

Interest in potential implications of necroptosis (Fig. 1)—i.e., a recently uncovered programmed cell death pathway—in neurodegenerative diseases has been growing over the recent years1,2. However, very few studies addressed its role in amyotrophic lateral sclerosis (ALS). Dominguez et al.3 recently brought some negative findings on this topic: (i) the genetic inactivation of receptor-interacting protein kinase (RIPK)1 did not protect against motor neuron degeneration in the superoxide dismutase (SOD)1 model of ALS and (ii) phosphorylated (p)-RIPK1 did not accumulate in the spinal cords of ALS compared to non-ALS patients. These findings raise doubts about the implication of necroptosis in the pathophysiology of ALS. In contrast, other studies detected robust activation of RIPK1, RIPK3, and mixed lineage kinases domain-like (MLKL) proteins in preclinical as well as clinical studies of ALS4–7. The discrepancies between these studies4,5 and others3,8 might be explained by (i) the use of western blotting in whole-tissue extracts4,5,8 versus in situ immunolabelling targeting motor neurons7, (ii) the removal of circulating blood cells—a rich source of necroptotic markers9—in some studies but not in others, (iii) the various time frame of tissue sampling, which matches or not the transient expression of necroptotic markers10, or (iv) technical limitations in the availability and specificity of in vivo tools of detection of necroptotic markers3–5,7,8.
Fig. 1. Schematic overview of necroptotic cell death.
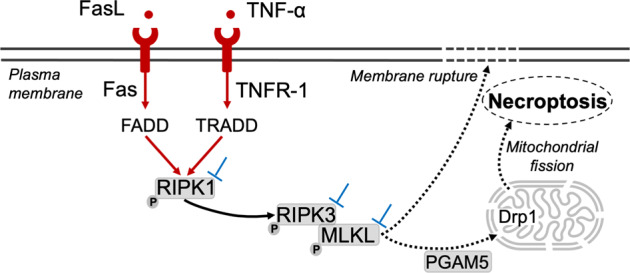
Necroptosis is triggered by inflammatory mediators, including TNF-α and FasL. Upon the recruitment of adapter proteins, the phosphorylation of RIPK1 is induced. The necrosome is composed of interaction and activation (i.e. phosphorylation) of RIPK1, RIPK3, and MLKL. Necroptosis of the cell is induced by mitochondrial fission through interaction of MLKL with mitochondrial phosphatase PGAM5 and Drp1 recruitment. Phosphorylated MLKL also triggers cell death through the disruption of the plasma membrane integrity1,2. Other mechanisms by which MLKL induces cell death remains to be elucidated. Pharmacological or genetic modulations (⊥) of necroptosis signaling can target RIPK1, RIPK3, MLKL, as well as their phosphorylated forms12–14. Drp1 dynamin-related protein 1, FADD Fas-associated protein with Death Domain, Fas-L Fas-ligand, MLKL mixed lineage kinases domain-like, p phosphorylated, PGAM5 phosphoglycerate mutase family member 5, RIPK receptor-interacting protein kinases, TNFα tumor necrosis factor α, TNFR1 tumor necrosis factor receptor 1, TRADD tumor necrosis factor receptor type 1-associated death.
Hence, the involvement of necroptosis in the pathophysiology of ALS remains questionable. However, the studies showing the activation of the necroptotic pathway are based on consistent results assessing all molecular steps of this cascade namely RIPK1, p-RIPK1, RIPK3, MLKL, p-MLKL, or p-RIPK3, and tumor necrosis factor (TNF)-α4,7, including in situ experiments focusing selectively on the cells of upmost interest, i.e. motor neurons of the anterior horn of the spinal cord7. In contrast, the studies showing the absence of activation of necroptosis assessed only one molecular step of the cascade (p-RIPK1 or unphosphorylated RIPK1)3,5 were often untargeted on specific neural cells, and not always targeting the phosphorylation of key necroptotic markers, which are fundamental to demonstrate the execution process of necroptosis (Fig. 1)11.
