

Abstract
Purpose:
Based on the recent discovery of mutations in Bruton’s tyrosine kinase (BTK) in Follicular Lymphoma (FL), we studied their functional properties.
Experimental Design:
We identified novel somatic BTK mutations in 7% of a combined total of 139 FL and 11 transformed FL cases, none of which had received prior treatment with B cell receptor (BCR) targeted drugs. We reconstituted WT and mutant BTK into various engineered lymphoma cell lines. We measured BCR-induced signal transduction events in engineered cell lines and primary human FL B cells.
Results:
We uncovered that all BTK mutants destabilized the BTK protein and some created BTK kinase-dead mutants. The PLCγ2 is a substrate of BTK but the BTK mutants did not alter PLCγ2 phosphorylation. Instead, we discovered that BTK mutants induced an exaggerated AKT phosphorylation phenotype in anti-immunoglobulin (IG) treated recombinant lymphoma cell lines. The shRNA-mediated knock-down of BTK expression in primary human non-malignant lymph node-derived B cells resulted in strong anti-IG-induced AKT activation, as did the degradation of BTK protein in cells lines using ibrutinib-based proteolysis targeting chimera (PROTAC). Finally, through analyses of primary human FL B cells carrying WT or mutant BTK, we detected elevated AKT phosphorylation following surface IG crosslinking in all FL B cells, including all BTK mutant FL. The augmented AKT phosphorylation following BCR crosslinking could be abrogated by pre-treatment with a PI3Kδ inhibitor.
Conclusions:
Altogether, our data uncover novel unexpected properties of FL-associated BTK mutations with direct implications for targeted therapy development in FL.
Keywords: Follicular lymphoma, BTK mutations, AKT activation
Introduction
Follicular lymphoma (FL) constitutes the most common indolent B-cell lymphoma in the US. FL remains incurable with conventional therapies and most patients receive multiple therapies during the course of their illness(1). The development of targeted FL therapies is desirable (2–4) but lags behind progress made in chronic lymphocytic leukemia (CLL) or mantle cell lymphoma (MCL) (5–7).
An expanding group of recurrently mutated genes has recently been shown to underlie the pathogenesis of FL (KMT2D/MLL2, CREBBP, EZH2, EP300, ARID1A, HIST1H1 B-E, STAT6, RRAGC, ATP6V1B2 and others) and some of these afford potential opportunities for novel targeted therapy developments (8–16). For example, studies into the role of EZH2 in germinal center development and activating mutations in EZH2 provided the rationale to target EZH2 with a small molecule in clinical trials (17–19), MTOR-activating mutations in RRAGC could delineate an FL group that may be responsive to MTOR inhibitors (20, 21), and autophagy activating mutation in vacuolar-type H+-ATPase (v-ATPase) could be amenable to transient autophagy inhibition (15).
Given the importance of the B cell receptor (BCR) and BCR signaling in B cell NHL, including FL, it is of interest that mutations in various genes linked to BCR signaling (BTK, CARD11 and others) have recently been described in FL by Krysiak et. al. (22). The mutations in BTK are of special interest given the highly successful therapeutic targeting of BTK in CLL, MCL and other B cell lympho-proliferative diseases using BTK targeted drugs (5, 6, 23, 24). However, the exact function of BTK in signaling pathways employed by the BCR in FL remains incompletely understood, which may in part explain why a mechanistic understanding of the lower efficaciousness of ibrutinib observed in clinical FL trials is lacking (25).
In this report, we summarize our results of detailed mechanistic studies into the properties of FL-associated mutations in BTK, for which no prior such studies are available. We detail the a priori somewhat surprising finding that FL-associated BTK mutations are not activating the tyrosine kinase, but instead destabilize the protein and for some mutants result in catalytically inactive BTK kinases. The FL-associated BTK mutations however augment the BCR-induced activation of AKT, as evidence by elevated AKT phosphorylation in multiple experimental settings. Further, through comparison of primary human FL B cells with non-malignant lymph node derived B cells, we identified augmented AKT signaling following BCR crosslinking in all studied FL, further supporting this as a common pathobiological abnormality in FL. Given the established importance of AKT activation and signaling in germinal center type B cell NHL, this finding provides a credible mechanism why FL would recurrently select for mutations in BTK (26, 27).
In aggregate, our data provide a mechanistic understanding why BTK protein kinase dead and destabilizing mutations occur in FL. These findings may also explain why FL patients that carry BTK mutations are unlikely to respond to BTK inhibitor therapies. Finally, these data allow for further improvements in personalized approaches to targeted therapies in FL.
Methods
Patient characteristics and lymphoma specimen source material
Results from the Sanger-based resequencing of BTK using template genomic DNA isolated from 139 flow-sorted FL (grades 1–3A) and 11 flow-sorted transformed FL (t-FL) are reported here. Methods for fluorescence-activated cell sorting of lymphoma specimens are as described previously (14). Ninety FL and 11 t-FL patients (cohort I) were enrolled in two separate lymphoma repositories at the University of Michigan Comprehensive Cancer Center (IRBMED #HUM00007985 and IRBMED #HUM00017055) after signing informed consent documents approved by the University of Michigan Institutional Review Board IRBMED. All investigations were performed in accordance with ethical guidelines outlined in the declaration of Helsinki. Genomic research on all specimens was approved through IRBMED #HUM00005467. We furthermore analyzed by Sanger sequencing DNA from 49 flow-sorted FL cases (cohort II) collected between 1990 and 2005 as previously described that were derived from de-identified leftover clinical material (28).
Exon resequencing of BTK in FL and RT-PCR based sequencing
Primers to amplify and sequence all coding exons and adjacent intronic sequences of BTK were designed using the primer 3 program (https://bioinfo.ut.ee/primer3-0.4.0/) and sequence information generated using direct sequencing. The sequence traces were analyzed using Mutation Surveyor® software (Softgenetics®) and by visual inspection. Mutations were confirmed to be somatically acquired using unamplified lymphoma cell-derived DNA and paired CD3 cell-derived DNA as templates isolated from highly pure flow-sorted cells. For sequence analysis of BTK cDNA, we used random priming (Invitrogen SuperScript® III system for RT-PCR # 18080–051) and RNA isolated from flow sorted FL B cells and dedicated BTK primers in separate exons covering known mutation sites.
BTK cDNA mutagenesis, materials, cell lines, lentiviral vector generation and cell transfection
Reagents and cDNA mutagenesisg:
A pEnter plasmid containing a BTK cDNA (cat#: CH864016; RefSeq# NM_000061) was purchased from Vigene Biosciences, and used as a template to generate mutant BTK cDNAs using the QuikChange® Lightning Site-Directed Mutagenesis Kit (Stratagene/Agilent, La Jolla, CA). Full-length WT and mutant HA-tagged BTK cDNAs were constructed using PCR and cloned into the PacI/NotI sites of the lentiviral vector FG9 (a gift from Dr. Colin Duckett, University of Michigan (29)). The same cloning strategies were used to construct WT and mutant HA-tagged BTK into the NotI/BamHI sites of the lentiviral vector pLVX-EF1α-IRES-mCherry Vector (Clontech, Mountain View, CA).
Antibodies:
Anti-beta-Actin (#A544) was from Sigma-Aldrich, RRID:SCR_008988. Anti-HA (C29F4) (#3724), anti-p-PLCγ2-Y1217 (#3871), anti-PLCγ2 (#3872), anti-p-BTK-Y223 (#5082), anti-BTK (#8547), anti-BLK (#3262), anti-p-AKT-S473 (#4060), anti-p-AKT-T308 (#4056), anti-AKT (#9272), anti-p-44/42-MAPK-T202/Y204 (ERK1/2) (#4370), anti-p44/42-MAPK (ERK1/2) (#4695) were from Cell Signaling Technology, RRID:SCR_004431. Anti-p-PLCγ2-Y759 (MAB7377) was from R&D System.
Lymphoma cell lines:
OCI-LY1 and OCI-LY7 cell lines were obtained from the University of Michigan in 2005 with permission of the cell line originators at the Ontario Cancer Institute. The SUDHL4 cell line was obtained from the University of Michigan in 2013 (Dr. Elenitoba-Johnson). The cell lines HBL1 and TMD8 were a gift from Dr. Guido Wendel (Memorial Sloan Kettering Cancer Center). Cell line authentication was performed through sequence analysis of six to eight gene mutations per line based on the COSMIC cell line project (catalogue of somatic mutations in cancer; http://cancer.sanger.ac.uk/cell_lines) or based on published mutations. The chicken DT40 BTK−/− cell line was a gift from Dr. Edward A. Clark (University of Washington in Seattle, WA). DT40 BTK−/− were cultured in RPMI 1640 supplemented with 10% FBS, 1% chicken serum, 50 µM β-mercaptoethanol, 1mM Sodium Pyruvate, 2% Tryptose phosphatase broth, and 1% PS. The original cell lines and recombinant cells lines were aliquoted and fresh aliquots thawed approximately monthly to conduct the described experiments. Test for mycoplasma were done periodically using the Venor™GeM Mycoplasma Detection Kit, PCR-based, Sigma-Aldrich # MP0025.
