

Summary
Memory T cells are thought to rely on oxidative phosphorylation and short-lived effector T cells on glycolysis. Here, we investigated how T cells arrive at these states during an immune response. To understand the metabolic state of rare, early activated T cells, we adapted mass cytometry to quantify metabolic regulators at single-cell resolution in parallel with cell signaling, proliferation, and effector function. We interrogated CD8+ T cell activation in vitro and in response to Listeria monocytogenes infection in vivo. This approach revealed a distinct metabolic state in early activated T cells characterized by maximal expression of glycolytic and oxidative metabolic proteins. Cells in this transient state were most abundant five days post-infection before rapidly decreasing metabolic protein expression. Analogous findings were observed in CAR T cells interrogated longitudinally in advanced lymphoma patients. Our study demonstrates the utility of single-cell metabolic analysis by mass cytometry to identify metabolic adaptations of immune cell populations in vivo and provides a resource for investigations of metabolic regulation of immune responses across a variety of applications.
Graphical Abstract
eTOC Blurb
Levine, Hiam-Galvez et al. develop a mass cytometry-based approach to quantify metabolic protein expression in single cells in vivo, revealing a distinct metabolic state early after CD8+ T cell activation characterized by simultaneous expression of glycolytic and oxidative proteins. This approach provides a resource for the study of metabolic regulation across a variety of applications.
Introduction
Understanding the regulatory mechanisms underlying immune responses is crucial to developing more rationally designed treatment strategies for acute and chronic infections, autoimmune diseases and malignancy (Buck et al., 2017). CD8+ T cells, when activated, expand and differentiate into potent short-lived effector cells (SLECs) as well as long-term memory cells, which can confer durable protection against re-infection and cancer relapse (Badovinac et al., 2007; Callahan et al., 2016; Restifo et al., 2012). The former mediate primary adaptive immune responses against pathogens through the release of cytotoxic granules and pro-inflammatory cytokines (Araki et al., 2010; Pearce et al., 2009). In contrast, long-lived memory cells remain quiescent until re-encountering antigen, upon which they rapidly mediate secondary immune responses (Gerriets and Rathmell, 2012). The field of immunometabolism provides critical insight into these processes, revealing a complex regulatory interplay of signaling, metabolic and epigenetic adaptations during CD8+ T cell differentiation (Olenchock et al., 2017; Zhang and Romero, 2017).
Upon activation, effector CD8+ T cells undergo clonal expansion, necessitating as many as 20 replication cycles to generate sufficient daughter cells to clear pathogens (Badovinac et al., 2007). This process is energetically costly and requires rapid ATP production for the biosynthesis of essential building blocks (Zhang and Romero, 2017). Previous studies suggest that the exit from quiescence is supported by a dramatic metabolic shift from oxidative phosphorylation (OXPHOS) in naïve cells, fueled by beta-oxidation of long chain fatty acids (LCFA), to aerobic glycolysis in SLECs, characterized by lactate production in the setting of adequate oxygen (Calderon et al., 2018; Menk et al., 2018; Wang et al., 2011). This metabolic conversion permits continued cycling through the pentose phosphate pathway and thus generation of intermediates necessary for nucleic acid and lipid biosynthesis. This adaptation also circumvents negative feedback induced by the accumulation of pyruvate and acetyl-CoA (Lee et al., 2014; Zhang and Romero, 2017). Additional feed-forward mechanisms supporting this process include the activation of transcription factors downstream of phosphoinositide 3-kinase (PI3K) signaling. For instance, hypoxia inducible factor 1 (HIF1α) mediates the increased expression of nutrient receptors including glucose transporter 1 (Glut1), the main point of entry for glucose into T cells (Wang et al., 2011).
Meanwhile, the transition to the memory T cell fate is associated with mitochondrial biogenesis driven by AMPK (Borges da Silva et al., 2018; Buck et al., 2016; D’Souza et al., 2007; MacIver et al., 2011; Pearce et al., 2009; Rolf et al., 2013), which is mediated by peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) (Andrejeva and Rathmell, 2017; Calderon et al., 2018). This tightly regulated metabolic shift results in an LCFA-fueled oxidative program characterized by increased mitochondrial mass (Buck et al., 2016). This property of memory cells confers additional oxidative potential, known as spare respiratory capacity (SRC), to permit more rapid recall during secondary immune responses (van der Windt et al., 2012).
While many studies of bulk T cell populations suggest that a reciprocal, tightly regulated relationship exists between OXPHOS and glycolysis and the signaling cascades that regulate these pathways, their precise interactions in individual cells have yet to be elucidated. Moreover, the regulation of metabolic machinery in rare, early activated T cells remains poorly understood. The early stages of infection lead to antigen specific CD8+ T cells acquiring transient cell states preceding differentiation into effector subsets, but precisely how these intermediate stages of differentiation metabolically orchestrate rapid proliferation and differentiation has remained technically challenging (Joshi et al., 2007; Kalia et al., 2010; Obar and Lefrangois, 2010). Advances in single-cell analysis have enabled studies of signaling and effector programs in T cells at high resolution (Krishnaswamy et al., 2014; Mingueneau et al., 2014). Analogous studies of T cell metabolic regulation would likely provide new insights. For instance, a study utilizing stable isotope tracing in activated T cells found that OXPHOS may be a prominent in effector T cells in vivo (Ma et al., 2019). However, in the absence of single-cell resolution, it remains unclear whether the same cells are responsible for both OXPHOS and glycolysis, or alternatively, whether individual cells already differentiate and preferentially utilize one pathway versus the other during the effector phase. Many of the regulatory mechanisms that govern cellular metabolism are post-transcriptional and therefore not directly measurable by RNA-sequencing (Andrejeva and Rathmell, 2017). Moreover, as gene expression is not always tightly associated with protein expression (Vogel and Marcotte, 2012), proteomic approaches afford unique opportunities to assess the integrated functional programs within individual cells.
Mass cytometry utilizes metal-tagged antibodies to directly measure over 40 proteins simultaneously in individual cells (Bandura et al., 2009; Bendall et al., 2011). This approach has permitted characterization of various aspects of cellular behavior including phenotype, signaling (Bodenmiller et al., 2012), proliferation (Good et al., 2019) and chromatin state (Cheung et al., 2018). Here, we adapted this platform to measure expression levels of enzymes and transporters involved in metabolic checkpoints. We integrated direct quantitative evaluation of the signaling cues thought to mediate their regulation along with proteins indicative of CD8+ T cell fate and function. This approach was used to interrogate key inflection points of the CD8+ T cell response to Listeria monocytogenes infection (Lm-OVA), a well-characterized model of CD8+ T cell differentiation (Ahmed and Gray, 1996; Badovinac et al., 2007; Harty and Badovinac, 2002; Kaech and Ahmed, 2001; McGregor et al., 1970; Shen et al., 1998; Wherry and Ahmed, 2004), revealing a distinct metabolic state in early activated T cells. The single-cell mass cytometry approach presented here constitutes a resource for investigating cellular metabolic adaptations in vivo across a broad array of applications.
Results
Mass cytometry permits high-dimensional quantification of metabolic regulators in single CD8+ T cells
T cell differentiation requires the coordinated interplay of signaling and metabolic pathways, including the increased expression of rate-limiting enzymes and regulatory switches. The transition to aerobic glycolysis in SLECs is mediated by co-stimulatory signaling through CD28 via the AKT/PI3K pathway (Pollizzi et al., 2015; Wang et al., 2011); therefore, we measured the downstream intermediates mTOR, pS6, p4EBP1, and HIF1α (Fig. 1A, Table S1). Signaling through this pathway promotes glucose uptake through the Glut1 receptor and the transcription of glycolytic enzymes (Dennis et al., 2012), including glyceraldehyde-3-phosphate dehydrogenase (GADPH) (Fig. 1B), a critical metabolic switch implicated in glycolytic activity, which we also quantified.
Figure 1. Querying the integrated functional program of CD8+ T cell activation.
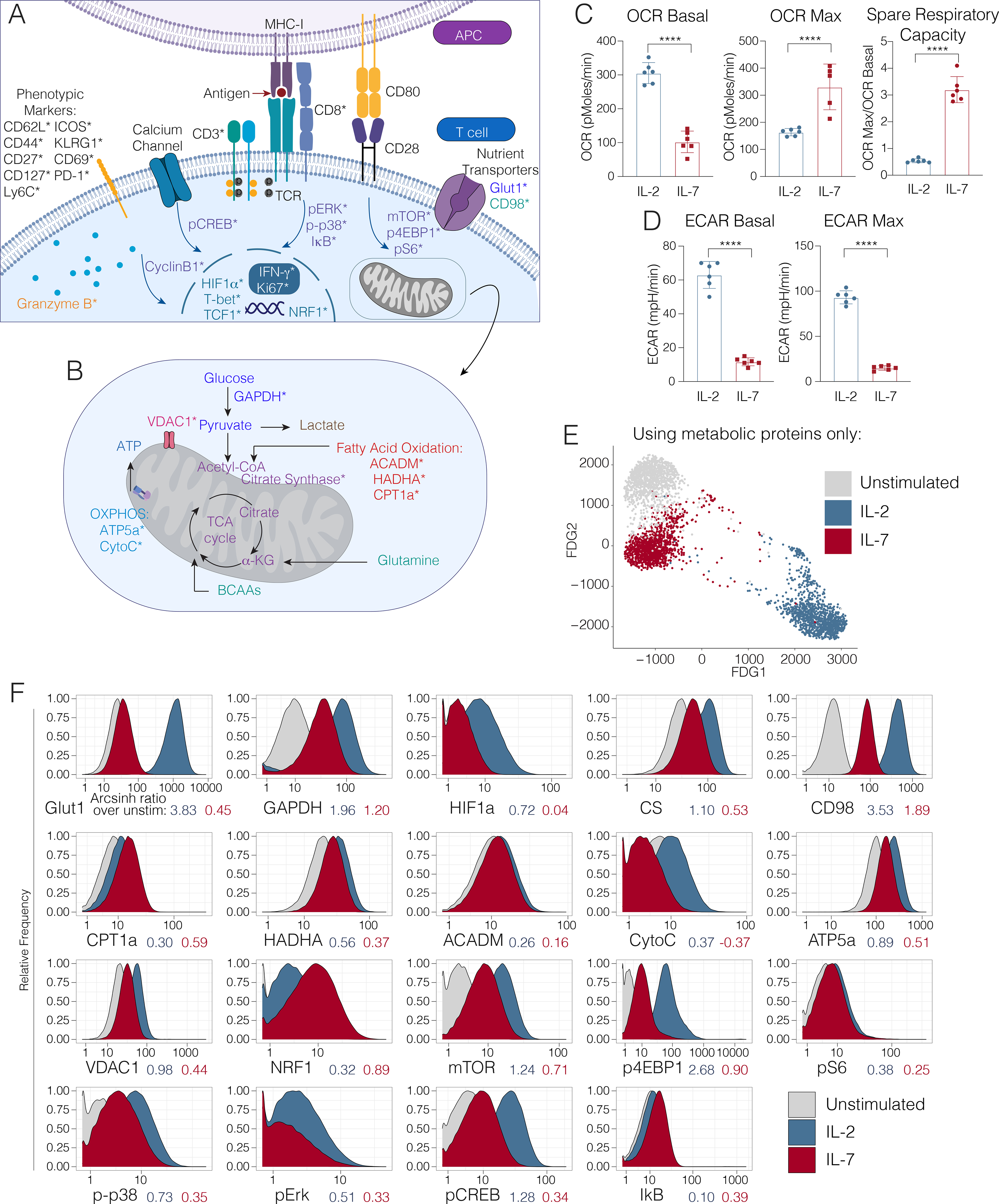
Panel schematic depicting signaling, metabolic, effector and phenotypic targets interrogated by mass cytometry. Cell surface, cytosolic and nuclear markers are depicted in (A) and mitochondrial markers are denoted in (B). Markers directly measured by mass cytometry are demarcated by an asterisk (*). (C) Extracellular acidification rate and (D) oxygen consumption rate of OT-1 transgenic CD8+ T cells, stimulated and polarized with IL-2 or IL-7, and quantified by Seahorse Assay. Significance analysis by student’s t-test (p<.001 ****). Error bars represent SEM. Data are representative of 3 independent experiments. (E) Single OT-1 T cells left unstimulated (gray) or stimulated and polarized with IL-2 (blue) or IL-7 (red) visualized using a force-directed graph, taking into account only the expression of metabolic proteins (Glutl, GAPDH, HIF1α, CS, CD98, CPT1a, HADHA, ACADM, CytoC, ATP5a, VDAC1, and NRF1). (F) Mass cytometry expression profiles of key metabolic enzymes in OT-1 T cells left unstimulated (gray) or stimulated and polarized with IL-2 (blue) or IL-7 (red). Arcsinh ratios between the median expression value of each stimulated condition compared to unstimulated cells are shown below the x-axis for each protein.
To investigate how the TCA cycle is regulated in activated T cells, we evaluated the expression of citrate synthase (CS) (Fig. 1B, Table S1), the first step of the cycle, which is directly regulated by NAD+ to NADH ratio, ADP/ATP ratio and succinyl-coA levels (Wiegand and Remington, 1986). As branched chain amino acid metabolism has been demonstrated to be critical for effective T cell activation (Ren et al., 2017a), we sought to understand this process by measuring the large neutral amino acid transporter (LAT1) chaperone CD98 (Fig. 1A, Table S1), a key mediator of the import of these essential nutrients (Hayashi et al., 2013; Nii et al., 2001; Sinclair et al., 2013).
Previous work has described a reciprocal relationship between aerobic glycolysis and OXPHOS, the latter of which is associated with memory T cell differentiation. Therefore, we sought to understand this regulation at the single-cell level by measuring carnitine palmitoyltransferase 1A (CPT1a), an enzyme that catalyzes the transport of LCFA from the cytoplasm to the mitochondria and that is critical for memory T cell function (van der Windt et al., 2012). Additionally, we measured the mitochondrial trifunctional complex, also known as hydroxyacyl-CoA dehydrogenase (HADHA), which catalyzes the final three steps of LCFA oxidation to acetyl-CoA in the mitochondria (Carpenter et al., 1992). As the role of β-oxidation of medium-chain fatty acids in T cell function has not been extensively evaluated (Howie et al., 2018), we also measured the expression of medium-chain acyl-CoA dehydrogenase (ACADM), an essential enzyme that catalyzes the initial step of this process (Fig. 1B, Table S1). Moreover, we measured key components of the electron transport chain, including cytochrome C (CytoC) and ATP synthase (ATP5a) (Fig. 1B, Table S1). To understand the counterregulatory processes governing OXPHOS activity and overall energy, we measured voltage-dependent ion channel 1 (VDAC1), a critical regulator controlling cytoplasmic-mitochondrial cross-talk (Fig. 1B, Table S1)(Cunningham et al., 2018; Tarze et al., 2007).
