

Summary
High expression levels of mitochondria-associated hexokinase-II (HKII) represent a hallmark of metabolically highly active cells such as fast proliferating cancer cells. Typically, the enzyme provides a crucial metabolic switch towards aerobic glycolysis. By imaging metabolic activities on the single-cell level with genetically encoded fluorescent biosensors, we here demonstrate that HKII activity requires intracellular K+. The K+ dependency of glycolysis in cells expressing HKII was confirmed in cell populations using extracellular flux analysis and nuclear magnetic resonance-based metabolomics. Reductions of intracellular K+ by gramicidin acutely disrupted HKII-dependent glycolysis and triggered energy stress pathways, while K+ re-addition promptly restored glycolysis-dependent adenosine-5′-triphosphate generation. Moreover, expression and activation of KV1.3, a voltage-gated K+ channel, lowered cellular K+ content and the glycolytic activity of HEK293 cells. Our findings unveil K+ as an essential cofactor of HKII and provide a mechanistic link between activities of distinct K+ channels and cell metabolism.
Subject Areas: Biochemistry, Biochemical Mechanism, Molecular Physiology
Graphical abstract
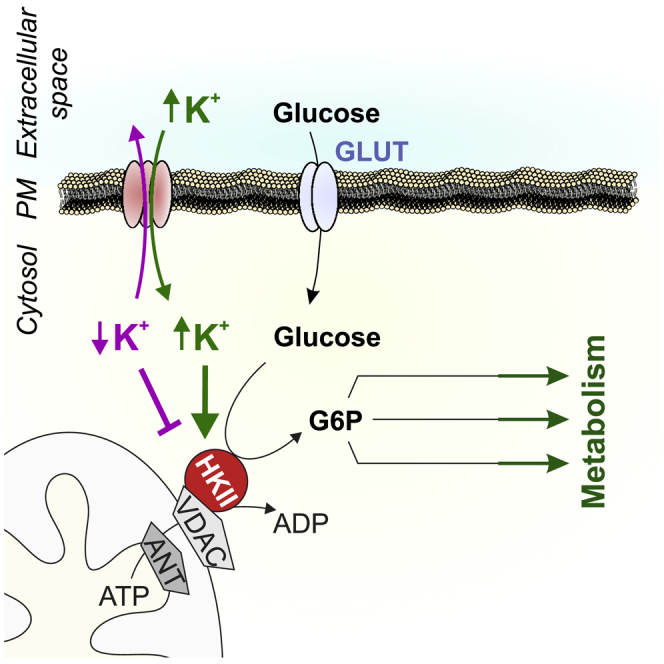
Highlights
-
•
HKII expression sensitizes cellular metabolism for intracellular K
-
•
Intracellular K+ depletion abates the metabolic activity of HKII-positive cells
-
•
(Re-)elevations of intracellular K+ restore glycolysis
Here, Bischof et al. report that the activity of hexokinase-II requires a high intracellular K+ concentration. Cells that rely on hexokinase-II activity for augmented aerobic glycolysis are thus vulnerable to intracellular K+ depletion.
Introduction
Cellular glucose uptake and the subsequent phosphorylation of glucose via hexokinase (HK) isoforms represent the initial and crucial step in glucose metabolism. From four different HK isoforms identified, hexokinase-I (HKI) and hexokinase-II (HKII) represent isoforms that are capable of associating with the voltage-dependent anion channel (VDAC) located at the outer mitochondrial membrane. The localization of HK isoforms at the surface of mitochondria facilitates adenosine-5′-triphosphate (ATP) supply of the enzymes and additionally protects cells from apoptosis (Abu-Hamad et al., 2008; Gottlob, 2001; Mathupala et al., 2009; Roberts and Miyamoto, 2015). Moreover, especially HKII, which is highly expressed in most cancer cells and has been shown to drive tumor initiation and progression in murine cancer models, is frequently associated with high cell metabolic activities (Anderson et al., 2017; Mathupala et al., 2006). Remarkably, potassium ions (K+), the most abundant intracellular cations (Palmer, 2015), exert a stabilizing effect on purified HKII in vitro, thereby preventing the thermal inactivation of the enzyme (Rose and Warms, 1982). Besides the high expression rate of cancer-associated HKII, cancer onset and progression are frequently associated with altered expression and activity of certain K+ channels that control and modulate abnormal cell proliferation (Huang and Jan, 2014; Mathupala et al., 2006; Mohr et al., 2020). Accordingly, pharmacological tools impairing the (sub-)cellular K+ homeostasis have shown potent anticancer properties in vitro and animal models. Interestingly, the activation of specific K+ channels or the inhibition of the sodium ion (Na+)/K+ adenosine triphosphatase represents effective anticancer and senolytic strategies (Guerrero et al., 2019; Lansu and Gentile, 2013; Sanderson et al., 2020; Steudel et al., 2017; Triana-Martínez et al., 2019). However, despite these known implications of targeting the cellular K+ homeostasis on (cancer) cell viability, the underlying mechanism mediating the cell death induction remains largely enigmatic. By exploiting single-cell imaging approaches to visualize subcellular ATP, glucose, K+, and lactate concentrations and dynamics, we here unveil that depletions of [K+]i reduce glycolysis and ATP generation in cells expressing HKII. Extracellular flux analysis of acidification and oxygen consumption and NMR metabolomics confirmed that HKII requires intracellular K+ for its full catalytic activity.
Results
Low [K+] impair HKII activity in vitro and aerobic glycolysis in cells
First, using the purified enzyme, we confirmed that high [K+] counteract the thermal inactivation of HKII with an EC50 of 18.98 (7.12–50.56) mM K+ (Figure S1A). Under these conditions, the enzymatic activity of HKI was not significantly affected (Figure S1B). To substantiate, whether K+ could directly affect HKII enzymatic activity, we investigated a crystallographic model of HKII (Figure 1A) (Nawaz et al., 2018). This analysis revealed several unidentified atoms or ions being near to amino acids known to feature strong K+ binding (Figure 1A) (Heaton and Armentrout, 2008). Such unidentified atoms or ions were absent in a crystallographic model of HKI (Figure 1B) (Rosano et al., 1999). Analysis of the protein sequences of HKI and HKII unveiled a high homology of the full-length protein sequences of the two HK isoforms of ~73.4% (Figure S1C). Further in-depth analysis of the potential amino acids interacting with the unidentified atoms in HKII demonstrated four positions of potential interaction that were different in HKII compared to HKI, comprising the amino acid transitions N89S, H422Q and K425R, Q471E and E821T, and E724H from HKII to HKI, respectively (Figure S1D). Interestingly, in three of the four positions, amino acids featuring strong K+ binding (Heaton and Armentrout, 2008) were found in HKII but not HKI. Also, the homology of HKI and HKII was reduced to ~65.7% in regions 10 amino acids upstream and downstream of the potential interaction sites. Non-homologous amino acids in these regions frequently comprised strongly K+ binding amino acids (Figures S1C and S1D). We hypothesized that these positions, which are additionally found close to the protein surface (Figure S1E), could sensitize HKII for K+.
