F1804), anti-myc mouse (1:2000; Invitrogen, 132500), anti–phospho-Thr172 mammalian AMPK (1:1000; Cell Signaling Technology, 2535S), anti-mouse horseradish peroxidase (HRP)–conjugated antibody (1:4000; Cell Signaling Technology, 7076S), and anti-rabbit HRP-conjugated antibody (1:4000; Cell Signaling Technology, 7074S). For chemiluminescence detection, WesternBright ECL HRP substrate (Advansta, K12045) or femtoLUCENT PLUS chemiluminescence detection reagent (G-Biosciences, 786-003) (for the Snf1-Kog1 interaction blots) was used. Protein band intensities were normalized to total protein (Coomassie-stained gel) and quantified using ImageJ (NIH). For Cat8 phosphorylation, band intensities were normalized to total Cat8 protein band intensity.
Immunoprecipitation and coimmunoprecipitation
For Kog1 immunoprecipitation, ~50 OD600 cells were collected by centrifugation at 1500g for 2 min and washed with water. The pellet was flash-frozen in liquid nitrogen and stored at −80°C. For Snf1 immunoprecipitation, ~50 OD600 cells were collected by rapid vacuum filtration with a 0.45-μm membrane filter. The cells were removed from the membrane with a spatula and flash-frozen. The frozen pellet was stored at −80°C. For cell lysis, pellet was resuspended in 400 μl of lysis buffer [50 mM HEPES buffer (pH 7.0), 50 mM NaF, 10% glycerol, 150 mM KCl, 1 mM EDTA, 2 mM sodium orthovanadate, 2 mM phenylmethylsulfonyl fluoride, 0.1 mM leupeptin, 2 mM pepstatin, and 0.25% Tween 20] and lysed by bead beating with intermittent ice bath cooling. Supernatant was collected by centrifugation (14,000 rpm/5 min at 4°C). This supernatant (lysate) was transferred to a fresh tube, and 30 μl of lysate sample was collected and boiled in sample buffer/SB [62.5 mM tris-HCl (pH 6.8), 2.5% SDS, 0.002% bromophenol blue, 0.7135 M (5%) 2-mercaptoethanol, and 10% glycerol] at 95°C for 5 min. Lysates were precleared by incubating with 3 μl of Dynabeads protein G beads (Invitrogen, 10004D) for 10 min at 4°C with gentle rotation. The beads were pulled down using DynaMag-2 (Invitrogen, 12321D), and precleared lysates were added to 15 μl of Dynabeads protein G beads conjugated with 2 μl of anti-Flag antibody (Sigma-Aldrich, F1804) in lysis buffer. The suspension was incubated for 45 min (for Kog1) or 4 hours (for Snf1) at 4°C with gentle rotation. The beads were pulled down, and the flow through was removed. The beads were washed with lysis buffer 4× (1- to 2-min wash). The immunoprecipitated proteins were eluted by adding 40 μl of a 3X-Flag peptide (0.5 mg/ml) in lysis buffer to the beads and incubated at room temperature for 40 min with intermittent mixing. The eluted fraction was collected and boiled in SB. For Kog1 immunopurification and silver staining, 20 μl of the boiled sample was loaded onto 4 to 12% bis-tris gel (Invitrogen, NP0336BOX) and run with MOPS buffer. For silver staining, a kit protocol (Invitrogen, LC6070) was used. For Snf1 immunopurification and Western blotting, 2 μl of boiled sample was loaded onto 4 to 12% bis-tris gel for Snf1 detection. For coimmunoprecipitation studies, 20 μl of immunopurified sample was loaded onto 4 to 12% bis-tris gel and run with MOPS buffer.
