

The multilevel fatigue resistance mechanism of tough and self-healing hydrogels with a hierarchical structure was revealed.
Abstract
We investigate the fatigue resistance of chemically cross-linked polyampholyte hydrogels with a hierarchical structure due to phase separation and find that the details of the structure, as characterized by SAXS, control the mechanisms of crack propagation. When gels exhibit a strong phase contrast and a low cross-linking level, the stress singularity around the crack tip is gradually eliminated with increasing fatigue cycles and this suppresses crack growth, beneficial for high fatigue resistance. On the contrary, the stress concentration persists in weakly phase-separated gels, resulting in low fatigue resistance. A material parameter, λtran, is identified, correlated to the onset of non-affine deformation of the mesophase structure in a hydrogel without crack, which governs the slow-to-fast transition in fatigue crack growth. The detailed role played by the mesoscale structure on fatigue resistance provides design principles for developing self-healing, tough, and fatigue-resistant soft materials.
INTRODUCTION
The soft and wet nature of hydrogels endows them with great application potentials in tissue engineering such as artificial soft tissues used for replacing damaged ones (1, 2). However, creating hydrogels with simultaneously excellent toughness, self-healing properties, and fatigue resistance that are similar to load-bearing soft biotissues remains a challenging task. Initiated by the research on double network gels (3–5), tremendous progress has been made in creating hydrogels with toughness comparable to that of cartilage by introducing covalent or noncovalent sacrificial bonds in polymer networks (6–10). However, recent studies on several synthetic tough hydrogels based on noncovalent sacrificial bonds show that these tough hydrogels exhibit a relatively poor fatigue resistance similar to that of a purely elastic network (11–13). This is in stark contrast with biological tissues that show excellent fatigue resistance along with high toughness (14, 15). These results suggest that the hierarchical structures, as seen in the biological tissues, could be beneficial for fatigue resistance. Recent studies have shown that the fatigue resistance of soft and stretchable materials is largely enhanced by embedding hard components, such as crystalline domains (16–18) and fibers (19–21). It is considered that the energy per unit area required to fracture the packed hierarchical structures is enhanced by pinning the crack tip at the hard component (22).
Recently, we found that tough and self-healing polyampholyte hydrogels (PA gels) with a hierarchical structure show a high fatigue resistance (23). The hierarchical structure of PA gels consists of reversible ionic bonds at the ~0.1-nm scale, a transient polymer network at the ~1-nm scale, a permanent polymer network at the ~10-nm scale, and a hard/soft microphase separated network at the ~100-nm scale (Fig. 1). The transient polymer network is formed by ionic bonds, and the permanent polymer network is formed by chemical cross-linking and trapped topological entanglements. The hard/soft microphase separated network consists of bicontinuous dense/sparse polymer phases in water. The polymer strands are in their collapsed globule conformation (24). The PA gels exhibit an extremely slow crack growth mode when the applied energy release rate (G) is above the fatigue threshold G0 and that slow propagation mode is maintained until a transition point Gtran that has a value several times that of G0, at which point a fast crack propagation mode is observed. The high fatigue resistance of the PA gels is attributed to such a multilevel fatigue behavior. This behavior of PA gels is very different from that of conventional hydrogels (13) in which the crack propagation rate increases rapidly with increasing G and only a fast crack propagation mode appears above the threshold value G0.
Fig. 1. Illustration of the hierarchical structure of tough and self-healing PA gels.
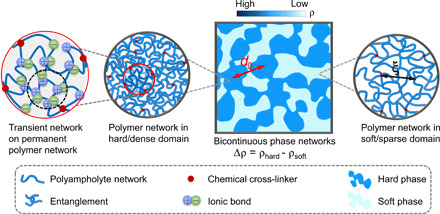
The gels contain reversible ionic bonds at the ~0.1-nm scale, a transient polymer network at the ~1-nm scale (indicated by black dotted circle), a permanent polymer network at the ~10-nm scale (indicated by red circle), and a bicontinuous hard/soft phase network at the ~100-nm scale. The polymer strands are in their globule conformation. The nanoscale mesh size of the permanent polymer network ξ depends on the effective cross-linking density of the polymer network νe from both chemical cross-linking and trapped topological entanglements. ξ can be tuned by the chemical cross-linker density CMBAA and monomer concentration Cm during the synthesis of the PA gels. The characteristic length of the phase network d0 and of the phase density contrast Δρ depend on ξ and on the dialysis temperature Tdial of the PA gels.
In this work, we intend to clarify why the slow crack propagation mode is observed for the materials with hierarchical structures and what determines the slow-to-fast propagation transition. We also intend to clarify the role of trapped topological entanglements on the intrinsic fatigue threshold G0. We assume that the bicontinuous hard/soft phase networks play the key role in suppressing the crack growth, whereas the hard/soft phase contrast might govern such multilevel fatigue resistance behavior. To test this hypothesis, in this work, we study the effect of the phase contrast of the mesoscale networks on the fatigue resistance of PA gels. The hard/soft phase contrast in the gels was tuned by varying the effective permanent cross-linking density of the nanoscale polymer networks of the gels during the synthesis step. Combining the results of small-angle x-ray scattering (SAXS) measurements, fatigue tests, and birefringence characterization, we will discuss the inherent correlations between the characteristic mechanical fatigue parameters and the hierarchical structure parameters. We will explore, in particular, the effect of the level of contrast of the microphase separated network on the ability to blunt the crack during propagation in fatigue, the existence of a material parameter controlling the transition from slow to fast crack propagation, and the effect of trapped topological entanglements on the fatigue threshold G0.
RESULTS
Tuning the mesoscale phase contrast
Our previous study showed that the level of phase contrast and characteristic phase length (d0) of PA gels depend on the effective chain density of permanent polymer network νe and temperature (Tdial) used to dialyze the small counterions from the gels during the sample preparation (24, 25). νe is due to the presence of chemical cross-links and trapped topological entanglements. In polymer solutions (26), the concentration of topological entanglements between polymer chains (Ce) scales with the polymer concentration C by Ce~C2.3. In the chemically cross-linked gels, the topological entanglements are permanently trapped and contribute to the elastic modulus similarly to the chemical cross-links (27). Accordingly, tuning the monomer concentration Cm at the gel synthesis step can change the polymer concentration and thereby the concentration of the trapped topological entanglements. In this work, we vary the chemical cross-linker density (CMBAA) to tune chemical cross-linking density and the total monomer concentration (Cm) to tune the topological entanglement density (24, 28). Two series of samples were prepared by one-step random copolymerization of the anionic monomer sodium p-styrenesulfonate (NaSS) and the cationic monomer methyl chloride quarternized N,N-dimethylamino ethylacrylate (DMAEA-Q). The sample series PA-2.5-CMBAA was prepared by varying CMBAA from 0 to 1.0 mole percent (mol %) relative to Cm at fixed Cm = 2.5 M; the sample series PA-Cm-0.1 was prepared by varying Cm from 1.75 to 2.5 M at a fixed CMBAA = 0.1 mol %. After copolymerization, the gels were immersed in deionized water until equilibrium was reached at Tdial = 30°C. During this process, the counterions were dialyzed away from the hydrogel, and the gels deswelled as a result of the Coulomb attraction between the opposite charges on polymer chains and hydrophobic interaction. The polymer volume fraction of the equilibrated PA gels after dialysis was 44.7 to 46.8 volume %, almost independent of the initial formulation, which indicates that the polymer strands are in their collapsed globule conformation (29). According to polymer theory (26, 29), for collapsed globule chains, the mesh size of the network (ξ) is related to the number of repeat units of a polymer strand between cross-linking points (Ne) through ξ ≅ bNe1/3, where b is the size of repeat unit. The polymer volume fraction in the gel ϕ is the ratio between the actual volume of a polymer strand and the volume it pervades. Therefore, ϕ ≅ (b3Ne)/ξ3 ~ (b3Ne)/(bNe1/3)3 ~ Ne0, independent of Ne. Therefore, in this collapsed conformation, the polymer volume fraction does not change with effective chain length. In the following experiments, the equilibrated gels were used. It is worthwhile to note that the structural and property changes of the PA gels reported in the following discussion are not due to the change in polymer volume fraction in the gel because it remains nearly the same for all the gels.