Cell transfection and lentivirus concentration in HEK293T cells:
HEK293T cells were transfected in 10 cm dishes with plasmids encoding either WT or mutant forms of BTK or various other constructs together with 1 µg of the plasmids REV, RRE and VSVG as indicated using polyethylenimine (PEI; Polyscience Inc., #23966). For lentivirus concentration, two 15 cm dishes of HEK293T were each transfected with 14 µg pLVX-EF1α-IRES-mCherry vector or vector expressing WT or various mutant forms of BTK, together with 2 µg of the plasmids REV, RRE and VSVG. A total of 80 ml virus-containing media were harvested at 48 and 72 h after transfection and centrifuged to remove HEK293T cells and debris. Afterwards, virus was pelleted by high-speed centrifugation at 41,656 x g (Sorvall LUNX 4000 centrifuge, Fiberlite™ F20–12 × 50 LEX Fixed-Angle Rotor) for 3 h at 4 °C. Supernatant was discarded and the virus pellets were gently resuspended using 2 ml of culture media.
Generation of OCI-LY7 BTK−/− and SUDHL4 BTK−/− cells using CRISPR-Cas9 gene targeting
Oligonucleotides encoding guide RNAs targeting the junction of the 5’ UTR and exon 1 of BTK were cloned into the pLentiCRISPRv2 (Addgene #52961) plasmid (26). HEK293T cells were transfected with recombinant pLentiCRISPRv2 together with viral packaging plasmids using PEI transfection. OCI-LY7 and SUDHL4 cell lines were transduced by spin inoculation at 30°C at 1,360 x g for 2 h using 8 µg/mL of polybrene before seeding into fresh medium and subsequent selection in puromycin (Sigma #P9620) at a concentration of 2 µg/mL for 3 d. Cells were subsequently plated in 96-well plates at an average density of three cells per well. After clonal expansion, recombinant cell clones were identified using diagnostic PCR performed directly on aliquots of 20,000 cells per well using the BTK forward primer: 5’- GAGCTCACAACATCGAACCA -3’ and reverse primer: 5’- ACTCCCCTCCTCCTACCAAC -3’ (30). PCR products were directly sequenced and analyzed using Mutation Surveyor® software and visual inspection of sequence traces. Positive clones were expanded in culture, and BTK disruption was confirmed by immunoblotting.
Generation of stable reconstituted OCI-LY7 BTK−/−, SUDHL4 BTK−/−, OCI-LY1, DT40 BTK−/−, HBL1 and TMD8 lymphoma cell lines expressing HA-tagged WT and mutant BTK
The cell lines OCI-LY1, OCI-LY7 BTK−/−, SUDHL4 BTK−/−, DT40 BTK−/−, HBL1 and TMD8 were transduced with FG9 lentivirus preparations carrying BTK WT or mutants by spin-inoculation as above before seeding into fresh medium. Enrichment for GFP-expressing cells was subsequently performed using FACS. Stably transfected cells were tested for expression levels of BTK protein using BTK- and HA-directed antibodies and immunoblotting.
BTK Immunoprecipitation followed by quantitative tandem mass spectrometry
5 × 107 DHL4 BTK−/−, BTK WT or mutant (p.K433T and p.P566L) reconstituted cells were starved in serum-free RMPI 1640 medium for 1 h followed by stimulation with 10 μg/ml anti-IgG for 10’. Cells were pelleted, washed and lysed on ice for 20’ in lysis buffer containing 0.2% NP-40 Surfact-Amps™ Detergent Solution (Thermo Fisher Scientific, RRID:SCR_008452; cat#85124), 100 mmol/L NaCl, 25 mmol/L Tris pH 8.0, 20 mmol/L NAF, 2 mmol/L EGTA, 2 mmol/L EDTA, supplemented with protease inhibitors (Sigma-Aldrich; #P8340) and phosphatase inhibitors (Sigma-Aldrich; #P0044), and sodium orthovanadate (Sigma-Aldrich; cat#450243), and PMSF. The detergent-soluble fraction of the cell lysates was obtained by centrifugation at 14,000 rpm for 10’. Two hundred μl of protein A agarose beads (Cell Signaling Technology; #9863,) were washed with PBS and incubated with 20 μl total mouse IgG-UNLB (Southern Biotech; #0107–01) for 2 h. The conjugated protein A agarose beads were washed with cold PBS and resuspended in lysis buffer. To pre-clear the protein samples, 50 μl conjugated protein A agarose beads were added to the protein lysate for 2 h at 4°C. Anti–HA-loaded beads (Sigma Aldrich; #A2095;) were blocked with 5% BSA/TBST for 2 h, followed by washing with lysis buffer. Forty μL of anti–HA-conjugated beads were added to pre-cleared cell lysates and incubated with rotation for 5 h at 4°C. The beads were washed 4 times with lysis buffer containing 150 mmol/L NACL, and pelleted by centrifugation at 2,000g for 5’. Afterward, half of each sample was processed by SDS-PAGE using a 10% Bis-Tris NuPage Mini-gel (Invitrogen) with the MES buffer system. The gel was run 2 cm. The mobility region was excised into 10 equally sized bands. Each band was processed by in-gel digestion with trypsin using a robot (ProGest, DigiLab) with the following protocol: Washed with 25mM ammonium bicarbonate followed by acetonitrile; Reduced with 10mM dithiothreitol at 60°C followed by alkylation with 50mM iodoacetamide at RT; Digested with sequencing grade trypsin (Promega) at 37°C for 4h; Quenched with formic acid and the supernatant was analyzed directly without further processing.
Half of each digested sample was analyzed by nano LC-MS/MS with a Waters M-Class HPLC system interfaced to a ThermoFisher Fusion Lumos mass spectrometer. Peptides were loaded on a trapping column and eluted over a 75µm analytical column at 350nL/min; both columns were packed with Luna C18 resin (Phenomenex). The mass spectrometer was operated in data-dependent mode, with the Orbitrap operating at 60,000 FWHM and 15,000 FWHM for MS and MS/MS respectively. The instrument was run with a 3s cycle for MS and MS/MS. 5 h of instrument time was used for the analysis of each sample.
The data were searched using a local copy of Mascot (Matrix Science) with the following parameters: Enzyme: Trypsin/P. Database: SwissProt Human (concatenated forward and reverse plus common contaminants). Fixed modification: Carbamidomethyl (C). Variable modifications: Oxidation (M), Acetyl (N-term), Pyro-Glu (N-term Q), Deamidation (N/Q). Mass values: Monoisotopic Peptide Mass Tolerance: 10 ppm. Fragment Mass Tolerance: 0.02 Da. Max Missed Cleavages: 2
Mascot DAT files were parsed into Scaffold (Proteome Software) for validation, filtering and to create a non-redundant list per sample. Data were filtered using at 1% protein and peptide FDR and requiring at least two unique peptides per protein.
BTK and PLCγ2 HEK293T cell phosphorylation assay
HEK293T cells were seeded at 3 × 105 per well in a 12 well plate. After 18 h, cells were transfected with plasmids encoding either FG9, WT or mutant forms of BTK with or without 0.5 µg of a PLCγ2 encoding plasmid (kindly provided by Dr. Edvard Smith, Karolinska Institute in Sweden) using PEI. Thirty-six hours later, cells were treated with or without ibrutinib at 1 μΜ for 1 h. Cells were then harvested, washed by PBS and lysed. Immunoblotting for BTK, p-BTK(223), PLCγ2, p-PLCγ2 (1217), PLCγ2 (759), and β-actin was performed as described.
Anti-IG treatment of cell lines, drug treatment and immunoblotting
Lymphoma cell lines were starved in serum-free RPMI 1640 for 1 h, followed by ibrutinib treatment. Subsequently, either anti-IgM (#2020–01) or anti-IgG (#2040–01) (Southern Biotech, Birmingham, AL) was added at 10 µg/ml for 10’. The cells were spun and pelleted at 4°C and lysates were prepared in lysis buffer containing: 1% Triton X-100 (#DSC41010; Dot Scientific), 150 mmol/L NaCl, 25 mmol/L Tris, pH 8.0, 20 mmol/L NaF, 2 mmol/L EGTA, 2 mmol/L EDTA, supplemented with protease inhibitors, phosphatase inhibitors, 1mM sodium orthovanadate and PMSF as above. The lysates were cleared by centrifugation at 14,000 rpm for 10’ and protein was fractioned by SDS-PAGE and prepared for immunoblotting using standard methods.
Immunoblotting for BTK, HA and actin was performed using: Anti-p-PLCγ2-Y1217, anti-PLCγ2, anti-p-BTK-Y223, anti-p-AKT-S473, anti-p-AKT-T308, anti-AKT, anti-p-p44/42-MAPK, anti-p44/42-MAPK antibodies were diluted in TBS-T with 5% BSA at 1:1000 for overnight incubation. Anti-p-PLCγ2-Y759 was diluted in TBS-T/ 5% dry milk at 1:500.