RNA isolation and sequencing
RNA was isolated from ~10 OD600 cells using established hot-acid phenol methods in biological replicates at two time points (2 and 4 hours after shift to SGE). Reagents used are as follows: TES buffer [10 mM tris-HCl (pH 7.5), 10 mM EDTA, and 0.5% SDS], acid phenol (Sigma-Aldrich, P4557), chloroform, 3 M sodium acetate (pH5.5) (Invitrogen, AM9740), ethanol, and nuclease-free water (Invitrogen, AM9937). RNA was checked for integrity with 2100 Bioanalyzer system (Agilent) using the Bioanalyzer RNA 6000 Nano Assay Kit (Agilent), and libraries were prepared using the TruSeq RNA Library Preparation Kit V2 (Illumina). The samples were run on Illumina Platform HiSeq 2500 run mode with 1 × 50–base pair sequencing format. Raw data are available with National Center for Biotechnology Information–Sequence Read Archive (NCBI-SRA) (accession number PRJNA645282).
Quantification and statistical analysis
RNA-seq analysis
The 50-nucleotide oligomer, single-end reads of RNA-seq experiments were mapped to the reference genome of S. cerevisiae S288C (from the Saccharomyces Genome Database (www.yeastgenome.org/). Burrows-Wheeler Aligner and SAMtools were used to map and process data. Mapped reads with the mapping quality of ≥20 were used for downstream analysis. Read count matrix containing the number of mapped reads corresponding to genes were generated and fed into EdgeR (a Bioconductor package) for differential expression analysis. Genes differentially expressed with the fold change of at least 1.5-fold and a P value of ≤10−4 were considered for further analysis. For gene overlap analysis of Snf1-dependent targets and Kog1-dependent targets (our dataset), the genes transcriptionally regulated by Snf1 were obtained in (20). The transcripts regulated by selected transcription factors were obtained from YEASTRACT+ database (41).
GO enrichment analysis
GO analysis was carried out using g: Profiler, biological processes enriched, with corrected P values of <0.05 reported (Benjamini-Hochberg correction). GO cluster function of GOplot R package (70) was used to visualize the selected GO terms, and the list of all the enriched GO terms is shown in table S3.
Data visualization and statistical test
Heatmaps were plotted using the pheatmap package of R (R. Kolde; https://cran.r-project.org/web/packages/pheatmap/index.html). Wilcox tests and Fisher’s exact tests were used to evaluate statistical significance of expression data and transcript overlap analysis, respectively. P values obtained are shown in the respective result sections.
For bar plots, all graphs were plotted and analyzed using GraphPad Prism 6. An unpaired Student’s t test was used to estimate statistical significance, unless otherwise specified. P values and n in bar plots have been specified in corresponding figure legends.
Acknowledgments
We thank J. Heitman, U. Kolthur, M. Carlson, and B. Tu for critical comments on the manuscript. We thank all SL lab members for fruitful discussions on the manuscript. We acknowledge instrument support from the NCBS/inStem/CCAMP Mass Spectrometry Facility for liquid chromatography–tandem mass spectrometry experiments, and the NCBS Next Generation Sequencing Facility for RNA-seq. Funding: Z.R. acknowledges support from inStem/DBT for fellowship funding. R.S. acknowledges a SERB-NPDF postdoctoral fellowship (PDF/2016/001877) from the Department of Science and Technology, Government of India. S.L. acknowledges support from a DBT-Wellcome Trust India Alliance fellowship (IA/I/14/2/501523) and institutional support from inStem and the Department of Biotechnology, Government of India. Author contributions: S.L. and Z.R. conceived the project. Z.R., R.S., and S.L. designed experiments. Z.R. and M.C. performed experiments. R.S. and Z.R. analyzed gene transcription data. Z.R. and S.L. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Raw RNA-seq data are available in NCBI-SRA under the accession number PRJNA645282. Further information and requests for resources and reagents should be directed to sunil@instem.res.in. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/16/eabe5544/DC1
REFERENCES AND NOTES