The cell signaling pathways that mediate mitochondrial fusion and biogenesis include MAP kinase and NFκB, which are activated during T cell priming (Calderon et al., 2018; Enamorado et al., 2018; Laforge et al., 2016); therefore, we measured the levels of phosphorylated (p) ERK and p-p38 MAP kinases in addition to the total levels of NFκB inhibitor alpha (IκBα). Calcium signaling, triggered by TCR ligation, has also been implicated in this process (Feske, 2007; Fracchia et al., 2013). Therefore, we additionally measured pCREB levels (Fig. 1A, Table S1).
It has been proposed that the activity of metabolic pathways induces epigenetic regulators such as Ezh2, which directly impact T cell fate and function (Chisolm et al., 2017; Gray et al., 2017). Therefore, we included a full range of well-characterized surface markers and transcription factors to subset T cells into naïve, central memory, effector memory and terminal effector populations. To measure the impact of all of these factors on T cell proliferation during clonal expansion, we measured expression of cyclinB1 and Ki67. Finally, to assess production of cytotoxic mediators, we also measured granzyme B (Fig. 1A, Table S1).
Mass cytometry recapitulates metabolic phenotypes of CD8+ T cell differentiation in vitro
In order to query the metabolic program underlying antigen-specific CD8+ T cell activation in vitro, we first stimulated TCR transgenic OT-1 splenocytes in the presence of their cognate antigen (the SIINFEKL peptide from ovalbumin) and IL-2 for 72 hours. After this initial priming period, antigen was removed, and cells were polarized in IL-2 or IL-7 for an additional 4 days to generate effector (OT-1eff) or central memory cells (OT-1mem), as described previously (Carrio et al., 2014; Pearce et al., 2009; van der Windt et al., 2012). We analyzed the resulting cells by mass cytometry and real-time metabolic profiling by Seahorse assay (Fig. 1C, S1B–D). In keeping with prior studies (Pearce et al., 2009; van der Windt et al., 2012), OT-1eff exhibited higher rates of extracellular acidification associated with glycolytic activity (Fig. 1C), while OT-1mem possessed marked spare respiratory capacity, though their basal oxygen consumption rate was lower (Fig. 1D) (Raud et al., 2018). Also consistent with previous reports (Raud et al., 2018), OT-1mem exhibited an increase in basal OCR in the presence of palmitate as measured by extracellular flux analysis (Fig. S1F–G), consistent with their ability to utilize fatty acid oxidation. While OT-1mem once again exhibited spare respiratory capacity, the increase in OCR after the addition of FCCP was largely independent of the presence of palmitate (Fig. S1F–G). These data indicated that while IL-7 polarized OT-1 T cells could indeed oxidize exogeneous palmitate, this substrate did not greatly alter the spare respiratory capacity of these cells (Raud et al., 2018).
We next characterized the metabolic programs of polarized OT-1 populations by mass cytometry. We first visualized these single-cell data via dimensionality reduction, only taking into account the expression of the metabolic proteins without the use of any phenotypic markers. This analysis showed that, purely on the basis of metabolic protein expression, cells polarized in different ways that were clearly unique (Fig. 1E). Moreover, to identify the relative contribution of each metabolic marker to clustering results, we repeated this analysis, each time leaving out one metabolic protein at a time (Fig. S1B). The integrity of the data structure was largely maintained with each omission, demonstrating that no single protein drives the entirety of the differences (Fig S1B).
Consistent with the results of our extracellular flux analyses, OT-1eff expressed elevated levels of glycolytic proteins at day 7 of activation, as evidenced by robust increase in expression of Glut1 and GAPDH (Fig. 1F, Fig S1A), suggestive of active glucose uptake and utilization. The expression of targets of the PI3K/mTORC1 pathway, including p4EBP1 and HIF1α, were likewise elevated in OT1eff (Fig. 1F), consistent with the promotion of aerobic glycolysis. Also in keeping with previous data (Ren et al., 2017a), the amino acid transporter CD98 was more highly expressed in OT1eff relative to OT-1mem (Fig. 1F). In contrast to their effector counterparts, OT-1mem did not demonstrate this glycolytic profile, but instead increased expression of CPT1a (Fig. 1F, S1A), the expression of which was therefore associated with increased SRC but not higher basal oxygen consumption under these conditions (Fig. 1D,F). Moreover, in keeping with previous reports (Raud et al., 2018), CPT1a expression was associated with the ability of OT1mem to metabolize exogenous palmitate as measured by extracellular flux analysis (Fig S1F–G), while the latter did not account for the observed spare respiratory capacity, underscoring the importance of both cellular adaptations and substrate availability for cellular metabolism.
Dynamic metabolic changes in canonical subsets of activated CD8+ T cells in vivo
To understand the metabolic changes during CD8+ T cell differentiation in a more physiologic context, we next evaluated the trajectory of the response to acute infection in vivo. C57BL/6 mice were infected with Listeria monocytogenes expressing whole cytoplasmic ovalbumin (Lm-OVA), a well-characterized model of CD8+ T cell metabolism (Buck et al., 2016; Pearce et al., 2009; van der Windt et al., 2012). Splenocytes were harvested daily over the first nine days post-infection for analysis by mass cytometry. We began by performing unsupervised clustering of the CD8+ T cells and visualizing them using dimensionality reduction (Fig. 2A, S2A). We assigned clusters to canonical T cell differentiation states based on their expression of classical markers of CD8+ T cell differentiation and investigated changes in metabolic enzyme and transporter expression over the course of the immune response. This analysis revealed considerable heterogeneity and dynamic functional changes across all major canonical T cell subsets over the course of the primary immune response to Listeria monocytogenes (Fig. 2A–B).
Figure 2. Single-cell analysis of the CD8+ T cell effector program in vivo.
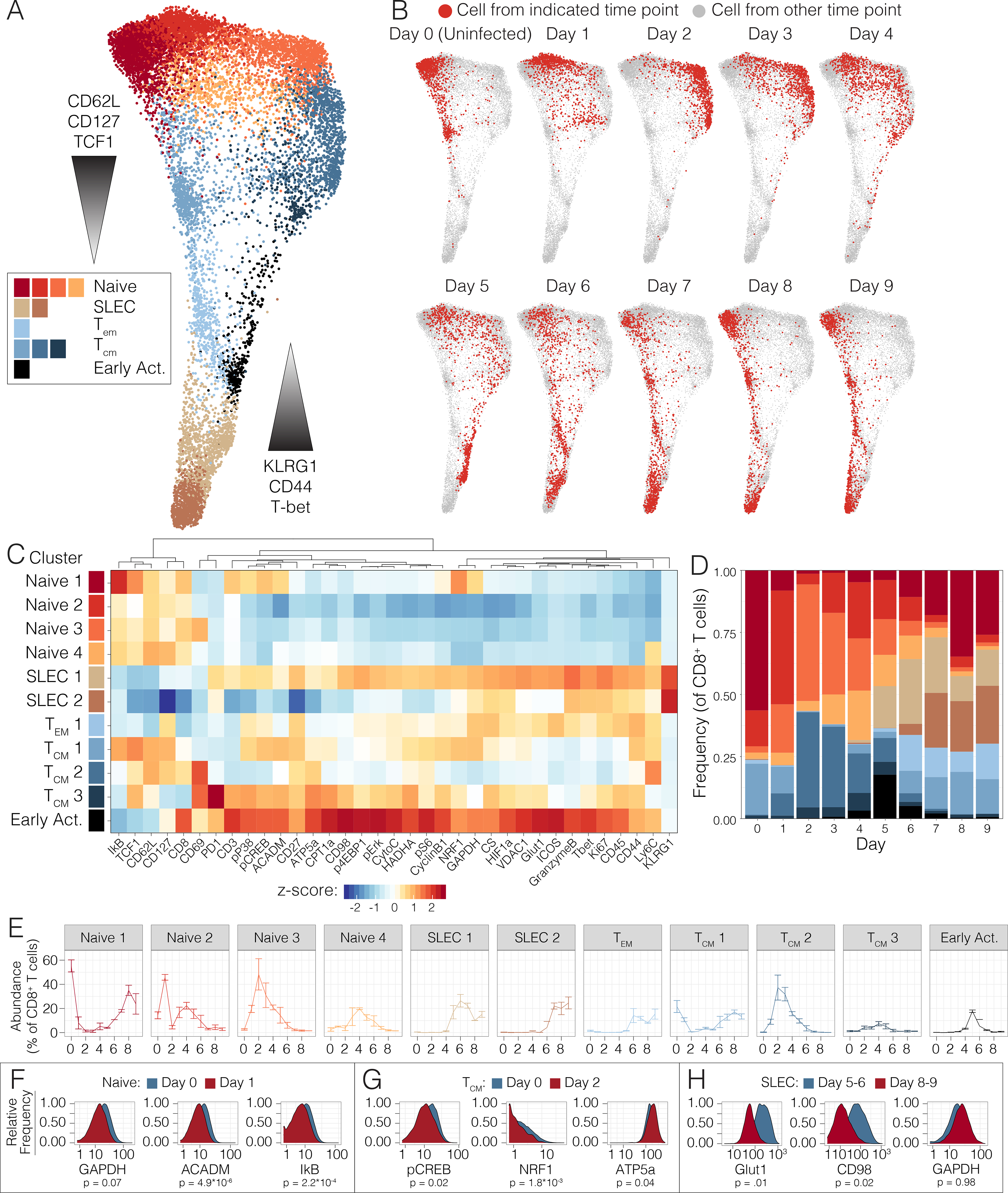
(A) Pooled CD8+ T cells from mice at days 0 to 9 of Lm-OVA infection (n=2–3 mice per time point) clustered by Phenograph and visualized by a force-directed graph. (B) Force-directed graphs indicating cellular distribution by time point. (C) Functional and phenotypic median expression profiles for each CD8+ T cell cluster. (D) Cluster proportion by time point. (E) Individual cluster frequency profiles at days 0 to 9 p.i. Error bars represent SEM. (F) Expression profiles of metabolic and signaling in naïve clusters between days 0 and 1 p.i. (G) Expression profiles of central memory cells between days 0 and 2 p.i. (H) Metabolic expression profiles of SLEC clusters between days 5–6 and 8–9 p.i. Significance analysis of all medians by two-tailed student’s t-test (p<0.05 *, p<0.01 **) is displayed.
At baseline, most naïve cells were predominantly contained within cluster Naïve 1, characterized by the expression of ACADM, pCREB, p-p38, NRF1 and weak expression of GAPDH (Fig. 2C). However, three clusters, Naïve 2 and Naïve 3, emerged at days 1 and 2 post infection (p.i) (Fig. 2D–E), all characterized by the decreased expression of all of the above metabolic and signaling markers (Fig. 2C, S2A). Interestingly, these clusters demonstrated low IκB expression, suggestive of signaling through NFκB pathway (Fig. 2C, 2F, S2A–B). While most naïve T cells were contained within the Naïve 2 cluster at day 1 post-infection (p.i.) (Fig. 2D–E), this gave way to a predominance of the Naïve 3 cluster and days 2 and 3 p.i. (Fig. 2D–E). By day 4 p.i., all these clusters as well as an additional cluster, Naïve 4, were present in similar proportions (Fig. 2D–E). Notably, the Naïve 1 cluster began to re-emerge at day 6 p.i., and ultimately dominated the naïve pools from day 7 p.i. onwards (Fig. 2D–E). This predominance was associated with the involution of clusters Naïve 2, Naïve 3, and Naïve 4, which became nearly undetectable by day 7 p.i (Fig. 2E). These findings are consistent with activation of both bystander and antigen specific T cells in the early stages of acute infection (Chu et al., 2013; Jiang et al., 2003) but reveal the metabolic adaptations that these cells undertake. Overall, these data support previous reports of a metabolically quiescent profile of naïve T cells while identifying transitions even within these cells.
Evaluation of the central memory cells over the course of infection revealed a similar pattern, starting with cluster TCM1, characterized by intermediate expression of expected markers of LCFA and OXPHOS including p-p38, pCREB, ACADM, HADHA, NRF1, and dim expression of ATP5a, CPT1a, pErk and CytoC (Fig. 2C). Interestingly, this cluster also expressed GAPDH and pS6 but dimly expressed HIF1α compared to effector subsets (Fig 2C, S2A). During days 1–2 p.i., cluster TCM2 emerged (Fig. 2D–E), which decreased expression all of these metabolic and signaling factors, with only weak expression of HADHA and pS6 and increased expression of ATP5a (Fig. 2C, 2G, S2C). This cluster predominated at days 2 through 4 p.i. Notably, day 2 p.i. was also marked the emergence of cluster TCM3 (Fig. 2D–E), defined by expression of enzymes of fatty acid oxidation (FAO), including CPT1a, HADHA, ACADM, along with oxidative proteins, such as CS, ATP5a, VDAC1, CytoC (Fig. 2C, S2A). These cells also expressed less pS6 and GAPDH (Fig. 2C). Commensurate with this oxidative profile, cells in this cluster also expressed p-p38, pErk, and pCREB (Fig. 2C, S2A). While cells in TCM3 also demonstrated expression of downstream intermediates of the PI3K cascade, such as p4EBP1 and pS6, along with transcription factors associated with aerobic glycolysis, such as HIF1α, these were associated with lower GADPH expression (Fig. 2C, S2A). This metabolically active TCM cluster was transient, completely regressing by day 7 p.i. (Fig. 2D–E). Notably TCM1 reemerged at day 5 p.i. and remained the predominant TCM cluster from days 6 through 9 p.i (Fig. 2D). These data confirmed the previously oxidative profile of central memory cells but also revealed dynamic metabolic changes within these subsets over the course of an immune response.