These gels show a similar dynamic modulus in the high-frequency regime from the ionic bonds, but the plateau modulus (μ) at low frequency (2 × 10−5 rad/s) increases with CMBAA and Cm following a relation of μ ~ (CMBAA + αCm2.3) for all the samples including that without chemical cross-linker (CMBAA = 0) (fig. S1 and table S1). The scaling exponent 2.3 is consistent with polymer solution theory (26) of Ce ~ Cm2.3, where Ce is the cross-linking concentration caused by trapped topological entanglements. This result indicates that the average number density of elastically effective chain density νe, which is proportional to the shear modulus, increases with increasing CMBAA and Cm. From the dynamic mechanical spectra, the characteristic relaxation time of the ionic bonds is on the millisecond scale (fig. S1). The mesh size ξ of the permanent polymer network, estimated by the relation νe ≅ ξ−3 (24), is in the range of 4 to 9 nm (table S1).
The phase structure is tuned by the chemical formulation. As an example, Fig. 2A presents the photo images of PA-2.5-CMBAA samples. The gels gradually change from semitransparent to transparent with increasing CMBAA. The two-dimensional (2D) SAXS patterns of PA-2.5-CMBAA gels are shown in Fig. 2B. For CMBAA < 0.5 mol %, an isotropic scattering pattern with a circular scattering ring can be noticed, demonstrating the isotropic mesoscale phase network structure of the undeformed gels. The intensity of the scattering ring decreases markedly with increasing CMBAA, and last, the ring disappears for PA-2.5-1.0, indicating that the phase contrast declines and the sample becomes homogeneous as CMBAA increases. A periodic peak is observed for CMBAA ≤ 0.5 mol % on azimuthally integrated 1D SAXS profiles (Fig. 2C). The peak position qm is related to the characteristic length scale of the phase structure (d0), by d0 = 2π/qm. The peak intensity of scattering Im for a two-phase system is proportional to V1V2(Δρ)2, where V1 and V2 are the volume fraction of phases 1 and 2, and Δρ is the electron density difference between them (30). In the case of PA gels, V1, V2, and Δρ can be considered as the volume fractions of soft and hard phases and their polymer volume density difference (also phase contrast), respectively. As the relative volume fractions of soft and hard phases weakly vary, the peak intensity Im is proportional to the phase contrast of hard and soft domains by Im ~ (Δρ)2 (24). The values of Im and d0 for the sample series PA-2.5-CMBAA and PA-Cm-0.1 dialyzed at 30°C are summarized in table S2. The remarkable decrease of Im and d0 with increasing CMBAA indicates a weakened phase separation as the result of competition between the free energy gain of polyion complexation and the entropic penalty of chain elasticity that increases with cross-link density (23, 24). Similarly, for the samples PA-Cm-0.1 with different monomer concentrations Cm but a fixed chemical cross-link density CMBAA, the peak intensity Im and d0 also decrease with increasing Cm (table S2), due to an increase in the topologically trapped entanglements that serve as effective permanent cross-linking density (24). For comparison, results of sample series PA-2.0-0.1 (Tdial) dialyzed at different Tdial [extracted from our previous work (25)] are also shown in table S2. A universal relation Im ~ d04 is observed for all the samples (with values of d0 depending on Cm, CMBAA, and Tdial) (Fig. 2D), consistent with our previous study (24). This relationship, combining with Im ~ (Δρ)2, shows that the phase contrast Δρ and size d0 are inherently correlated by Δρ ~ d02 in the PA hydrogel system. This result, showing that the stronger the phase contrast, the larger the phase structure, should be attributed to the competition between the phase separations and the elasticity of the network. It is worthwhile to note that the changes of Im, d0, and the elasticity are not due to the change of overall polymer volume fraction in water, because it remains nearly the same for all the gels.
Fig. 2. PA gels with a tuned bicontinuous hard/soft phase network structure.
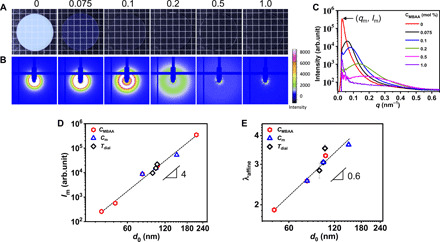
(A) Photographs of the representative gels PA-2.5-CMBAA with different chemical cross-linker densities, and the variable CMBAA is shown at the top of each picture. (B) Corresponding 2D SAXS patterns of (A). (C) Corresponding 1D scattering intensity profiles of (B). (D) Logarithmic plot of SAXS peak intensity Im and characteristic length of the phase structure (d0) estimated from qm for three series of samples, i.e., series PA-2.5-CMBAA and series PA-Cm-0.1 dialyzed at 30°C, and series PA-2.0-0.1(Tdial) dialyzed at different temperatures Tdial (detailed sample information can be found in tables S1 and S2) at undeformed state. (E) Logarithmic plot of maximum stretch ratio for affine deformation (λaffine) of the phase networks in uniaxial tensile test as a function of d0 for the three series of samples. The data of series PA-2.0-0.1(Tdial) are extracted from (25). The error bars are SEs from two to three measurements.