- 1.Broach J. R., Nutritional control of growth and development in yeast. Genetics 192, 73–105 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palm W., Thompson C. B., Nutrient acquisition strategies of mammalian cells. Nature 546, 234–242 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.González A., Hall M. N., Nutrient sensing and TOR signaling in yeast and mammals. EMBO J. 36, 397–408 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hardie D. G., AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 8, 774–785 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Liu G. Y., Sabatini D. M., mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 21, 183–203 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loewith R., Jacinto E., Wullschleger S., Lorberg A., Crespo J. L., Bonenfant D., Oppliger W., Jenoe P., Hall M. N., Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 10, 457–468 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Reinke A., Anderson S., McCaffery J. M., Yates J. III, Aronova S., Chu S., Fairclough S., Iverson C., Wedaman K. P., Powers T., TOR complex 1 includes a novel component, Tco89p (YPL180w), and cooperates with Ssd1p to maintain cellular integrity in saccharomyces cerevisiae. J. Biol. Chem. 279, 14752–14762 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Adami A., García-Álvarez B., Arias-Palomo E., Barford D., Llorca O., Structure of TOR and its complex with KOG1. Mol. Cell 27, 509–516 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Stracka D., Jozefczuk S., Rudroff F., Sauer U., Hall M. N., Nitrogen source activates TOR (Target of Rapamycin) complex 1 via glutamine and independently of Gtr/Rag proteins. J. Biol. Chem. 289, 25010–25020 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hughes Hallett J. E., Luo X., Capaldi A. P., State transitions in the TORC1 signaling pathway and information processing in Saccharomyces cerevisiae. Genetics 198, 773–786 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hedbacker K., Carlson M., SNF1/AMPK pathways in yeast. Front. Biosci. 13, 2408–2420 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCartney R. R., Schmidt M. C., Regulation of Snf1 kinase. Activation requires phosphorylation of threonine 210 by an upstream kinase as well as a distinct step mediated by the Snf4 subunit. J. Biol. Chem. 276, 36460–36466 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Hong S. P., Carlson M., Regulation of Snf1 protein kinase in response to environmental stress. J. Biol. Chem. 282, 16838–16845 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Wilson W. A., Hawley S. A., Hardie D. G., Glucose repression/derepression in budding yeast: SNF1 protein kinase is activated by phosphorylation under derepressing conditions, and this correlates with a high AMP:ATP ratio. Curr. Biol. 6, 1426–1434 (1996). [DOI] [PubMed] [Google Scholar]
- 15.Mayer F. V., Heath R., Underwood E., Sanders M. J., Carmena D., McCartney R. R., Leiper F. C., Xiao B., Jing C., Walker P. A., Haire L. F., Ogrodowicz R., Martin S. R., Schmidt M. C., Gamblin S. J., Carling D., ADP regulates SNF1, the saccharomyces cerevisiae homolog of AMP-activated protein kinase. Cell Metab. 14, 707–714 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nicastro R., Tripodi F., Guzzi C., Reghellin V., Khoomrung S., Capusoni C., Compagno C., Airoldi C., Nielsen J., Alberghina L., Coccetti P., Enhanced amino acid utilization sustains growth of cells lacking Snf1/AMPK. Biochim. Biophys. Acta 1853, 1615–1625 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Tripodi F., Fraschini R., Zocchi M., Reghellin V., Coccetti P., Snf1/AMPK is involved in the mitotic spindle alignment in Saccharomyces cerevisiae. Sci. Rep. 8, 5853 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Portillo F., Mulet J. M., Serrano R., A role for the non-phosphorylated form of yeast Snf1: Tolerance to toxic cations and activation of potassium transport. FEBS Lett. 579, 512–516 (2005). [DOI] [PubMed] [Google Scholar]