Cycloheximide pulse chase protein stability assay
5 × 105 OCI-LY7 BTK−/− cells reconstituted with BTK WT or various BTK mutants were seeded in 1ml of RPMI medium supplemented with 10% FCS. Starting at 0 h, 50 µg/ml cycloheximide was added into parallel cultures at 2 h intervals for a total of 8 h. Cells were harvested, detergent lysates made and prepared for immunoblotting to detect and measure BTK and β-actin from duplicate gel loadings. BTK protein density was quantitated using ImageJ, RRID:SCR_003070, version 2.0.0, NIH. The BTK protein density measurements at each longitudinal time point (2, 4, 6 and 8 h) were divided by the corresponding values at 0 h treatment and further normalized to actin.
General Methods for Chemistry
Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker Advance 400 MHz spectrometer. 1H NMR spectra were reported in parts per million (ppm) downfield from tetramethylsilane (TMS). In reported spectral data, the format (δ) chemical shift (multiplicity, J values in Hz, integration) was used with the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet. MS analyses were carried out with a Waters UPLC-mass spectrometer. The final compounds were all purified by C18 reverse phase preparative HPLC column with solvent A (0.1% TFA in H2O) and solvent B (0.1% TFA in MeCN) as eluents. The purity of all the final compounds was confirmed to be >95% by UPLC analysis (10% to 100% MeCN in H2O containing 0.1% CF3COOH in 10 min).
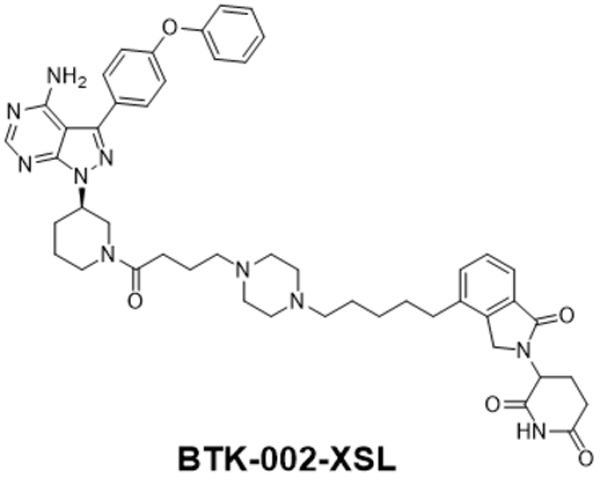
BTK-002-XSL
3-(4-(5-(4-(4-((R)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)-4-oxobutyl)piperazin-1-yl)pentyl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione.
1H NMR (400 MHz, MeOD) δ 8.40–8.39 (m, 1H), 7.70–7.64 (m, 3H), 7.49–7.47 (m, 2H), 7.44–7.40 (m, 2H), 7.21–7.16 (m, 3H), 7.12–7.10 (m, 2H), 5.20–5.16 (m, 1H), 4.99–4.93 (m, 1H), 4.59 (d, J = 11.2 Hz, 1H), 4.54–4.43 (m, 2H), 4.36–4.13 (m, 1H), 3.96–3.92 (m, 1H), 3.82–3.77 (m, 1H), 3.50–3.44 (m, 1H), 3.42–3.32 (m, 5H), 3.10–2.96 (m, 5H), 2.92–2.87 (m, 1H), 2.81–2.80 (m, 1H), 2.76–2.72 (m, 2H), 2.68–2.56 (m, 2H), 2.54–2.47 (m, 1H), 2.40–2.31 (m, 1H), 2.26–2.23 (m, 1H), 2.21–2.16 (m, 1H), 2.11–2.07 (m, 1H), 2.00–1.94 (m, 2H), 1.78–1.70 (m, 5H), 1.47–1.41 (m, 2H); ESI-MS calculated for C48H56N10O5 [M + H]+ = 853.44, found: 853.22; UPLC purity: 99.28%, retention time: 3.71 min.
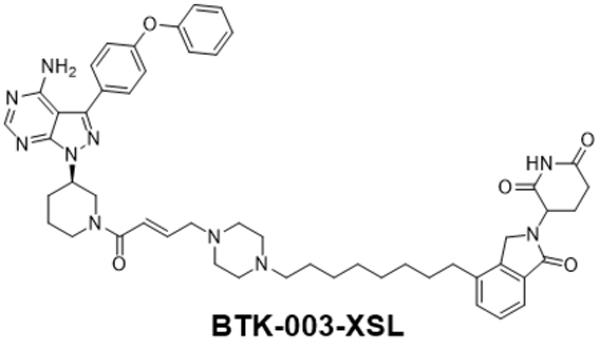
BTK-003-XSL
3-(4-(8-(4-((E)-4-((R)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)-4-oxobut-2-en-1-yl)piperazin-1-yl)octyl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione.
1H NMR (400 MHz, MeOD) δ 8.39 (s, 1H), 7.70–7.64 (m, 3H), 7.47–7.46 (m, 2H), 7.44–7.40 (m, 2H), 7.21–7.10 (m, 5H), 6.78–6.57 (m, 2H), 5.20–5.16 (m, 1H), 4.56–4.54 (m, 1H), 4.50–4.43 (m, 2H), 4.23–4.17 (m, 1H), 4.07–3.92 (m, 1H), 3.63–3.57 (m, 1H), 3.44–3.41 (m, 1H), 3.38–3.33 (m, 2H), 3.26–3.17 (m, 2H), 3.10–3.02 (m, 3H), 2.97–2.88 (m, 2H), 2.81–2.66 (m, 5H), 2.58–2.47 (m, 2H), 2.42–2.34 (m, 2H), 2.28–2.23 (m, 1H), 2.21–2.04 (m, 3H), 1.74–1.62 (m, 5H), 1.41–1.30 (m, 8H); ESI-MS calculated for C51H60N10O5 [M + H]+ = 893.47, found: 893.28. UPLC purity: 98.64%, retention time: 4.23 min.
BTK degrader (PROTAC) treatment of lymphoma cell lines
OCI-LY7, SUDHL4 and OCI-LY1 cells were treated with the BTK degraders #002 and #003 at 500 ng/ml for 24 h. The cells were subsequently starved in serum-free medium for 1 h before IG stimulation at 10 µg/ml for 10’. The cells were harvested and detergent cell lysates prepared for immunoblotting using indicated antibodies. In order to detect BTK expression in OCI-LY1 cells, concentrated cell lysates were prepared for immunoblotting using high sensitivity ECL reagents.
Ibrutinib drug kill and cell viability assay
Stable transduced HBL1 or TMD8 cells (3 × 105 cells per well) expressing either WT or mutant BTK were seeded in a 96 well plate in 100 μl medium containing ibrutinib at different concentrations. On days 2 and 4, 50 μl culture media containing 1x ibrutinib was added to replenish nutrients and ibrutinib. CellTiter-Glo reagent mix (Promega, Madison, WI) was added into each well on day 6 for HBL1 cells, or day 3 for TMD8 cells. Fluorescent signal intensity was measured using a GloMax®-Multi Detection System (Promega, Madison, WI).
Cell lines drug treatment
OCI-LY7 BTK−/− or BTK−/− / BLK−/− and SUDHL4 BTK−/− or BTK−/− / BLK−/− cell lines were starved in RPMI 1640 for 1 h, followed by treatment with SU6656, dasatinib or saracatinib at 5 μM for 1 h. Subsequently, either anti-IgM (#2020–01) or anti-IgG (#2040–01) (Southern Biotech, Birmingham, AL) was added at 10 µg / ml for 10’ followed by pelleting through centrifugation at 4°C.
OCI-LY7, SUDHL4, HBL1 and TMD8 cell lines were starved in RPMI 1640 for 1 h, followed by treatment with ibrutinib and acalabrutinib at 1 μM for 1, 2, 4 or 6 hours, followed by anti-IgM or anti-IgG stimulation at 10 µg / ml for 10’, followed by pelleting through centrifugation at 4°C.
Purification of FL B cells from lymph node biopsies and anti-IG stimulation
Cryopreserved single cell suspensions derived from FL biopsies were thawed, washed, and resuspended in degassed BSA/EDTA (1X PBS, 0.5%BSA, and 1 mmol/L EDTA) buffer at an approximate concentration of 107 cells per 80 µL. Cells were treated with 20 μL/107 cells of Miltenyi CD3 magnetic microbeads (human cat#130050101) and 20 μL/107 cells of Miltenyi CD14 magnetic microbeads (human cat#130050201), and incubated at 4°C for 25’. Cells were washed with BSA/EDTA and resuspended in 500 µL of BSA/EDTA and passed through Miltenyi LS columns (cat#130042401) loaded onto a QuadriMax magnet as per the manufacturer’s recommendations. The flow-through fraction containing CD3 and CD14 double negative enriched FL B cells was centrifuged and resuspended in RPMI1640 medium supplemented with 10% FBS and cultured in 24-well tissue culture plates (Fisher, Nunc Low Cell Binding, cat#145387). Subsequently, 4×105 cells were starved in serum-free RMPI-1640 for 1 h and stimulated with or without anti-IgM for 10’ followed by pelleting through centrifugation at 4°C. The cells were harvested and detergent cell lysates prepared for immunoblotting using indicated antibodies.
