

Abstract
Purpose:
In a head and neck squamous cell carcinoma (HNSCC) “window of opportunity” clinical trial, we reported that trametinib reduced MEK-Erk1/2 activation and resulted in tumor responses in a subset of patients. Here, we investigated resistance to trametinib and molecular correlates in HNSCC cell lines and patient samples.
Experimental design:
HNSCC cell lines were treated with trametinib to generate resistant lines. Candidate bypass pathways were assessed using immunoblotting, CRISPR knockout, and survival assays. Effectiveness of combined trametinib and verteporfin targeting was evaluated. Patient-derived xenografts (PDX) from responder patients were treated with trametinib and resistant tumors were analyzed. Window trial clinical samples were subjected to whole exome and RNA sequencing.
Results:
HNSCC cell lines developed resistance (CAL27-TR and HSC3-TR) after prolonged trametinib exposure. Downstream effectors of the Hippo pathway were activated in CAL27-TR and HSC3-TR, and combined trametinib and verteporfin treatment resulted in synergistic treatment response. We defined the Hippo pathway effector Yap1 as an induced survival pathway promoting resistance to trametinib in HSC3-TR. Yap1 was necessary for HSC3-TR trametinib resistance, and constitutively active Yap1 was sufficient to confer resistance in parental HSC3. Analysis of trametinib neoadjuvant trial patient tumors indicated canonical MEK-Erk1/2 pathway activating mutations were infrequent, and Yap1 activity increased following trametinib treatment. Trametinib treatment of a PDX from a responder patient resulted in evolution of resistance with increased Yap1 expression and activity.
Conclusions:
These studies identify a Yap1 dependent resistance to trametinib therapy in HNSCCs. Combined Yap1 and MEK targeting may represent a strategy to enhance HNSCC response.
Keywords: trametinib resistance, YAP1, window clinical trial
INTRODUCTION
Annually, over 500,000 people worldwide receive the diagnosis of head and neck squamous cell carcinoma (HNSCC) (1,2), and 60% succumb to their disease within 5 years (3). Of the reported head and neck cancer cases, approximately half were primary oral cavity squamous cell carcinomas (OCSCC) (2). Despite significant advances in detection, surgical methods and adjuvant radiation and chemotherapy, the prognosis for HNSCC has remained stable for years (1,2,4).
Genomic analysis has identified multiple signaling and regulatory proteins participating in HNSCC development and progression. These include aberrations in TP53, CDKN2A, PIK3CA, PTEN, and NOTCH1 (5,6). In addition, upregulation of the Ras/Raf/MEK/Erk1/2 (MAPK) signaling pathway has been reported in OCSCC (7–11). The MAPK pathway regulates a breadth of fundamental cellular differentiation, proliferation and migratory pathways, and as a result its dysregulation is a major driver in cancer progression and development (12). In other malignancies, oncogenic activation of the MAPK pathway often occurs through activating mutations in BRAF or RAS genes (13,14). However, data from The Cancer Genome Atlas (TCGA) analysis has shown that Ras and Raf proteins are uncommonly mutated in HNSCC. HRAS activating mutations are observed in 4-8% of cases and NRAS, KRAS, or BRAF mutations are only rarely identified (15,16).
Despite infrequent RAS/RAF mutation in HNSCC, the presence of an activated MAPK pathway is common. Several groups have reported immunohistochemical analysis of phosphorylated Erk1/2 (p-Erk1/2) in primary HNSCCs (7–11). These include two detailed HNSCC studies showing that 98 and 91% of tumors were positive for p-Erk1/2 staining, respectively (7,17). In our previous study, we identified increased p-Erk1/2 activation to be associated with more aggressive tumor growth in a carcinogen-induced mouse model of OCSCC (18). Interestingly, p-Erk1/2 staining in HNSCC patients was associated with advanced nodal stages and was increased in recurrent tumors compared to the tumor at initial presentation (7,17), providing a rationale for therapeutically targeting the MEK pathway in HNSCCs. In a completed “window-of-opportunity” clinical trial, our group performed a neoadjuvant (preoperative) trial to determine the biomarker and tumor response of OCSCC to the allosteric MEK1/2 inhibitor trametinib (19). We demonstrated that brief trametinib treatment resulted in significant reduction in Erk1/2 pathway activation and both clinical and metabolic tumor responses in patients with OCSCC (19).
In BRAF-mutant melanoma, therapies targeting the MAPK pathway (Raf and MEK inhibitors) are transiently effective (13,14), with eventual development of resistance. Numerous studies have described multiple resistance mechanisms to MAPK inhibition in Ras/Raf mutant cancers: reactivation of the MAPK pathway leading to sustained Erk activation in the presence of BRAF inhibition (20), increased RTK-mediated signaling of alternative survival pathways such as the PI3K-Akt-mTOR pathway (21), increased activation of the Src/FAK/signal transducers and activators of transcription-3 (STAT3) signaling axis (22,23), and upregulation of the Hippo effector Yap1 (14). However, the mechanistic details of trametinib response and resistance have yet to be evaluated in HNSCC.
In the present study, we generated HNSCC models of acquired trametinib resistance. Importantly, we identified that in cell line and HNSCC patient-derived xenograft (PDX) models, increased Yap1 expression and activity contributed to trametinib resistance without reactivation of the MAPK pathway. In addition, genetic or pharmacologic inhibition of Yap1 re-sensitized resistant cells to trametinib. Combination therapy using trametinib and the Yap1 inhibitor verteporfin synergized to enhance treatment response in HNSCC resistant models. These studies support further clinical evaluation of combination MEK and Yap1 inhibitors in HNSCC therapy.
MATERIALS AND METHODS
Cell culture
The HSC3 (JCRB Cat# JCRB0623, Research Resource Identifier (RRID):CVCL_1288), CAL27 (ATCC Cat# CRL-2095, RRID:CVCL_1107) and Detroit562 (ATCC Cat# CCL-138, RRID:CVCL_1171) lines were authenticated by short tandem repeat (STR) analysis, and experiments using HSC3, CAL27 and Detroit562 cells were from the same stocks that were validated by STR analysis. HSC3 cells were grown in RPMI-1640 (Life Technologies). Detroit562 and CAL27 were grown in DMEM/F12 supplemented with glutaMAX (Life Technologies). HEK293T (NCBI_Iran Cat# C498, RRID:CVCL_0063) cells were grown in DMEM (Life Technologies). All cell lines were cultured in media supplemented with 10% heat-inactivated FBS (Hyclone) and penicillin/streptomycin (Lonza). All cell lines tested negative for mycoplasma.
Patient sample acquisition
The protocols for tumor sample acquisition and correlative studies were approved by the Washington University Human Research Protection Office and Animal Studies Committee. Samples were obtained, following informed patient consent, from either surgical biopsies (prior to trametinib treatment) or resections (following trametinib treatment) from patents enrolled in a neoadjuvant clinical trial with trametinib (NCT01553851; WU IRB:201205124) (19). All patients provided written, signed, informed consent to participate. This study followed ethical guidelines of the Declaration of Helsinki, Belmont Report and the US Common Rule.
Generation of trametinib-resistant HNSCC models
Cell lines:
The HSC3 and CAL27 trametinib-resistant (HSC3-TR, CAL27-TR) lines were generated by culturing HSC3 and CAL27 parental lines with sequentially increasing concentrations of trametinib (Selleckchem) up to 1 µM trametinib. Simultaneously, HSC3 and CAL27 parental cells were cultured in media containing DMSO (0.01%, Sigma Aldrich) to generate control lines (HSC3-C and CAL27-C).
PDXs:
Tumor biopsies from an HNSCC trametinib neoadjuvant Phase II clinical trial (NCT01553851) were obtained under institutionally approved protocol (WU IRB 201205124). Primary xenografts were generated as previously described (27). Briefly, patient tumor biopsies were collected in sterile DMEM containing 10% Fetal Calf Serum and 1% amphotericin. Biopsies were sectioned into multiple pieces with a portion used specifically for xenograft generation and another for next generation sequencing analysis.
PDXs were generated as described, and tumor pieces from PDX2233 (P0 passage) (19,24) were engrafted to a 5-week-old female NSG mouse (Jackson Laboratory, Bar Harbor, ME). This PDX was further passaged into ten NSG mice to generate PDX2233 P1. Once tumor volumes reached 500 mm3, trametinib (3 mg/kg) was administered to tumor-bearing NSG mice by daily oral gavage. Trametinib (Selleckchem, Houston, TX) was dissolved in DMSO (10 mg/mL) and diluted into an aqueous 100 μl dose containing 0.5% hypromellose (Sigma-Aldrich, St. Louis, MO) and 2% Tween-80 (Sigma-Aldrich, St. Louis, MO). Vehicle treated mice received this same carrier solution (100 μl) administered daily by oral gavage in order to generate comparable control PDXs (PDX2233-C). Daily treatment began 7 days after implantation and continued for 150 days when tumor re-growth was typically observed. Tumor dimensions were measured twice weekly. All animal studies were performed under protocols approved by The Institutional Animal Care and Use Committees (IACUCs) of Washington University and the Dana-Farber Cancer Institute.
Immunoblots
Cells (500,000) were plated in 6-well plates 24 hours before drug treatment. Cells were collected in lysis buffer (RIPA buffer, Cell Signaling Technologies, Beverly, MA) supplemented with protease inhibitors (Sigma Aldrich, St. Louis, MO) and phosphatase inhibitor (EMD Millipore, Burlington, MA), and lysates were clarified by centrifugation. PDX tissues were pestle homogenized in lysis buffer and then clarified. Equal amounts of protein were separated by SDS-PAGE (4-12 %) and transferred onto nitrocellulose membranes (Life Technologies, Carlsbad, CA) for immunoblot analysis. Membranes were blocked with TBS blocking buffer (LI-COR) for an hour, then incubated with primary antibody overnight at 4o C, washed and incubated with secondary antibodies (LI-COR). Membranes were developed using a fluorescence system (LI-COR), and bands were quantified using LI-COR Image Studio Software. Primary antibody information is listed in Supplementary Material and Methods.
CellTiter-Glo assay
HNSCC cells were plated in 96-well cell culture plates (Corning) at 1,500 cells/well and incubated for 24 hours prior to treatment with varying doses of trametinib alone, verteporfin (Sigma Aldrich) alone, or combination for 96 hours. As a surrogate for cell viability, ATP concentration was assessed by CellTiter-Glo® Luminescent Cell Viability Assay (Promega, Madison, WI) according to the manufacturer’s instructions. Luminescent signal was measured using a TECAN plate reader according to the manufacturer’s protocol. Cell metabolic data were fitted to variable slope normalized dose-response curves with media without cells representing maximum effect as has been reported elsewhere (25). IC50 values were calculated using GraphPad Prism (GraphPad Software). All experiments were performed at least three times with three technical replicates.