Figure 1.

Intracellular K+ depletion induces disturbances of cellular energy homeostasis in HEK293 cells.
(A and B) HKII (A) and HKI (B) structure with potential K+ binding amino acids in blue and unknown atoms or ions found in the crystallographic model of HKII in red.
(C) IF images of HEK293 cells. Scale bar shows 20 μm, n = 7.
(D) [GLU]cyto over-time of single HEK293 cells in response to gramicidin. n = 4.
(E) Average [GLU]cyto over-time ± standard deviation (SD) of HEK293 cells kept in a cytosol-like buffer and an ionophore mixture. Ions were added or removed as indicated. n = 4.
(F) [ATP]mito responses of single HEK293 cells in response to gramicidin. n = 4.
(G and H) [ATP]mito (G) and [GLU]cyto (H) over [K+]. Both panels show average ± standard error of mean of 3 independent experiments.
(I) 13C-lactate production (LP)/13C-glucose uptake (GU) ratio of HEK293 cells either treated with DMSO (ctrl, white) or gramicidin (GRAM, magenta). Data represent each replicate and average ± SD. n = 6, ∗∗p ≤ 0.01, Mann-Whitney test.
(J) [LAC]cyto over-time (left) and statistics (right) of HEK293 cells treated with gramicidin. Single cell responses and average ± SD are shown, ∗∗∗p ≤ 0.001, Wilcoxon matched-pairs signed rank test, n = 4.
(K) ECAR/OCR ratios of HEK293 cells treated with DMSO (ctrl) or gramicidin (GRAM). ∗∗p ≤ 0.01, Mann-Whitney test. n = 4. Boxes indicate the median and the first and third quartile. Lower and upper whiskers indicate 5-95 percentile; outliers are indicated as black dots.
(L) Responses of single HEK293 cells expressing AMPKAR upon DMSO (grey) or gramicidin (magenta) treatment. n = 3.
See also Figures S1 and S2.
To elucidate, whether K+ controls HKII activity also in its cellular environment, we aimed to manipulate [K+]i. We proved its efficiency using K+ biosensors (Bischof et al., 2017) expressed in different human cell lines including HEK293 (human embryonic kidney cells) and the cancer cell lines HeLa (human cervical cancer cells), INS-1 832/13 (a rat insulinoma cell line), MCF-7, and MDA-MB-453 (human breast cancer cell lines). Administration of gramicidin efficiently lowered cytosolic (Figure S2A) and mitochondrial [K+] ([K+]cyto, [K+]mito) (Figure S2B), respectively, within few minutes in all cell types tested. We next monitored the cytosolic glucose concentration ([GLU]cyto) over-time as a measure of glycolytic activity, using a fluorescence resonance energy transfer (FRET)-based glucose sensor (Bermejo et al., 2010), while depleting [K+]i in HEK293 cells, as these cells show high endogenous HKII expression levels (Figure 1C) (Peng et al., 2008). [K+]i reduction yielded immediate elevations of [GLU]cyto (Figure 1D), indicating hampered glucose phosphorylation, causing an increase of [GLU]cyto due to ongoing glucose import in HEK293 cells, when intracellular K+ is depleted. To scrutinize a potential role of other physiologically relevant metal ions (Bischof et al., 2019), we permeabilized HEK293 cells using a mixture of different ionophores and gramicidin, followed by manipulating the concentrations of calcium (Ca2+), magnesium (Mg2+), sodium (Na+) ions, and K+, respectively (Figure 1E). These experiments unveiled that solely K+ removal induced a pronounced increase of [GLU]cyto in HKII-expressing HEK293 cells, supporting the idea of a K+-modulated HKII activity.
We next monitored the mitochondrial concentration of adenosine-5′-triphosphate ([ATP]mito) using an approved ATP biosensor targeted to the mitochondrial matrix (Imamura et al., 2009). [K+]i depletion reduced [ATP]mito over-time in HEK293 cells (Figure 1F), similar to extracellular glucose removal (Figure S2C). These results imply the necessity of high [K+]i for maintaining [ATP]mito supply and glycolytic activity in HEK293 cells.
To estimate the sensitivity of cell metabolism for K+, we analyzed [GLU]cyto and [ATP]mito in HEK293 cells upon their permeabilization with gramicidin in the presence of different [K+]ex. [K+]cyto strictly followed [K+]ex as measured by NES lc-LysM GEPII 1.0 (Figure S2D), again indicating the suitability of gramicidin to freely manipulate [K+]cyto. Next, we analyzed [ATP]mito under these conditions. These experiments unveiled a decrease of [ATP]mito, which well correlated with decreasing [K+]cyto (Figures 1G and S2E). As expected, [ATP]mito of gramicidin-treated HEK293 cells decreased in a [K+]-dependent manner (Figures 1G and S2E). Thereby, the decrease of [ATP]mito upon decreasing [K+]cyto seemed biphasic, which would indicate multiple K+ binding sites with different affinities of HKII (Figures 1G and S2E). The sensitivities of [ATP]mito for K+ were observed at a half-maximal effective concentration (EC50) of ~3.0 mM K+ and a second EC50 of ≥70.0 mM (Figure 1G). Similar results were obtained by analyzing [GLU]cyto in a HEK293 cell population upon gramicidin treatment in the presence of distinct [K+]ex, which also unveiled an EC50 of [GLU]cyto for K+ of 3.63 mM (2.12 mM–6.23 mM) (Figure 1H). Under these conditions, a second EC50 could not be determined yet (Figure 1H). Together, these data indicate that the full depletion of [ATP]mito and maximal accumulation of [GLU]cyto requires a strong intracellular K+ depletion. Nevertheless, our analysis also indicates that physiological intracellular K+ fluctuations in the higher mM range might modulate HKII-dependent cell metabolic activity.