Lentiviral shRNA-mediated knockdown of BTK in non-malignant lymph node derived purified human B cells
Cryopreserved non-malignant human lymph node cell suspensions were depleted of CD3+ T cells and CD14+ cells and purified B cells cultured in B cell medium (Celprogen Catalog#: M33001–02S) for 2 h. Concentrated lentiviruses carrying BTK shRNA1 or shRNA2 (Millipore Sigma # TRCN0000009935 and TRCN0000009936) or scrambled shRNA were prepared by ultracentrifugation of 80 ml of virus-containing medium at 41,656 x g for 3 h followed by resuspension of the pellets in 2 ml. These lentiviral preparations were used to infect purified B cells by spin-inoculation at 30ºC at 1,360 x g for 2 h. Following viral infection, the cells were cultured in B cells medium for 36 h and starved for 1 h before anti-IG stimulation at 10 µg/ml for 10’ with or without pre-treatment for 1 h with 1 µM ibrutinib. The cells were harvested and detergent cell lysates prepared for immunoblotting using indicated antibodies.
Results
Identification of novel somatic mutations in BTK in follicular lymphoma
Based on the recent report of mutations in BTK in FL by Krysiak et al, (22) we re-sequenced all BTK coding exons and adjacent intronic regions using Sanger sequencing at 97% coverage in highly pure FL B cell DNA isolated from flow sorted FL B cells in a combined total of 139 FL grades 1–3A and 11 transformed FL cases. We confirmed the somatic nature of all sequence variants using paired sorted lymph node derived CD3+ T cell DNA. We identified 7% (10/150) of FL cases with non-synonymous clonal BTK mutations with high mutant fractions (BTK is located on the X-chromosome) (see Figure 1A–B and Supplementary Table 1 for details). We also confirmed expression of mutant BTK mRNA in five FL cases with available mRNA/cDNA and detected predominantly mutant mRNA expression in each case (Supplementary Figure 1). The mutations published by Krysiak et al (22) and the novel BTK mutations described here did not overlap suggesting that the full complement of BTK mutations in FL has not yet been identified.
Figure 1: Follicular lymphoma-associated BTK mutations are distributed over the BTK coding region and cluster on the protein surface.
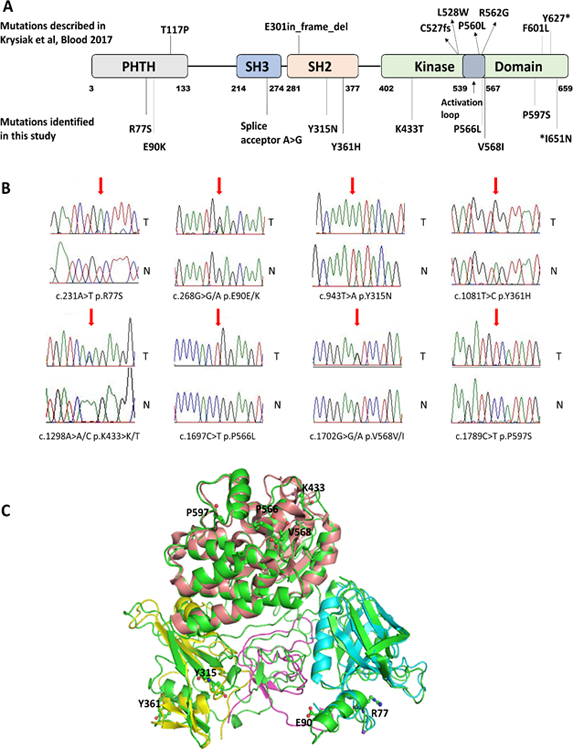
A: Location of BTK mutations identified by Krysiak et al Blood 2017 and in this study in a linear schema of BTK (22). B: Sanger sequence traces of paired sorted FL B cell (T) and paired CD3 T cell (N) DNA. Mutated residues are indicated by a red arrow. C: Composite 3-D model of BTK with protein domains and location of identified BTK missense mutations.
Of the BTK mutations reported here, three were detected in males, three in females while the sex of four patients with mutations detected in samples from the de-identified FL cohort II is unknown. Further, four BTK mutations were detected in biopsies from untreated FL patients and two in relapsed patients, while the clinical and treatment status of four patients with mutations detected in samples from the de-identified FL cohort II is unknown. Importantly, none of the patients had been treated with ibrutinib or idelalisib prior to lymph node biopsies.
FL-associated mutations in BTK occurred in all domains of the protein, including the PH, SH3, SH2 and catalytic domains (see Figure 1A–B). Of note, while most mutations detected were missense mutations, occasional splice-acceptor and non-sense mutations were detected as well. No hotspot for mutations was identified but some clustering of mutations in the BTK kinase domain was noted.
We modeled the location of the BTK missense mutations identified in this study onto a composite 3D model of BTK. As can be seen in Figure 1C, the mutations are distributed over the BTK protein surface within various BTK domains.
FL-associated BTK mutant proteins are destabilizing the BTK protein.
To study the properties of mutant BTK in lymphoma cell lines devoid of WT BTK, we disrupted the BTK gene using CRISPR-Cas9 targeting of the BTK intron/coding exon 1 boundary in the GC-DLBCL cell lines OCI-LY7 and SUDHL4. Following single cell cloning and BTK sequencing, we confirmed absent BTK expression in multiple independently derived clones per line by immunoblotting (see Supplementary Figure 2A–B, lanes 1 labelled vector). Next, we stably reconstituted these engineered BTK−/− lines with HA-tagged BTK WT and BTK mutant cDNAs using lentiviral gene transduction. We also created stable OCI-LY1 cell lines (OCI-LY1 has low endogenous BTK expression visible on longer blot exposures) expressing these BTK cDNAs (Supplementary Figure 2C) and reconstituted previously described chicken DT40 BTK−/− cell lines (Supplementary Figure 2D) (31).
Summarizing results across all four lymphoid cell lines, we detected lower expression of most of the BTK mutant proteins than for the reconstituted BTK WT protein; this was especially prominent for the mutations located in the BTK kinase domain. Qualitatively similar findings were obtained in transiently transfected HEK293T cells (Supplementary Figure 2E).
To confirm the unexpected BTK protein destabilizing properties of the BTK mutations, we performed serial BTK protein immunoblotting for up to 8 h in BTK WT and mutant reconstituted OCI-LY7 BTK−/− cells following inhibition of protein translation with cycloheximide. As can be seen in Figure 2, all eight BTK mutant proteins were substantially less stable than the BTK WT protein. Combined, the data provided support to the hypothesis that a reduction in BTK protein mass and/or kinase activity underlies the biological effects of FL-associated BTK mutations.
Figure 2: Follicular lymphoma-associated BTK mutations destabilize the mutant BTK proteins.
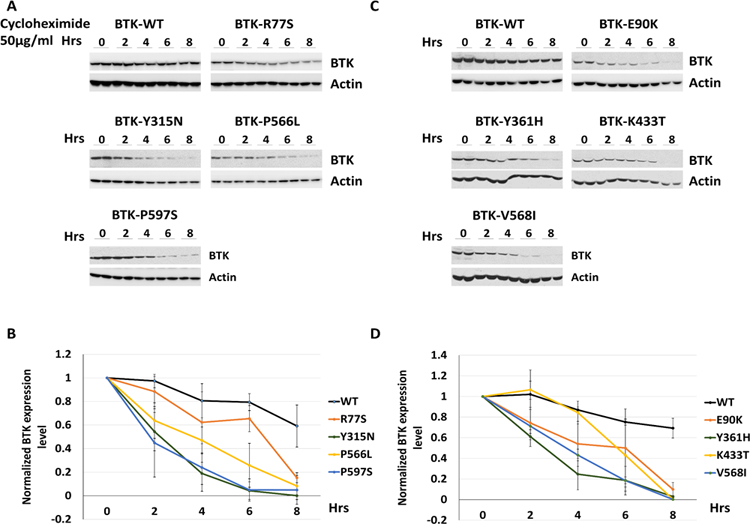
A, C: Reconstituted OCI-LY7 BTK−/− cells generated through CRISPR-Cas9 targeting of BTK were reconstituted with BTK WT or BTK mutant cDNAs using lentiviral transduction. Cells were cultured in the presence of 50 µg/ml Cycloheximide for the indicated time periods. Immunoblotting for BTK and actin was performed. B, D: Densitometry quantification of BTK band intensities based on triplicate experiments with immunoblotting of two lanes per samples normalized to measurements of BTK WT or individual BTK mutants at 0 h and to actin.