Combination therapy experiments were performed using a series of constant-ratio drug combinations with several concentration points above and below the IC50 values. The combination index (CI) was determined by the Chou-Talalay method using Calcusyn (Biosoft) software as previously described (26,27). CI values were (26,28) for synergy, additivity or antagonism of the drug combinations tested (27). All experiments were performed at least three times with at least 2 technical replicates.
Cell viability assay
Cells were plated 1500 cells/well into 96-well culture plates and incubated for 24 hours prior to treatment. Varying doses of trametinib were then added, and treatment continued for 96 hours. Cell toxicity was assayed using CellTox Green™ (Promega G8472) according to the manufacturer’s instructions for an endpoint method. Fluorescence was measured using a TECAN plate reader. Dose-response cell viability was analyzed using a variable slope (GraphPad Software). All experiments were performed at least two times with three technical replicates.
Cell proliferation assay
Prior to plating, a 96-well xCelligence plate with 50 μL of media per well was placed in the xCelligence RTCA SP instrument for 30 minutes to obtain a background reading. HNSCC cells were then added at 5,000 cells/well. The cell index was measured at 15-minute intervals for more than 120 hours to obtain a complete growth curve. Doubling times were calculated using the xCelligence RTCA software beginning 24 hours after plating and ending 10 hours before the plateau of the cell index. All experiments were performed at least 3 times with a minimum of three technical replicates.
RNA isolation, cDNA synthesis and quantitative real-time RT-PCR
Total RNA was extracted from cells using the RNeasy Mini Kit (Qiagen, Germantown, MD). The cDNA was transcribed from total RNA using High Capacity RNA-to-cDNA Kit (Applied Biosystems), per manufacturer’s protocol. The cDNA was used as a template for real-time PCR detection using TaqMan Fast Advanced Master Mix on an Applied Biosystems Step One Plus (Applied Biosystems). Expression of human YAP1, ANKRD1, NPPB, CTGF genes and endogenous control gene GAPDH were detected using Taqman probesets (Life Technologies). Relative target gene expression levels normalized to GAPDH were determined by ΔΔC(t) method (29).
Luciferase reporter assay
The 8 x GTIIC-luciferase plasmid, a synthetic TEAD luciferase reporter plasmid (RRID:Addgene_34615), and Renilla luciferase pRL-TK (RRID:Addgene_11313) were co-transfected into cell lines in a 6-well plate format. Plasmids were transfected using FuGENE HD Transfection Reagent (Promega) according to the manufacturer’s protocol. After 48-72 hours, transfected cells were harvested, and lysates were analyzed using the Dual-Luciferase Reporter Assay Kit (Promega).
Lentiviral infection
pLenti-CRISPRv2 containing sgNT (non-targeting guide RNA), sgYAP1 #1-;5 pLKO.1 containing scramble-shRNA control, shYAP1 #1-5 pLKO.1 containing YAP1 targeting sequences (see Supplementary Table 1), pLX304-empty vector (RRID:Addgene_25890), pLX304-human YAP1-WT, and pLX304-human YAP1-5SA were generously provided by Dr. David Barbie (Dana-Farber Cancer Institute). HEK293T cells were transfected with 1 μg lentivirus-based expression vectors and packaging plasmids using X-tremeGENE 9 transfection reagent (Roche) according to the manufacturer’s instructions. After 48-72 hrs incubation, the media containing lentivirus particles was collected, passed through a 0.45μm filter and added to the target cells in the presence of 8 μg/mL polybrene. For selection of virally infected cells, 1-2 μg/ml of puromycin (pLKO.1 (RRID:Addgene_8453) and pLenti-CRISPRv2 guide-puro vectors) or 5 μg/ml of blasticidin (pLX304 vectors) was used 48 hours post transfection. Infected cell lines were selected for one week before being used for further experiments and were maintained in selection media.
In vivo treatment efficacy studies
HSC3-C and HSC3-TR cells (2 x 106 in PBS) were injected subcutaneously into the flanks of 6-8 week old NSG mice. For PDX treatment studies, equal-sized pieces from 2233-TR sgNT and 2233-TR sgYAP1 #2 PDXs were implanted subcutaneously into NSG mice. Once tumors reached 100 mm3, trametinib was administered to tumor-bearing NSG mice by daily oral gavage. DMSO in hypromellose (0.5%) and 2% Tween-80 (250 uL) was used as vehicle solution. Digital caliper measurements of the tumors were taken twice weekly, and tumor volume (0.5 × length × width2) was calculated. These in vivo studies were performed using protocols approved by the Dana-Farber Cancer Institute IACUC.
Sequencing and data analysis
Nucleic acid extraction was performed by the Siteman Cancer Center Tissue Processing Core and sequencing was performed at the McDonnell Genome Institute at Washington University School of Medicine (St. Louis, MO), as previously described (24).
Whole exome sequencing
Genomic DNA was isolated from either tumor tissue, xenograft samples, or blood samples using the DNeasy Blood and Tissue Kit (Qiagen, Venlo, Netherlands), and fragmented using the Covaris E210 DNA Sonicator (Covaris, WoBurn, MA). Dual-indexed whole exome sequencing (WES) libraries were constructed according to the manufacturer’s instructions using the Kapa Auto Illumina (Kapa Biosystems, Woburn, MA) library preparation kit and captured using the NimbleGen SeqCap EZ Human Exome Library v3.0 Kit (Roche NimbleGen, Madison, WI). WES libraries were pooled and sequenced on the Illumina HiSeq 4000 platform (2×150bp reads). Sequencing data derived from xenograft samples was filtered in silico to remove reads that aligned to the mouse reference genome (mm10) by Xenome (30). The Genome Modeling System (GMS) was used for all analysis, including reference genome alignment and somatic variant detection, as previously described (24,31). We designed a custom capture reagent (NimbleGen SeqCap EZ Choice), targeting all SNVs and Indels detected by WES in tumors and xenografts. Validation capture sequencing was performed on variants in all WES libraries (20 normal blood, 20 baseline tumor biopsies, all xenograft samples); custom capture libraries were pooled and sequenced on the Illumina Novaseq platform (S4 flow cell). Data visualization was performed in R v3.3.2 using the ggplot2 R package (v2.2.1) (32) and GenVisR v1.8.0 (33).
RNA sequencing
Total RNA was isolated from tumor and xenograft samples using the Qiagen RNeasy kits, and single-indexed RNA sequencing (RNAseq) libraries were prepared using the Illumina TruSeq Stranded Total RNA kit with 500 ng of starting material according to the manufacturer’s recommendations as previously described (24) and sequenced on the Illumina HiSeq 4000 platform (2×150bp reads). RNAseq reads derived from xenograft samples were aligned competitively against the human reference genome (NCBI build 38, GRCh38) and the mouse reference genome (Genome Reference Consortium Mouse Build 38, mm10) using Xenome (v1.0.0) to filter out murine-specific reads (30). Gene expression levels were quantified using Cufflinks v2.1.1 (RRID:SCR_014597)(34) and HTSeq-count v0.5.4p1(RRID:SCR_011867) (35). Differential expression analysis and statistics were performed on gene expression raw counts (from HTSeq) using the DESeq2 R package (RRID:SCR_000154)(36). Subsequent gene set enrichment analysis (GSEA) and statistical analysis was performed on the log2 fold change (from DESeq2) in gene expression using the fgsea R package (37) across the Hallmark, KEGG (RRID:SCR_012773), and Reactome gene sets (38–40).
Data availability:
Genomic data have been deposited with dbGaP under study accession phs001623.
Statistics
Statistical analyses were performed as indicated in the figure legends as either an unpaired t-test or a one-way ANOVA with Dunnet’s multiple comparisons test. Asterisks were used to denote significance as follows: p<0.05 (*); p<0.01 (**); p<0.001 (***); p<0.0001 (****). Variable slope dose-response curves were used to fit cell metabolic activity data and to determine IC50 values. All calculations were carried out using GraphPad Prism (RRID:SCR_002798) software. Details of the statistics and number of experimental replicates can be found in figure legends.
RESULTS
Generation of trametinib resistant HNSCC models
To delineate mechanisms of HNSCC trametinib response and resistance, we assessed the trametinib sensitivity of seven HNSCC lines (Figure 1A). Out of the seven HNSCC lines tested, HSC3, CAL27, and SCC25 were the most sensitive to trametinib, having IC50s less than 30 nM. Additionally, we determined that 100 nM trametinib treatment suppressed Erk1/2 phosphorylation in all seven HNSCC cell lines (Figure 1B), while 10 nM trametinib treatment was insufficient to suppress Erk1/2 phosphorylation in HSC3, CAL27 and SCC25 dose response experiments (Figure 1C). HSC3 and SCC25 do not harbor canonical RAS/BRAF activating mutations while CAL27 harbors non-hot spot NRAS D92N and R68T mutations (41). To generate acquired trametinib-resistant cell line models as tools to define mechanisms of resistance, HSC3, CAL27, and SCC25 were subjected to prolonged trametinib treatment. Progressively increasing doses of trametinib (increased every 2-3 weeks) were applied to these 3 lines over a period of 6-12 months until they displayed little to no sensitivity to the drug. Resistance to trametinib developed in HSC3 (HSC3-TR) after 6 months and CAL27 (CAL27-TR) after 12 months, and their response to trametinib was compared to their respective control cells (HSC3-C and CAL27-C) (Figure 1D). Note that SCC25 did not develop resistance despite 12 months of exposure (not shown). HSC3 displayed a baseline IC50 of 0.027 nM which increased to 9.06 μM, whereas CAL27 had a baseline IC50 of 0.023 nM which increased to 2.29 μM. Cell growth doubling times did not differ between HSC3-C and HSC3-TR nor between CAL27-C and CAL27-TR (Figure 1E). Thus, 2 of 3 lines displayed acquired resistance to trametinib.
Figure 1. Development of acquired trametinib resistant HNSCC models.