Effector memory cells (TEM) uniformly constituted cluster TEM1, which emerged at day 5 p.i. and maintained stable frequency through day 9 p.i (Fig. 2D–E). These cells demonstrated a more glycolytic metabolic profile, with increased expression of GAPDH, Glut1 and HIF1a, and dim oxidative and FAO marker expression (Fig. 2C, S2A). Meanwhile, SLECs comprised clusters SLEC1 and SLEC2 and emerged at days 5 and 6 p.i., respectively (Fig. 2D–E). These two clusters demonstrated distinctive metabolic phenotypes. The first population to appear, SLEC1, demonstrated expression of p4EBP1, pS6, HIF1α, Glut1, and GAPDH suggestive of a glycolytic profile (Fig. 2C, 2H, S2A, S2D). Previous studies have demonstrated that early effector cells continue TCA cycle engagement fueled by the uptake of amino acids and LCFA (O’Sullivan et al., 2014; Ren et al., 2017b); consistently, cells in this cluster also expressed HADHA, CD98, CS, VDAC1 (Fig. 2C, 2H, S2A, S2D). However, ATP5a and CPT1a levels in this cluster were lower than those observed in the more active TCM clusters, such as TCM3, distinguishing them from these more classically oxidative pools (Fig. 2C, S2A). In comparison, cluster SLEC2 demonstrated a more muted metabolic profile, decreasing expression of all metabolic mediators except HIF1α, GAPDH and CS, taking on the terminal glycolytic state observed in previous studies (Fig. 2C, 2H, S2A, S2D). As expected, the more metabolically active cells in cluster SLEC1 expressed higher levels of Ki67 and granzyme B compared to cluster SLEC2 (Fig. 2C, S2A). Taken together, these findings agreed with previous reports of a predominantly glycolytic terminal effector state.
Early activated T cells exhibit maximal expression of glycolytic and oxidative proteins
In addition to these well-characterized cell subsets, unsupervised high-dimensional analysis also revealed a group of early activated T cells that emerged at day 4 post-infection (Fig. 2D–E). These cells had high expression of Ki67, indicative of proliferation, and expressed high levels of CD44, CD27 and ICOS but low levels of CD62L (Fig. 2C, S2A). This early activated cluster was most abundant at day 5, when it comprised nearly 20% of the CD8+ T cell population, and it nearly completely disappeared by day 7 (Fig. 2D–E). As ICOS has been found to signal through the PI3K cascade (Zeng et al., 2016), we anticipated that this population would be glycolytic. Indeed, these early activated cells expressed the highest levels of Glut1 and GAPDH across all CD8+ T cells (Fig. 2C). However, these cells simultaneously exhibited peak expression of oxidative markers, including CPT1a, HADHA, ACADM, and ATP5a (Fig. 2C, S2A). Commensurate with this observation, the signaling program of this population was marked by maximal expression of both pS6 and pCREB as well as minimal expression of IκB, reflecting simultaneous activity of both the PI3K/mTORC1 and NFκB pathways (Fig. 2C, S2A). In contrast to SLEC and memory cells, these early activated cells also expressed maximal expression of the amino acid transporter CD98 (Fig. 2C, S2A).
Given the unique metabolic expression profile of these early activated cells, we sought to confirm these observations through direct inspection of the primary data. We undertook further phenotypic analysis of these cells, which identified the high-affinity IL-2 receptor subunit, CD25, as another surface marker co-expressed by this population of interest, consistent with their recent activation (Fig. 3A). Consistent with the clustering analysis, we found that these manually gated cells peaked in frequency at day 5 followed by a rapid decline in abundance (Fig. 3B). Moreover, in comparison to all other CD8+ T cells present in the animals at day 5, these cells clearly expressed elevated levels of both glycolytic and oxidative proteins (Fig. 3C, S3A). Therefore, early activated T cells exhibited peak expression of metabolic mediators of oxidative and glycolytic pathways.
Figure 3. Early activated T cells exhibit a distinctive metabolic profile characterized by peak oxidative and glycolytic activity.
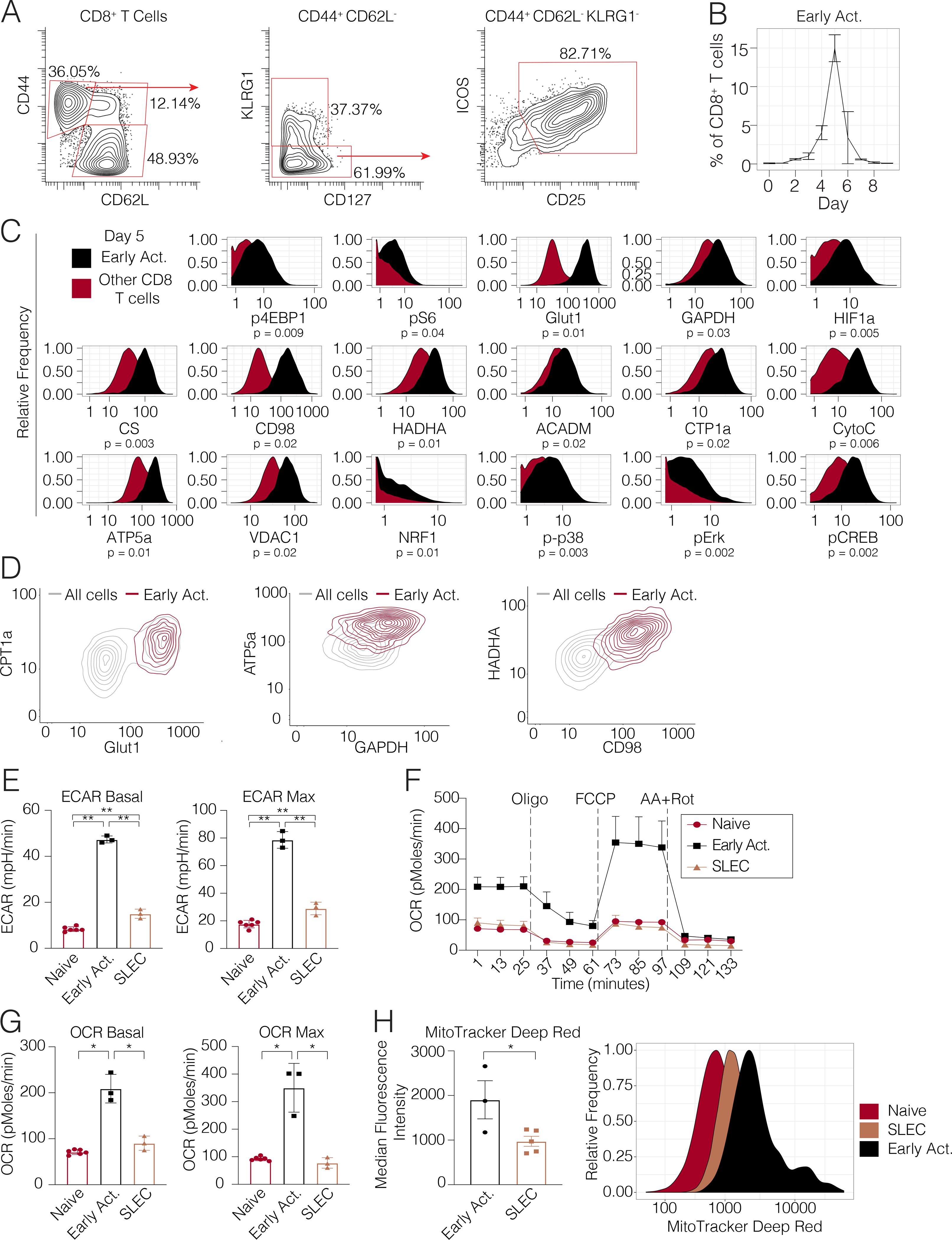
(A) Biaxial scatter plots indicating surface marker expression profile of the early activated T cell pool. (B) Frequency of early activated cells during days 0 to 9 p.i. (C) Metabolic expression profiles of metabolic and signaling markers in early activated T cells in comparison to all other CD8+ T cells during days 0 to 9 p.i. as depicted by histograms. Significance analysis of all medians by paired two-tailed student’s t-test (p<0.05 *, p<0.01 **) is displayed. (D) Scatterplots denoting the expression of proteins involved in glycolysis, long-chain fatty acid oxidation, oxidative phosphorylation and branched-chain amino acid metabolism in early activated CD8+ T cells on day 5 p.i. (E-G) CD8+ T cell subsets of interest were sorted on days 5 (naïve cells, early activated cells) and 8 (SLECs) p.i. and analyzed by Mitochondrial Stress Test (n=5 mice per time point). (E) Basal ECAR and maximal ECAR measured upon oligomycin administration. (F) OCR over time. (G) Basal and maximal OCR readings obtained upon FCCP administration. (H) MitoTracker signal in each subset (n=5 mice per subset). Significance analysis by paired two-tailed student’s t-test (p<0.05 *, p<0.01 **). Error bars represent SEM. Data are representative of at least 2 independent experiments.
A key advantage of any single-cell approach is the ability to resolve novel insights of cellular behavior and function by revealing heterogeneity within canonical populations. Therefore, to confirm the co-expression of metabolic proteins across key metabolic pathways within individual early activated CD8+ cells, we visualized glycolytic and oxidative markers by scatter plots. This visualization approach clearly demonstrated that early activated CD8+ T cells co-expressed CPT1a and Glut1 compared to all other CD8+ T cells at Day 5 p.i. Similar co-expression profiles between GAPDH and ATP5a were also noted, and confirmed that glycolytic and oxidative proteins are co-expressed at the single-cell level in early activated T cells. Similar co-expression profiles were noted between HADHA and CD98, demonstrating co-expression of proteins involved in branched-chain amino acid import and long-chain fatty acid oxidation in during this phase of T cell activation (Fig. 3D). To further evaluate co-expression of metabolic proteins of interest with single-cell resolution, we took another complementary approach by computing pairwise correlations for each metabolic protein across single early activated CD8+ T cells. Consistent with the prior results, all studied metabolic proteins were positively correlated, without evidence of negative correlations between oxidative and glycolytic markers (Fig. S3B). Taken together, these results indicated that oxidative and glycolytic proteins were indeed co-expressed in individual early activated CD8+ T cells. They also demonstrated the ability to resolve metabolic marker expression at the single-cell level by mass cytometry.
Early activated T cells demonstrate peak glycolytic activity and increased mitochondrial activity and mass
Since these early activated T cells were distinguished by simultaneously elevated levels of glycolytic and oxidative enzymes, we posited that this expression profile would translate to greater metabolic activity along these pathways when compared to their SLEC counterparts. To assess real-time bioenergetic flux through oxidative and glycolytic pathways, we sorted naïve, early activated, and SLEC T cells for analysis by Seahorse assay (Fig S3B). As expected, SLECs demonstrated significantly higher baseline and maximum ECAR compared to their naïve counterparts (Fig. 3D), confirming a predominantly glycolytic program driving the terminal effector state in vivo. However, in accordance with their enzymatic expression profile by mass cytometry, early activated T cells exhibited significantly higher basal and maximal ECAR even compared to SLECs (Fig. 3D).
Moreover, baseline and maximal OCR did not significantly differ between the SLEC and naive pools, as described previously (van der Windt et al., 2012) (Fig. 3E–F). However, early activated T cells did indeed exhibit significantly higher oxidative activity compared to both the SLEC and naïve cells (Fig. 3E–F). Since these cells exhibited maximal expression of electron transport complexes, we hypothesized that that this population would possess SRC. Indeed, while neither the naïve or SLEC pools were capable of surpassing their baseline OCR upon FCCP administration, the OCR of early activated T cells nearly doubled (Fig. 3E–F).
We next determined whether these cells could utilize exogenous fatty acids as an energy source. Consistent with a prior study (Raud et al., 2018), naïve T cells exhibited higher OCR in the presence of palmitate. In comparison, early activated cells also exhibited an increase in OCR in the presence of palmitate to levels that also exceeded those of naïve cells, confirming their ability to oxidize fatty acids. As we observed from cells activated in vitro, SRC was largely independent of the presence of exogenous palmitate (Fig. S3C–D). As SRC has been associated with greater mitochondrial mass (Buck et al., 2016), we sought to quantify the mitochondrial content of these cells using MitoTracker Deep Red, a fluorescent dye staining mitochondria in live cells. Consistent with our mass cytometry and Seahorse data, the early activated T cells contained significantly more mitochondrial mass than the SLEC or naïve pools (Fig. 3G). Overall, these observations confirmed the unique, simultaneously oxidative and glycolytic profile of early activated T cells.
Antigen-specific CD8+ T cells transit through the early activation state commensurate with the onset of proliferation
In order to query the antigen-specificity of these metabolic adaptations of early T cell activation, we adoptively transferred OT-1 T cells pooled from 3 naïve donors into congenic hosts, which were then infected with Lm-OVA. Splenocytes from recipient mice were analyzed daily from days 3 through 7 p.i for metabolic analysis by mass cytometry (Fig. 4A–B, S4A). A reserved aliquot of the initial OT-1 T cells that were transferred was also analyzed in parallel as a baseline sample. Indeed, unsupervised clustering analysis of adoptively transferred OT-1 T cells revealed early activated cells with an analogous state of metabolic activity, arising in small numbers at day 3 p.i. and peaking at day 4 before rapidly regressing by day 5 (Fig. 4A–B, S4A). The kinetics of the emergence of this cluster were slightly earlier compared to the previously characterized endogenous cells, perhaps a result of higher TCR affinity or increased frequency of antigen-specific precursor cells. Consistent with our findings in endogenous CD8+ T cells (Fig. 2C, 3C), cells comprising this cluster exhibited simultaneous peak expression of markers of glycolysis, OXPHOS, and LCFA oxidation (Fig. 4C, S4A). We hypothesized that these metabolic adaptations were undertaken in support of clonal expansion of antigen-specific populations. Therefore, we assessed the proliferation of CFSE-labeled adoptively transferred OT-1 T cells on days 3 through 7 p.i. by flow cytometry. Commensurate with the emergence of this early activated metabolic state, the first antigen-specific T cells to divide did so at day 4 p.i. (Fig. 4D). By day 5 p.i., all adoptively transferred cells had divided multiple times (Fig. 4D). Consistent with this finding, the total number of OT1 T cells only modestly increased between days 3 and 4 p.i., but they subsequently rapidly expanded between days 4 and 5 p.i before plateauing thereafter (Fig. 4E). These observations collectively suggested that early activated antigen-specific CD8+ T cells underwent a transition to a metabolic state characterized by peak OXPHOS and glycolytic activity at the same time as they began blasting, supporting the dramatic expansion of these cells during productive immune responses.