We further analyzed the lactate production (LP)/glucose uptake (GU) ratio of HEK293 cells by NMR metabolomics. These experiments confirmed the importance of [K+]i for aerobic glycolysis in HEK293 cells, as LP/GU ratio significantly decreased in cells treated with gramicidin for a depletion of [K+]i compared to intact control cells (Figure 1I). The decline of cytosolic lactate concentrations ([LAC]cyto) upon lowering [K+]i in HEK293 cells was also observed using a lactate biosensor (San Martín et al., 2013) (Figure 1J). Extracellular flux analysis confirmed a reduced glycolytic activity of HEK293 cells by low [K+]i (Figure 1K). In line with our expectations, the analysis of adenosine monophosphate-activated protein kinase (AMPK) activity, a central cell energy stress sensor (Hardie et al., 2012; Najafov et al., 2020), using an AMPK activity reporter (Tsou et al., 2011), revealed increasing AMPK activity upon reducing [K+]i in HEK293 cells (Figure 1L), confirming the induction of energy stress by cellular K+ depletion. Altogether, our results indicate an abated aerobic glycolysis activity when [K+]i is low in HKII-expressing HEK293 cells (Figure S2F).
K+ controls glycolysis in (cancer) cell lines with a Warburg setting
We further aimed to identify, whether [K+]i also modulates the metabolism of other (cancer) cell lines. Therefore, we assessed [GLU]cyto and [ATP]mito upon lowering [K+]i in HeLa, INS-1 832/13, MCF-7, and MDA-MB-453 cells (Figures 2A, S2G, and S2H). Interestingly, we observed declining [ATP]mito in all cell lines tested, except INS-1 832/13 cells, and increasing [GLU]cyto in HEK293, MCF-7 and MDA-MB-453, while [GLU]cyto remained unaffected in HeLa and INS-1 832/13 cells upon gramicidin administration (Figure 2A). Besides cell treatment with gramicidin for depleting [K+]i, we tested other metabolic interventions, including the removal of extracellular glucose, the depolarization of mitochondria, or inhibition of HKII by 3-bromo-2-oxopropionic acid (3-BP) (Figure 2A). Interestingly, the effect of depleting [K+]i on [GLU]cyto and [ATP]mito differed from all other metabolic interventions in all cell lines, confirming their differential nutrient requirements (Figure 2A). Thereby, high [K+]i is essential for maintaining energy homeostasis in cells with a Warburg setting (Figures 2A and 2B), as also confirmed by NMR metabolomics and extracellular flux analysis. While LP/GU ratio decreased in HeLa cells (Figure 2C) and ECAR/OCR ratio decreased in HeLa, MCF-7, and MDA-MB-453 cells upon reducing [K+]i (Figure 2D), LP/GU ratio and ECAR/OCR ratio remained unaffected in INS-1 832/13 cells (Figures 2C and 2D), possessing the high oxidative and low glycolytic activity typical for an anti-Warburg setting (Figure 2B).
Figure 2.

Intracellular K+ depletion affects energy homeostasis in cells with a metabolic Warburg setting.
(A) Average [GLU]cyto and [ATP]mito of different cell lines upon metabolic interventions as indicated, n ≥ 3.
(B) Basal ECAR/OCR ratios of different cell lines. High ratios indicate the metabolic Warburg setting. Boxes indicate the median and the first and third quartile. Lower and upper whiskers indicate 5-95 percentile; outliers are indicated as black dots. n = 4 for all. ∗∗∗p ≤ 0.001, Friedman test followed by Dunn's MC test.
(C) 13C-lactate production (LP)/13C-glucose uptake (GU) ratio of HeLa and INS-1 832/13 cells either treated with DMSO (ctrl, white) or gramicidin (GRAM, magenta). Data represent each replicate and average ± standard deviation. n = 6, ∗∗p ≤ 0.01, Mann-Whitney test.
(D) ECAR/OCR ratios of different cell lines treated with DMSO (ctrl, white) or gramicidin (GRAM, magenta). Boxes indicate the median and the first and third quartile. Lower and upper whiskers indicate 5-95 percentile; outliers are indicated as black dots. ∗p ≤ 0.05, ∗∗∗p ≤ 0.001, Mann-Whitney test. n = 4 for all.
See also Figure S2.
Restoring high [K+]i instantly re-establishes glucose metabolism
Our subsequent experiments aimed to identify the reversibility of the reduced metabolic activity upon [K+]i depletion. Both [K+]cyto (Figure S3A) and [K+]mito (Figure S3B) were restored within minutes upon extracellular re-addition of 140.0 mM K+. Restoring high [K+]i resulted in a recovery of AMPK activity (Figure 3A), [ATP]mito (Figure 3B), and [GLU]cyto (Figure 3C) in all cell lines investigated. The rescue of the metabolic activity was furthermore confirmed by extracellular flux analysis (Figures 3D, S3C, and S3D) and NMR metabolomics (Figures 3E and S3E–S3G), while INS-1 832/13 cells remained again unaffected (Figures S3H–S3M). In sum, our data demonstrate that glucose metabolism is re-established by restoring high [K+]i (Figure 3F).
Figure 3.

Restoring high [K+]i restores aerobic glycolysis.
(A) HEK293 cells expressing AMPKAR were analyzed upon glucose removal and gramicidin pre-treatment. At the time point indicated, [K+]ex was increased from 0 mM to 140.0 mM. n = 4.
(B) [ATP]mito over-time of gramicidin-treated cells upon addition of 140.0 mM K+ as indicated. n ≥ 3 for all.
(C) [GLU]cyto over-time of HEK293 cells pre-stimulated with gramicidin in the presence of glucose and the absence of K+. At the time point indicated, 140.0 mM K+ was added. n = 3.
(D and E) (D) 140.0 mM K+ (HK)/0 mM K+ (LK) ECAR/OCR ratios and (E) HK/LK 13C-LP/13C-GU ratios of cells treated with DMSO (ctrl, white) or gramicidin (GRAM, green). Data are either represented as boxes with 5-95 percentile and outliers as black dots (D) or each replicate and average ± standard deviation (E). n = 4 for (D) and n ≥ 5 for (E).
(F) Simplified illustration of K+-modulated glycolysis.
See also Figure S3.
[K+]i is required for full glycolytic activity
We next investigated whether the K+ dependency of glycolysis is also given when mitochondria are depolarized. Interestingly, the inhibition of mitochondrial respiration drastically reduced [GLU]cyto in the strongly glycolytically active cell lines HEK293 (Figure 4A) and HeLa (Figure 4B), eventually due to increasing glycolytic (Figure S4A) and decreasing respiratory activity (Figure S4B). Glucose removal reduced [GLU]cyto (Figures 4A and 4B), which remained low upon glucose re-addition, suggesting that glucose turnover rates exceed the velocity of glucose uptake. Strikingly, also under these conditions, a lowering of [K+]i induced strong [GLU]cyto accumulation (Figures 4A and 4B), emphasizing that [K+]i depletion also abolishes propelled glucose turnover (Figure S4C).