(A) Seven HNSCC lines were treated with various concentrations of trametinib for 96 hours. Cell Titer Glo cell metabolic assay IC50 and maximal growth inhibition results are shown. Data shown are representative of at least three independent experiments. (B) Immunoblot analysis of YAP1, p-Erk1/2 and total Erk levels in seven HNSCC cell lines treated with vehicle control or 100 nM trametinib for 24 hours (Upper panel). Fold-change in the ratio of p-Erk1/2 to total Erk with trametinib was quantified and normalized for each cell line to the ratio of p-Erk1/2 to total Erk with vehicle control treatment. Graphed data were obtained from the quantification of three independent immunoblots. Normalized p-Erk1/2 level following trametinib treatment was compared to vehicle treatment level for each cell line (n=3, **** p<0.0001, ***<0.0005, and ** p<0.01, unpaired t-test) (C) Immunoblot analysis of CAL27, HSC3 and SCC25 parental for p-Erk1/2 and total Erk1/2 with increasing trametinib concentrations. The figure is representative of three independent experiments. (D) Cell ATP concentration, measured by Cell Titer Glo, IC50 and maximal inhibition for HSC3-C and HSC3-TR (left panel) and CAL27-C, and CAL-27 TR (right panel) (n=3). Cell lines were treated with increasing concentrations of drug for 96 hours. Treatment with increasing concentration of trametinib over time led to the emergence of trametinib-resistant HSC3 (HSC3-TR) and CAL27 (CAL27-TR) with indicated IC50s. (E) Cell doubling times did not differ between HSC3-C and HSC3-TR nor between CAL27-C and CAL27-TR.
Acquired trametinib-resistant HNSCC model reveals upregulation of Yap1 expression and activity
Multiple mechanisms of acquired resistance to MAPK inhibition have been reported in cancers harboring RAS/RAF mutations. These include reactivation of the MAPK pathway (20), activation of parallel signaling pathways such as the PI3K-AKT-mTOR pathway (21), increased activation of the Src/FAK/STAT3 signaling axis (22,23), and upregulation of the Hippo effector Yap1 (13,14). Based on these findings, we used a candidate approach to ask whether any of these alterations were present in the models we developed. We evaluated the activity of the MAPK, PI3K-AKT-mTOR and STAT3 signaling pathways and expression of Yap1 by immunoblot. We noted unabated activation of the MAPK pathway in the presence of trametinib only in CAL27-TR cells, as p-Erk1/2 levels were sustained in this line following trametinib treatment (Figure 2A). As previously reported (23), we observed increased STAT3 phosphorylation following MEK inhibition in CAL27-C and HSC3-C (Figure 2A). This increase in p-STAT3 was more modest in trametinib-resistant model cell line HSC3-TR compared to HSC3-C, but this difference was not statistically significant (Figure 2A). Phospho-AKT (p-AKT) expression was reduced in CAL27-C but sustained in CAL27-TR following trametinib treatment, while increased in HSC3-C and generally unchanged in HSC3-TR lines following trametinib treatment. (Figure 2A). Despite sustained p-AKT levels in CAL27-TR compared to CAL27-C, we did not observe elevated activity of the downstream effectors of the AKT pathway such as phosphorylated ribosomal protein S6 kinase (p-S6) (42), and phospho 4E-BP1 (p-4E-BP1) (43) in CAL27-TR as compared to CAL27-C (Figure S1).
Figure 2. Acquired trametinib-resistant HSC3 displays enriched YAP1 expression and activity.
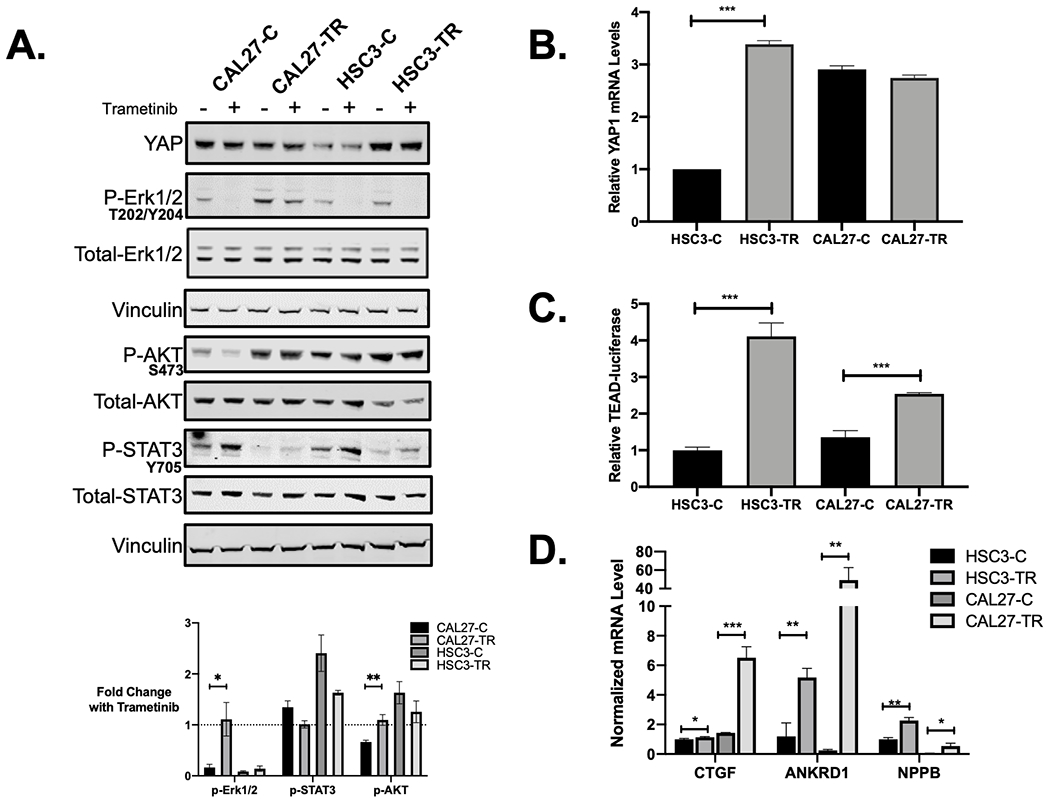
(A) Lysates from CAL27-C, CAL27-TR, HSC3-C and HSC3-TR treated with either trametinib (100nM) or DMSO for 24 hours were resolved by SDS-PAGE and immunoblotted for YAP1, p-Erk1/2, total Erk1/2, p-AKT, total AKT, p-STAT3 and total STAT3. Vinculin was used as a loading control. Ratios of phospho- to total proteins were quantified, and ratios following trametinib treatment were normalized to the respective cell line treated with vehicle control (lower panel). Quantifications were from at least 3 independent immunoblots. Fold change in the normalized phosphoprotein levels with trametinib treatment were compared between CAL27-C and CAL27-TR and between HSC3-C and HSC3-TR (** p<0.01 and * p<0.05, unpaired t-test). (B) Fold change in YAP1 mRNA in HSC3-TR was compared to its control line counterpart (**** p<0.0001, unpaired t-test) and in CAL27-TR compared to its control line counterpart (not statistically significant (ns)) with human GAPDH as control. (C) Fold change in the TEAD luciferase reporter expression in HSC3-TR was compared to its control line HSC3-C. TEAD luciferase reporter expression was also quantified in CAL27-C and CAL27-TR and compared to HSC3-C. HSC3-C, HSC3-TR, CAL27-C and CAL27-TR lines were transfected with 8X GTIIC TEAD luciferase reporter, in which eight TEAD-YAP binding sites can drive the promoter with expression of firefly luciferase and pRL-TK renilla luciferase as control. Reporter luciferase activity was normalized to Renilla luciferase (*** p<0.001, unpaired t-test). (D) qPCR analysis of YAP-target gene expression in HSC3-C, HSC3-TR, CAL27-C and CAL27-TR (n=3, **** p<0.0001, ***<0.0005, ** p<0.01 and * p<0.05, unpaired t-test). Figures (A), (B), (C), and (D) are representative of at least three independent experiments.
HSC3-TR did not show increased p-STAT3, reactivation of the MAPK pathway, nor increased activity of the PI3K-AKT-mTOR pathways compared to HSC3-C (Figure 2A, Supplementary Figure 1). Conversely, HSC3-TR showed a distinct pattern where we observed increased Yap1 protein expression in HSC3-TR (Figure 2A) as compared to parental HSC-3. This increased Yap1 protein expression occurred with a 3-fold increase in YAP1 mRNA expression in HSC3-TR as compared to HSC3-C (Figure 2B). We next asked whether elevated YAP1 expression was associated with increased functional activity using a TEAD-Yap1 luciferase-based gene expression reporter assay (44). Consistent with increased protein expression, HSC3-TR cells showed enhanced Yap1 activity compared to the control HSC3 (Figure 2C, p<0.0001). Furthermore, we found that transcripts of Yap1 target genes, including CTGF, ANKRD1 and NPPB (45) were increased in HSC3-TR (Figure 2D). CAL27-TR, which did exhibit sustained MAPK signaling in the presence of trametinib, did not have increased Yap1 protein nor mRNA expression compared to CAL27-C (Figures 2A and B). However, TEAD luciferase activity and expression of Yap1 target genes CTGF, ANKRD1 and NPPB were significantly increased in CAL27-TR compared to CAL27-C (Figure 2C and 2D), suggesting the possibility of Yap1 activation in CAL27-TR. We decided to pursue the significance of increased YAP pathway activation in mediating acquired resistance to trametinib in these models.
Pharmacological inhibition of Yap1 is synergistic with trametinib in acquired and intrinsic trametinib resistant HNSCC models.