BTK mutations and their effects on Phospholipase C gamma 2 (PLCγ2) phosphorylation following BCR crosslinking in stable transduced lymphoid cell lines
The PLCγ2 is a critical mediator of BCR signaling and one of a very few well-characterized direct BTK substrates (31, 32). To deduce the catalytic properties of BTK mutant kinases within cells, we proceeded to study the phosphorylation state of PLCγ2 at tyrosine residues 1217 and 759 in the BTK WT or mutant reconstituted OCI-LY7 BTK−/− and SUDHL4 BTK−/− cell lines described above. Following IgM (OCI-LY7) or IgG (SUDHL4) crosslinking, we detected substantial increased PLCγ2-Y1217 or p-PLCγ2-Y759 phosphorylation in both cell lines; however, this increase was neither BTK dependent (see lanes labelled FG9; empty vector) nor was it substantially altered in any of the reconstituted mutant BTK cell lines (Supplementary Figures 3 and 4).
The pre-treatment with ibrutinib substantially blunted the anti-IgG induced increase in PLCγ2-Y1217 phosphorylation in reconstituted SUDHL4 BTK−/− cell lines, implicating multiple ibrutinib sensitive kinases in phosphorylating PLCγ2. Ibrutinib is known to inhibit a limited number of kinases involved in BCR signaling including the SRC family kinase BLK (33). We proceeded to generate OCI-LY7 BTK−/−BLK−/− and SUDHL4 BTK−/−BLK−/− cell lines using CRISPR-Cas9 targeting of BLK in BTK−/− cells followed by single cell cloning. Interestingly, in OCI-LY7 BTK−/−BLK−/− mutant cell lines (see Supplementary Figure 5 clones G1-G8 or G2-E4), the anti-IgM crosslinking induced phosphorylation of PLCγ2 was preserved.
Next, we employed small molecule kinase inhibitors with varying target profiles in the OCI-LY7 BTK−/−/BLK−/− and SUDHL4 BTK−/−/BLK−/− cell lines. Pre-treatment using the multi targeted SRC family kinase inhibitors SU6656, dasatinib or saracatinib substantially inhibited or abolished anti-IG stimulation induced PLCγ2 phosphorylation (Supplementary Figure 6).
In aggregate, these findings implicate multiple tyrosine kinases in PLCγ2 phosphorylation at amino acid residues 759 and 1217 in anti-IG treated lymphoma cell lines. The data do not support that FL-associated BTK mutations aberrantly regulate the phosphorylation state of PLCγ2.
BTK mutations targeting the kinase domain inactivate the BTK kinase
We proceeded to further study the kinase properties of BTK mutants in HEK293 cells transiently co-transfected with various plasmids encoding WT or mutant BTK and PLCγ2. We found that all BTK kinase domain mutations inactivated the BTK kinase resulting in kinase dead BTK; a finding that was corroborated in reconstituted lymphoma cells (see below). The mutations in other BTK domains retained the ability of BTK to phosphorylate PLCγ2 and to autophosphorylate at BTK p-223 (Figure 3).
Figure 3: BTK kinase domain mutations inactivate the BTK kinase.
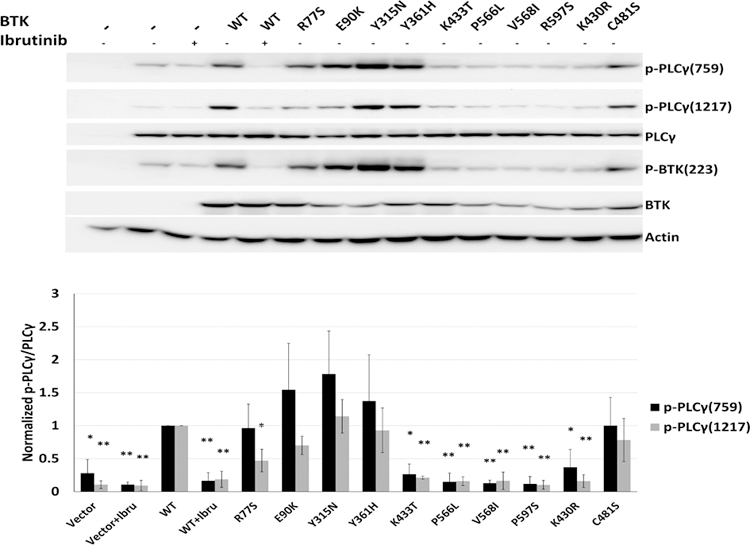
HEK293T cells were co-transfected with BTK WT or BTK mutant plasmids and PLCγ2. Some cells were pre-treated with ibrutinib for 1 h followed by anti-Ig treatment for 10’. Detergent cell lysates were made, fractioned by SDS-Page and prepared for immunoblotting with antibodies targeting the indicated epitopes. Displayed are composite data from three independent experiments per cell line. Lane 1: no plasmid transfection control; lanes 2 and 3: PLCγ2 encoding plasmid only. Vector: cells transfected with PLCγ2 plasmid only (paired two-sample t-test; * p<0.05; ** p<0.01).
BTK mutations augment AKT phosphorylation following BCR crosslinking
We broadened the search for effects of BTK mutant proteins on signaling pathways by measuring the degree of BTK-223 auto-phosphorylation, the phosphorylation state of AKT at the mTORC2- (AKT-473) and PDK1- (AKT-308) dependent sites as well as p-ERK-202/204. As expected, following BCR crosslinking, we detected increased p-BTK-223 in the OCI-LY7 BTK−/− and SUDHL4 BTK−/− cell lines reconstituted with BTK WT, which was inhibited by ibrutinib pre-treatment. The p-BTK-223 auto-phosphorylation was absent however in the cell lines with reconstituted BTK kinase domain mutants (BTK-P566L, V568I and P597S) confirming that this class of mutation abolishes BTK kinase function. Most other BTK mutations preserved some or all of p-BTK-223 (Figure 4A–D).
Figure 4: BTK mutations enhance anti-IG induced AKT phosphorylation.
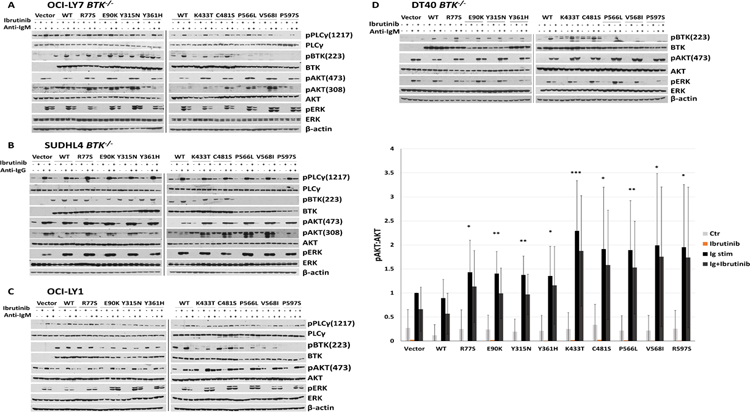
A–D: Engineered OCI-LY7 BTK−/−, SUDHL4 BTK−/−, or DT40 BTK−/− cell lines as well as OCI-LY1 cells expressing low level endogenous BTK were reconstituted with HA-tagged BTK WT or mutant cDNAs and stable cell lines generated. The cells were rested for 1 h in serum-free medium and cell aliquots pre-treated for 1 h with 1 µM of ibrutinib followed by stimulation for 10’ with 10 µg / ml of anti-IG antibodies. Detergent cell lysates were made, fractioned by SDS-PAGE and prepared for immunoblotting with antibodies targeting the indicated epitopes. Displayed are immunoblotting data from representative experiments (vector: cells infected with empty FG9 lentivirus). Displayed are composite densitometry data from three independent experiments per cell line for the p-AKT473:AKT data (paired two-sample t-test; * p<0.05; ** p<0.01; *** p<0.001).
Next, we studied the phosphorylation states of AKT and ERK following BCR crosslinking in four separate cell lines expressing WT or mutant BTK. We detected substantially increased AKT phosphorylation in most cell lines carrying the various BTK mutants (Figure 4A–D; quantitative data from three intendent experiments per cell line for p-AKT-473:AKT are displayed and corresponding data for p-AKT-308:AKT are shown in Supplementary Figure 7). In contrast, across multiple replicate experiments in all cell lines studied, p-ERK remained largely unchanged.
In summary, enhanced phosphorylation of AKT was the most prominent BCR signaling change imparted by BTK mutants, a finding that is in line with the important role served by AKT in chronic B cell receptor signaling in DLBCL (26, 27).
BTK WT or mutants do not directly interact with AKT or differentially interact with known AKT regulators.