Figure 4. Antigen-specific CD8+ T cells transit through the early activated metabolic state.
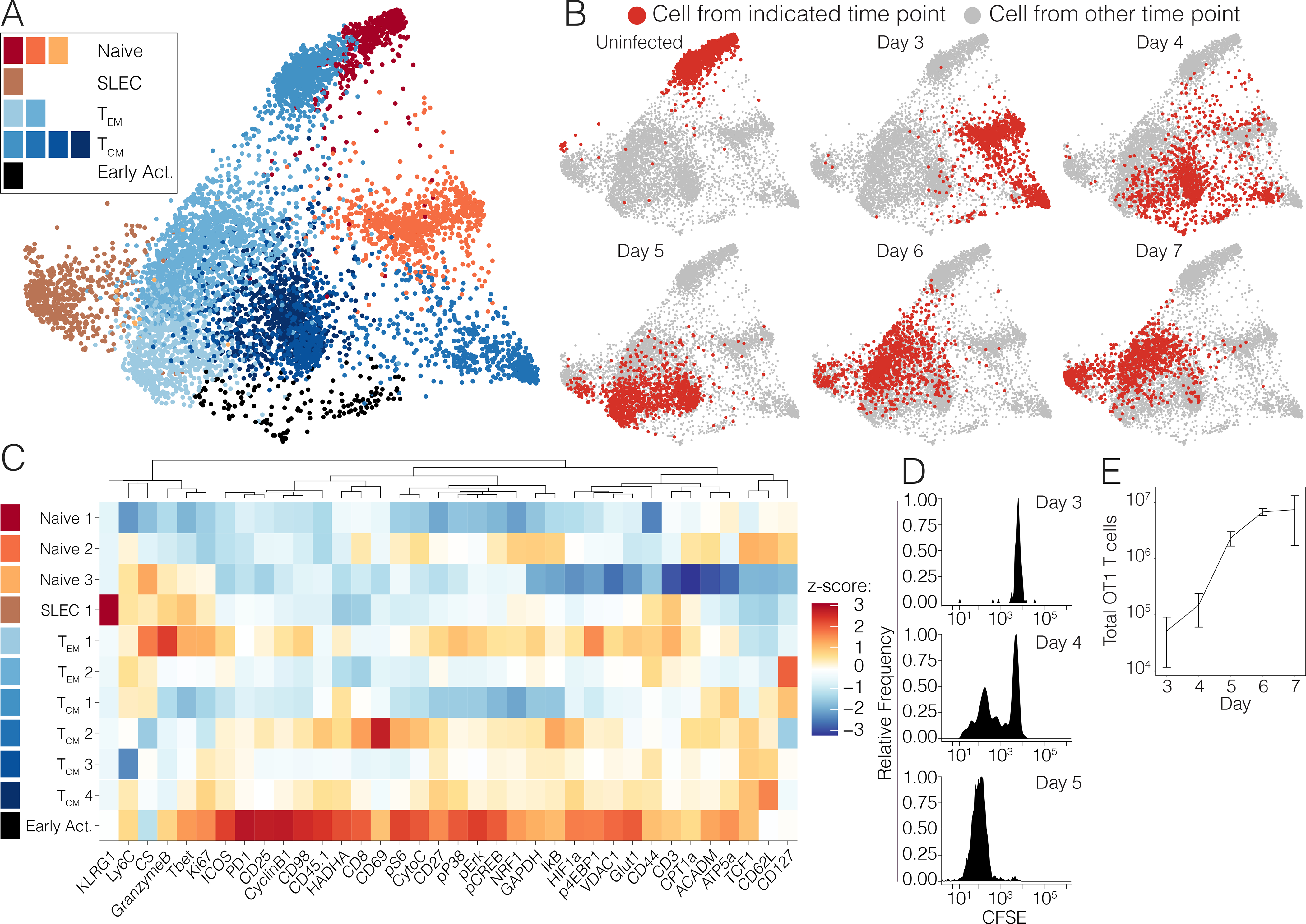
OT-1 T cells pooled from 3 naive mice were adoptively into congenic hosts, which were then infected with 5*104 CFU Lm-OVA. Splenocytes were harvested daily on days 3 through 7 p.i for metabolic analysis by mass cytometry. (A) Pooled OT1 cells from uninfected mice or after transfer into recipient mice and on days 3 to 7 after Lm-OVA infection (n=3 mice per time point except day 3 (n=1)) clustered by Phenograph and visualized by a force-directed graph. (B) Force-directed graphs indicating cellular distribution by time point. (C) Functional and phenotypic median expression profiles for each CD8+ T cell cluster. (D) Proliferation of adoptively transferred OT1s as measured by CFSE dilution at days 3–5 p.i. and (E) absolute cell counts at days 3–7 p.i.
The early activated state is transient and precedes the development of both effector and memory cells
As early activated CD8+ T cells with peak metabolic protein expression were highly transient, only detectable for a few days during the immune response, we investigated the changes that take place in these cells thereafter. We sorted early activated T cells and transferred them into congenic hosts before isolating splenic T cells four days later (Fig. 4A). At the end of this period of time, the transferred early activated cells had given rise to a mixture of cells with phenotypes consistent with SLECs (KLRG1+CD127−), double-positive cells (KLRG1+CD127+), and memory cells (KLRG1-CD127+). Further analysis of the CD127+KLRG1− memory fraction indicated that most of these cells lack expression of CD62L, consistent with an effector memory phenotype, while a smaller fraction expressed CD62L, consistent with a central memory phenotype (Fig. 4B). In addition, we investigated the expression of both CD27 and TCF1 in these subsets, both markers associated with memory cells (Fig. 4C). Consistent with our expectations, the memory (CD127+KLRG1−) and double-positive (CD127+KLRG1+) cells expressed higher levels of these proteins. We also observed that the memory cells did not express CD69. Given the transient, elevated expression of cell cycle markers by the early activated cells (Fig. 2C), we hypothesized that these cells would proliferate upon adoptive transfer. Indeed, these cells expanded but lost expression of Ki67 over the course of the four days (Fig. 5, S5A). These early activated T cells also decreased expression of CD25 and ICOS as well as Granzyme B by this later time point (Fig. 5D S5B). Consistent with a transient burst of metabolic activity, these cells exhibited markedly lower expression of both glycolytic and oxidative markers at this later time point compared to the early activated T cells from which they originated (Fig. 5D, S5B). Collectively, these data indicated that during CD8+ T cell differentiation, early activated T cells underwent a transient period of peak metabolic activity. Thereafter, decreased expression of glycolytic and oxidative pathways took place coordinate with differentiation into short-lived or memory cells.
Figure 5. The early activated state is transient and differentiates into effector and memory cells.
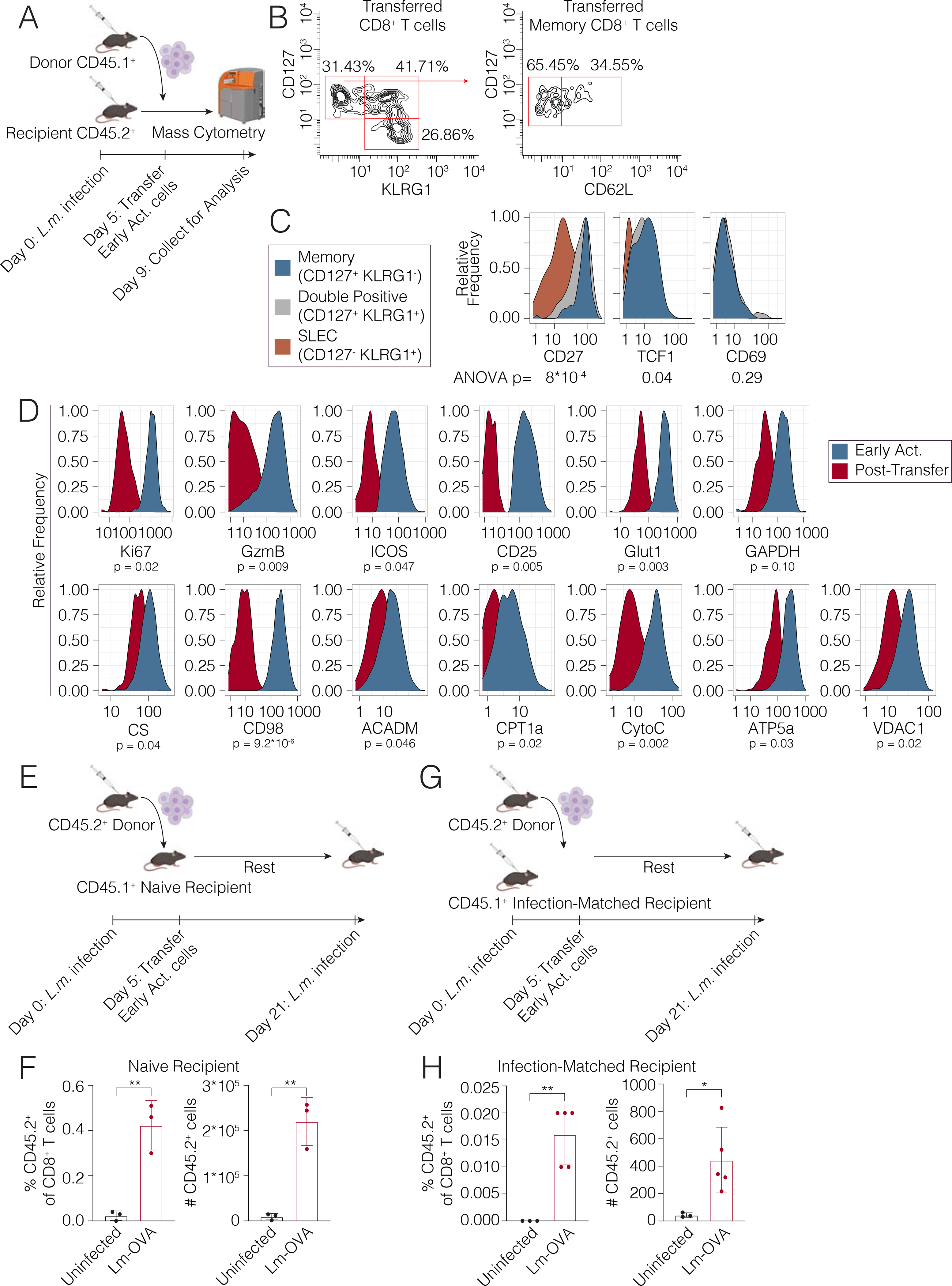
(A) Early activated T cells were sorted from CD45.1+ mice on day 5 p.i. and transferred into infected CD45.2+ hosts on day 5 p.i (n=2 per group) before sacrifice 4 days later for analysis by mass cytometry. (B) Differentiation state of early activated cells determined by CD44 and KLRG1 expression at day 9 p.i. (C) Histograms of memory markers expressed by adoptively transferred early activated T cells at day 9 p.i. (D) Metabolic and signaling marker profiles before and after transfer at days 5 and 9 p.i. are represented by histograms. (E) Early activated T cells isolated from CD45.2 mice at day 5 p.i. were adoptively transferred into naive CD45.1+ recipient mice. Recipient animals were either infected with 1.0 ×105 CFU Lm-OVA or left unchallenged (n = 3 mice per group). (F) Percentages and absolute cell counts of CD45.2+ CD8+ T cells in uninfected and challenged recipients. (G) Early activated T cells isolated from CD45.2 mice at day 5 p.i. were adoptively transferred into infection-matched CD45.1+ recipient mice. Recipient animals were either infected with 1.0 ×106 CFU Lm-OVA or left unchallenged (n = 3–5 mice per group). (H) Percentages and absolute cell counts of CD45.2+ CD8+ T cells in uninfected and rechallenged recipients. Significance analysis by unpaired student’s t-test (*p<0.05, **p<0.005).
To better understand the potential of the early activated T cell pool to generate functional memory responses, we transferred early activated T cells at day 5 p.i. from CD45.2+ mice into naïve CD45.1+ congenic recipient mice (Fig. 5E). These recipient mice were subsequently infected with Lm-OVA 21 days after the initial infection event. As expected, CD45.2+ cells were nearly undetectable in unchallenged recipients, consistent with a contracted state (Fig. 5F). However, upon challenge with Lm-OVA, CD45.2+ transferred cells expanded significantly as a percent of CD8+ T cells and in absolute numbers, consistent with the re-expansion of a memory population (Fig. 5F). As an alternative approach, we also adoptively transferred CD45.2+ early activated T cells at day 5 p.i. into infection-matched CD45.1+ recipient mice, which were subsequently re-challenged with Lm-OVA 21 days after the initial infection (Fig. 5G). CD45.2+ transferred cells expanded significantly upon rechallenge once again (Fig. 5H). Taken together, these data confirmed that early activated cells can differentiate into memory cells capable of mounting functional recall responses.
Single-cell metabolic dynamics of CAR T cells in response to malignancy
A study utilized mass cytometry to assess changes in metabolic protein expression in human immune cells stimulated in vitro (Hartmann et al., 2020). Therefore, we extended that approach to understand the metabolic dynamics of antigen-specific T cells over the course of a human immune response in vivo. We assessed the metabolic adaptations in chimeric antigen receptor (CAR) T cells over time. Axicabtagene ciloleucel CAR T cell products were sampled from two patients with advanced refractory DLBCL at the time of infusion, and PBMCs were also isolated from these same patients on days 7, 14, 21, 28 and 90 after CAR T cell infusion for functional analysis by mass cytometry (Fig. 6A; Table S1). Dimensionality reduction revealed that CD8+ CAR T cells at time of infusion were distinct as compared to cells from all other timepoints (Fig. 6B). At the time of infusion, adoptively transferred CAR T-cells exhibited analogous metabolic dynamics to the early activated T cells in mice, with peak oxidative and glycolytic marker expression (Fig. 6C). Most metabolic proteins were subsequently diminished by day 7 of treatment, with decreasing expression thereafter (Fig. 6C). Notably, Spearman correlation analysis demonstrated that there was a positive association between oxidative phosphorylation and glycolytic proteins at time of infusion, suggesting that these processes are not mutually exclusive (Fig. 6D). Moreover, consistent with our mouse models of infection, ICOS was also uniquely expressed by cells in this early activated state (Fig. 6E).