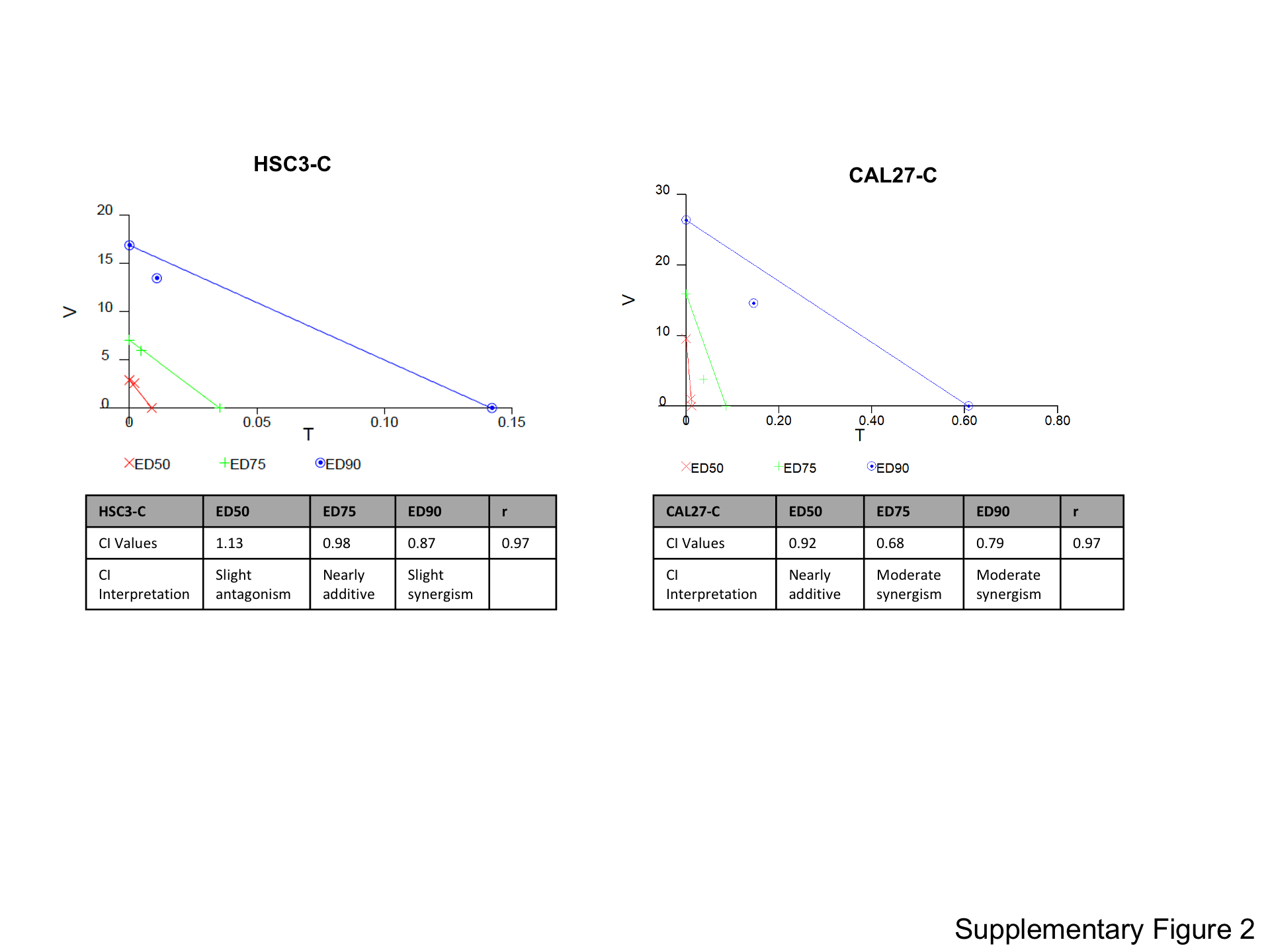
Because Yap1 was implicated in trametinib resistance, we sought to define whether treatment with the Yap inhibitor verteporfin in combination with MEK targeting could bypass trametinib resistance in the HSC3-TR and CAL27-TR HNSCC models of acquired resistance. We determined the verteporfin IC50 in HSC3-TR and CAL27-TR to be 4.93 μM and 6.54 μM, respectively. We then assessed trametinib and verteporfin synergy using the Chou-Talalay method (26). The resulting isobolograms and CIs (Figure 3A and 3B) provided evidence of synergy between trametinib and verteporfin in these two HNSCC models of acquired trametinib resistance. We also assessed verteporfin and trametinib synergy in HSC3-C and CAL27-C using the same methodology (Figure 2). Verteporfin IC50s were determined to be 5.25 μM and 4.41 μM for HSC3-C and CAL27-C, respectively. The ED50 and ED75 CIs for HSC3-C were 1.13 and 0.98, indicating slight antagonism and nearly additive effects, respectively. This was in contrast to the ED50 and ED75 CIs for HSC3-TR, which were 0.87 and 0.68, indicating slight synergism and synergism, respectively. These analyses support the acquisition of greater synergy of targeting Yap and MEK in HSC3-TR compared to HSC3-C. CAL27-TR and CAL27-C had more similar trametinib-verteporfin synergies, as both lines displayed moderate synergism at ED75 and ED90, while the ED50 CIs indicated verteporfin and trametinib had slight synergism in CAL27-TR and were nearly additive in CAL27-C. We attempted to extend these studies to the in vivo setting for testing trametinib resistance with Yap1 co-targeting by engrafting the HSC3-TR cells into NSG mice. However, resistance to trametinib was lost upon engraftment, with tumors displaying drug re-sensitization, negating our ability to assess in vivo combination effects (data not shown).
Figure 3. Trametinib and verteporfin have synergistic treatment response in models of acquired and intrinsic trametinib resistance.
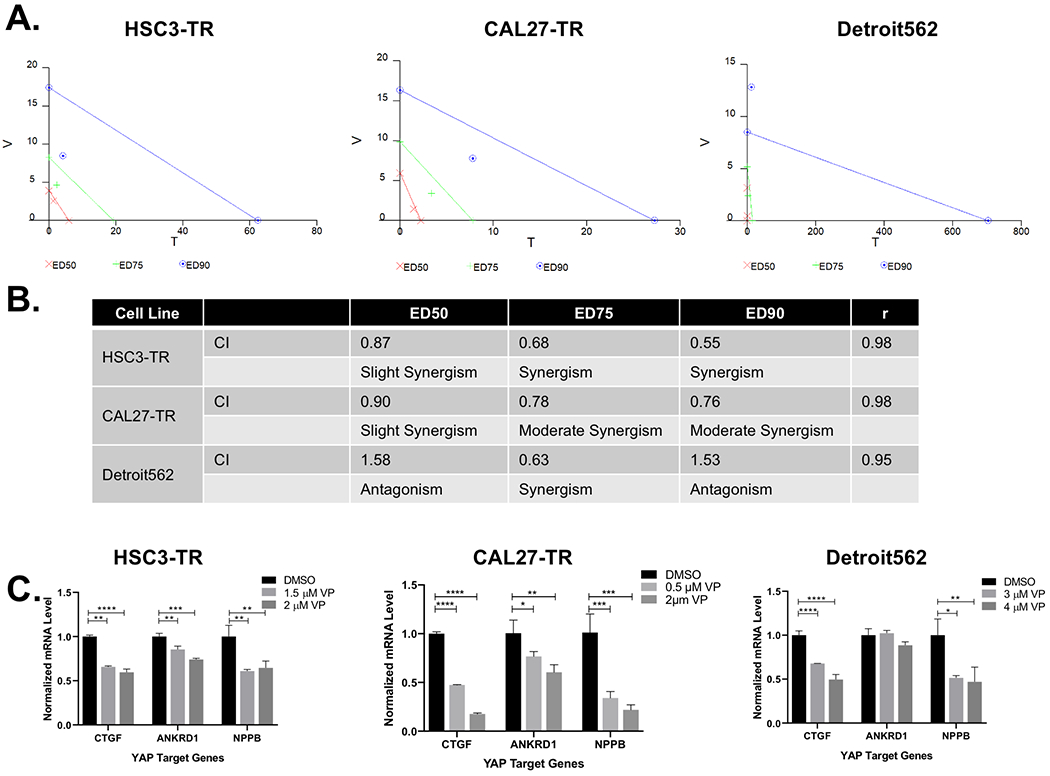
(A) Isobolograms from verteporfin (V) and trametinib (T) treatment combinations of HSC3-TR (left panel), CAL27-TR (middle panel) and Detroit562 (right panel) from Calcusyn analysis of Cell Titer Glo data. Cells were treated for 96 hours with vehicle, verteporfin alone, trametinib alone or combinations of the two drugs at a fixed ratio for each cell line. (B) The Combination index (CI) was determined using Calcusyn software, which employs the Chou-Talalay method for assessing synergy, additivity or antagonism. (C) Inhibition of YAP1 target gene expression in HSC3-TR (left panel), CAL27-TR (middle panel) and Detroit562 (right panel) treated with DMSO as control or increasing concentration of verteporfin (VP) for 24 hours. Data were normalized to DMSO (**** p≤0.0001, ** p<0.005 One-way ANOVA with Dunnett’s multiple comparisons test). Figure (A), (B) and (C) are representative of at least two independent experiments.
Confirming Yap1 targeting with verteporfin treatment in vitro, we noted a significant reduction in CTGF, ANKRD1 and NPPB mRNAs upon verteporfin treatment in HSC3-TR and CAL27-TR (Figure 3C) . These data confirmed appropriate “on target” activity of verteporfin in these lines.
Subsequently, we sought to determine whether Yap1 contributed to intrinsic trametinib resistance in distinct HNSCC cell lines. Yap1 expression levels were evaluated using a panel of 12 HNSCC cell lines, including HSC3-C, HSC3-TR and CAL27. Detroit562 had the highest level of Yap1 protein (Supplementary Figure S3A) and displayed reduced sensitivity to trametinib (IC50=1.89 μM), compared to 3 selected HNSCC cell lines, SNU899, HSC2 and BICR56 (Supplementary Figure S3B). Of note, 100 nM trametinib treatment inhibited Erk1/2 phosphorylation in Detroit562 (Supplementary Figure S3C). Trametinib and verteporfin IC50s in Detroit562 cells were determined to be 1.89 μM and 5.16 μM, respectively. Performing a series of constant-ratio drug combinations with several concentration points above and below the IC50, we determined the CIs for verteporfin and trametinib. Only the ED75 CI demonstrated synergy in Detroit562 (Figure 3A, right panel and 3B). The CIs at ED50 and ED90 indicated antagonism, suggesting that the synergism in Detroit562 occurred within a more narrow dosing range than observed with HSC3-TR and CAL27-TR. We confirmed that verteporfin treatment of Detroit562 resulted in reduced expression of the Yap target genes CTGF and NPPB, while expression of ANKRD1 was not significantly reduced following verteporfin treatment at the doses evaluated (Figure 3C). Together, our results suggest that pharmacological Yap1 targeting can counteract intrinsic and acquired trametinib resistance in these HNSCC models.
Yap1 mediates trametinib resistance in HNSCC models
As verteporfin has targets in addition to Yap1, we next asked whether Yap1 was specifically necessary for mediating trametinib resistance in these models. We employed multiple approaches to deplete Yap1 expression. First, we used CRISPR/Cas9 to knockout YAP1 in HSC3-TR using five distinct YAP1 sgRNAs or a nontargeting sgRNA (sgNT). All 5 YAP1 sgRNAs were found to attenuate Yap1 levels. sgYAP1 #2 and sgYAP1 #4 resulted in the most effective Yap1 reduction and were, thus, selected for subsequent functional studies (Figure 4A). Using these Yap1-deficient lines, we assessed Yap1 functional activity using the TEAD-Yap1 reporter and observed decreased activity in HSC3-TR-sgYAP1 #2 and HSC3-TR-sgYAP1 #4 cell lines as compared with the control line HSC3-TR-sgNT (Figure 4B, p<0.0001 and p<0.0005, respectively). Expression of Yap1 target genes ANKRD1 and NPPB in HSC3-TR Yap1-depleted cell lines was significantly diminished (Figure 4C, p<0.0001). These data confirmed effective knockdown of Yap1 expression and activity in the HSC3-TR line.