Tensile and fracture behaviors
We first studied the uniaxial tensile behavior and the microscopic deformation of the phase network by in situ time-resolved SAXS during the tensile test (fig. S2). The results of series PA-2.5-CMBAA and series PA-Cm-0.1 at the stretch rate of 1 s−1 are shown in fig. S3. Gels with a relatively strong and sharp phase separation (Im > 1000 arb. unit for series PA-2.5-CMBAA with CMBAA = 0 to 0.1 mol % and series PA-Cm-0.1) exhibit little differences in uniaxial tensile behavior, regardless of the notably different phase contrast (fig. S3, A and E). Furthermore, the fracture behavior in pure shear geometry of these gels was also very similar (fig. S3D). Using the SAXS data, we characterized the correlation between the macroscopic deformation λ of the sample and the microscopic deformation of the phase network in the direction parallel (d///d0) and perpendicular (d⊥/d0) to the stretching direction (fig. S2). For incompressible materials, if d///d0 = λ and d⊥/d0 = λ−1/2, the deformation is called affine. We observed that the phase network deforms affinely up to a quite large bulk stretch ratio λ. The maximum stretch ratio for affine deformation of the phase networks (λaffine), which was interpreted as the onset of the damage of hard phase network (23), increases as Im and d0 increase (table S2). This result suggests that coarser structures can bear larger deformation due to larger d0. It should be noted that the purely physical gel without chemical cross-linking, PA-2.5-0, shows the largest Im and d0, but we could not accurately measure λaffine of this sample because of the detection limit of SAXS. We also could not measure λaffine of samples PA-2.5-CMBAA with CMBAA = 0.5 and 1.0 mol % because of a too weak scattering intensity. From the measured results, we found that λaffine ~ d00.6 at the observing loading rate (Fig. 2E). Because the stretched length of the phase network at the maximum affine deformation (d//,m) is expressed as λaffine = d//,m/d0, we get a relation d0 ~ d//,m0.6. This power law relation between the phase mesh size in its undeformed state (d0) and in its stretched state (d//,m) at the maximum affine deformation looks similar to the power law relationship between the end-to-end distance (ξ) and its fully stretched length (contour length Lc) of a self-avoiding polymer chain, ξ ~ Lc3/5 (26). Accordingly, the strands of phase network could be effectively considered as self-avoiding chains and the strands of hard phase break at λ = λaffine with the maximum stretching length d//,m at the observation time scale.
Fatigue fracture behaviors
Fatigue experiments were carried out with prenotched samples in the pure shear geometry at a nominal loading rate 1 s−1. A triangular loading profile was applied during cyclic loading. The maximum stretch ratio was kept constant at a preset value λmax, while the minimum stretch ratio was kept at 1 (Fig. 3A). The crack propagation in fatigue test starts when λmax reaches a threshold, denoted as λ0. For λmax < λ0, no measurable crack growth was observed over 30,000 cycles. As an example, the crack propagation behaviors of samples with the high phase contrast (strongly phase-separated) PA-2.5-0.075 (Im = 19.6k) and the low phase contrast (weakly phase-separated) PA-2.5-0.5 (Im = 0.26k) are shown in Fig. 3 (B and C). λ0 is found to be 1.98 for PA-2.5-0.075 and 1.2 for PA-2.5-0.5. Above λ0, the two gels with different phase structures show notably different fatigue behaviors. For PA-2.5-0.075 with a strongly phase-separated structure, two characteristic regimes of λmax were observed: At small λmax > λ0, after an initial growth over thousands of cycles, the crack length c hardly propagates with increasing cycle number N, resulting in an extremely slow crack propagation rate Δc/ΔN in steady state, and such propagation delaying phenomenon is observed below a second threshold λmax = λtran(=3.14 ± 0.2). When λmax > λtran, the crack length c increases rapidly with cycle number N and the whole sample ruptures quickly (Fig. 3B), resulting in a fast crack propagation rate Δc/ΔN, as observed in our previous work (25). For the weakly phase-separated PA-2.5-0.5, the crack length c always increases linearly with N for λmax > λ0, even at an extremely small λmax (Fig. 3C).
Fig. 3. Crack propagation behaviors of the representative strongly and weakly phase-separated gels under fatigue test.
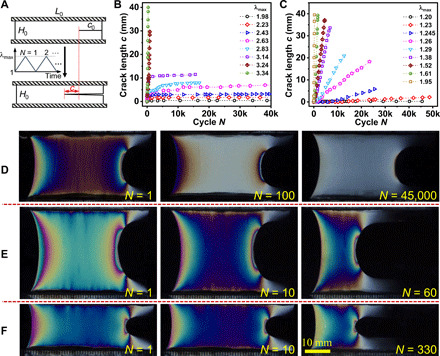
(A) Pure shear geometry (L0 = 50 mm, H0 = 10 mm, and c0 = 10 mm) and loading profile for fatigue test. During cyclic loading, the length of crack propagation was recorded as c. (B and C) Crack propagation behavior at different λmax for the strongly phase-separated gel PA-2.5-0.075 (B) and the weakly phase-separated gel PA-2.5-0.5 (C). (D and E) Typical evolutions of sample birefringence images with cycle number N for the representative strongly phase-separated gels at λmax < λtran (D) and λmax > λtran (E). Sample PA-2.5-0.075 at λmax = 2.64 and at λmax = 3.44 are taken as examples in (D) and (E), respectively. (F) Typical evolutions of sample birefringence images with cycle number N for the representative weakly phase-separated gel at a small λmax. Sample PA-2.5-0.5 at λmax = 1.81 is taken as an example.
Fracture mechanics theory shows that the deformation field around the crack tip is usually much larger than in the bulk far from the crack due to the presence of a singularity in both stress and strain (31–34). To observe the stress distribution around the crack tip during the fatigue test, the birefringence (circular polarization) was monitored during cyclic loading. Figure 3 (D and E) shows the birefringence patterns of a representative strongly phase-separated gel PA-2.5-0.075 at λmax < λtran and λmax > λtran, respectively. For λmax < λtran, a blunted crack tip with an arc-shaped stress concentration zone was observed, and the distribution of birefringence colors around the crack tip became substantially weaker with increasing cycle numbers (Fig. 3D), demonstrating that the stress concentration is reduced with increasing fatigue cycles. It seems even that the singularity around the crack tip can be nearly eliminated after tens of thousands of fatigue cycles for the strongly phase-separated gels (N ~ 45,000 in Fig. 3D and movie S1), resulting in the deceleration or even arrest of crack advancement. While a severe butterfly-like stress concentration always exists around the crack tip for increasing cycle number N at λmax > λtran (Fig. 3E and movie S2), which suggests that λtran corresponds to the critical bulk stretch above which stress concentration does not vanish under cyclic loading. This butterfly-like stress concentration is also always observed on the weak phase-separated gels PA-2.5-0.5 even at small λmax as shown in Fig. 3F and movie S3, indicating that stress concentration around the crack tip is not alleviated with the increase in fatigue cycles even at a small λmax for the weakly phase-separated gels. It should be noted that, regardless of whether it occurs for the strongly phase separated gels or for the weakly phase separated gels, once the butterfly-like stress concentration appears for increasing cycle numbers N, a tremendous crack extension occurs until the fracture of the whole sample (see N = 1 to N = 60 in Fig. 3E and N = 1 to N = 330 in Fig. 3F).