Altogether, the final conclusion about the role of necroptosis in ALS and the interplay between the various molecular mediators and cell subtypes involved would deserve further descriptive clinical findings on larger cohorts of ALS patients, as well as preclinical studies. Such research should combine pharmacological or genetic modulations of necroptotic signaling in ALS models to uncover the pathophysiological mechanisms potentially at play in the occurrence of ALS and define the key check points of putative intervention12–14.
Acknowledgements
We would like to thank Randy Pinsky for having edited the manuscript.
Funding
Research reported in this publication was supported by grants from the Canadian Institute of Health Research (CIHR), The Foundation of Stars, The Montreal Children’s Hospital Foundation, La Région Auvergne-Rhône-Alpes and l’Université Jean Monnet (France). M.C. is supported by a scholarship from the Fonds de Recherche du Québec en Santé (FRQ-S).
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Zhang S, Tang M-B, Luo H-Y, Shi C-H, Xu Y-M. Necroptosis in neurodegenerative diseases: a potential therapeutic target. Cell Death Dis. 2017;8:e2905. doi: 10.1038/cddis.2017.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dhuriya YK, Sharma D. Necroptosis: a regulated inflammatory mode of cell death. J. Neuroinflamm. 2018;15:199. doi: 10.1186/s12974-018-1235-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dominguez, S. et al. Genetic inactivation of RIP1 kinase does not ameliorate disease in a mouse model of ALS. Cell Death Differ. 10.1038/s41418-020-00625-7 (2020). [DOI] [PMC free article] [PubMed]
- 4.Ito Y, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 2016;353:603–608. doi: 10.1126/science.aaf6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dermentzaki, G. et al. Deletion of ripk3 prevents motor neuron death in vitro but not in vivo. Eneuro6. 10.1523/ENEURO.0308-18.2018 (2019). [DOI] [PMC free article] [PubMed]
- 6.Xu D, et al. TBK1 suppresses RIPK1-driven apoptosis and inflammation during development and in aging. Cell. 2018;174:1477–1491.e19. doi: 10.1016/j.cell.2018.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chevin M, Sébire G, Deltenre P, Kadhim H. Necroptotic neuronal cell death in amyotrophic lateral sclerosis: a relevant hypothesis with potential therapeutic implication? Med. Hypotheses. 2020;144:110295. doi: 10.1016/j.mehy.2020.110295. [DOI] [PubMed] [Google Scholar]
- 8.Wang T, et al. Necroptosis is dispensable for motor neuron degeneration in a mouse model of ALS. Cell Death Differ. 2020;27:1728–1739. doi: 10.1038/s41418-019-0457-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakazawa D, et al. Activated platelets induce MLKL-driven neutrophil necroptosis and release of neutrophil extracellular traps in venous thrombosis. Cell Death Discov. 2018;4:6. doi: 10.1038/s41420-018-0073-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Samson AL, et al. MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat. Commun. 2020;11:3151. doi: 10.1038/s41467-020-16887-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuan J, Amin P, Ofengeim D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 2019;20:19–33. doi: 10.1038/s41583-018-0093-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Degterev A, Linkermann A. Generation of small molecules to interfere with regulated necrosis. Cell Mol. Life Sci. 2016;73:2251–2267. doi: 10.1007/s00018-016-2198-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Degterev A, Ofengeim D, Yuan J. Targeting RIPK1 for the treatment of human diseases. Proc. Natl Acad. Sci. USA. 2019;116:9714–9722. doi: 10.1073/pnas.1901179116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gagliardi D, Comi GP, Bresolin N, Corti S. MicroRNAs as regulators of cell death mechanisms in amyotrophic lateral sclerosis. J. Cell. Mol. Med. 2019;23:1647–1656. doi: 10.1111/jcmm.13976. [DOI] [PMC free article] [PubMed] [Google Scholar]