Figure 4. YAP is necessary for trametinib resistance in HSC3-TR and Detroit562 HNSCC cell lines.
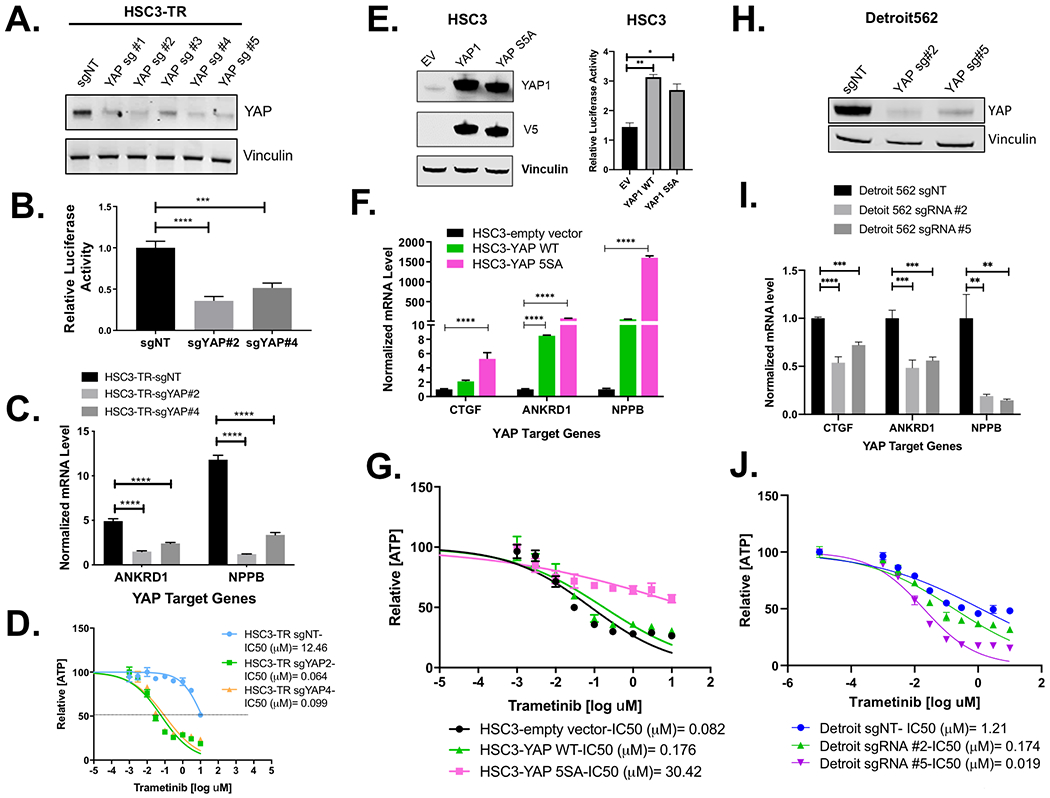
(A)Western blot showing decrease in YAP1 expression following gene silencing by CRISPR/Cas9 in HSC3-TR. (B, C) Reduced YAP1 activity in CRISPR/Cas9-mediated YAP1 knock-out HSC3-TR cells. (B) Expression of TEAD luciferase reporter in HSC-TR transduced with the indicated guide RNA. HSC3-TR cells were transfected with 8X GTIIC TEAD luciferase reporter and pRL-TK renilla luciferase. Renilla luciferase was used as an internal control. Data was normalized to HSC3-TR-sgNT (**** p<0.0001, *** p<0.0005, One-way ANOVA with Dunnett’s multiple comparisons test) for (B). (C) qPCR analysis of YAP-target gene expression in HSC3-TR infected with the indicated guide RNA. Data was normalized to HSC3-TR-sgYAP#2, the cell line that showed the least values for both genes (**** p<0.0001, One-way ANOVA with Dunnett’s multiple comparisons test). (D) HSC3-TR-sgNT, HSC3-TR-sgYAP#2 and HSC3-TR-sgYAP#4 cells were treated with various concentrations of trametinib for 96 hours. IC50 and maximal inhibition results are shown. (E), (F), and (G) Enhanced YAP1 activity promotes trametinib resistance in HSC3. Increased YAP1 expression (E, left panel), TEAD activity (E, right panel) and YAP1 downstream target expression (F) in HSC3 cells overexpressing wild-type (YAP WT) or constitutively active YAP1 (YAP 5SA) compared to empty vector (EV). (G) HSC3-YAP WT, HSC3-YAP 5SA and HSC3-EV were treated with various concentrations of trametinib for 96 hours, and IC50 estimates are shown (n=3). (H) Western blot showing decrease in YAP1 expression following gene editing by CRISPR/Cas9 in the Detroit562 line. (I) Reduction in YAP target genes in CRISPR mediated YAP1 deleted Detroit562 line. Data was normalized to Detroit562-sgNT (*** p<0.001, ** p<0.005, unpaired t-test). (J) Detroit562-sgNT, Detroit562 sgYAP#2 and Detroit sgYAP#5 cells were treated with various concentrations of trametinib for 96 hours. IC50 and maximal inhibition results are shown. All data in Figure 4 are representative of at least three independent experiments.
We next assessed whether YAP1 knock-out restored trametinib sensitivity in HSC3-TR. CRISPR/Cas9 YAP1 knockout (Figure 4D) resulted in increased sensitivity to trametinib, with sgYAP1 #2 showing an IC50=0.064 μM and sgYAP1 #4 IC50=0.099 μM compared to an sgNT IC50=12.46 μM in HSC3-TR cells. Doubling times in the YAP1 knockdown lines did not differ from the NT control (Supplementary Figure S4A). Knockdown of YAP with sgYAP#1, sgYAP#3 and sgYAP#5 also resulted in increased HSC3-TR sensitivity to trametinib, though not to the same extent as sgYAP#2 and sgYAP#4 (Supplementary Figure S4B). In parallel, we also utilized shRNAs to knock down Yap1 in HSC3-TR cells and determined that YAP1 knockdown by shRNA increased sensitivity to trametinib, although to a lesser degree (Supplementary Figure S4C and S4D). Conversely, we tested whether enforced expression of wild type (YAP1 WT) or constitutively active YAP (YAP1 5SA) were sufficient to increase trametinib resistance in trametinib-sensitive HNSCC lines (Figure 4E, F, G). Interestingly, we found that although Yap1 WT and 5SA were similarly overexpressed and similarly enhanced TEAD activity (Figure 4E), only stable expression of constitutively active Yap 5SA substantially increased expression of all three Yap target genes and decreased sensitivity to trametinib in HSC3 cells, resulting in an IC50=30.42 μM as compared to HSC3 infected with its empty vector control IC50=0.082 μM (Figure 4G). Stable overexpression of wild type Yap1 did not decrease trametinib sensitivity in HSC3 (IC50=0.18 μM) (Figure 4G). Together, these data confirm the contribution of Yap1 activity to trametinib sensitivity in HSC3 cells.
Similarly, we evaluated whether YAP1 knock-out restored trametinib sensitivity in CAL27-TR cells (Supplementary Figure S5). YAP1 knock-out with sgYAP1 #1, sgYAP1 #2 and sgYAP1 #5 all yielded reduced Yap1 protein by immunoblot (Supplementary Figure S5A). However, trametinib sensitivity in the CAL27-TR YAP1 knock-downs did not differ from the parental line (Supplementary Figure S5B). CAL27-TR transduced with the NT sgRNA had substantially increased trametinib IC50 compared to the parental line, 19.48 μM and 2.95 μM, respectively, possibly reflecting off-target effects of the NT sgRNA in these cells. Consistent with YAP1 knock-down, we observed Yap1 downstream target genes had reduced expression in CAL27-TR with sgYAP #1, sgYAP1 #2 and sgYAP1 #5 compared to the parental CAL27-TR and CAL27TR-NT sgRNA controls (Supplementary Figure S5C). ANKRD1 expression remained 4-fold, 15-fold and 20-fold higher than CAL27-C expression levels in CAL27-TR expressing YAP1sg #2, YAP1sg #5 and YAP1sg #1, respectively. We did not test sgYAP#3 or sgYAP#4 in the CAL27 models.
To investigate whether Yap1 contributed to Detroit562 response to trametinib, we employed CRISPR/Cas9 to deplete YAP1 (Figure 4H). In Detroit562 sgYAP1 #2 and sgYAP#5 there was significant downregulation of Yap1-target genes as compared with the control Detroit 562-sgNT (Figure 4I) and a concomitant 6-fold and 60-fold reduction in IC50 for sgRNA #2 (IC50=0.25μM) and sgRNA #5 (IC50=0.019 μM), respectively, compared to the non-targeting control (IC50=1.211μM) (Figure 4J). Together, these data indicate that Yap1 modulates trametinib resistance in HSC3 and Detroit562 HNSCC models.
All studies of in vitro response to trametinib and/or verteporfin described above were performed using ATP concentration as a surrogate readout for cell viability. In order to directly assess cell cytotoxicity in response to drug treatment and/or Yap1 knockdown, we measured cytotoxicity directly in the HSC3-C and HSC3-TR models using Celltox green (Supplementary Figure 6). We observed trametinib treatment induced a dose-dependent cytotoxicity in HSC3-C cells (IC50= 2.13 μM), which was higher than the IC50 based on cellular ATP levels. HSC3-TR cells exhibited only modest cytotoxicity in response to high doses of trametinib (IC50=26.06 μM) (Supplementary Figure S6A). Neither verteporfin in combination with trametinib nor Yap1 knockdown in combination with trametinib induced cytotoxicity in HSC3-TR by these assays (Supplementary Figure S6B, S6C and S6D). These data indicate that although targeting Yap1 in combination with trametinib reduced cellular ATP concentration, this combined targeting did not induce cell death in this model of acquired trametinib resistance.