Figure 4.

HKII status determines K+ sensitivity of cell metabolism.
(A and B) [GLU]cyto over-time (left) and statistics (right) of HEK293 (A) and HeLa cells (B) in response to oligomycin-A and antimycin-A, glucose removal, or gramicidin treatment as indicated. Single cell responses and average ± standard deviation (SD) are shown. ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, Friedman test followed by Dunn's MC test. n = 3 for HEK293, n = 6 for HeLa.
(C) [ATP]mito dynamics of HeLa cells in response to gramicidin, mannose, and high [K+]ex. n = 4.
(D) Quantification of HKI (left) and HKII expression (right) in HEK293, HeLa, and INS-1 832/13 cells using quantitative PCR. Data represent each replicate and average ± standard error of mean. n = 3.
(E) Representative IF images of different cell lines. HKI or HKII (magenta), Hoechst (blue), and α-tubulin (green) are shown. Scale bar in the lower left image represents 10 µm. n ≥ 6.
(F) Quantification of HKI (left) and HKII expression (right) from images as shown in (E). Whiskers extend to the 5-95 percentile. Outliers are indicated as black dots. n ≥ 6.
(G–I) Average [GLU]cyto over-time ± standard error of mean (left) and corresponding statistics ± SD of HEK293 cells treated with siRNA against HKI (siHKI, light grey) or HKII (siHKII, black) (F), or HeLa (G), and INS-1 832/13 cells (H) either expressing HKI-mRuby3 (+HKI-mR3, light grey) or HKII-mRuby3 (+HKII-mR3, black), respectively. n ≥ 4. ∗p ≤ 0.05, ∗∗∗p ≤ 0.001, Repeated measures one-way analysis of variance (ANOVA) test with Tukey's MC test (G, H), # and ∗p ≤ 0.05, Unpaired t test.
See also Figure S4.
We next analyzed [ATP]mito of HeLa cells upon treatment with mannose (Figure S4D), a hexose sugar that is phosphorylated by HKs to mannose-6-phosphate (M6P) but is not further processed to generate ATP within this cell type. As observed earlier (Depaoli et al., 2018), mannose addition rapidly reduced [ATP]mito due to active ATP consumption by HKs and the lack of ATP de novo synthesis (Figures 4C, S4E, and S4F). [ATP]mito transiently increased upon mannose removal from control cells (Figures S4E and S4F), possibly due to mitochondrial HKs catalyzing the reverse reaction, thereby generating ATP from M6P and adenosine-5′-diphosphate (Depaoli et al., 2018), while [K+]i-depleted cells did not show [ATP]mito elevations until [K+]i was restored (Figures 4C and S3F). These results also confirm a K+ dependency of HKII in HeLa cells.
HKI and HKII expression levels determine the [K+]i sensitivity of glucose metabolism
To investigate whether the K+ sensitivity of glucose metabolism correlates with the HK expression, we manipulated the HKI/HKII ratio of HEK293, HeLa, and INS-1 832/13 cells, showing highly differential HK expression patterns (Figures 4D–4F). To this end, treatment with siRNAs against HKI or HKII, respectively, strongly reduced their expression levels in HEK293 cells (Figure S4G). Next, we analyzed [GLU]cyto and [ATP]mito of HEK293 cells treated with the different siRNAs. We found higher basal [GLU]cyto upon knockdown of HKII compared to HKI (Figure 4G). [K+]i depletion increased [GLU]cyto in siHKI-treated cells, while the effect was reduced in siHKII-treated HEK293 cells (Figure 4G). Re-addition of high K+ reduced [GLU]cyto to basal levels, and glucose removal depleted [GLU]cyto under both conditions (Figure 4G). The potential K+-sensitive activity of HKII was confirmed by analyzing [ATP]mito under these conditions (Figures S4H–S4K). Interestingly, treatment of HEK293 cells with siHKII yielded lower basal [ATP]mito compared to siHKI (Figures S4H–S4J). Administration of gramicidin strongly depleted [ATP]mito of siHKI, but to a lesser extent of siHKII-treated HEK293 cells (Figure S4K). Restoring [K+]i re-established basal [ATP]mito (Figures S4H and S4I). Altogether, these findings strongly point to a K+-sensitive activity of HKII in HEK293 cells.
Vice versa, we addressed whether increasing HKII levels introduce a K+-sensitive glucose utilization. Therefore, we expressed HKI-mRuby3 or HKII-mRuby3 (Figure S4K) in HeLa and INS-1 832/13 cells (Figure S4L), respectively. Indeed, [GLU]cyto of HeLa (Figure 4H) and INS-1 832/13 cells (Figure 4I) expressing HKII-mRuby3 were lower compared to cells expressing HKI-mRuby3 under resting conditions. Expression of HKII-mRuby3, but not HKI-mRuby3, sensitized [GLU]cyto of both cell lines for [K+]i (Figures 4H and 4I), indicating that solely HKII mediates the K+ sensitivity of cell metabolism.
Mitochondrial localization of HKII is required for its K+ sensitivity
To further investigate whether the mitochondrial association of HKII is required for its K+ sensitive activity, we expressed a truncated HKII-GFP (trHKII-GFP) variant lacking the first 21 amino acids (Figure S4M), which encode for the N-terminal mitochondrial binding domain (Sun et al., 2008). As expected, trHKII-EGFP was found throughout the cytosol of HeLa cells expressing the construct (Figure S4M). We further aimed to identify whether also the expression of trHKII-GFP could sensitize [GLU]cyto of HeLa cells to alterations of [K+], as observed for the full-length construct (Figure 4H). Interestingly, basal [GLU]cyto of HeLa cells expressing trHKII was similar to wild-type HeLa cells (Figure 5A). Neither the administration of gramicidin in the absence of [K+]ex nor the re-addition of 140.0 mM K+ significantly affected [GLU]cyto of WT or trHKII-EGFP-expressing HeLa cells (Figure 5A). Removal of extracellular glucose, however, yielded [GLU]cyto depletion under both conditions (Figure 5A). These results emphasize that the modulatory effect of K+ on HKII activity requires the ability of HKII to associate with the outer mitochondrial membrane.
Figure 5.

HKII remains mitochondria associated upon [K+]i depletion and its mitochondrial association is required for the K+-sensitive activity.