- 19.González A., Hall M. N., Lin S. C., Hardie D. G., AMPK and TOR: The Yin and Yang of cellular nutrient sensing and growth control. Cell Metab. 31, 472–492 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Shirra M. K., McCartney R. R., Zhang C., Shokat K. M., Schmidt M. C., Arndt K. M., A chemical genomics study identifies Snf1 as a repressor of GCN4 translation. J. Biol. Chem. 283, 35889–35898 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Staschke K. A., Dey S., Zaborske J. M., Palam L. R., McClintick J. N., Pan T., Edenberg H. J., Wek R. C., Integration of general amino acid control and Target of Rapamycin (TOR) regulatory pathways in nitrogen assimilation in yeast. J. Biol. Chem. 285, 16893–16911 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bertram P. G., Choi J. H., Carvalho J., Chan T.-F., Ai W., Zheng X. F. S., Convergence of TOR-nitrogen and Snf1-glucose signaling pathways onto Gln3. Mol. Cell. Biol. 22, 1246–1252 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamada Y., Yoshino K., Kondo C., Kawamata T., Oshiro N., Yonezawa K., Ohsumi Y., Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol. Cell. Biol. 30, 1049–1058 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yi C., Tong J., Lu P., Wang Y., Zhang J., Sun C., Yuan K., Xue R., Zou B., Li N., Xiao S., Dai C., Huang Y., Xu L., Li L., Chen S., Miao D., Deng H., Li H., Yu L., Formation of a Snf1-Mec1-Atg1 module on mitochondria governs energy deprivation-induced autophagy by regulating mitochondrial respiration. Dev. Cell 41, 59–71.e4 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Hughes Hallett J. E., Luo X., Capaldi A. P., Snf1/AMPK promotes the formation of Kog1/Raptor-bodies to increase the activation threshold of TORC1 in budding yeast. eLife 4, e09181 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prouteau M., Desfosses A., Sieben C., Bourgoint C., Mozaffari N. L., Demurtas D., Mitra A. K., Guichard P., Manley S., Loewith R., TORC1 organized in inhibited domains (TOROIDs) regulate TORC1 activity. Nature 550, 265–269 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Orlova M., Kanter E., Krakovich D., Kuchin S., Nitrogen availability and TOR regulate the Snf1 protein kinase in Saccharomyces cerevisiae. Eukaryot. Cell 5, 1831–1837 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nomura W., Maeta K., Kita K., Izawa S., Inoue Y., Methylglyoxal activates Gcn2 to phosphorylate eIF2α independently of the TOR pathway in Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 86, 1887–1894 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Kingsbury J. M., Sen N. D., Cardenas M. E., Branched-chain aminotransferases control TORC1 signaling in Saccharomyces cerevisiae. PLOS Genet. 11, e1005714 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang J., Vaga S., Chumnanpuen P., Kumar R., Vemuri G. N., Aebersold R., Nielsen J., Mapping the interaction of Snf1 with TORC1 in Saccharomyces cerevisiae. Mol. Syst. Biol. 7, 545 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pezze P. D., Ruf S., Sonntag A. G., Langelaar-Makkinje M., Hall P., Heberle A. M., Navas P. R., van Eunen K., Tölle R. C., Schwarz J. J., Wiese H., Warscheid B., Deitersen J., Stork B., Fäßler E., Schäuble S., Hahn U., Horvatovich P., Shanley D. P., Thedieck K., A systems study reveals concurrent activation of AMPK and mTOR by amino acids. Nat. Commun. 7, 13254 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang C.-S., Jiang B., Li M., Zhu M., Peng Y., Zhang Y.-L., Wu Y.-Q., Li T. Y., Liang Y., Lu Z., Lian G., Liu Q., Guo H., Yin Z., Ye Z., Han J., Wu J.-W., Yin H., Lin S.-Y., Lin S.-C., The lysosomal v-ATPase-ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 20, 526–540 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Sabatini D.-M., Kim D.-H., Sarbassov D. D., Ali S. M., King J. E., Latek R. R., Erdjument-Bromage H., Tempst P., mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Nakashima A., Maruki Y., Imamura Y., Kondo C., Kawamata T., Kawanishi I., Takata H., Matsuura A., Lee K. S., Kikkawa U., Ohsumi Y., Yonezawa K., Kamada Y., The yeast Tor signaling pathway is involved in G2/M transition via polo-kinase. PLOS ONE 3, e2223 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Urban J., Soulard A., Huber A., Lippman S., Mukhopadhyay D., Deloche O., Wanke V., Anrather D., Ammerer G., Riezman H., Broach J. R., De Virgilio C., Hall M. N., Loewith R., Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol. Cell 26, 663–674 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Beck T., Hall M. N., The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature 402, 689–692 (1999). [DOI] [PubMed] [Google Scholar]