An HNSCC PDX with acquired trametinib resistance displays increased Yap1 expression
Having examined cell line model response and resistance, we next queried whether PDX models derived from patient tumors from our clinical trial retained sensitivity to trametinib. As previously reported, of 40 tumor biopsies (20 baseline; 20 post-treatment surgical specimens), 22 (55%) showed successful engraftment and displayed genomic fidelity with primary tumors (24). As our window trial was limited to only a 2-week period, we utilized PDX models from the baseline biopsy of two clinical trial patients for an extended treatment approach. We previously described one of these PDXs, which showed a partial response to trametinib, paralleling the clinical response observed in the patient (24). A second sample, PDX2233, was derived from a patient whose tumor displayed attenuation of Erk1/2 phosphorylation during the window phase (Supplementary Figure 7) and significant metabolic tumor response (19). Mice bearing PDX2233 treated with vehicle showed progressive tumor growth, whereas daily trametinib treatment resulted in near complete tumor regression. However, after extended daily trametinib treatment, resistant tumor outgrowth was observed (Figure 5A–B). Tumors were harvested and analyzed by immunoblotting, RNA-Seq, and whole exome sequencing (WES). When we interrogated for specific candidate pathways, 4/4 acquired trametinib-resistant PDX2233 (PDX2233-TR) escape PDXs showed increased Yap1 levels whereas 1/3 vehicle-treated, trametinib-sensitive tumors showed modest Yap1 expression (Figure 5C). Note that the PDX2233-TR tumor samples showed sustained p-Erk1/2 levels. To ask whether this sustained Erk1/2 phosphorylation could be due to in vivo trametinib levels and timing of tumor harvest (Figure 5C), we generated transient cultures from PDX2233-TR for sustained trametinib exposure. Following four hours of trametinib treatment in vitro, primary cell lines derived from PDX2233-TR displayed ablation of p-Erk1/2 activity (Supplementary Figure 8A and 8B). Similar to HSC3-TR, PDX2233-TR did not show increased PI3K-AKT or STAT3 signaling nor reactivation of the MAPK pathway activity (Figure 5C and D). Furthermore, we transiently cultured PDX2233-TR to knock-out YAP1 using CRISPR/Cas9 (Supplementary Figure 8C and 8D). YAP1 depletion in PDX2233-TR (PDX2233-TR-sgYAP #2) resulted in decreased in vivo growth compared to the control PDX2233-TR-sgNT (Supplementary Figure 8E). Therefore, we were unable to assess the impact of trametinib in these modified lines.
Figure 5. Increased YAP1 expression is associated with acquired resistance to trametinib in HNSCC patient-derived xenograft (PDX) models.
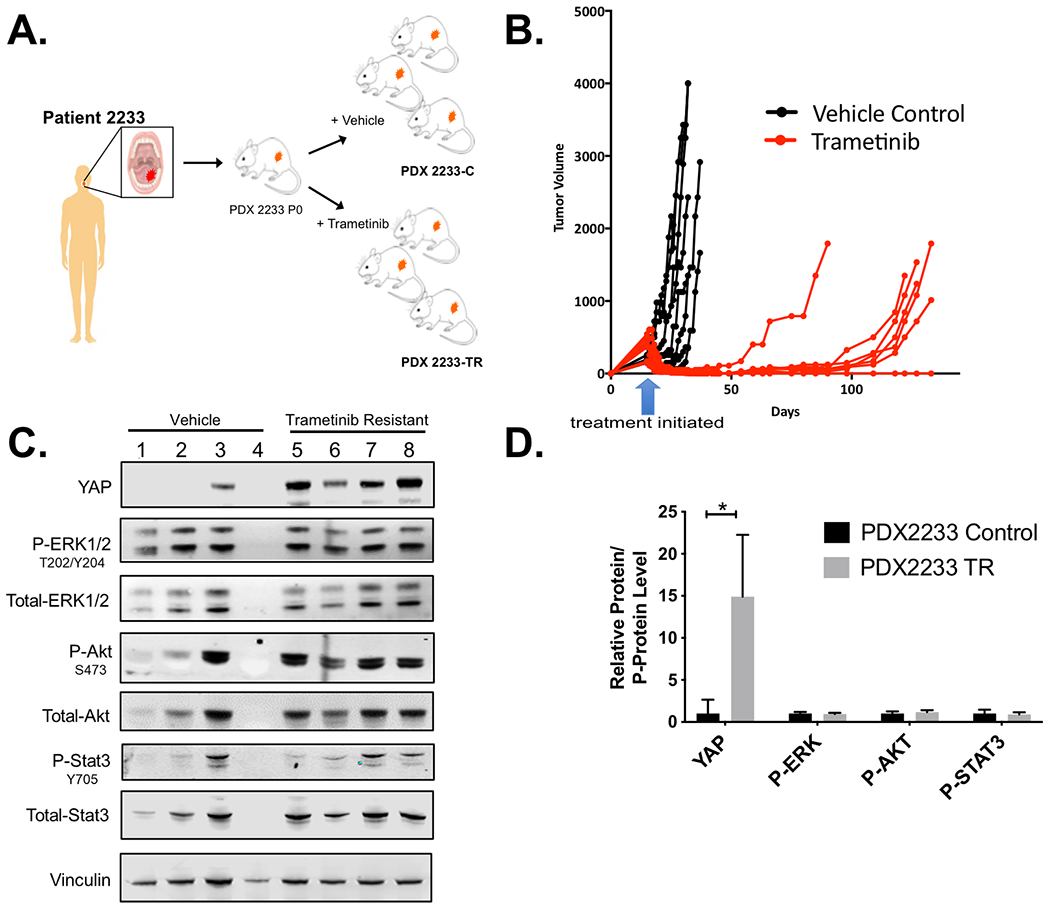
(A) Generation of trametinib resistant PDX2233. Passage 1 PDX2233 were treated with either HPMC vehicle as control or trametinib (3 mg/kg) by daily oral gavage. (B) Daily administration of trametinib led to the emergence of trametinib resistant PDX2233 (PDX2233-TR). (C) Lysates from four different PDX2233 vehicle tumors and four different PDX2233 trametinib resistant tumors were resolved by SDS-PAGE for protein expression analysis as in Figure 1A. Vinculin was used as a loading control. (D) Quantification of YAP1 protein levels (YAP), ratio of phospho-to-total Erk1/2 (p-Erk1/2), ratio of phospho-to-total AKT (p-AKT), and phospho-to-total STAT3 (p-STAT3) was performed for immunoblot lanes containing vehicle treated tumors #1, #2 and #3 and the four trametinib resistant tumors in C. Vehicle and trametinib resistant tumor lysates were quantified from two independent blots. A representative blot is provided (* p<0.05, unpaired t-test).
To explore potential mechanisms by which PDX2233-TR was no longer responding to trametinib, we completed whole exome sequencing (WES), high-depth custom-capture validation sequencing, and RNA sequencing (RNAseq) to explore both genomic and expression-based mechanisms driving this escape phenotype in vivo. There were 94 somatic single nucleotide variants (SNVs) and small insertions or deletions (indels) detected in PDX2233 (passage 0, PDX2233-P0) derived from this patient; 90 of these variants were also detected in the primary tumor (Supplementary Table 2). The 4 variants specific to PDX2233-P0 were present at 10.5-30.1% DNA VAF (missense mutations in ZNF326, FRG1, POU6F2, and MAGEC1). PDX2233-TR samples (n=3; passage 3) shared 92-93 variants with the P0 xenograft; PDX2233-C samples (n=3; passage 3) shared 91-92 variants with the P0 xenograft. There were 7-9 (median 7) new variants in PDX2233-C samples and 7-10 (median 9) new variants in PDX2233-TR samples. Three variants were shared by PDX2233-C and PDX2233-TR samples, and remaining variants were present at lower VAF, suggesting that these were acquired over passaging since PDX2233-P0 (Supplementary Table 2) and not associated with acquired resistance to trametinib. In addition, de novo variants in PDX2233-TR (passage 3) did not have a clear biological association with the YAP pathway. Collectively, these results suggest that trametinib resistance may not be driven by a genetic event.
Differentially expressed gene sets in PDX2233-TR samples, compared to PDX2233-C samples, were associated with general transcriptional regulation (REACTOME GENERIC TRANSCRIPTION PATHWAY, Wald test; p=0.001, FDR=0.077) or xenobiotic metabolism (REACTOME PHASE 1 FUNCTIONALIZATION OF COMPOUNDS; Wald test, p=0.001, FDR=0.077). PDX2233-C samples showed upregulation of cell cycle processes, compared to PDX2233-TR samples (Supplementary Table 3–4), consistent with the rapid growth observed in these xenografts. YAP1 was not differentially expressed between the PDX2233-TR and PDX2233-C xenografts, and neither were its downstream effectors CTGF and CYR61. However, its effector ANKRD1 was more highly expressed in the PDX2233-TR (Wald test, Log2FC=3.39, p=1.44e-5, FDR=0.003; Supplementary Table 3).
Genomic correlates from HNSCC trametinib window trial
We investigated whether HNSCC clinical trial patients displayed patterns of YAP1 activity with relation to trametinib treatment (19), including whether patients exhibit patterns of YAP1 pathway activity at baseline or following treatment that may suggest intrinsic or acquired resistance evolution to trametinib. WES was performed on baseline tumor biopsies (n=20) and RNAseq analysis on paired baseline tumor biopsies (n=16) and post-treatment surgical resections (n=15; 10 matched). Tumor DNA was sequenced with an average of 69X coverage, and matched normal DNA was sequenced with an average of 72X coverage across the targeted exome. SNVs and indels were detected by comparing tumor samples to matched normal blood; there were a total of 1,853 nonsilent SNVs and indels detected across the 20 primary tumor samples (average 101 variants per patient; Supplementary Table 5). SNVs and indels were validated by high-depth sequencing in all samples (average 544X coverage across targeted regions). The genomic landscape of our cohort was highly concordant with the genomic landscape of HPV-negative HNSCC, including recurrent mutations in TP53, CDKN2A, USH2A, NOTCH1, and CASP8 (16) (Figure 6). Known activating mutations were observed in PIK3CA (E545K, responder Patient GSK2899 and non-responder GSK4795) and HRAS (G13I, patient GSK1978, response not evaluable). There were no clear differences in the genomic profiles between responders and non-responders.
Figure 6. YAP pathway expression and genomic correlates in patient samples and translational models.