(A) [GLU]cyto over-time of HeLa WT cells (left panel, grey curve) or HeLa cells expressing trHKII-EGFP (middle panel, green curve) in response to gramicidin treatment in the absence of [K+]ex, upon [K+]ex re-addition and upon glucose removal. Data show average ± standard error of mean, n = 4 for both conditions. Right panel demonstrates corresponding statistics, representing the basal FRET-ratio signal of FLII12Pglu-700μδ6 expressed in HeLa WT cells or HeLa cells expressing trHKII-EGFP. n = 4 for both, data show each single cell and average ±standard deviation (SD).
(B and C) array confocal laser scanning microscope (ACLSM) images (C) and cytosolic fluorescence intensity ratios ± standard error of mean over-time (B) of regions of interest as demonstrated in (C). HeLa cells expressing HKI-mRuby3 (red, upper images) and HKII-EGFP (green, lower images) prior and after stimulation with DMSO (left), 3-BP (middle), or gramicidin (right) are shown. Scale bars show 10 μm. n ≥ 4 independent experiments for each condition.
(D) Left panel displays representative thresholded immunofluorescence images of HeLa cells stained for endogenous HKI (red, left panel, left images) and Hoechst for nuclear staining (blue, left panel, middle images). Right images demonstrate an overlay of HKI (red) and Hoechst staining (blue). HeLa cells were either untreated (ctrl, upper images) or treated with 3-BP (3-BP, middle images) or gramicidin (+GRAM) prior to fixation. Right panel represents corresponding statistics as Pearson correlation of HKI and Hoechst colocalization of every image acquired and mean ± SD of n = 3 independent experiments for each condition. Scale bar in the lower right image represents 10 μm.
(E) Left panel displays representative thresholded immunofluorescence images of HeLa cells stained for endogenous HKII (red, left panel, left images) and Hoechst for nuclear staining (blue, left panel, middle images). Right images demonstrate an overlay of HKII (red) and Hoechst staining (blue). HeLa cells were either untreated (ctrl, upper images) or treated with 3-BP (3-BP, middle images) or gramicidin (+GRAM) prior to fixation. Right panel represents corresponding statistics as Pearson correlation of HKII and Hoechst colocalization of every image acquired and mean ± SD of n = 3 independent experiments for each condition. ∗p ≤ 0.05, ∗∗∗p ≤ 0.001, one-way ANOVA test with Tukey's MC test. Scale bar in the lower right image represents 10 μm.
See also Figure S4.
HKII remains mitochondria associated upon lowering [K+]i
Along these lines, we investigated whether low [K+]i causes the inhibitory effect on HKII by affecting its mitochondrial association. Therefore, we expressed HKI-mRuby3 (Figure S4K) and HKII-EGFP (Figure S4N) simultaneously in HeLa cells. While both HK-FP fusion constructs (Figures S4K and S4N) remained mitochondria associated over-time under control conditions (Figures 5B and 5C), inhibition of HKII via 3-BP induced translocation of HKII-EGFP from the outer mitochondrial membrane (Figures 5B and 5C). We next depleted [K+]i, which did not affect HKI/II-FP localization ratio (Figures 5B and 5C). These results were furthermore confirmed by analyzing endogenous HKI and HKII localization by immunofluorescence. While the localization of HKI in HeLa cells did neither alter upon the administration of 3-BP nor upon treatment with gramicidin (Figures 5D and S4O), we found a reduced mitochondrial association of HKII upon 3-BP treatment (Figures 5E and S4P). Localization of HKII was, however, not affected upon depleting [K+]i (Figures 5E and S4P). These data emphasize that low [K+]i do not modulate HKII activity via inducing its translocation from mitochondria. Rather, K+ has a direct effect on the catalytic activity of mitochondria-associated HKII in its physiologic environment.
Expression and activation of KV1.3 abates glucose metabolism in HEK293 cells
Our findings imply that the presence and activity of specific K+ channels in the plasma membrane (PM) should modulate HKII-dependent metabolism. To test this assumption, we expressed RFP-tagged KV1.3 variants in HEK293 cells (Figures 6A and S5A). Patch-clamp experiments unveiled the functionality of TagRFP-KV1.3, while TagRFP-KV1.3 PD, possessing the single point mutation W388F (Figure S5A), showed less conductance (Figures 6B and S5B). A large portion of TagRFP-KV1.3 was, however, found within intracellular structures (Figure S5C). Notably, the abundance of TagRFP-KV1.3 at the PM was improved by unspecific K+ channel inhibition using tetraethylammonium chloride (TEA), while the subcellular localization of TagRFP-Kv1.3 PD remained unchanged under these conditions (Figure S5C). TEA removal increased the K+ currents of TagRFP-KV1.3 across the PM (Figure S5D), drastically reduced [K+]cyto (Figures 6C and 6D), and yielded [GLU]cyto accumulation (Figure 6E), indicative for a reduced glucose turnover upon K+ efflux via TagRFP-KV1.3. These signals were less pronounced in cells expressing TagRFP-KV1.3 PD (Figures 6C and 6E). Under these conditions, [K+]i and [GLU]cyto were restored upon administration of 30.0 mM K+ to the extracellular space (Figures 6C and 6E). We furthermore analyzed the ECAR/OCR ratio of HEK293 cells stably expressing these KV1.3 variants (Figure S5E). In the presence of PAP-1, a specific inhibitor of KV1.3 (Schmitz et al., 2005) (Figure 6F), increased ECAR/OCR ratios of HEK293 cells stably expressing TagRFP-KV1.3 were recorded (Figures 6G and S5F), emphasizing a promoted glycolytic activity by inhibition of cellular K+ efflux. In contrast, PAP-1 neither affected ECAR nor OCR levels in HEK293 cells expressing TagRFP-KV1.3 PD (Figure S5G). These data emphasize that the presence of specific K+ channels, e.g. KV1.3, may modulate cellular metabolic activity, especially of HKII-positive cells, and are in line with a recent report, demonstrating an implication of KV1.3 inhibition on obesity and insulin resistance (Upadhyay et al., 2013).
Figure 6.

Consequences of voltage-gated K+ channel expression on metabolism.
(A and B) Representative ACLSM images (A) and currents (B) of HEK293 cells either expressing TagRFP-KV1.3 (upper image and black traces) or TagRFP-KV1.3 PD (lower image and red traces). Scale bar shows 10 μm. n ≥ 12.
(C and E) (C) Average (bold) and single cell (faint) [K+]cyto and (E) [GLU]cyto over-time of HEK293 cells either expressing TagRFP-KV1.3 (black) or TagRFP-KV1.3 PD (red). At the time point indicated, TEA was removed, followed by a re-addition of 30.0 mM K+. n ≥ 3.
(D) Correlation of [K+]cyto loss with TagRFP-KV1.3 expression in HEK293 cells. n = 3.