As a result, the crack propagation rate Δc/ΔN, obtained from the slope of the plots of crack length c versus cycle N in steady state, shows a slow mode and a fast mode for the strongly phase-separated gels, while only the fast mode is observed for the weakly phase-separated gels above the threshold value λ0. Figure 4A shows Δc/ΔN versus λmax for PA-2.5-CMBAA with varying CMBAA content. The threshold λ0 decreases from 2.14 to 1.18 as CMBAA increases from 0 to 1.0 mol %. For the strongly phase-separated samples (CMBAA ≤ 0.1 mol %) at λmax > λ0, Δc/ΔN first increases slowly with λmax, showing a plateau-like mode with Δc/ΔN < 0.1 μm per cycle. Then, Δc/ΔN jumps to a fast crack growth mode when λmax > λtran. λtran shifts to a higher value, and the amplitude of the jump in Δc/ΔN increases for gels with stronger hard/soft phase contrast. The sample series PA-Cm-0.1 prepared with different monomer concentrations Cm shows a similar correlation between the microstructure and the fatigue behavior (Fig. 4B). That is, the fast crack propagation in fatigue is delayed to a larger value of λtran for the materials with a high hard/soft phase contrast and large d0. Δc/ΔN at λmax > λtran is orders of magnitude larger than that at λ0 < λmax < λtran, indicating that the crack propagation in cyclic fatigue is substantially suppressed at λmax < λtran. However, for the weakly phase-separated gels (CMBAA > 0.1 mol %), no slow crack propagation mode is observed, and the fast crack propagation mode occurs directly once λmax becomes slightly larger than λ0 (Fig. 4A). It is noticed that although the sample with CMBAA = 0.2 mol % shows a similar fracture toughness as that of the samples with CMBAA ≤ 0.1 mol % (fig. S3D), it only shows the fast crack propagation mode in fatigue test. This suggests that the phase network is responsible for the suppression of the fast crack growth in fatigue tests.
Fig. 4. Fatigue behavior of PA gels with different mesoscale phase contrasts.
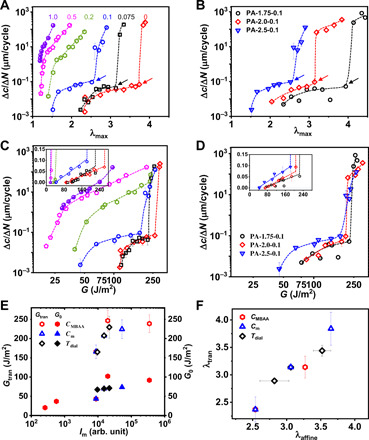
(A and B) Crack propagation rate Δc/ΔN in steady state as a function of λmax for sample series PA-2.5-CMBAA (A) and PA-Cm-0.1 (B). The numbers beside each curve in (A) represent CMBAA (mol %). The dashed lines are a guide for the eyes. The arrows in (A) and (B) indicate the transition points of slow-to-fast fatigue crack growth. (C and D) Crack propagation rate Δc/ΔN as a function of energy release rate G corresponding to (A) and (B), respectively. Inset shows the initial linear regime in the linear scale. Symbols in (C) are the same as those in (A). (E) Energy release rate at transition points of slow-to-fast fatigue crack growth (Gtran) (left y axis) and the threshold G0 (right y axis) extracted from (C) and (D) versus Im for the three series of samples. (F) Correlation between λtran for slow-to-fast mode transition in the fatigue test and the maximum stretch ratio λaffine for affine deformation of the phase structure in the tensile test for the three series of samples. The data of series PA-2.0-0.1(Tdial) are extracted from (25). The error bars are SEs from two to four measurements. Note that in these PA gels, G0 and λtran are characteristic material parameters, while Gtran is geometry dependent (see Fig. 5). The measurements were performed with samples of pure shear geometry (L0 = 50 mm, H0 = 10 mm, and c0 = 10 mm) and nominal strain rate 1 s−1.
In fracture mechanics of soft elastic materials (35), the crack growth is considered to be driven by the release of the stored elastic energy, and crack propagation rate Δc/ΔN is a function of the energy release rate (G). The Δc/ΔN versus G profiles allow, in principle, a comparison of the fatigue behaviors between samples with different moduli and different geometries, while the Δc/ΔN versus λmax profiles do not allow such comparisons. In the pure shear geometry, G can be calculated from G = Wel(λmax)H0, where Wel(λmax) is the stored elastic energy and H0 is the initial sample height. For purely elastic materials, Wel is equal to the input work Wex. However, for viscoelastic PA gels containing dynamic bonds, the input work Wex is partially dissipated during loading, and there is no well-established method to calculate G. For simplicity, we use the method presented in (33) to estimate Wel from Wex and the mechanical hysteresis area Uhys in steady state of the fatigue test (fatigue cycle N ~ 3000 cycles), using the relation Wel = Wex − Uhys (see the Supplementary Materials and fig. S4E).
Figure 4 (C and D) shows Δc/ΔN versus G plots of series PA-2.5-CMBAA and PA-Cm-0.1, respectively, from which the fatigue threshold G0 and the energy release rate Gtran, corresponding to λ0 and λtran, respectively, can be obtained. G0 and Gtran are plotted in Fig. 4E as a function of the scattering intensity Im. Only strongly phase-separated gels (Im > 1000 arb. unit) show values of Gtran, which are several times larger than that of G0, while the weakly phase-separated gels (Im < 1000 arb. unit) only show a well-defined and low value of G0 (<50 J/m2). The series PA-2.0-0.1(Tdial) also obeys the same trend as shown in Fig. 4E. When compared at the same applied G = 150 J/m2, the strongly phase-separated gels are in a slow propagation mode, while weakly phase-separated gels are in a fast mode of crack growth. Note that for all the samples, the ratio between hysteresis energy and the total input work Uhys/Wex is approximately the same (ca. 0.4) in steady-state (accommodated) conditions (fig. S4H). This also supports the notion that the differences in crack propagation rate Δc/ΔN observed for different gels are not directly related to the viscoelastic character of the gels but rather to their differences in phase structure. It should also be noted that the fast self-recovery of large hysteresis loop during fatigue test (fig. S4), caused by the fast reforming of broken ionic bonds, enhances fatigue resistance, in comparison with purely elastic gel. As shown in fig. S5, compared with the as-prepared gel that is purely elastic in the observation time scale, the equilibrated gel with large hysteresis loop shows a larger fatigue threshold G0 and a slower crack extension above G0. The slowdown phenomenon of crack propagation is also observed in other self-recovery systems with noncovalent interactions, as indicated by Bai et al. (11).
Origin of the controlling structural parameter for the slow-to-fast fatigue transition
As clarified above, the two regimes (slow and fast modes) of crack propagation only occur for the PA gels with a strong phase contrast, corresponding to a high modulus contrast between the hard and soft phase networks. We hypothesize that the delayed crack growth above the threshold λ0 is due to the weak stress concentration at the crack tip (strong crack blunting) (25). Therefore, the following two questions arise: What determines the value of λtran and the transition to a fast crack growth rate? Is λtran or the corresponding energy release rate Gtran an intrinsic material parameter determining the slow-to-fast crack transition?