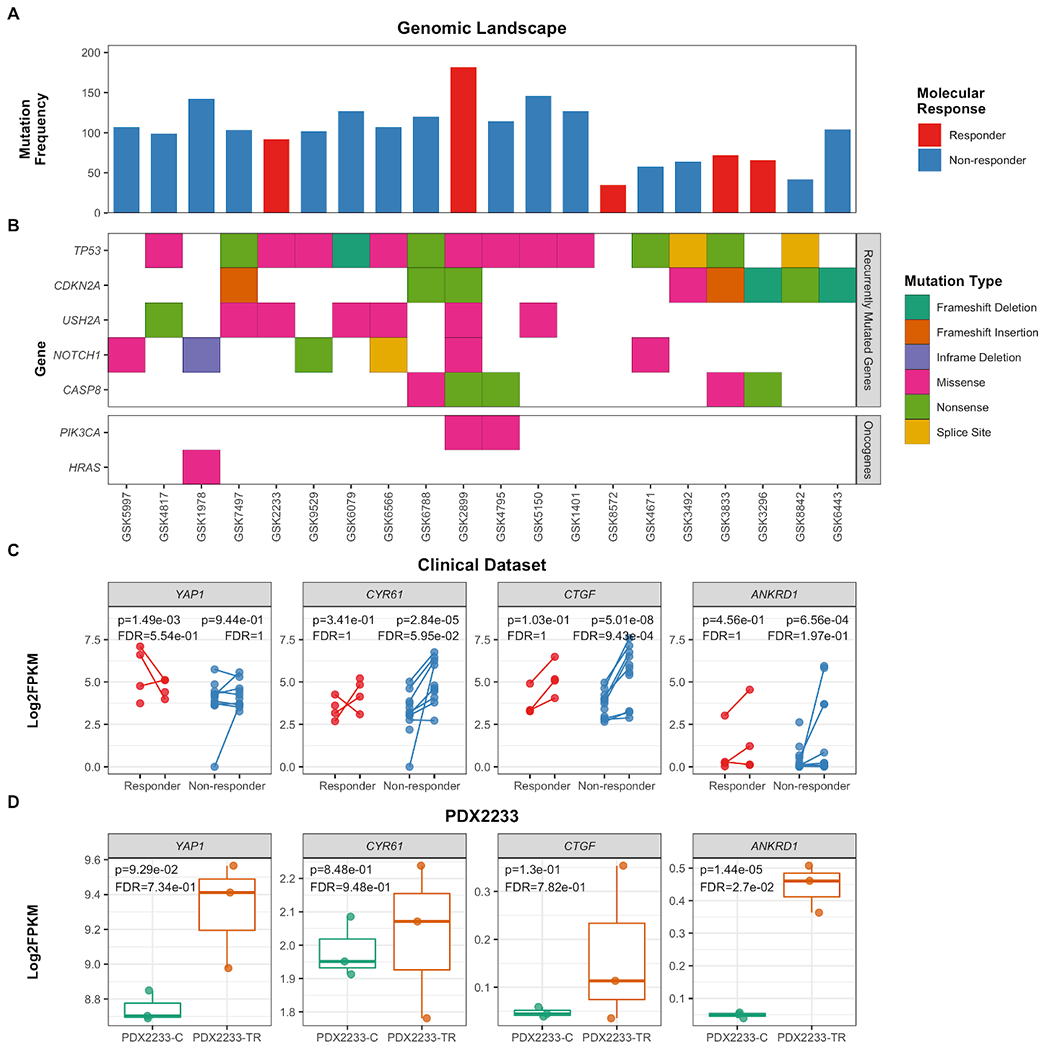
(A) Baseline (non-silent) mutation frequency for each patient, colored by molecular response. Responders are defined by those that exhibited a decrease in pErk staining following two weeks of daily trametinib treatment. (B) Waterfall depiction of recurrently mutated genes described in HNSCC and other relevant oncogenes. (C) Expression of YAP1 and its target genes in RNAseq data derived from baseline tumor biopsies (n=16) and post-treatment tumor resections (n=15). Paired samples are indicated by a connecting line, and samples are colored based upon molecular response. (D) Expression of YAP1 and its target genes in RNAseq data derived from PDX2233 models (PDX2233-C [n=3], PDX2233-TR [n=3]; indicated on the x-axis).. Statistical comparisons are indicated in each plot, indicating the Wald’s test p-value and Benamini-Hochberg-corrected FDR value.
YAP1 pathway activity in patient samples
Based upon our observations in preclinical models, we evaluated whether patient samples depicted patterns of either intrinsic or acquired resistance evolution to trametinib via the YAP1 pathway by evaluating the expression of YAP1 and its downstream targets. Interestingly, YAP1 was more highly expressed at baseline in trametinib responders (n=4), compared to non-responders (n=12; log2FC=1.89, p=1.73e-6, q=0.002; Supplementary Table 6); however, this significance was strongly driven by 2 of the 4 patients (Figure 6A–C). Yap targets CTGF, CYR61, and ANKRD1 were not differentially expressed at baseline between responders and non-responders. When pre- and post-treatment samples were compared, YAP1 expression was not significantly modulated in either responders or non-responders. All three Yap1 targets were significantly higher in non-responders post-treatment (p<0.05, q<0.20 for all genes), and responders showed trends of increased Yap1-target expression post-treatment (p=0.103-0.456; q=1). Together, these results suggest that this gene program may be increased due to trametinib treatment.
Patient GSK2233 and its corresponding PDX samples consistently exhibited high expression of YAP1, compared to the rest of the cohort. Only ANKRD1 was significantly upregulated in PDX2233-TR, compared to PDX2233-C (p=1.44e-05, FDR=0.0027; Figure 6E); CTGF and CYR61 were not differentially expressed between PDX2233-C and PDX2233-TR.
DISCUSSION
Although the RAS/MAPK/Erk pathway is infrequently mutated in HNSCCs, increased pathway activity in tumor cells highlight this pathway as a potential therapeutic target. To address validity of targeting of this pathway in patients, we completed a window of opportunity clinical trial (NCT01553851) with trametinib and a p-Erk1/2 biomarker endpoint (19). Interestingly, we identified several patients who not only showed biomarker responses but also metabolic and clinical activity in response to trametinib. Here, we further explored these findings in laboratory models to begin to define response and resistance mechanisms to MEK inhibition in HNSCCs. We observed increased Yap1 activity as a shared mechanism of trametinib resistance in models of both intrinsic (Detroit562) and acquired resistance (HSC3-TR). In these settings, resistance was established or acquired by a mechanism other than sustained Erk signaling. We found that Yap1 function was necessary and sufficient for this activity as enforced expression of constitutively active Yap1 conferred resistance in a sensitive model and genetic ablation resulted in sensitivity in resistant models. Confirming that Yap1 acts as a bypass pathway for MEK inhibition, pharmacologic blockade of Yap1 resulted in synergy with trametinib on resistant cell lines. Extending these data to a PDX derived from a patient who exhibited a clinical response to trametinib, we observed development of acquired resistance accompanied by increased Yap1 expression and activity. Patient tumor baseline YAP1 expression was not associated with expression of its target genes (ANKRD1, CTGF, CYR61); however, the expression of Yap1 targets were overall increased following 14 days of trametinib treatment. These findings confirm the importance of YAP1 pathway activity in mediating trametinib resistance and suggest inhibition of Yap1 as a potential strategy to enhance the efficacy of trametinib in HNSCC patients.
Despite the activation of Hippo pathway effectors, we were unable to establish Yap1 dependency for trametinib resistance in CAL27-TR, whereby resistance was acquired concomitantly with maintenance of Erk signaling. Sustained Erk activity despite the presence of trametinib suggests CAL27-TR specific compensatory signaling pathway(s) that differ from the other trametinib resistant models presented, which bypass Erk signaling with Yap1 dependency. It remains plausible that a Hippo pathway signaling component other than Yap1, e.g. TAZ, is a primary mediator of trametinib resistance dependence. At present, the mechanism for acquired trametinib resistance in this model remains undefined.
TCGA analysis identified 5% of HNSCC tumors with canonical HRAS mutations (codon 12, 13 or 61) with very low mutation rates in KRAS, NRAS or BRAF genes. Genomic analysis of our 20-patient cohort showed that 19 patient tumors did not bear any canonical drivers of MEK dependency and one patient tumor (5%) harbored an HRAS mutation. This latter patient did not complete the trial and thus was not evaluable. Similarly, we were not able to identify driver(s) of MEK dependency in HSC3, CAL27 or Detroit562. However, published proteomic analysis (7,17) shows increased Erk1/2 phosphorylation in HNSCCs consistent with other drivers impinging on this pathway. Similar to breast cancer, where again RAS pathway mutations are rare, this activation is likely the result of aberrant signaling from receptor tyrosine kinases (46). These data formed the basis of our initial studies targeting MEK as a central and important node in key signaling pathways in HNSCCs.
Toward further defining mechanisms of resistance, we used a candidate approach and identified increased YAP1 expression and activity in HSC3-TR and a similar correlation in the escape tumors from a PDX derived from a trametinib responder. Yap1 was identified as a bypass pathway for BRAF inhibitor therapy in BRAF mutant cells using an unbiased signaling pathway shRNA library approach (9). Our present work in non-RAS/RAF mutant HNSCC models and a responder patient is consistent with YAP1 expression and pathway/target activation as drivers of acquired resistance to MEK inhibition. Of note, our results show that elevated Yap1 protein expression and activity is supported by its pathway/target activation without necessarily its mRNA upregulation in trametinib resistant HNSCC models. Additionally, WES did not reveal selective growth of tumor subclones in PDXs or specific protein coding alterations to explain development of resistance. RNA-seq showed shifts in expression, but these general transcription pathways are modulated by a variety of transcription factors. The epigenetic landscape was not queried in this study, but our results suggest that resistance is mediated by non-genetic mechanisms.
Several questions remain regarding the mechanism by which Yap1 induces trametinib resistance in HNSCC. Previous reports have shown that Yap1 signaling reduces apoptosis and promotes resistance in RAS/RAF mutant malignancies (14,47). As a result, Yap1 inhibition re-sensitized resistant models to MAPK inhibitors by inducing greater apoptosis (14,47). Furthermore, YAP1 has been implicated in the epithelial to mesenchymal transition (EMT) program, a critical regulator of the cancer stem cell phenotype (CSC) (48). CSC resistance to chemotherapeutic agents has been attributed to increased expression of antiapoptotic proteins, elevated levels of ATP-binding cassette transporters, transmembrane protein transporters that are important for drug efflux and, thus, result in multidrug resistant cancer cells (48). Bromodomain containing protein-4 (BRD4) has been shown to mediate cetuximab resistance by upregulating the transcription of various RTKs in HNSCC (49). Since Yap1 is known to physically interact with BRD4 (50), this association may confer a transcriptional advantage of genes that mediate trametinib resistance in HNSCC.