(F) IV curves ± standard error of mean of HEK293 cells stably expressing TagRFP-KV1.3, either in the absence (ctrl, white) or presence of 1 μM PAP-1 (+PAP-1, grey). n ≥ 13.
(G) Basal ECAR/OCR ratios of HEK293 cells expressing TagRFP-KV1.3, either in the presence of DMSO (white) or PAP-1 (grey). Whiskers extend to the 5-95 percentile. Outliers are indicated as black dots. n = 3, ∗∗∗p ≤ 0.001, Unpaired t test.
See also Figure S5.
Discussion
Within this study, we visualized in single cells how [K+]i fluctuations tightly and promptly affect intracellular levels of glucose, lactate, and ATP most likely as a consequence of controlling HKII-dependent cell metabolism primarily. By exploiting the first genetically encoded fluorescent biosensors for imaging [K+]i in intact cells (Bischof et al., 2017), we established and tested protocols for efficient manipulation of [K+]i using gramicidin, a well-characterized ionophoretic antibiotic peptide (Szule and Rand, 2003). Based on these protocols, we could correlate cellular K+ depletion and restorations with subcellular metabolic and AMPK activities, demonstrating that only in cells with high HKII expression, K+ is mandatory to maintain high aerobic glycolysis activities.
Our finding may especially gain importance in cancer biology due to the high expression of HKII in many cancer cells (Mathupala et al., 2006; Patra et al., 2013), particularly of those in tumors with bad prognosis (Anderson et al., 2017; Gong et al., 2012; Ogawa et al., 2015). In the light of several studies that point to K+ channels and the cellular K+ homeostasis as important determinants of tumorigenesis, tumor progression, and tumor malignancy (Lansu and Gentile, 2013; Mohr et al., 2019, 2020; Steudel et al., 2017; Urrego et al., 2014), our study linking [K]i to tumor cell metabolism is particularly interesting as it has the potency to lead to the development of novel anticancer strategies. Importantly, while we here provide different EC50 values ranging from 3 mM to 70 mM for [K+]i to alter [GLU]cyto and [ATP]mito in HKII-expressing single cells, our study is less conclusive if unphysiological strong or already moderate physiological [K+]i fluctuations significantly modulate cell metabolism and thus the proliferation rates of HKII-expressing cells. However, it is tempting to speculate that buffering extracellular K+ specifically within the tumor microenvironment, in which [K+]ex compared to healthy tissues is significantly increased (Eil et al., 2016), might attenuate the metabolic activity of HKII-positive tumor cells. Notably, here we could demonstrate that the same voltage-gated K+ channel, KV1.3, which rescued T cells from high [K+]ex-mediated immune suppression (Eil et al., 2016), affected [K+]i and consequently controlled [GLU]cyto most likely by regulating HKII activity. Much more research is essential to further validate if this finding can be exploited therapeutically. However, we here highlight that the heterologous expression of voltage-gated K+ channel associates with metabolic alterations. Nonetheless, such an approach would require a high cancer cell specificity without severely affecting the function of other tissues or organs, as HKII is also found in insulin-sensitive tissues, muscle cells, or adipocytes (Mathupala et al., 2006; Roberts and Miyamoto, 2015; Wilson, 2003). Also, in these aspects, our study asks for further research to investigate whether global or local K+ fluctuations control the metabolic activity of single cells, organs, or even whole animals, as K+ might represent an essential regulatory component of mitochondria-associated HKII.
Our results strongly emphasize that [K+]i acts as an important and direct modulator of HKII activity in multiple cell lines, independent of their origin. However, additional research needs to be conducted to identify the K+-sensitive structures of the enzyme and to understand the exact molecular mechanism by which K+ might control the enzymatic activity. Our predictions of several positions within a model of HKII that may directly bind K+ might be helpful in this context. However, it remains unclear if the thermal inactivation of HKII in vitro, which is under the control of K+ (Nawaz et al., 2018), is based on the same or similar mechanisms as the loss of HKII activity in K+-depleted cells. In this context, it would be also important to understand if K+ might alter the inhibitory effect of glucose-6-phosphate, i.e. the product inhibition of HKII. Our experiments exclude that low K+ affects the mitochondrial localization of HKII, which is based on the N-terminus of the enzyme and facilitates ATP supply to the enzyme (Sun et al., 2008). However, by exploiting a truncated version of HKII, which is unable to localize to mitochondria, we show evidence that the localization of HKII to mitochondria is essential to control glycolysis and ATP generation by [K+]i. Thus, it is tempting to speculate that not only global, but also particularly local subcellular K+ signals on the surface of mitochondria might regulate cell metabolism in subdomains of a cell by controlling the activity of HKII. Such microdomains might, for example, be formed at the close vicinity to VDAC due to its cation gating function, consequently impacting on the activity of VDAC-associated HKII (Pastorino and Hoek, 2008). Following this study, targeted fluorescent biosensors (Bischof et al., 2017; Burgstaller et al., 2019; Imamura et al., 2009; Takanaga et al., 2008), informative protocols, and experimental settings including in vivo animal models might help to identify and unravel local K+-dependent metabolic settings in health and disease.
Limitations of the study
Our data were generated in well-established cell lines. Hence, whether and how cellular K+ fluctuations and [K+]i alterations modulate HKII activity in isolated primary cells or even living animals remains elusive and will be part of future studies. Moreover, our study does not address whether increases of [K+]ex, as found within the tumor microenvironment, promote tumor cell proliferation by stimulating HKII-dependent glycolysis and if the manipulation of [K+]i and/or [K+]ex represents a feasible therapeutic strategy to fight cancers by altering their metabolic activity.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Helmut Bischof (helmut.bischof@uni-tuebingen.de).
Materials availability
Plasmids generated in this study are available from the lead contact upon request.
Data and code availability
Original/source data for all figures in the paper are available from the lead contact upon request.
Methods
All methods can be found in the accompanying transparent methods supplemental file.