- 37.Schmidt A., Beck T., Koller A., Kunz J., Hall M. N., The TOR nutrient signalling pathway phosphorylates NPR1 and inhibits turnover of the tryptophan permease. EMBO J. 17, 6924–6931 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huber A., Bodenmiller B., Uotila A., Stahl M., Wanka S., Gerrits B., Aebersold R., Loewith R., HuberBernd A., Characterization of the rapamycin-sensitive phosphoproteome reveals that Sch9 is a central coordinator of protein synthesis. Genes Dev. 23, 1929–1943 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lillie S. H., Pringle J. R., Reserve carbohydrate metabolism in Saccharomyces cerevisiae: Responses to nutrient limitation. J. Bacteriol. 143, 1384–1394 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeLuna A., Avendaño A., Riego L., González A., NADP-glutamate dehydrogenase isoenzymes of Saccharomyces cerevisiae: Purification, kinetic properties, and physiological roles. J. Biol. Chem. 276, 43775–43783 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Monteiro P. T., Oliveira J., Pais P., Antunes M., Palma M., Cavalheiro M., Galocha M. O., Godinho C. P., Martins L. C., Bourbon N., Mota M. N., Ribeiro R. A., Viana R., Sá-Correia I., Teixeira M. C., YEASTRACT+: A portal for cross-species comparative genomics of transcription regulation in yeasts. Nucleic Acids Res. 48, D642–D649 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Treitel M. A., Kuchin S., Carlson M., Snf1 protein kinase regulates phosphorylation of the Mig1 repressor in Saccharomyces cerevisiae. Mol. Cell. Biol. 18, 6273–6280 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Randez-Gil F., Bojunga N., Proft M., Entian K. D., Glucose derepression of gluconeogenic enzymes in Saccharomyces cerevisiae correlates with phosphorylation of the gene activator Cat8p. Mol. Cell. Biol. 17, 2502–2510 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shashkova S., Wollman A. J. M., Leake M. C., Hohmann S., The yeast Mig1 transcriptional repressor is dephosphorylated by glucose-dependent and -independent mechanisms. FEMS Microbiol. Lett. 364, fnx133 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Estruch F., Treitel M. A., Yang X., Carlson M., N-terminal mutations modulate yeast SNF1 protein kinase function. Genetics 132, 639–650 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Owen O. E., Kalhan S. C., Hanson R. W., The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 277, 30409–30412 (2002). [DOI] [PubMed] [Google Scholar]
- 47.Zimmermann F. K., Kaufmann I., Rasenberger H., Haubetamann P., Genetics of carbon catabolite repression in Saccharomycess cerevisiae: Genes involved in the derepression process. Mol. Gen. Genet. 151, 95–103 (1977). [DOI] [PubMed] [Google Scholar]
- 48.Araki T., Uesono Y., Oguchi T., Toh-e A., LAS24/KOG1, a component of the TOR complex 1 (TORC1), is needed for resistance to local anesthetic tetracaine and normal distribution of actin cytoskeleton in yeast. Genes Genet. Syst. 80, 325–343 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Hara K., Maruki Y., Long X., Yoshino K.-i., Oshiro N., Hidayat S., Tokunaga C., Avruch J., Yonezawa K., Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110, 177–189 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Kim K., Qiang L., Hayden M. S., Sparling D. P., Purcell N. H., Pajvani U. B., MTORC1-independent Raptor prevents hepatic steatosis by stabilizing PHLPP2. Nat. Commun. 7, 10255 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fan S. J., Snell C., Turley H., Li J. L., McCormick R., Perera S. M. W., Heublein S., Kazi S., Azad A., Wilson C., Harris A. L., Goberdhan D. C. I., PAT4 levels control amino-acid sensitivity of rapamycin-resistant mTORC1 from the Golgi and affect clinical outcome in colorectal cancer. Oncogene 35, 3004–3015 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takeda E., Jin N., Itakura E., Kira S., Kamada Y., Weisman L. S., Noda T., Matsuura A., Vacuole-mediated selective regulation of TORC1-Sch9 signaling following oxidative stress. Mol. Biol. Cell 29, 510–522 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sturgill