There are several limitations of this study including the number of cell line models tested and patient samples available, definition of the mechanism of Yap1 mediated bypass activity and limited transcriptomic data with respect to matched patient samples. We subjected 3 HNSCC cell lines to extended trametinib treatment based on the number of lines we were able to identify with baseline drug sensitivity. We tested 2 PDXs for extended trametinib treatment (PDX2233 shown in this work and PDX4817 (24)) again as we focused on PDXs that were available from responders in our clinical trial. We tested multiple potential Yap1 mediated resistance pathways (negative data, not shown) and our future work aims to further explore potential non-genetic mechanisms. Finally, in vivo validation of combination Yap1 and MEK targeting in HSC3-TR was not feasible as resistance was not maintained when resistant xenografts were re-implanted into untreated mice. The cytostatic impact with reduced cellular ATP without cytotoxicity limits combination therapeutic targeting clinically and is commonly seen with other targeted therapeutics (51). For PDX2233, YAP1 was required for growth thus limiting a CRISPR based approach (Supplementary Figure 8S). This rapid reversibility of resistance may suggest against permanent genetic changes in these models as an underlying mechanism. This may be addressed by targeting resistance as it evolves for example in the PDX2233 model. We will integrate this approach in future work as we aim to further delineate Yap1 contribution to HNSCC progression and resistance to therapy.
In conclusion, as trametinib therapy resulted in clinical responses, we extended these findings and identified Yap1 as a resistance mechanism in HNSCCs. These data are consistent with findings in BRAF/RAS mutant tumors and our work extends these data to HNSCC where RAS mutations are less common. Our current work is aimed at defining mechanistic aspects of Yap1 mediated trametinib resistance and approaches integrating MEK targeting with immunotherapy in HNSCCs.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Statement of Translational Relevance.
Targeting of the MEK/Erk pathway results in biomarker and clinical responses in only a subset of HNSCC patients. Defining adaptive resistance mechanisms that evolve in response to trametinib therapy may thus inform therapeutic approaches for these patients. Prolonged trametinib exposure resulted in evolution of resistance in cell line models, and YAP1 was identified as a pathway contributing to this resistance. Combination Yap1 and MEK targeting resulted in synergistic activity in a resistant model. Analysis of patient-derived samples and xenografts revealed upregulation of YAP1 pathways in response to trametinib. Our results highlight implications of a combinatorial MEK and YAP1 therapeutic strategy approach in HNSCC patients.
Acknowledgements:.
RU was supported by NIH/NIDCR DE024403, DE027736 and a V Foundation Translational Research Award. Additional funding was from the National Comprehensive Cancer Network (NCCN) Oncology Research Program from general research support provided by Novartis Pharmaceutical Corporation (Novartis). OLG was supported by the National Cancer Institute (NIH NCI K22CA188163, NIH NCI U01CA209936 and NIH NCI U24CA237719) and a Cancer Research Foundation Young Investigator Award. We thank S. Kitajima and D. Barbie for YAP1 CRISPR and shRNA plasmids
Disclosures: EKB has equity, intellectual property, and is a board member of Geneoscopy LLC. KMC is a shareholder in Geneoscopy LLC and is a consultant for PACT Pharma, Inc. All other authors disclose no potential conflicts of interest.
Footnotes
Conflict of interest: The authors declared no potential conflicts of interest with respect to the work presented in this manuscript.
REFERENCES
- 1.Haddad RI, Shin DM. Recent advances in head and neck cancer. N Engl J Med. 2008;359:1143–54. [DOI] [PubMed] [Google Scholar]
- 2.Kademani D Oral cancer. Mayo Clin Proc. 2007;82:878–87. [DOI] [PubMed] [Google Scholar]
- 3.Ferlay J, Shin H-R, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–917. [DOI] [PubMed] [Google Scholar]
- 4.Rogers SN, Brown JS, Woolgar JA, Lowe D, Magennis P, Shaw RJ, et al. Survival following primary surgery for oral cancer. Oral Oncol. 2009;45:201–11. [DOI] [PubMed] [Google Scholar]
- 5.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Albanell J, Codony-Servat J, Rojo F, Del Campo JM, Sauleda S, Anido J, et al. Activated extracellular signal-regulated kinases: association with epidermal growth factor receptor/transforming growth factor alpha expression in head and neck squamous carcinoma and inhibition by anti-epidermal growth factor receptor treatments. Cancer Res. 2001;61:6500–10. [PubMed] [Google Scholar]
- 8.Bancroft CC, Chen Z, Dong G, Sunwoo JB, Yeh N, Park C, et al. Coexpression of proangiogenic factors IL-8 and VEGF by human head and neck squamous cell carcinoma involves coactivation by MEK-MAPK and IKK-NF-κB signal pathways. Clin Cancer Res. AACR; 2001;7:435–42. [PubMed] [Google Scholar]
- 9.Hoover AC, Strand GL, Nowicki PN, Anderson ME, Vermeer PD, Klingelhutz AJ, et al. Impaired PTPN13 phosphatase activity in spontaneous or HPV-induced squamous cell carcinomas potentiates oncogene signaling through the MAP kinase pathway. Oncogene. 2009;28:3960–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu S-L, Loss of transforming growth factor-beta type II receptor promotes metastatic head-and-neck squamous cell carcinoma [Internet]. Genes & Development. 2006. page 1331–42. Available from: 10.1101/gad.1413306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molinolo AA, Amornphimoltham P, Squarize CH, Castilho RM, Patel V, Gutkind JS. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol. 2009;45:324–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–90. [DOI] [PubMed] [Google Scholar]
- 13.Welsh SJ, Rizos H, Scolyer RA, Long GV. Resistance to combination BRAF and MEK inhibition in metastatic melanoma: Where to next? Eur J Cancer. 2016;62:76–85. [DOI] [PubMed] [Google Scholar]
- 14.Lin L, Sabnis AJ, Chan E, Olivas V, Cade L, Pazarentzos E, et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat Genet. 2015;47:250–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hammerman PS, Hayes DN, Grandis JR. Therapeutic insights from genomic studies of head and neck squamous cell carcinomas. Cancer Discov. 2015;5:239–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Søland TM, Husvik C, Koppang HS, Boysen M, Sandvik L, Clausen OPF, et al. A study of phosphorylated ERK1/2 and COX-2 in early stage (T1-T2) oral squamous cell carcinomas. J Oral Pathol Med. 2008;37:535–42. [DOI] [PubMed] [Google Scholar]
- 18.Judd NP, Winkler AE, Murillo-Sauca O, Brotman JJ, Law JH, Lewis JS Jr, et al. ERK1/2 regulation of CD44 modulates oral cancer aggressiveness. Cancer Res. 2012;72:365–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uppaluri R, Winkler AE, Lin T, Law JH, Haughey BH, Nussenbaum B, et al. Biomarker and Tumor Responses of Oral Cavity Squamous Cell Carcinoma to Trametinib: A Phase II Neoadjuvant Window-of-Opportunity Clinical Trial. Clin Cancer Res. 2017;23:2186–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Long GV, Fung C, Menzies AM, Pupo GM, Carlino MS, Hyman J, et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat Commun. 2014;5:5694. [DOI] [PubMed] [Google Scholar]
- 21.Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4:80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagathihalli NS, Castellanos JA, Lamichhane P, Messaggio F, Shi C, Dai X, et al. Inverse Correlation of STAT3 and MEK Signaling Mediates Resistance to RAS Pathway Inhibition in Pancreatic Cancer. Cancer Res. 2018;78:6235–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vultur A, Villanueva J, Krepler C, Rajan G, Chen Q, Xiao M, et al. MEK inhibition affects STAT3 signaling and invasion in human melanoma cell lines. Oncogene. 2014;33:1850–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Campbell KM, Lin T, Zolkind P, Barnell EK, Skidmore ZL, Winkler AE, et al. Oral Cavity Squamous Cell Carcinoma Xenografts Retain Complex Genotypes and Intertumor Molecular Heterogeneity. Cell Rep. 2018;24:2167–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41:D955–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chou T-C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–6. [DOI] [PubMed] [Google Scholar]
- 27.Bijnsdorp IV, Giovannetti E, Peters GJ. Analysis of drug interactions. Methods Mol Biol. 2011;731:421–34. [DOI] [PubMed] [Google Scholar]
- 28.Chou T-C, Talalay P. Analysis of combined drug effects: a new look at a very old problem. Trends Pharmacol Sci. Elsevier Current Trends; 1983;4:450–4. [Google Scholar]
- 29.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 30.Conway T, Wazny J, Bromage A, Tymms M, Sooraj D, Williams ED, et al. Xenome—a tool for classifying reads from xenograft samples. Bioinformatics. Oxford University Press; 2012;28:i172–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Griffith M, Griffith OL, Smith SM, Ramu A, Callaway MB, Brummett AM, et al. Genome Modeling System: A Knowledge Management Platform for Genomics. PLoS Comput Biol. 2015;11:e1004274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wickham H ggplot2: Elegant Graphics for Data Analysis. 1st ed. Springer-Verlag New York; 2009. [Google Scholar]
- 33.Skidmore ZL, Wagner AH, Lesurf R, Campbell KM, Kunisaki J, Griffith OL, et al. GenVisR: Genomic Visualizations in R. Bioinformatics. 2016;32:3012–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sergushichev AA. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation [Internet]. bioRxiv. 2016. [cited 2019 Apr 26]. page 060012. Available from: https://www.biorxiv.org/content/10.1101/060012v1.abstract [Google Scholar]
- 38.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019;569:503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gingras AC, Raught B, Gygi SP, Niedzwiecka A, Miron M, Burley SK, et al. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 2001;15:2852–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–83. [DOI] [PubMed] [Google Scholar]
- 45.Stein C, Bardet AF, Roma G, Bergling S, Clay I, Ruchti A, et al. YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers. PLoS Genet. 2015;11:e1005465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagaria TS, Shi C, Leduc C, Hoskin V, Sikdar S, Sangrar W, et al. Combined targeting of Raf and Mek synergistically inhibits tumorigenesis in triple negative breast cancer model systems. Oncotarget. 2017;8:80804–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016. page 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14:611–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leonard B, Brand TM, O’Keefe RA, Lee ED, Zeng Y, Kemmer JD, et al. BET Inhibition Overcomes Receptor Tyrosine Kinase–Mediated Cetuximab Resistance in HNSCC. Cancer Res. American Association for Cancer Research; 2018;78:4331–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zanconato F, Battilana G, Forcato M, Filippi L, Azzolin L, Manfrin A, et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat Med. 2018;24:1599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Subbiah V, Baik C, Kirkwood JM. Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer Res. 2020;6:797–810. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genomic data have been deposited with dbGaP under study accession phs001623.