λtran is found to be positively correlated to the limit of affine deformation of the phase networks λaffine for the three sets of gels prepared at different CMBAA, different Cm, or different Tdial (Fig. 4F). It suggests that the maximum bulk stretch λmax, where crack blunting is observed during a fatigue test, is correlated to an intrinsic damage of the structure in the bulk region (away from the crack tip) during the tensile deformation. However, this is in clear contradiction with fracture mechanics theory because it suggests that the crack propagation is controlled by the stretch in the bulk and not at the crack tip. In other words, a larger pure shear sample would apply a larger G at the same bulk stretch than a smaller sample. In turn, this larger G should cause a larger local stretch at the crack tip and, hence, a larger dc/dN. To verify this, we compared the fatigue results of sample PA-2.0-0.1(Tdial = 5°C) measured with samples with a different initial height (H0) deformed at a nominal stretch rate = 1 s−1 while keeping the aspect ratios L0/H0 = 5 and c0/H0 = 1 unchanged (Fig. 5, A and B). The value of λtran for the two different H0 only changes by about 3% (λtran = 2.79 ± 0.05 for H0 = 15 mm and λtran = 2.89 ± 0.05 for H0 = 10 mm), while the corresponding Gtran changes by about 30% (234.9 ± 6.9 J/m2 for H0 = 15 mm and 165 ± 9 J/m2 for H0 = 10 mm). On the other hand, G0 values measured with different H0 are almost the same (inset of Fig. 5B), confirming that G0 is an intrinsic material parameter in this viscoelastic system. A comparison between samples with larger height differences (H0 = 10 versus 20 mm, at a nominal stretch rate = 0.8 s−1) is shown in fig. S6 using a different batch of PA-2.0-0.1 gels. In this case, λtran for the two different H0 changes about 10% (λtran = 2.35 ± 0.05 for H0 = 20 mm and λtran = 2.6 ± 0.10 for H0 = 10 mm), while the corresponding Gtran changes by about 30% (Gtran = 202.3 ± 9.1 J/m2 for H0 = 20 mm and Gtran = 142.1 ± 15.3 J/m2 for H0 = 10 mm). Furthermore, we observed that the value of λtran determined from samples with a rectangular geometry (the geometry used for SAXS measurements) is very close to the value of λtran determined from the samples in the pure shear geometry (fig. S7). These results strongly suggest that λtran is a better material parameter that governs the slow-to-fast mode transition, while Gtran, showing a relatively strong geometry dependence, is not an intrinsic material parameter in this strong crack blunting system with hierarchical structures. One should be, however, a bit careful because in the limit of very large samples, G should become again a better material parameter. Our results, however, show that for reasonable laboratory-scale geometries, the blunting process at the crack tip nearly eliminates the stress concentration and effectively nearly stops crack propagation in fatigue. Only when the bulk stretch exceeds a critical value, the stress concentration persists and the crack grows.
Fig. 5. Geometry effect of fatigue behaviors.
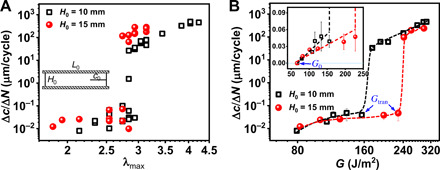
(A) Comparison of fatigue crack propagation rate Δc/ΔN in steady state versus λmax for a sample tested at different initial height H0 of pure shear geometry. The aspect ratios L0/H0 = 5 and c0/H0 = 1 were kept the same when changing H0 (H0 = 10 and 15 mm). (B) Crack propagation rate Δc/ΔN versus energy release rate G in logarithmic scale. Inset shows the initial regime in the linear scale, and the fatigue threshold G0 (indicated by arrow) is obtained by linearly extrapolating the results to the horizontal axis. The Gtran corresponding to the energy release rate at λtran is also indicated by arrows. Sample PA-2.0-0.1(Tdial = 5°C) was used. The data for H0 = 10 mm are extracted from (25). The error bars are SEs from (A).
Why does λtran correlate to λaffine for the tensile behavior with no crack? We explain this result from two features of the hierarchical structure. One feature is the different force-carrying abilities of the hard and soft phase networks. At λmax < λaffine, the two-phase networks are maintained intact during loading in the bulk region, and the hard phase network should carry most of the stress, and the soft network should redistribute the stress homogeneously, similar to the role played by the hard and soft constituents in other fatigue resistant materials (21) or to the role played by the first network and the second network in tough double network materials (3, 4, 36–38). Another feature is the formation of an oriented structure of the phase networks due to the presence of ionic bonds. At the fast stretch rate used here (~1 s−1), the deformation only partially recovers upon unloading, as seen by the large residual strain at zero stress (fig. S4C), and the phase networks in the bulk gradually adapt to form an oriented metastable structure under the cyclic loading, which is detected by SAXS (25). This orientation of the phase networks causes a blunting of the crack tip and suppresses crack advance. This explains why at the beginning of the cyclic loading, the crack tip is more highly stressed than the bulk, consistent with a relatively fast crack extension. With increasing number of cycles, the crack gradually blunts as a result of competition between high stress-induced crack growth and the formation of a highly oriented structure at the crack tip, which substantially decreases the stress concentration and results in an extremely slow crack propagation mode. At λmax > λaffine, the hard phase in the bulk begins to damage and the soft phase network (made from regions with a sparse polymer density) cannot bear the load to form a sufficiently strong oriented structure with cyclic loading, and the stress concentration persists at the crack tip, resulting in rapid crack growth. The shift of λtran to a higher value and the increase in the amplitude of the jump in Δc/ΔN for gels with a stronger hard/soft phase contrast could be explained by the intrinsic correlation Im ~ d04 (Fig. 2D). The increase of the mesh size d0 results in the increase of λaffine through λaffine ~ d00.6 (Fig. 2E), and above λaffine, the internal damage of the hard phase network results in faster crack propagation in fatigue. The increase of Im corresponds to the increase of the relative strength of the hard and soft phase network, so crack growth per cycle is accelerated above the λtran threshold.
Note that although the weakly phase-separated gel PA-2.5-0.2 shows a value of λaffine = 1.9, no slow crack propagation mode is observed. This indicates that the fatigue crack growth can only be mitigated when the hard phase is sufficiently stronger than the soft phase, i.e., when the stiffness contrast between hard and soft phase is high enough, a general design principle of crack- and fatigue-resistant materials (21, 39).