Figure 6. CAR T cells exhibit an analogous transient early activated program upon infusion into advanced non-Hodgkin Lymphoma patients.
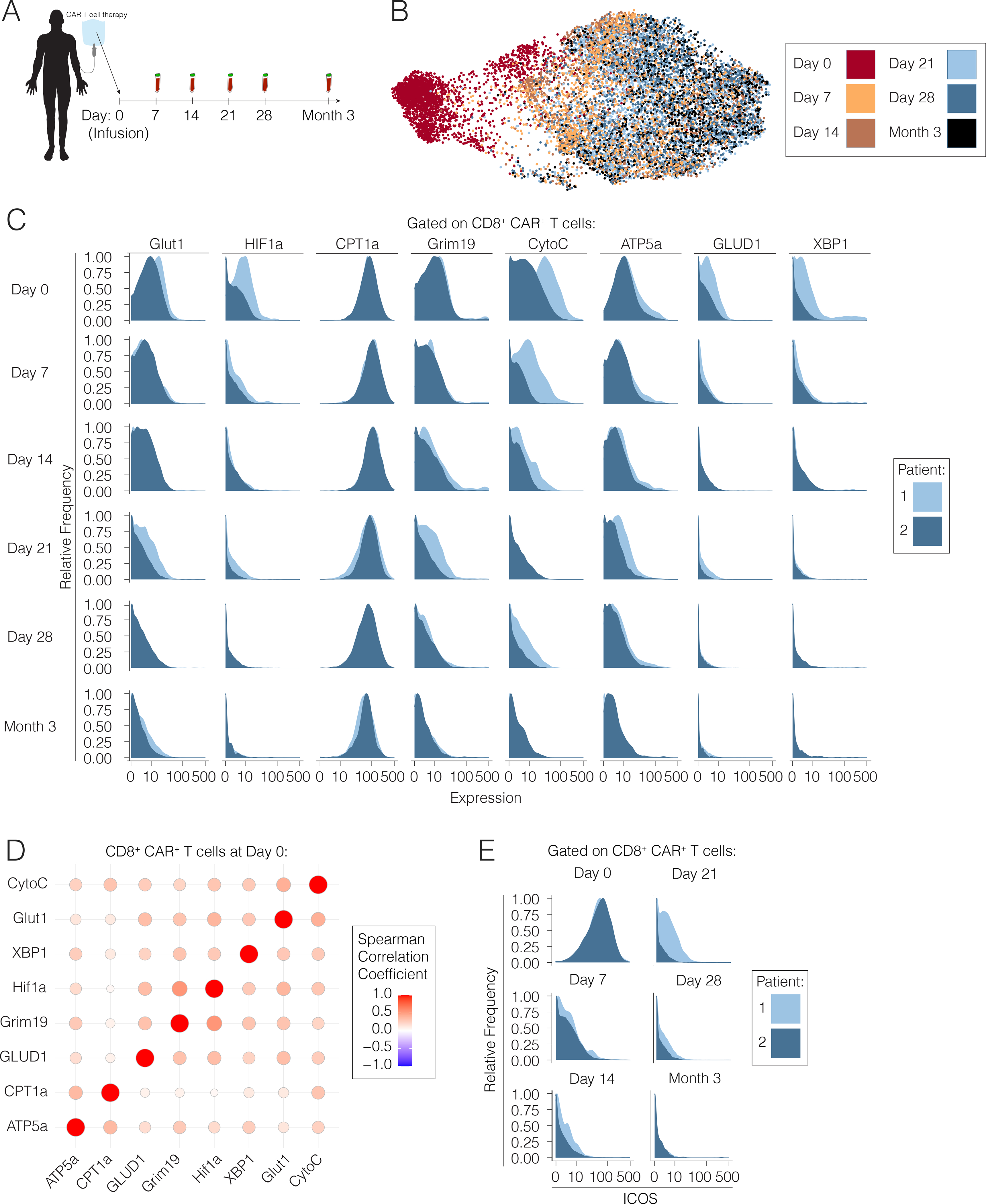
(A) Axicabtagene ciloleucel chimeric antigen receptor T cells from two advanced non-Hodgkin Lymphoma patients were sampled at the time of infusion, and PBMCs were isolated from the same patients on days 7, 14, 21, 28 and month 3 post-infusion for analysis by mass cytometry. (B) Pooled CD8+ CAR T cells from all time points post-infusion visualized by a force-directed graph. (C) Expression levels of key metabolic markers by CAR T cells at each time point post-infusion by patient. (D) Spearman correlation matrix of CAR T cells at time of infusion (D0) into advanced lymphoma patients. (E) ICOS expression in individual patients at each time point post-infusion represented by histograms.
Discussion
Mass cytometry permits broad-spectrum characterization of immune responses in healthy and diseased states (Spitzer and Nolan, 2016). To date, this approach has been used to query the phenotypic and signaling adaptations undertaken by cells during differentiation (Bendall et al., 2014; Zunder et al., 2015). However, the coordinated downstream metabolic cues supporting these programs in vivo have remained incompletely understood at the single-cell level. Here, we directly measured the expression levels of essential nutrient receptors, enzymes, signaling intermediates, and markers of cellular differentiation and effector function at the proteomic level. This allowed us to more thoroughly characterize CD8+ T cell responses during acute infection, highlighting the metabolic adaptations of canonical T cell subsets and capturing a unique metabolic state in rare, early activated T cells.
The field of immunometabolism is rapidly advancing, with novel techniques accelerating our capability to resolve metabolism at the single-cell level (Artyomov and Van den Bossche, 2020). Cytometry-based assays have been used to study the metabolic programs of human immune cells in vitro (Ahl et al., 2020; Hartmann et al., 2020). Here, we have adapted mass cytometry to study the metabolic trajectories of both murine and human antigen-specific immune responses in vivo. Moreover, we have leveraged these tools to probe context-dependent metabolic heterogeneity, identifying and validating transitional metabolic states arising during CD8+ T cell differentiation. We propose that this discovery-driven approach may be used by investigators to identify and understand metabolic states of various cell types in vivo across a wide range of contexts.
Previous models based on bulk data have proposed a reliance of effector T cells on aerobic glycolysis (Sukumar et al., 2013; Wang et al., 2011). However, a prior study of intracellular flux in activated T cells has reported that effector T cells may utilize oxidative phosphorylation in vivo (Ma et al., 2019). Additionally, it has been observed that effector T cells engage in more active LCFA uptake than their memory cell counterparts, which instead have been shown to mobilize these substrates from lysosomal triglycerides (Sullivan et al., 2014). It is therefore feasible that fatty acid uptake may provide additional substrate for OXPHOS early in the course of T cell activation. Our data unify these observations, supporting a coordinated program in which glycolysis and OXPHOS are maintained simultaneously in individual cells during an earlier stage of T cell activation.
Our approach to metabolic profiling by mass cytometry affords investigators the opportunity to functionally characterize the metabolic adaptations of rare cellular populations, such as antigen-specific T cells. These cells would be otherwise difficult to analyze by current standard metabolomics assays due to the prohibitively large number of cells, extensive processing, and ex vivo culture techniques required for these studies (Cantor et al., 2017; van der Windt et al., 2016). Here, we were able to characterize the metabolic, signaling and phenotypic progeny of adoptively transferred cells with single-cell resolution. This approach revealed a diversification during CD8+ T cell differentiation in the context of acute infection, with a highly metabolically active and proliferative state in T cells early in the course of their response, which later decrease expression of specific metabolic pathways as they differentiate into both memory and terminal effector cells.
The maximal expression of metabolic proteins early after T cell activation suggests a potential role for TCR ligation and/or co-stimulation during CD8+ T cell priming. Notably 4–1BB ligation during co-stimulation has been shown to induce mitochondrial fusion via TRAF2-mediated signaling through p38 and PGC1α (Calderon et al., 2018; Enamorado et al., 2018). Similarly, CD28 ligation has been demonstrated to induce CPT1a expression in vitro (Klein Geltink et al., 2017). Whether these signals potentiate the observed spare respiratory capacity and increased mitochondrial mass in early activated T cells will be important to determine. As Drp1-mediated mitochondrial fission has been described in effector cells during metabolic reprogramming to the aerobic glycolytic program (Buck et al., 2016), it is possible that the absence of co-stimulation and loss of IL-2 signaling upon pathogen clearance may result in mitophagy and/or mitochondrial fission, repressing oxidative activity in terminal effector subsets.
As the importance of metabolism to immune cell fate and function is increasingly appreciated, methods to evaluate these pathways in models of productive and dysregulated immune responses will be critical. The approach presented here may be adapted to any cell type of interest, including both immune cells and non-immune cells, such as interacting epithelial tissues or tumors. This methodology should enable investigators to study the functional programs underlying the development of the full spectrum of immune cell lineages and their altered states in the context of autoimmunity or malignancy. Additionally, integrated functional analysis of rare cellular subsets will permit simultaneous evaluation of the effects of various treatments on rare populations, such as tumor infiltrating lymphocytes or neoantigen-specific T cells.
Limitations of the study
While our approach enables single-cell characterization of cellular metabolism, limitations to the current study afford opportunities to further validate and build on this work. While we elected to pursue an integrated analysis of signaling, metabolism and effector function, a more refined survey of additional enzymes along specific pathways, with particular emphasis on anabolic processes would provide additional insights into the mechanisms governing T cell activation and differentiation. Moreover, direct measurement of epigenetic regulators of these metabolic processes by either mass cytometry or ATAC-seq would further facilitate understanding of the concerted processes underlying the transition to the early activated state reported in our study. As metabolic state is driven by substrate availability in addition to cellular adaptations, additional orthogonal methods such as metabolic flux analysis will continue to determine the relative contributions of pathways of interest.
STAR Methods Text
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Matthew Spitzer (matthew.spitzer@ucsf.edu).
Materials Availability
This study did not generate new unique materials. Information regarding antibody conjugates is presented in Table S1.
Data and Code Availability
Mass cytometry data will be made publicly available on Mendeley Data at https://data.mendeley.com/drafts/cnfkpk2t8m. No new code or algorithms were developed during this study; however, code will be provided upon request without limitations.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All mice were housed in an American Association for the Accreditation of Laboratory Animal Care-accredited animal facility and maintained in specific pathogen-free conditions. Animal experiments were approved and conducted in accordance with AN157618. Wild-type female C57BL/6 mice and BoyJ CD45.1 between 8–10 weeks old were purchased from The Jackson Laboratory and housed at our facility. TCR Transgenic OT-I CD45.1 mice and heterozygous CD45.2/CD45.1 mice were bred at our facility. Animals were housed under standard SPF conditions with typical light/dark cycles and standard chow.
Human Subjects
Samples were collected from two advanced Non-Hodgkin Lymphoma patients receiving Axicabtagene ciloleucel therapy as standard of care. Patient CAR T cell sample collection was approved by the Vanderbilt University Medical Center IRB (#171340).
Infectious Agents
Listeria monocytogenes strain 10403s expressing whole cytoplasmic OVA (Lm-OVA) was kindly provided by Shomyseh Sanjabi (UCSF). Lm-OVA stocks frozen at −80°C were grown overnight at 37°C in BHI broth supplemented with 5μg/ml Erythromycin (Bio Basic, Amherst, New York). Then, overnight cultures were sub-cultured by diluting into fresh BHI broth supplemented with 5μg/ml Erythromycin and grown for 4 hours. Bacteria CFU was then quantified by measuring optical density at 600 nm. For primary infections, bacterial culture was then diluted to 5×104 CFU/100μl in sterile 1X PBS and 100μl was injected per mouse i.v. via the retroorbital vein. For rechallenge infections, bacteria were diluted to 1×105 CFU/100μl if the host mice were naive or 1×106 CFU/100μl if the host mice were previously infected.
METHOD DETAILS
Mass Cytometry Antibodies
All mass cytometry antibodies and concentrations used for analysis can be found in Table S1. Primary conjugates of mass cytometry antibodies were prepared using the MaxPAR antibody conjugation kit (Fluidigm, South San Francisco, CA) according to the manufacturer’s recommended protocol sourcing metals from Fluidigm (Fluidigm, South San Francisco, CA) or Trace Sciences International (Richmond Hill, Canada). Following labeling, antibodies were diluted in Candor PBS Antibody Stabilization solution (Candor Bioscience GmbH, Wangen, Germany) supplemented with 0.02% NaN3 to between 0.1 and 0.3 mg/mL and stored long-term at 4°C. Each antibody clone and lot was titrated to optimal staining concentrations using primary murine samples with all appropriate positive and negative controls: polyclonal murine CD8+ T cells purified by positive selection kit (Stem Cell Technologies, Vancouver, Canada) stimulated with PMA/Ionomycin via eBioscience Cell Stimulation Cocktail (ThermoFisher Scientific, Waltham, Massachusetts) for 15 minutes, 3 hours and 6 hours or plate-bound CD3 (145–2C11) and soluble CD28 (37.51) antibodies (UCSF Monoclonal Antibody Core, San Francisco) for 3 days, OT-1 splenocytes at day 7 of IL-2 or IL-7 polarization as below, and appropriate CD8+ T cell subsets (Naive, Short-lived Effector and Central Memory) at Day 8 of Lm-OVA infection. Titration results were cross-referenced to the literature as described in the text.
In vitro OT1 Stimulation and Polarization
OT-1 polarizations were carried out as previously described (Carrio et al, 2004). Briefly, splenocytes from OT-1 mice were cultured at 1×106 cells/mL in 24 well-plates of complete RPMI (UCSF Media Core facility) supplemented with 10% FBS (Omega Scientific, Tarzana, California), 100U/mL penicillin-streptomycin (Fisher Scientific, Hampton, New Hampshire), 2mM L-glutamine (Sigma-Aldrich, St. Louis, Missouri) and 50μM β-mecaptoethanol (Thermo Fisher Scientific, Waltham, Massachusetts ) and 10mM HEPES (UCSF Media Core Facility) in the presence of OVA257–264 peptide (0.1nM) (Invivogen, San Diego, California) and IL-2 (100U/ml) (Teceleukin) kindly provided by NCI Frederick. After 3 days in culture, activated cells were washed 3 times with RPMI 1640 and recultured in T25 culture flasks at 1×105 cells/mL in the presence of either IL-7, IL-15 (Biolegend, San Diego, California) or IL-2 (Teceleukin) kindly provided by NCI Frederick. (all cytokines 10ng/ml) After 2 additional days in culture, cells were passaged and recultured under the same conditions without peptide for an additional two days for total of 7 days in culture. Viability was confirmed by trypan blue exclusion (Thermo Fisher, Waltham, Massachusetts) or mass cytometry as described below.