Acknowledgments
We thank C.B. Newgard, Department of Pharmacology and Cancer Biology, Duke University School of Medicine, USA, for INS-1 832/13 cells. This work was funded by the Alexander von Humboldt Foundation (H.B.), the Austrian Infrastructure Program 2016/2017 (T.M.), the Austrian Research Promotion Agency (FFG), project numbers 864690 and 870454 (T.M.), the Austrian Science Fund (FWF), project numbers I3716 (R.M.), P3326 (K.G.), P28701 (R.S.), P28854 and I3792 (T.M.), and W1226 (T.M., W.F.G.), BioTechMed Graz (Flagship project) (T.M.), the Brain & Behavior Research Foundation NARSAD Young Investigator Grant 20748 (L.M.), the German Research Foundation (DFG), project numbers RL 1490/10-1 (R.L.), MA 8113/2-1 (L.M.), and DFG PL 249 15-1 (N.P.), the Germany's Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy – ID 390857198) (N.P.), the ICEPHA Graduate Program “Membrane-associated Drug Targets in Personalized Cancer Medicine” (R.L.), the Integrative Metabolism Research Center Graz (T.M.), the Styrian Government (Zukunftsfond) (T.M.), and the Wilhelm Schuler Stiftung (L.M.). T.R. and T.S. are doctoral fellows (Molecular Medicine, Medical University of Graz). O.A.B. is a doctoral fellow of the doctoral program Metabolic and Cardiovascular Disease (MCD).
Author contributions
H.B., R.L., W.F.G., and R.M., were involved in conceptualization; H.B., S.B., A.S., L.M., and T.S. were involved in data curation; H.B., S.B., A.S., L.M., O.A.B., and T.M. assisted in formal analysis; H.B., K.G., R.S., T.M., N.P., R.L., W.F.G., and R.M. acquired financial support; H.B., S.B., A.S., L.M., and T.S. were involved in investigations; H.B., S.B., R.S., T.M., R.L., and R.M. designed the methodology; H.B., R.L., W.F.G., and R.M. performed project administration; L.M., K.G., R.S., T.M., N.P., R.L., W.F.G., and R.M. provided resources; R.L., W.F.G., and R.M. supervised the project, H.B. and S.B. visualized data, H.B., R.L., W.F.G., and R.M. wrote the original draft. All authors critically reviewed the manuscript and stated comments.
Declaration of interests
The authors declare no competing interests.
Published: April 23, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102346.
Contributor Information
Helmut Bischof, Email: helmut.bischof@uni-tuebingen.de.
Roland Malli, Email: roland.malli@medunigraz.at.
Supplemental information
References
- Abu-Hamad S., Zaid H., Israelson A., Nahon E., Shoshan-Barmatz V. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1. J. Biol. Chem. 2008;283:13482–13490. doi: 10.1074/jbc.M708216200. [DOI] [PubMed] [Google Scholar]
- Anderson M., Marayati R., Moffitt R., Yeh J.J. Hexokinase 2 promotes tumor growth and metastasis by regulating lactate production in pancreatic cancer. Oncotarget. 2017;8:56081–56094. doi: 10.18632/oncotarget.9760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermejo C., Haerizadeh F., Takanaga H., Chermak D., Frommer W.B. Dynamic analysis of cytosolic glucose and ATP levels in yeast using optical sensors. Biochem. J. 2010;432:399–406. doi: 10.1042/BJ20100946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof H., Rehberg M., Stryeck S., Artinger K., Eroglu E., Waldeck-Weiermair M., Gottschalk B., Rost R., Deak A.T., Niedrist T. Novel genetically encoded fluorescent probes enable real-time detection of potassium in vitro and in vivo. Nat. Commun. 2017;8:1422. doi: 10.1038/s41467-017-01615-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof H., Burgstaller S., Waldeck-Weiermair M., Rauter T., Schinagl M., Ramadani-Muja J., Graier W.F., Malli R. Live-cell imaging of physiologically relevant metal ions using genetically encoded FRET-based probes. Cells. 2019;8:492. doi: 10.3390/cells8050492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgstaller S., Bischof H., Gensch T., Stryeck S., Gottschalk B., Ramadani-Muja J., Eroglu E., Rost R., Balfanz S., Baumann A. pH-lemon, a fluorescent protein-based pH reporter for acidic compartments. ACS Sens. 2019;4:883–891. doi: 10.1021/acssensors.8b01599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depaoli M.R., Karsten F., Madreiter-Sokolowski C.T., Klec C., Gottschalk B., Bischof H., Eroglu E., Waldeck-Weiermair M., Simmen T., Graier W.F. Real-time imaging of mitochondrial ATP dynamics reveals the metabolic setting of single cells. Cell Rep. 2018;25:501–512.e3. doi: 10.1016/j.celrep.2018.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eil R., Vodnala S.K., Clever D., Klebanoff C.A., Sukumar M., Pan J.H., Palmer D.C., Gros A., Yamamoto T.N., Patel S.J. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature. 2016;537:539–543. doi: 10.1038/nature19364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong L., Cui Z., Chen P., Han H., Peng J., Leng X. Reduced survival of patients with hepatocellular carcinoma expressing hexokinase II. Med. Oncol. 2012;29:909–914. doi: 10.1007/s12032-011-9841-z. [DOI] [PubMed] [Google Scholar]
- Gottlob K. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15:1406–1418. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero A., Herranz N., Sun B., Wagner V., Gallage S., Guiho R., Wolter K., Pombo J., Irvine E.E., Innes A.J. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 2019;1:1074–1088. doi: 10.1038/s42255-019-0122-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie D.G., Ross F.A., Hawley S.A. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton A.L., Armentrout P.B. Experimental and theoretical studies of potassium cation interactions with the acidic amino acids and their amide derivatives. J. Phys. Chem. B. 2008;112:12056–12065. doi: 10.1021/jp802427n. [DOI] [PubMed] [Google Scholar]
- Huang X., Jan L.Y. Targeting potassium channels in cancer. J. Cell Biol. 2014;206:151–162. doi: 10.1083/jcb.201404136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura H., Huynh Nhat K.P., Togawa H., Saito K., Iino R., Kato-Yamada Y., Nagai T., Noji H. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. PNAS. 2009;106:15651–15656. doi: 10.1073/pnas.0904764106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansu K., Gentile S. Potassium channel activation inhibits proliferation of breast cancer cells by activating a senescence program. Cell Death Dis. 2013;4:e652. doi: 10.1038/cddis.2013.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathupala S.P., Ko Y.H., Pedersen P.L. Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006;25:4777–4786. doi: 10.1038/sj.onc.1209603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathupala S.P., Ko Y.H., Pedersen P.L. Hexokinase-2 bound to mitochondria: cancer’s stygian link to the “Warburg effect” and a pivotal target for effective therapy. Semin. Cancer Biol. 2009;19:17–24. doi: 10.1016/j.semcancer.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr C.J., Gross D., Sezgin E.C., Steudel F.A., Ruth P., Huber S.M., Lukowski R. KCa3.1 channels confer radioresistance to breast cancer cells. Cancers (Basel) 2019;11:1285. doi: 10.3390/cancers11091285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr C.J., Schroth W., Mürdter T.E., Gross D., Maier S., Stegen B., Dragoi A., Steudel F.A., Stehling S., Hoppe R. Subunits of BK channels promote breast cancer development and modulate responses to endocrine treatment in preclinical models. Br. J. Pharmacol. 2020 doi: 10.1111/bph.15147. [DOI] [PubMed] [Google Scholar]