Origin of the controlling structural parameter for the fatigue threshold G0
Last, we discuss what determines the fatigue threshold G0 for the viscoelastic PA gels. According to the recent studies by Suo and colleagues (11, 40), G0 of tough hydrogels containing dynamic bonds is related to the permanent polymer network that is chemically cross-linked, regardless of the transient network physically cross-linked by reversible sacrificial bonds. Here, we correlate the fatigue threshold G0 with the mesh size ξ between effective cross-linking points of the permanent network formed by both chemical cross-links and trapped physical entanglements in PA gels. We observe that all the data from three different series of samples with varied CMBAA, Cm, and dialysis temperature Tdial fall on the same curve, which gives a scaling relation of G0 ~ ξ2.7 (Fig. 6), indicating that the threshold G0 is positively correlated to the mesh size of the permanent network, consistent with previous reports on chemically cross-linked single and double network gels without the microphase separation (41–43). From Fig. 6, we know that both the chemical cross-links and the physically trapped topological entanglements act similarly as permanent cross-links to control the fatigue threshold in our PA system. On the other hand, considering the globule conformation of the polymer strands in the PA gels, we would expect a relatively weak scaling relation of G0 ~ ξ, using the Lake-Thomas model (see section S3) (44). The experimentally observed much stronger relation between G0 and ξ than the scaling relation deduced from Lake-Thomas model suggests that the physical bonds might also influence G0. As shown in fig. S4H, the dissociation and reforming times of ionic bonds for all of the strongly and weakly phase separated gels are much shorter than the observation time scale (Uhys/Wex is ~40% in steady state for all the samples). For the PA gels with such fast ionic bond reformation, it is difficult to exclude all the contribution from ionic bonds, while in the Lake-Thomas model only chemical bonds were considered.
Fig. 6. Fatigue threshold determined by primary network structure.
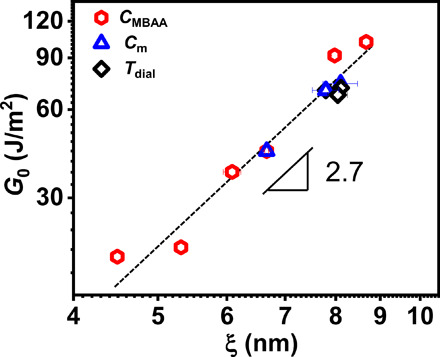
Logarithmic plot of fatigue threshold G0 as a function of mesh size ξ between permanent cross-linking points from both chemical cross-linking and trapped topological entanglements for three series of samples. The error bars are SEs from three measurements.
Because G0 is only related to the chemically cross-linked structure, independent of the existence of physical bonds and entanglements as suggested in the classic theory of threshold (44), a purely physically cross-linked gel would be expected not to show a finite fatigue threshold G0 when fractured by a chain pull-out mechanism without bond scission (45). However, we also observed a multilevel fatigue resistance behavior in the physical gel PA-2.5-0 made without chemical cross-linker. This indicates that the relatively strong ionic bonds prevent chain pullout at the observation time scale, and the topologically entangled chains [~16 entanglements per chain (see section S4)] behave as being permanently cross-linked and show a finite fatigue threshold G0 at the observation time scale.
DISCUSSION
Our study highlighted the role of multiscale structures in multimode fatigue resistance, which is reflected by two essential fatigue parameters, i.e., the threshold energy release rate G0 and the critical stretch ratio λtran for the slow-to-fast crack growth transition. The nanoscale permanent polymer network structure, effectively cross-linked by both chemical cross-linking and topological entanglement, controls G0. Above G0, the fatigue crack growth behavior notably depends on the phase contrast of the mesoscale bicontinuous networks. When the phase contrast is sufficiently high, that is, the hard phase is sufficiently denser than the soft phase, it is presumably also stronger and better able to bear the stress and the slow mode appears at a small stretch ratio and then jumps to a fast mode at λmax > λtran. Because the crack propagation rate is lower than 0.1 μm per cycle at λmax < λtran, the gels have a strong fatigue resistance through a crack blunting mechanism. The jump to the fast mode for λmax > λtran is directly correlated to the onset of damage of the hard phase network in the bulk of the material (λaffine), which is itself related to the characteristic size of the phase separated network through λaffine∝d00.6. On the other hand, when the phase contrast is weak, the fast crack growth mode occurs immediately above the threshold G0 (<50 J/m2), and the gels exhibit a poor fatigue resistance.
It is interesting to point out that for our laboratory-size macroscopic samples, λtran is a characteristic material parameter for the slow-to-fast crack growth mode transition and not Gtran. This is highly unusual and is a direct consequence of the ability of the material to blunt the crack and eliminate the stress concentration by local plastic strain at the crack tip. The correlation between λtran and the onset of deviation from affine deformation λaffine in the PA gels without crack indicates that the bulk structure change influences the fatigue crack growth through formation of metastable oriented structure under cyclic loading by ionic bond formation. Although λtran and λaffine should depend on the stretch rate due to the dynamic nature of the ionic bonds, the detailed stretch rate dependences of λtran and λaffine are beyond the scope of this work and should be studied in a separate work. The mechanism for enhancing multilevel fatigue resistance of PA hydrogels in this work provides not only understanding of fatigue resistance of materials with hierarchical structures but also a promising strategy for developing highly extensible, self-healing, and fatigue-resistant soft materials with hard and soft hierarchical components.
MATERIALS AND METHODS
Materials
The anionic monomer NaSS, cationic monomer DMAEA-Q, ultraviolet (UV) initiator α-ketoglutaric acid (α-keto), and chemical cross-linker N,N-methylenebis(acrylamide) (MBAA) were purchased from Wako Pure Chemical Industries Ltd. and used as received. Deionized water was applied in all experiments.
Preparation of the PA gels
PA gels were prepared by free radical copolymerization of NaSS and DMAEA-Q with a composition around the point of charge balance in the presence of a chemical cross-linker MBAA following the procedure previously reported (7, 46). Briefly, a mixed precursor aqueous solution of NaSS, DMAEA-Q, MBAA, and UV initiator α-keto was polymerized by UV irradiation in argon atmosphere. To tune the microstructure of the gels, the total monomer concentration Cm = (NaSS) + (DMAEA-Q) was varied from 1.75 to 2.5 M, and the MBAA content was varied from 0 to 1.0 mol % (relative to Cm). After polymerization, the gels were dialyzed in 30°C deionized water for 3 weeks to remove the counterions and deswell to reach equilibrium, during which the phase separation formed (24). The equilibrated gels were used in all experiments, unless otherwise mentioned. A summary of the equilibrated gels is shown in table S1.
SAXS measurement
SAXS measurements were carried out at the Synchrotron Radiation Facility (BL19U2, Shanghai, China). The x-ray wavelength used was 1.03 Å. A 2D detector (Pilatus 1M with a resolution of 981 × 1043 pixels and pixel size of 172 μm, Dectris Co. Ltd.) was used to record data. The sample-to-detector distance was 5730 mm. To characterize the bicontinuous hard/soft phase networks of undeformed gels, the 2D SAXS patterns were acquired with an exposure time of 2 s. To detect the deformation of bicontinuous phase networks during loading, in situ time-resolved SAXS measurements were performed, and unnotched rectangular samples (7.5 mm × 16 mm × 1.65 mm, L0 × H0 × t0) were applied. 2D SAXS patterns were acquired at a rate of 0.25 s per frame. The nominal loading stretch rate was 1 s−1.