Cell Preparation
All tissue preparations were performed simultaneously from each individual mouse, as previously reported (Spitzer et al. 2017). After euthanasia by CO2 inhalation, spleens were collected and homogenized in PBS + 5mM EDTA at 4°C. All tissues were washed with PBS/EDTA and re-suspended 1:1 with PBS/EDTA and 100mM Cisplatin (Enzo Life Sciences, Farmingdale, NY) for 60s before quenching 1:1 with PBS/EDTA + 0.5% BSA to determine viability as previously described (Spitzer et al., 2015). Cells were centrifuged at 500 g for 5 min at 4°C and re-suspended in PBS/EDTA/BSA at a density between 1–10×106 cells/ml. Care was taken to maintain all samples at 4°C during all phases of tissue harvest and preparation except viability staining and fixation. Suspensions were fixed for 10 min at RT using 1.6% PFA (Fisher Scientific, Hampton, New Hampshire) and frozen at −80°C.
For experiments with adoptively transferred OT1 T cells, immunomagnetic enrichment was performed to facilitate the detection of extremely rare cells before proliferation. Following lysis of red blood cells with ACK lysis buffer (ThermoFisher Scientific, Waltham, Massachusetts), EasySep Streptavidin Negative Selection was used with the following biotinylated antibodies: MHCII (AF6–120.1),CD11b (M1/70), Ly6C (RB6–8C5), B220 (RA3–6B2), CD4 (GK1.5), Ter119 (TER-119).
For Seahorse experiments with primary mouse T cell populations, cells were sorted by FACS from splenocytes harvested from naïve or Lm infected WT C57Bl/6 mice. For palmitate Seahorse experiments, CD8+ T cells were immunomagnetically enriched prior to sorting by EasySep Streptavidin Negative Selection using the following biotinylated antibodies: MHCII (AF6–120.1), CD11b (M1/70), Ly6G (1A8), B220 (RA3–6B2), CD4 (GK1.5), Ter119 (TER-119). Then, viable sorted cells were counted by hemocytometer and Trypan blue staining before proceeding to extracellular flux preparation and analysis.
For experiments with human samples, blood and was obtained from two advanced Non-Hodgkin Lymphoma patients receiving axicabtagene ciloleucel therapy as standard of care. Residual CAR T product was rinsed from infusion bags immediately following infusion completion and frozen until analysis. Within two hours of blood collection, patient PBMCs were isolated by centrifugation in CPT tubes (BD, Franklin Lakes, NJ) and frozen at −80°C until analysis.
Mass-Tag Cellular Barcoding
Mass-tag cellular barcoding was performed as previously described (Zunder et al., 2015). Briefly, 1 *106 cells from each animal were barcoded with distinct combinations of stable Pd isotopes in 0.02% saponin in PBS. Samples from any given tissue from each mouse per experiment group were barcoded together. Cells were washed once with cell staining media (PBS with 0.5% BSA and 0.02% NaN3), and once with 1X PBS, and pooled into a single FACS tube (BD Biosciences, San Jose, California). After data collection, each condition was deconvoluted using a single-cell debarcoding algorithm (Zunder et al., 2015).
Mass Cytometry Staining and Measurement
Cells were resuspended in cell staining media (PBS with 0.5% BSA and 0.02% NaN3), and antibodies against CD16/32 (BioLegend, San Diego, California) were added at 20 mg/ml for 5 min at RT on a shaker to block Fc receptors. Surface marker antibodies were then added, yielding 500 uL final reaction volumes and stained for 30 min at RT on a shaker. Following staining, cells were washed 2 times with cell staining media, then permeabilized with methanol for at 10 min at 4°C. Cells were then washed twice in cell staining media to remove remaining methanol, and stained with intracellular antibodies in 500 uL for 1 hour at RT on a shaker. Cells were washed twice in cell staining media and then stained with 1mL of 1:4000 191/193Ir DNA intercalator (Fluidigm, South San Francisco, CA) diluted in PBS with 4% PFA overnight. Cells were then washed once with cell staining media, once with 1X PBS and once with Cell Acquisition Solution (Fluidigm, South San Francisco, CA). Care was taken to assure buffers preceding analysis were not contaminated with metals in the mass range above 100 Da. Mass cytometry samples were diluted in Cell Acquisition Solution containing bead standards (Fluidigm, South San Francisco, CA) to approximately 106 cells per mL and then analyzed on a Helios mass cytometer (Fluidigm, South San Francisco, CA) equilibrated with Cell Acquisition Solution. We analyzed 1–5*105 cells per animal per time point, consistent with generally accepted practices in the field. For adoptive transfer experiments, 1–4*106 cells per animal were analyzed.
PBMCs isolated from advanced non-Hodgkin Lymphoma patients as above were thawed, washed in RPMI media supplemented with 10% FBS, glutamine, HEPES, and beta mercaptoethanol, and rested for 15min in media at 37°C, 5% CO2. Cells were counted, and then dispensed into a 96-well plate for staining. Cells were stained in 200nM cisplatin, washed, stained with surface antibodies, washed, fixed with 1.6% PFA, washed, permeabilized in methanol, washed, stained with intracellular antibodies, washed, and resuspended with 191/193Ir DNA intercalator overnight at 4°C as above. Mass cytometry samples were washed, resuspended to 500,000 cells/mL containing bead standards for acquisition on a Helios mass cytometer.
Mass Cytometry Bead Standard Data Normalization
Data normalization was performed as previously described (Spitzer et al., 2017). Briefly, just before analysis, the stained and intercalated cell pellet was resuspended in freshly prepared Cell Acquisition Solution containing the bead standard at a concentration ranging between 1 and 2*104 beads/ml. The mixture of beads and cells were filtered through a filter cap FACS tubes (BD Biosciences) before analysis. All mass cytometry files were normalized together using the mass cytometry data normalization algorithm (Finck et al., 2013), which uses the intensity values of a sliding window of these bead standards to correct for instrument fluctuations over time and between samples.
Adoptive T Cell Transfer
For adoptive transfer of early activated (EA) cells and SLECs, T cells were sorted by FACS from splenocytes harvested from WT CD45.2 C57Bl/6 mice or CD45.1 BoyJ mice 5 days post-infection. For memory rechallenge experiments, CD8+ T cells were immunomagnetically enriched prior to sorting by EasySep Streptavidin Negative Selection using the following biotinylated antibodies: MHCII (AF6–120.1), CD11b (M1/70), Ly6G (1A8), B220 (RA3–6B2), CD4 (GK1.5), Ter119 (TER-119). Then, viable sorted cells were counted by hemocytometer and Trypan blue staining, resuspended in sterile PBS and transferred into infection-matched congenic mice intravenously via the retroorbital vein.
For memory rechallenge experiments, 3 × 105 EA cells were adoptively transferred into infection matched or naïve congenic recipient mice. Recipient mice were then infected 21 days following the primary infection. Recipient mice were then euthanized 5 days post rechallenge for harvesting of spleens and quantification of donor adoptively transferred cells by flow cytometry.
For adoptive transfer of pathogen specific T cells to validate the antigen specificity of the early activated cells, CD8+ T cells were immunomagnetically enriched from the pooled spleens of three CD45.1+ OT1 TCR transgenic mice with EasySep Streptavidin Negative Selection using the following biotinylated antibodies: MHCII (AF6–120.1), CD11b (M1/70), CD11c (N418), Gr1 (RB6–8C5), B220 (RA3–6B2), CD4 (GK1.5), Ter119 (TER-119). Viable cells were quantified by counting on a hemocytometer with Trypan blue staining. 1×106 Cells were then resuspended in sterile PBS and transferred into naïve WT CD45.2+ mice intravenously via the retroorbital vein.
Flow Cytometry and Cell Sorting
Cells were stained for viability with Zombie-NIR dye. Cell surface staining was performed in cell staining media (PBS with 0.5% BSA and 0.02% NaN3) for 30 minutes at room temperature. The following anti-mouse antibodies were used: TCRβ – APC (H57–597), CD8+ – PE (53–5.8), CD62L - BV421 (MEL-14), KLRG1 – BV510 (2F1/KLRG1), CD44 – PE-Cy7 (IM7), CD25 – FITC (3C7), CD19 – APC-Cy7 (1D3/CD19), F480 APC-Cy7 (BM8). Stained cells were analyzed with an LSR II flow cytometer (BD Biosciences). MitoTracker Deep Red (Thermo Fisher, Waltham, Massachusetts) staining was performed per manufacturer’s instructions and as previously (Scharping et al, 2016). For MitoTracker Deep Red experiments, Zombie-UV dye was used (Biolegend, San Diego, California).
For sorting experiments, cells were prepared as described for flow cytometry and then sorted into FBS containing media (RPMI 1640, 20% FBS, 1% HEPES, 100 mg/mL penicillin/streptomycin) on a FACSAria II (BD Biosciences).
Extracellular Flux Analysis
Seahorse Assays were carried out utilizing Agilent Mitochondrial Stress Test kit as previously (van der Windt et al, 2012) and per the manufacturer’s instructions. For both in vitro and in vivo experiments, 2.5 ×105 cells per well were plated for analysis utilizing Cell-Tak adhesive per manufacturer’s instructions. Oxygen consumption rates (OCR) and extracellular acidification rates (ECAR) were measured in XF media (non-buffered RPMI 1640 containing 10 mM glucose, 2mM L-glutamine, and 1 mM sodium pyruvate) under basal conditions and in response to 1 μM oligomycin, 1 μM fluoro-carbonyl cyanide phenylhydrazone (FCCP) and 100 nM rotenone + 1 μM antimycin A (all from Agilent, Santa Clara, California) using a 96 well XF Extracellular Flux Analyzer (EFA) (Agilent, Santa Clara, California).
For palmitate oxidation assays OT-I T cells stimulated with IL-2 and SIINFEKL for 3 days followed by polarization in 10ng/ml IL-7 prior to incubation in FAO media (111 mM NaCl, 4.7mM KCl, 1.25mM CaC12, 2.0mM MgSO4, 1.2mM Na2HPO4, 2.5mM glucose, 0.5mM carnitine, and 5mM HEPES) for 15 minutes with the addition of either palmitate-BSA or BSA for extracellular analysis in the presence of 1.5 μM Oligomycin, 1μM FCCP, and 0.5μM Antimcyin A/Rotenone as previously (Raud et al., 2018).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
All significance analysis of Seahorse data and cellular frequency was performed by paired two-sided student’s t-test with error bars representing SEM in Prism v8. (GraphPad, San Diego, California). Analysis of median protein expression was performed by paired or unpaired (as indicated) two-sided student’s t-test in R. Spearman correlation matrices were generated using the Hmisc package in R.
Unsupervised Clustering Analysis
Cell clusters were identified using the Phenograph algorithm as implemented in the ‘cytofkit’ package in R (Levine et al., 2015). Standard settings were utilized (with k = 30 for endogenous CD8+ T cells and k = 100 for OT1 T cells).