We performed quantitative immunoprecipitation (IP)-mass spectrometry experiments out of BTK−/− (control) and HA-BTK WT and mutant (K433T and P566L) reconstituted DHL4 cells to detect and measure BTK WT and mutant binding proteins. We found that all BTK proteins bound the well-characterized HSP90-associated co-chaperone protein CDC37. The BTK WT and the kinase-active mutant K433T but not the kinase-dead mutant P566L also bound PLCγ2 consistent with active kinase substrate interactions. Overall, we noticed a substantially reduced number of proteins to co-IP with the kinase-dead BTK mutant P566L compared with BTK WT or the BTK mutant K433T even after correction for the amount of immunoprecipitated BTK. Of note, we did not detect AKT or known AKT regulators in the immunoprecipitates. The only possible exception was RAC2, which precipitated equally with WT and MUT BTK. These data are summarized in Supplementary Table 2 and the embedded Venn diagram.
Acute chemical inhibition of BTK does not enhance AKT phosphorylation.
How BTK participates in the regulation of AKT in normal or malignant B cells is largely unknown. The increased AKT phosphorylation at critical regulatory sites measured in cells carrying mutants BTK proteins reported here could be a consequence of activation of a feedback loop that senses reduced BTK kinase activity or alternatively be due to reduced BTK protein mass. To differentiate between these scenarios, we incubated four lymphoma cell lines (OCI-LY7, SUDHL4, HBL1 and TMD8) with ibrutinib or the more targeted BTK inhibitor acalabrutinib for various times (0–6 h) and measured p-AKT-473 as well as p-BTK-223 and p-ERK202/204 following BCR-crosslinking (Supplementary Figure 8). Both drugs effectively inhibited phosphorylation of p-BTK-223, but only ibrutinib modestly reduced phosphorylation of p-AKT473. Acalabrutinib pre-treatment did not inhibit or augment phosphorylation of p-AKT473.
The proteolysis targeting chimera (PROTAC)-mediated degradation of BTK protein results in augmentation of BCR-crosslinking induced AKT phosphorylation.
To study the effects of reductions in BTK protein expression on anti-IG induced AKT phosphorylation, we incubated the cell lines OCI-LY7, SUDHL4 and OCI-LY1 with 500 nM of two distinct PROTAC molecules (#002 and #003) comprised of ibrutinib chemically linked to lenalidomide (see methods). Following 24 h of incubation and starvation of 1 h and subsequent stimulation with anti-IG for 10’, we measured BTK, p-AKT473, AKT and actin expression by immunoblotting in detergent cell lysates. We found that the very potent degrader #002 resulted in complete loss of BTK expression, while the degrader #003 resulted in a substantial reduction of BTK expression (Supplementary Figure 9A–F) without loss in cell viability. Following anti-IG treatment, we subsequently detected substantially elevated p-AKT473 in all cell lines with experimentally reduced BTK expression (Supplementary Figure 9A–F). In contrast, cells treated with lenalidomide alone did not show any such changes and no loss in viability. The results also show that a partial reduction in BTK expression was sufficient for the observed phenotypes. This data supports the emerging model that the expression levels of BTK protein regulates anti-IG induced AKT473 phosphorylation.
The lentiviral shRNA-mediated knock-down of BTK expression in primary human lymph node-derived non-malignant B cells results in strong augmentation of sIG-crosslinking induced AKT phosphorylation.
To provide additional evidence that the expression levels of BTK protein influences anti-IG induced AKT phosphorylation, we resorted to lentiviral-mediated BTK-targeting shRNA delivery into primary human lymphnode-derived purified non-malignant B cells. These experiments were technically enabled through use of highly concentrated lentiviral preparations. In separate experiments using purified B cells isolated from four non-malignant lymphnode specimens, we found that knock-down of BTK expression resulted in very strong sIG-induced AKT-473 phosphorylation (Figure 5A–E).
Figure 5: The lentiviral shRNA-mediated knock-down of BTK expression in primary human lymph node-derived B cells results in strong augmentation of sIG-crosslinking induced AKT phosphorylation.
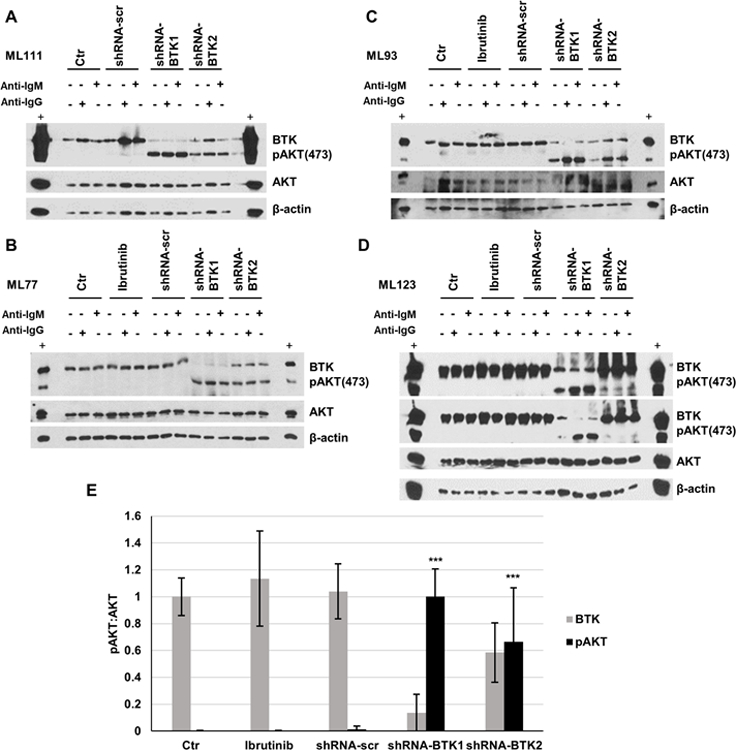
Non-malignant human lymph node derived B cells were purified from LN biopsies via depletion of CD3+ and CD14+ cells. Purified B cells were cultured and transduced using highly concentrated lentiviral preparations of pLKO-BTK targeted or a scrambled shRNA. Following 36 h culture in B cell medium, cells were rested for 1 h in serum-free medium followed by treatment with anti-IG at 10 µg/ml for 10’. Detergent cell lysates were made, fractioned by SDS-PAGE and prepared for immunoblotting with antibodies targeting the indicated epitopes. A–D: Displayed are immunoblotting data based on four separate lymph node biopsies (Ctr: uninfected; shRNA-scr: scrambled shRNA; panel D: long and short pAKT exposures are shown). E: Displayed are composite densitometry data for BTK and p-AKT473:AKT from four experiments shown in A–D. (unpaired t-test; *** p<0.001).
All primary human FL B cells are characterized by augmented AKT signaling following surface IG crosslinking.
We proceeded to purify FL B cells from cryopreserved LN biopsies from FL patients carrying WT (N=9) or mutant BTK (N=3; limited by the availability of cryopreserved specimens. L117: BTK p.E90K; L119: BTK p.K433T; ML162: BTK p.I651N). The BTK p.I651N variant demonstrated the protein instability phenotype detected in other BTK kinase domain mutations (see Figure 2). We also purified B cells from human LN biopsies (N=5) from subjects without NHL (non-malignant LNs). Purified FL B cells were cultured in serum-free RPMI medium for 1 h and left unstimulated or stimulated for 10’ with anti-IG followed by cell lysis and immunoblotting for various indicated epitopes.
At baseline we detected elevated p-AKT-473 in most of the FL B cells when compared with non-malignant B cells (Figure 6; left panel and Supplementary Figure 10). Following surface IG crosslinking, all FL cases demonstrated substantially elevated p-AKT-473 when compared with non-malignant B cells (Figure 6; right panel; black label). This included all BTK mutant FL cases, which demonstrated very strong anti-IG induced AKT phosphorylation (Figure 6; right panel; red label). Data support that substantially elevated p-AKT-473 following BCR crosslinking is a general characteristic of FL B cells and that BTK mutations are one contributing genetic factor.
Figure 6: Primary human FL B cells have augmented AKT phosphorylation following surface IgM crosslinking:
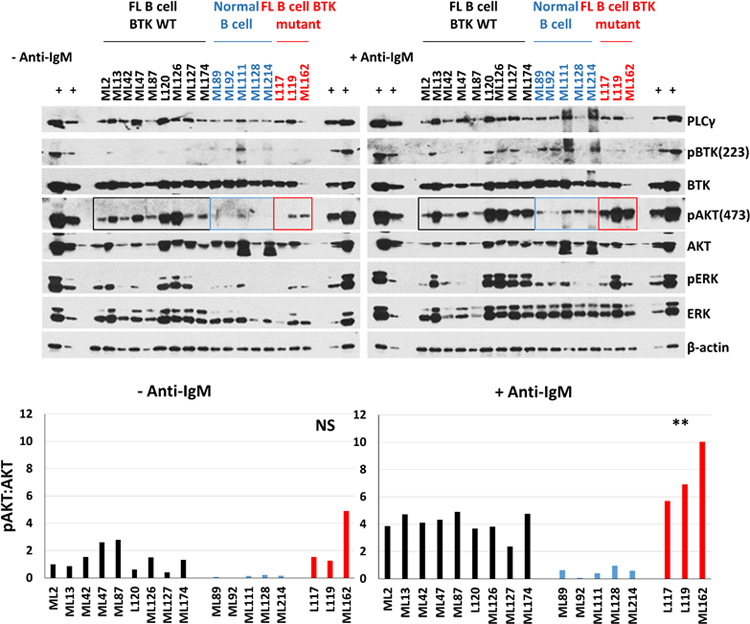
Upper panels: Primary cryopreserved human FL B cells carrying WT BTK (N=9; black) or mutant BTK (N=3; red) and primary human lymph node derived non-malignant B cells (N=5; blue) were purified over Miltenyi columns to deplete CD3+ and CD14+ cells. Purified FL B cells were cultured for 1 h in serum-free RPMI1640 medium followed by surface IgM crosslinking for 10’. Cell lysates were prepared and protein fractioned by SDS-PAGE and prepared for immunoblotting to the indicated epitopes. Lower panels: The mean densitometry result for p-AKT473:AKT from two separate immunoblots (technical replicates) of the same samples are shown (please also see Supplementary figure 10). ** P=0.015 using unpaired t test with Welch’s correction comparing BTK MUT cases versus BTK WT.