- Najafov A., Luu H.S., Mookhtiar A.K., Mifflin L., Xia H., Amin P.P., Ordureau A., Wang H., Yuan J. RIPK1 promotes energy sensing by the mTORC1 pathway. Mol. Cell. 2020;81:370–385.e7. doi: 10.1016/j.molcel.2020.11.008. [DOI] [PubMed] [Google Scholar]
- Nawaz M.H., Ferreira J.C., Nedyalkova L., Zhu H., Carrasco-López C., Kirmizialtin S., Rabeh W.M. The catalytic inactivation of the N-half of human hexokinase 2 and structural and biochemical characterization of its mitochondrial conformation. Biosci. Rep. 2018;38 doi: 10.1042/BSR20171666. BSR20171666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa H., Nagano H., Konno M., Eguchi H., Koseki J., Kawamoto K., Nishida N., Colvin H., Tomokuni A., Tomimaru Y. The combination of the expression of hexokinase 2 and pyruvate kinase M2 is a prognostic marker in patients with pancreatic cancer. Mol. Clin. Oncol. 2015;3:563–571. doi: 10.3892/mco.2015.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer B.F. Regulation of potassium homeostasis. CJASN. 2015;10:1050–1060. doi: 10.2215/CJN.08580813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino J.G., Hoek J.B. Regulation of hexokinase binding to VDAC. J. Bioenerg. Biomembr. 2008;40:171–182. doi: 10.1007/s10863-008-9148-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra K.C., Wang Q., Bhaskar P.T., Miller L., Wang Z., Wheaton W., Chandel N., Laakso M., Muller W.J., Allen E.L. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell. 2013;24:213–228. doi: 10.1016/j.ccr.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng S.-Y., Lai P.-L., Pan H.-W., Hsiao L.-P., Hsu H.-C. Aberrant expression of the glycolytic enzymes aldolase B and type II hexokinase in hepatocellular carcinoma are predictive markers for advanced stage, early recurrence and poor prognosis. Oncol. Rep. 2008;19:1045–1053. [PubMed] [Google Scholar]
- Roberts D.J., Miyamoto S. Hexokinase II integrates energy metabolism and cellular protection: akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015;22:248–257. doi: 10.1038/cdd.2014.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosano C., Sabini E., Rizzi M., Deriu D., Murshudov G., Bianchi M., Serafini G., Magnani M., Bolognesi M. Binding of non-catalytic ATP to human hexokinase I highlights the structural components for enzyme–membrane association control. Structure. 1999;7:1427–1437. doi: 10.1016/s0969-2126(00)80032-5. [DOI] [PubMed] [Google Scholar]
- Rose I.A., Warms J.V.B. Stability of hexokinase II in Vitro and in ascites tumor cells. Arch. Biochem. Biophys. 1982;213:625–634. doi: 10.1016/0003-9861(82)90592-6. [DOI] [PubMed] [Google Scholar]
- San Martín A., Ceballo S., Ruminot I., Lerchundi R., Frommer W.B., Barros L.F. A genetically encoded FRET lactate sensor and its use to detect the Warburg effect in single cancer cells. PLoS One. 2013;8:e57712. doi: 10.1371/journal.pone.0057712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson S.M., Xiao Z., Wisdom A.J., Bose S., Liberti M.V., Reid M.A., Hocke E., Gregory S.G., Kirsch D.G., Locasale J.W. 2020. The Na+/K+ ATPase Regulates Glycolysis and Modifies Immune Metabolism in Tumors (Cancer Biology) [Google Scholar]
- Schmitz A., Sankaranarayanan A., Azam P., Schmidt-Lassen K., Homerick D., Hänsel W., Wulff H. Design of PAP-1, a selective small molecule Kv1.3 blocker, for the suppression of effector memory T cells in autoimmune diseases. Mol. Pharmacol. 2005;68:1254–1270. doi: 10.1124/mol.105.015669. [DOI] [PubMed] [Google Scholar]
- Steudel F.A., Mohr C.J., Stegen B., Nguyen H.Y., Barnert A., Steinle M., Beer-Hammer S., Koch P., Lo W.-Y., Schroth W. SK4 channels modulate Ca2+ signalling and cell cycle progression in murine breast cancer. Mol. Oncol. 2017;11:1172–1188. doi: 10.1002/1878-0261.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Shukair S., Naik T.J., Moazed F., Ardehali H. Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. MCB. 2008;28:1007–1017. doi: 10.1128/MCB.00224-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szule J.A., Rand R.P. The effects of gramicidin on the structure of phospholipid assemblies. Biophys. J. 2003;85:1702–1712. doi: 10.1016/S0006-3495(03)74600-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takanaga H., Chaudhuri B., Frommer W.B. GLUT1 and GLUT9 as major contributors to glucose influx in HepG2 cells identified by a high sensitivity intramolecular FRET glucose sensor. Biochim. Biophys. Acta. 2008;1778:1091–1099. doi: 10.1016/j.bbamem.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triana-Martínez F., Picallos-Rabina P., Da Silva-Álvarez S., Pietrocola F., Llanos S., Rodilla V., Soprano E., Pedrosa P., Ferreirós A., Barradas M. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 2019;10:4731. doi: 10.1038/s41467-019-12888-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou P., Zheng B., Hsu C.-H., Sasaki A.T., Cantley L.C. A fluorescent reporter of AMPK activity and cellular energy stress. Cell Metab. 2011;13:476–486. doi: 10.1016/j.cmet.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyay S.K., Eckel-Mahan K.L., Mirbolooki M.R., Tjong I., Griffey S.M., Schmunk G., Koehne A., Halbout B., Iadonato S., Pedersen B. Selective Kv1.3 channel blocker as therapeutic for obesity and insulin resistance. Proc. Natl. Acad. Sci. 2013;110:E2239–E2248. doi: 10.1073/pnas.1221206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urrego D., Tomczak A.P., Zahed F., Stuhmer W., Pardo L.A. Potassium channels in cell cycle and cell proliferation. Philos. Trans. R. Soc. B Biol. Sci. 2014;369:20130094. doi: 10.1098/rstb.2013.0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson J.E. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J. Exp. Biol. 2003;206:2049–2057. doi: 10.1242/jeb.00241. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original/source data for all figures in the paper are available from the lead contact upon request.