Rheological test
The dynamic rheological test of the PA gels was performed with an Advanced Rheometric Expansion System rheometer (Rheometric Scientific Inc.). A disk-shaped sample with diameter and thickness of 20 and ~1.65 mm, respectively, was fixed between two metal plates. To prevent water evaporating from the gel, the sample was kept surrounded by water during the whole procedure of test. A rheological frequency sweep from 0.05 to 100 Hz was performed at a shear strain of 0.1% at temperatures from 0.1 to 95°C. By following the principle of time-temperature superposition, master curves of storage modulus (G′), loss modulus (G′′), and loss factor tan δ over a wide frequency range were constructed at a reference temperature of 25°C.
Tensile and toughness test
Tensile test was performed on an Instron 5566 universal tensile tester. Unnotched rectangular sample (7.5 mm × 16 mm × 1.65 mm, L0 × H0 × t0, see inset of fig. S3A) was stretched to fracture. The fracture energy of gels was obtained from a pure shear test using sample in the pure shear geometry (50 mm × 10 mm × 1.65 mm, L0 × H0 × t0, the initial notch c0 was 10 mm). During the tensile and pure shear tests, the water vapor was sustainably supplied around the gels. The experiment temperature was kept constant at 24°C, and the nominal loading stretch rate was 1 s−1.
Fatigue test and birefringence test
The fatigue test was performed mainly in the pure shear geometry (50 mm × 10 mm × 1.65 mm, L0 × H0 × t, the initial notch c0 was 10 mm) using a Shimadzu tensile tester (AG-X, Shimadzu Corporation) in a humidity chamber. To verify that λtran for slow-to-fast crack growth mode transition is geometry independent, fatigue tests have also been performed for samples with different initial height H0, while the aspect ratios L0/H0 = 5 and c0/H0 = 1 were kept the same. The fatigue test for sample with H0 = 10 and 15 mm was performed at a nominal strain rate of 1 s−1. The fatigue test for samples with H0 = 20 mm could only be performed at a maximal stretch rate of 0.8 s−1 because of the limits of the crosshead speed of the tensile machine. Fatigue test was also performed for sample with rectangular geometry (7.5 mm × 16 mm × 1.65 mm, L0 × H0 × t, the initial notch c0 was 1 mm) at a nominal strain rate of 1 s−1. During the test, the temperature was kept constant at 24°C. The value of energy release rate Gtran, characterizing the energy required to cause the slow-to-fast transition of crack propagation for strongly phase separated gels, is only comparable for gels with a fixed geometry. A homemade circular polarizing optical system (47) was applied to observe the stress distribution during fatigue tests. Further details on the methods are available in the Supplementary Materials.
Acknowledgments
The SAXS experiments were performed at NCPSS BL19U2 beam line at Shanghai Synchrotron Radiation Facility (SSRF), China. The Hefei Puliang Technology Co. Ltd. is appreciated for providing the tensile machine with well-controlled humidity and temperature for time-resolved SAXS measurement. Institute for Chemical Reaction Design and Discovery (ICReDD) was established by World Premier International Research Initiative (WPI), MEXT, Japan. Funding: This work was supported by JSPS KAKENHI (grant nos. JP17H06144 and JP19K23617) and by the ImPACT Program of Council for Science, Technology and Innovation (Cabinet Office, Government of Japan). C.C. had received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program under Grant Agreement AdG No. 695351. Author contributions: X.L., K.C., and J.P.G. conceived the idea and designed the study. X.L. synthesized samples and performed mechanical characterization and fatigue test. X.L., K.C., Y.N.Y., and C.Y. performed SAXS measurements. All the authors analyzed and interpreted the results. X.L. and J.P.G. drafted the manuscript, and all the authors revised it. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/16/eabe8210/DC1
REFERENCES AND NOTES
- 1.Green J. J., Elisseeff J. H., Mimicking biological functionality with polymers for biomedical applications. Nature 540, 386–394 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haque M. A., Kurokawa T., Gong J. P., Super tough double network hydrogels and their application as biomaterials. Polymer 53, 1805–1822 (2012). [Google Scholar]
- 3.Gong J. P., Katsuyama Y., Kurokawa T., Osada Y., Double-network hydrogels with extremely high mechanical strength. Adv. Mater. 15, 1155–1158 (2003). [Google Scholar]
- 4.Gong J. P., Why are double network hydrogels so tough? Soft Matter 6, 2583 (2010). [Google Scholar]
- 5.Gong J. P., Materials both tough and soft. Science 344, 161–162 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y. S., Khademhosseini A., Advances in engineering hydrogels. Science 356, eaaf3627 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun T. L., Kurokawa T., Kuroda S., Ihsan A. B., Akasaki T., Sato K., Haque M. A., Nakajima T., Gong J. P., Physical hydrogels composed of polyampholytes demonstrate high toughness and viscoelasticity. Nat. Mater. 12, 932–937 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Sun J.-Y., Zhao X., Illeperuma W. R., Chaudhuri O., Oh K. H., Mooney D. J., Vlassak J. J., Suo Z., Highly stretchable and tough hydrogels. Nature 489, 133–136 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dai X., Zhang Y., Gao L., Bai T., Wang W., Cui Y., Liu W., A mechanically strong, highly stable, thermoplastic, and self-healable supramolecular polymer hydrogel. Adv. Mater. 27, 3566–3571 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Wang W., Zhang Y., Liu W., Bioinspired fabrication of high strength hydrogels from non-covalent interactions. Prog. Polym. Sci. 71, 1–25 (2017). [Google Scholar]
- 11.Bai R., Yang J., Morelle X. P., Yang C., Suo Z., Fatigue fracture of self-recovery hydrogels. ACS Macro Lett. 7, 312–317 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Bai R., Yang Q., Tang J., Morelle X. P., Vlassak J., Suo Z., Fatigue fracture of tough hydrogels. Extr. Mech. Lett. 15, 91–96 (2017). [Google Scholar]
- 13.Bai R., Yang J., Suo Z., Fatigue of hydrogels. Eur. J. Mech. Solid 74, 337–370 (2019). [Google Scholar]