Data Visualization
Unsupervised force-directed graphs were generated as previously reported (Spitzer et al., 2015) with the following modifications. Single cells were downsampled to n = 1,000 cells from each time point by randomly selecting cells across the biological replicates. All cells were combined in a single graph with edge weights defined as the cosine similarity between the vectors of marker values of each cell. All the pairwise distances were calculated and for each node only the 10 edges of highest weight were retained. The graph was then laid out using the ForceAtlas2 algorithm in Gephi.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mass cytometry antibodies are found in Table S1. | This paper | N/A |
| Anti-mouse TCRβ-Brilliant Violet 421 clone H57–59] | BioLegend | Cat#109229; RRID: AB_10933263 |
| Anti-mouse TCRβ-APC clone H57–597 | BioLegend | Cat#109211; RRID: AB_313434 |
| Anti-mouse CD8-PE clone 53–6.7 | BioLegend | Cat#100708; RRID: AB_312747 |
| Anti-mouse CD8-Brilliant Violet 650 clone 53–6.7 | BioLegend | Cat#100741; RRID: AB_11204079 |
| Anti-mouse CD62L- Brilliant Violet 421 clone MEL-14 | BioLegend | Cat#104435; RRID: AB_10900082 |
| Anti-mouse CD62L- PE Dazzle clone MEL-14 | BioLegend | Cat#104447; RRID: N/A |
| Anti-mouse KLRG1-PE/Cy7 clone MAFA | BioLegend | Cat#138415; RRID: N/A |
| Anti-mouse KLRG1- Brilliant Violet 510 clone 2F1/KLRG1 | BioLegend | Cat#138421; RRID: N/A |
| Anti-mouse CD44-PE/Cy7 clone IM] | BioLegend | Cat#103029; RRID: N/A |
| Anti-mouse CD44-PerCP/Cy5.5 clone IM7 | BioLegend | Cat#138418; RRID: N/A |
| Anti-mouse CD25-FITC clone 3C7 | BioLegend | Cat#102006; RRID: AB_961210 |
| Anti-mouse CD25-APC clone 3C7 | BioLegend | Cat#101909; RRID: AB_2280288 |
| Anti-mouse CD45.1-PE clone A20 | BioLegend | Cat#110708; RRID: AB_313496 |
| Anti-mouse CD45.2-APC clone 104 | BioLegend | Cat#109813; RRID: AB_389210 |
| Anti-mouse I-ab-Pacific Blue clone AF6–120.1 | BioLegend | Cat#116421; RRID: AB_10613291 |
| Anti-mouse I-ab-PE/Cy7 clone AF6–120.1 | BioLegend | Cat#116419; RRID: AB_10575904 |
| Anti-mouse F4/80-APC/Cy7 clone BM8 | BioLegend | Cat#123117; RRID: AB_893489 |
| Anti-mouse CD19-APC/Cy7 clone 1D3/CD19 | BioLegend | Cat#115529; RRID: N/A |
| Anti-mouse Ter119-Biotin clone TER119 | BioLegend | Cat#116204; RRID: AB_313704 |
| Anti-mouse B220-Biotin clone RA3-6B2 | BioLegend | Cat#103204; RRID: AB_312988 |
| Anti-mouse Ly6G-Biotin clone 1A8 | BioLegend | Cat#127603; RRID: AB_1186105 |
| Anti-mouse I-ab-Biotin clone AF6–120.1 | BioLegend | Cat#116404; RRID: AB_313722 |
| Anti-mouse CD11b-Biotin clone M1/70 | BioLegend | Cat# 101204; RRID: AB_312787 |
| Anti-mouse CD11c-Biotin clone N418 | BioLegend | Cat# 117303; RRID: AB_313772 |
| Anti-mouse CD4-Biotin clone GK1.5 | BioLegend | Cat#100404; RRID: AB_312689 |
| Biological Samples | ||
| PMBCs from non-Hodgkin Lymphoma patients | Vanderbilt University Medical Center | https://vumc.org |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Mouse IL-7 | Biolegend | Cat# 577802; RRID: N/A |
| Recombinant human IL-2 (Tecaleukin) | NCI Frederick | Cat# N/A RRID: N/A |
| MitoTracker Deep Red | Thermo Fisher Scientific | Cat# M22426; RRID: N/A |
| Zombie UV | BioLegend | Cat# 423108; RRID: N/A |
| Zombie NIR | BioLegend | Cat# 423105; RRID: N/A |
| OVA(257–264) SIINFEKL peptide | Invivogen | Cat#vac-sin RRID: N/A |
| Benzonase | Sigma-Aldrich | Cat# E8263-25KU; RRID: N/A |
| Cisplatin | Sigma-Aldrich | Cat#P439425MG; RRID: N/A |
| Intercalator-Ir | Fluidigm | Cat# 201192B; RRID: N/A |
| Calibration beads, EQ™ Four Element | Fluidigm | Cat#201078; RRID: N/A |
| UltraComp eBeads beads | eBioscience | Cat# 01-2222-42; RRID: N/A |
| Seahorse XF Base RPMI pH 7.4 | Agilent Technologies | Part#103576-100; RRID: N/A |
| TrueStain FcX (anti-mouse CD16/32 antibody (clone 93) | BioLegend | Cat#101320; RRID: N/A |
| Cell Acquisition Solution | Fluidigm | Cat#201240; RRID: N/A |
| Glutamine (0.2M), 100X | UCSF Media Production Core Facility | Cat#CCFGB002 RRID: N/A |
| 1M HEPES pH 7.4 | UCSF Media Production Core Facility | Cat#CCFGL002 RRID: N/A |
| Fetal Bovine Serum | Omega | Cat#FB-01 RRID: N/A |
| Gibco 2-Mercaptoethanol | Thermo Fisher Scientific | Cat# 21985023 RRID: N/A |
| XF 100mL Pyruvate Solution | Agilent Technologies | Cat# 103578-100 RRID: N/A |
| XF 1.0 Glucose Solution | Agilent Technologies | Cat#103577-100 RRID: N/A |
| XF 200mM Glutamine Solution | Agilent Technologies | Cat#103579-100 RRID: N/A |
| Critical Commercial Assays | ||
| MaxPar Antibody Conjugation Kit | Fluidigm | Cat#201300; RRID: N/A |
| Seahorse XF Palmitate Oxidation Stress Test Kit | Agilent Technologies | Cat#103693-100 RRID: N/A |
| Seahorse XF Mito Stress Test Kit | Agilent Technologies | Cat#103015-100 RRID: N/A |
| Seahorse XF Flux Pack | Agilent Technologies | Cat# |
| EasySep Mouse Streptavidin RapidSpheres Isolation Kit | Stem Cell Technologies | Cat#19860 RRID: N/A |
| Deposited Data | ||
| Mass cytometry data | This paper | |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6-Tg(TcraTcrb)1100Mjb/J (OT-1) | Jackson Laboratory | Cat# 003831; RRID: IMSR_JAX:003831 |
| Mouse: B6.SJL-Ptprca Pepcb/BoyJ (BoyJ) | Jackson Laboratory | Cat# 002014; RRID: IMSR_JAX002014 |
| Mouse: C57BL/6J | Jackson Laboratory | Cat#000664; RRID: IMSR_JAX000664 |
| Listeria Monocytogenes strain 10403s expressing whome cytoplasmic OVA (Lm-OVA) | Shomyseh Sanjabi (UCSF) | Cat# N/A RRID:N/A |
| Recombinant DNA | ||
| Sequence-Based Reagents | ||
| Software and Algorithms | ||
| Cytobank analysis software | Cytobank, Inc | https://cytobank.org RRID: SCR_014043 |
| Cellengine analysis software | Primitybio | https://primitybio.com/cellengine.html RRID: N/A |
| Normalizer | Finck et al., 2013 | https://github.com/nolanlab/bead-normalization, RRID: N/A |
| RPhenograph | Levine et al., 2015 | https://github.com/JinmiaoChenLab/Rphenograph RRID: N/A |
| Gephi | Bastian et al., 2009 | https://gephi.org RRID:SCR_004293 |
| Prism v9 | GraphPad | https://graphpad.com/scientific-software/prism/ RRID: SCR_005375 |
| R environment | R Development Core Team, 2008 | https://www.r-project.org/, RRID:SCR_001905 |
| Other | ||
| Helios mass cytometer | Fluidigm | Cat# N/A |
| FACSAria II | BD | Cat# NA |
| LSR II Fortessa | BD | Cat# N/A |
| XF96 Seahorse Analyzer | Agilent | Cat# N/A |
TABLE WITH EXAMPLES FOR AUTHOR REFERENCE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Snail | Cell Signaling Technology | Cat#3879S; RRID: AB_2255011 |
| Mouse monoclonal anti-Tubulin (clone DM1A) | Sigma-Aldrich | Cat#T9026; RRID: AB_477593 |
| Rabbit polyclonal anti-BMAL1 | This paper | N/A |
| Biological Samples | ||
| Healthy adult BA9 brain tissue | University of Maryland Brain & Tissue Bank; http://medschool.umaryland.edu/btbank/ | Cat#UMB1455 |
| Human hippocampal brain blocks | New York Brain Bank | http://nybb.hs.columbia.edu/ |
| Patient-derived xenografts (PDX) | Children’s Oncology Group Cell Culture and Xenograft Repository | http://cogcell.org/ |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MK-2206 AKT inhibitor | Selleck Chemicals | S1078; CAS: 1032350-13-2 |
| SB-505124 | Sigma-Aldrich | S4696; CAS: 694433-59-5 (free base) |
| Picrotoxin | Sigma-Aldrich | P1675; CAS: 124-87-8 |
| Human TGF-β | R&D | 240-B; GenPept: P01137 |
| Activated S6K1 | Millipore | Cat#14-486 |
| GST-BMAL1 | Novus | Cat#H00000406-P01 |
| Critical Commercial Assays | ||
| EasyTag EXPRESS 35S Protein Labeling Kit | Perkin-Elmer | NEG772014MC |
| CaspaseGlo 3/7 | Promega | G8090 |
| TruSeq ChIP Sample Prep Kit | Illumina | IP-202-1012 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE63473 |
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Experimental Models: Cell Lines | ||
| Hamster: CHO cells | ATCC | CRL-11268 |
| D. melanogaster: Cell line S2: S2-DRSC | Laboratory of Norbert Perrimon | FlyBase: FBtc0000181 |
| Human: Passage 40 H9 ES cells | MSKCC stem cell core facility | N/A |
| Human: HUES 8 hESC line (NIH approval number NIHhESC-09-0021) | HSCI iPS Core | hES Cell Line: HUES-8 |
| Experimental Models: Organisms/Strains | ||
| Streptococcus pyogenes: M1 serotype strain: strain SF370; M1 GAS | ATCC | ATCC:700294 |
| C. elegans: Strain BC4011: srl-1(s2500) II; dpy-18(e364) III; unc-46(e177)rol-3(s1040) V. | Caenorhabditis Genetics Center | WB Strain: BC4011; WormBase: WBVar00241916 |
| D. melanogaster: RNAi of Sxl: y[1] sc[*] v[1]; P{TRiP.HMS00609}attP2 | Bloomington Drosophila Stock Center | BDSC:34393; FlyBase: FBtp0064874 |
| S. cerevisiae: Strain background: W303 | ATCC | ATTC: 208353 |
| Mouse: R6/2: B6CBA-Tg(HDexon1)62Gpb/3J | The Jackson Laboratory | JAX: 006494 |
| Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtml.1Wsy/J | The Jackson Laboratory | RRID: IMSR_JAX:008471 |
| Zebrafish: Tg(Shha:GFP)t10: t10Tg | Neumann and Nuesslein-Volhard, 2000 | ZFIN: ZDB-GENO-060207-1 |
| Arabidopsis: 35S::PIF4-YFP, BZR1-CFP | Wang et al., 2012 | N/A |
| Arabidopsis: JYB1021.2: pS24(AT5G58010)::cS24:GFP(-G):NOS #1 | NASC | NASC ID: N70450 |
| Recombinant DNA | ||
| pLVX-Tight-Puro (TetOn) | Clonetech | Cat#632162 |
| Plasmid: GFP-Nito | This paper | N/A |
| cDNA GH111110 | Drosophila Genomics Resource Center | DGRC:5666; FlyBase:FBcl0130415 |
| AAV2/1-hsyn-GCaMP6- WPRE | Chen et al., 2013 | N/A |
| Mouse raptor: pLKO mouse shRNA 1 raptor | Thoreen et al., 2009 | Addgene Plasmid #21339 |
| Sequence-Based Reagents | ||
| siRNA targeting sequence: PIP5K I alpha #1: ACACAGUACUCAGUUGAUA | This paper | N/A |
| Primers for XX, see Table SX | This paper | N/A |
| Primer: GFP/YFP/CFP Forward: GCACGACTTCTTCAAGTCCGCCATGCC | This paper | N/A |
| Morpholino: MO-pax2a GGTCTGCTTTGCAGTGAATATCCAT | Gene Tools | ZFIN: ZDB-MRPHLNO-061106-5 |
| ACTB (hs01060665_g1) | Life Technologies | Cat#4331182 |
| RNA sequence: hnRNPA1_ligand: UAGGGACUUAGGGUUCUCUCUAGGGACUUAGGGUUCUCUCUAGGGA | This paper | N/A |
| Software and Algorithms | ||
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| Other | ||
| Sequence data, analyses, and resources related to the ultra-deep sequencing of the AML31 tumor, relapse, and matched normal. | This paper | http://aml31.genome.wustl.edu |
| Resource website for the AML31 publication | This paper | https://github.com/chrisamiller/aml31SuppSite |
Highlights.
A mass cytometry approach quantifies metabolic proteins in single cells in vivo
Early activated T cells exhibit simultaneous peak oxidative and glycolytic activity
CD8+ T cells transit through this transient state prior to differentiation
CAR T cells exhibit an analogous transient program upon infusion into patients
Acknowledgements:
We would like to thank the members of the Spitzer lab, Vinh Ngyuen, Stanley Tamaki, Sagar Bapat, Adil Daud, Lawrence Fong, Andrei Goga, Rushika Perera for their experimental contributions and helpful discussions. Listeria monocytogenes strain 10403s expressing OVA (Lm-OVA) was kindly provided by Shomyseh Sanjabi (UCSF). We acknowledge the Parnassus Flow Cytometry Core Facility supported in part by NIH Grants P30DK063720, S10OD018040, S10OD018040 and S10OD021822. Recombinant human IL-2 (IL-2; TECIN; Teceleukin) was provided by the National Cancer Institute.
This work was supported by NIH grants DP5OD023056 to M.H.S., R01DK105550 and R01HL136664 to J.C.R., and M.Z.M was supported by F30CA239367. L.S.L. was supported by Conquer Cancer Foundation Young Investigator Award grant CA-0122026. M.H.S. is a Chan Zuckerberg Biohub investigator and a Parker Institute for Cancer Immunotherapy investigator.