The enhanced phosphorylation of AKT caused by BTK mutations is fully blocked by phosphoinositol 3 kinase delta (PI3Kδ) inhibition.
The major upstream regulator of AKT in hematopoietic cells is PI3Kδ (34, 35). We therefore tested if PI3Kδ regulated the augmented AKT activation measured in lymphoma cells carrying mutant BTK proteins. We pre-treated OCI-LY7 BTK−/− and SUDHL4 BTK−/− cell lines reconstituted with BTK WT or selected BTK mutants (including the experimental kinase-inactive BTK mutant K430R) with the clinically approved PI3Kδ inhibitor CAL101 followed by IG crosslinking and immunoblotting as above. We found that CAL101 almost completely blocked sIG-induced AKT phosphorylation in all cell lines tested, implicating activation of canonical PI3Kδ signaling in sIG-induced AKT activation by BTK mutant proteins (Supplementary Figure 11).
We also tested expression levels of surface IG by flow cytometry in the various cell lines reconstituted with WT or mutant forms of BTK and detected no differences, excluding upregulated surface IG as the cause for elevated AKT activation (36).
A subset of FL-associated BTK mutants cause ibrutinib resistance
During ibrutinib therapy some patients with CLL and Waldenstrom’s macroglobulinemia acquire mutations in BTK (most commonly p.BTK-C481S), which affects ibrutinib binding and causes acquired resistance to ibrutinib (37). We asked if FL-associated BTK mutations, which we detected in biopsies from patients never exposed to BCR-targeted drugs affected responsiveness to ibrutinib (38).
The lymphoma cell lines HBL1 and TMD8 are highly sensitive to ibrutinib treatment with IC50 values in the low nanomolar range (39). We generated stable HBL1 and TMD8 lymphoma cell lines expressing WT or mutant forms of BTK (Supplementary Figure 12A, C). We treated these lines with ibrutinib in culture and measured cell viability. As previously reported, the p.BTK-C481S mutation caused ibrutinib resistance in these lines (Supplementary Figure 12B, D; displayed are summary data for N=3 independent experiments; please note the log10-scale). Of interest, two of the FL-associated BTK mutations (p.Y315N and p.P597S) also caused ibrutinib resistance, while all other BTK mutations resembled BTK WT.
Discussion
In this manuscript, we present the discovery and functional characterization of novel mutations in Bruton’s tyrosine kinase (BTK) in FL. Similar but non-overlapping mutations in BTK have previously been reported in patients with FL (22). Unlike mutations in BTK that are detected in patients with CLL relapsing on ibrutinib therapy (37), the mutations in BTK in FL were detected in FL biopsies from patients that were never treated with BCR-targeted drugs.
We report on the unexpected novel finding that all mutations in BTK destabilize the BTK protein. In addition, a subset of the BTK mutations located in the BTK catalytic domain inactivate the BTK kinase activity. Subsequently, we found that the FL-associated BTK mutations had no effect on the phosphorylation state of PLCγ2, a major well-studied substrate of BTK (32). In contrast, we discovered that FL-associated BTK mutations cause augmented AKT phosphorylation following BCR crosslinking as measured in 1) multiple lymphoma cell lines engineered to express BTK mutants; 2) multiple lymphoma cell lines treated with BTK protein degraders, 3) primary human lymph node-derived non-malignant B cells following shRNA-mediated BTK knock-down, and, 4) in primary human FL B cells, when compared with non-malignant lymph node-derived primary human B cells. Therefore, BTK mutations constitute one genetic cause for increased AKT phosphorylation following surface IG crosslinking in FL, which we found is characteristic of the pathobiology of FL (40). The finding that treatment with a PI3Kδ inhibitor abolished the increased AKT phosphorylation following BCR crosslinking in BTK mutant lymphoma cell lines suggests that AKT phosphorylation remains fully under the control of canonical activated PI3Kδ signaling. This finding provides a potential therapeutic solution to counter the elevated BCR-induced AKT activity in FL that carry BTK mutations (20, 41, 42).
Mechanistically, very little is known about the regulation of AKT by BTK. Using IP-mass spectrometry we did not detect a physical BTK - AKT interaction and also did not detect differential interactions of BTK with AKT regulatory proteins. BTK and AKT, like other PH-domain containing signaling molecules, bind to membrane-bound phosphoinositol triphosphate (PIP3) that is generated following PI3Kδ activation (43, 44), and such binding is part of the activation mechanism of these molecules. We surmise that BTK protein destabilization facilitates AKT activation through facilitated AKT interactions with membrane bound phosphoinositol triphosphate (45) but other possibilities remain (46–48).
Further, regarding various MAPK signaling pathways (for instance the BCR/ RAS/ RAF/ MAP3K/ MEK/ ERK1/2 pathway), we focused on ERK1/2 as the terminal kinase without a more detailed analysis of upstream MAPK signaling components. We found that the phosphorylation of ERK1/2 was not altered by BTK mutant proteins.
The BTK inhibitor ibrutinib is in clinical development in FL and is in clinical use in various other B cell malignancies. The reported response rate in relapsed FL was 37%, which indicates single agent activity but also a majority of patients with primary resistance (25). Our combined experimental findings offer a molecular explanation for some of these resistant patients: the reduction of BTK expression by destabilizing BTK mutants and the discovery of kinase-dead BTK mutants. This discovery and related properties reported for CARD11 mutations and likely other FL-associated gene mutations motivates a future strategy of screening for specific gene mutations prior to initiation of ibrutinib-based therapies in FL (39). Such a strategy would increase the probability of response to ibrutinib in the remaining patients.
In summary, we report on unexpected loss-of-function and gain-of-function phenotypes for FL-associated BTK mutations that result in augmented BCR-activated AKT signaling. Our findings have direct implications for the optimized use of small molecules targeting BTK, PI3Kδ and possible AKT in FL, thus motivating further in-depth studies of other FL-associated mutated signaling molecules for novel phenotypes and therapeutic implications (49).
Supplementary Material
TRANSLATIONAL RELEVANCE.
Follicular Lymphoma (FL) constitutes the second most common non-Hodgkin’s lymphoma (NHL) in the United States, with over 100,000 patients living with the disease. Almost all patients with FL need therapy within years from diagnosis, and most patients receive multiple chemo- or immunotherapies over their lifetimes. Despite recent progress, the development of targeted therapies in FL is in early stages. Here, we report the surprising novel finding that mutations in BTK in FL inactivate the BTK kinase or destabilize the BTK protein, thus demonstrating that some FL do not need functional BTK for growth or survival. We furthermore show that in the setting of mutant BTK, B cell receptor induced AKT phosphorylation is augmented and that this phenotype can be ablated with PI3K inhibitors. These findings are directly relevant to the ongoing clinical development of small molecule therapeutics in FL.
ACKNOWLEDGEMENTS
We are grateful for services provided by the genomics, bioinformatics and flow cytometry cores of the University of Michigan Rogel Comprehensive Cancer Center. We are also grateful to the contribution of the hematological malignancy group of the University of Michigan.
FUNDING SOURCES
This work was supported in part by R01CA190384, Janssen R&D and Pharmacyclics 16-PAF02251, a University of Michigan Rogel Cancer Center Scholar award (all to SNM) and the Weatherhall foundation (to MK).
Footnotes
Presented as part of a podium presentation at the American Society of Hematology Meeting, Atlanta, Georgia, 2017.