- 14.Meyers M. A., McKittrick J., Chen P.-Y., Structural biological materials: Critical mechanics-materials connections. Science 339, 773–779 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Wegst U. G., Bai H., Saiz E., Tomsia A. P., Ritchie R. O., Bioinspired structural materials. Nat. Mater. 14, 23–36 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Lin S., Liu X., Liu J., Yuk H., Loh H.-C., Parada G. A., Settens C., Song J., Masic A., McKinley G. H., Zhao X., Anti-fatigue-fracture hydrogels. Sci. Adv. 5, eaau8528 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin S., Liu J., Liu X., Zhao X., Muscle-like fatigue-resistant hydrogels by mechanical training. Proc. Natl. Acad. Sci. U.S.A. 116, 10244–10249 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu J., Lin S., Liu X., Qin Z., Yang Y., Zang J., Zhao X., Fatigue-resistant adhesion of hydrogels. Nat. Commun. 11, 1071 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li C., Yang H., Suo Z., Tang J., Fatigue-resistant elastomers. J. Mech. Phys. Solids 134, 103751 (2020). [Google Scholar]
- 20.Su G., Cao J., Zhang X., Zhang Y., Yin S., Jia L., Guo Q., Zhang X., Zhang J., Zhou T., Human-tissue-inspired anti-fatigue-fracture hydrogel for a sensitive wide-range human–machine interface. J. Mater. Chem. A 8, 2074–2082 (2020). [Google Scholar]
- 21.Xiang C., Wang Z., Yang C., Yao X., Wang Y., Suo Z., Stretchable and fatigue-resistant materials. Mater. Today 34, 7–16 (2020). [Google Scholar]
- 22.Zhao X., EML webinar overview: Extreme mechanics of soft materials for merging human-machine intelligence. Extr. Mech. Lett. 39, 100784 (2020). [Google Scholar]
- 23.Cui K., Sun T. L., Liang X., Nakajima K., Ye Y. N., Chen L., Kurokawa T., Gong J. P., Multiscale energy dissipation mechanism in tough and self-healing hydrogels. Phys. Rev. Lett. 121, 185501 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Cui K., Ye Y. N., Sun T. L., Yu C., Li X., Kurokawa T., Gong J. P., Phase separation behavior in tough and self-healing polyampholyte hydrogels. Macromolecules 53, 5116–5126 (2020). [Google Scholar]
- 25.Li X., Cui K., Sun T. L., Meng L., Yu C., Li L., Creton C., Kurokawa T., Gong J. P., Mesoscale bicontinuous networks in self-healing hydrogels delay fatigue fracture. Proc. Natl. Acad. Sci. U.S.A. 117, 7606–7612 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.M. Rubinstein, R. H. Colby, Polymer Physics (Oxford Univ. Press, 2003). [Google Scholar]
- 27.Yoo S. H., Yee L., Cohen C., Effect of network structure on the stress-strain behaviour of endlinked PDMS elastomers. Polymer 51, 1608–1613 (2010). [Google Scholar]
- 28.Sun T. L., Luo F., Kurokawa T., Karobi S. N., Nakajima T., Gong J. P., Molecular structure of self-healing polyampholyte hydrogels analyzed from tensile behaviors. Soft Matter 11, 9355–9366 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Higgs P. G., Joanny J. F., Theory of polyampholyte solutions. J. Chem. Phys. 94, 1543–1554 (1991). [Google Scholar]
- 30.Boldon L., Laliberte F., Liu L., Review of the fundamental theories behind small angle X-ray scattering, molecular dynamics simulations, and relevant integrated application. Nano Rev. 6, 25661 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mai T.-T., Okuno K., Tsunoda K., Urayama K., Crack-tip strain field in supershear crack of elastomers. ACS Macro Lett. 9, 762–768 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Zhang T., Lin S., Yuk H., Zhao X., Predicting fracture energies and crack-tip fields of soft tough materials. Extr. Mech. Lett. 4, 1–8 (2015). [Google Scholar]
- 33.Mzabi S., Berghezan D., Roux S., Hild F., Creton C., A critical local energy release rate criterion for fatigue fracture of elastomers. J. Polym. Sci. B 49, 1518–1524 (2011). [Google Scholar]
- 34.Long R., Hui C.-Y., Gong J. P., Bouchbinder E., The fracture of highly deformable soft materials: A tale of two length scales. Annu. Rev. Cond. Matter Phys. 12, 104028 (2020). [Google Scholar]
- 35.Rivlin R. S., Thomas A. G., Rupture of rubber. I. Characteristic energy for tearing. J. Polym. Sci. 10, 291–318 (1953). [Google Scholar]
- 36.Matsuda T., Kawakami R., Nakajima T., Gong J. P., Crack tip field of a double-network gel: Visualization of covalent bond scission through mechanoradical polymerization. Macromolecules 53, 8787–8795 (2020). [Google Scholar]
- 37.Ducrot E., Chen Y., Bulters M., Sijbesma R. P., Creton C., Toughening elastomers with sacrificial bonds and watching them break. Science 344, 186–189 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Millereau P., Ducrot E., Clough J. M., Wiseman M. E., Brown H. R., Sijbesma R. P., Creton C., Mechanics of elastomeric molecular composites. Proc. Natl. Acad. Sci. U.S.A. 115, 9110–9115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cui W., King D. R., Huang Y., Chen L., Sun T. L., Guo Y., Saruwatari Y., Hui C. Y., Kurokawa T., Gong J. P., Fiber-reinforced viscoelastomers show extraordinary crack resistance that exceeds metals. Adv. Mater. 32, e1907180 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Zhang W., Hu J., Tang J., Wang Z., Wang J., Lu T., Suo Z., Fracture toughness and fatigue threshold of tough hydrogels. ACS Macro Lett. 8, 17–23 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Zhou Y., Zhang W., Hu J., Tang J., Jin C., Suo Z., Lu T., The stiffness-threshold conflict in polymer networks and a resolution. J. Appl. Mech. 87, 031002 (2020). [Google Scholar]
- 42.Zhang W., Liu X., Wang J., Tang J., Hu J., Lu T., Suo Z., Fatigue of double-network hydrogels. Eng. Fract. Mech. 187, 74–93 (2018). [Google Scholar]
- 43.Creton C., 50th anniversary perspective: Networks and gels: Soft but dynamic and tough. Macromolecules 50, 8297–8316 (2017). [Google Scholar]
- 44.Lake G., Thomas A., The strength of highly elastic materials. Proc. R. Soc. Lond. A Math. Phys. Sci. 300, 108–119 (1967). [Google Scholar]
- 45.Naassaoui I., Ronsin O., Baumberger T., A poroelastic signature of the dry/wet state of a crack tip propagating steadily in a physical hydrogel. Extr. Mech. Lett. 22, 8–12 (2018). [Google Scholar]
- 46.Ihsan A. B., Sun T. L., Kurokawa T., Karobi S. N., Nakajima T., Nonoyama T., Roy C. K., Luo F., Gong J. P., Self-healing behaviors of tough polyampholyte hydrogels. Macromolecules 49, 4245–4252 (2016). [Google Scholar]
- 47.Luo F., Sun T. L., Nakajima T., Kurokawa T., Zhao Y., Ihsan A. B., Guo H. L., Li X. F., Gong J. P., Crack blunting and advancing behaviors of tough and self-healing polyampholyte hydrogel. Macromolecules 47, 6037–6046 (2014). [Google Scholar]
- 48.Gao Z., Lister K., Desai J. P., Constitutive modeling of liver tissue: Experiment and theory. Ann. Biomed. Eng. 38, 505–516 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Long R., Hui C.-Y., Fracture toughness of hydrogels: Measurement and interpretation. Soft Matter 12, 8069–8086 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Dobrynin A. V., Colby R. H., Rubinstein M., Polyampholytes. J. Polym. Sci. B 42, 3513–3538 (2004). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/16/eabe8210/DC1