Declaration of Interests:
O.O.O.: honoraria from Kite Pharmaceuticals; consulting for Kite, Pfizer, Spectrum, Curio science and Bayer; research funding from Kite. J.C.R.: founder, member of the scientific advisory board of Sitryx Therapeutics; member of the scientific advisory boards of Caribou Biosciences, Istesso Ltd; research funding from Kadmon, Incyte, Tempest, and Calithera; honoraria from Merck and Pfizer. M.H.S.: founder and board member of Teiko Bio; consulting for Five Prime Therapeutics, Earli, Ono Pharmaceutical and January, Inc.; research funding from Roche/Genentech, Pfizer, Valitor Inc., and Bristol-Myers Squibb.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahl PJ, Hopkins RA, Xiang WW, Au B, Kaliaperumal N, Fairhurst A-M, and Connolly JE (2020). Met-Flow, a strategy for single-cell metabolic analysis highlights dynamic changes in immune subpopulations. Commun. Biol 3, 305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed R, and Gray D (1996). Immunological Memory and Protective Immunity: Understanding Their Relation. Science (80-. ). 272, 54 LP–60. [DOI] [PubMed] [Google Scholar]
- Andrejeva G, and Rathmell JC (2017). Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab 26, 49–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, and Ahmed R (2010). HHS Public Access. 460, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artyomov MN, and Van den Bossche J (2020). Immunometabolism in the Single-Cell Era. Cell Metab 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badovinac VP, Haring JS, and Harty JT (2007). Initial T Cell Receptor Transgenic Cell Precursor Frequency Dictates Critical Aspects of the CD8+ T Cell Response to Infection. Immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandura DR, Baranov VI, Ornatsky OI, Antonov A, Kinach R, Lou X, Pavlov S, Vorobiev S, Dick JE, and Tanner SD (2009). Mass Cytometry: Technique for Real Time Single Cell Multitarget Immunoassay Based on Inductively Coupled Plasma Time-of-Flight Mass Spectrometry. Anal. Chem 81, 6813–6822. [DOI] [PubMed] [Google Scholar]
- Bendall SC, Simonds EF, Qiu P, Amir EAD, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, et al. (2011). Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science (80-. ). 332, 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Davis KL, Amir ED, Tadmor MD, Simonds EF, Chen TJ, Shenfeld DK, Nolan GP, and Pe’er D (2014). Single-Cell Trajectory Detection Uncovers Progression and Regulatory Coordination in Human B Cell Development. Cell 157, 714–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenmiller B, Zunder ER, Finck R, Chen TJ, Savig ES, Bruggner RV, Simonds EF, Bendall SC, Sachs K, Krutzik PO, et al. (2012). Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators. Nat. Biotechnol 30, 858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges da Silva H, Beura LK, Wang H, Hanse EA, Gore R, Scott MC, Walsh DA, Block KE, Fonseca R, Yan Y, et al. (2018). The purinergic receptor P2RX7 directs metabolic fitness of long-lived memory CD8(+) T cells. Nature 559, 264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck MD, Sowell RT, Kaech SM, and Pearce EL (2017). Metabolic Instruction of Immunity. Cell 169, 570–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck MDD, O’Sullivan D, Klein Geltink RII, Curtis JDD, Chang CH, Sanin DEE, Qiu J, Kretz O, Braas D, van der Windt GJJW, et al. (2016). Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 166, 63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon MJ, Scharping NE, Dunstane D, Watkins SC, Menk AV, Watson MJ, Delgoffe GM, and Rivadeneira DB (2018). 4–1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J. Exp. Med 215, 1091–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan MK, Postow MA, and Wolchok JD (2016). Targeting T Cell Co-receptors for Cancer Therapy. Immunity 44, 1069–1078. [DOI] [PubMed] [Google Scholar]
- Cantor JR, Abu-Remaileh M, Kanarek N, Freinkman E, Gao X, Louissaint A, Lewis CA, and Sabatini DM (2017). Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 169, 258–272.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter K, Pollitt RJ, and Middleton B (1992). Human liver long-chain 3-hydroxyacyl-coenzyme a dehydrogenase is a multifunctional membrane-bound beta-oxidation enzyme of mitochondria. Biochem. Biophys. Res. Commun 183, 443–448. [DOI] [PubMed] [Google Scholar]
- Carrio R, Bathe OF, and Malek TR (2014). Initial Antigen Encounter Programs CD8+ T Cells Competent to Develop into Memory Cells That Are Activated in an Antigen-Free, IL-7- and IL-15-Rich Environment. J. Immunol 172, 7315–7323. [DOI] [PubMed] [Google Scholar]
- Cheung P, Vallania F, Warsinske HC, Donato M, Schaffert S, Chang SE, Dvorak M, Dekker CL, Davis MM, Utz PJ, et al. (2018). Single-Cell Chromatin Modification Profiling Reveals Increased Epigenetic Variations with Aging. Cell 173, 1385–1397.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisolm DA, Savic D, Moore AJ, Ballesteros-Tato A, León B, Crossman DK, Murre C, Myers RM, and Weinmann AS (2017). CCCTC-Binding Factor Translates Interleukin 2- and α-Ketoglutarate-Sensitive Metabolic Changes in T Cells into Context-Dependent Gene Programs. Immunity 47, 251–267.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu T, Tyznik AJ, Roepke S, Berkley AM, Woodward-Davis A, Pattacini L, Bevan MJ, Zehn D, and Prlic M (2013). Bystander-Activated Memory CD8 T Cells Control Early Pathogen Load in an Innate-like, NKG2D-Dependent Manner. Cell Rep [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CA, Hoppins S, and Fink PJ (2018). Cutting Edge: Glycolytic Metabolism and Mitochondrial Metabolism Are Uncoupled in Antigen-Activated CD8 + Recent Thymic Emigrants. J. Immunol 201, 1627–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza AD, Parikh N, Kaech SM, and Shadel GS (2007). Convergence of multiple signaling pathways is required to coordinately up-regulate mtDNA and mitochondrial biogenesis during T cell activation. Mitochondrion 7, 374–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MK, Field AS, Burai R, Ramesh C, Whitney K, Bologa CG, Oprea TI, Yamaguchi Y, Hayashi S, Sklar L a, et al. (2012). Postranscriptional control of T cell effector function by aerobic glycolysis. Cell 127, 358–366. [Google Scholar]
- Enamorado M, Labiano S, Melero I, Rouzaut A, Teijeira A, Bolaños E, Garasa S, Sancho D, Santamaría E, Azpilikueta A, et al. (2018). Mitochondrial Morphological and Functional Reprogramming Following CD137 (4–1BB) Costimulation. Cancer Immunol. Res 6, 798–811. [DOI] [PubMed] [Google Scholar]
- Feske S (2007). Calcium signalling in lymphocyte activation and disease. Nat. Rev. Immunol 7, 690–702. [DOI] [PubMed] [Google Scholar]
- Fracchia KM, Pai CY, and Walsh CM (2013). Modulation of T Cell Metabolism and Function through Calcium Signaling. Front. Immunol 4, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerriets VA, and Rathmell JC (2012). Metabolic pathways in T cell fate and function. Trends Immunol 33, 168–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good Z, Borges L, Gonzalez NV, Sahaf B, Samusik N, Tibshirani R, Nolan GP, and Bendall SC (2019). Proliferation tracing with single-cell mass cytometry optimizes generation of stem cell memory-like T cells. Nat. Biotechnol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray SM, Amezquita RA, Guan T, Kleinstein SH, and Kaech SM (2017). Polycomb Repressive Complex 2-Mediated Chromatin Repression Guides Effector CD8+ T Cell Terminal Differentiation and Loss of Multipotency. Immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann FJ, Mrdjen D, McCaffrey E, Glass DR, Greenwald NF, Bharadwaj A, Khair Z, Verberk SG, Baranski A, Baskar R, et al. (2020). Single-cell metabolic profiling of human cytotoxic T cells. Nat. Biotechnol (in print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harty JT, and Badovinac VP (2002). Influence of effector molecules on the CD8+ T cell response to infection. Curr. Opin. Immunol 14, 360–365. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Jutabha P, Endou H, Sagara H, and Anzai N (2013). LAT1 Is a Critical Transporter of Essential Amino Acids for Immune Reactions in Activated Human T Cells. J. Immunol 191, 4080 LP–4085. [DOI] [PubMed] [Google Scholar]
- Howie D, Ten Bokum A, Necula AS, Cobbold SP, and Waldmann H (2018). The Role of Lipid Metabolism in T Lymphocyte Differentiation and Survival. Front. Immunol 8, 1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Lau LL, and Shen H (2003). Selective Depletion of Nonspecific T Cells During the Early Stage of Immune Responses to Infection. J. Immunol [DOI] [PubMed] [Google Scholar]
- Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, and Kaech SM (2007). Inflammation Directs Memory Precursor and Short-Lived Effector CD8+ T Cell Fates via the Graded Expression of T-bet Transcription Factor. Immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, and Ahmed R (2001). Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naïve cells. Nat. Immunol 2, 415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, and Ahmed R (2010). Prolonged Interleukin-2Rα Expression on Virus-Specific CD8+ T Cells Favors Terminal-Effector Differentiation In Vivo. Immunity 32, 91–103. [DOI] [PubMed] [Google Scholar]
- Klein Geltink RI, O’Sullivan D, Corrado M, Bremser A, Buck MD, Buescher JM, Firat E, Zhu X, Niedermann G, Caputa G, et al. (2017). Mitochondrial Priming by CD28. Cell 171, 385–397.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnaswamy S, Spitzer MH, Mingueneau M, Bendall SC, Litvin O, Stone E, Pe’er D, and Nolan GP (2014). Conditional density-based analysis of T cell signaling in single-cell data. Science (80-. ). 346, 1250689–1250689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laforge M, Rodrigues V, Silvestre R, Gautier C, Weil R, Corti O, and Estaquier J (2016). NF-κB pathway controls mitochondrial dynamics. Cell Death Differ 23, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Walsh MC, Hoehn KL, James DE, Wherry EJ, and Choi Y (2014). Regulator of fatty acid metabolism, acetyl coenzyme a carboxylase 1, controls T cell immunity. J. Immunol 192, 3190–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma EH, Verway MJ, Johnson RM, Roy DG, Steadman M, Hayes S, Williams KS, Sheldon RD, Samborska B, Kosinski PA, et al. (2019). Metabolic Profiling Using Stable Isotope Tracing Reveals Distinct Patterns of Glucose Utilization by Physiologically Activated CD8+ T Cells. Immunity 1–15. [DOI] [PubMed] [Google Scholar]
- MacIver NJ, Blagih J, Saucillo DC, Tonelli L, Griss T, Rathmell JC, and Jones RG (2011). The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J. Immunol 187, 4187–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGregor DD, Koster FT, and Mackaness GB (1970). Biological sciences: The short lived small lymphocyte as a mediator of cellular immunity. Nature 228, 855–856. [DOI] [PubMed] [Google Scholar]
- Menk AV, Scharping NE, Moreci RS, Zeng X, Guy C, Salvatore S, Bae H, Xie J, Young HA, Wendell SG, et al. (2018). Early TCR Signaling Induces Rapid Aerobic Glycolysis Enabling Distinct Acute T Cell Effector Functions. Cell Rep 22, 1509–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingueneau M, Krishnaswamy S, Spitzer MH, Bendall SC, Stone EL, Hedrick SM, Pe’er D, Mathis D, Nolan GP, and Benoist C (2014). Single-cell mass cytometry of TCR signaling: Amplification of small initial differences results in low ERK activation in NOD mice. Proc. Natl. Acad. Sci 111, 16466–16471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nii T, Segawa H, Taketani Y, Tani Y, Ohkido M, Kishida S, Ito M, Endou H, Kanai Y, Takeda E, et al. (2001). amino acid transporter LAT1 during T-cell activation. Reactions 704, 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan D, vanderWindt GWJ, Huang SCC, Curtis JD, Chang CH, Buck MDL, Qiu J, Smith AM, Lam WY, DiPlato LM, et al. (2014). Memory CD8+ T Cells Use Cell-Intrinsic Lipolysis to Support the Metabolic Programming Necessary for Development. Immunity 41, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obar JJ, and Lefrançois L (2010). Early Signals during CD8 + T Cell Priming Regulate the Generation of Central Memory Cells. J. Immunol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olenchock BA, Rathmell JC, and Vander Heiden MG (2017). Biochemical Underpinnings of Immune Cell Metabolic Phenotypes. Immunity 46, 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, and Choi Y (2009). Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 460, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollizzi KN, Patel CH, Sun I-H, Oh M-H, Waickman AT, Wen J, Delgoffe GM, and Powell JD (2015). mTORC1 and mTORC2 selectively regulate CD8+ T cell differentiation. J. Clin. Invest 125, 2090–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raud B, Roy DG, Divakaruni AS, Tarasenko TN, Franke R, Ma EH, Samborska B, Hsieh WY, Wong AH, Stuve P, et al. (2018). Etomoxir Actions on Regulatory and Memory T Cells Are Independent of Cpt1a-Mediated Fatty Acid Oxidation. Cell Metab 28, 504–515.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren W, Liu G, Yin J, Tan B, Wu G, Bazer FW, Peng Y, and Yin Y (2017a). Amino-acid transporters in T-cell activation and differentiation. Cell Death Dis 8, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren W, Liu G, Yin J, Tan B, Wu G, Bazer FW, Peng Y, and Yin Y (2017b). Amino-acid transporters in T-cell activation and differentiation. Cell Death Dis [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restifo NP, Dudley ME, and Rosenberg SA (2012). Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol 12, 269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolf J, Zarrouk M, Finlay DK, Foretz M, Viollet B, and Cantrell DA (2013). AMPKα1: a glucose sensor that controls CD8 T-cell memory. Eur. J. Immunol 43, 889–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Miller JF, Fan X, Kolwyck D, Ahmed R, and Harty JT (1998). Compartmentalization of Bacterial Antigens: Differential Effects on Priming of CD8 T Cells and Protective Immunity. Cell 92, 535–545. [DOI] [PubMed] [Google Scholar]
- Sinclair LV, Rolf J, Emslie E, Shi Y-B, Taylor PM, and Cantrell DA (2013). Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol 14, 500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer MH, and Nolan GP (2016). Mass Cytometry: Single Cells, Many Features. Cell 165, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, Roychoudhuri R, Palmer DC, Muranski P, Karoly ED, et al. (2013). Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Invest 123, 4479–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan DO, Windt G.J.W. Van Der, Huang SC, Jonathan D, Chang C, Buck MD, Qiu J, Smith AM, Lam WY, Diplato LM, et al. (2014). Memory CD8+ T Cells use cell intrinsic lipolysis. Immunity 41, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarze A, Deniaud A, Le Bras M, Maillier E, Molle D, Larochette N, Zamzami N, Jan G, Kroemer G, and Brenner C (2007). GAPDH, a novel regulator of the pro-apoptotic mitochondrial membrane permeabilization. Oncogene 26, 2606–2620. [DOI] [PubMed] [Google Scholar]
- Vogel C, and Marcotte EM (2012). Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet 13, 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, et al. (2011). The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation. Immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, and Ahmed R (2004). Memory CD8 T-cell differentiation during viral infection. J. Virol. 78, 5535–5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand G, and Remington SJ (1986). CITRATE SYNTHASE: Structure, Control, and Mechanism. Annu. Rev. Biophys. Biophys. Chem 15, 97–117. [DOI] [PubMed] [Google Scholar]
- van der Windt GJW, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, and Pearce EL (2012). Mitochondrial Respiratory Capacity Is a Critical Regulator of CD8+T Cell Memory Development. Immunity 36, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Windt GJW, Chang C-H, and Pearce EL (2016). Measuring Bioenergetics in T Cells Using a Seahorse Extracellular Flux Analyzer. Curr. Protoc. Immunol 113, 3.16B.1–3.16B.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Cohen S, Guy C, Shrestha S, Neale G, Brown SA, Cloer C, Kishton RJ, Gao X, Youngblood B, et al. (2016). mTORC1 and mTORC2 Kinase Signaling and Glucose Metabolism Drive Follicular Helper T Cell Differentiation. Immunity 45, 540–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, and Romero P (2017). Metabolic Control of CD8 + T Cell Fate Decisions and Antitumor Immunity. Trends Mol. Med 24. [DOI] [PubMed] [Google Scholar]
- Zunder ER, Lujan E, Goltsev Y, Wernig M, and Nolan GP (2015). A continuous molecular roadmap to iPSC reprogramming through progression analysis of single-cell mass cytometry. Cell Stem Cell 16, 323–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mass cytometry data will be made publicly available on Mendeley Data at https://data.mendeley.com/drafts/cnfkpk2t8m. No new code or algorithms were developed during this study; however, code will be provided upon request without limitations.