T. W., Cohen A., Diefenbacher M., Trautwein M., Martin D. E., Hall M. N., TOR1 and TOR2 have distinct locations in live cells. Eukaryot. Cell 7, 1819–1830 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brown C. R., Hung G. C., Dunton D., Chiang H. L., The TOR complex 1 is distributed in endosomes and in retrograde vesicles that form from the vacuole membrane and plays an important role in the vacuole import and degradation pathway. J. Biol. Chem. 285, 23359–23370 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hatakeyama R., Péli-Gulli M. P., Hu Z., Jaquenoud M., Garcia Osuna G. M., Sardu A., Dengjel J., De Virgilio C., Spatially distinct pools of TORC1 balance protein homeostasis. Mol. Cell 73, 325–338.e8 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Pessina S., Tsiarentsyeva V., Busnelli S., Vanoni M., Alberghina L., Coccetti P., Snf1/AMPK promotes S-phase entrance by controlling CLB5 transcription in budding yeast. Cell Cycle 9, 2189–2200 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Hedbacker K., Carlson M., Regulation of the nucleocytoplasmic distribution of Snf1-Gal83 protein kinase. Eukaryot. Cell 5, 1950–1956 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McCartney R. R., Garnar-Wortzel L., Chandrashekarappa D. G., Schmidt M. C., Activation and inhibition of Snf1 kinase activity by phosphorylation within the activation loop. Biochim. Biophys. Acta 1864, 1518–1528 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Simpson-Lavy K. J., Johnston M., SUMOylation regulates the SNF1 protein kinase. Proc. Natl. Acad. Sci. 110, 17432–17437 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wilson M. A., Koutelou E., Hirsch C., Akdemir K., Schibler A., Barton M. C., Dent S. Y. R., Ubp8 and SAGA regulate Snf1 AMP kinase activity. Mol. Cell. Biol. 31, 3126–3135 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Coccetti P., Nicastro R., Tripodi F., Conventional and emerging roles of the energy sensor Snf1 / AMPK in Saccharomyces cerevisiae. Microb. Cell 5, 482–494 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ahuatzi D., Riera A., Peláez R., Herrero P., Moreno F., Hxk2 regulates the phosphorylation state of Mig1 and therefore its nucleocytoplasmic distribution. J. Biol. Chem. 282, 4485–4493 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Tripodi F., Castoldi A., Nicastro R., Reghellin V., Lombardi L., Airoldi C., Falletta E., Maffioli E., Scarcia P., Palmieri L., Alberghina L., Agrimi G., Tedeschi G., Coccetti P., Methionine supplementation stimulates mitochondrial respiration. Biochim. Biophys. Acta 1865, 1901–1913 (2018). [DOI] [PubMed] [Google Scholar]
- 64.Gupta R., Walvekar A. S., Liang S., Rashida Z., Shah P., Laxman S., A tRNA modification balances carbon and nitrogen metabolism by regulating phosphate homeostasis. eLife 8, e44795 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Magasanik B., Ammonia assimilation by Saccharomyces cerevisiae. Eukaryot. Cell 2, 827–829 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Spinelli J. B., Yoon H., Ringel A. E., Jeanfavre S., Clish C. B., Haigis M. C., Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 358, 941–946 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hess D. C., Lu W., Rabinowitz J. D., Botstein D., Ammonium toxicity and potassium limitation in yeast. PLOS Biol. 4, e351 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.J. P. Van Dijken, J. Bauer, L. Brambilla, P. Duboc, J. M. Francois, C. Gancedo, M. L. F. Giuseppin, J. J. Heijnen, M. Hoare, H. C. Lange, E. A. Madden, P. Niederberger, J. Nielsen, J. L. Parrou, T. Petit, D. Porro, M. Reuss, N. Van Riel, M. Rizzi, H. Y. Steensma, C. T. Verrips, J. Vindeløv, J. T. Pronk, Enzyme and Microbial Technology (Elsevier Science Inc., 2000), vol. 26, pp. 706–714. [DOI] [PubMed] [Google Scholar]
- 69.Walvekar A., Rashida Z., Maddali H., Laxman S., A versatile LC-MS/MS approach for comprehensive, quantitative analysis of central metabolic pathways. Wellcome Open Res. 3, 122 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Walter W., Sánchez-Cabo F., Ricote M., GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics 31, 2912–2914 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/16/eabe5544/DC1