Conflict of Interest
SNM owns stock equity in Abbvie
REFERENCES
- 1.Friedberg JW. Progress in Advanced-Stage Follicular Lymphoma. J Clin Oncol. 2018;36:2363–5. [DOI] [PubMed] [Google Scholar]
- 2.Dave SS, Wright G, Tan B, Rosenwald A, Gascoyne RD, Chan WC, et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N Engl J Med. 2004;351:2159–69. [DOI] [PubMed] [Google Scholar]
- 3.Stevenson FK, Stevenson GT. Follicular lymphoma and the immune system: from pathogenesis to antibody therapy. Blood. 2012;119:3659–67. [DOI] [PubMed] [Google Scholar]
- 4.Kridel R, Sehn LH, Gascoyne RD. Pathogenesis of follicular lymphoma. J Clin Invest. 2012;122:3424–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369:507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farooqui MZ, Valdez J, Martyr S, Aue G, Saba N, Niemann CU, et al. Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemia with TP53 aberrations: a phase 2, single-arm trial. Lancet Oncol. 2015;16:169–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yildiz M, Li H, Bernard D, Amin NA, Ouillette P, Jones S, et al. Activating STAT6 mutations in follicular lymphoma. Blood. 2015;125:668–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes AB, et al. Genetics of follicular lymphoma transformation. Cell Rep. 2014;6:130–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okosun J, Bodor C, Wang J, Araf S, Yang CY, Pan C, et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet. 2014;46:176–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Green MR. Chromatin modifying gene mutations in follicular lymphoma. Blood. 2018;131:595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H, Kaminski MS, Li Y, Yildiz M, Ouillette P, Jones S, et al. Mutations in linker histone genes HIST1H1 B, C, D, and E; OCT2 (POU2F2); IRF8; and ARID1A underlying the pathogenesis of follicular lymphoma. Blood. 2014;123:1487–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang F, Gatica D, Ying ZX, Peterson LF, Kim P, Bernard D, et al. Follicular lymphoma-associated mutations in vacuolar ATPase ATP6V1B2 activate autophagic flux and mTOR. J Clin Invest. 2019;129:1626–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carbone A, Roulland S, Gloghini A, Younes A, von Keudell G, Lopez-Guillermo A, et al. Follicular lymphoma. Nat Rev Dis Primers. 2019;5:83. [DOI] [PubMed] [Google Scholar]
- 17.Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890–6. [DOI] [PubMed] [Google Scholar]
- 19.Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013;23:677–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ying ZX, Jin M, Peterson LF, Bernard D, Saiya-Cork K, Yildiz M, et al. Recurrent Mutations in the MTOR Regulator RRAGC in Follicular Lymphoma. Clin Cancer Res. 2016;22:5383–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okosun J, Wolfson RL, Wang J, Araf S, Wilkins L, Castellano BM, et al. Recurrent mTORC1-activating RRAGC mutations in follicular lymphoma. Nat Genet. 2016;48:183–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krysiak K, Gomez F, White BS, Matlock M, Miller CA, Trani L, et al. Recurrent somatic mutations affecting B-cell receptor signaling pathway genes in follicular lymphoma. Blood. 2017;129:473–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burger JA, Tedeschi A, Barr PM, Robak T, Owen C, Ghia P, et al. Ibrutinib as Initial Therapy for Patients with Chronic Lymphocytic Leukemia. N Engl J Med. 2015;373:2425–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Treon SP, Tripsas CK, Meid K, Warren D, Varma G, Green R, et al. Ibrutinib in previously treated Waldenstrom’s macroglobulinemia. N Engl J Med. 2015;372:1430–40. [DOI] [PubMed] [Google Scholar]
- 25.Bartlett NL, Costello BA, LaPlant BR, Ansell SM, Kuruvilla JG, Reeder CB, et al. Single-agent ibrutinib in relapsed or refractory follicular lymphoma: a phase 2 consortium trial. Blood. 2018;131:182–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Havranek O, Xu J, Kohrer S, Wang Z, Becker L, Comer JM, et al. Tonic B-cell receptor signaling in diffuse large B-cell lymphoma. Blood. 2017;130:995–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463:88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ross CW, Ouillette PD, Saddler CM, Shedden KA, Malek SN. Comprehensive analysis of copy number and allele status identifies multiple chromosome defects underlying follicular lymphoma pathogenesis. Clin Cancer Res. 2007;13:4777–85. [DOI] [PubMed] [Google Scholar]
- 29.Galban S, Hwang C, Rumble JM, Oetjen KA, Wright CW, Boudreault A, et al. Cytoprotective effects of IAPs revealed by a small molecule antagonist. Biochem J. 2009;417:765–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu N, Malek SN. Gene Disruption Using CRISPR-Cas9 Technology. Methods Mol Biol. 2019;1881:201–9. [DOI] [PubMed] [Google Scholar]
- 31.Takata M, Kurosaki T. A role for Bruton’s tyrosine kinase in B cell antigen receptor-mediated activation of phospholipase C-gamma 2. J Exp Med. 1996;184:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watanabe D, Hashimoto S, Ishiai M, Matsushita M, Baba Y, Kishimoto T, et al. Four tyrosine residues in phospholipase C-gamma 2, identified as Btk-dependent phosphorylation sites, are required for B cell antigen receptor-coupled calcium signaling. J Biol Chem. 2001;276:38595–601. [DOI] [PubMed] [Google Scholar]
- 33.Berglof A, Hamasy A, Meinke S, Palma M, Krstic A, Mansson R, et al. Targets for Ibrutinib Beyond B Cell Malignancies. Scand J Immunol. 2015;82:208–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chantry D, Vojtek A, Kashishian A, Holtzman DA, Wood C, Gray PW, et al. p110delta, a novel phosphatidylinositol 3-kinase catalytic subunit that associates with p85 and is expressed predominantly in leukocytes. J Biol Chem. 1997;272:19236–41. [DOI] [PubMed] [Google Scholar]
- 35.Vanhaesebroeck B, Welham MJ, Kotani K, Stein R, Warne PH, Zvelebil MJ, et al. P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc Natl Acad Sci U S A. 1997;94:4330–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drennan S, Chiodin G, D’Avola A, Tracy I, Johnson PW, Trentin L, et al. Ibrutinib Therapy Releases Leukemic Surface IgM from Antigen Drive in Chronic Lymphocytic Leukemia Patients. Clin Cancer Res. 2019;25:2503–12. [DOI] [PubMed] [Google Scholar]
- 37.Woyach JA, Ruppert AS, Guinn D, Lehman A, Blachly JS, Lozanski A, et al. BTKC481S-Mediated Resistance to Ibrutinib in Chronic Lymphocytic Leukemia. J Clin Oncol. 2017;35:1437–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen JG, Liu X, Munshi M, Xu L, Tsakmaklis N, Demos MG, et al. BTK(Cys481Ser) drives ibrutinib resistance via ERK1/2 and protects BTK(wild-type) MYD88-mutated cells by a paracrine mechanism. Blood. 2018;131:2047–59. [DOI] [PubMed] [Google Scholar]
- 39.Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med. 2015;21:922–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Myklebust JH, Brody J, Kohrt HE, Kolstad A, Czerwinski DK, Walchli S, et al. Distinct patterns of B-cell receptor signaling in non-Hodgkin lymphomas identified by single-cell profiling. Blood. 2017;129:759–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paul J, Soujon M, Wengner AM, Zitzmann-Kolbe S, Sturz A, Haike K, et al. Simultaneous Inhibition of PI3Kdelta and PI3Kalpha Induces ABC-DLBCL Regression by Blocking BCR-Dependent and -Independent Activation of NF-kappaB and AKT. Cancer Cell. 2017;31:64–78. [DOI] [PubMed] [Google Scholar]
- 42.Gopal AK, Kahl BS, de Vos S, Wagner-Johnston ND, Schuster SJ, Jurczak WJ, et al. PI3Kdelta inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med. 2014;370:1008–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li T, Rawlings DJ, Park H, Kato RM, Witte ON, Satterthwaite AB. Constitutive membrane association potentiates activation of Bruton tyrosine kinase. Oncogene. 1997;15:1375–83. [DOI] [PubMed] [Google Scholar]
- 44.Saito K, Scharenberg AM, Kinet JP. Interaction between the Btk PH domain and phosphatidylinositol-3,4,5-trisphosphate directly regulates Btk. J Biol Chem. 2001;276:16201–6. [DOI] [PubMed] [Google Scholar]
- 45.Manning BD, Toker A. AKT/PKB Signaling: Navigating the Network. Cell. 2017;169:381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Honda F, Kano H, Kanegane H, Nonoyama S, Kim ES, Lee SK, et al. The kinase Btk negatively regulates the production of reactive oxygen species and stimulation-induced apoptosis in human neutrophils. Nat Immunol. 2012;13:369–78. [DOI] [PubMed] [Google Scholar]
- 47.Chung JK, Nocka LM, Decker A, Wang Q, Kadlecek TA, Weiss A, et al. Switch-like activation of Bruton’s tyrosine kinase by membrane-mediated dimerization. Proc Natl Acad Sci U S A. 2019;116:10798–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Q, Pechersky Y, Sagawa S, Pan AC, Shaw DE. Structural mechanism for Bruton’s tyrosine kinase activation at the cell membrane. Proc Natl Acad Sci U S A. 2019;116:9390–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Erdmann T, Klener P, Lynch JT, Grau M, Vockova P, Molinsky J, et al. Sensitivity to PI3K and AKT inhibitors is mediated by divergent molecular mechanisms in subtypes of DLBCL. Blood. 2017;130:310–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.